[level-membership-for-pathology-category]
Glaucoma
Normal Anatomy (Figs. 16.1–16.3)
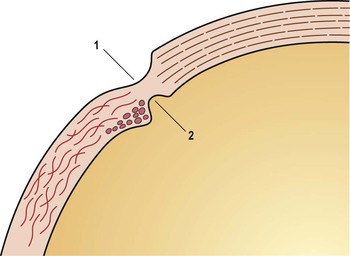
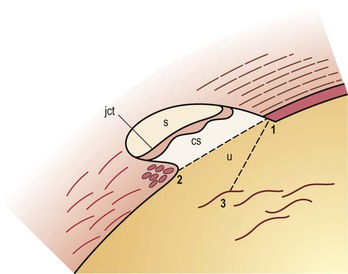
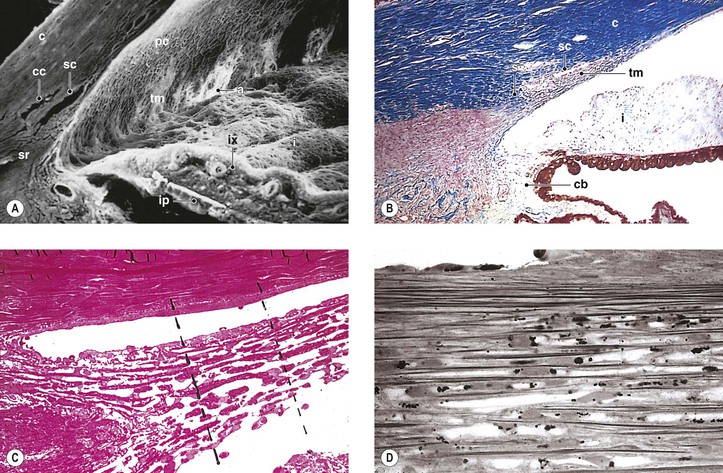
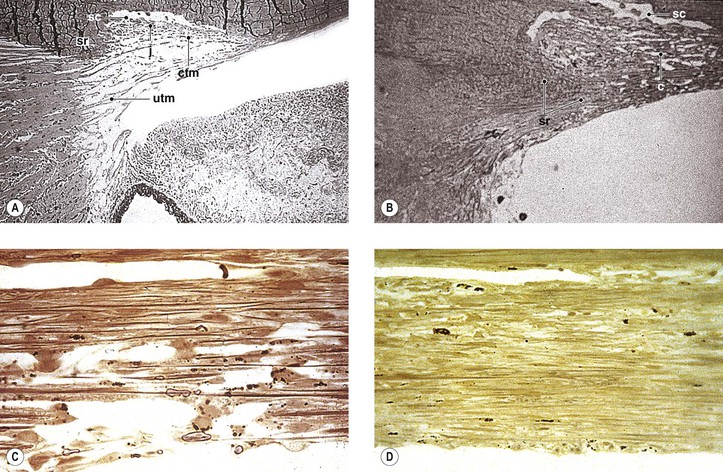
I. The outermost or corneoscleral layer of the eye can be separated into corneal and scleral portions by two circumferential grooves—a shallow outer one, the outer scleral sulcus, and a deeper inner one, the inner scleral sulcus.
C. Deep within this inner sulcus and applied closely to the collagenous tissue of the corneosclera lies the large vessel called the canal of Schlemm.
3. The inner wall rests on a thinner or patchy basement membrane that is associated with a zone of delicate connective tissue, the juxtacanalicular connective tissue.
4. Pores are present in the wall of Schlemm’s canal.
B. The meshwork may be easily and usefully separated into two parts by an imaginary line extending from the scleral roll to the end of Descemet’s membrane (see Fig. 16.2).
Introduction
The cell and molecular biology and gene rearrangement aspects of the glaucomas are fascinating, but an in-depth analysis of these matters is beyond the scope of our discussion. For example, genome-wide expression profiling of patients with primary open-angle glaucoma (POAG) identified 563 genes that were significantly dysregulated in POAG compared with normal controls. These genes impacted numerous functions, including nucleoside, nucleotide, and nucleic acid metabolism; the mitogen-activated protein kinase kinase kinase (MAPKKK) cascade; apoptosis; protein synthesis; cell cycle; intracellular signaling cascade; and nervous system development and function. Therefore, we highlight only a few salient facts regarding these areas.
Currently, 15 chromosome loci, which are designated GLC1A to GLC10, are associated with POAG. Candidate genes include myocilin (GLC1A), WD40-repeat36 (GLC1G), optineurin (GLC1E), and neurotrophin-4 (NTF-4) (optineurin is discussed with normal pressure glaucoma later in this chapter). Nevertheless, it has been estimated that mutations in known glaucoma genes account for less than 15% of cases. The prevalence of heterozygous cytochrome p450 1B1 (CY1B1) gene changes in a mixed glaucoma population suggests that the pathogenesis of glaucoma is genetically heterogeneous and may be polygenic. Single nucleotide polymorphisms (SNPs) between the CAV1 and CAV2 genes on chromosome 7q31 code for two members of the caveolin family of proteins are associated with POAG in white Americans, particularly women. These proteins impact modulation of endothelial cell membranes, which could alter the process of ocular aqueous fluid drainage. In general, however, SNPs are not discussed in any detail, but they have been described elsewhere (see Bibliography). Gene copy number variations also may play a role in the development of POAG. For example, homozygous deletions that reduce galactosylceramidase activity may increase the risk of POAG.
I. Glaucoma is characterized by an intraocular pressure (IOP) sufficient to produce ocular tissue damage, either transient or permanent.
A. Glaucoma is a “family” of diseases having in common a type of optic atrophy called optic nerve head cupping or excavation.
B. Although most individuals associate glaucoma with an elevated IOP, the pressure may, in fact, be within the statistically “normal” range and still cause ocular tissue damage in normal-tension (improperly called low-tension) glaucoma.
1. IOP is a risk factor for glaucoma, and the higher the pressure, the greater the probability of the development of the disorder.
a. The accurate measurement of IOP is vital to the proper diagnosis and treatment of glaucoma.
c. CCT is increased in children with ocular hypertension.
2. Normal-tension glaucoma probably accounts for approximately one-third of all cases of POAG.
Optineurin
Currently, 15 gene loci, designated GLC1A to GLC10, are associated with POAG. The optinuerin gene is associated with several disorders, including glaucoma, amyotrophic lateral sclerosis, other neurodegenerative disorders, and Paget’s disease of bone. A glaucoma-causing gene has been identified at GLC1E, and sequence variations in this optineurin (OPTN) gene on GLC1E have been found to be associated with the development of normal-tension glaucoma. The gene is located on chromosome 10. The glaucoma associated with optineurin is not characterized by marked IOP elevation. Rather, the E50K mutation in the optineurin gene is associated with increased severity of normal-tension glaucoma in white and Latino populations. There may be racial differences in glaucoma-associated optineurin genotypes. Its primary effect may be to increase susceptibility of retinal ganglion cells to premature cell death. Thus, optineurin may serve an optic nerve protective effect that is lost through mutation. Optineurin gene alterations do not appear to have a significant role in typical POAG.
II. Glaucoma suspect
B. POAG accounts for approximately two-thirds of all glaucoma seen in white patients.
Impaired Outflow
Congenital Glaucoma
A. The rate of congenital glaucoma is from 1 : 5000 to 1 : 10,000 live births.
C. Approximately 60–70% of affected children are boys.
D. The disease is bilateral in 64–88% of cases.
II. Pathogenesis (many theories)
C. An “embryonic” anterior chamber angle that results from faulty cleavage of tissue during embryonic development of the eye prevents the aqueous from leaving the anterior chamber.
