Chapter 53 Generalized Seizures
As reviewed in Chapter 50, seizures are classified into two basic groups: partial and generalized [Commission on Classification and Terminology of the International League Against Epilepsy, 1981]. The International League Against Epilepsy (ILAE) Commission on Classification and Terminology has proposed a modification of the classification system of seizures and epilepsy [Berg et al., 2010]. Generalized seizures are those defined as occurring in bilaterally distributed networks whereas focal (partial) seizures involve networks limited to one hemisphere with either discretely localized or more widely distributed disturbances. While the proposed classification system has some advantages over the 1981 classification, it has not yet been widely used or endorsed by clinicians and in this chapter the older classification system will be used. Partial seizures involve only a portion of the brain at the onset but can spread, resulting in generalized tonic-clonic seizures. Primary generalized seizures are those in which the first clinical changes indicate initial synchronous involvement of both hemispheres without clinical, electroencephalographic (EEG), or other evidence of focal or partial onset. Impairment of consciousness is usual during generalized seizures, although some seizures, such as myoclonic, may be so brief that the level of consciousness cannot be assessed. The generalized seizures span a wide range of clinical presentations, ranging from the rather benign-appearing absence seizure to the dramatic and frightening generalized tonic-clonic seizure.
In most epidemiologic studies, primary generalized seizures are reported to be less common than partial seizures [Hauser and Kurland, 1975; Juul-Jensen and Foldspang, 1983]. For example, Hauser and Kurland [1975] and Juul-Jensen and Foldspang [1983] found that primary generalized seizures accounted respectively for 40.5 percent and 45.8 percent of the seizure types. Studies confined to children have been more variable. Todt [1984], in a study of children with seizures ranging in age from 3 to 16 years, found that 61.5 percent had primary generalized seizures, compared with only 26.6 percent of children between 2 months and 14 years of age in a series reported by Sofijanov [1982]. Of the primary generalized seizures, generalized tonic-clonic are the most common, followed by absence and myoclonic seizures [Hauser and Kurland, 1975].
These epidemiologic studies may be somewhat misleading. With improvement of diagnostic techniques in recent decades, especially with long-term video monitoring using EEG, it has become clear that the prevalence of primary generalized tonic-clonic epilepsy has been overestimated. In fact, most generalized tonic-clonic seizures begin as partial seizures and then generalize [Schmidt et al., 1983; Jobst et al., 2001].
This chapter discusses generalized tonic-clonic seizures and absence, clonic, tonic, and atonic seizures, along with the syndromes in which these are the predominant seizure type. Myoclonic seizures, which are generalized seizures, are reviewed in Chapter 56 and partial seizures in Chapter 54.
Generalized Tonic-Clonic Seizures
Clinical Features
As indicated by the name, generalized tonic-clonic seizures have two distinct phases: tonic and clonic [Gastaut and Broughton, 1972]. Loss of consciousness usually occurs simultaneously with the onset of a generalized stiffening of flexor or extensor muscles – the tonic phase. The loss of consciousness usually is complete, and it is rare for patients to have any partial awareness of what happened during the seizure. During the tonic phase, prolonged extension of the back, neck, and all limbs often occurs. The eyes remain open, and a cry or yell is common. The tonic phase typically lasts 10–30 seconds and is followed by the clonic phase, which usually starts with a rapid tremor and then slows to massive jerks of the extremities and trunk. A decrescendo pattern to the frequency (although not necessarily the strength) of the jerks is seen as the seizure ceases. The clonic phase typically lasts 30–60 seconds.
In a number of syndromes, generalized seizures are a predominant feature. Epilepsy with so-called grand mal seizures on awakening is a syndrome in which the generalized tonic-clonic seizures occur only on awakening [Betting et al., 2006; Unterberger et al., 2001; Beghi et al., 2006]. In generalized epilepsy with febrile seizures plus, patients initially have febrile seizures; later, as the condition evolves, they experience afebrile generalized tonic-clonic or other types of generalized seizures [Abou-Khalil et al., 2001; Baulac et al., 2004; Scheffer and Berkovic, 1997; Wallace et al., 2001, 2002]. Generalized tonic-clonic or clonic-tonic-clonic seizures often can be a prominent component of juvenile myoclonic epilepsy, a condition discussed in Chapter 56. Children with Lennox–Gastaut syndrome may have generalized tonic-clonic seizures, although tonic seizures are the core seizure type in this syndrome.
