[level-membership-for-opthalmology-category]
Chapter 31 Genetic Mechanisms of Retinal Disease
Introduction
The purpose of this chapter is to provide an overview of concepts underlying our current understanding of the genetic basis of inherited retinal diseases (iRDs). iRDs are perhaps the best understood of human hereditary disorders. In part this is because diseases that affect vision are easily recognized and the retina is an accessible and well-characterized tissue. In many ways, though, we are still at an early stage of understanding the causes and consequences of these diseases. In fact, the causes of iRDs are highly varied: many different types of retinal disease are known, many different genes are involved, and there may be dozens of disease-causing mutations reported within a single gene. For example, currently at least 220 genes are known which can cause one or another form of retinal disease,1 and over 5000 mutations have been reported, in total, in these genes.2 In spite of the underlying complexity, it is now possible to identify the disease-causing gene and mutation, or mutations, in a substantial fraction of affected individuals and families.3,4
Therefore the cause of disease in an individual with an inherited condition such as RP is “simple,” in the sense that only one gene is affected (and usually affected in an obvious way), whereas there may be multiple contributory factors in an individual with AMD and the differences may be subtle. We already known exceptions to this rule – for example, there are digenic forms of RP with two affected genes5 – but the exceptions are rare.
This chapter focuses on genetic differences that are single-gene in nature and have a direct cause-and-effect relationship with disease, that is, inherited diseases of the retina. Genetic factors contributing to AMD are discussed in Chapter 64 (AMD: Etiology, genetics, and pathogenesis).
Basic concepts in human genetics
Inheritance
Figure 31.1 shows pedigrees illustrating autosomal dominant, autosomal recessive, and X-linked recessive inheritance (see Nussbaum et al.6 for details).
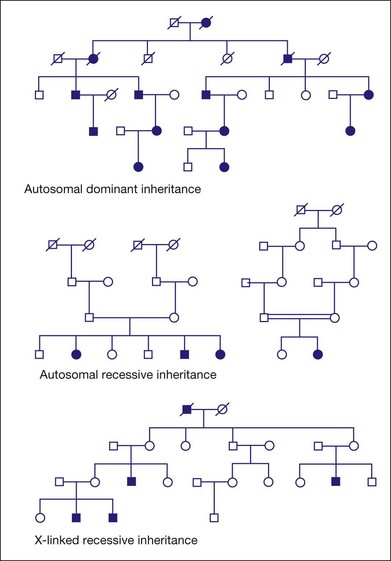
Fig. 31.1 Pedigrees illustrating autosomal dominant, autosomal recessive, and X-linked recessive inheritance.
Autosomal dominant inheritance
Variable clinical expression means that individuals with the same mutation may vary in onset, progression, or severity of disease or, in some cases, may have distinctly different clinical findings. Autosomal dominant RP is notoriously variable in expression. For example, mutations in one autosomal gene, PRPH2 (also known as RDS), can cause dominant RP, dominant macular degeneration, or dominant panretinal maculopathy, even among members of the same family.7–11
Incomplete penetrance, or nonpenetrance, means that some individuals with a disease-causing mutation will not be affected. For instance, 20% of individuals with a dominant-acting mutation in PRPF31 will have normal vision by age 60 even though relatives with the same mutation may have RP by age 20.12–15 One indicator of nonpenetrance in a multigenerational family is a “skipped generation,” that is, an unaffected individual with an affected parent and an affected child. This is often seen in families with PRPF31 mutations.
Autosomal recessive inheritance
Examples of autosomal recessive retinopathies include Leber congenital amaurosis and Usher syndrome.
X-linked or sex-linked inheritance
The disease status of female carriers is more complex, though. Although females have two Xs, one of the Xs, selected at random in each cell, is inactivated in most tissues. This is X-inactivation or lyonization, named for Mary Lyon, who first described the phenomenon.16,17 Lyonization increases the likelihood that a female carrier will be affected since some cells will express only the mutant protein. In fact, many female carriers of X-linked RP mutations show clinical symptoms. Females are less severely affected than males with the same mutation, but female carriers of X-linked RP mutations may have significant loss of vision by midlife or earlier.18–22
One consequence of clinical disease in carrier females is that families with X-linked RP may appear to have autosomal dominant RP if several females are affected.23 This is an example of complexities that arise in determining the mode of inheritance of iRDs.