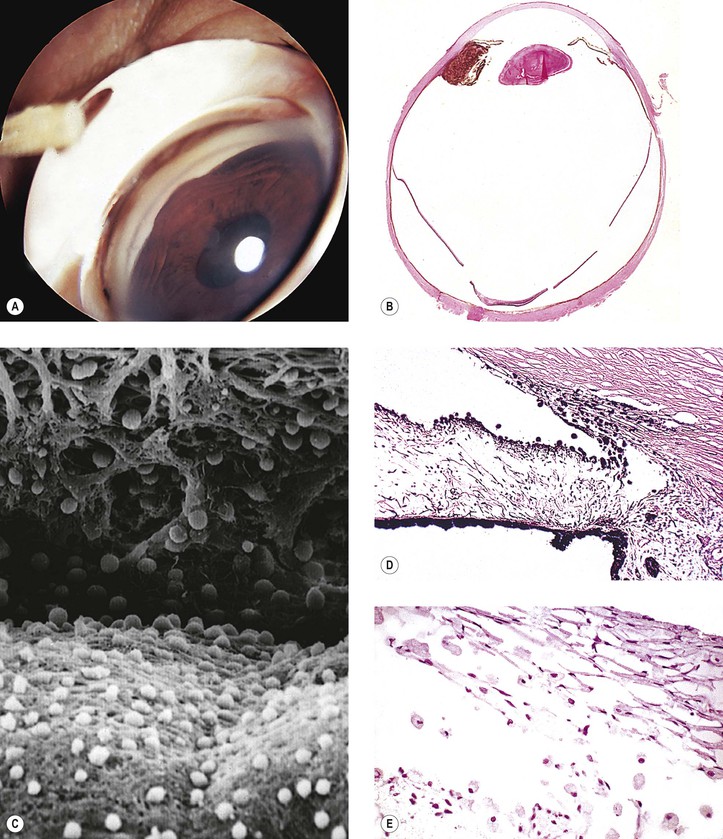
1. Histologically, the angle shows an anterior “insertion” of the iris root, anteriorly displaced ciliary processes, insertion of the ciliary meridional muscles into the trabecular meshwork instead of into (or over) the scleral roll, and mesenchymal tissue in the anterior chamber angle (Fig. 16.4).
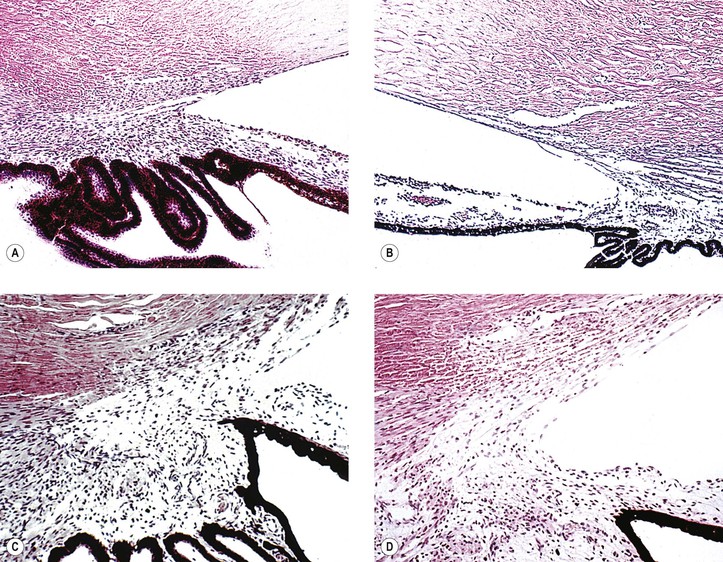
2. Many nonglaucomatous infant eyes show a similar anterior chamber angle structure.
3. To interpret angle histology accurately, it is necessary to study truly meridional sections through the anterior chamber angle.
a. Tangential sectioning makes interpretation difficult (see Figs. 16.4C and 16.4D).
D. The actual cause or causes of congenital glaucoma probably remain unknown.
III. Associated diseases and conditions
A. Iris anomalies (see Chapter 9)
1. Hypoplasia of the iris (“aniridia”) and iris coloboma may be associated with congenital glaucoma.
a. The PAX6 point mutation defect (1630A > T) on band p13 of chromosome 11 has been associated with some cases of aniridia. PAX6 mutations result in alterations in corneal cytokeratin expression, cell adhesion, and glycoconjugate expression. There is also corneal stem cell deficiency, which contributes to associated keratopathy.
B. Axenfeld’s anomaly and Rieger’s syndrome (see Chapter 8)
C. Peters’ anomaly (see Chapters 2 and 8). Peters’ anomaly and primary congenital glaucoma may share a common molecular pathophysiology. Both of these disorders can be associated with mutation in the cytochrome P4501B1 (CYP1B1) gene (discussed previously).
D. Phakomatoses
1. Sturge–Weber syndrome (see Chapter 2)
2. Neurofibromatosis (see Chapter 2)
E. Lowe’s syndrome (see Chapter 10)
H. Marfan’s syndrome (see Chapter 10)
I. Homocystinuria (see Chapter 10)
J. Microcornea (see Chapter 8)
K. Spherophakia (see Chapter 10)
L. Chromosomal abnormalities (e.g., trisomy 13; see Chapter 2)
M. Persistent hyperplastic primary vitreous (see Chapter 18)
N. Retinopathy of prematurity (see Chapter 18)
O. Retinoblastoma (see Chapter 18)
P. Juvenile xanthogranuloma (see Chapter 9)
V. Neurofibromatosis type 1 should be excluded in newborns with unilateral congenital glaucoma.
IV. Secondary histologic ocular effects in young eyes (<10 years of age)
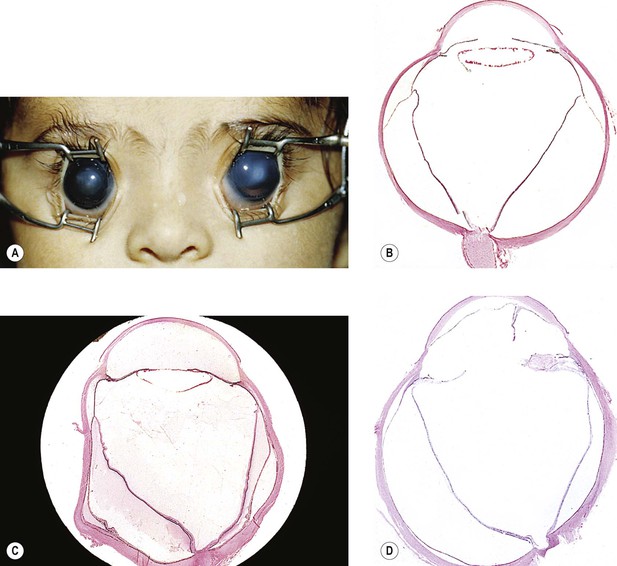
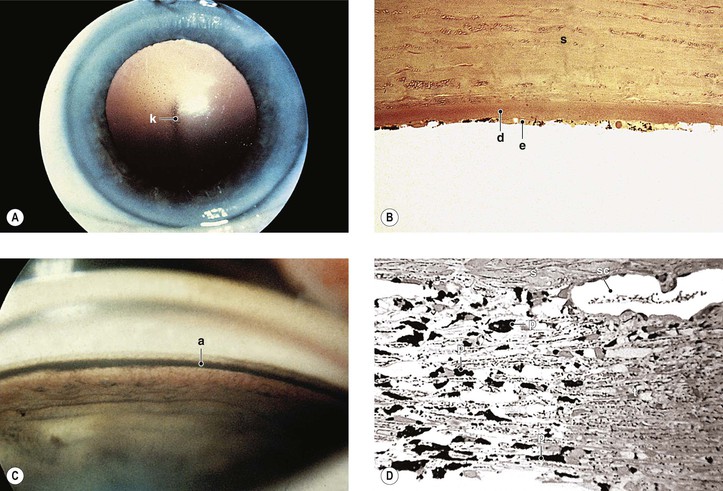
A. Buphthalmos (“large eye”) is caused by an enlargement, stretching, and thinning of the coats of the eye, especially marked in the anterior segment, resulting in a deep anterior chamber (Fig. 16.5). Subluxated lenses may develop in these enlarged eyes.
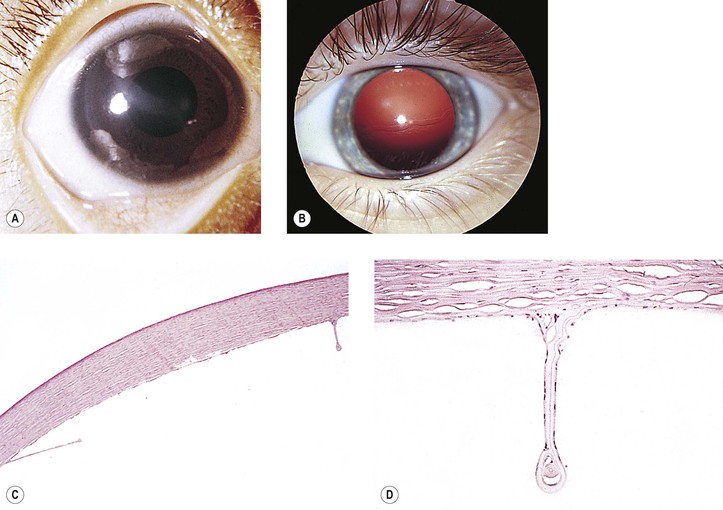
B. Ruptures of Descemet’s membrane (Haab’s striae) may be found in the enlarged corneas (Fig. 16.6), are usually horizontal in the central cornea but concentric toward the limbus, are located mainly in the lower half, and are often associated with corneal edema.
