[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 3
Glucocorticoid Receptors
Their Mechanisms of Action and Glucocorticoid Resistance
Robert H. Oakley, Laura J. Lewis-Tuffin, Carl D. MaLchoff, Diana Mark Malchoff and John A. Cidlowski
Glucocorticoid Receptors*
Glucocorticoids are a class of endogenous and synthetic steroid hormones that affect virtually every aspect of human physiology. Currently they are one of the most commonly prescribed classes of drugs.1 The physiologic actions of glucocorticoids include highly effective antiinflammatory and immunomodulatory actions that are exploited for the treatment of diseases such as asthma, arthritis, allergic rhinitis, and leukemia/lymphoma.2,3 In addition, glucocorticoids have important roles in development of the lung and nervous systems, skeletal growth, behavior, reproduction, and intermediary metabolism. Ultimately, glucocorticoids act to maintain homeostasis in the face of stressful stimuli. The broad actions of glucocorticoids account for the serious side effects commonly experienced with chronic glucocorticoid treatment. These include the development of glucocorticoid resistance in diseased tissues, osteoporosis, growth retardation in children, muscle atrophy, and signs of the metabolic syndrome.1,4,5 The physiologic and pharmacologic actions of glucocorticoids are mediated by the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of proteins that regulate gene transcription in a ligand-dependent manner.6 In this section, we review the basic mechanisms of glucocorticoid action with an emphasis on how these mechanisms contribute to the antiinflammatory and immunomodulatory effects of glucocorticoids.
The Glucocorticoid Receptor
Like other members of the nuclear receptor superfamily, GR is a modular protein comprising an amino-terminal transactivation domain (NTD); a central, two-zinc-finger DNA-binding domain (DBD); and a carboxyl-terminal ligand-binding domain (LBD) (Fig. 3-1A).7 Separating the DBD and LBD is a flexible region of the molecule termed the hinge region. The receptor contains two regions involved in activating transcription, one in the NTD (AF1) which can act independently of ligand binding, and a second in the LBD (AF2) whose function is dependent on glucocorticoid binding. The LBD also possesses a nuclear localization signal and sites for interaction with other transcription factors, coregulators, and protein chaperones. An additional nuclear localization signal spans the junction of the DBD and hinge region. The GR protein is also a substrate for various types of posttranslational modifications including phosphorylation, ubiquitination, sumoylation, and acetylation that regulate GR signaling by modulating the expression level and/or transcriptional activity of the receptor.8
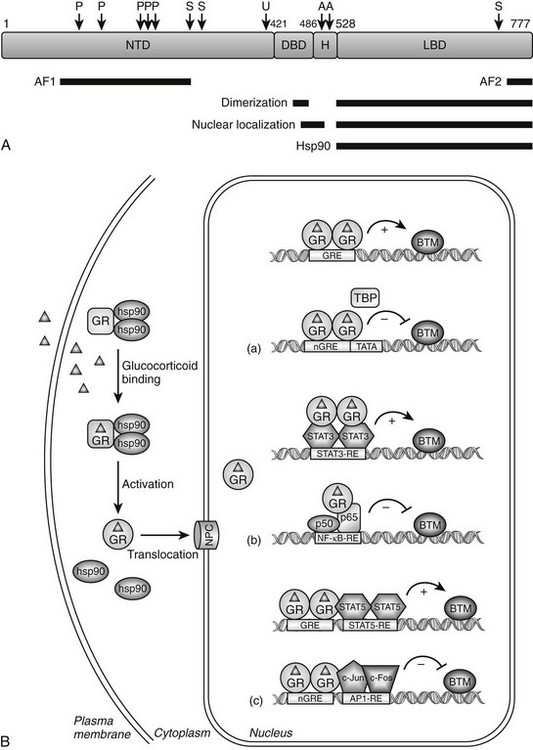
FIGURE 3-1 Glucocorticoid receptor (GR) domain structure and signaling pathway. A, GR is composed of an amino-terminal transactivation domain (NTD), DNA-binding domain (DBD), hinge region (H), and ligand-binding domain (LBD). Regions involved in transactivation (AF1 and AF2), dimerization, nuclear localization, and hsp90 binding are indicated. The location of residues posttranslationally modified by phosphorylation (P) (Ser-113, Ser-141, Ser-203, Ser-211, and Ser-226); ubiquitination (U) (Lys-419); sumoylation (S) (Lys-277, Lys-293, and Lys-703); and acetylation (A) (Lys-494 and Lys-495) are also depicted. Numbers are for the human glucocorticoid receptor. B, The unliganded GR resides in the cytoplasm of cells in a complex with chaperone proteins. Upon binding glucocorticoids (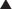 ), the receptor undergoes a change in conformation, dissociates from accessory proteins, and translocates into nucleus via the nuclear pore complex (NPC). Nuclear GR regulates gene expression in three primary ways: (a) GR binds directly to DNA and enhances or inhibits transcription of target genes. (b) GR interacts with DNA-bound transcription factors without itself binding to DNA and enhances or inhibits transcription of target genes. (c) GR binds directly to DNA and interacts with transcription factors bound to neighboring sites and enhances or inhibits transcription of target genes. BTM, Basal transcription machinery; TBP, TATA-box binding protein.
), the receptor undergoes a change in conformation, dissociates from accessory proteins, and translocates into nucleus via the nuclear pore complex (NPC). Nuclear GR regulates gene expression in three primary ways: (a) GR binds directly to DNA and enhances or inhibits transcription of target genes. (b) GR interacts with DNA-bound transcription factors without itself binding to DNA and enhances or inhibits transcription of target genes. (c) GR binds directly to DNA and interacts with transcription factors bound to neighboring sites and enhances or inhibits transcription of target genes. BTM, Basal transcription machinery; TBP, TATA-box binding protein.
GR Regulation of Transcription via Direct Binding to DNA
Unliganded GR is located in the cytoplasm of cells as part of a multiprotein complex that includes two molecules of heat shock protein 90 (hsp90) (see Fig. 3-1B).9 These chaperone proteins maintain the receptor in a transcriptionally inactive state that favors high-affinity ligand binding. Upon binding glucocorticoids, GR undergoes a conformational change resulting in the dissociation of hsp90, exposure of the nuclear localization signals, and translocation of the receptor into the nucleus via the nuclear pore. The receptor then regulates gene transcription by binding directly to specific DNA sequences known as glucocorticoid response elements (GREs) and/or by binding other transcription factors and modulating their activity. Global gene expression analyses indicate that up to 20% of the genome is induced or repressed by glucocorticoids.5,10
The consensus GRE is an imperfect palindrome, GGTACAnnnTGTTCT, that is usually found in the promoter region of target genes.11,12 The interaction of ligand-bound GR with the GRE stimulates the transcription of numerous genes, including the metabolic enzymes tyrosine amino transferase, phosphoenolpyruvate carboxykinase, and glucose-6-phosphatase (see Fig. 3-1B[a], upper scheme). High-affinity GRE binding requires receptor dimerization and is short lived because the receptor rapidly cycles on and off target sites every few seconds.13,14 Upon binding DNA, GR undergoes a conformational change that results in the coordinated recruitment of three general types of coactivators necessary for stimulating transcription of the target gene: the ATP-dependent complex BRG1 (SWI/SNF) that mediates large noncovalent disruptions in chromatin structure; CBP, p300, and members of the SRC/p160 family of proteins that modify chromatin structure locally through their intrinsic histone acetyltransferase activity; and components of the DRIP/TRAP complex that assist in the recruitment of the basal transcription machinery.15 The alterations in chromatin structure result in DNA unwinding, thereby permitting promoter access to transcription factors and cofactors that enhance target gene expression. The specific coactivators recruited by GR determine the gene induction profile, and this assembly is dependent on the promoter context, the bound glucocorticoid, and the availability and activity of the coactivators themselves.
