Head and spinal injuries
HEAD INJURY
The severity of head injury is often assessed using the Glasgow coma scale (Table 11.1). This yields scores of 3 (the worst score) to 15 (the best score) based on an assessment of ocular, verbal, and motor responses.
Table 11.1
| Best eye response (maximum = 4) | |||
| 1. | No eye opening | 3. | Eyes opening to verbal command |
| 2. | Eye opening to pain | 4. | Eyes open spontaneously |
| Best verbal response (maximum = 5) | |||
| 1. | No verbal response | 4. | Confused |
| 2. | Incomprehensible sounds | 5. | Oriented |
| 3. | Inappropriate words | ||
| Best motor response (maximum = 6) | |||
| 1. | No motor response | 4. | Withdrawal from pain |
| 2. | Extension to pain | 5. | Localizing to pain |
| 3. | Flexion to pain | 6. | Obeys commands |
| Coma score | Clinical correlate |
| 13 or above | Mild brain injury |
| 9–12 | Moderate brain injury |
| 8 or less | Severe brain injury |
From: Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974; 2:81–84.
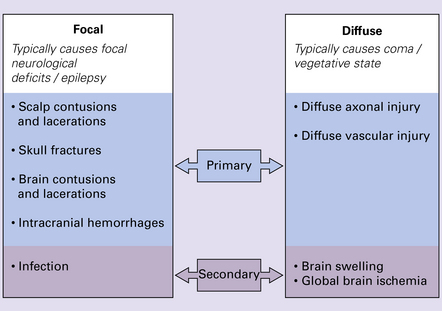
NATURE OF LESIONS IN HEAD INJURY
Type of head injury




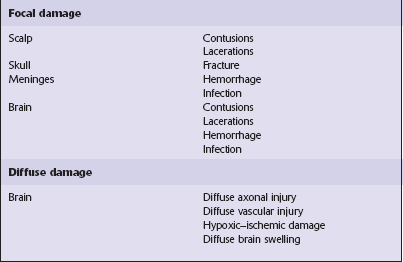
Time course
Brain damage following trauma can be viewed as occurring in two phases (Fig. 11.1):
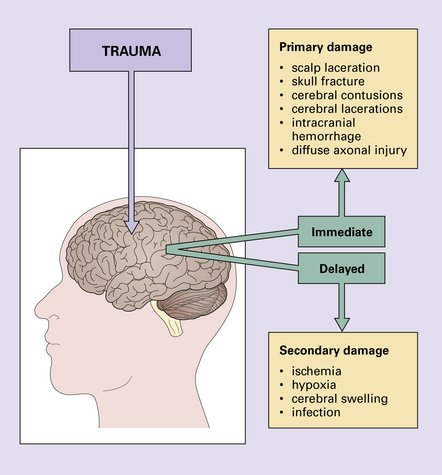


Progressive neurologic deterioration over many years has been noted in 15% of patients who have suffered severe head trauma (Fig. 11.2).
 HEAD INJURY
HEAD INJURY



NON-MISSILE HEAD INJURY
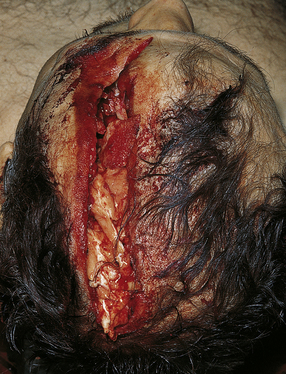
Laceration or bruising of the scalp is a common form of focal injury and a good indicator of the site of the trauma (Fig. 11.3).
FOCAL LESIONS OF THE SKULL
 Contact with a flat surface tends to produce closed fissure fractures, which often extend into the base of the skull.
Contact with a flat surface tends to produce closed fissure fractures, which often extend into the base of the skull.


 MECHANISMS OF PRIMARY DAMAGE IN NON-MISSILE HEAD INJURY
MECHANISMS OF PRIMARY DAMAGE IN NON-MISSILE HEAD INJURY
Acceleration or deceleration damage
Applying force to the head causes the skull to accelerate in a linear or rotational fashion. Due to inertia, the movement of the brain lags behind that of the skull and damage may result from the following mechanisms:
 Contact of the inner surface of the moving skull with the stationary brain may produce contusions, particularly in places where the skull protrudes internally. When the moving head is abruptly halted, the brain continues briefly to move due to its inertia. Impact of the moving brain against the stationary skull may again produce contusions.
Contact of the inner surface of the moving skull with the stationary brain may produce contusions, particularly in places where the skull protrudes internally. When the moving head is abruptly halted, the brain continues briefly to move due to its inertia. Impact of the moving brain against the stationary skull may again produce contusions.
 Traction on the bridging veins may cause them to tear, resulting in subdural hemorrhage.
Traction on the bridging veins may cause them to tear, resulting in subdural hemorrhage.

The risk of intracranial hematoma is significantly increased in patients with skull fractures.
Fractures of the base of the skull predispose to:
 Infection, as they often pass through the petrous temporal bone or the anterior cranial fossa and cause leakage of cerebrospinal fluid (CSF) through the ear, nose, or nasopharynx. These breaks in the integrity of the skull are also routes for the spread of infection.
Infection, as they often pass through the petrous temporal bone or the anterior cranial fossa and cause leakage of cerebrospinal fluid (CSF) through the ear, nose, or nasopharynx. These breaks in the integrity of the skull are also routes for the spread of infection.

Depressed fractures may tear the dura and associated blood vessels, leading to intracranial hemorrhage (see p. 274).
MACROSCOPIC APPEARANCES
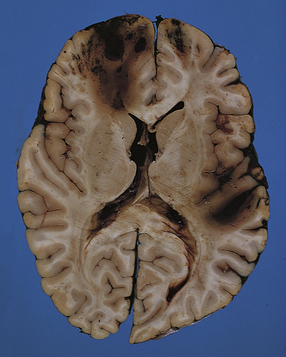
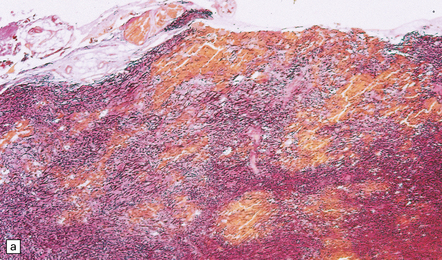
Acutely, contusions appear as superficial hemorrhagic areas associated with some hemorrhage into the overlying leptomeninges and variable brain swelling (Figs 11.4–11.6). Over subsequent days and weeks their color changes to brown or orange, and involved gyri become indented or superficially cavitated as necrotic tissue is resorbed. In brain slices, old contusions often have a triangular shape, with the point in the depths of the cortex or underlying white matter and a wide base at the surface of the crest of the gyrus (Fig. 11.7, see also Fig. 11.31). Contusions can be distinguished from foci of old ischemic damage, which are almost invariably more severe within the depths of sulci.
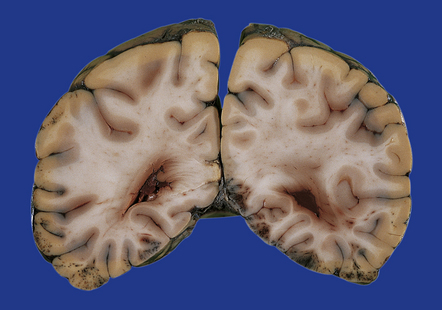
11.4 Superficial contusions on the underside of the occipital lobes.
The contusions in the left occipital lobe are mostly superficial whereas those on the right are more hemorrhagic and involve the full thickness of the cortex.
11.5 Severe cerebral contusions.
Severe frontal and temporal lobe contusions associated with extensive hemorrhage into the overlying subarachnoid space.
11.6 Superficial contusions of orbital frontal cortex.
The contusions are more extensive in the right frontal lobe.
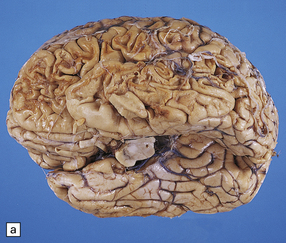
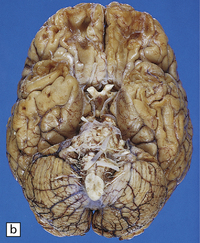
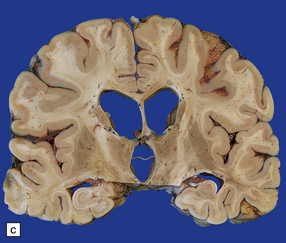
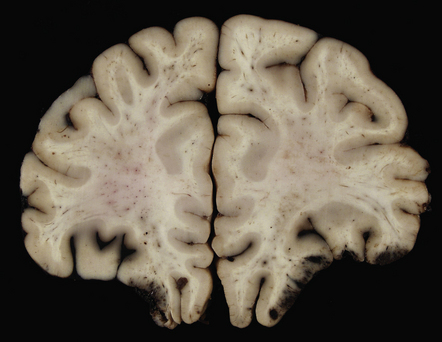
11.7 Old cerebral contusions.
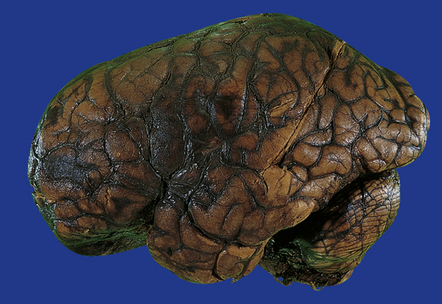
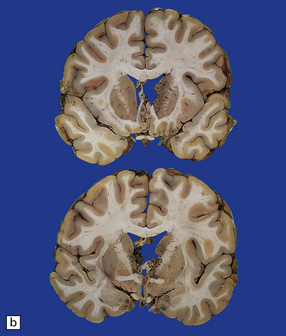
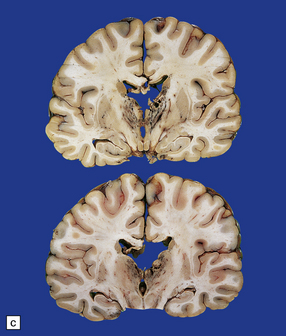
(a) Old contusions over the lateral aspect of frontal, temporal and parietal lobes. These appear as brown or orange indentations in the crests of gyri. (b) Old contusions on inferior aspect of frontal lobes (c) Old wedge-shaped contusion of the left inferior temporal gyrus (arrow). There is also evidence of old diffuse axonal injury: the ventricles are dilated, the corpus callosum is thinned and yellow, and the temporal and right parasagittal frontal white matter show gray discoloration.
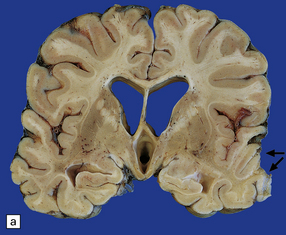
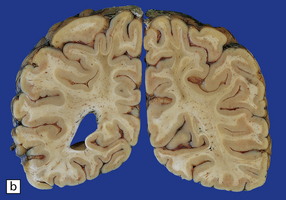
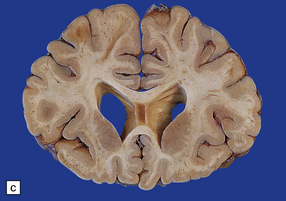
11.31 Long-term effects of DAI.
(a) Old trauma with contusions of the right middle and superior temporal gyri (arrows) and ventricular dilatation. Note the ill-defined gray discoloration of the white matter due to DAI. This is most marked in the temporal lobes (including the non-contused left temporal lobe), but also involves the corpus callosum and internal capsule. (b) Gray discoloration of the parasagittal white matter in the posterior part of the parietal lobes. (c) The gray-brown streaks in the parasagittal white matter in this brain are old gliding contusions.
Contusions may occur in the region of impact (a form of contact damage), particularly if this causes fracturing, but also occur elsewhere over the brain in a stereotyped distribution that tends to be much the same whatever the site of the original injury. The contusions involve the crests of gyri that come into contact with protuberances within the skull (Figs 11.8, 11.9). Contusions tend therefore, to be located in the following regions:
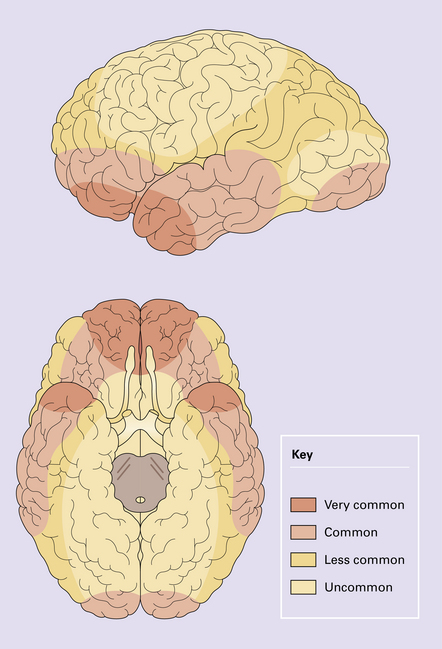
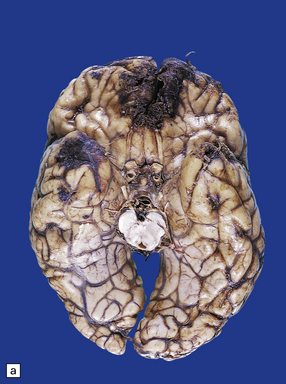
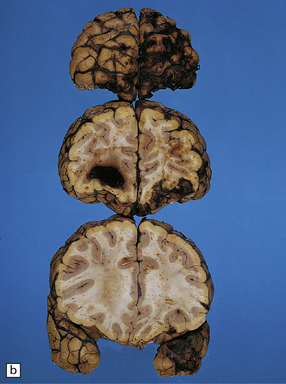
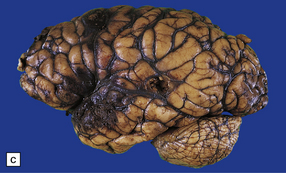
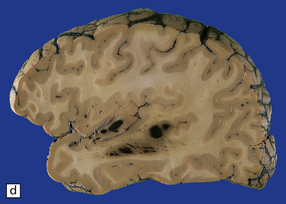
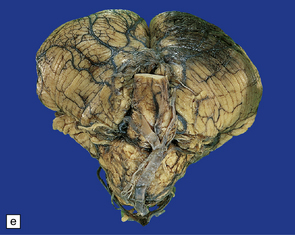
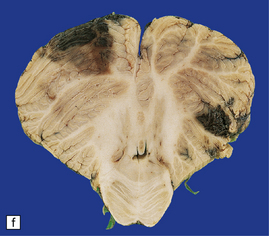
11.9 Common sites for contusions.
(a) Contusions of the orbital surface of the frontal lobes and tips of the temporal lobes. (b) Cut surface of brain showing contusions of the orbital surfaces of frontal lobes and tip of left temporal lobe. There is a hematoma in the right frontal lobe. (c) and (d) Contusions on either side of the sylvian fissure. (e) Contusions of the right cerebellar hemisphere. (f) A horizontal slice through the cerebellum shows that the contusions extend into the cerebellar white matter.