C. The limbal region becomes stretched and thin, with a resultant limbal ectasia (see Fig. 16.5).
E. Continued high IOP may cause atrophy of the ciliary body, choroid, and retina; cupping of the optic disc (see Fig. 16.5); and atrophy of the optic nerve.
Primary Glaucoma (Closed- and Open-Angle)
II. Optical coherence tomography and ultrasound biomicroscopy can be utilized to evaluate the anatomic configuration of the anterior chamber angle and adjacent structures for the classification of the pathophysiology of the glaucomas.
A. Specific features with such imaging studies that may contribute to angle-closure include:
3. Smaller anterior chamber width, area, and volume
4. Dynamic increase or lesser reduction in iris volume during dilation
5. Choroidal expansion accompanying angle-closure
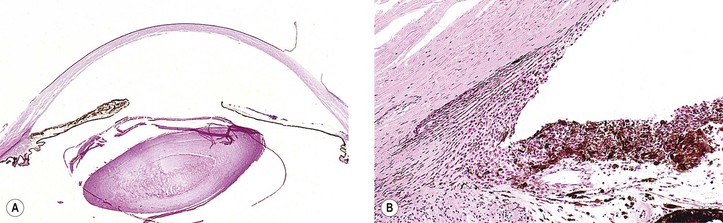
III. Closed-angle (narrow-angle; angle-closure; acute congestive) glaucoma (Figs. 16.7 and 16.8)
A. In anatomically predisposed eyes, primary closed-angle glaucoma develops.
2. Small, hypermetropic eyes are especially vulnerable to angle-closure.
A founder gene effect is probably related to two families of the Faroe Islands with hereditary high hyperopia, angle-closure glaucoma, uveal effusion, cataract, esotropia, and amblyopia.
3. The lens is normal-sized or large.
C. A sudden rise in IOP results from the peripheral iris being in apposition to the filtering trabecular meshwork from the pupillary block mechanisms except in the plateau iris syndrome, which in its pure form does not involve pupillary block.
The association of typical acute closed-angle glaucoma with increasing age is due, in part, to progressive pupillary block from the increase in lens size as it adds layers of lens fibers over time.
F. Histology
b. Segmental iris atrophy is usually seen in the upper half of the iris in a sector configuration.
2. Irregular pupil results from necrosis of the dilator and sphincter muscles.
a. Histologically, segments of the dilator muscle or its entire length are absent.
b. The sphincter muscle shows varying degrees of atrophy.
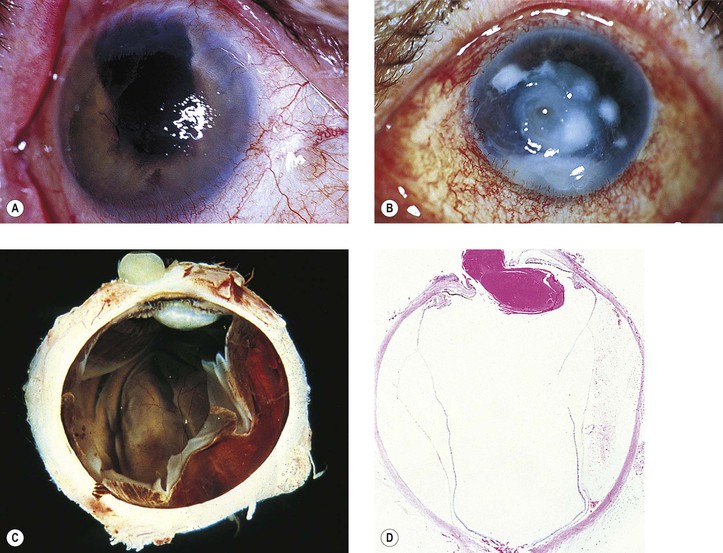
3. Glaukomflecken (cataracta disseminata subcapsularis glaukomatosa; see Fig. 16.8)
a. Glaukomflecken probably results from interference with the normal metabolism of the anterior lens cells due to a stagnation of aqueous humor that contains toxic products of necrosis or from foci of pressure necrosis (see Chapter 10).
b. Anterior subcapsular, multiple, tiny gray-white lenticular opacities are seen.
b. Irreversible vision impairment after an acute attack is mainly caused by optic nerve damage.
G. Failure to reverse the initial attack of angle closure can result in chronic angle-closure glaucoma.
1. This entity can be confused clinically with chronic open-angle glaucoma unless careful gonioscopy is performed routinely on all suspected cases of glaucoma.
Chronic angle-closure glaucoma is associated with the necessity of continued treatment and subsequent procedures even in the presence of a patent iridotomy.
2. Histology
There is disorganization of the trabecular architecture, narrowing or loss of trabecular spaces, scarring of the trabecular beams, trabecular endothelial cell loss, deposition of banded fibrillar material, and melanin pigment deposition.
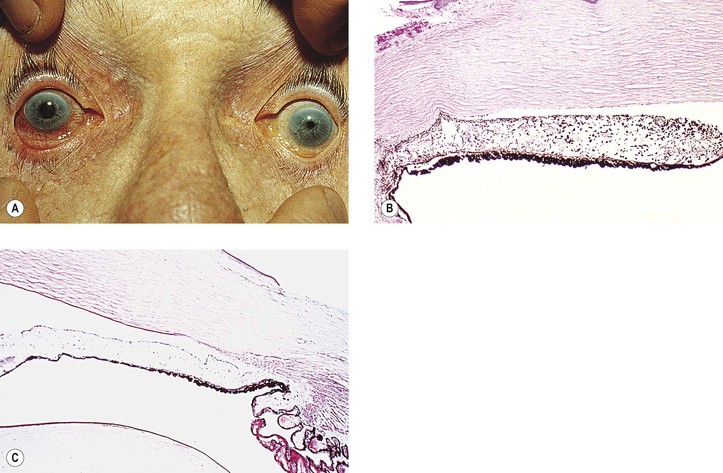
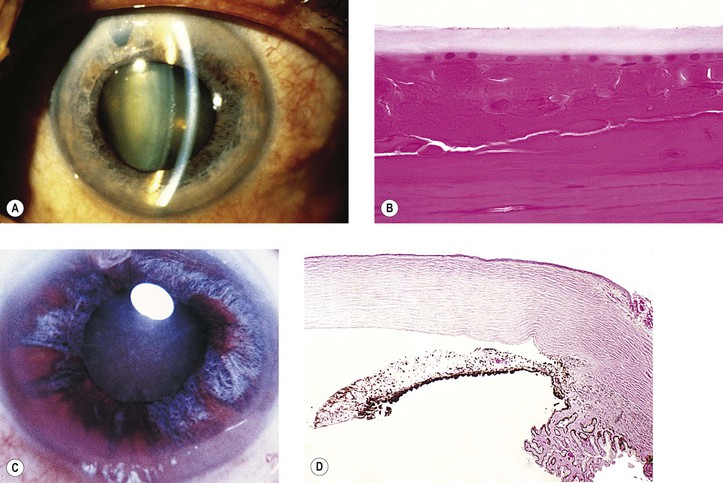
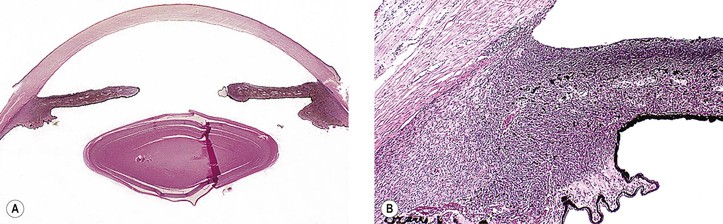
IV. Chronic open-angle (chronic simple) glaucoma (POAG) (Figs. 16.9 and 16.10)
A. The angle appears normal gonioscopically.
C. The condition is most often bilateral.
1. Glaucoma may develop in one eye months to years before the fellow eye.
2. One eye may be more severely affected than the other eye.
D. Prevalence (see section Introduction)
1. In most cases, the condition is probably inherited as an autosomal-recessive trait.
2. Myocilin
Myocilin was the first gene to be associated with glaucoma. Myocilin is the most frequently mutated gene in POAG patients worldwide. There are myocilin genotype–phenotype correlations, such as age of diagnosis, maximum IOP, and response to medical therapy. Mutations in the myocilin gene are present in 1–4% of POAG patients but not in patients with pseudoexfoliation. Myocilin gene is located on chromosome 1, localized to the olfactomedin domain, and was the first gene identified for POAG in the GLC1A locus. Its associated glaucoma is transmitted as an autosomal-dominant trait. There appears to be compromised stability of the protein, which categorizes the resulting disorder as a disease of protein misfolding. There is strong evidence that myocilin polymorphisms are related to POAG susceptibility with significant racial variation such that for whites Q368x is most important, whereas T353I is not important for Asians. The Gln48His mutation is unique to Indian patients. There is a low prevalence of myocilin mutations in African-Americans with POAG.