Digenic and polygenic inheritance
Nearly all iRDs are monogenic, with only one gene affected per person. This is based on empirical observation, but it may be misleading since more complex forms of inheritance are hard to prove. Two counter examples are known for iRDs. First, one form of RP is caused by a combination of one mutation in the PRPH2 (RDS) gene and another mutation in the ROM1 gene.5,22 These two mutations are benign alone but pathogenic in combination. This is digenic inheritance. Secondly, Bardet–Biedl syndrome (BBS), a form of RP combined with congenital abnormalities, is in most instances a recessive disease with mutations in any one of at least 15 known BBS genes.1,24 Some cases of BBS, though, require a third mutation in a second BBS gene for disease expression.25,26 This is called trigenic or triallelic inheritance. Whether these examples of polygenic inheritance of iRDs are just rare anomalies or hint at greater complexity of retinal diseases is unclear.
DNA, RNA, and proteins
Figure 31.2 shows the steps in DNA duplication, RNA translation, and protein synthesis.27
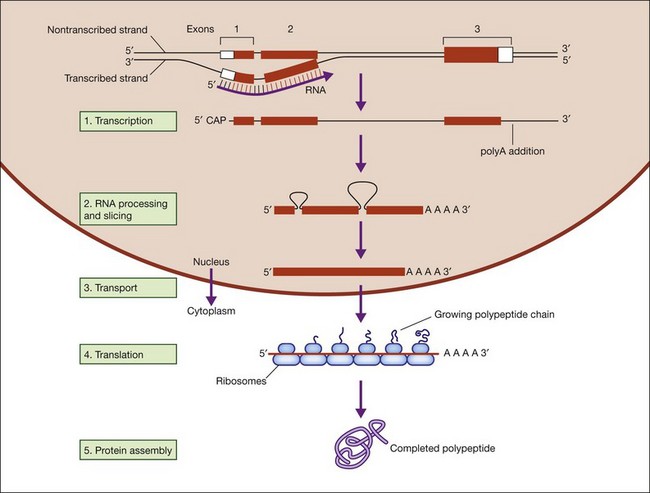
Fig. 31.2 Steps in DNA duplication, RNA translation, and protein synthesis.
(Reproduced from Nussbaum RL, McInness RR, Willard HF. Thompson and Thompson’s genetics in medicine, 7th ed. Philadelphia, PA: Saunders Elsevier; 2007, p. 31, with permission from Elsevier.)
DNA function is called the central dogma of DNA in recognition of the landmark explanation of DNA structure and function by Watson and Crick in 1953, and subsequent unraveling of the genetic code over the next decade.28,29 DNA is comprised of a phosphate backbone with nucleotide bases, A, T, G, or C, in linear array along the backbone. The backbone is conventionally drawn from the 5’ phosphate on one end to the 3’ phosphate on the other end. The opposite strand forms by pairing of cognate bases, A to T and G to C, on the parent strand. The opposite strand naturally aligns in a helical, antiparallel fashion, from 3’ to 5’ phosphates. This arrangement essentially explains inheritance in all living things.