Contusions may also involve the cerebellar hemispheres (Fig. 11.9e,f).
Lacerations develop when the severity of trauma has been sufficient to cause tearing of the pia. This may occur in association with a depressed skull fracture. The most severe pattern of cerebral laceration is embraced by the term ‘burst lobe’. This is often associated with a skull fracture and most commonly involves the frontal and temporal lobes, which show confluent intracerebral and subarachnoid bleeding (sometimes even extending into the subdural or extradural space), associated with massive disruption of the affected lobe (Fig. 11.10).
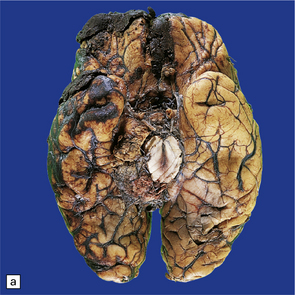
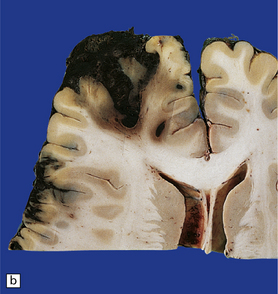
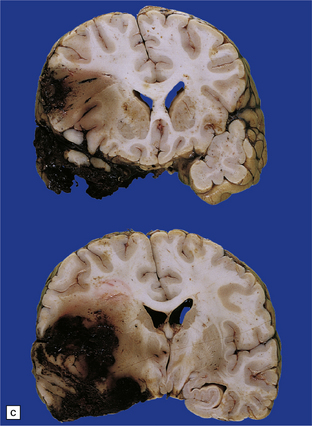
11.10 Severe contusion.
(a) Severe contusion of the right frontal and temporal poles with blood clot over the frontal pole suggesting cerebral laceration. (b) Sectioning confirms laceration of the frontal lobe associated with severe contusional damage and extension of bleeding into the subarachnoid and subdural spaces. (c) Another common site of ‘burst lobe’ is the temporal region (as shown here).
MICROSCOPIC APPEARANCES
The appearance of contusions evolves with time through several stages:
 Initially the contusion is visible as microscopic regions of perivascular hemorrhage (Fig. 11.11a) following the track of small vessels in the cortex and usually running perpendicular to the cortical surface. The hemorrhage occurs within seconds or minutes of the injury.
Initially the contusion is visible as microscopic regions of perivascular hemorrhage (Fig. 11.11a) following the track of small vessels in the cortex and usually running perpendicular to the cortical surface. The hemorrhage occurs within seconds or minutes of the injury.
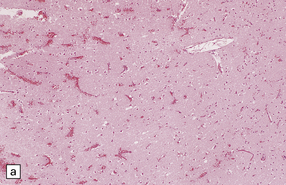
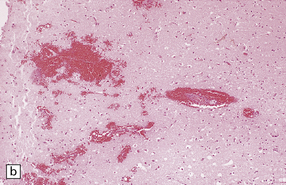
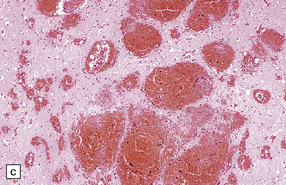
11.11 Microscopic appearance of early contusions.
(a) Early contusions with small perivascular hemorrhages. (b) Early contusions showing extravasation of blood into the cortex associated with acute degeneration of the surrounding neurons. (c) Later evolution leads to more extensive perivascular hemorrhages in the cortex and superficial white matter.
 Over several hours blood continues to seep into the adjacent cortex, which shows local swelling and confluent hemorrhage. If the contusion is severe there is extension into the underlying white matter. During this phase, neurons in the immediate vicinity begin to degenerate (Fig. 11.11b,c). Blood vessels within the contused tissue may show margination by neutrophils (Fig. 11.12), which later infiltrate the adjacent parenchyma.
Over several hours blood continues to seep into the adjacent cortex, which shows local swelling and confluent hemorrhage. If the contusion is severe there is extension into the underlying white matter. During this phase, neurons in the immediate vicinity begin to degenerate (Fig. 11.11b,c). Blood vessels within the contused tissue may show margination by neutrophils (Fig. 11.12), which later infiltrate the adjacent parenchyma.
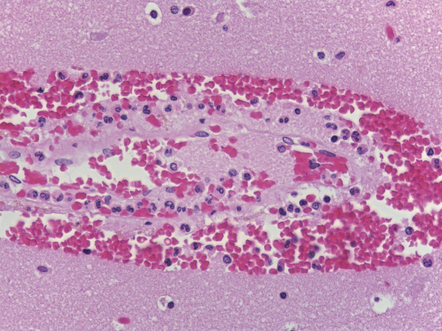

 What remains eventually is a superficial, roughly wedge-shaped, region of neuronal loss and glial scarring (Fig. 11.13). Hemosiderin pigment is found in scattered residual macrophages and astrocytes. The remaining neurons may become mineralized. The subjacent white matter is rarefied and gliotic.
What remains eventually is a superficial, roughly wedge-shaped, region of neuronal loss and glial scarring (Fig. 11.13). Hemosiderin pigment is found in scattered residual macrophages and astrocytes. The remaining neurons may become mineralized. The subjacent white matter is rarefied and gliotic.
INTRACRANIAL HEMORRHAGES
Extradural hematoma
Extradural hematomas are relatively localized and may accumulate slowly over a period of hours because of the adherence between the dura and the inner aspect of the calvarium (Fig. 11.14). They are evident macroscopically as a biconvex accumulation of clotted blood between the skull and the dura (Fig. 11.15).
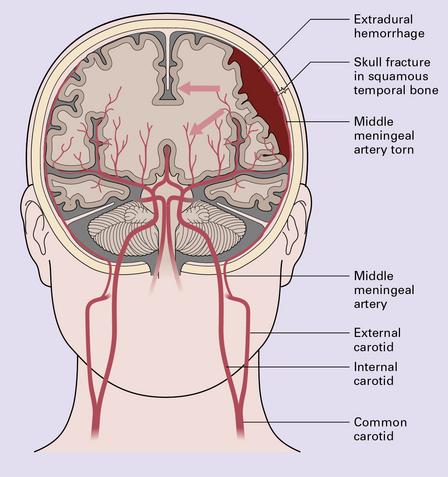
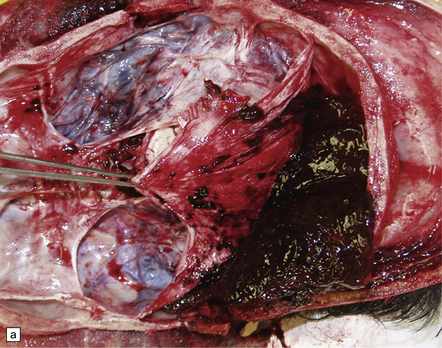
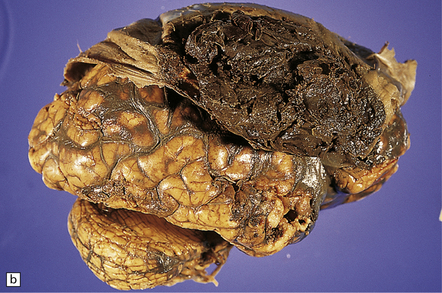
11.15 Extradural hematoma.
(a) Fracture of the right squamous temporal bone with an associated extradural hematoma. Extradural blood is visible through the dura in the left middle and anterior cranial fossae. (b) Extradural hematoma in right frontoparietal region. ((b) Courtesy of Professor Michael A. Farrell, Dublin, Ireland.)
 LUCID INTERVAL
LUCID INTERVALSubdural hematoma
Subdural hematoma usually results from tearing of bridging veins that cross the subdural space, especially those related to the superior sagittal sinus. Blood from the ruptured vessels spreads freely through the subdural space and can envelop the entire hemisphere (Fig. 11.16).
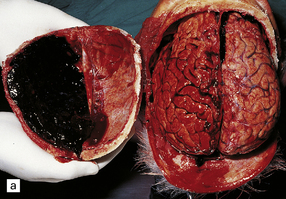
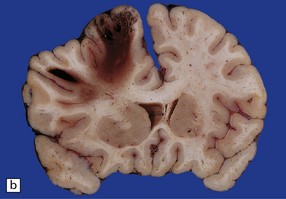
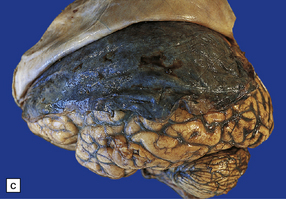
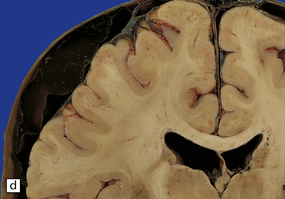
11.16 Subdural hematoma.
(a) Removal of the calvarium at necropsy reveals a left subdural hematoma that has compressed the underlying brain. The arachnoid surface of the left cerebral hemisphere is blood-stained and the gyri are flattened. (b) Indentation of the left cerebral hemisphere due to a subdural hematoma. Note the contusion, hemorrhage, and focal infarction in the underlying brain tissue. (c) Chronic subdural hematoma enclosed in a thin ‘membrane’ of granulation tissue. Although the hematoma was known to be more than 2 weeks old, it consisted of clotted blood. (d) Subdural hematoma showing compression of brain.
Chronic subdural hematomas become surrounded by a ‘membrane’ of organizing granulation tissue (Fig. 11.17). This is usually evident on the dural aspect of the hematoma within about 1 week, and later on its deep surface. Progressive enlargement of the hematoma may occur, mainly due to recurrent bleeding from friable blood vessels in the granulation tissue, although an osmotic effect of blood breakdown products may also contribute. Attempted dating of subdural hematomas by histologic examination of the ‘membranes’ has proven unreliable.
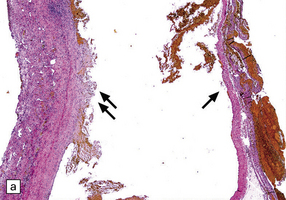
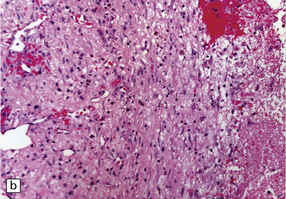
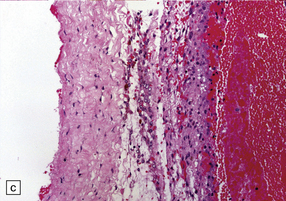
11.17 Chronic subdural hematoma.
(a) This hematoxylin-van Gieson-stained section through a chronic subdural hematoma illustrates the differing thickness of the outer (twin arrows) and inner walls (single arrow). The collagen within the hematoma walls is stained red. Very little blood (stained orange) remains within the hematoma cavity, which macroscopic examination had shown to contain a mixture of loose blood clot and turbid fluid. (b) A higher-magnification view of the outer wall (here stained with H&E) shows it to consist of collagenous connective tissue within which are numerous thin-walled blood vessels, some with adjacent microscopic foci of hemorrhage. The tissue also includes scattered lymphocytes and hemosiderin-laden macrophages. (c) The outer wall is much thinner and less vascular. In this case there has also been more recent hemorrhage into the underlying subarachnoid space (to the right of the image). (d) The accumulation of hemosiderin pigment in the outer membrane is well demonstrated in a section stained by Perls’ method.
Traumatic subarachnoid hemorrhage
There are several possible sources of subarachnoid bleeding:
 Subarachnoid blood may derive from severe contusions and lacerations (Fig. 11.18, see also Figs 11.5, 11.10); the hemorrhage is usually localized to the region of cortical damage.
Subarachnoid blood may derive from severe contusions and lacerations (Fig. 11.18, see also Figs 11.5, 11.10); the hemorrhage is usually localized to the region of cortical damage.
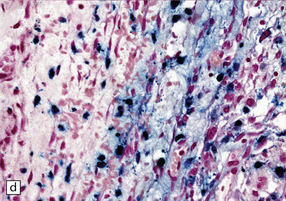
11.18 Traumatic subarachnoid hemorrhage.
Blood is present over the cerebral convexity and in the sylvian fissure.
 Fractures of the skull base can tear large vessels at the base of the brain.
Fractures of the skull base can tear large vessels at the base of the brain.
 Even in the absence of basal fractures, cranial trauma occasionally causes rupture or dissection of the vertebral arteries, resulting in arterial occlusion or subarachnoid hemorrhage; the tear usually originates near where the arteries pass through the dura into the cranial cavity (Fig. 11.19). More rarely, subarachnoid hemorrhage results from a tear in the intraosseous part of the vertebral arteries within the cervical spine (Fig. 11.20).
Even in the absence of basal fractures, cranial trauma occasionally causes rupture or dissection of the vertebral arteries, resulting in arterial occlusion or subarachnoid hemorrhage; the tear usually originates near where the arteries pass through the dura into the cranial cavity (Fig. 11.19). More rarely, subarachnoid hemorrhage results from a tear in the intraosseous part of the vertebral arteries within the cervical spine (Fig. 11.20).
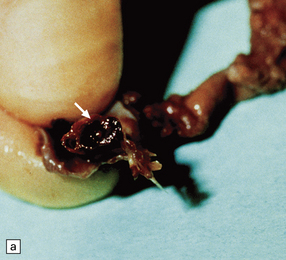
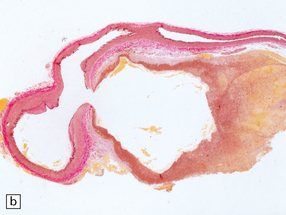
11.19 Traumatic rupture of the vertebral artery in the region of entry into the cranial cavity.
(a) A pseudoaneurysm (arrow) is present at the site of vertebral artery rupture after head injury. The patient presented with delayed subarachnoid hemorrhage several days later. (b) The pseudoaneurysm is clearly visible in an elastic/van Gieson-stained section. (Courtesy of Dr Hugh White, North Bristol NHS Trust, UK.)
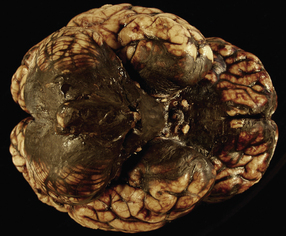