F. Normal-tension (improperly called low-tension) glaucoma is a subdivision of POAG (see section Introduction in this chapter).
G. Histology and pathophysiology
1. Optic nerve (see later in this chapter)
2. Little is known about the early histologic changes that take place in the region of the drainage angle because the number of human eyes that have well-characterized early open-angle glaucoma available for histologic examination is small (see Fig. 16.9).
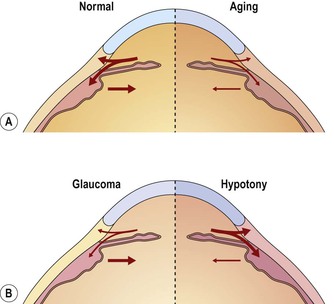
4. Aging changes in the drainage angle of the anterior chamber
d. The degree of obstruction is a quantitative problem; two extremes of excessive obstruction are:
e. The two major aging changes—each may be seen in almost pure form, or they may be combined.
1) They may also be associated with proliferations of trabecular endothelial cells in the meshwork.
2) COAG depends on the amount of aqueous inflow and the degree of obstruction to outflow caused by the aging changes (see Fig. 16.10).
f. Other changes in the trabecular meshwork in POAG include loss of cells, increased accumulation of extracellular matrix, changes in cytoskeleton, cellular senescence, and the process of subclinical inflammation.
Oxidative stress is likely to be one important mechanism in the pathogenesis of POAG.
Secondary Closed-Angle Glaucoma
Causes
I. Chronic primary angle-closure glaucoma
B. Histologically, peripheral anterior synechiae are seen, sometimes broad based.
II. Phacomorphic
V. Iridocorneal endothelial (ICE) syndrome
1. All three entities share a basic corneal endothelial defect and iris involvement.
3. Glaucoma occurs in approximately 50% of cases.
5. Each variant is described separately, but all are considered to belong to the ICE syndrome.
B. Iris nevus syndrome (Figs. 16.11 and 16.12)
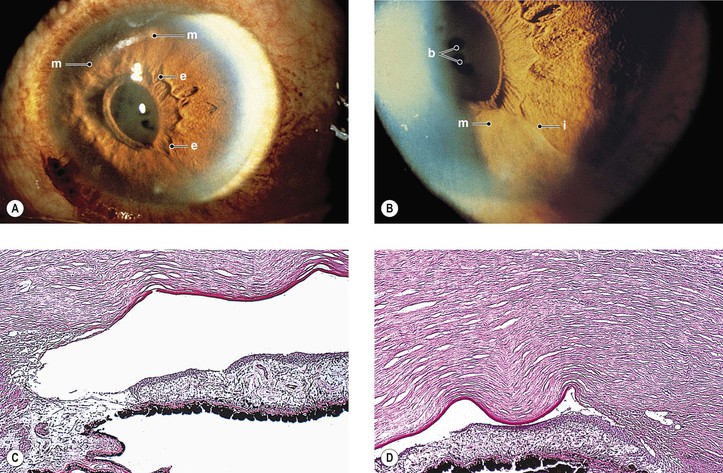
C. Chandler’s syndrome (Fig. 16.13)
2. Endothelial dystrophy causes corneal edema to develop at a slightly elevated or normal IOP.
3. Small peripheral anterior synechiae and mild pupillary distortion are found.
4. Small areas of iris stromal thinning may be found, but through-and-through holes are rare.
5. Histology
a. The iris stroma is atrophic.
b. In the areas of iris hole formation, the stroma and pigment epithelium are absent.
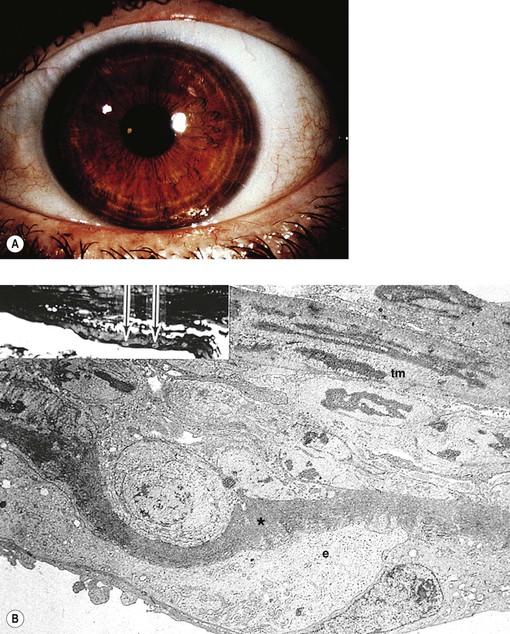
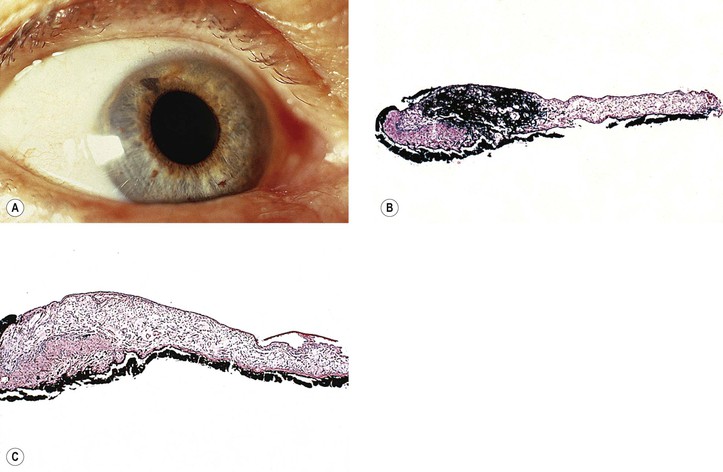
E. Essential iris atrophy (Fig. 16.14)
1. Essential iris atrophy is usually unilateral, is found most often in women, and is of unknown cause.
2. The onset is usually in the third decade.
3. Corneal edema often develops when IOP is slightly elevated or even normal.
The corneal endothelium shows a fine, hammered-silver appearance, similar to cornea guttata but less coarse.
4. The initial event is the formation of a peripheral anterior synechia distorting the pupil to that side.
a. The pupil becomes more distorted, sometimes with the development of an ectropion uveae.
b. Through-and-through holes develop in the iris, usually opposite to the distorted pupil.
5. Peripheral anterior synechiae increase circumferentially, and an intractable glaucoma develops.
Corneal endothelial overgrowth is a feature common to all three variants included in the ICE syndrome.
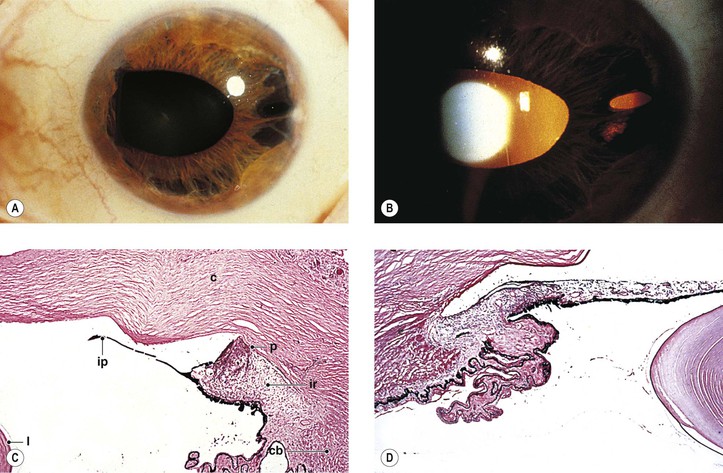
B. The pupil is not displaced and remains reactive.
C. The anterior iris stromal layers separate widely from the deeper layers, resembling spaghetti.
1. The lower half of the iris is most frequently involved.
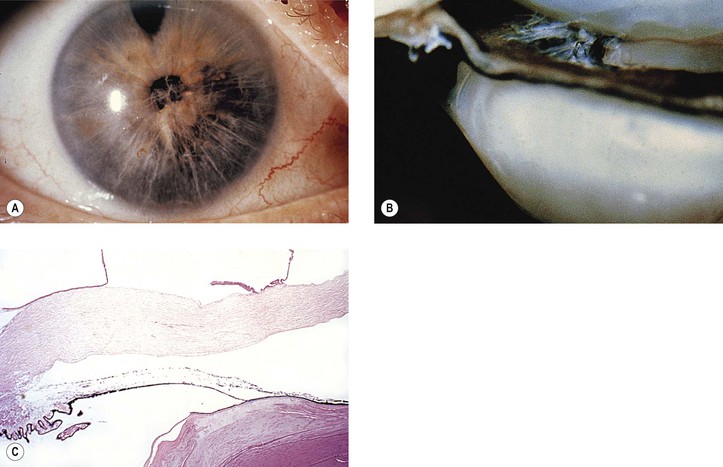
VII. Anterior uveitis (see Figs. 3.11 and 3.12)—anterior uveitis from any cause [e.g., trauma, infection, “allergy,” sympathetic uveitis (phacoanaphylactic endophthalmitis)] may result in posterior synechiae, iris bombé, and, finally, peripheral anterior synechiae.