Gene structure
Figure 31.3 shows gene structure based on the relationship between the protein sequence, mRNA intermediate, and original DNA gene sequence.27
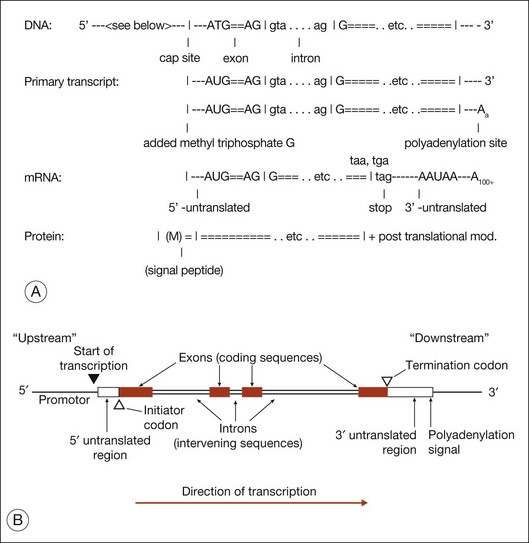
The evolutionary significance of splicing is still disputed, but its functional consequence is clear: it vastly increases the number of distinct proteins. This is because when splicing occurs, alternate combinations of introns may be removed. Alternate splicing is the norm in human genes, not the exception, and usually results in alternate mRNAs and alternate protein isoforms – all from a “single” gene. There are many examples of alternately spliced retinal genes producing multiple protein isoforms.30,31
Mitosis, meiosis, and linkage
Figure 31.4 shows the steps in meiosis.6
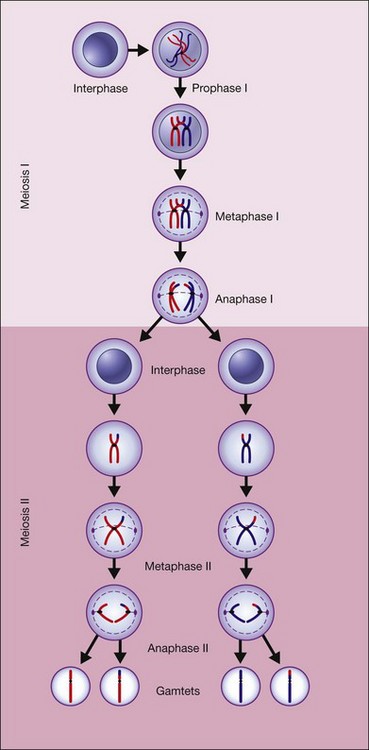
Fig. 31.4 The steps in meiosis.
(Reproduced from Nussbaum RL, McInness RR, Willard HF. Thompson and Thompson’s genetics in medicine, 7th ed. Philadelphia, PA: Saunders Elsevier; 2007, p. 19, with permission from Elsevier.)
To put this in context, the average chromosome is 100 Mb in length. Genes that are 50 Mb apart are unlinked, and genes within 10 Mb of each other show linkage. If genes are within 1 Mb of each other the chance of recombination is less than 1%. There are roughly 5–20 genes per Mb in the human genome, so hundreds of contiguous genes on a chromosome may show linkage to each other.32
Evolution
Theodosius Dobzhansky said “nothing in biology makes sense except in the light of evolution.”33 This observation serves to emphasize the fundamental organizing principal of biology. Without evolution, biology is largely a random collection of facts and principles; with evolution, biology (and medicine) is a coherent science. Evolution explains why the central dogma is true for all species, why all eukaryotic cells share a common architecture, why animal models are useful in understanding and treating human diseases, why human DNA variants are evaluated by comparison to other species. Evolution is so central to biology and medicine that we often forget its impact, but modern, 21st-century biomedicine is unimaginable without it.
The human genome
Overview
In one of the greatest scientific achievements in modern history, the Human Genome Project produced the first human genome sequences in 2001.34,35 Since then hundreds of complete human genomes have been sequenced, and the genomes of thousands of other species are known.36
Polymorphisms
Several broad classes of polymorphism are found in the human genome
Single-nucleotide polymorphisms
Single-nucleotide polymorphisms (SNPs) are sites at which more than one nucleotide is found in a population. There are usually only two alleles at a SNP locus, e.g., an A or a T. Millions of SNP sites have been identified in humans.37 Some are found in only one major population, such as Africans, but many are polymorphic in all major human groups. A SNP site at which the nucleotide substitution leads to an amino acid substitution, or otherwise affects a protein, is a coding SNP (cSNP). There are tens of thousands of cSNPs, at least one in almost every gene.
Mutations
Figure 31.5 illustrates several of the types of mutation that cause iRDs.