Cerebral and cerebellar hematomas
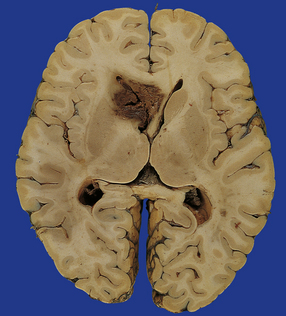
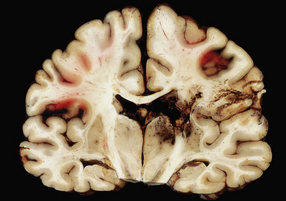
Superficial lobar hematomas are generally related to overlying contusional damage and are mainly seen in the frontal and temporal lobes (Fig. 11.21). They also occur in association with contusions in the cerebellum (Fig. 11.22). Deep hematomas tend to be related to the basal ganglia and thalamus (Fig. 11.23, see also Fig. 11.30a) and are often associated with diffuse axonal injury.
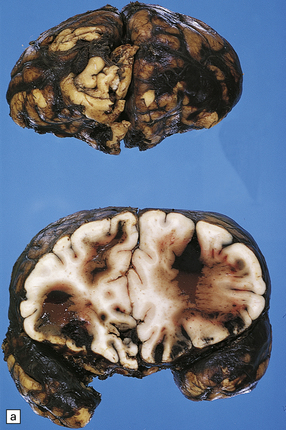
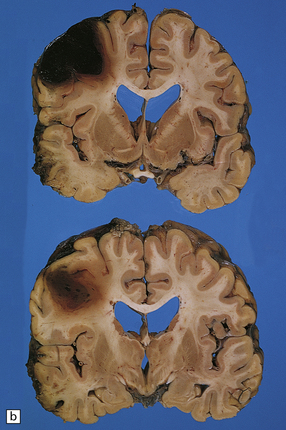
11.21 Cerebral hematomas.
(a) Hematomas in the white matter of the frontal lobe associated with severe contusions. (b) Superficial lobar hematoma in the posterior frontal region. The hematoma was related to a cortical laceration associated with a depressed skull fracture.
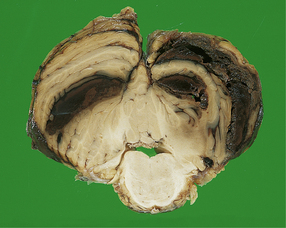
11.22 Cerebellar hematoma.
Hematomas in the cerebellum are usually associated with severe contusional damage to the cerebellar hemispheres, which is often related to a skull fracture in the posterior fossa.
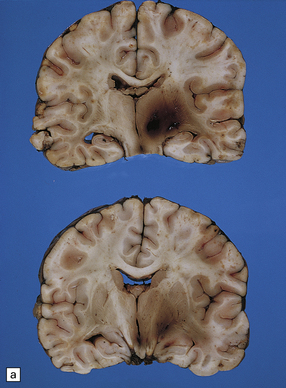
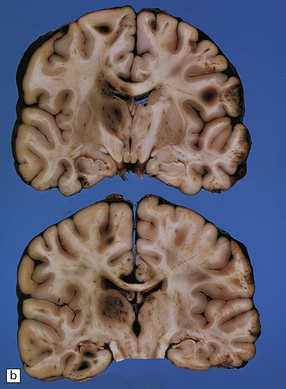
11.23 Hematomas of the basal ganglia and thalamus.
(a) Traumatic hematoma in the right basal ganglia associated with mass effect and compression of the ventricular system. The appearance resembles that of a primary intracerebral bleed. Histologic examination revealed diffuse axonal injury in a wide distribution. (b) A cluster of hematomas in the left thalamus caused by head injury. There is other damage in the form of cortical contusions, and a small hematoma in the hippocampus. A hemorrhagic lesion in the corpus callosum is indicative of diffuse axonal injury.
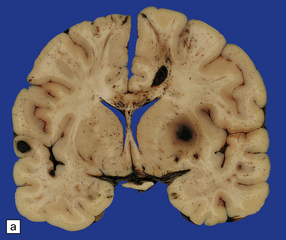
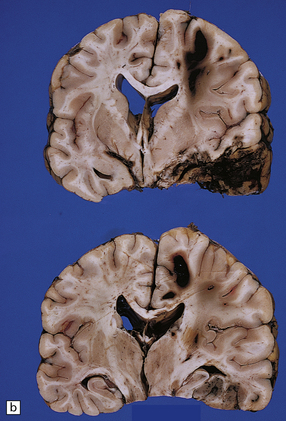
11.30 Gliding contusions.
(a) Bilateral gliding contusions in the parasagittal white matter. The one on the left is no more than a perivascular streak of hemorrhagic white matter discoloration. The one on the right includes several hemorrhagic foci and a larger hematoma. There are also typical DAI-related lesions in the corpus callosum, contusions of both unci, and several other hematomas, including one in the right putamen. (b) Large lesions extending through the parasagittal white matter towards the corpus callosum. In this case there is also severe contusion of the temporal lobe and petechial hemorrhages are present in the corpus callosum in the lower slice.
 DISTINGUISHING PRIMARY AND TRAUMATIC INTRACEREBRAL HEMATOMAS
DISTINGUISHING PRIMARY AND TRAUMATIC INTRACEREBRAL HEMATOMAS


UNCOMMON TYPES OF FOCAL BRAIN DAMAGE
Uncommon types of focal brain damage include:
 Ischemic brain damage due to traumatic arterial dissection and thrombosis resulting from stretching of the vertebral or carotid arteries by hyperextension of the neck (Fig. 11.24).
Ischemic brain damage due to traumatic arterial dissection and thrombosis resulting from stretching of the vertebral or carotid arteries by hyperextension of the neck (Fig. 11.24).
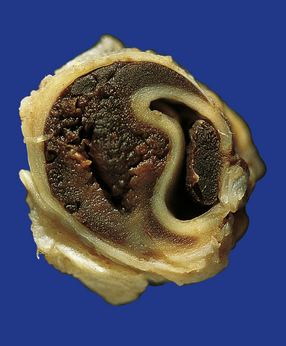
11.24 Traumatic arterial dissection.
Traumatic dissection and associated thrombosis of the vertebral artery in a patient who developed brain stem infarction after head injury.

 A pontomedullary rent resulting from severe injury associated with hyperextension of the neck (Fig. 11.25).
A pontomedullary rent resulting from severe injury associated with hyperextension of the neck (Fig. 11.25).
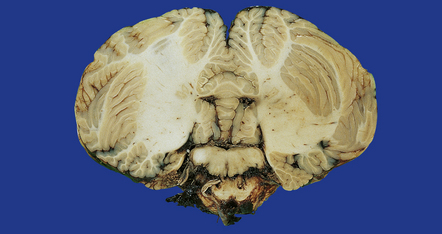
11.25 Pontomedullary rent.
Tearing of the brain stem at the junction between the upper medulla and lower pons has produced a hemorrhagic lesion termed a pontomedullary rent. In this case the injury resulted from a high-speed motor-cycle collision in which the patient suffered a hyperextension injury to the neck. At necropsy there was a large quantity of subarachnoid blood in the posterior fossa, but no clear site of origin of the hemorrhage. Once the brain had been fixed and sliced it became clear that the site of bleeding had been this injury to the brain stem.

INFECTION
Infection is predominantly a complication of skull fractures:
 Fractures of the calvarium or skull base can provide a route for bacteria to pass from the major air sinuses into the subarachnoid space and cause meningitis. Such fractures are often associated with a leakage of CSF or the formation of an aerocele.
Fractures of the calvarium or skull base can provide a route for bacteria to pass from the major air sinuses into the subarachnoid space and cause meningitis. Such fractures are often associated with a leakage of CSF or the formation of an aerocele.

The incidence of brain abscesses is, however, increased even after closed head injuries, presumably because the devitalized tissues are prone to colonization in the event of a transient bacteremia (see Chapter 15).
 NON-ACCIDENTAL INJURIES IN CHILDREN
NON-ACCIDENTAL INJURIES IN CHILDREN
 Non-accidental injuries in children are associated with distinct patterns of head injury, reflecting the types of trauma (often involving shaking, with or without impact), the flexibility and thinness of the scalp and skull, the immaturity of the brain, the relative size and weight of the head (12.5% of the weight of a young infant compared with 2.5% of that of an adult), and relative weakness of the neck muscles in infants.
Non-accidental injuries in children are associated with distinct patterns of head injury, reflecting the types of trauma (often involving shaking, with or without impact), the flexibility and thinness of the scalp and skull, the immaturity of the brain, the relative size and weight of the head (12.5% of the weight of a young infant compared with 2.5% of that of an adult), and relative weakness of the neck muscles in infants.
 There may be impact damage, with focal lesions of the scalp and skull.
There may be impact damage, with focal lesions of the scalp and skull.
 Even in the absence of impact damage, vigorous shaking of the child can cause:
Even in the absence of impact damage, vigorous shaking of the child can cause:
• subdural hematomas, which are often bilateral and interhemispheric and may only be thin films
• scanty subarachnoid hemorrhage
• retinal and optic nerve sheath hemorrhages
• traumatic axonal injury, particularly in the lower medulla or upper cervical cord (Fig. 11.26); there may also be hemorrhage into paravertebral strap muscles and adjacent soft tissue, vertebral injury including avulsion of spinous processes, and epidural hemorrhage around the cervical cord
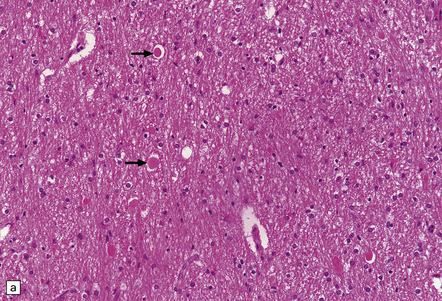
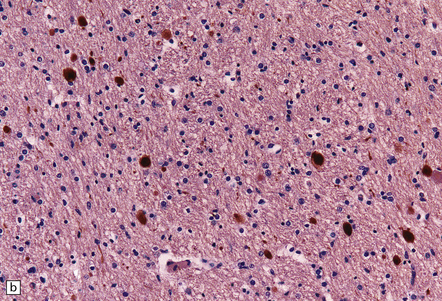
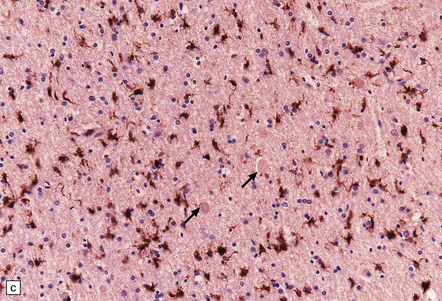
11.26 Non-accidental injury in a 6-week-old child who died 6 days after being shaken.
(a) Scattered axonal swellings (arrows) are visible in the medullary pyramids near the cervicomedullary junction. (b) The axonal swellings are more readily visualized by immunohistochemistry for β-amyloid precursor protein (APP). (c) By 6 days, there is already a prominent microglial reaction in the region of traumatic axonal injury, as revealed by immunostaining for CD68. Note the unlabeled axonal swellings (arrows).
• diffuse brain swelling with secondary ischemic injury is a frequent finding; the development of brain swelling may be delayed by several hours
In early childhood, shaking or impact damage can produce a distinctive lesion called a contusional tear, which is characterized by slit-like separation of the cortex from the underlying white matter (Fig. 11.27). This may become hemorrhagic or be associated with localized swelling. Non-hemorrhagic lesions are very hard to see macroscopically, but can be found on histologic examination, which reveals the axonal tearing and local glial reaction.
11.27 Contusional tear in the frontal lobe of an infant aged 2 months who had a sustained non-accidental shaking injury.
This distinctive pattern of damage is due to differential movement of the cerebral cortex and white matter and may be related to the poor myelination of the white matter at this age. Non-hemorrhagic contusional tears are difficult to see and may be misinterpreted as artefacts produced during removal of the brain. Histologic examination is needed to resolve this: a vital reaction is seen even in recent contusional tears and is characterized by swelling of astrocytes and, later, infiltration by macrophages.
DIFFUSE DAMAGE
TAI is the term given to axonal damage that is directly attributable to trauma, usually involving acceleration and deceleration of the head. There is a spectrum of severity from the mildest, subclinical TAI, involving only a few, scattered axons, to the widespread axonal damage that is seen in diffuse axonal injury (DAI) (Table 11.3). Patients who have sustained DAI are typically unconscious from the moment of impact, do not experience a lucid interval, and remain unconscious, vegetative, or at least severely disabled until death. Lesser degrees of TAI are compatible with recovery of consciousness, with or without persisting neurologic deficits of varying severity.
Table 11.3
Traumatic axonal injury – terminology
| Axonal injury | A non-specific term referring to damage to axons of any etiology |
| Traumatic axonal injury (TAI) | Damage to axons caused by trauma. This may vary from small foci of axonal injury to more widespread brain damage |
| Diffuse TAI | Originally termed ‘DAI’, this is the most severe form of traumatic axonal damage |
| Diffuse axonal injury (DAI) | First described as a clinicopathologic syndrome in which there is widespread traumatic axonal damage throughout the brain, including the brain stem. However, because axonal injury may be caused by other pathological processes, the etiology of the axonal damage should always be indicated when the term DAI is used as a neuropathologic diagnosis |
From: Geddes JF, Whitwell HL, Graham DI. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol 2000; 26:105–116.
MACROSCOPIC APPEARANCES
 Focal lesions in the corpus callosum, which appear as clusters of petechial hemorrhages or soft hemorrhagic foci (Fig. 11.28). The lesions may disrupt the interventricular septum, in which case there is often associated intraventricular hemorrhage.
Focal lesions in the corpus callosum, which appear as clusters of petechial hemorrhages or soft hemorrhagic foci (Fig. 11.28). The lesions may disrupt the interventricular septum, in which case there is often associated intraventricular hemorrhage.
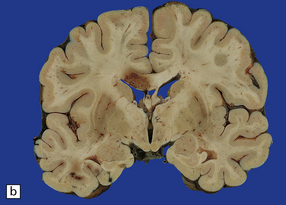
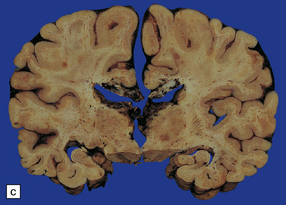
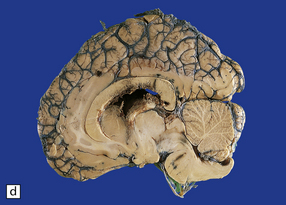
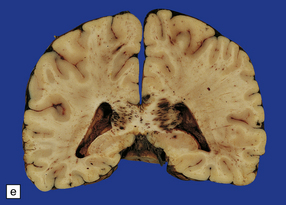
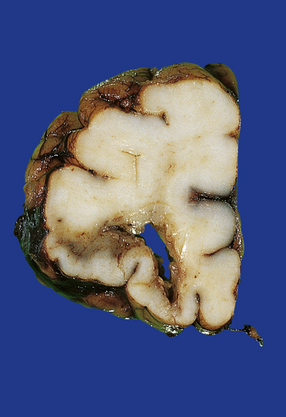
11.28 Lesions in the corpus callosum in DAI.
These may vary from small petechiae to larger foci of hemorrhagic discoloration and softening and extensive hemorrhagic disruption. (a) Small petechiae. (b) Larger focus of hemorrhagic discoloration and softening. (c) Extensive hemorrhagic disruption. (d) In the sagittal plane lesions typically involve the corpus callosum for several centimeters along its length. (e) The lesions may occur anywhere along the corpus callosum, but there is a tendency for them to be concentrated in the splenium.
 Focal lesions in the dorsolateral quadrants of the rostral brain stem. Small lesions appear as discrete foci of petechial hemorrhage in and adjacent to the superior cerebellar peduncles. More severe damage results in hemorrhagic softening of the dorsal part of the midbrain and rostral pons (Fig. 11.29).
Focal lesions in the dorsolateral quadrants of the rostral brain stem. Small lesions appear as discrete foci of petechial hemorrhage in and adjacent to the superior cerebellar peduncles. More severe damage results in hemorrhagic softening of the dorsal part of the midbrain and rostral pons (Fig. 11.29).