VIII. Retinopathy of prematurity
B. Closed-angle glaucoma may result, sometimes years after the initial damage.
IX. Spherophakia (Weill–Marchesani syndrome; see Chapter 10)
X. Persistent hyperplastic primary vitreous (see Chapter 18)
A. Repeated hemorrhages result in organization and iridocorneal synechiae.
B. Less often, swelling of the lens or iris bombé can produce a closed angle.
XI. Epithelialization of anterior chamber angle (see Chapter 5)
XII. Endothelialization of anterior chamber angle (see Fig. 5.34)
XIII. Neovascularization of anterior surface of the iris (clinically termed rubeosis iridis; see Figs. 9.13, 9.14, and 15.5)
The many causes include diabetes mellitus, central retinal vein or artery occlusion, branch retinal artery occlusion, diffuse retinal vascular disease, carotid artery ischemia, retinoblastoma, malignant melanoma of uvea (Table 16.2), long-standing retinal detachment, any chronic retinal disease, penetrating or contusive ocular injuries, metastatic tumors to the retina and vitreous including cutaneous melanoma, and Fuchs’ heterochromic iridocyclitis (see Chapter 3).
TABLE 16.2
Histopathologic Mechanisms Producing Secondary Glaucoma in Eyes Containing Uveal Malignant Melanomas

(Modified from Yanoff M: Glaucoma mechanisms in ocular malignant melanomas. Am J Ophthalmol 70:898. © Elsevier 1970.)
XIV. Cysts of iris and anterior ciliary body (see Figs. 9.9 and 9.10)
A. Multiple cysts of the iris and ciliary epithelium can cause both secondary acute and chronic closed-angle glaucoma.
B. Histologically, a proliferation of the posterior layer of the iris pigment epithelium or of the inner layer of ciliary epithelium lines the cyst (see Chapter 9).
XV. Juvenile xanthogranuloma (see Chapter 9)
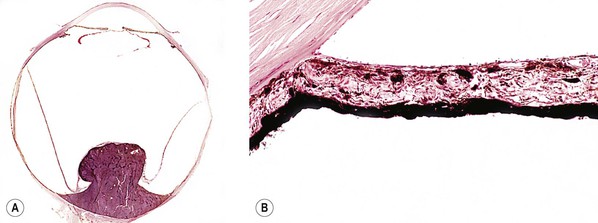
XVI. Secondary to uveal malignant melanoma (Figs. 16.16–16.18)
A. Posterior synechiae and iris bombé (see Fig. 16.16)
A large posterior malignant melanoma and a total neural retinal detachment may combine to displace the iris lens diaphragm anteriorly, resulting in posterior synechiae and iris bombé followed by secondary peripheral anterior synechiae. Similar changes may occur with a large posterior metastatic neoplasm.
B. Neovascularization of iris (see Fig. 16.17)
Neovascularization of the iris may occur with a large posterior choroidal malignant melanoma and cause peripheral anterior synechias. Similar changes may occur with a large posterior metastatic lesion.
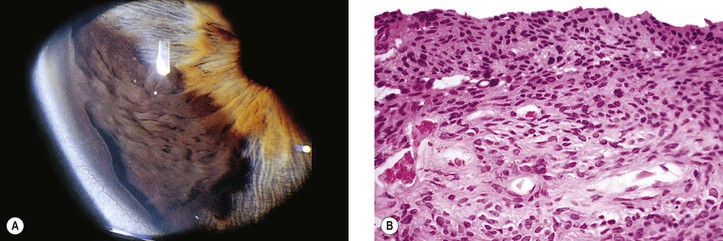
C. Diffuse iris malignant melanoma (see Fig. 16.18)
A diffuse iris malignant melanoma, or even a diffuse iris nevus, may induce peripheral anterior synechiae, although diffuse melanomas do not usually present with such changes. The condition may simulate the ICE syndrome.
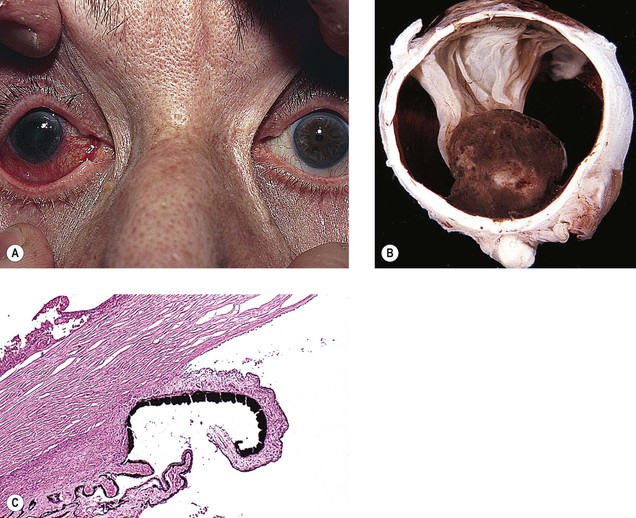
XIX. Snake bite is an unusual cause of bilateral angle-closure glaucoma.
Secondary Open-Angle Glaucoma
I. Secondary to cells or debris in angle
B. Uveitis
1. Cyclitis (or iridocyclitis) may lead to excessive cellular production that obstructs the open angle.
2. Glaucomatocyclitic crisis (Posner–Schlossman syndrome)
c. Little or no reaction occurs in the aqueous humor, and the angle usually appears normal.