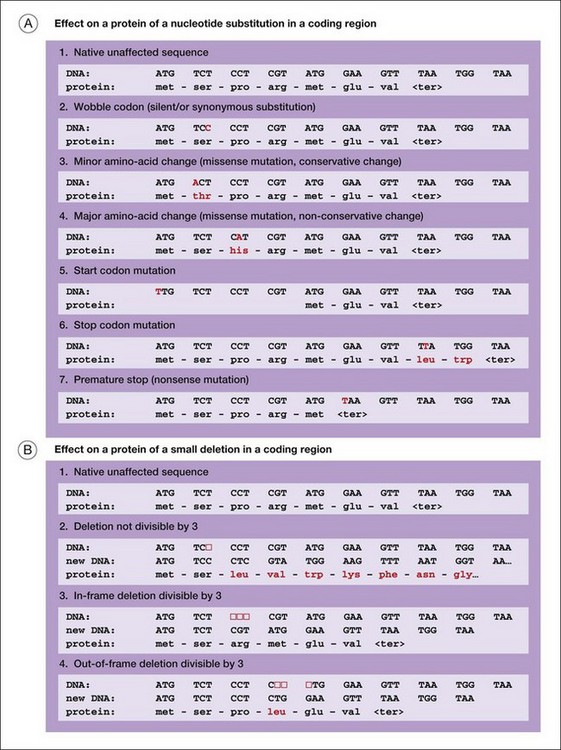
Another way to categorize mutations is to work from DNA to mRNA to proteins:
1. DNA deletions or rearrangements may result in the absence of a protein or a critical part of a protein. Surprisingly, humans harbor large, polymorphic deletions, some 100 kb in length or longer.38 In apparently healthy people, these are in a heterozygous state: one chromosome segment is deleted and the matching segment on the homologous chromosome is intact. Large, homozygous deletions are severely deleterious. Smaller deletions, usually the size of a gene or less, affecting one or a few proteins, may cause autosomal dominant, autosomal recessive, or X-linked retinal diseases.14,39,40
2. DNA changes 5’ to the start of transcription may block synthesis of mRNA or may affect timing or amounts of mRNA. These are promoter or expression mutations. Further, any changes to the canonical nucleotides that define the donor or acceptor slice sites of introns may profoundly affect mRNA splicing. These are splice site mutations. Splice site mutations typically result in a structurally abnormal protein or no protein at all.
3. Finally, mutations can alter proteins directly in a myriad of ways. Mutations in DNA that directly affect proteins are broadly classed into nucleotide substitutions in contrast to small insertions or deletions, typically 1–15 bp in length, called indels. A nucleotide substitution which causes replacement of one amino acid with another is a missense mutation. Missense mutations may alter protein function, or produce a toxic protein or a dominant negative protein. (A nucleotide substitution that changes a codon but does not alter an amino acid is a silent substitution. Most silent substitutions are benign.) An indel that alters the order of codons is a frameshift mutation or an inframe amino acid deletion. A nucleotide substitution that introduces a signal to stop translation of a message prior to the normal stop is a nonsense mutation or a premature-stop mutation. Indels and premature-stop mutations produce a severely abnormal protein or no protein.
Genetic testing methods
The primary purpose of genetic testing for iRDs is to determine the underlying cause of disease in an affected individual and in his or her family. A broader goal is to use genetic testing for research purposes, e.g., to find novel mutations, to discover new disease-causing genes, to identify patients for clinical trials, or to study the natural history of disease. There is no clear demarcation between diagnostic testing and research, but there are practical distinctions. Diagnostic testing is often limited to testing affected individuals, screening known genes only, and is usually conducted in a certified laboratory. (Diagnostic laboratories in the USA have Clinical Laboratory Improvement Amendments (CLIA)41 and/or College of American Pathologists (CAP)42 certification.) Research testing may involve other family members, may screen novel genes, and is often conducted in facilities which are not certified.