11.29 Brain stem lesions associated with DAI.
These consist of clusters of petechial hemorrhages or foci of hemorrhagic softening in the dorsolateral part of the midbrain and dorsolateral part of the rostral pons. (a) The right dorsolateral part of the midbrain contains petechiae. (b) The dorsolateral part of the rostral pons includes petechiae bilaterally.
 ‘Gliding contusions,’ which are hemorrhagic lesions affecting the parasagittal white matter in the superior part of the cerebral hemispheres. Small lesions occur close to the junction between the white matter and the overlying cortex, but larger lesions curve downwards through the parasagittal white matter towards the corpus callosum (Fig. 11.30). Gliding contusions are often bilateral, but are not usually symmetric.
‘Gliding contusions,’ which are hemorrhagic lesions affecting the parasagittal white matter in the superior part of the cerebral hemispheres. Small lesions occur close to the junction between the white matter and the overlying cortex, but larger lesions curve downwards through the parasagittal white matter towards the corpus callosum (Fig. 11.30). Gliding contusions are often bilateral, but are not usually symmetric.
The brains of long-term survivors of DAI typically show moderate to marked cerebral atrophy with dilatation of the lateral and third ventricles, thinning of the corpus callosum (sometimes marked), ill-defined gray discoloration of the cerebral white matter, and, in some cases, atrophy of the cerebral peduncles, base of the pons, and medullary pyramids (Fig. 11.31). In the absence of other injuries such as cerebral contusions the cortical ribbon usually appears normal.
MICROSCOPIC APPEARANCES
Histology is needed to substantiate the diffuse damage to axons. The main regions affected are:
 superior parasagittal cerebral white matter
superior parasagittal cerebral white matter

 subcortical fiber tracts (fornix, internal and external capsules)
subcortical fiber tracts (fornix, internal and external capsules)


The time course of histologic changes is as follows:
 Within about 2–4 hours there are focal axonal accumulations of β-amyloid precursor protein (APP) and other anterogradely transported proteins. These can be identified immunohistochemically at a time when conventional tinctorial stains do not show definite abnormalities (Fig. 11.32).
Within about 2–4 hours there are focal axonal accumulations of β-amyloid precursor protein (APP) and other anterogradely transported proteins. These can be identified immunohistochemically at a time when conventional tinctorial stains do not show definite abnormalities (Fig. 11.32).
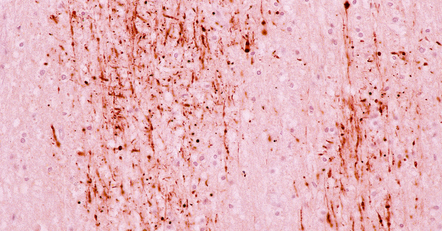
11.32 Clusters of axons with abnormal accumulation of APP early after head injury.
A few axons show focal swelling, but this is still mild and many of the axons appear to be intact.
 By 12–24 hours axonal varicosities may be evident on conventional histologic examination (Fig. 11.33).
By 12–24 hours axonal varicosities may be evident on conventional histologic examination (Fig. 11.33).
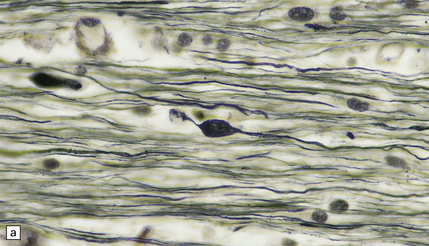
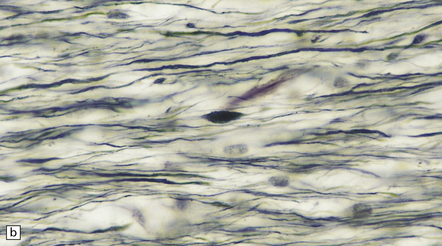
11.33 Microscopic changes 12–24 hours after head injury with DAI.
(a) and (b) Silver impregnation may reveal axonal varicosities, as shown in these illustrations.
 From 24 hours to 2 months after the injury conventional histology reveals axonal swellings. These are eosinophilic (Fig. 11.34a,c), but are best demonstrated by silver impregnation techniques (Fig. 11.34b,d) or immunohistochemistry for APP (Fig. 11.35, see also Fig. 11.26). Axonal swellings can also be detected by immunohistochemistry for ubiquitin, APP, or neurofilament protein, but these methods are less sensitive at later times.
From 24 hours to 2 months after the injury conventional histology reveals axonal swellings. These are eosinophilic (Fig. 11.34a,c), but are best demonstrated by silver impregnation techniques (Fig. 11.34b,d) or immunohistochemistry for APP (Fig. 11.35, see also Fig. 11.26). Axonal swellings can also be detected by immunohistochemistry for ubiquitin, APP, or neurofilament protein, but these methods are less sensitive at later times.
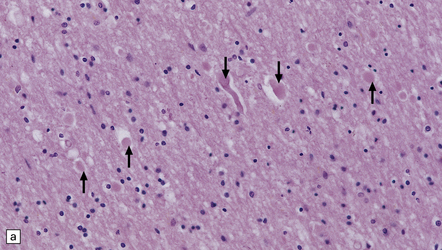
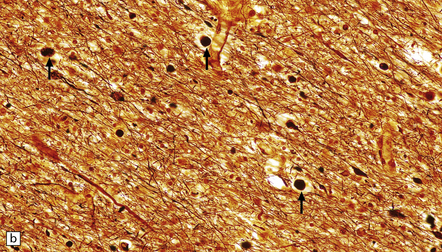
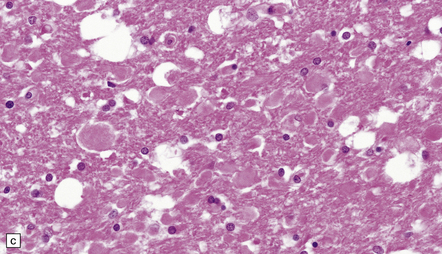
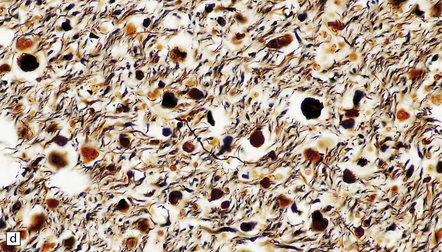
11.34 Microscopic changes after head injury with DAI.
In sections stained with hematoxylin and eosin, the axonal swellings are generally small and inconspicuous, but they are well visualized by silver impregnation. (a) Hematoxylin and eosin section. The axonal swellings (arrows) are small and inconspicuous. (b) Silver impregnation highlights the axonal swellings (arrows). (c) The axonal swellings may be obvious in a hematoxylin and eosin section in severe DAI, particularly if the patient survives for several days as here. (d) Palmgren silver impregnation of section adjacent to (c) showing large axonal swellings.
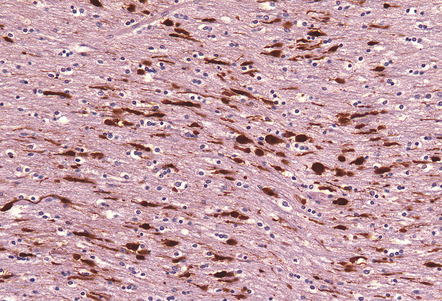
11.35 Demonstration of traumatic axonal injury by immunostaining for APP.
In this case the patient had DAI, the pathologic manifestations of which included the presence of numerous APP-immunopositive axons and axonal swellings in the corpus callosum, as illustrated.
 Two weeks to 5 months after the injury clusters of microglia are seen in the affected regions (Fig. 11.36).
Two weeks to 5 months after the injury clusters of microglia are seen in the affected regions (Fig. 11.36).
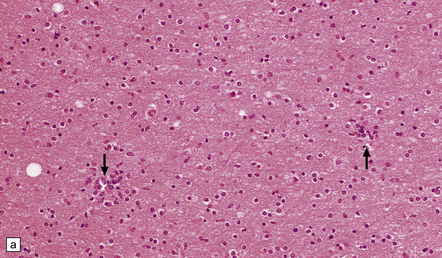
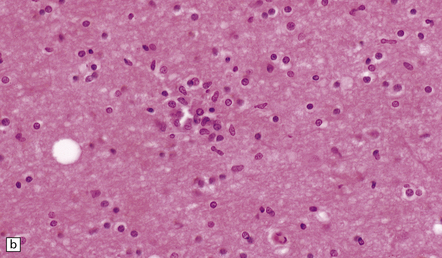
11.36 Clustering of microglial cells after head injury with DAI. From about 2 weeks after the injury and persisting for many months, the white matter contains numerous clusters of microglial cells.
(a) Clustering of cells, seen at low magnification (arrows). This is particularly well demonstrated in thick sections cut at 20 μm, as here. (b) At higher magnification, the clusters are seen to consist of microglia.
 From 2 months to years after the injury wallerian degeneration leads to loss of myelinated fibers (Fig. 11.37).
From 2 months to years after the injury wallerian degeneration leads to loss of myelinated fibers (Fig. 11.37).
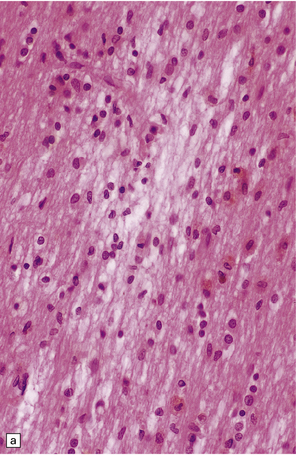
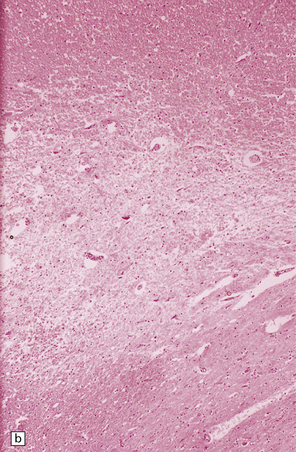
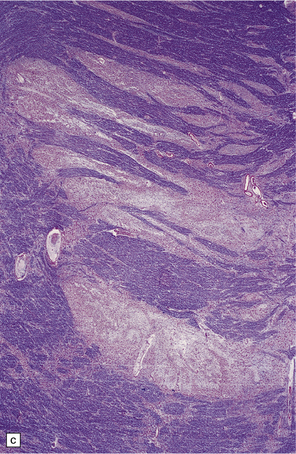
11.37 Long-term microscopic appearance of head injury with DAI.
(a) Focal glial scarring in a long-term survivor of head injury with DAI. (b) Focal rarefaction of the white matter in a long-term survivor of head injury with DAI. (c) Patchy depletion of corticospinal fibers in the pons of a patient who survived several months with severe DAI.
Grading of DAI is shown in Table 11.4.
Table 11.4

From: Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definitions, diagnosis and grading. Histopathology 1989; 15:49–59.
 PATHOGENESIS OF TAI
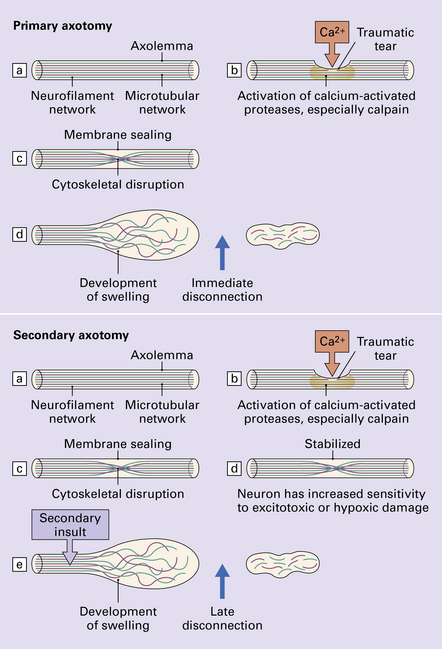
PATHOGENESIS OF TAI Clinical and experimental studies suggest that there are two mechanisms of traumatic axonal injury (Fig. 11.38): primary axotomy, which is almost immediate and secondary axotomy, which occurs over a period of hours after the injury.
Clinical and experimental studies suggest that there are two mechanisms of traumatic axonal injury (Fig. 11.38): primary axotomy, which is almost immediate and secondary axotomy, which occurs over a period of hours after the injury.