Although their disorder currently is not classified as a syndrome, a distinct group of patients with photosensitive epilepsy have seizures provoked by flickering lights and have a photoconvulsive response to strobe light on EEG. The photoconvulsive response is characterized by spike-and-wave and multiple spike-and-wave complexes that are bilaterally synchronous, symmetric, and widespread (Figure 53-1). These should be contrasted with spikes that are time-linked with the photic stimulation and confined to the occipital region, which in children are normal. The photoconvulsive response may be self-limited and cease during stimulation or continue beyond the stimulation [Reilly and Peters, 1973]. The most effective frequency of the flash is 10–20 Hz.
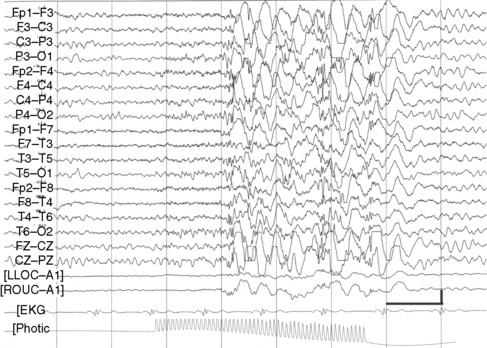
The seizures evoked by photic stimulation usually are primary generalized in type: generalized tonic-clonic, absence, or myoclonic seizures. Although patients with partial seizures may have photoconvulsive responses during an EEG study, it is unusual for a partial seizure to be precipitated by photic stimulation [Jayakar and Chiappa, 1990]. Generalized epileptiform discharges during photic stimulation correlate with generalized seizures, whereas focal discharges are more commonly associated with partial seizures [Gilliam and Chiappa, 1995].
Photosensitive epilepsy can be classified into two major groups: (1) pure photosensitive epilepsy, in which clinical seizures occur only when the patient is exposed to the photic stimulus, and (2) photosensitive epilepsy, in which spontaneous seizures occur in addition to those induced by light stimulation [Binnie and Jeavons, 1992]. Precipitating stimuli that can produce a seizure include sunlight reflected from water, sunlight viewed through leaves of trees in a breeze or while driving along tree-lined streets, discotheque lighting, faulty fluorescent lamp (“nervous ballast”), television, and video games. Photosensitive epilepsy usually appears around puberty, with a mean age at onset of 14 years and a higher incidence in girls than in boys [Jeavons, 1982].
Differential Diagnosis
Difficulty is rarely encountered in correctly diagnosing generalized tonic-clonic seizures (formerly termed grand mal seizures). Generalized tonic-clonic seizures can be divided into those that are primarily generalized from onset and those that begin as partial seizures and then secondarily generalize [Schmidt et al., 1983; Jobst et al., 2001]. Differentiating features are listed in Table 53-1. On very rare occasions, generalized tonic-clonic seizures begin as another generalized seizure type such as absence [Niedermeyer, 1976] before evolving into tonic-clonic activity.
Table 53-1 Comparison of Primary and Secondary Generalized Tonic-Clonic Seizures
| Primary GTC | Secondary GTC | |
|---|---|---|
| Prodrome | Occasionally | Occasionally |
| Aura | Never | Frequently |
| Usual duration | 1–3 min | 1–3 min |
| Family history for seizures | Frequently | Occasionally |
| Acquired lesions | Rarely | Frequently |
| EEG Interictal Photoconvulsive response Hyperventilation |
Normal or generalized spike-wave Frequently May activate spike-wave discharges |
Normal or focal or multifocal spikes Rarely Usually does not activate epileptiform discharges |
Breath-holding attacks, pseudoseizures (nonepileptic events), and syncope occasionally may resemble generalized tonic-clonic seizures. These are discussed in Chapters 64 and 65.
Etiology
The etiology of seizures in children who have secondary generalized tonic-clonic seizures is the same as those described for partial seizures. Patients with primary generalized tonic-clonic seizures usually do not have structural lesions and often have a familial disorder, such as juvenile myoclonic epilepsy. The genetics of generalized tonic-clonic seizures are discussed in Chapter 52.
Comorbidities Associated with Generalized Seizures
While a major goal in the treatment of epilepsy is stopping the seizures, it is not the only treatment goal. Patients with epilepsy are a risk for a number of comorbidities. Comorbidity refers to the co-occurrence of two supposedly separate conditions that occur together more than by chance (also see Chapter 62). Depression occurs more frequently in patients with epilepsy than in the normal population, so that epilepsy and depression are comorbidities. Comorbidities are not necessarily causal. For example, because epilepsy and depression are comorbidities does not mean that epilepsy caused the depression or depression caused the epilepsy. Rather, it is possible that both conditions have a common biological substrate or that another independent variable triggers one of the comorbidities. For example, epilepsy often leads to drug therapy, which could cause depression independently of the epilepsy.