Screening known genes and mutations
For each category of disease there is a set of known, possible disease-causing genes and a set of known mutations within each gene. Among the known genes and mutations, some are more common causes of disease than others. For example, mutations in 22 genes are known to cause autosomal dominant RP1 and more than 680 mutations have been reported in these genes.2 However, mutations in one gene, rhodopsin, account for at least 25% of cases of autosomal dominant RP in the USA.4 Also, more than 150 mutations have been reported in rhodopsin alone, but six of these mutations account for more than half of all cases.22
As a consequence, the natural next step is to test the most likely disease-causing genes in an affected individual, based on clinical findings and family history. Testing is usually done by polymerase chain reaction-based di-deoxy chain termination cycle sequencing, also called Sanger sequencing, the “gold standard” of DNA sequencing.43,44 Sequencing laboratories and services in the USA for iRDs include commercial facilities, university-based services, and federally supported programs.45 A current list of testing facilities in Europe and the USA is maintained by GeneTests.46
An alternative to DNA sequencing is to detect known mutations only. One approach is to use microarrays with short single-stranded DNA sequences that bind to the region containing the targeted mutation and detect the presence or absence of the mutation. An example of this technology is arrayed primer extension.47,48 The advantage of DNA sequencing is that it detects both known and unknown mutations in the genes tested. The advantage of testing known mutations only is that it is much less expensive than sequencing.
Additional possible tests include sequencing all the known disease-causing genes, even the rare causes of disease, and using methods other than sequencing to detect mutations. An example of the latter is the use of multiplex ligation-dependent probe amplification to detect large deletions.14,49,50
Linkage and homozygosity mapping
In practice, linkage testing is most useful in large families with autosomal dominant or X-linked disease since more affected family members are available for testing. Currently, linkage testing is often done using microarrays that assay up to a million SNPs, made by Affymetrix, among others.51,52 Several computer programs are available for analysis of linkage data.53–55 Once linkage is established, genes in the linkage region are sequenced, by a variety of methods, looking for mutations. Determining whether a rare variant in the linkage region is pathogenic can be challenging.51
In some cases, linkage can be useful in small recessive families, too. One effective approach is homozygosity mapping.56 If the mutations in a family with recessive disease are IBD, then not only are the mutations themselves identical, but DNA sequences within thousands of basepairs of the mutations are also identical, because of linkage. This manifests as long chromosomal segments surrounding the disease gene that contain no variant sites, for example, no heterozygous SNP sites. Long, homozygous DNA sequences are rare in humans, and often indicative of IBD. They can be detected using the same SNP-testing arrays used for conventional linkage testing. Once the region containing the disease gene is located by homozygosity mapping, strategies to identify mapped genes can be applied. This has been a fruitful approach to finding genes causing recessive iRDs.57,58
High-throughput DNA sequencing
In recent years, novel methods for very rapid DNA sequencing have become available. This is referred to as next-generation sequencing or deep sequencing.59,60 These approaches can be up to 10 000 times as fast as conventional di-deoxy cycle sequencing. The principle is to shear human DNA into short fragments and to sequence these fragments, up to 1 million simultaneously, in micron-sized wells or slots. The isolated sequences are then assembled computationally by comparison to the reference human genome. This is called “shotgun sequencing” because many short, random fragments are sequenced and later assembled into long, useful sequences. Next-generation sequencing is more error-prone than conventional sequencing, but it more than compensates by the extremely high sequencing rate.
With next-generation sequencing it is practical and inexpensive to sequence large tracts of the genome in individual patients. One application is to sequence the coding regions of all human genes. This is referred to as whole-exome sequencing – the “exome” is all known exons. Another application is whole-genome sequencing, that is, sequencing the entire genome of an individual. Since exons are only 1.5% of the genome, whole-exome sequencing is currently faster and cheaper than whole-genome sequencing, but whole-genome sequencing will be common within a few years. Already, whole-exome and whole-genome sequencing have shown promise in finding novel iRD genes and mutations.51,61–63
1 RetNet. The Retinal Information Network. Available online at http://www.sph.uth.tmc.edu/RetNet/
2 HGMD. Human Gene Mutation Database. Available online at http://www.hgmd.cf.ac.uk/
3 Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–375.
4 Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–158.
5 Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;38:1972–1982.
6 Nussbaum RL, McInnes RR, Williard HF. Thompson and Thompson genetics in medicine, 7th ed. Philadelphia, PA: Saunders Elsevier; 2007.