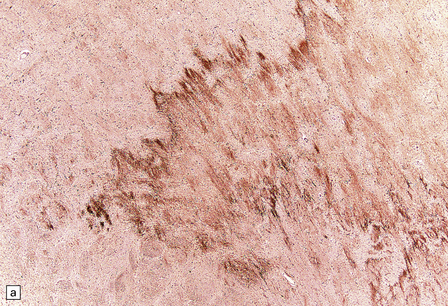
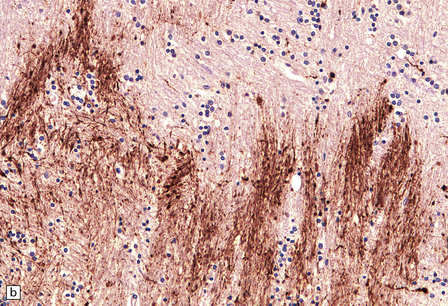
 DIFFERENTIATION OF ACUTE ISCHEMIC FROM TRAUMATIC AXONAL INJURY
DIFFERENTIATION OF ACUTE ISCHEMIC FROM TRAUMATIC AXONAL INJURY
Appearances that favor a diagnosis of ischemic axonal damage include:







DIFFUSE VASCULAR INJURY
In a small proportion of patients who die within minutes after head injury, the only discernible structural abnormalities are petechial hemorrhages, mostly in the frontal and temporal white matter, diencephalon and brainstem (Fig. 11.40). This diffuse vascular damage probably results from acute deformation, stretching and tearing of small blood vessels.
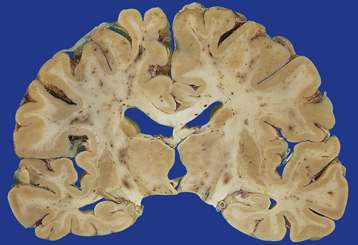
11.40 Diffuse vascular injury.
Diffuse vascular injury has produced numerous petechial hemorrhages in this brain from a patient who died within minutes after severe head injury.
 DIFFUSE VASCULAR INJURY AND FAT EMBOLISM
DIFFUSE VASCULAR INJURY AND FAT EMBOLISM
 Diffuse vascular injury should be distinguished from CNS fat embolism, which is probably underdiagnosed in patients with head injury and usually manifests clinically with dyspnea, hypoxia and confusion only 2–3 days after trauma, but can present earlier if severe.
Diffuse vascular injury should be distinguished from CNS fat embolism, which is probably underdiagnosed in patients with head injury and usually manifests clinically with dyspnea, hypoxia and confusion only 2–3 days after trauma, but can present earlier if severe.
 Most patients with fat embolism will have sustained multiple injuries, including long bone fractures, and will have radiologic and pathologic evidence of pulmonary fat emboli (which can be demonstrated in touch preparations of fresh lung tissue), although fat embolism can also occur in other clinical contexts (see Chapter 10).
Most patients with fat embolism will have sustained multiple injuries, including long bone fractures, and will have radiologic and pathologic evidence of pulmonary fat emboli (which can be demonstrated in touch preparations of fresh lung tissue), although fat embolism can also occur in other clinical contexts (see Chapter 10).
Macroscopic features of fat embolism
 If death occurs within two days the brain may appear macroscopically normal.
If death occurs within two days the brain may appear macroscopically normal.
 After survival of three or four days the brain contains numerous petechial hemorrhages and small perivascular foci of gray discoloration (Fig. 11.41). With longer premortem intervals, these appear as scattered small foci of necrosis. The lesions are most prominent in the cerebral white matter, but may also involve the cerebral and cerebellar cortex, deep gray nuclei, and brain stem.
After survival of three or four days the brain contains numerous petechial hemorrhages and small perivascular foci of gray discoloration (Fig. 11.41). With longer premortem intervals, these appear as scattered small foci of necrosis. The lesions are most prominent in the cerebral white matter, but may also involve the cerebral and cerebellar cortex, deep gray nuclei, and brain stem.
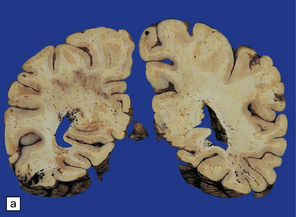
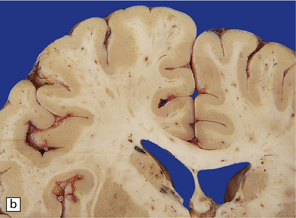
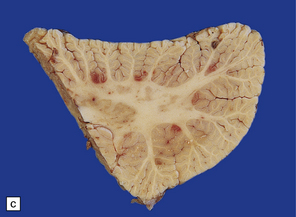
11.41 Slices of brain from patients who had sustained direct head injury but also developed fat embolism.
The lesions may appear as petechial hemorrhages or small perivascular foci of gray discoloration. Both gray and white matter are usually involved, although lesions in the white matter are easier to see on macroscopic examination. (a) Petechial hemorrhages and foci of gray discoloration in the white matter of the occipital lobes. (b) Perivascular foci of gray discoloration in the frontal lobe. (c) Petechial hemorrhages and foci of gray discoloration in the cortex and white matter of the cerebellum.
Microscopic features of fat embolism
 Histology reveals scattered capillaries surrounded by small ring or ball hemorrhages (Fig. 11.42).
Histology reveals scattered capillaries surrounded by small ring or ball hemorrhages (Fig. 11.42).
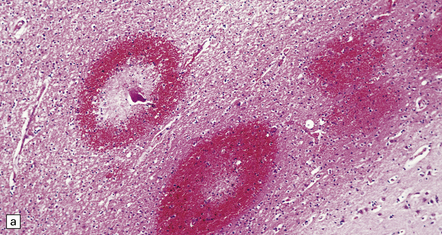
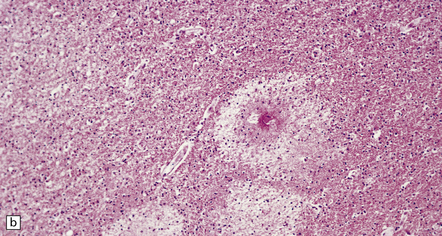
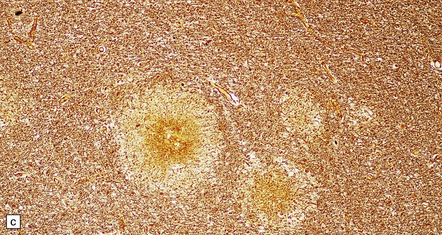
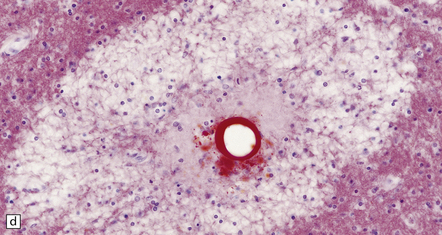
11.42 Microscopic features of fat embolism.
(a) Hemorrhagic lesions due to fat embolism, some of which are obviously centered on capillaries showing fibrinoid necrosis. (b) Non-hemorrhagic lesions, some of which are also centered on capillaries showing fibrinoid necrosis. (c) Silver impregnation reveals fragmentation of axons immediately adjacent to the necrotic capillaries. (d) In a frozen section lipid globules can be demonstrated in and around the necrotic foci by oil red O staining.
 Some of the capillaries show fibrinoid necrosis.
Some of the capillaries show fibrinoid necrosis.


 Later there is infiltration by macrophages and astrocytic gliosis.
Later there is infiltration by macrophages and astrocytic gliosis.
BRAIN SWELLING AND RAISED INTRACRANIAL PRESSURE
 cerebral vasodilatation and an increase in cerebral blood volume (i.e. congestive brain swelling)
cerebral vasodilatation and an increase in cerebral blood volume (i.e. congestive brain swelling)

 an increase in the water content of neurons and glia (i.e. cytotoxic cerebral edema)
an increase in the water content of neurons and glia (i.e. cytotoxic cerebral edema)
Three patterns of brain swelling are encountered in patients who have sustained a head injury:
 Swelling adjacent to contusions. Physical disruption of blood vessels in the surrounding brain tissue results in increased capillary permeability and a loss of normal blood flow regulation at the arteriolar level.
Swelling adjacent to contusions. Physical disruption of blood vessels in the surrounding brain tissue results in increased capillary permeability and a loss of normal blood flow regulation at the arteriolar level.
 Diffuse swelling of one cerebral hemisphere. This is especially common after evacuation of an ipsilateral subdural hematoma (Fig. 11.43), which may also be followed by delayed intracerebral hemorrhage, probably due to reperfusion of infarcted tissue.
Diffuse swelling of one cerebral hemisphere. This is especially common after evacuation of an ipsilateral subdural hematoma (Fig. 11.43), which may also be followed by delayed intracerebral hemorrhage, probably due to reperfusion of infarcted tissue.
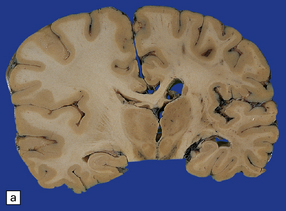
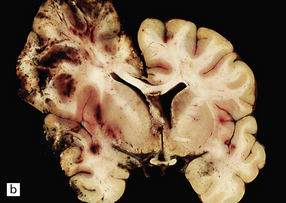
11.43 Brain swelling after evacuation of subdural hematoma.
(a) Swelling of the left cerebral hemisphere after evacuation of an ipsilateral subdural hematoma. (b) Brain swelling during evacuation of a subdural hematoma has led to external herniation of brain through the craniotomy, and focal parenchymal hemorrhage and infarction.
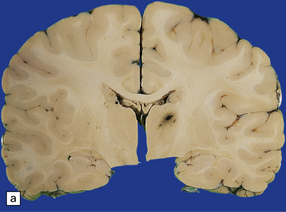
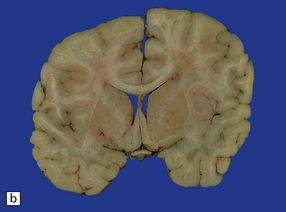
 Diffuse swelling of both cerebral hemispheres (Fig. 11.44). Children and adolescents are particularly susceptible and may develop brain swelling after relatively minor head injury. The swelling is thought to result from a global increase in brain blood volume. It may be associated with epileptic activity and even status epilepticus. In children the onset of swelling may occasionally be delayed for several hours.
Diffuse swelling of both cerebral hemispheres (Fig. 11.44). Children and adolescents are particularly susceptible and may develop brain swelling after relatively minor head injury. The swelling is thought to result from a global increase in brain blood volume. It may be associated with epileptic activity and even status epilepticus. In children the onset of swelling may occasionally be delayed for several hours.
HERNIATION
The cranial cavity is subdivided by the relatively rigid tentorium and falx cerebri into three compartments with limited capacity to accommodate accumulations of blood or swelling due to edema without an increase in pressure. Differences in pressure between two adjacent intracranial compartments, or between an intracranial compartment and the spinal canal, cause displacement of the soft substance of the brain into the lower-pressure compartment (i.e. internal herniation). There are three sites where this tends to occur, resulting in subfalcine, tentorial, or tonsillar herniation (Figs 11.45–11.48). Tentorial herniation may involve the medial part of the temporal lobe (the uncus and sometimes the parahippocampal gyrus), or diencephalic structures (the inferior part of the hypothalamus and thalamus); the latter type of tentorial herniation is also known as diencephalic or central transtentorial herniation. Raised intracranial pressure can also lead to external herniation of brain tissue through a skull fracture or craniotomy.
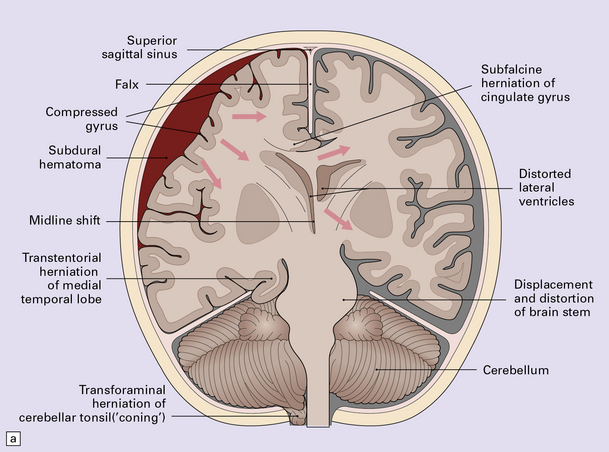
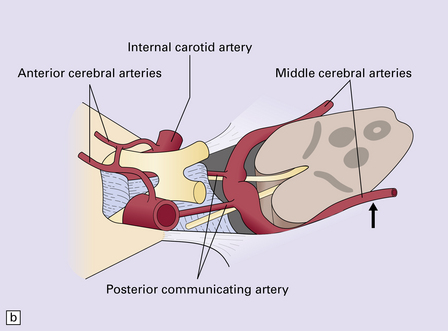
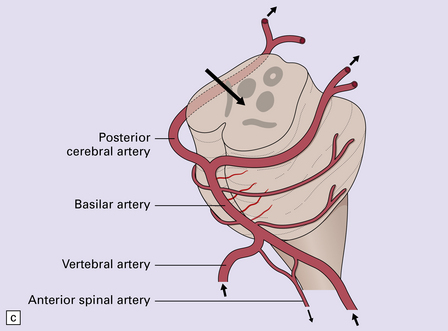
11.45 Internal brain herniation.
(a) Space occupancy by an expanding lesion causes herniation at several sites. (b) Transtentorial herniation of the medial part of the temporal lobe may compress the posterior cerebral artery where it crosses the free edge of the tentorium and cause infarction (usually hemorrhagic) in the corresponding perfusion territory. (c) Downward displacement of the brain stem causes deformity and traction of perforating branches (shown in light red) of the basilar artery and this may result in foci of infarction and hemorrhage (Duret hemorrhages).
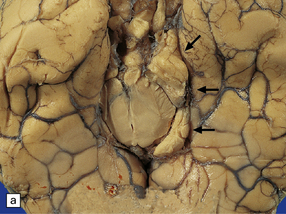
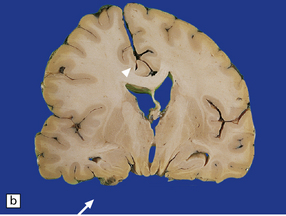
11.46 Transtentorial herniation.
(a) Herniation of the parahippocampal gyrus through the tentorial hiatus. The free edge of the tentorium cerebelli has indented the cerebrum (arrows) along the margin of the herniated brain tissue. (b) Herniation contusions (arrow) may be visible where the uncus or parahippocampal gyrus has been pressed against the edge of the tentorium, in this case by a subdural hematoma (not visible in figure). Histologic examination of the contusions reveals focal necrosis and small hemorrhages. The subdural hematoma has also displaced midline structures to the right, caused subfalcine herniation of the left cingulate gyrus (arrowhead) and downward displacement of the floor of the third ventricle (central transtentorial herniation).
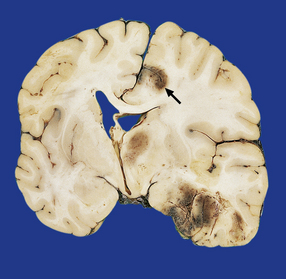
11.47 Cerebral herniation.
In this case, contusional damage and hemispheric swelling have caused subfalcine herniation of the right cingulate gyrus. This has resulted in adjacent hemorrhagic infarction (arrow). Note also the shift of midline structures and herniation of the right uncus.