C. Phacolytic glaucoma (see Chapter 10)
D. Nondenatured lens material-induced glaucoma usually follows a very recent traumatic rupture of the lens.
1. If glaucoma develops after needling of a soft cataract, it occurs within the first week.
2. After penetrating ocular injury
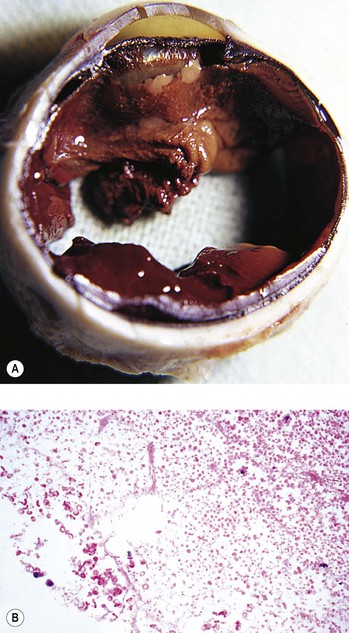
E. Hemolytic (ghost cell) glaucoma (Figs. 16.19 and 16.20)
1. Hemolytic glaucoma presents as an acute open-angle glaucoma.
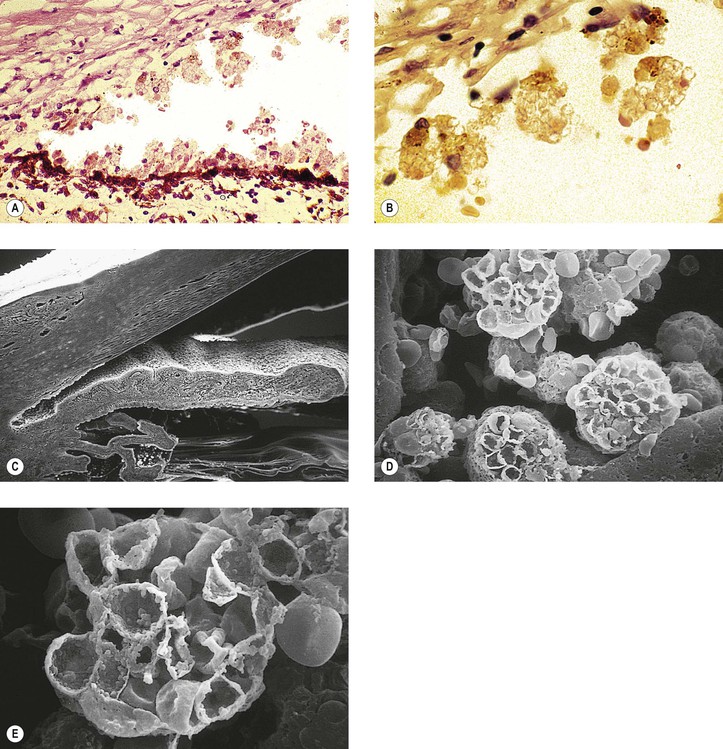
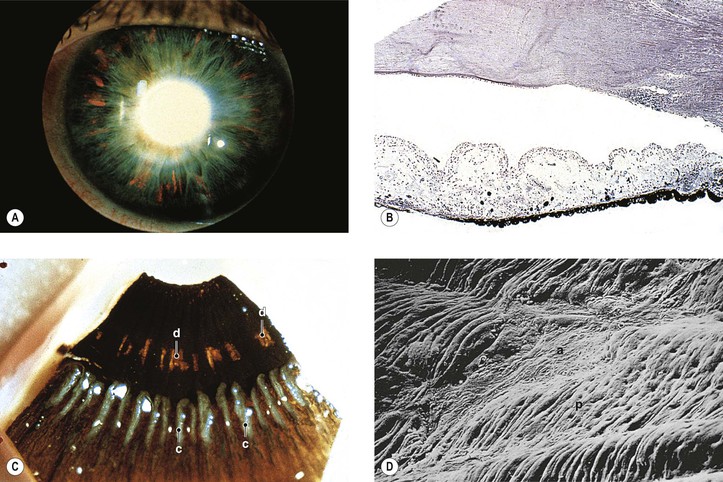
F. Pigment dispersion syndrome (pigmentary glaucoma; Figs. 16.21 and 16.22)
1. The pigment dispersion syndrome is found most often in young, myopic, adult, white men.
3. Iridodonesis may be present.
5. The pigment is deposited on the iris surface, lens, zonules, and in the trabecular meshwork.
8. Histologically, the posterior layer of iris pigment epithelium, mainly at the junction of middle and peripheral thirds of the iris, atrophies in foci that correspond to the clinically observed peripheral foci of increased iris transillumination.
a. The dilator muscle may be dysplastic, present in excessive amounts, atrophic, or absent.
b. The adjacent iris stroma contains pigment-filled macrophages.
9. Familial occurrence of pigment dispersion syndrome has been reported.
G. Pseudoexfoliation syndrome (see Chapter 10)
1. LOXL1 (lysyl oxidase-like 1) promoter haplotype is associated with pseudoexfoliation syndrome and pseudoexfoliation glaucoma in the U.S. white population, which is similar to most non-African populations.
2. Polymorphisms in elastin are not associated with pseudoexfoliation syndrome and glaucoma.
H. Secondary to uveal malignant melanomas (Figs. 16.23–16.25; see Table 16.2)
1. Seeded malignant melanoma cells (see Fig. 16.23) may block the anterior chamber angle.
2. A ring malignant melanoma (see Fig. 16.24) may directly invade the anterior chamber angle structures and block the open angle. Therefore, glaucoma may be the presenting finding.
3. Melanomalytic glaucoma (see Fig. 16.25)
b. The liberated melanin induces phagocytosis by macrophages.
c. The melanin-laden macrophages then obstruct the open angle of the anterior chamber.
4. Epithelialization or endothelialization of anterior chamber angle (see Chapter 5)
II. Secondary to damaged outflow channels
A. Old uveitis may result in “scarring” of the tissues in the drainage angle.
C. Repeated hyphema may damage the aqueous outflow tissue.
E. Trauma
1. It may have a direct effect on the tissues of the drainage angle by inducing scarring (sclerosis) of the trabecular meshwork, or it may cause a postcontusion deformity of the anterior chamber angle (see Chapter 5).
F. Cornea guttata [Fuchs (see Chapter 8)]
IV. Unknown mechanisms (usually reversible)
Tissue Changes Caused by Elevated Intraocular Pressure
Cornea (Figs. 16.26–16.28; see also Fig. 8.55)
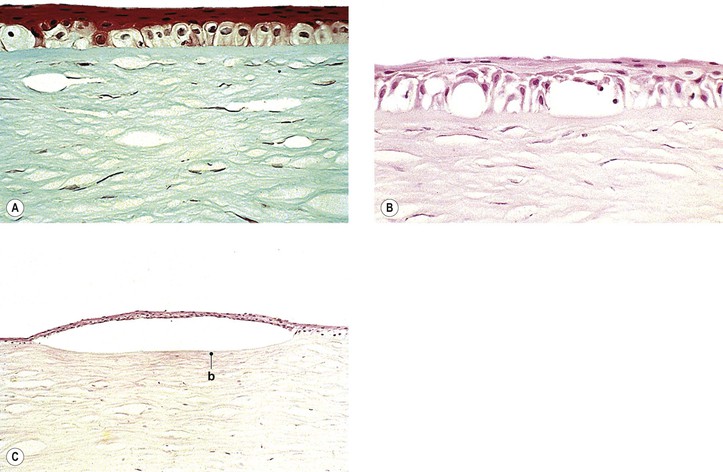
I. Edema of stroma and epithelium (see Fig. 16.26)
II. Epithelial bullae (bullous keratopathy; see Fig. 16.26)
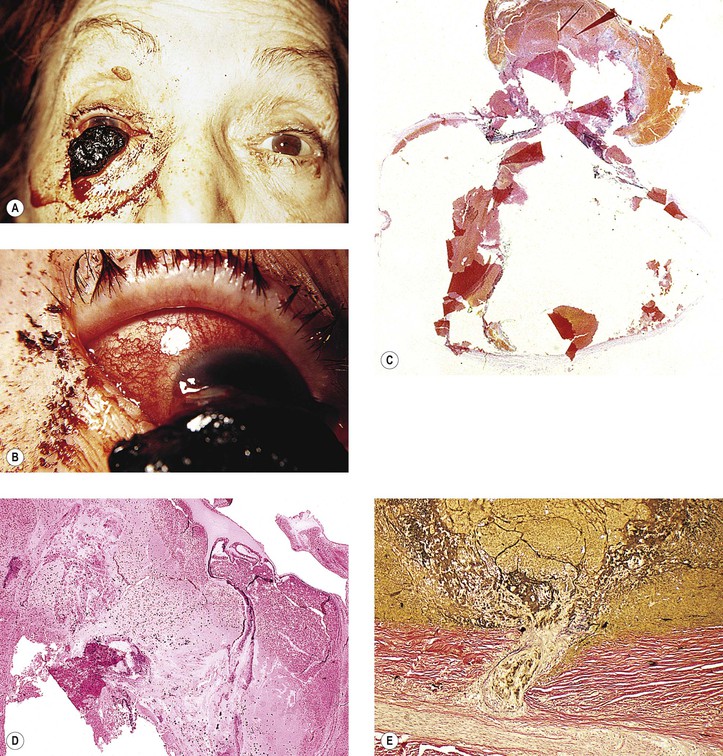
III. Corneal ulcer
B. The corneal ulcer can result in corneal perforation, and even in an expulsive hemorrhage (see Figs. 10.5, 16.27, and 16.28).
IV. Degenerative subepithelial pannus
C. The edema then spreads to overlying (anterior) epithelial cells.
D. Further accentuation of the edema ruptures the cell membranes, and macrocysts or blebs result.
E. At the same time, the epithelium is lifted off the underlying Bowman’s membrane by collections of fluid.
1. The overlying epithelium then appears irregular with areas of atrophy and hypertrophy.
2. The basement membrane of the epithelium is usually irregular.
V. Atrophy of epithelium and endothelium
Anterior Chamber Angle
Lens
Cataract, especially after glaucoma surgery or after an acute attack of glaucoma (e.g., glaukomflecken with acute closed-angle glaucoma)
Sclera
Ectasia (thinning) or, if lined by uvea, staphyloma (Fig. 16.30)
Neural Retina (Fig. 16.31)
Optic Nerve
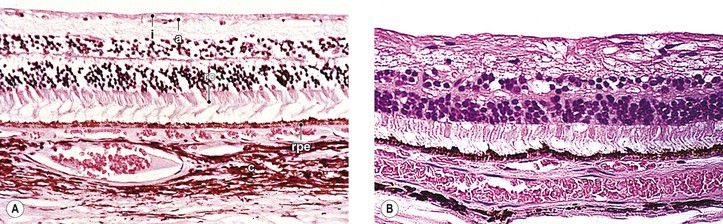
II. Optic nerve atrophy results from a loss of the nerve fibers of the inner neural retina and optic nerve.
A. Whether the neural damage is caused by local or distant astrocytic damage or by vascular insufficiency is not known.
Astrocyte metabolism is altered in glaucoma or in cells cultured at elevated IOP.