7 Weleber RG, Carr RE, Murphey WH, et al. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111:1531–1542.
8 Wells J, Wroblewski J, Keen J, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3:213–218.
9 Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat. 1997;10:301–309.
10 Sears JE, Aaberg TA, Sr., Daiger SP, et al. Splice site mutation in the peripherin/RDS gene associated with pattern dystrophy of the retina. Am J Ophthalmol. 2001;132:693–699.
11 Boon CJ, van Schooneveld MJ, den Hollander AI, et al. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007;91:1504–1511.
12 Birch DG, Wheaton DH, Locke KG, et al. Clinical characterization of RP11 in five families with identified PRPF31 mutations. Invest Ophthalmol Vis Sci. 47, 2006. E-Abstract 1037
13 Rivolta C, McGee TL, Frio TR, et al. Variation in retinitis pigmentosa-11 (PRPF31 or RP11) gene expression between symptomatic and asymptomatic patients with dominant RP11 mutations. Hum Mutat. 2006;27:644–653.
14 Sullivan LS, Bowne SJ, Seaman CR, et al. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:4579–4588.
15 Vithana EN, Abu-Safieh L, Pelosini L, et al. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest Ophthalmol Vis Sci. 2003;44:4204–4209.
16 Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–373.
17 Lyon MF. The Lyon and the LINE hypothesis. Semin Cell Dev Biol. 2003;14:313–318.
18 Fahim AT, Bowne SJ, Sullivan LS, et al. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS ONE. 2011;6:ie23021.
19 Pelletier V, Jambou M, Delphin N, et al. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype–phenotype correlations and impact on genetic counseling. Hum Mutat. 2007;28:81–91.
20 Rozet JM, Perrault I, Gigarel N, et al. Dominant X linked retinitis pigmentosa is frequently accounted for by truncating mutations in exon ORF15 of the RPGR gene. J Med Genet. 2002;39:284–285.
21 Sharon D, Sandberg MA, Rabe VW, et al. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73:1131–1146.
22 Sullivan LS, Bowne SJ, Birch DG, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa (adRP): a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–3064.
23 Bowne SJ, Sullivan LS, Koboldt DC, et al. Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest Ophthalmol Vis Sci. 2010;52:494–503.
24 Chen J, Smaoui N, Hammer MB, et al. Molecular analysis of Bardet–Biedl syndrome families: report of 21 novel mutations in 10 genes. Invest Ophthalmol Vis Sci. 2011;52:5317–5324.
25 Badano JL, Leitch CC, Ansley SJ, et al. Dissection of epistasis in oligogenic Bardet–Biedl syndrome. Nature. 2006;439:326–330.
26 Katsanis N. The oligogenic properties of Bardet–Biedl syndrome. Hum Mol Genet. 2004;13:R65–R71.
27 Lewin B, Genes X. Sudbury. MA: Jones and Bartlett; 2011.
28 Watson JD, Crick FHC. A structure for deoxyribose nucleic acid. Nature. 1953;171:737–738.
29 Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563.
30 Bowne SJ, Liu Q, Sullivan LS, et al. Why do mutations in the ubiquitously expressed housekeeping gene IMPDH1 cause retina-specific photoreceptor degeneration? Invest Ophthalmol Vis Sci. 2006;47:3754–3765.
31 Schmid F, Glaus E, Cremers FP, et al. Mutation- and tissue-specific alterations of RPGR transcripts. Invest Ophthalmol Vis Sci. 2010;51:1628–1635.
32 Matise TC, Chen F, Chen W, et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786.
33 Dobzhansky T. Nothing in biology makes sense except in the light of evolution. Am Biol Teacher. 1973;35:125–129.
34 Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
35 Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351.
36 Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197.
37 dbSNP. dbSNP Short genetic variations. Available online at http://www.ncni.nlm.nih.gov/projects/SNP/
38 Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455.
39 Stone EM. Leber congenital amaurosis – a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811.
40 Travaglini L, Brancati F, Attie-Bitach T, et al. Expanding CEP290 mutational spectrum in ciliopathies. Am J Med Genet A. 2009;149A:2173–2180.