Negative GREs (nGREs) are the transcription-repressing counterpart of positive GREs.16 These response elements bear little resemblance to positive GREs, are highly variable, and a consensus sequence has not been determined. How ligand-bound GR represses transcription via nGREs is unclear and likely involves multiple mechanisms dependent upon the promoter context. For some genes, such as osteocalcin, DNA-bound GR may sterically interfere with binding of positively acting transcription factors to elements that overlap the nGRE (see Fig. 3-1B[a], lower scheme).17 The situation is more complex for other repressed genes, since both binding to DNA and interacting with neighboring transcription factors appear to be required of the receptor (see Fig. 3-1B[c], lower scheme). Referred to as a composite GRE, nGREs of this nature are found in the promoters of the proliferin and corticotropin-releasing hormone (CRH) genes. Here, DNA-bound GR interacts with the activator protein-1 (AP-1) transcription factor occupying an adjacent site to repress transcription.18,19 Finally, since nGREs correspond poorly to the GRE consensus sequence, negative regulation may be achieved at some target genes by GR interacting with DNA-bound positive transcription factors without itself binding to the promoter. This “tethering” mechanism of repression (described in more detail later) appears to mediate the glucocorticoid-dependent inhibition of gonadotropin-releasing hormone receptor and pro-opiomelanocortin expression via the interaction of GR with the promoter-bound Oct1 and Nur77 transcription factors, respectively.20,21
GR Regulation of Transcription via Protein-Protein Interactions
In addition to transcriptional regulation by direct binding to DNA, ligand-bound GR can also interact with other transcription factors and modulate their activity on glucocorticoid-responsive promoters without itself binding to DNA. The two most studied examples of this form of regulation involve the transcription factors AP-1 and nuclear factor κB (NF-κB). These two proteins are central mediators of the inflammatory and immune responses, and their inhibition by GR is thought to underlie the major antiinflammatory and immunosuppressive actions of glucocorticoids.22,23 When activated by stress signals such as proinflammatory cytokines, bacterial and viral infectious agents, or pro-apoptotic stimuli, AP-1 and NF-κB bind their cognate response elements and induce the expression of many proinflammatory genes, including cytokines, cell adhesion molecules, and enzymes involved in tissue destruction. Glucocorticoids indirectly antagonize the actions of AP-1 and NF-κB at multiple levels by inducing the expression of other regulatory proteins. Activated GR stimulates expression of the IκB protein, which sequesters NF-κB in the cytoplasm,24 MAPK phosphatase 1, which dephosphorylates c-Jun N-terminal kinase to prevent activation of AP-1,25 and tristetraprolin, which destabilizes the mRNA of many AP-1 and NF-κB-induced genes.26
The primary way, however, by which hormone-bound GR represses the activity of AP-1 and NF-κB is through a direct interaction with the c-Jun subunit of AP-1 and the p65 subunit of NF-κB (see Fig. 3-1B[b], lower scheme).22,23 Interestingly, the antagonism is reciprocal; the association of these proteins with GR represses its activity on target genes. Multiple mechanisms appear to underlie the antagonism, suggesting the mode of action may vary in a promoter, cell-type, and/or signal-dependent manner. In early studies, the receptor was reported to sequester AP-1 and NF-κB in the cytoplasm and/or nucleus and prevent DNA binding. More recent findings, however, suggest the receptor is tethered to DNA-bound AP-1 and NF-κB and alters the subsequent assembly and/or activity of recruited transcriptional proteins. For example, on several toll-like receptor genes, GR represses NF-κB by disrupting the interaction of p65 with the promoter-specific coactivator IRF3 (interferon regulatory factor 3).27 At the interleukin 8 (IL-8) and intercellular adhesion molecule 1 (ICAM-1) promoters, the association of GR with NF-κB impairs the phosphorylation of the C-terminal domain of RNA polymerase II.28 GR-mediated repression of the AP-1-responsive collagenase-3 promoter and the NF-κB-responsive IL-8 promoter have both been shown to be potentiated by receptor-dependent recruitment of the coactivator GRIP1, indicating coactivators can actually function as corepressors in the appropriate configuration with GR.29 Finally, GR has been reported to interact with histone deacetylase 2 (HDAC2) and to repress the histone acetyltransferase activity of NF-κB, suggesting that GR antagonizes NF-κB by effects on chromatin structure.30
In contrast to AP-1 and NF-κB, the physical association of GR with the signal transducer and activator of transcription (STAT) family of proteins enhances their activity on target genes.31 STAT transcription factors are activated by a range of cytokines through induction of the Janus kinase pathway (JAK) and tyrosine phosphorylation. Upon binding their cognate response elements, STATs regulate genes involved in the immune response, differentiation, survival, and apoptosis. GR has been shown to interact with several members of the STAT family, including STAT3 and STAT5, and to synergistically enhance their activity on target genes in a promoter-dependent fashion.31 The association of GR with STAT3 at the γ-fibrinogen and α2-macroglobulin promoters, which lack identifiable GREs, super-induces their expression (see Fig. 3-1B[b], upper scheme).32–34 Similarly, the synergistic activation of the β-casein and toll-like receptor-2 genes results from the association of GR with promoter-bound STAT5.35–38 However, the observed synergy at these STAT5-responsive genes may also require GR binding to DNA and more accurately reflect a composite GRE (see Fig. 3-1B[c], upper scheme).37,38 How GR synergistically activates STAT-regulated genes is poorly understood but may involve GR enhancing STAT nuclear localization,39 prolonging the promoter occupancy of STAT by inhibiting its tyrosine dephosphorylation32,40 and/or promoting the co-utilization of certain coactivators.31 Interestingly, the synergism of these two transcription factors is not always mutual, since GR activity is inhibited or stimulated depending on the particular STAT isoform employed.34,35,41,42
Multiple GR Isoforms Derived from Single Gene
The human GR gene is located on chromosome 5q31-32 and comprises 9 exons.43–46 Alternative splicing in exon 9 generates two receptor isoforms, termed GRα and GRβ, that are identical through to amino acid 727 but then diverge at their carboxyl-termini (Fig. 3-2).45–47 The classic, full-length GRα contains an additional 50 amino acids, whereas GRβ has an additional, nonhomologous 15 amino acids. Because of its unique carboxyl-terminus, GRβ does not bind glucocorticoids and resides constitutively in the nucleus of cells.45,48 GRβ has been shown to function as a dominant negative inhibitor and repress the transcriptional activity of GRα45,49–51; therefore, alterations in GRβ expression may contribute to changes in glucocorticoid responsiveness. Indeed, expression of GRβ is selectively increased over GRα in cells exposed to proinflammatory cytokines and microbial superantigens, leading to reduced sensitivity to glucocorticoids.52–56 In addition, glucocorticoid-resistant forms of inflammatory diseases (e.g., asthma, rheumatoid arthritis, and ulcerative colitis) have been associated with elevated expression of GRβ.50 Conversely, methotrexate, an effective drug for treating autoimmune and inflammatory diseases, promotes a selective increase in GRα at the expense of GRβ and improves glucocorticoid sensitivity of lymphocytes.57
FIGURE 3-2 Alternative processing of the single glucocorticoid receptor (GR) gene generates multiple GR isoforms. The human GR gene comprises 9 exons. Alternative splicing at the 3′ end of the primary transcript generates GRα and GRβ mRNAs, which encode GRα and GRβ proteins differing only at their carboxyl-terminus. Alternative translation initiation from 8 different AUG start codons derived from exon 2 generates additional protein isoforms with progressively shorter NTDs. Numbers shown denote the first and last residues for the human GR isoforms. For simplicity, only the most proximal of 9 alternate exon 1s (1H) is shown.
Elevated GRβ levels also result from a naturally occurring ATTTA to GTTTA polymorphism (A3669G) in the 3′ untranslated region of GRβ. This nucleotide substitution disrupts an mRNA destabilization motif and leads to increased stability of the GRβ mRNA and enhanced protein expression.58,59 The A3669G allele has been associated with reduced central obesity in women and a more favorable lipid profile in men,60 suggesting the increase in GRβ might antagonize some of the undesirable effects of GRα on fat distribution and lipid metabolism. The A3669G-directed rise in GRβ may also compromise the immunosuppressive actions of GRα. Persons harboring the A3669G allele have an elevated risk of the autoimmune disease rheumatoid arthritis and a reduced risk of bacterial nasal infection.58,61 Moreover, A3669G carriers exhibit an increased risk of myocardial infarction and coronary heart disease, two pathologies with inflammatory underpinnings.62
A broader role for GRβ in cell signaling has recently emerged with the demonstration that this isoform can modulate gene expression apart from its effects on GRα. In a genome-wide microarray analysis performed in cells selectively expressing GRβ, the isoform was found to alter the expression of over 5000 genes.63 Less than 20% of the genes were commonly regulated by ligand-activated GRα, indicating that GRβ possesses its own unique gene-regulatory profile. GRβ was also shown to bind the glucocorticoid antagonist mifepristone (RU486), and binding of this ligand abolished most of the GRβ-mediated changes in gene expression.63 As a bona fide transcription factor, GRβ may contribute to alterations in glucocorticoid responsiveness in healthy and diseased tissues by genomic effects independent of its dominant negative activity on GRα.
Alternative translation initiation of the GR mRNA produces an additional cohort of receptor subtypes (see Fig. 3-2).10,64,65 Eight GR isoforms with progressively shorter NTDs are generated from one GRα mRNA transcript via different AUG start codons: GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, and GRα-D3. GRα-A is the classic, full-length 777 amino acid protein that is generated from the first initiator AUG codon. The GRβ mRNA also contains the identical start codons and would be expected to give rise to a similar complement of subtypes. The GRα translational isoforms show a widespread tissue distribution; however, the relative levels of the subtypes differ both between and within tissues.64,66 Functionally, the isoforms bind glucocorticoids with similar affinity and bind GREs with similar capacity.66 Additionally, all eight isoforms occupy the nucleus of cells following glucocorticoid treatment. In the absence of hormone, however, the subcellular distribution of the subtypes differ, with the GRα-D isoforms residing predominantly in the nucleus and the others predominantly in the cytoplasm.64
Marked differences have also been reported in the transcriptional properties of the GRα translational isoforms.64,66 On GRE-containing reporter and endogenous genes, the GRα-C3 isoform was the most active stimulating gene expression, whereas the GRα-D subtypes were the most deficient. These isoform-selective effects have been attributed to differences among the subtypes in their ability to recruit various transcriptional factors and coregulators, such as CBP and RNA polymerase II, to the promoter. In contrast to these divergent effects on gene induction, no significant differences have been observed so far in the ability of the GRα isoforms to repress NF-κB.66 In a whole-genome microarray analysis performed on U2OS osteosarcoma cells selectively expressing the individual receptor isoforms, each subtype was found to regulate both a common and a unique set of genes.66 Of the approximately 6500 genes regulated by at least one GRα isoform, less than 500 were regulated commonly by all the receptor isoforms. Thus the majority of glucocorticoid-responsive genes were selectively regulated by different GRα subtypes. These isoform-unique gene regulatory profiles were further shown to produce functional differences in cellular responsiveness to glucocorticoids; the GRα translational isoforms exhibited distinct capabilities to induce apoptosis.66 Cells expressing GRα-C3 were the most sensitive to the apoptosis-inducing actions of glucocorticoids, whereas cells expressing GRα-D3 were the most resistant. Isoform-selective differences in the induction of the proapoptotic enzymes granzyme A and caspase-6 may account for the observed phenotype.