III. Atrophy results in loss of substance from the optic nerve head, leading to cupping (Fig. 16.32) or, if the loss is extensive, to excavation of the optic nerve head. Cup enlargement, in turn, results in increased visibility of lamina cribrosa pores.
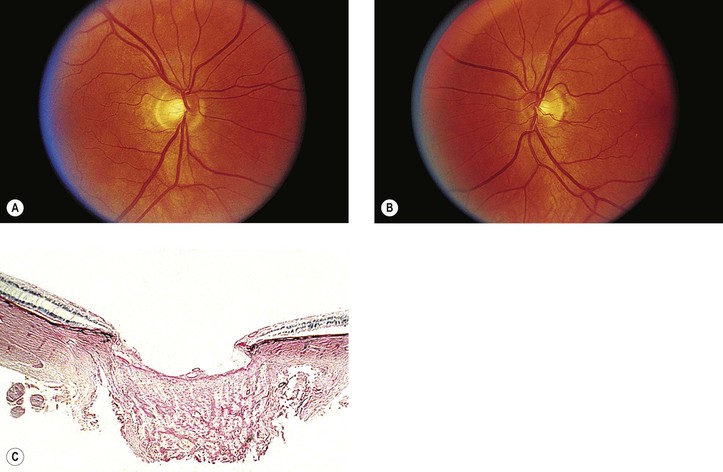
IV. Cavernous (Schnabel’s) optic atrophy (Fig. 16.33) consists of cystoid spaces, usually posterior to scleral lamina cribrosa. The cystoid spaces are filled with hyaluronic acid (see Chapter 13).
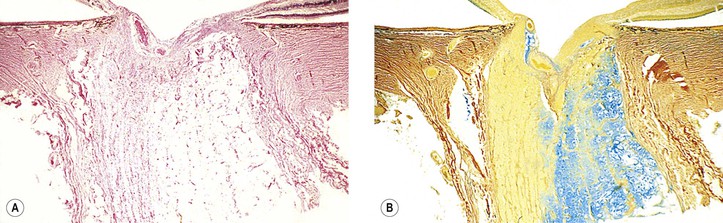
V. Parapapillary chorioretinal atrophy is associated with glaucoma.
A. Alpha parapapillary chorioretinal atrophy shows irregular hypopigmentation and hyperpigmentation.
Access the complete reference list online at 
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Glaucoma
Normal Anatomy (Figs. 16.1–16.3)
I. The outermost or corneoscleral layer of the eye can be separated into corneal and scleral portions by two circumferential grooves—a shallow outer one, the outer scleral sulcus, and a deeper inner one, the inner scleral sulcus.
C. Deep within this inner sulcus and applied closely to the collagenous tissue of the corneosclera lies the large vessel called the canal of Schlemm.
3. The inner wall rests on a thinner or patchy basement membrane that is associated with a zone of delicate connective tissue, the juxtacanalicular connective tissue.
4. Pores are present in the wall of Schlemm’s canal.
B. The meshwork may be easily and usefully separated into two parts by an imaginary line extending from the scleral roll to the end of Descemet’s membrane (see Fig. 16.2).
Introduction
The cell and molecular biology and gene rearrangement aspects of the glaucomas are fascinating, but an in-depth analysis of these matters is beyond the scope of our discussion. For example, genome-wide expression profiling of patients with primary open-angle glaucoma (POAG) identified 563 genes that were significantly dysregulated in POAG compared with normal controls. These genes impacted numerous functions, including nucleoside, nucleotide, and nucleic acid metabolism; the mitogen-activated protein kinase kinase kinase (MAPKKK) cascade; apoptosis; protein synthesis; cell cycle; intracellular signaling cascade; and nervous system development and function. Therefore, we highlight only a few salient facts regarding these areas.
Currently, 15 chromosome loci, which are designated GLC1A to GLC10, are associated with POAG. Candidate genes include myocilin (GLC1A), WD40-repeat36 (GLC1G), optineurin (GLC1E), and neurotrophin-4 (NTF-4) (optineurin is discussed with normal pressure glaucoma later in this chapter). Nevertheless, it has been estimated that mutations in known glaucoma genes account for less than 15% of cases. The prevalence of heterozygous cytochrome p450 1B1 (CY1B1) gene changes in a mixed glaucoma population suggests that the pathogenesis of glaucoma is genetically heterogeneous and may be polygenic. Single nucleotide polymorphisms (SNPs) between the CAV1 and CAV2 genes on chromosome 7q31 code for two members of the caveolin family of proteins are associated with POAG in white Americans, particularly women. These proteins impact modulation of endothelial cell membranes, which could alter the process of ocular aqueous fluid drainage. In general, however, SNPs are not discussed in any detail, but they have been described elsewhere (see Bibliography). Gene copy number variations also may play a role in the development of POAG. For example, homozygous deletions that reduce galactosylceramidase activity may increase the risk of POAG.
I. Glaucoma is characterized by an intraocular pressure (IOP) sufficient to produce ocular tissue damage, either transient or permanent.
A. Glaucoma is a “family” of diseases having in common a type of optic atrophy called optic nerve head cupping or excavation.
B. Although most individuals associate glaucoma with an elevated IOP, the pressure may, in fact, be within the statistically “normal” range and still cause ocular tissue damage in normal-tension (improperly called low-tension) glaucoma.
1. IOP is a risk factor for glaucoma, and the higher the pressure, the greater the probability of the development of the disorder.
a. The accurate measurement of IOP is vital to the proper diagnosis and treatment of glaucoma.
c. CCT is increased in children with ocular hypertension.
2. Normal-tension glaucoma probably accounts for approximately one-third of all cases of POAG.
Optineurin
Currently, 15 gene loci, designated GLC1A to GLC10, are associated with POAG. The optinuerin gene is associated with several disorders, including glaucoma, amyotrophic lateral sclerosis, other neurodegenerative disorders, and Paget’s disease of bone. A glaucoma-causing gene has been identified at GLC1E, and sequence variations in this optineurin (OPTN) gene on GLC1E have been found to be associated with the development of normal-tension glaucoma. The gene is located on chromosome 10. The glaucoma associated with optineurin is not characterized by marked IOP elevation. Rather, the E50K mutation in the optineurin gene is associated with increased severity of normal-tension glaucoma in white and Latino populations. There may be racial differences in glaucoma-associated optineurin genotypes. Its primary effect may be to increase susceptibility of retinal ganglion cells to premature cell death. Thus, optineurin may serve an optic nerve protective effect that is lost through mutation. Optineurin gene alterations do not appear to have a significant role in typical POAG.
II. Glaucoma suspect
B. POAG accounts for approximately two-thirds of all glaucoma seen in white patients.
Impaired Outflow
Congenital Glaucoma
A. The rate of congenital glaucoma is from 1 : 5000 to 1 : 10,000 live births.
C. Approximately 60–70% of affected children are boys.
D. The disease is bilateral in 64–88% of cases.
II. Pathogenesis (many theories)
C. An “embryonic” anterior chamber angle that results from faulty cleavage of tissue during embryonic development of the eye prevents the aqueous from leaving the anterior chamber.
1. Histologically, the angle shows an anterior “insertion” of the iris root, anteriorly displaced ciliary processes, insertion of the ciliary meridional muscles into the trabecular meshwork instead of into (or over) the scleral roll, and mesenchymal tissue in the anterior chamber angle (Fig. 16.4).
2. Many nonglaucomatous infant eyes show a similar anterior chamber angle structure.
3. To interpret angle histology accurately, it is necessary to study truly meridional sections through the anterior chamber angle.
a. Tangential sectioning makes interpretation difficult (see Figs. 16.4C and 16.4D).
D. The actual cause or causes of congenital glaucoma probably remain unknown.
III. Associated diseases and conditions
A. Iris anomalies (see Chapter 9)
1. Hypoplasia of the iris (“aniridia”) and iris coloboma may be associated with congenital glaucoma.
a. The PAX6 point mutation defect (1630A > T) on band p13 of chromosome 11 has been associated with some cases of aniridia. PAX6 mutations result in alterations in corneal cytokeratin expression, cell adhesion, and glycoconjugate expression. There is also corneal stem cell deficiency, which contributes to associated keratopathy.
B. Axenfeld’s anomaly and Rieger’s syndrome (see Chapter 8)
C. Peters’ anomaly (see Chapters 2 and 8). Peters’ anomaly and primary congenital glaucoma may share a common molecular pathophysiology. Both of these disorders can be associated with mutation in the cytochrome P4501B1 (CYP1B1) gene (discussed previously).