Comorbidities associated with epilepsy include depression [Camfield and Camfield, 2009; Dunn et al., 1999], suicidality [Bridge et al., 2007; Tellez-Zenteno et al., 2007; Donner et al., 2001; McGregor and Wheless, 2006], attention-deficit hyperactivity disorder (ADHD) [Dunn and Kronenberger, 2005], conduct disorders [Dunn et al., 2009], anxiety [Caplan et al., 2005], cognitive impairment and learning disabilities [Fastenau et al., 2004; Aldenkamp et al., 2005], and migraine [Pellock, 2004].
Medical Treatment
One of the issues is that, in many studies, partial seizures with secondary generalization are included along with primary generalized tonic-clonic seizures. In the United States, AEDs that have an indication for monotherapy or adjunctive therapy of generalized seizures include phenytoin and carbamazepine. However, while both drugs are effective in partial seizures, they can exacerbate primary generalized seizures [Perucca et al., 1998; Guerrini et al., 1998]. Studies examining efficacy and safety of AEDs in children with primary generalized epilepsy are very limited. Although a number of the newer AEDs are being used for the treatment of primary generalized tonic-clonic seizures, even in adults definitive evidence for their effectiveness is lacking [Faught, 2003; French et al., 2004]. In addition, data for adults and children often are combined when studies are reported. Antiepileptic drug studies done solely in children with primary generalized tonic-clonic seizures are limited. Although most of the newer antiepileptic drugs have been used in the treatment of generalized tonic-clonic seizures, their usefulness in children has not yet been established [Holmes, 2003; Jarrar and Buchhalter, 2003].
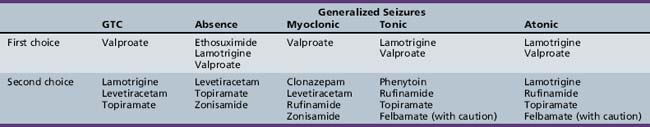
The major drugs typically used to treat primary generalized tonic-clonic seizures include valproate [Murphy and Delanty, 2000; Ramsay and DeToledo, 1997; Marson et al., 2007], lamotrigine [Mikati and Holmes, 1997; Trevathan et al., 2006; Marson et al., 2007], levetiracetam [Rosenfeld et al., 2009;Noachtar et al., 2008;Wheless, 2007], and topiramate [Biton et al., 1999; Marson et al., 2007]. Although phenobarbital is effective in the treatment of primary generalized tonic-clonic seizures, the adverse side effects profile has reduced its use as initial therapy for primary generalized tonic-clonic seizures [Taylor et al., 2001].
The four drugs most commonly used for primary generalized tonic-clonic seizures are reviewed briefly here. Additional information about AEDs is available in Chapter 59.
Valproate
Valproate is approximately 90 percent protein-bound. Protein binding varies with drug plasma concentration, and the free fraction increases with increasing plasma concentration [Davis et al., 1994]. Therefore, with high blood levels of valproate, the free, or active, portion of valproate becomes greater, and toxicity may occur. In addition, drugs or endogenous substances, such as free fatty acids, can alter protein binding. The primary metabolism is by hepatic hydroxylation (mitochondrial beta oxidation) and conjugation with glucuronide. Eighty percent of an administered dose of valproic acid is metabolized through these two pathways and then excreted by the kidney [Levy et al., 2002]. The remainder of a dose is excreted in other oxidized metabolites. Drug interactions are common with valproate [Scheyer, 2002].
Gastrointestinal toxicity includes anorexia, nausea, and indigestion. These symptoms may be reduced with the divalproex sodium preparations. Dose-related toxicities are action tremor (seen in 40 percent of adults, less frequently in children), elevated plasma transaminase (usually transient but a possible harbinger of serious hepatic disease), and hyperammonemia. Idiosyncratic toxicity includes hepatic necrosis (treatable with l-carnitine), thrombocytopenia, pancreatitis (in 0.5 percent, sometimes fatal), teratogenicity, and stupor and coma. The risk of hepatic fatality is greatest in children younger than 2 years and in patients taking valproic acid in combination with other AEDs [Dreifuss et al., 1987, 1989]. Toxic effects with long-term use are weight gain, hair loss, and platelet dysfunction. Routine monitoring of liver function tests, complete blood count, and valproate blood level determination is recommended.