GR Control of Inflammation and Immune Response
There are two general mechanisms by which ligand-activated GR controls inflammation and the immune response. First, GR protects cells at sites of injury or inflammation from undergoing inflammation-induced apoptosis. This is accomplished by both positive and negative regulation of gene transcription and protein expression. GR stimulates production of antiinflammatory proteins such as secretory leukocyte protease inhibitor, IL-1 receptor antagonist, IL-10, and neutral endopeptidase.2 In addition, by regulating the activity of NF-κB, AP-1, and STATs, GR inhibits the expression of a variety genes important to the control of inflammation and the immune response, including proinflammatory cytokines (e.g., IL-2, IL-3, IL-6, and TNF-α), chemokines that attract inflammatory cells to sites of inflammation, nitric oxide synthase (NOS), and cyclooxygenase 2 (COX-2), among others.2 The second mechanism by which GR controls inflammation and the immune response is by inducing programmed cell death in immune cells that underlie inflammation. Glucocorticoids reduce the survival of eosinophils, T lymphocytes, mast cells, and dendritic cells.2 Although the mechanisms and target proteins responsible for GR-induced apoptosis are not well understood, GR-dependent control of transcription has been reported to be involved in the initiation of programmed cell death in lymphocytes.67
Glucocorticoid Receptor Resistance*
History
Vingerhoeds et al. first described generalized glucocorticoid resistance, also known as primary cortisol resistance, in 1976.68 It is quite rare; fewer than 30 separate probands have been reported.68–79 It is sometimes caused by functionally abnormal hGRs. The clinical presentation of glucocorticoid resistance varies from asymptomatic hypercortisolism to clinical syndromes of mineralocorticoid or adrenal androgen excess. Since the first description of a pathogenetic hGR mutation in 1990,80,81 nine other pathogenetic hGR mutations have been identified.69,77–79,82–85
Pathogenesis
GGR may be caused by abnormal hGR. This was first demonstrated in assays of hGR function.70–76 More recent studies have validated this expectation by identifying hGR mutations69,77–85 that decrease hGR-mediated gene transcription.
The clinical findings are a consequence of impaired hGR function. The HPA axis and its negative feedback regulation of cortisol production are described elsewhere. The glucocorticoid sensitivity of all tissues is decreased in GGR. The entire HPA axis is reset (Fig. 3-3). At the pituitary and hypothalamus, serum cortisol concentrations, which otherwise would be considered normal, are insufficient to suppress corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) secretion. Consequently, ACTH secretion is increased. ACTH stimulates the adrenal glands to produce greater-than-normal amounts of cortisol, adrenal androgens, and mineralocorticoids. In the peripheral tissues, the glucocorticoid resistance is equal to that of the pituitary and hypothalamus; however, sensitivity to androgens and mineralocorticoids is normal. Hence, the clinical findings are not those of glucocorticoid excess but rather those of mineralocorticoid or androgen excess. The glucocorticoid circadian rhythm and response to stress are maintained.
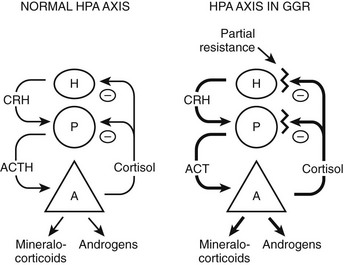
FIGURE 3-3 Hypothalamic-pituitary-adrenal (HPA) axis in normal and in generalized glucocorticoid resistance subjects. Normally, corticotropin-releasing hormone (CRH) from the hypothalamus (H) stimulates the pituitary (P) to produce adrenocorticotropic hormone (ACTH). ACTH stimulates the adrenal (A) to produce of mineralocorticoids, cortisol, and adrenal androgens. Cortisol inhibits (−) secretion of CRH and ACTH from the hypothalamus and pituitary, respectively. In GGR, there is partial blockade of the negative feedback at the pituitary and hypothalamus. This causes increased secretion of CRH and ACTH. ACTH stimulates the adrenal gland to make excess glucocorticoids, mineralocorticoids, and androgens. The HPA axis is qualitatively normal but quantitatively reset at higher hormone concentrations than normal. (Adapted from Javier EC, Reardon GE, Malchoff CD: Glucocorticoid resistance and its clinical presentations. Endocrinologist 1:141–148, 1991.)
The resistance to cortisol is partial, and plasma ACTH concentrations can be suppressed by high doses of exogenous glucocorticoids. Complete glucocorticoid resistance may be incompatible with life, as suggested by animal models.86
Glucocorticoid Receptor Abnormalities
Functionally abnormal hGRs with mutations in the primary hGR alpha structure cause GGR. However, some individuals with glucocorticoid resistance have apparently normal hGR primary structure.79,87,88 Functional changes include decreased receptor number,70,73 decreased receptor affinity for glucocorticoids,70–72,84,85 decreased DNA binding,74 thermolability,75 delayed translocation into the nucleus,89 and impaired coactivator interaction.78
No assay is completely sensitive for all hGR functional abnormalities. Binding of [3H] dexamethasone to fresh mononuclear leukocytes is used most commonly, but this is normal in some subjects.70 Dexamethasone induction of aromatase in cultured skin fibroblasts90 and dexamethasone suppression of mitogen-stimulated thymidine incorporation into mononuclear leukocytes70 have been used as assays for glucocorticoid resistance.
Pathogenetic mutations of the hGR gene have been found in some subjects. A splice-site microdeletion, which interferes with hGR messenger RNA (mRNA) processing and reduces the hGR number by 50%, causes glucocorticoid resistance in one kindred.69 Fig. 3-4 summarizes the nine missense point mutations that are known to cause glucocorticoid resistance. The original subject of Vingerhoeds et al., who presented with hypertension and hypokalemia, is homozygous for an aspartate-to-valine change at amino acid 641.82 The heterozygous I559N and I747M mutations cause more severe glucocorticoid resistance than would be expected for a single allele defect, suggesting a dominant negative effect.78,83,89 Most hGR mutations causing glucocorticoid resistance are located in the LBD and interfere with ligand binding.77,79–85 Only one has been described within the DNA-binding domain.79 Some hGR mutations are silent polymorphisms.87 Therefore, identification of an hGR gene mutation is not conclusive evidence of GGR. In some subjects, no hGR mutation is identified, and the cause remains unknown.79
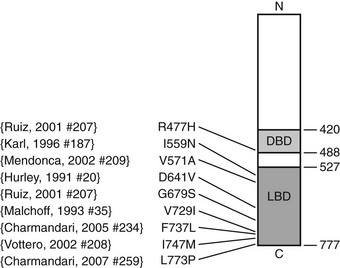
FIGURE 3-4 Diagrammatic representation of the human glucocorticoid receptor (hGR) point mutations in generalized glucocorticoid resistance. The DNA-binding domain (light gray) and ligand-binding domain (dark gray) are two of the major functional units of the receptor. Point mutations have been identified at the indicated amino acids (aa).77–79,81–85 Not shown is a splice-site microdeletion that disrupts processing of the hGR messenger RNA.69
At high concentrations, the beta isoform of the glucocorticoid receptor (hGRβ), which does not bind ligand, interferes with the action of the alpha isoform of the hGR.49 Increased expression of the hGRβ combined with markedly decreased expression of the hGR is suggested to be the cause of GGR in one subject.91
In large population studies, the relatively common R23K hGR polymorphism is associated with decreased glucocorticoid sensitivity,92,93 whereas the N363S polymorphism is associated with increased glucocorticoid sensitivity94 in peripheral tissues. This suggests that the effects of these polymorphisms are not precisely equal in the pituitary and in different peripheral tissues. In support of this hypothesis, the wild-type and N363S hGR were found to mediate different gene expression profiles, as determined by microarray analysis, even though they demonstrated similar glucocorticoid sensitivity in reporter gene assays.95
Clinical Features
The most common characteristic is hypercortisolism without features of glucocorticoid excess. It was this finding that led to the first description of the syndrome,68 and occasionally it is discovered during the investigation of other problems unrelated to glucocorticoid resistance.73 However, the diagnosis of glucocorticoid resistance may not be straightforward. Hypercortisolism is most frequently characterized by an elevated urinary cortisol excretion. However, this may not be a universal finding; some subjects demonstrate resistance only upon testing with dexamethasone.70 It may be difficult to differentiate glucocorticoid resistance from Cushing’s syndrome based on clinical features, since many clinical features of Cushing’s syndrome are nonspecific.
Although hypercortisolism without cushingoid features usually suggests the diagnosis, this constellation of features usually does not bring the patient to clinical attention. The presenting clinical features are secondary to excess androgens or mineralocorticoids. The most common clinical presentation is evidence of increased androgens in women. This was the presentation in four of the six probands described by Lamberts et al.70 and in two probands with hGR mutations described by Ruiz et al.79 Characteristics include hirsutism, acne, and menstrual irregularities. This presentation is caused by the increased adrenal androgens. In children, isosexual precocious pseudopuberty was the clinical presentation in a 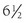 -year-old boy71 but has not been described in others. Although heterosexual precocious pseudopuberty has not been described in a girl, a female newborn did present with ambiguous genitalia.77 It is not clear why presentations in childhood are not more common. The clinical features may be overlooked because they are subtle. Infertility in men has been reported and is secondary to the increased adrenal androgen production and subsequent suppression of the hypothalamic-pituitary-testicular axis.83 Hypertension and hypokalemia were the presenting clinical findings of the first subject described by Vingerhoeds et al.68 Hypertension with severe thiazide-induced hypokalemia occurred in another patient.70 Other subjects have been found to be hypertensive without hypokalemia.70,73,74 In these latter cases, the contribution of GGR to the hypertension is less clear, since essential hypertension is common.
-year-old boy71 but has not been described in others. Although heterosexual precocious pseudopuberty has not been described in a girl, a female newborn did present with ambiguous genitalia.77 It is not clear why presentations in childhood are not more common. The clinical features may be overlooked because they are subtle. Infertility in men has been reported and is secondary to the increased adrenal androgen production and subsequent suppression of the hypothalamic-pituitary-testicular axis.83 Hypertension and hypokalemia were the presenting clinical findings of the first subject described by Vingerhoeds et al.68 Hypertension with severe thiazide-induced hypokalemia occurred in another patient.70 Other subjects have been found to be hypertensive without hypokalemia.70,73,74 In these latter cases, the contribution of GGR to the hypertension is less clear, since essential hypertension is common.
At least one subject with glucocorticoid resistance developed nodular adrenal hyperplasia.70 The frequency of this event in all affected subjects has not been ascertained.