D. Phakomatoses
1. Sturge–Weber syndrome (see Chapter 2)
2. Neurofibromatosis (see Chapter 2)
E. Lowe’s syndrome (see Chapter 10)
H. Marfan’s syndrome (see Chapter 10)
I. Homocystinuria (see Chapter 10)
J. Microcornea (see Chapter 8)
K. Spherophakia (see Chapter 10)
L. Chromosomal abnormalities (e.g., trisomy 13; see Chapter 2)
M. Persistent hyperplastic primary vitreous (see Chapter 18)
N. Retinopathy of prematurity (see Chapter 18)
O. Retinoblastoma (see Chapter 18)
P. Juvenile xanthogranuloma (see Chapter 9)
V. Neurofibromatosis type 1 should be excluded in newborns with unilateral congenital glaucoma.
IV. Secondary histologic ocular effects in young eyes (<10 years of age)
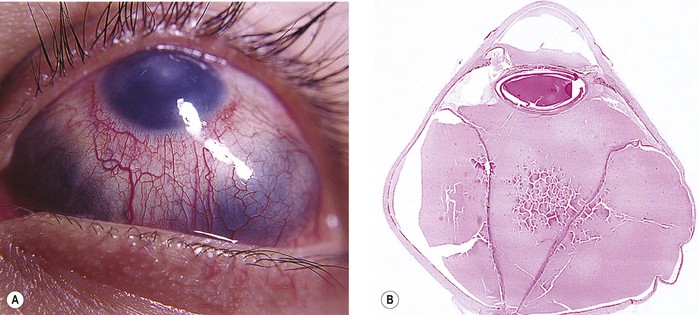
A. Buphthalmos (“large eye”) is caused by an enlargement, stretching, and thinning of the coats of the eye, especially marked in the anterior segment, resulting in a deep anterior chamber (Fig. 16.5). Subluxated lenses may develop in these enlarged eyes.
B. Ruptures of Descemet’s membrane (Haab’s striae) may be found in the enlarged corneas (Fig. 16.6), are usually horizontal in the central cornea but concentric toward the limbus, are located mainly in the lower half, and are often associated with corneal edema.
C. The limbal region becomes stretched and thin, with a resultant limbal ectasia (see Fig. 16.5).
E. Continued high IOP may cause atrophy of the ciliary body, choroid, and retina; cupping of the optic disc (see Fig. 16.5); and atrophy of the optic nerve.
Primary Glaucoma (Closed- and Open-Angle)
II. Optical coherence tomography and ultrasound biomicroscopy can be utilized to evaluate the anatomic configuration of the anterior chamber angle and adjacent structures for the classification of the pathophysiology of the glaucomas.
A. Specific features with such imaging studies that may contribute to angle-closure include:
3. Smaller anterior chamber width, area, and volume
4. Dynamic increase or lesser reduction in iris volume during dilation
5. Choroidal expansion accompanying angle-closure
III. Closed-angle (narrow-angle; angle-closure; acute congestive) glaucoma (Figs. 16.7 and 16.8)
A. In anatomically predisposed eyes, primary closed-angle glaucoma develops.
2. Small, hypermetropic eyes are especially vulnerable to angle-closure.
A founder gene effect is probably related to two families of the Faroe Islands with hereditary high hyperopia, angle-closure glaucoma, uveal effusion, cataract, esotropia, and amblyopia.
3. The lens is normal-sized or large.
C. A sudden rise in IOP results from the peripheral iris being in apposition to the filtering trabecular meshwork from the pupillary block mechanisms except in the plateau iris syndrome, which in its pure form does not involve pupillary block.
The association of typical acute closed-angle glaucoma with increasing age is due, in part, to progressive pupillary block from the increase in lens size as it adds layers of lens fibers over time.
F. Histology
b. Segmental iris atrophy is usually seen in the upper half of the iris in a sector configuration.
2. Irregular pupil results from necrosis of the dilator and sphincter muscles.
a. Histologically, segments of the dilator muscle or its entire length are absent.
b. The sphincter muscle shows varying degrees of atrophy.
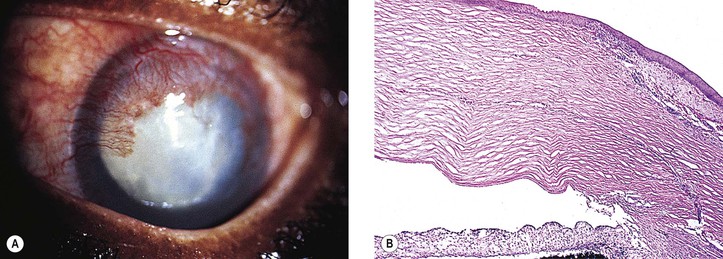
3. Glaukomflecken (cataracta disseminata subcapsularis glaukomatosa; see Fig. 16.8)
a. Glaukomflecken probably results from interference with the normal metabolism of the anterior lens cells due to a stagnation of aqueous humor that contains toxic products of necrosis or from foci of pressure necrosis (see Chapter 10).
b. Anterior subcapsular, multiple, tiny gray-white lenticular opacities are seen.
b. Irreversible vision impairment after an acute attack is mainly caused by optic nerve damage.
G. Failure to reverse the initial attack of angle closure can result in chronic angle-closure glaucoma.
1. This entity can be confused clinically with chronic open-angle glaucoma unless careful gonioscopy is performed routinely on all suspected cases of glaucoma.
Chronic angle-closure glaucoma is associated with the necessity of continued treatment and subsequent procedures even in the presence of a patent iridotomy.
2. Histology
There is disorganization of the trabecular architecture, narrowing or loss of trabecular spaces, scarring of the trabecular beams, trabecular endothelial cell loss, deposition of banded fibrillar material, and melanin pigment deposition.
IV. Chronic open-angle (chronic simple) glaucoma (POAG) (Figs. 16.9 and 16.10)
A. The angle appears normal gonioscopically.
C. The condition is most often bilateral.
1. Glaucoma may develop in one eye months to years before the fellow eye.
2. One eye may be more severely affected than the other eye.
D. Prevalence (see section Introduction)
1. In most cases, the condition is probably inherited as an autosomal-recessive trait.
2. Myocilin
Myocilin was the first gene to be associated with glaucoma. Myocilin is the most frequently mutated gene in POAG patients worldwide. There are myocilin genotype–phenotype correlations, such as age of diagnosis, maximum IOP, and response to medical therapy. Mutations in the myocilin gene are present in 1–4% of POAG patients but not in patients with pseudoexfoliation. Myocilin gene is located on chromosome 1, localized to the olfactomedin domain, and was the first gene identified for POAG in the GLC1A locus. Its associated glaucoma is transmitted as an autosomal-dominant trait. There appears to be compromised stability of the protein, which categorizes the resulting disorder as a disease of protein misfolding. There is strong evidence that myocilin polymorphisms are related to POAG susceptibility with significant racial variation such that for whites Q368x is most important, whereas T353I is not important for Asians. The Gln48His mutation is unique to Indian patients. There is a low prevalence of myocilin mutations in African-Americans with POAG.
F. Normal-tension (improperly called low-tension) glaucoma is a subdivision of POAG (see section Introduction in this chapter).
G. Histology and pathophysiology
1. Optic nerve (see later in this chapter)
2. Little is known about the early histologic changes that take place in the region of the drainage angle because the number of human eyes that have well-characterized early open-angle glaucoma available for histologic examination is small (see Fig. 16.9).
4. Aging changes in the drainage angle of the anterior chamber
d. The degree of obstruction is a quantitative problem; two extremes of excessive obstruction are:
e. The two major aging changes—each may be seen in almost pure form, or they may be combined.
1) They may also be associated with proliferations of trabecular endothelial cells in the meshwork.
2) COAG depends on the amount of aqueous inflow and the degree of obstruction to outflow caused by the aging changes (see Fig. 16.10).
f. Other changes in the trabecular meshwork in POAG include loss of cells, increased accumulation of extracellular matrix, changes in cytoskeleton, cellular senescence, and the process of subclinical inflammation.
Oxidative stress is likely to be one important mechanism in the pathogenesis of POAG.
Secondary Closed-Angle Glaucoma
Causes
I. Chronic primary angle-closure glaucoma
B. Histologically, peripheral anterior synechiae are seen, sometimes broad based.
II. Phacomorphic
V. Iridocorneal endothelial (ICE) syndrome
1. All three entities share a basic corneal endothelial defect and iris involvement.
3. Glaucoma occurs in approximately 50% of cases.
5. Each variant is described separately, but all are considered to belong to the ICE syndrome.