11.48 Tonsillar herniation and necrosis.
In this case head injury has resulted in severe tonsillar herniation and necrosis. The medulla is partly ensheathed by the necrotic tonsillar tissue.
 RAISED INTRACRANIAL PRESSURE
RAISED INTRACRANIAL PRESSURE




Internal herniation may result in compression or stretching of blood vessels, leading to secondary ischemic damage (Figs 11.49, 11.50). It is important to recognize the secondary nature of these lesions and not attribute any bleeding to primary hemorrhage. Compression of the oculomotor nerves by downwards displacement of the posterior cerebral arteries can cause focal nerve contusion.
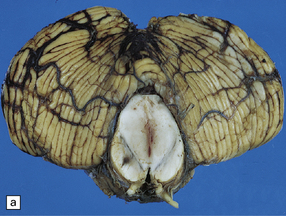
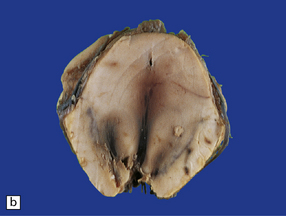
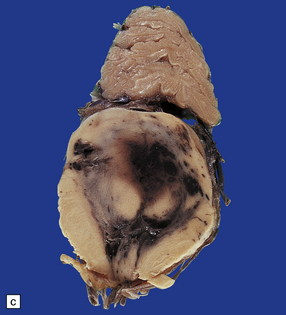
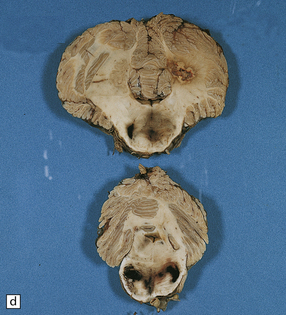
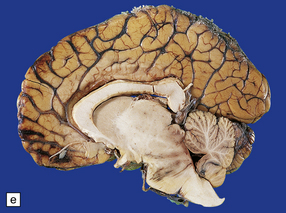
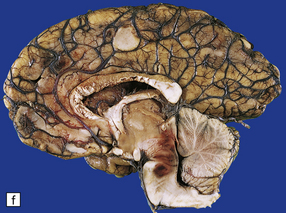
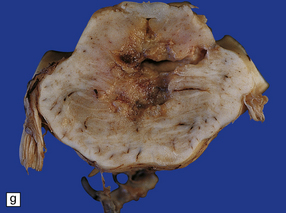
11.49 Effects of internal herniation.
With severe persisting herniation there is downwards displacement of diencephalic structures and descent of the pons, resulting in anteroposterior kinking or buckling of the brain stem with traction on the extramedullary segments of penetrating arteries and compression of their intramedullary segments. This results in foci of necrosis, and hemorrhages termed Duret hemorrhages, in the midbrain and upper pons. (a) Small Duret hemorrhage appearing as a linear area of midline hemorrhage. (b) A more severe lesion associated with hemorrhage in the midline and substantia nigra. (c) Duret hemorrhages appearing as extensive foci of hemorrhagic necrosis. (d) Marked hemorrhage and necrosis in the pons. (e) and (f) Sagittal sections, which demonstrate the buckling of the upper brain stem and the distribution of hemorrhagic lesions. (e) Early Duret hemorrhages. (f) Later, more extensive Duret hemorrhages. (g) Although internal herniation and brain stem hemorrhage are usually terminal events, rarely survival is long term, in which case the lesions in the midbrain and pons undergo organization and appear as soft cystic areas of cavitation.

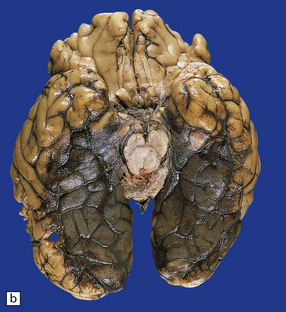
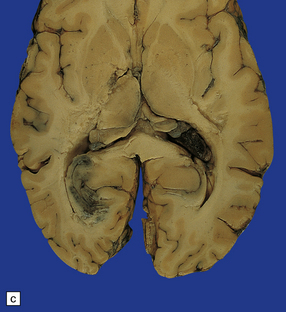
11.50 Posterior cerebral infarction due to herniation of the parahippocampal gyrus.
During herniation of the parahippocampal gyrus the posterior cerebral artery is compressed against the free edge of the tentorium. This can cause occipital lobe infarction, in some cases largely confined to the primary visual cortex in the calcarine fissure, but much more extensive in others. (a) Extensive infarction of inferomedial part of the left occipital lobe and a small region of the right occipital lobe. (b) Bilateral inferomedial occipital and adjacent temporal infarction. (c) Cut surface of brain showing medial occipital infarction.
Focal contusion or hemorrhagic infarction of the hippocampus or parahippocampal gyrus may result from compression of these structures against the edge of the tentorium during transtentorial herniation (Figs 11.46, 11.47). Similar damage may result from subfalcine herniation of the cingulate gyrus (Fig. 11.47). Transtentorial herniation also produces distortion of the midbrain and compression of the cerebral peduncles (Fig. 11.51). In some cases this causes necrosis of the contralateral cerebral peduncle due to its compression against the edge of the tentorium or of the ipsilateral cerebral peduncle due to its compression by the herniated uncus.
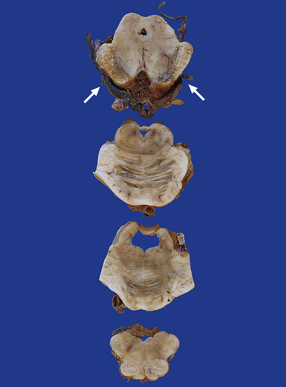
11.51 Long-term damage to brain stem after transtentorial herniation.
Sections through the brain stem of a patient who survived several months after head injury with bilateral uncal herniation. There is bilateral necrosis of the cerebral peduncles (arrows), and associated gray discoloration and focal cavitation of the descending fiber tracts in the base of the pons and medullary pyramids.
ISCHEMIC DAMAGE

 hypotension with a systolic blood pressure less than 80 mmHg for at least 15 minutes
hypotension with a systolic blood pressure less than 80 mmHg for at least 15 minutes
 episodes of raised intracranial pressure (i.e. over 30 mmHg).
episodes of raised intracranial pressure (i.e. over 30 mmHg).
Evidence of hypoxic–ischemic damage may be confined to the hippocampus (Fig. 11.52) or associated with more widespread changes in the cerebral cortex (Fig. 11.53) and deep gray matter. Cortical damage is often accentuated in the border zones between the major cerebral arterial territories, particularly between the anterior and middle cerebral arteries (Fig. 11.54), but may be diffuse. The damage is bilateral in most cases. Infarction may be restricted to the territory supplied by a single artery, but this is comparatively rare.
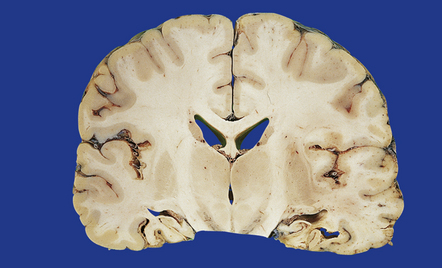
11.52 Ischemic brain damage.
Bilateral hippocampal necrosis caused by ischemic damage resulting from head injury.
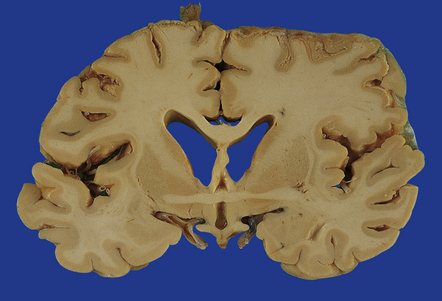
11.53 Ischemic damage caused by head injury.
Appearance of bilateral cortical necrosis in the perfusion territories of the middle cerebral arteries after survival of several weeks.
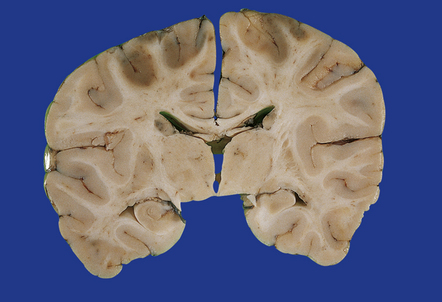
11.54 Ischemic damage caused by head injury.
Acute boundary zone infarction in the watershed between the anterior and middle cerebral arterial territories bilaterally.
Internal herniation may be complicated by secondary infarction, especially of the occipital lobes (Fig. 11.50) and upper brain stem (Fig. 11.49).
MISSILE HEAD INJURY
In perforating injuries, the missile (usually a bullet) enters and exits the skull, in most cases passing through the brain (Figs 11.55–11.57).
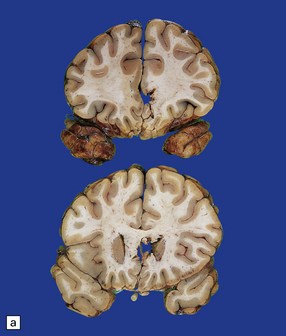
11.55 Penetrating injury due to bullet from a small-bore handgun.
Note that the slices are viewed from the front. The entry wound was in the forehead in the midline. The exit wound was in the occipital region of the skull. (a) The missile has passed through the corpus callosum and head of caudate nucleus on the left. (b) There is hemorrhagic disruption of the caudate nucleus and interventricular septum. (c) There is also bilateral thalamic damage.
11.56 Suicide resulting from discharge of a small-bore handgun in the mouth.
The bullet passed through the nasopharynx and base of skull and produced a hemorrhagic lesion in the caudate nucleus and corpus callosum.
11.57 Penetrating injury due to bullet from a small-bore handgun.
The bullet entered the skull in the right temporoparietal region, traversed both cerebral hemispheres, and exited through the left posterior temporal region of the skull. This brain slice shows the hemorrhagic disruption caused by the bullet in the right parietal cortex and white matter, the posterior part of the right putamen, internal capsule and thalamus.
 Low-velocity missiles are more likely to fragment or ricochet within the skull, producing a penetrating rather than a perforating injury, and to drive fragments of cloth, bone, and scalp into the brain.
Low-velocity missiles are more likely to fragment or ricochet within the skull, producing a penetrating rather than a perforating injury, and to drive fragments of cloth, bone, and scalp into the brain.

CHRONIC TRAUMATIC ENCEPHALOPATHY
PUNCH-DRUNK SYNDROME (DEMENTIA PUGILISTICA)
 PUNCH-DRUNK SYNDROME
PUNCH-DRUNK SYNDROME



MACROSCOPIC AND MICROSCOPIC APPEARANCES

 degeneration of the substantia nigra with neuronal loss
degeneration of the substantia nigra with neuronal loss
 neuronal loss from the cortex and cerebellum
neuronal loss from the cortex and cerebellum
 neurofibrillary tangle formation in the cerebral cortex (Fig. 11.58).
neurofibrillary tangle formation in the cerebral cortex (Fig. 11.58).
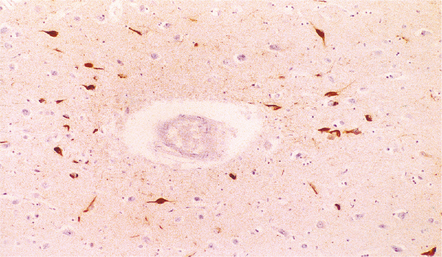
11.58 Punch-drunk syndrome.
Neurofibrillary tangles in the cerebral cortex of a 23-year-old boxer. The tangles were most numerous in the vicinity of the cortical blood vessels and were immunoreactive for tau protein, as shown here. In older boxers tangles may be present in large numbers in the cerebral cortex and brain stem. (Courtesy of Dr J. Geddes, Royal London Hospital.)