41 CLIA. Clinical Laboratory Improvement Amendments (CLIA). https://www.cms.gov/clia/, 2011. US Departments of Health and Human Services, Centers for Medicare and Medical Services
42 CAP. College of American Pathologists (CAP) Accreditation. Available online at http://www.cap.org/accreditation/
43 Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomics. 2009;93:105–111.
44 Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467.
45 EyeGENE. National Ophthalmic Disease Genotyping Network. Available online at http://www.nei.nih.gov/resources/eyegene.asp
46 GeneTests. geneTests. Available online at http://www.genetests.org/
47 Clark GR, Crowe P, Muszynska D, et al. Development of a diagnostic genetic test for simplex and autosomal recessive retinitis pigmentosa. Ophthalmology. 2010;117:2169–2177.
48 Jaakson K, Zernant J, Kulm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403.
49 Aguirre-Lamban J, Riveiro-Alvarez R, Maia-Lopes S, et al. Molecular analysis of the ABCA4 gene for reliable detection of allelic variations in Spanish patients: identification of 21 novel variants. Br J Ophthalmol. 2009;93:614–621.
50 Pieras JI, Barragan I, Borrego S, et al. Copy-number variations in EYS: a significant event in the appearance of arRP. Invest Ophthalmol Vis Sci. 2011;52:5625–5631.
51 Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant-acting mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19:1074–1081.
52 Affymetrix. Available online at http://2www.affymetrix.com/
53 Abecasis GR, Cherny SS, Cookson WO, et al. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101.
54 Lathrop GM, Lalouel JM, Julier C, et al. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci U S A. 1984;81:3443–3446.
55 Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575.
56 Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570.
57 Collin RW, van den Born LI, Klevering BJ, et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Invest Ophthalmol Vis Sci. 2011;52:2227–2239.
58 Littink KW, Koenekoop RK, van den Born LI, et al. Homozygosity mapping in patients with cone–rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci. 2010;51:5943–5951.
59 Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402.
60 Schuster SC. Next-generation sequencing transforms today’s biology. Nat Methods. 2008;5:16–18.
61 Ozgul RK, Siemiatkowska AM, Yucel D, et al. Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am J Hum Genet. 2011;89:253–264.
62 Tucker BA, Scheetz TE, Mullins RF, et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2011;108:E569–E576.
63 Zuchner S, Dallman J, Wen R, et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am J Hum Genet. 2011;88:201–206.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 31 Genetic Mechanisms of Retinal Disease
Introduction
The purpose of this chapter is to provide an overview of concepts underlying our current understanding of the genetic basis of inherited retinal diseases (iRDs). iRDs are perhaps the best understood of human hereditary disorders. In part this is because diseases that affect vision are easily recognized and the retina is an accessible and well-characterized tissue. In many ways, though, we are still at an early stage of understanding the causes and consequences of these diseases. In fact, the causes of iRDs are highly varied: many different types of retinal disease are known, many different genes are involved, and there may be dozens of disease-causing mutations reported within a single gene. For example, currently at least 220 genes are known which can cause one or another form of retinal disease,1 and over 5000 mutations have been reported, in total, in these genes.2 In spite of the underlying complexity, it is now possible to identify the disease-causing gene and mutation, or mutations, in a substantial fraction of affected individuals and families.3,4
Therefore the cause of disease in an individual with an inherited condition such as RP is “simple,” in the sense that only one gene is affected (and usually affected in an obvious way), whereas there may be multiple contributory factors in an individual with AMD and the differences may be subtle. We already known exceptions to this rule – for example, there are digenic forms of RP with two affected genes5 – but the exceptions are rare.
This chapter focuses on genetic differences that are single-gene in nature and have a direct cause-and-effect relationship with disease, that is, inherited diseases of the retina. Genetic factors contributing to AMD are discussed in Chapter 64 (AMD: Etiology, genetics, and pathogenesis).
Basic concepts in human genetics
Inheritance
Figure 31.1 shows pedigrees illustrating autosomal dominant, autosomal recessive, and X-linked recessive inheritance (see Nussbaum et al.6 for details).