In general, individuals with the greatest degree of hypercortisolism produce the most androgens and mineralocorticoids and subsequently have the most obvious clinical findings. Variability of clinical characteristics within a kindred may be due in part to a gene dose effect. Homozygotes are affected to a greater degree than heterozygotes. In addition, some mutations have a dominant negative effect, so that heterozygotes are more severely affected than expected for inactivation of a single allele.89 Women seem to be affected more frequently than men. This may be an ascertainment bias, since women are most likely to present with clinical features of increased adrenal androgens. It is not clear why androgen effects predominate in some patients, whereas the mineralocorticoid effects predominate in others.
One subject with severe GGR and a missense hGR mutation developed Cushing’s syndrome due to an ACTH-secreting pituitary adenoma.83 Presumably, the hGR mutation was the initial tumorigenic abnormality, which combined with other somatic pituitary gene mutations to effect the adenoma phenotype.
The possibility of tissue-specific or localized glucocorticoid resistance is in early stages of investigation. It does occur in some neoplasms. In ACTH-dependent Cushing’s syndrome, the pituitary or the ectopic ACTH source is well known to be more resistant to glucocorticoids than are the normal tissues. The molecular basis of this resistance is being probed; hGR mutations, gene deletions, or both, occur in some circumstances.83,94,96,97 Some neoplasms of hematopoietic origin demonstrate glucocorticoid resistance to apoptosis, and in some cell lines derived from these neoplasms, hGR mutations have been shown to cause glucocorticoid resistance.98,99 It is attractive to speculate that differences in tissue sensitivity to glucocorticoids could contribute to the development of clinical disorders such as glucocorticoid-resistant asthma,100 rheumatoid arthritis,101 obesity,102 hypertension,103 insulin resistance, and diabetes.104 It is also possible that infection with the HIV virus may increase105,106 or decrease107 glucocorticoid sensitivity. Studies of these possible relationships and their mechanisms are evolving.
Inheritance Pattern
Initial studies suggested an autosomal-dominant transmission of GGR. The inheritance pattern may vary depending upon the exact hGR mutation. It is clearly autosomal dominant in the kindred, with a splice-site microdeletion of one allele.69 In the kindred described by Vingerhoeds et al.68 and investigated further in collaboration with Loriaux and Chrousos,72,108 the proband, who is most severely affected, is homozygous for a mutation of the hGR ligand-binding domain. The son and nephew, who are mildly affected, are both heterozygous.68,72,82,108 Therefore, in this kindred, the disorder is recessive, or there is a gene dose effect. The hGR mutation in one family arose as a new mutation, so that both parents were unaffected.83 However, this individual had more severe glucocorticoid resistance than would be expected from an abnormality of a single allele, and subsequent in vitro studies indicate that the mutant receptor had a dominant negative effect.89 Extensive family studies noting genotype-phenotype correlations have not been performed. In summary, inheritance patterns vary, and biochemical investigation of familial members may not identify all the subjects carrying the mutant hGR allele.
Biochemical Characteristics
Hypercortisolism, usually measured as increased urinary cortisol excretion, was found in most affected individuals.68,70,72–74,76 Occasionally, urinary cortisol excretion is normal, and resistance to dexamethasone suppression is the only abnormality.70 In contrast, in the most severely affected patients, urinary cortisol excretion may exceed the upper-normal limit by 200-fold.68,70 This considerable variability suggests heterogeneity in the degree of resistance and in the pathogenetic receptor abnormality.
Hypercortisolism of GGR is distinguished from that of Cushing’s syndrome by the presence of a diurnal variation in serum cortisol. As discussed before, the HPA axis is qualitatively normal but quantitatively reset with higher-than-normal hormone concentrations. Serum cortisol concentrations usually decrease by at least 50% from 8 am to 8 pm in normal subjects109 and in glucocorticoid-resistant subjects,70–72 whereas in Cushing’s syndrome, there is loss of the diurnal variation.110 An example of the diurnal variation of serum cortisol reset at higher-than-normal concentrations in a subject with generalized glucocorticoid resistance is shown in Fig. 3-5.
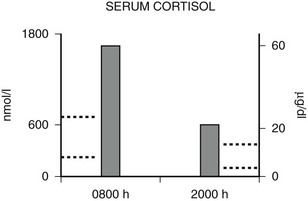
FIGURE 3-5 Diurnal variation of serum cortisol in a subject with generalized glucocorticoid resistance. Serum cortisol concentrations are represented in nanomoles per liter on the left ordinate and in micrograms per deciliter on the right ordinate. The 8 am and 8 pm serum cortisol concentrations from a patient with glucocorticoid resistance are shown. The results are the mean of three separate determinations. The normal range of serum cortisol at 8 am is shown by the dotted lines on the left ordinate, and the normal range at 8 pm is shown by the dotted lines on the right ordinate. (Adapted from Malchoff CD, Javier EC, Malchoff DM et al: Primary cortisol resistance presenting as isosexual precocity. J Clin Endocrinol Metab 70:503–507, 1990. © The Endocrine Society.)
Serum cortisol concentrations can be increased up to eightfold above normal but may be normal if the resistance is mild.68,70,72 Comparison of cortisol concentrations must be made with controls from the same time of day. The 8 am serum cortisol is normally not suppressed by 1 mg of dexamethasone given orally at 11 pm the previous evening.70 Cortisol production is ACTH-dependent, and plasma ACTH concentrations can be suppressed by supraphysiologic glucocorticoid doses.71,72 Plasma ACTH concentrations are less frequently increased above the normal range than are cortisol concentrations but tend to be the highest in individuals with the greatest resistance.
Adrenal androgens (androstenedione, dehydroepiandrosterone, and dehydroepiandrosterone sulfate) are increased in most children and women.68,70–74,76 These increases range from mild to fivefold above the upper normal limit, and they can be suppressed by high doses of glucocorticoids.
The mineralocorticoids have been less extensively studied. However, when they have been measured, deoxycorticosterone (DOC) and corticosterone are usually increased by two- to fivefold above the upper-normal limit, aldosterone concentrations are low, and plasma renin activity is suppressed.71–74 Production of DOC and corticosterone is ACTH-dependent.111 The high concentrations of DOC and/or cortisol cause hypokalemia and volume-dependent hypertension by activating the aldosterone receptor. Volume expansion suppresses plasma renin activity.
Diagnosis
It can be difficult to distinguish cortisol resistance from Cushing’s syndrome, since no single test is completely discriminatory. Individuals with glucocorticoid resistance lack clinical features of glucocorticoid excess. However, many features of glucocorticoid excess can be subtle, and others are nonspecific. The usual tests used to distinguish Cushing’s syndrome subjects from normal subjects do not distinguish Cushing’s syndrome subjects from GGR subjects. In both Cushing’s syndrome and GGR, urinary cortisol excretion is elevated, dexamethasone suppression tests are abnormal, and (presumably) late-night salivary cortisol will be elevated. Studies that demonstrate the qualitative integrity of the HPA axis help to distinguish Cushing’s syndrome from GGR. The diurnal rhythm is found in glucocorticoid resistance but is absent in Cushing’s syndrome (see Fig. 3-5). There is a normal response to hypoglycemic stress in GGR71 but not in Cushing’s syndrome.112 These tests have been useful in a limited number of patients, but cortisol concentrations in Cushing’s syndrome may vary throughout the day110 and mimic a diurnal variation. Clinical judgment and repeated evaluations over time are essential.
In children, the growth curves may help to distinguish subjects with GGR from those with Cushing’s syndrome. In Cushing’s syndrome, growth is slowed. Although the growth curves in most GGR subjects have not been carefully examined, it seems unlikely that growth would be slowed. In one GGR child presenting with isosexual precocious pseudopuberty, growth was accelerated compared to the expected normal rate. This accelerated growth was associated with advanced bone age, and both were caused by increased adrenal androgens.71
Other diagnostic considerations include increased cortisol-binding globulin (CBG) and pharmaceuticals that interfere with the cortisol assays. Increased CBG may imitate glucocorticoid resistance, since it causes high serum cortisol concentrations with a diurnal variation. Radioimmunoassays for CBG are commercially available, and CBG is normal in glucocorticoid resistance.71,72 Carbamazepine interferes with testing of the HPA axis in multiple ways. It increases the rate of dexamethasone metabolism, so patients may appear dexamethasone resistant. Patients taking carbamazepine demonstrate increased circulating cortisol concentrations and increased urinary cortisol excretion, although usually not out of the normal range, as occurs in glucocorticoid resistance.113 Finally, both carbamazepine and fenofibrate or their metabolites may interfere with some of the commercial high-performance liquid chromatography (HPLC) assays for cortisol and produce a false elevation.114,115
If the clinical and biochemical studies suggest the diagnosis of glucocorticoid resistance, then direct hGR functional studies may be confirmatory. As discussed, a number of functional receptor abnormalities have been reported.70–76 Unfortunately, these different studies require fresh or cultured mononuclear leukocytes or fibroblasts and are not commercially available. In addition, there may be other causes of abnormal ligand-binding assays. The apparent receptor number may be slightly decreased in Cushing’s syndrome,116 and in patients with AIDS, the hGR may have a decreased affinity for glucocorticoids.117 Finally, there are some glucocorticoid-resistant subjects with normal hGR ligand binding. Therefore, even this research tool is not completely sensitive or specific.