In some cases, patients have also developed a widespread TDP-43 (TAR DNA-binding protein of approximately 43 kd) proteinopathy (see Chapters 27, 28 and 31), associated with numerous glial and neuronal inclusions in the cerebral cortex, basal ganglia, diencephalon, and brainstem. A few patients have developed a motor neuron disease resembling amyotrophic lateral sclerosis and have had many TDP-43-positive inclusions in the primary motor cortex and spinal cord.
SPINAL INJURY
Included in this chapter are injuries to the spinal cord resulting from trauma and degenerative disease of the spinal column (Fig. 11.59). Neoplastic, ischemic, infectious, toxic, and metabolic disorders that may affect the spinal cord are considered in the appropriate chapters elsewhere in this book.
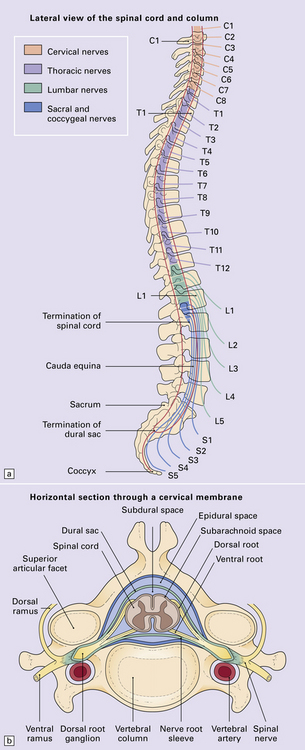
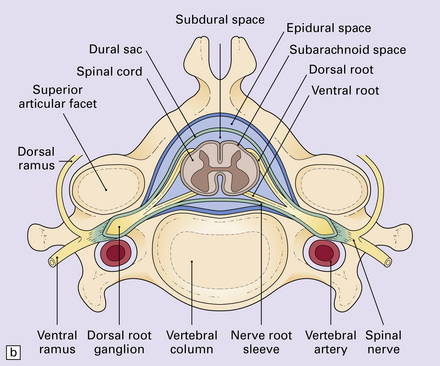
11.59 Spinal anatomy.
(a) Lateral view of the spinal cord and column. (b) Horizontal section through a cervical membrane.
TRAUMATIC SPINAL CORD INJURY
NATURE OF LESIONS


Distribution
The nature and site of injury vary according to the underlying etiology:





MACROSCOPIC APPEARANCES
 CLINICAL FEATURES OF SPINAL CORD INJURY
CLINICAL FEATURES OF SPINAL CORD INJURY


In most cases the soft tissues around the vertebral column are hemorrhagic and the fracture or fracture–dislocation is visible or at least palpable on external examination of the column. The site of the fracture is obvious on dividing the column (Figs 11.60, 11.61). Blood is usually visible in the epidural space and may also be present in the subarachnoid space around the cord. The macroscopic damage to the adjacent cord varies from mild focal indentation to severe hemorrhagic disruption that may extend several segments above and below the level of the fracture (Fig. 11.62). Even in the absence of obvious hemorrhage, the injured cord is usually swollen and congested.
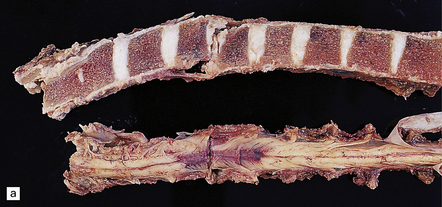
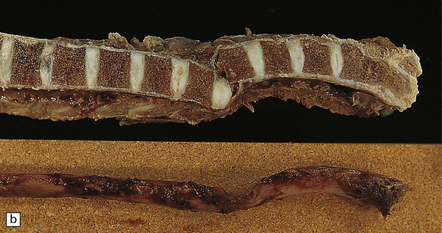
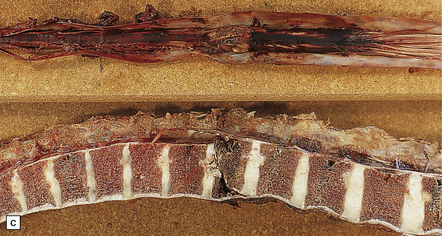
11.60 Fracture–dislocations demonstrated by sawing midsagittally through the vertebral bodies.
(a) Fracture–dislocation of the cervical column. The anterior longitudinal ligament is torn, but despite the displacement, the posterior longitudinal ligament is still intact and there is only scanty hemorrhage into the soft tissues around the spinal column or extradural space. The horizontal slice through the cord was made during the course of the examination. (b) Fracture–dislocation of the cervical column. The marked displacement has caused severe compression of the cord and is associated with more marked soft tissue and extradural hemorrhage than in (a). (c) Fracture–dislocation of the lumbar column. The fractured vertebral body has compressed and lacerated the cord and is associated with extensive extradural and subarachnoid hemorrhage. (Courtesy of Dr T. Moss, Frenchay Hospital, Bristol.)
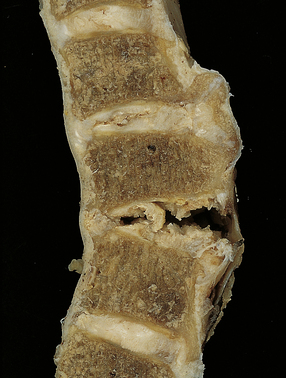
11.61 Hyperextension injury of the cervical cord.
In this case, the patient had degenerative spinal disease with anterior osteophytes. As a result of a fall, he sustained a cervical hyperextension injury causing cord compression. There has been tearing of the anterior longitudinal ligament and intervertebral disc space, but the posterior longitudinal ligament is intact and there is no horizontal displacement of the column.

11.62 Hemorrhagic cord lesion.
Hemorrhagic disruption extending several segments above the level of cord compression at the site of the vertebral fracture–dislocation. The rostral and caudal ends of the hemorrhagic lesion have a typical tapering spindle shape. (Courtesy of Dr T. Moss, Frenchay Hospital, Bristol.)
MICROSCOPIC APPEARANCES
During the acute phase after spinal injury, histology reveals variable combinations of edema, disruption of long tracts with prominent axonal swellings, hemorrhage, and foci of infarction (Figs 11.63–11.65). Over the next few weeks there is infiltration by macrophages, and gradual removal of myelin and neuronal debris (Figs 11.66, 11.67). Rostral and caudal segments of cord show degeneration of the ascending and descending long tracts that were damaged at the level of cord injury.
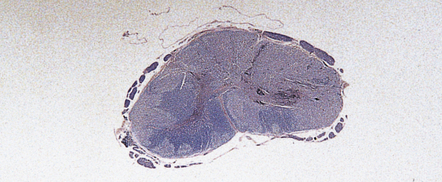
11.63 Acute cord injury.
Hemorrhagic necrosis of gray matter and disruption and extensive infarction of white matter in the compressed cord at the level of a cervical fracture–dislocation.
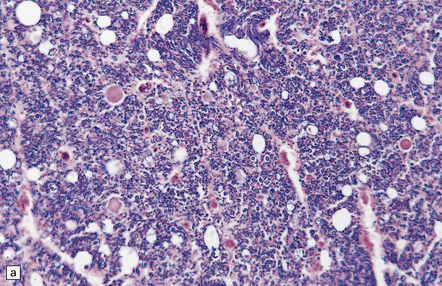
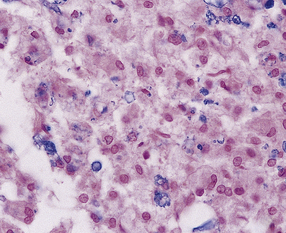
11.64 Microscopic appearance of spinal cord in a patient who survived several days after cord injury.
(a) There are scattered axonal spheroids in the lateral columns. (b) The axonal spheroids are well demonstrated by Palmgren silver impregnation.
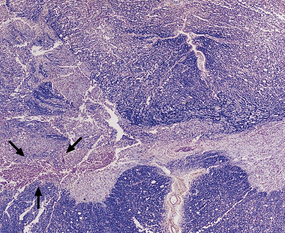
11.65 Microscopic appearance of spinal cord one week after cord injury.
At the level of injury the cervical cord is disrupted and there are foci of gray matter necrosis with infiltration by macrophages (arrows).
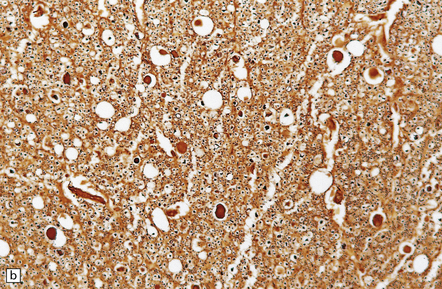
11.66 Infiltration of injured cord by macrophages.
Removal of myelin debris from the posterior columns of an injured cord by infiltrating macrophages.
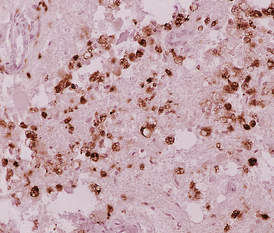
11.67 Infiltration of injured cord by macrophages.
CD68-immunoreactive macrophages in an injured spinal cord.
 ASSESSMENT OF THE SPINAL CORD AFTER VERTEBROSPINAL TRAUMA OR OTHER COMPRESSIVE CORD LESIONS
ASSESSMENT OF THE SPINAL CORD AFTER VERTEBROSPINAL TRAUMA OR OTHER COMPRESSIVE CORD LESIONS



The gray matter at the level of injury may become necrotic and, later, cavitated (Fig. 11.68), and often shows prominent proliferation and later hyaline thickening of small blood vessels. With time there is often infiltration by fibroblasts and associated collagenous fibrosis. Frequently, cavitation also involves the anterior part of the posterior columns. Such cavitation is usually maximal at the level of gray matter injury, but can extend several segments above and below (post-traumatic syringomyelia). The cavitation may continue to extend rostrally and caudally within the cord over many years.
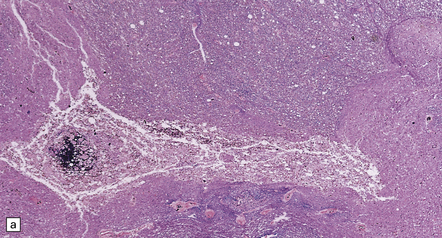
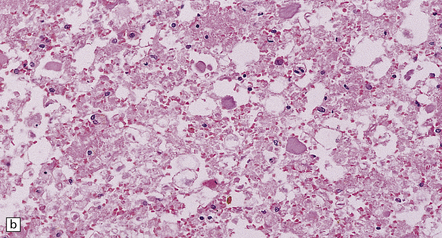
11.68 Necrosis of cord after trauma.
(a) Necrosis of central gray matter and immediately adjacent part of the posterior columns above the level of a spinal fracture. (b) Higher magnification of a hematoxylin and eosin-stained section reveals necrotic hemorrhagic tissue, scattered macrophages, and a few axonal spheroids.
NON-TRAUMATIC SPINAL CORD INJURY
 NON-TRAUMATIC CAUSES OF CORD COMPRESSION
NON-TRAUMATIC CAUSES OF CORD COMPRESSION
Craniocervical/atlantoaxial deformities or subluxation
 Odontoid hypoplasia or dysplasia and resulting atlantoaxial instability causing compression due to atlantoaxial subluxation or associated thickening of the dura mater in the craniocervical region: Down syndrome; Morquio’s syndrome (mucopolysaccharidosis type IVA); rarely other mucopolysaccharidoses or mucolipidoses; primary bone dysplasias; achondroplasia.
Odontoid hypoplasia or dysplasia and resulting atlantoaxial instability causing compression due to atlantoaxial subluxation or associated thickening of the dura mater in the craniocervical region: Down syndrome; Morquio’s syndrome (mucopolysaccharidosis type IVA); rarely other mucopolysaccharidoses or mucolipidoses; primary bone dysplasias; achondroplasia.


 Chiari malformation (type I, or type II/Arnold–Chiari malformation) (see Chapter 3).
Chiari malformation (type I, or type II/Arnold–Chiari malformation) (see Chapter 3).



Vertebral expansion or collapse


 Osteoporosis (Fig. 11.69). In some cases, the onset of neurologic signs and symptoms may be delayed by weeks or months after an osteoporotic fracture as fragments of bone are slowly displaced posteriorly.
Osteoporosis (Fig. 11.69). In some cases, the onset of neurologic signs and symptoms may be delayed by weeks or months after an osteoporotic fracture as fragments of bone are slowly displaced posteriorly.
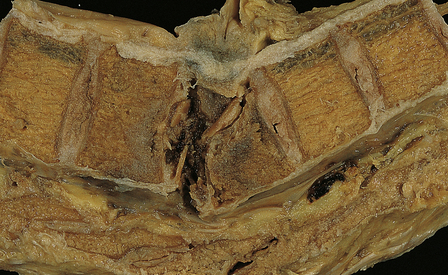
11.69 Osteoporotic vertebral collapse.
Little remains of the collapsed, wedge-shaped vertebra. In some patients with osteoporotic vertebral collapse, symptoms and signs of cord compression are delayed several weeks as the fragments of bone are gradually extruded posteriorly.
 Osteonecrosis: usually associated with corticosteroid administration or radiotherapy (Fig. 11.70).
Osteonecrosis: usually associated with corticosteroid administration or radiotherapy (Fig. 11.70).