Fig. 31.1 Pedigrees illustrating autosomal dominant, autosomal recessive, and X-linked recessive inheritance.
Autosomal dominant inheritance
Variable clinical expression means that individuals with the same mutation may vary in onset, progression, or severity of disease or, in some cases, may have distinctly different clinical findings. Autosomal dominant RP is notoriously variable in expression. For example, mutations in one autosomal gene, PRPH2 (also known as RDS), can cause dominant RP, dominant macular degeneration, or dominant panretinal maculopathy, even among members of the same family.7–11
Incomplete penetrance, or nonpenetrance, means that some individuals with a disease-causing mutation will not be affected. For instance, 20% of individuals with a dominant-acting mutation in PRPF31 will have normal vision by age 60 even though relatives with the same mutation may have RP by age 20.12–15 One indicator of nonpenetrance in a multigenerational family is a “skipped generation,” that is, an unaffected individual with an affected parent and an affected child. This is often seen in families with PRPF31 mutations.
Autosomal recessive inheritance
Examples of autosomal recessive retinopathies include Leber congenital amaurosis and Usher syndrome.
X-linked or sex-linked inheritance
The disease status of female carriers is more complex, though. Although females have two Xs, one of the Xs, selected at random in each cell, is inactivated in most tissues. This is X-inactivation or lyonization, named for Mary Lyon, who first described the phenomenon.16,17 Lyonization increases the likelihood that a female carrier will be affected since some cells will express only the mutant protein. In fact, many female carriers of X-linked RP mutations show clinical symptoms. Females are less severely affected than males with the same mutation, but female carriers of X-linked RP mutations may have significant loss of vision by midlife or earlier.18–22
One consequence of clinical disease in carrier females is that families with X-linked RP may appear to have autosomal dominant RP if several females are affected.23 This is an example of complexities that arise in determining the mode of inheritance of iRDs.
Digenic and polygenic inheritance
Nearly all iRDs are monogenic, with only one gene affected per person. This is based on empirical observation, but it may be misleading since more complex forms of inheritance are hard to prove. Two counter examples are known for iRDs. First, one form of RP is caused by a combination of one mutation in the PRPH2 (RDS) gene and another mutation in the ROM1 gene.5,22 These two mutations are benign alone but pathogenic in combination. This is digenic inheritance. Secondly, Bardet–Biedl syndrome (BBS), a form of RP combined with congenital abnormalities, is in most instances a recessive disease with mutations in any one of at least 15 known BBS genes.1,24 Some cases of BBS, though, require a third mutation in a second BBS gene for disease expression.25,26 This is called trigenic or triallelic inheritance. Whether these examples of polygenic inheritance of iRDs are just rare anomalies or hint at greater complexity of retinal diseases is unclear.
DNA, RNA, and proteins
Figure 31.2 shows the steps in DNA duplication, RNA translation, and protein synthesis.27
Fig. 31.2 Steps in DNA duplication, RNA translation, and protein synthesis.
(Reproduced from Nussbaum RL, McInness RR, Willard HF. Thompson and Thompson’s genetics in medicine, 7th ed. Philadelphia, PA: Saunders Elsevier; 2007, p. 31, with permission from Elsevier.)
DNA function is called the central dogma of DNA in recognition of the landmark explanation of DNA structure and function by Watson and Crick in 1953, and subsequent unraveling of the genetic code over the next decade.28,29 DNA is comprised of a phosphate backbone with nucleotide bases, A, T, G, or C, in linear array along the backbone. The backbone is conventionally drawn from the 5’ phosphate on one end to the 3’ phosphate on the other end. The opposite strand forms by pairing of cognate bases, A to T and G to C, on the parent strand. The opposite strand naturally aligns in a helical, antiparallel fashion, from 3’ to 5’ phosphates. This arrangement essentially explains inheritance in all living things.
Gene structure
Figure 31.3 shows gene structure based on the relationship between the protein sequence, mRNA intermediate, and original DNA gene sequence.27
[/not-level-membership-for-opthalmology-category]