Treatment
The most common approach to therapy is the use of exogenous glucocorticoids (Fig. 3-6, strategy A). It has been used for patients with hypertension and hypokalemia,70,118 with sexual precocity,111 and for women with hirsutism.70,76 The goal is to suppress adrenal androgen and mineralocorticoid concentrations without using so much dexamethasone as to cause Cushing’s syndrome. Dexamethasone, which does not interfere with radioimmunoassays for cortisol, is chosen so that serum cortisol concentrations can be monitored and titrated into the normal range.70,76,111 With this therapy, androgens and mineralocorticoids fall, and the clinical features improve.70,76,111,118
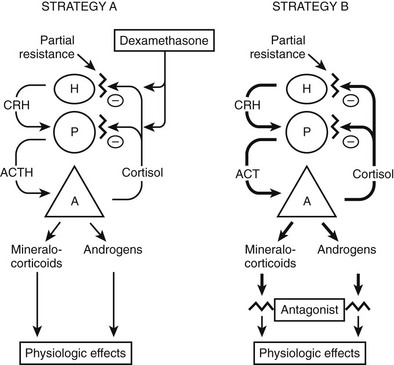
FIGURE 3-6 Treatment strategies for generalized glucocorticoid resistance. Two strategies have been proposed. In the first strategy (A), exogenous glucocorticoids (usually dexamethasone) are administered to decrease adrenocorticotropic hormone (ACTH) secretion and subsequently decrease the secretion of adrenal mineralocorticoids and androgens. The dose is adjusted to titrate cortisol into the normal range. In the second (B), mineralocorticoid and androgen antagonists block the peripheral effects of the hormones, producing clinical effects. CRH, Corticotropin-releasing hormone; H, hypothalamus; P, pituitary; A, adrenal gland. (Adapted from Malchoff CD, Malchoff DM, Reardon G: Glucocorticoid resistance. In Bardin CW (ed): Current Therapy in Endocrinology and Metabolism, 5th ed. St. Louis: Mosby-Year Book, 1994, pp 167–171.)
An alternative approach (see Fig. 3-6, strategy B) is to use mineralocorticoid or androgen antagonists. For example, hypertension and hypokalemia should be treatable with spironolactone, which blocks the mineralocorticoid receptor, or with amiloride, which blocks the sodium-potassium exchange in the distal tubule.
References
1. Rhen, T, Cidlowski, JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723.
2. Barnes, PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998;94:557–572.
3. Sapolsky, RM, Romero, LM, Munck, AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89.
4. Miner, JN, Hong, MH, Negro-Vilar, A. New and improved glucocorticoid receptor ligands. Expert Opin Investig Drugs. 2005;14:1527–1545.
5. Chrousos, GP, Kino, T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 48, 2005.
6. Evans, RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895.
7. Kumar, R, Thompson, EB. Gene regulation by the glucocorticoid receptor:structure:function relationship. J Steroid Biochem Mol Biol. 2005;94:383–394.
8. Duma, D, Jewell, CM, Cidlowski, JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21.
9. Pratt, WB, Toft, DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360.
10. Lu, NZ, Cidlowski, JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006;16:301–307.
11. Beato, M. Gene regulation by steroid hormones. Cell. 1989;56:335–344.
12. Freedman, LP. Anatomy of the steroid receptor zinc finger region. Endocr Rev. 1992;13:129–145.
13. Drouin, J, Sun, YL, Tremblay, S, et al. Homodimer formation is rate-limiting for high affinity DNA binding by glucocorticoid receptor. Mol Endocrinol. 1992;6:1299–1309.
14. McNally, JG, Muller, WG, Walker, D, et al. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265.
15. Lonard, DM, O’Malley, BW. Expanding functional diversity of the coactivators. Trends Biochem Sci. 2005;30:126–132.
16. Dostert, A, Heinzel, T. Negative glucocorticoid receptor response elements and their role in glucocorticoid action. Curr Pharm Des. 2004;10:2807–2816.
17. Meyer, T, Gustafsson, JA, Carlstedt-Duke, J. Glucocorticoid-dependent transcriptional repression of the osteocalcin gene by competitive binding at the TATA box. DNA Cell Biol. 1997;16:919–927.
18. Diamond, MI, Miner, JN, Yoshinaga, SK, et al. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249:1266–1272.
19. Malkoski, SP, Dorin, RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol. 1999;13:1629–1644.
20. Chandran, UR, Warren, BS, Baumann, CT, et al. The glucocorticoid receptor is tethered to DNA-bound Oct-1 at the mouse gonadotropin-releasing hormone distal negative glucocorticoid response element. J Biol Chem. 1999;274:2372–2378.
21. Martens, C, Bilodeau, S, Maira, M, et al. Protein-protein interactions and transcriptional antagonism between the subfamily of NGFI-B/Nur77 orphan nuclear receptors and glucocorticoid receptor. Mol Endocrinol. 2005;19:885–897.
22. Necela, BM, Cidlowski, JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc. 2004;1:239–246.
23. Newton, R, Holden, NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol. 2007;72:799–809.
24. De Bosscher, K, Vanden Berghe, W, Haegeman, G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522.
25. Caelles, C, Gonzalez-Sancho, JM, Munoz, A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 1997;11:3351–3364.
26. Smoak, K, Cidlowski, JA. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol Cell Biol. 2006;26:9126–9135.
27. Ogawa, S, Lozach, J, Benner, C, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721.
28. Nissen, RM, Yamamoto, KR. The glucocorticoid receptor inhibits NF-kappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000;14:2314–2329.
29. Rogatsky, I, Luecke, HF, Leitman, DC, et al. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci U S A. 2002;99:16701–16706.
30. Ito, K, Barnes, PJ, Adcock, IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903.
31. Rogatsky, I, Ivashkiv, LB. Glucocorticoid modulation of cytokine signaling. Tissue Antigens. 2006;68:1–12.
32. Lerner, L, Henriksen, MA, Zhang, X, et al. STAT3-dependent enhanceosome assembly and disassembly: synergy with GR for full transcriptional increase of the alpha 2-macroglobulin gene. Genes Dev. 2003;17:2564–2577.
33. Takeda, T, Kurachi, H, Yamamoto, T, et al. Crosstalk between the interleukin 6 (IL-6)-JAK-STAT and the glucocorticoid-nuclear receptor pathway: synergistic activation of IL-6 response element by IL-6 and glucocorticoid. J Endocrinol. 1998;159:323–330.
34. Zhang, Z, Jones, S, Hagood, JS, et al. STAT3 acts as a co-activator of glucocorticoid receptor signaling. J Biol Chem. 1997;272:30607–30610.
35. Stocklin, E, Wissler, M, Gouilleux, F, et al. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–728.
36. Stoecklin, E, Wissler, M, Moriggl, R, et al. Specific DNA binding of Stat5, but not of glucocorticoid receptor, is required for their functional cooperation in the regulation of gene transcription. Mol Cell Biol. 1997;17:6708–6716.
37. Lechner, J, Welte, T, Tomasi, JK, et al. Promoter-dependent synergy between glucocorticoid receptor and Stat5 in the activation of beta-casein gene transcription. J Biol Chem. 1997;272:20954–20960.
38. Hermoso, MA, Matsuguchi, T, Smoak, K, et al. Glucocorticoids and tumor necrosis factor alpha cooperatively regulate toll-like receptor 2 gene expression. Mol Cell Biol. 2004;24:4743–4756.
39. Cella, N, Groner, B, Hynes, NE. Characterization of Stat5a and Stat5b homodimers and heterodimers and their association with the glucocorticoid receptor in mammary cells. Mol Cell Biol. 1998;18:1783–1792.
40. Wyszomierski, SL, Yeh, J, Rosen, JM. Glucocorticoid receptor/signal transducer and activator of transcription 5 (STAT5) interactions enhance STAT5 activation by prolonging STAT5 DNA binding and tyrosine phosphorylation. Mol Endocrinol. 1999;13:330–343.
41. Biola, A, Lefebvre, P, Perrin-Wolff, M, et al. Interleukin 2 inhibits glucocorticoid receptor transcriptional activity through a mechanism involving STAT5 (signal transducer and activator of transcription 5) but not AP-1. Mol Endocrinol. 2001;15:1062–1076.
42. De Miguel, F, Lee, SO, Onate, SA, et al. Stat3 enhances transactivation of steroid hormone receptors. Nucl Recept. 2003;1:3.
43. Encio, IJ, Detera-Wadleigh, SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991;266:7182–7188.
44. Francke, U, Foellmer, BE. The glucocorticoid receptor gene is in 5q31-q32 [corrected]. Genomics. 1989;4:610–612.
45. Oakley, RH, Sar, M, Cidlowski, JA. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem. 1996;271:9550–9559.
46. Theriault, A, Boyd, E, Harrap, SB, et al. Regional chromosomal assignment of the human glucocorticoid receptor gene to 5q31. Hum Genet. 1989;83:289–291.
47. Hollenberg, SM, Weinberger, C, Ong, ES, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641.
48. Oakley, RH, Webster, JC, Sar, M, et al. Expression and subcellular distribution of the beta-isoform of the human glucocorticoid receptor. Endocrinology. 1997;138:5028–5038.
49. Bamberger, CM, Bamberger, AM, de Castro, M, et al. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95:2435–2441.
50. Lewis-Tuffin, LJ, Cidlowski, JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9.
51. Oakley, RH, Jewell, CM, Yudt, MR, et al. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274:27857–27866.
52. Hauk, PJ, Hamid, QA, Chrousos, GP, et al. Induction of corticosteroid insensitivity in human PBMCs by microbial superantigens. J Allergy Clin Immunol. 2000;105:782–787.
53. Orii, F, Ashida, T, Nomura, M, et al. Quantitative analysis for human glucocorticoid receptor alpha/beta mRNA in IBD. Biochem Biophys Res Commun. 2002;296:1286–1294.
54. Strickland, I, Kisich, K, Hauk, PJ, et al. High constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med. 2001;193:585–593.
55. Tliba, O, Cidlowski, JA, Amrani, Y. CD38 expression is insensitive to steroid action in cells treated with tumor necrosis factor-alpha and interferon-gamma by a mechanism involving the up-regulation of the glucocorticoid receptor beta isoform. Mol Pharmacol. 2006;69:588–596.
56. Webster, JC, Oakley, RH, Jewell, CM, et al. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A. 2001;98:6865–6870.
57. Goecke, IA, Alvarez, C, Henriquez, J, et al. Methotrexate regulates the expression of glucocorticoid receptor alpha and beta isoforms in normal human peripheral mononuclear cells and human lymphocyte cell lines in vitro. Mol Immunol. 2007;44:2115–2123.