Developmental or idiopathic spinal stenosis
Connective tissue, intervertebral disc, and joint disease
 Rheumatoid arthritis (rarely ankylosing spondylitis or psoriatic arthritis) can cause C1/C2 (atlantoaxial), C2/C3 (subaxial), or much less commonly, C3/C4 subluxation (Fig. 11.71).
Rheumatoid arthritis (rarely ankylosing spondylitis or psoriatic arthritis) can cause C1/C2 (atlantoaxial), C2/C3 (subaxial), or much less commonly, C3/C4 subluxation (Fig. 11.71).
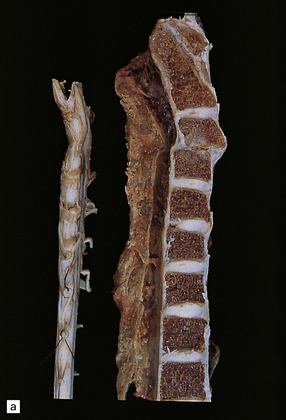
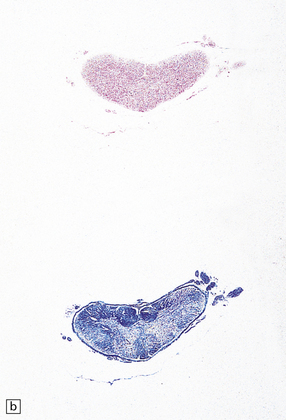
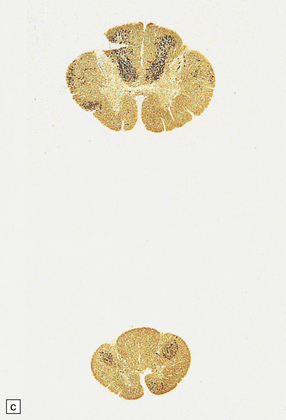
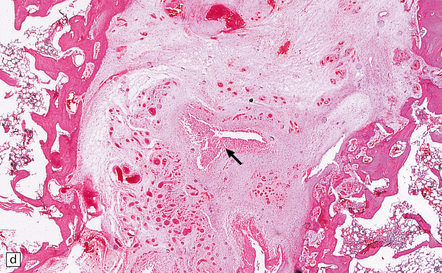
11.71 Cord damage due to rheumatoid arthritis.
(a) C3/C4 subluxation due to rheumatoid arthritis with resulting cord compression. (b) There is extensive white matter damage at the level of cord compression. (c) Marchi preparations reveal symmetric posterior column degeneration at the C2 level (top section) and pyramidal tract degeneration in the thoracic cord (bottom section). (d) The affected intervertebral disc space is replaced by vascular fibrous connective tissue with occasional foci of fibrinoid necrosis (arrow). (e) Higher magnification through a focus of fibrinoid necrosis shows peripheral palisading by fibroblasts and macrophages.
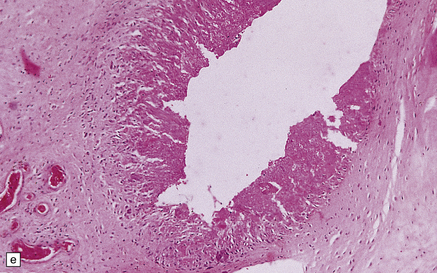
 Acute disc prolapse (Fig. 11.72).
Acute disc prolapse (Fig. 11.72).
11.72 Acute disc prolapse.
Indentation of the thoracic cord due to acute anterolateral disc prolapse.
 Spondylosis (Fig. 11.73). The changes in the spinal cord are due to several factors: the direct effects of compression and indentation; ischemic injury caused by vascular compression; root damage due to entrapment; and possibly injury caused by traction of the denticulate ligament. There is often associated fibrous thickening of the leptomeninges and of the walls of blood vessels within the cord. Occasionally the cord is invaded by peripheral nerve fibers as in other forms of chronic cord injury.
Spondylosis (Fig. 11.73). The changes in the spinal cord are due to several factors: the direct effects of compression and indentation; ischemic injury caused by vascular compression; root damage due to entrapment; and possibly injury caused by traction of the denticulate ligament. There is often associated fibrous thickening of the leptomeninges and of the walls of blood vessels within the cord. Occasionally the cord is invaded by peripheral nerve fibers as in other forms of chronic cord injury.
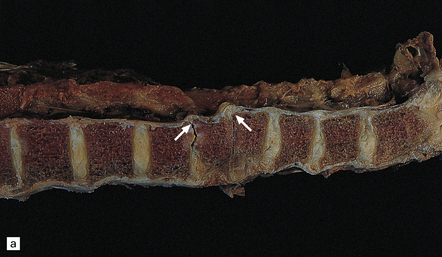
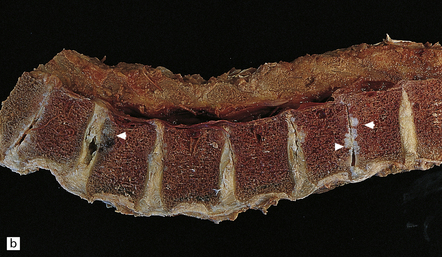
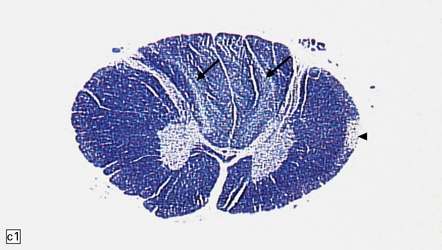
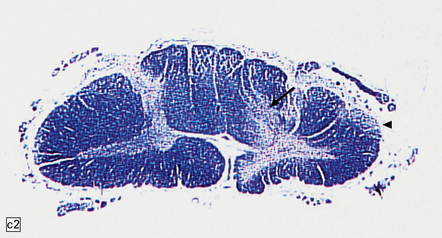
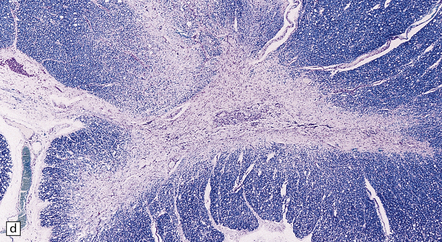
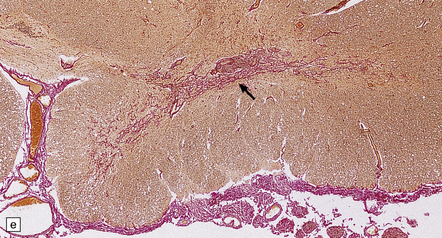
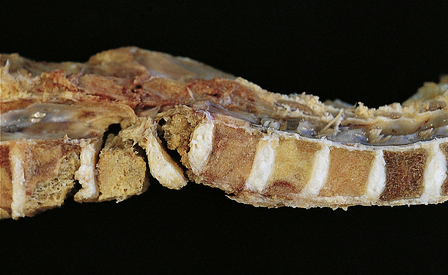
11.73 Cord injury due to spondylosis.
(a) Cervical and (b) lumbar spondylosis, with loss of intervertebral disc material at multiple levels. Nodules of herniated disc material (Schmorl’s nodes) are visible in some of the vertebral bodies immediately adjacent to some of the disc spaces (arrowheads). Posterior osteophytic bars (arrows) have compressed the adjacent cord. The cord at (ci) and above (cii) the level of compression contains wedge-shaped zones of fiber degeneration (arrow) in the posterior column, due to entrapment damage to the posterior nerve roots. There is also a small subpial zone of fiber degeneration in the lateral column (arrowhead). This relatively common finding in cervical spondylotic cord disease may be due to traction of the denticulate ligament against the displaced cord, causing focal distortion or ischemia. (d) Higher magnification of the cord at the level of compression reveals gray matter and anterior column degeneration, which is probably ischemic, and collagenous scarring. (e) Section adjacent to (d), stained with hematoxylin/van Gieson to show the collagenous scarring (arrow). (f) Invasion of the scarred spinal gray matter by peripheral nerve fibers.

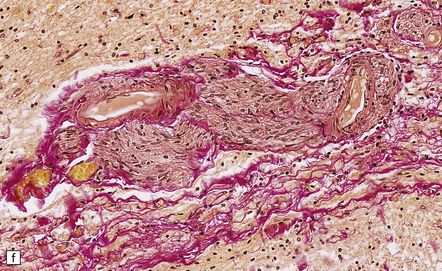
 Amyloidosis, particularly due to β2-microglobulin deposition in patients on chronic hemodialysis (Fig. 11.74). This can cause an arthropathy as well as thickening of the posterior longitudinal ligament and ligamentum flavum.
Amyloidosis, particularly due to β2-microglobulin deposition in patients on chronic hemodialysis (Fig. 11.74). This can cause an arthropathy as well as thickening of the posterior longitudinal ligament and ligamentum flavum.
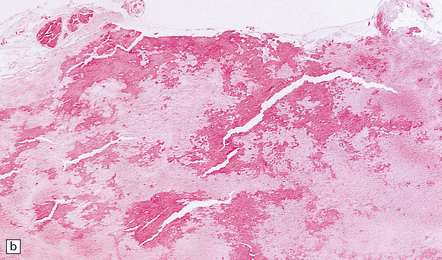

11.74 Cord compression due to amyloid.
Adjacent sections showing deposits of β2-microglobulin amyloid in and adjacent to the ligamentum flavum in a patient on chronic hemodialysis. (a) Van Gieson/Weigert’s elastic stain. (b) Sirius red viewed with unpolarized light. (c) Sirius red viewed with polarized light.

Epidural space-occupying lesions (see also Chapters 16, 17, 18, 46, 47)
 Tumors: primary including lipoma and angiolipoma; secondary, especially lymphoma.
Tumors: primary including lipoma and angiolipoma; secondary, especially lymphoma.
 Epidural abscess (tuberculous or pyogenic).
Epidural abscess (tuberculous or pyogenic).

 Hemorrhage (Fig. 11.75): spontaneous or due to a bleeding diathesis, most commonly after the administration of streptokinase or tissue plasminogen activator for treatment of a myocardial infarct.
Hemorrhage (Fig. 11.75): spontaneous or due to a bleeding diathesis, most commonly after the administration of streptokinase or tissue plasminogen activator for treatment of a myocardial infarct.

11.75 Spinal epidural hematoma.
Posterior compression of the spinal cord due to an epidural (extradural) hematoma in a patient who had been receiving streptokinase for treatment of a myocardial infarct.
 Extramedullary hemopoiesis, especially in thalassemia or myelofibrosis.
Extramedullary hemopoiesis, especially in thalassemia or myelofibrosis.
Subdural and subarachnoid space-occupying lesions (see also Chapters 16, 17, 18, 46, 47)



REFERENCES
Adams, J.H., Jennett, B., Murray, L.S., et al. Neuropathological findings in disabled survivors of a head injury. J Neurotrauma.. 2011;28:701–709.
Bramlett, H.M., Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab.. 2004;24:133–150.
Contostavlos, D.L. Isolated basilar traumatic subarachnoid hemorrhage: an observer’s 25 year re-evaluation of the pathogenetic possibilities. Forensic Sci Int.. 1995;73:61–74.
Dowling, G., Curry, B. Traumatic basal subarachnoid hemorrhage. Report of six cases and review of the literature. Am J Forensic Med Pathol.. 1988;9:23–31.
Elder, G.A., Cristian, A. Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mt Sinai J Med.. 2009;76:111–118.
Eriksson, E.A., Pellegrini, D.C., Vanderkolk, W.E., et al. Incidence of pulmonary fat embolism at autopsy: an undiagnosed epidemic. J Trauma.. 2011;71:312–315.
Finnie, J.W., Blumbergs, P.C. Traumatic brain injury. Vet Pathol.. 2002;39:679–689.
Fusco, M.R., Harrigan, M.R. Cerebrovascular dissections: a review. Part II: blunt cerebrovascular injury. Neurosurgery.. 2011;68:517–530;. [discussion 530].
Geddes, J.F., Whitwell, H.L. Head injury in routine and forensic pathological practice. Curr Top Pathol.. 2001;95:101–124.
Geddes, J.F., Whitwell, H.L., Graham, D.I. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol.. 2000;26:105–116.
Graham, D.I., Smith, C., Reichard, R., et al. Trials and tribulations of using β-amyloid precursor protein immunohistochemistry to evaluate traumatic brain injury in adults. Forensic Sci Int.. 2004;146:89–96.
Gross, A. Traumatic basal subarachnoid hemorrhages: autopsy material analysis. Forensic Sci Int.. 1990;45:53–61.
Johnson, V.E., Stewart, W., Smith, D.H. Widespread tau and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2011. [[E-pub ahead of print]].
Kocsis, J.D., Tessler, A. Pathology of blast-related brain injury. J Rehabil Res Dev.. 2009;46:667–672.
Larson, P.S., Reisner, A., Morassutti, D.J., et al. Traumatic intracranial aneurysms. Neurosurg Focus.. 2000;8:e4.
Maxwell, W.L., Povlishock, J.T., Graham, D.L. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma.. 1997;14:419–440.
McKee, A.C., Cantu, R.C., Nowinski, C.J., et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol.. 2009;68:709–735.
McKee, A.C., Gavett, B.E., Stern, R.A., et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929.
Oehmichen, M., Meissner, C., Konig, H.G. Brain injury after gunshot wounding: morphometric analysis of cell destruction caused by temporary cavitation. J Neurotrauma.. 2000;17:155–162.
Oehmichen, M., Meissner, C., Konig, H.G. Brain injury after survived gunshot to the head: reactive alterations at sites remote from the missile track. Forensic Sci Int.. 2001;115:189–197.
Oehmichen, M., Meissner, C., Konig, H.G., et al. Gunshot injuries to the head and brain caused by low-velocity handguns and rifles. A review. Forensic Sci Int.. 2004;146:111–120.
Saing, T., Dick, M.C., Nelson, P.T., et al. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma.. 2011. [[E-pub ahead of print]].
Accidental and non-accidental pediatric brain injury
Case, M.E. Accidental traumatic head injury in infants and young children. Brain Pathol.. 2008;18:583–589.
Case, M.E. Inflicted traumatic brain injury in infants and young children. Brain Pathol.. 2008;18:571–582.
Geddes, J.F., Hackshaw, A.K., Vowles, G.H., et al. Neuropathology of inflicted head injury in children. I. Patterns of brain damage. Brain.. 2001;124:1290–1298.
Geddes, J.F., Vowles, G.H., Hackshaw, A.K., et al. Neuropathology of inflicted head injury in children. II. Microscopic brain injury in infants. Brain.. 2001;124:1299–1306.
Matschke, J., Voss, J., Obi, N., et al. Nonaccidental head injury is the most common cause of subdural bleeding in infants < 1 year of age. Pediatrics.. 2009;124:1587–1594.
Oehmichen, M., Schleiss, D., Pedal, I., et al. Shaken baby syndrome: re-examination of diffuse axonal injury as cause of death. Acta Neuropathol.. 2008;116:317–329.
Squier, W. The “Shaken Baby” syndrome: pathology and mechanisms. Acta Neuropathol.. 2011;122:519–542.
Traumatic and non-traumatic spinal cord injury
Cornish, R., Blumbergs, P.C., Manavis, J., et al. Topography and severity of axonal injury in human spinal cord trauma using amyloid precursor protein as a marker of axonal injury. Spine (Phila Pa 1976). 2000;25:1227–1233.
Henderson, F.C., Geddes, J.F., Crockard, H.A. Neuropathology of the brainstem and spinal cord in end stage rheumatoid arthritis: implications for treatment. Ann Rheum Dis.. 1993;52:629–637.
Ito, T., Oyanagi, K., Takahashi, H., et al. Cervical spondylotic myelopathy. Clinicopathologic study on the progression pattern and thin myelinated fibers of the lesions of seven patients examined during complete autopsy. Spine (Phila Pa 1976).. 1996;21:827–833.
Kakulas, B.A. A review of the neuropathology of human spinal cord injury with emphasis on special features. J Spinal Cord Med.. 1999;22:119–124.
Kakulas, B.A. Neuropathology: the foundation for new treatments in spinal cord injury. Spinal Cord.. 2004;42:549–563.
O’Brien, M.F., Casey, A.T., Crockard, A., et al. Histology of the craniocervical junction in chronic rheumatoid arthritis: a clinicopathologic analysis of 33 operative cases. Spine (Phila Pa 1976).. 2002;27:2245–2254.