58. Derijk, RH, Schaaf, MJ, Turner, G, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388.
59. Schaaf, MJ, Cidlowski, JA. AUUUA motifs in the 3′ UTR of human glucocorticoid receptor alpha and beta mRNA destabilize mRNA and decrease receptor protein expression. Steroids. 2002;67:627–636.
60. Syed, AA, Irving, JA, Redfern, CP, et al. Association of glucocorticoid receptor polymorphism A3669G in exon 9beta with reduced central adiposity in women. Obesity (Silver Spring). 2006;14:759–764.
61. van den Akker, EL, Nouwen, JL, Melles, DC, et al. Staphylococcus aureus nasal carriage is associated with glucocorticoid receptor gene polymorphisms. J Infect Dis. 2006;194:814–818.
62. van den Akker, EL, Koper, JW, van Rossum, EF, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168:33–39.
63. Lewis-Tuffin, LJ, Jewell, CM, Bienstock, RJ, et al. Human glucocorticoid receptor β binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27:2266–2282.
64. Lu, NZ, Cidlowski, JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–342.
65. Yudt, MR, Cidlowski, JA. Molecular identification and characterization of a and b forms of the glucocorticoid receptor. Mol Endocrinol. 2001;15:1093–1103.
66. Lu, NZ, Collins, JB, Grissom, SF, et al. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol Cell Biol. 2007;27:7143–7160.
67. Distelhorst, CW. Recent insights into the mechanism of glucocorticosteroid-induced apoptosis. Cell Death Differ. 2002;9:6–19.
68. Vingerhoeds, A, Thijssen, J, Shwarz, F. Spontaneous hypercortisolism without Cushing’s syndrome. J Clin Endocrinol Metab. 1976;43:1128–1133.
69. Karl, M, Lamberts, SWJ, Detera-Wadleigh, SD, et al. Familial glucocorticoid resistance caused by a splice site deletion in the human glucocorticoid receptor gene. J Clin Endocrinol Metab. 1993;76:683–689.
70. Lamberts, SWJ, Koper, JW, Biemond, P, et al. Cortisol receptor resistance: the variability of its clinical presentation and response to treatment. J Clin Endocrinol Metab. 1992;74:313–321.
71. Malchoff, C, Javier, E, Malchoff, D, et al. Primary cortisol resistance presenting as isosexual precocity. J Clin Endocrinol Metab. 1990;70:503–507.
72. Chrousos, GP, Vingerhoeds, ACM, Brandon, D, et al. Primary cortisol resistance in man: a glucocorticoid receptor-mediated disease. J Clin Invest. 1982;69:1261–1269.
73. Iida, S, Gomi, M, Moriwaki, K, et al. Primary cortisol resistance accompanied by a reduction in glucocorticoid receptors in two members of the same family. J Clin Endocrinol Metab. 1985;60:967–971.
74. Nawata, H, Sekiya, K, Higuchi, K, et al. Decreased deoxyribonucleic acid binding of glucocorticoid-receptor complex in cultured skin fibroblasts from a patient with glucocorticoid resistance syndrome. J Clin Endocrinol Metab. 1987;65:219–226.
75. Bronnegard, M, Werner, S, Gustafsson, J. Primary cortisol resistance associated with a thermolabile glucocorticoid receptor in a patient with fatigue as the only symptom. J Clin Invest. 1986;78:1270–1278.
76. Lamberts, SWJ, Poldermans, D, Zweens, M, et al. Familial cortisol resistance: differential diagnostic and therapeutic aspects. J Clin Endocrinol Metab. 1986;63:1328–1333.
77. Mendonca, B, Leite, M, de Castro, M, et al. Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab. 2002;87:1805–1809.
78. Vottero, A, Kino, T, Combe, H, et al. A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab. 2002;87:2658–2667.
79. Ruiz, M, Lind, U, Gafvels, M, et al. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol. 2001;55:363–371.
80. Brufsky, A, Malchoff, D, Javier, E, et al. A glucocorticoid receptor mutation in a subject with primary cortisol resistance. Trans Assoc Am Phys. 1990;103:53–63.
81. Malchoff, DM, Brufsky, A, Reardon, G, et al. A point mutation of the human glucocorticoid receptor in primary cortisol resistance. J Clin Invest. 1993;91:1918–1925.
82. Hurley, D, Accili, D, Stratakis, C, et al. Point mutation causing a single amino acid substitution in the hormone binding domain of the glucocorticoid receptor in familial glucocorticoid resistance. J Clin Invest. 1991;87:680–686.
83. Karl, M, Lamberts, SW, Koper, JW, et al. Cushing’s disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Phys. 1996;108:296–307.
84. Charmandari, E, Raji, A, Kino, T, et al. A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of HGR LBD in conferring transactivational activity. J Clin Endocrinol Metab. 2005;90:3696–3705.
85. Charmandari, E, Kino, T, Ichijo, T, et al. A novel pint mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol and Metab. 2007;92:3986–3990.
86. Cole, TJ, Blendy, JA, Monaghan, AP, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621.
87. Koper, JW, Stolk, RP, de Lange, P, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet. 1997;99:663–668.
88. Huizenga, N, de Lange, P, Koper, J, et al. Five patients with biochemical and/or clinical generalized glucocorticoid resistance without alterations in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 2000;85:2076–2081.
89. Kino, T, Stauber, RH, Resau, JH, et al. Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: importance of the ligand-binding domain for intracellular GR trafficking. J Clin Endocrinol Metab. 2001;86:5600–5608.
90. Berkovitz, GD, Carter, KM, Levine, MA, et al. Abnormal induction of aromatase activity by dexamethasone in fibroblasts from a patient with cortisol resistance. J Clin Endocrinol Metab. 1990;70:1608–1611.
91. Shahadi, H, Vottero, A, Stratakis, C, et al. Imbalanced expression of the glucocorticoid receptor isoforms in cultured lymphocytes from a patient with systemic glucocorticoid resistance and chronic lymphocytic leukemia. Biochem Biophys Res Commun. 1999;254:559–565.
92. van Rossum, E, Koper, J, Huizenga, N, et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes. 2002;51:3128–3134.
93. van Rossum, E, Voorhoeve, P, te Velde, S, et al. The ER2/23EK polymorphism in the glucocorticoid receptor gene is associated with a beneficial body composition and muscle strength in young adults. J Clin Endocrinol and Metab. 2004;89:4004–4009.
94. Huizenga, NA, de Lange, P, Koper, JW, et al. Human adrenocorticotropin-secreting pituitary adenomas show frequent loss of heterozygosity at the glucocorticoid receptor gene locus. J Clin Endocrinol Metab. 1998;83:917–921.
95. Jewell, CM, Cidlowski, JA. Molecular evidence for a link between the N363S glucocorticoid receptor polymorphism and altered gene expression. J Clin Endocrinol Metab. 2007;92:3268–3277.
96. Karl, M, Von Wichert, G, Kempter, E, et al. Nelson’s syndrome associated with a somatic frame shift mutation in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 1996;81:124–129.
97. Dahia, PL, Honegger, J, Reincke, M, et al. Expression of glucocorticoid receptor gene isoforms in corticotropin-secreting tumors. J Clin Endocrinol Metab. 1997;82:1088–1093.
98. Strasser-Wozak, EM, Hattmannstorfer, R, Hala, M, et al. Splice site mutation in the glucocorticoid receptor gene causes resistance to glucocorticoid-induced apoptosis in a human acute leukemic cell line. Cancer Res. 1995;55:348–353.
99. Hala, M, Hartmann, BL, Bock, G, et al. Glucocorticoid-receptor gene defects and resistance to glucocorticoid-induced apoptosis in human leukemic cell lines. Int J Cancer. 1996;68:663–668.
100. Sher, ER, Leung, DY, Surs, W, et al. Steroid resistant asthma. Cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J Clin Invest. 1994;93:33–39.
101. Schlaghecke, R, Kornely, E, Wollenhaupt, J, et al. Glucocorticoid receptors in rheumatoid arthritis. Arthritis Rheum. 1992;35:740–744.
102. Huizenga, NA, Koper, JW, de Lange, P, et al. A polymorphism in the glucocorticoid receptor gene may be associated with an increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab. 1998;83:144–151.
103. Panarelli, M, Holloway, CD, Fraser, R, et al. Glucocorticoid receptor polymorphism, skin vasoconstriction, and other metabolic intermediate phenotypes in normal human subjects. J Clin Endocrinol Metab. 1998;83:1846–1852.
104. Nyirenda, MJ, Lindsay, RS, Kenyon, CJ, et al. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest. 1998;101:2174–2181.
105. Guo, WX, Antakly, T, Cadotte, M, et al. Expression and cytokine regulation of glucocorticoid receptors in Kaposi’s sarcoma. Am J Pathol. 1996;148:1999–2008.
106. Refaeli, Y, Levy, DN, Weiner, DB. The glucocorticoid receptor type II complex is a target of the HIV-1 vpr gene product. Proc Natl Acad Sci U S A. 1995;92:3621–3625.
107. Norbiato, G, Bevilacqua, M, Vago, T, et al. Glucocorticoids and interferon-alpha in the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1996;81:2601–2606.
108. Chrousos, G, Vingerhoeds, A, Loriaux, D, et al. Primary cortisol resistance: a family study. J Clin Endocrinol Metab. 1983;56:1243–1245.
109. Rivest, R, Schulz, P, Lustenberger, S, et al. Differences between circadian and ultradian organization of cortisol and melatonin rhythms during activity and rest. J Clin Endocrinol Metab. 1989;68:721–729.
110. Van Cauter, E, Refetoff, S. Evidence for two subtypes of Cushing’s disease based on the analysis of episodic cortisol secretion. N Engl J Med. 1985;312:1343–1349.
111. Malchoff, CD, Reardon, G, Javier, EC, et al. Dexamethasone therapy for isosexual precocious pseudopuberty caused by generalized glucocorticoid resistance. J Clin Endocrinol Metab. 1994;79:1632–1636.
112. Besser, G, Edwards, C. Cushing’s syndrome. Clin Endocrinol Metab. 1972;1:451–490.
113. Perini, G, Devinsky, O, Hauser, P, et al. Effects of carbamazepine on pituitary-adrenal function in health volunteers. J Clin Endocrinol Metab. 1992;74:406–412.
114. Findling, J, Pinkstaff, SM, Shaker, J, et al. Pseudohypercortisoluria: spurious elevation of urinary cortisol due to carbamazepine. Endocrinologist. 1998;8:51–54.
115. Meikle, A, Findling, J, Kushnir, M, et al. Pseudo-Cushing’s syndrome caused by fenofibrate interference with urinary cortisol assayed by high-performance liquid chromatography. J Clin Endocrinol Metab. 2003;88:3521–3524.
116. Kontula, K, Pelkonen, R, Andersson, L, et al. Glucocorticoid receptors in adrenocorticoid disorders. J Clin Endocrinol Metab. 1980;51:654–657.
117. Norbiato, G, Bevilacqua, M, Vago, T, et al. Cortisol resistance in acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1992;74:608–613.
118. Lipsett, M, Tomita, M, Brandon, D, et al. Cortisol resistance in man. Adv Exp Med Biol. 1986;196:97–110.
*The section titled “Glucocorticoid Receptors” was written by Robert H. Oakley, Laura J. Lewis-Tuffin, and John A. Cidlowski.
*The section titled “Glucocorticoid Receptor Resistance” was written by Carl D. Malchoff and Diana Malchoff.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 3
Glucocorticoid Receptors
Their Mechanisms of Action and Glucocorticoid Resistance
Robert H. Oakley, Laura J. Lewis-Tuffin, Carl D. MaLchoff, Diana Mark Malchoff and John A. Cidlowski
Glucocorticoid Receptors*
Glucocorticoids are a class of endogenous and synthetic steroid hormones that affect virtually every aspect of human physiology. Currently they are one of the most commonly prescribed classes of drugs.1 The physiologic actions of glucocorticoids include highly effective antiinflammatory and immunomodulatory actions that are exploited for the treatment of diseases such as asthma, arthritis, allergic rhinitis, and leukemia/lymphoma.2,3 In addition, glucocorticoids have important roles in development of the lung and nervous systems, skeletal growth, behavior, reproduction, and intermediary metabolism. Ultimately, glucocorticoids act to maintain homeostasis in the face of stressful stimuli. The broad actions of glucocorticoids account for the serious side effects commonly experienced with chronic glucocorticoid treatment. These include the development of glucocorticoid resistance in diseased tissues, osteoporosis, growth retardation in children, muscle atrophy, and signs of the metabolic syndrome.1,4,5 The physiologic and pharmacologic actions of glucocorticoids are mediated by the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of proteins that regulate gene transcription in a ligand-dependent manner.6 In this section, we review the basic mechanisms of glucocorticoid action with an emphasis on how these mechanisms contribute to the antiinflammatory and immunomodulatory effects of glucocorticoids.
The Glucocorticoid Receptor
Like other members of the nuclear receptor superfamily, GR is a modular protein comprising an amino-terminal transactivation domain (NTD); a central, two-zinc-finger DNA-binding domain (DBD); and a carboxyl-terminal ligand-binding domain (LBD) (Fig. 3-1A).7 Separating the DBD and LBD is a flexible region of the molecule termed the hinge region. The receptor contains two regions involved in activating transcription, one in the NTD (AF1) which can act independently of ligand binding, and a second in the LBD (AF2) whose function is dependent on glucocorticoid binding. The LBD also possesses a nuclear localization signal and sites for interaction with other transcription factors, coregulators, and protein chaperones. An additional nuclear localization signal spans the junction of the DBD and hinge region. The GR protein is also a substrate for various types of posttranslational modifications including phosphorylation, ubiquitination, sumoylation, and acetylation that regulate GR signaling by modulating the expression level and/or transcriptional activity of the receptor.8
FIGURE 3-1 Glucocorticoid receptor (GR) domain structure and signaling pathway. A, GR is composed of an amino-terminal transactivation domain (NTD), DNA-binding domain (DBD), hinge region (H), and ligand-binding domain (LBD). Regions involved in transactivation (AF1 and AF2), dimerization, nuclear localization, and hsp90 binding are indicated. The location of residues posttranslationally modified by phosphorylation (P) (Ser-113, Ser-141, Ser-203, Ser-211, and Ser-226); ubiquitination (U) (Lys-419); sumoylation (S) (Lys-277, Lys-293, and Lys-703); and acetylation (A) (Lys-494 and Lys-495) are also depicted. Numbers are for the human glucocorticoid receptor. B, The unliganded GR resides in the cytoplasm of cells in a complex with chaperone proteins. Upon binding glucocorticoids (), the receptor undergoes a change in conformation, dissociates from accessory proteins, and translocates into nucleus via the nuclear pore complex (NPC). Nuclear GR regulates gene expression in three primary ways: (a) GR binds directly to DNA and enhances or inhibits transcription of target genes. (b) GR interacts with DNA-bound transcription factors without itself binding to DNA and enhances or inhibits transcription of target genes. (c) GR binds directly to DNA and interacts with transcription factors bound to neighboring sites and enhances or inhibits transcription of target genes. BTM, Basal transcription machinery; TBP, TATA-box binding protein.
GR Regulation of Transcription via Direct Binding to DNA
Unliganded GR is located in the cytoplasm of cells as part of a multiprotein complex that includes two molecules of heat shock protein 90 (hsp90) (see Fig. 3-1B).9 These chaperone proteins maintain the receptor in a transcriptionally inactive state that favors high-affinity ligand binding. Upon binding glucocorticoids, GR undergoes a conformational change resulting in the dissociation of hsp90, exposure of the nuclear localization signals, and translocation of the receptor into the nucleus via the nuclear pore. The receptor then regulates gene transcription by binding directly to specific DNA sequences known as glucocorticoid response elements (GREs) and/or by binding other transcription factors and modulating their activity. Global gene expression analyses indicate that up to 20% of the genome is induced or repressed by glucocorticoids.5,10
The consensus GRE is an imperfect palindrome, GGTACAnnnTGTTCT, that is usually found in the promoter region of target genes.11,12 The interaction of ligand-bound GR with the GRE stimulates the transcription of numerous genes, including the metabolic enzymes tyrosine amino transferase, phosphoenolpyruvate carboxykinase, and glucose-6-phosphatase (see Fig. 3-1B[a], upper scheme). High-affinity GRE binding requires receptor dimerization and is short lived because the receptor rapidly cycles on and off target sites every few seconds.13,14 Upon binding DNA, GR undergoes a conformational change that results in the coordinated recruitment of three general types of coactivators necessary for stimulating transcription of the target gene: the ATP-dependent complex BRG1 (SWI/SNF) that mediates large noncovalent disruptions in chromatin structure; CBP, p300, and members of the SRC/p160 family of proteins that modify chromatin structure locally through their intrinsic histone acetyltransferase activity; and components of the DRIP/TRAP complex that assist in the recruitment of the basal transcription machinery.15 The alterations in chromatin structure result in DNA unwinding, thereby permitting promoter access to transcription factors and cofactors that enhance target gene expression. The specific coactivators recruited by GR determine the gene induction profile, and this assembly is dependent on the promoter context, the bound glucocorticoid, and the availability and activity of the coactivators themselves.
Negative GREs (nGREs) are the transcription-repressing counterpart of positive GREs.16 These response elements bear little resemblance to positive GREs, are highly variable, and a consensus sequence has not been determined. How ligand-bound GR represses transcription via nGREs is unclear and likely involves multiple mechanisms dependent upon the promoter context. For some genes, such as osteocalcin, DNA-bound GR may sterically interfere with binding of positively acting transcription factors to elements that overlap the nGRE (see Fig. 3-1B[a], lower scheme).17 The situation is more complex for other repressed genes, since both binding to DNA and interacting with neighboring transcription factors appear to be required of the receptor (see Fig. 3-1B[c], lower scheme). Referred to as a composite GRE, nGREs of this nature are found in the promoters of the proliferin and corticotropin-releasing hormone (CRH) genes. Here, DNA-bound GR interacts with the activator protein-1 (AP-1) transcription factor occupying an adjacent site to repress transcription.18,19 Finally, since nGREs correspond poorly to the GRE consensus sequence, negative regulation may be achieved at some target genes by GR interacting with DNA-bound positive transcription factors without itself binding to the promoter. This “tethering” mechanism of repression (described in more detail later) appears to mediate the glucocorticoid-dependent inhibition of gonadotropin-releasing hormone receptor and pro-opiomelanocortin expression via the interaction of GR with the promoter-bound Oct1 and Nur77 transcription factors, respectively.20,21
GR Regulation of Transcription via Protein-Protein Interactions
In addition to transcriptional regulation by direct binding to DNA, ligand-bound GR can also interact with other transcription factors and modulate their activity on glucocorticoid-responsive promoters without itself binding to DNA. The two most studied examples of this form of regulation involve the transcription factors AP-1 and nuclear factor κB (NF-κB). These two proteins are central mediators of the inflammatory and immune responses, and their inhibition by GR is thought to underlie the major antiinflammatory and immunosuppressive actions of glucocorticoids.22,23 When activated by stress signals such as proinflammatory cytokines, bacterial and viral infectious agents, or pro-apoptotic stimuli, AP-1 and NF-κB bind their cognate response elements and induce the expression of many proinflammatory genes, including cytokines, cell adhesion molecules, and enzymes involved in tissue destruction. Glucocorticoids indirectly antagonize the actions of AP-1 and NF-κB at multiple levels by inducing the expression of other regulatory proteins. Activated GR stimulates expression of the IκB protein, which sequesters NF-κB in the cytoplasm,24 MAPK phosphatase 1, which dephosphorylates c-Jun N-terminal kinase to prevent activation of AP-1,25 and tristetraprolin, which destabilizes the mRNA of many AP-1 and NF-κB-induced genes.26
The primary way, however, by which hormone-bound GR represses the activity of AP-1 and NF-κB is through a direct interaction with the c-Jun subunit of AP-1 and the p65 subunit of NF-κB (see Fig. 3-1B[b], lower scheme).22,23 Interestingly, the antagonism is reciprocal; the association of these proteins with GR represses its activity on target genes. Multiple mechanisms appear to underlie the antagonism, suggesting the mode of action may vary in a promoter, cell-type, and/or signal-dependent manner. In early studies, the receptor was reported to sequester AP-1 and NF-κB in the cytoplasm and/or nucleus and prevent DNA binding. More recent findings, however, suggest the receptor is tethered to DNA-bound AP-1 and NF-κB and alters the subsequent assembly and/or activity of recruited transcriptional proteins. For example, on several toll-like receptor genes, GR represses NF-κB by disrupting the interaction of p65 with the promoter-specific coactivator IRF3 (interferon regulatory factor 3).27 At the interleukin 8 (IL-8) and intercellular adhesion molecule 1 (ICAM-1) promoters, the association of GR with NF-κB impairs the phosphorylation of the C-terminal domain of RNA polymerase II.28 GR-mediated repression of the AP-1-responsive collagenase-3 promoter and the NF-κB-responsive IL-8 promoter have both been shown to be potentiated by receptor-dependent recruitment of the coactivator GRIP1, indicating coactivators can actually function as corepressors in the appropriate configuration with GR.29 Finally, GR has been reported to interact with histone deacetylase 2 (HDAC2) and to repress the histone acetyltransferase activity of NF-κB, suggesting that GR antagonizes NF-κB by effects on chromatin structure.30
In contrast to AP-1 and NF-κB, the physical association of GR with the signal transducer and activator of transcription (STAT) family of proteins enhances their activity on target genes.31 STAT transcription factors are activated by a range of cytokines through induction of the Janus kinase pathway (JAK) and tyrosine phosphorylation. Upon binding their cognate response elements, STATs regulate genes involved in the immune response, differentiation, survival, and apoptosis. GR has been shown to interact with several members of the STAT family, including STAT3 and STAT5, and to synergistically enhance their activity on target genes in a promoter-dependent fashion.31 The association of GR with STAT3 at the γ-fibrinogen and α2-macroglobulin promoters, which lack identifiable GREs, super-induces their expression (see Fig. 3-1B[b], upper scheme).32–34 Similarly, the synergistic activation of the β-casein and toll-like receptor-2 genes results from the association of GR with promoter-bound STAT5.35–38 However, the observed synergy at these STAT5-responsive genes may also require GR binding to DNA and more accurately reflect a composite GRE (see Fig. 3-1B[c], upper scheme).37,38 How GR synergistically activates STAT-regulated genes is poorly understood but may involve GR enhancing STAT nuclear localization,39 prolonging the promoter occupancy of STAT by inhibiting its tyrosine dephosphorylation32,40 and/or promoting the co-utilization of certain coactivators.31 Interestingly, the synergism of these two transcription factors is not always mutual, since GR activity is inhibited or stimulated depending on the particular STAT isoform employed.34,35,41,42
Multiple GR Isoforms Derived from Single Gene
The human GR gene is located on chromosome 5q31-32 and comprises 9 exons.43–46 Alternative splicing in exon 9 generates two receptor isoforms, termed GRα and GRβ, that are identical through to amino acid 727 but then diverge at their carboxyl-termini (Fig. 3-2).45–47 The classic, full-length GRα contains an additional 50 amino acids, whereas GRβ has an additional, nonhomologous 15 amino acids. Because of its unique carboxyl-terminus, GRβ does not bind glucocorticoids and resides constitutively in the nucleus of cells.45,48 GRβ has been shown to function as a dominant negative inhibitor and repress the transcriptional activity of GRα45,49–51; therefore, alterations in GRβ expression may contribute to changes in glucocorticoid responsiveness. Indeed, expression of GRβ is selectively increased over GRα in cells exposed to proinflammatory cytokines and microbial superantigens, leading to reduced sensitivity to glucocorticoids.52–56 In addition, glucocorticoid-resistant forms of inflammatory diseases (e.g., asthma, rheumatoid arthritis, and ulcerative colitis) have been associated with elevated expression of GRβ.50 Conversely, methotrexate, an effective drug for treating autoimmune and inflammatory diseases, promotes a selective increase in GRα at the expense of GRβ and improves glucocorticoid sensitivity of lymphocytes.57
FIGURE 3-2 Alternative processing of the single glucocorticoid receptor (GR) gene generates multiple GR isoforms. The human GR gene comprises 9 exons. Alternative splicing at the 3′ end of the primary transcript generates GRα and GRβ mRNAs, which encode GRα and GRβ proteins differing only at their carboxyl-terminus. Alternative translation initiation from 8 different AUG start codons derived from exon 2 generates additional protein isoforms with progressively shorter NTDs. Numbers shown denote the first and last residues for the human GR isoforms. For simplicity, only the most proximal of 9 alternate exon 1s (1H) is shown.
Elevated GRβ levels also result from a naturally occurring ATTTA to GTTTA polymorphism (A3669G) in the 3′ untranslated region of GRβ. This nucleotide substitution disrupts an mRNA destabilization motif and leads to increased stability of the GRβ mRNA and enhanced protein expression.58,59 The A3669G allele has been associated with reduced central obesity in women and a more favorable lipid profile in men,60 suggesting the increase in GRβ might antagonize some of the undesirable effects of GRα on fat distribution and lipid metabolism. The A3669G-directed rise in GRβ may also compromise the immunosuppressive actions of GRα. Persons harboring the A3669G allele have an elevated risk of the autoimmune disease rheumatoid arthritis and a reduced risk of bacterial nasal infection.58,61 Moreover, A3669G carriers exhibit an increased risk of myocardial infarction and coronary heart disease, two pathologies with inflammatory underpinnings.62
A broader role for GRβ in cell signaling has recently emerged with the demonstration that this isoform can modulate gene expression apart from its effects on GRα. In a genome-wide microarray analysis performed in cells selectively expressing GRβ, the isoform was found to alter the expression of over 5000 genes.63 Less than 20% of the genes were commonly regulated by ligand-activated GRα, indicating that GRβ possesses its own unique gene-regulatory profile. GRβ was also shown to bind the glucocorticoid antagonist mifepristone (RU486), and binding of this ligand abolished most of the GRβ-mediated changes in gene expression.63 As a bona fide transcription factor, GRβ may contribute to alterations in glucocorticoid responsiveness in healthy and diseased tissues by genomic effects independent of its dominant negative activity on GRα.
Alternative translation initiation of the GR mRNA produces an additional cohort of receptor subtypes (see Fig. 3-2).10,64,65 Eight GR isoforms with progressively shorter NTDs are generated from one GRα mRNA transcript via different AUG start codons: GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, and GRα-D3. GRα-A is the classic, full-length 777 amino acid protein that is generated from the first initiator AUG codon. The GRβ mRNA also contains the identical start codons and would be expected to give rise to a similar complement of subtypes. The GRα translational isoforms show a widespread tissue distribution; however, the relative levels of the subtypes differ both between and within tissues.64,66 Functionally, the isoforms bind glucocorticoids with similar affinity and bind GREs with similar capacity.66 Additionally, all eight isoforms occupy the nucleus of cells following glucocorticoid treatment. In the absence of hormone, however, the subcellular distribution of the subtypes differ, with the GRα-D isoforms residing predominantly in the nucleus and the others predominantly in the cytoplasm.64
Marked differences have also been reported in the transcriptional properties of the GRα translational isoforms.64,66 On GRE-containing reporter and endogenous genes, the GRα-C3 isoform was the most active stimulating gene expression, whereas the GRα-D subtypes were the most deficient. These isoform-selective effects have been attributed to differences among the subtypes in their ability to recruit various transcriptional factors and coregulators, such as CBP and RNA polymerase II, to the promoter. In contrast to these divergent effects on gene induction, no significant differences have been observed so far in the ability of the GRα isoforms to repress NF-κB.66 In a whole-genome microarray analysis performed on U2OS osteosarcoma cells selectively expressing the individual receptor isoforms, each subtype was found to regulate both a common and a unique set of genes.66 Of the approximately 6500 genes regulated by at least one GRα isoform, less than 500 were regulated commonly by all the receptor isoforms. Thus the majority of glucocorticoid-responsive genes were selectively regulated by different GRα subtypes. These isoform-unique gene regulatory profiles were further shown to produce functional differences in cellular responsiveness to glucocorticoids; the GRα translational isoforms exhibited distinct capabilities to induce apoptosis.66 Cells expressing GRα-C3 were the most sensitive to the apoptosis-inducing actions of glucocorticoids, whereas cells expressing GRα-D3 were the most resistant. Isoform-selective differences in the induction of the proapoptotic enzymes granzyme A and caspase-6 may account for the observed phenotype.
GR Control of Inflammation and Immune Response
There are two general mechanisms by which ligand-activated GR controls inflammation and the immune response. First, GR protects cells at sites of injury or inflammation from undergoing inflammation-induced apoptosis. This is accomplished by both positive and negative regulation of gene transcription and protein expression. GR stimulates production of antiinflammatory proteins such as secretory leukocyte protease inhibitor, IL-1 receptor antagonist, IL-10, and neutral endopeptidase.2 In addition, by regulating the activity of NF-κB, AP-1, and STATs, GR inhibits the expression of a variety genes important to the control of inflammation and the immune response, including proinflammatory cytokines (e.g., IL-2, IL-3, IL-6, and TNF-α), chemokines that attract inflammatory cells to sites of inflammation, nitric oxide synthase (NOS), and cyclooxygenase 2 (COX-2), among others.2 The second mechanism by which GR controls inflammation and the immune response is by inducing programmed cell death in immune cells that underlie inflammation. Glucocorticoids reduce the survival of eosinophils, T lymphocytes, mast cells, and dendritic cells.2 Although the mechanisms and target proteins responsible for GR-induced apoptosis are not well understood, GR-dependent control of transcription has been reported to be involved in the initiation of programmed cell death in lymphocytes.67