Lamotrigine
Lamotrigine is a broad-spectrum AED that has a number of attractive features for childhood use. However, there is a risk of rash and the drug must be used with care. Lamotrigine has broad-spectrum efficacy, demonstrating efficacy in partial seizures, absences, tonic, atonic, and generalized tonic-clonic seizures [Besag et al., 1995; Eriksson et al., 1998; Duchowny et al., 2002]. It is indicated as adjunctive therapy for adults and in children with primary generalized seizures and generalized seizures in the Lennox–Gastaut syndrome in children over the age of 2 years. There are indications that the drug is useful in absence seizures.
Lamotrigine is generally well tolerated in children [Duchowny et al., 2002]. The drug is not associated with behavioral or cognitive side effects. With high doses, particularly when used as adjunctive therapy, dizziness, diplopia, headache, ataxia, tremor, and nausea may occur.
Data from clinical trials with lamotrigine indicate that the risk of serious rash in pediatric patients is higher than in adults. The incidence of rash associated with hospitalization among adults treated with lamotrigine is 0.3 percent and the incidence among children is 1.0 percent [Messenheimer, 1998; Hirsch et al., 2006]. The incidence of cases reported as possible Stevens–Johnson syndrome is 0.1 percent for adult patients and 0.5 percent for pediatric patients. The risk of rash is higher in children who are taking valproate, presumably because it decreases the metabolism of lamotrigine. Most rashes occur during the first 6 weeks of therapy. The risk of rash is reduced by slow upward titration of dose.
Levetiracetam
Levetiracetam is a broad-spectrum drug that is widely used in both children and adults for partial and generalized seizures [Carreno, 2007; Wheless, 2007]. It is rapidly and almost completely absorbed after oral administration. The absorption and elimination of levetiracetam are linear. Levetiracetam is not extensively metabolized in humans; the majority of the drug is excreted unchanged in the urine. The major metabolic pathway of levetiracetam is enzymatic hydrolysis of the acetamide group. Levetiracetam produces no inhibition of cytochrome P450 isoforms, epoxide hydrolase, or UDP-glucuronide enzymes. There are no reports of drug interactions between levetiracetam and AEDs or other drugs. The metabolites have no known pharmacological activity and are excreted in the urine. The elimination half-life is 4–8 hours in children. The pharmacodynamic effects of levetiracetam are longer than would be predicted by half-life and the drug can be given twice daily.
There are no reports of serious toxicity with levetiracetam. The drug is well tolerated in children [Wheless and Ng, 2002]. In children, headache, anorexia, somnolence, and mild infection (otitis media, pharyngitis, and gastroenteritis) have been reported [Glauser et al., 2002]. These usually occur early in treatment and can be reduced by slow drug initiation and/or dose reduction. Levetiracetam has caused irritability and aggressiveness [Khurana et al., 2007]. Reversible treatment-emergent psychosis has been associated with levetiracetam therapy [Kossoff et al., 2001]. No hematological or hepatic disturbances have been reported and routine laboratory studies are not recommended.
Topiramate
Topiramate has a broad spectrum of efficacy in children [Ormrod and McClellan, 2001], and has been found to be beneficial in the treatment of medically refractory primary generalized tonic-clonic seizures [Biton et al., 1999; French et al., 2004; Wheless, 2000].
Topiramate is rapidly absorbed and has very low protein binding [Bourgeois, 2000]. In the absence of enzyme-inducing drugs, 80 percent of a dose is excreted unchanged in the urine, with an elimination half-life of 20–30 hours. In the presence of enzyme-inducing drugs, 50–80 percent of a dose is excreted unchanged in the urine, with an elimination half-life of 12–15 hours in teenagers. The metabolic products of topiramate are formed in the liver and do not appear to be biologically active. Prepubescent children have a higher clearance and shorter elimination half-life than in adults [Bourgeois, 2000].
The most common side effects with topiramate are central nervous system-related: drowsiness, dizziness, decreased attention or impaired concentration, paresthesia, nervousness, confusion, and impaired memory. These side effects usually are mild to moderate, develop during the first weeks of therapy, and may decline over time. The central nervous system side effects appear to be fewer when topiramate is used for monotherapy than when it is used in polytherapy, and are less common in children than in adults. Weight loss [Ormrod and McClellan, 2001] and acidosis [Izzedine et al., 2004; Takeoka et al., 2001] can occur with topiramate therapy. Renal stones [Kuo et al., 2002; Lamb et al., 2004] and acute myopia [Coats, 2003] associated with secondary angle-closure glaucoma [Browne et al., 1974] have been reported. These side effects appear to be very rare in children.
Table 53-2 summarizes the AEDs used in the treatment of generalized tonic-clonic seizures and other generalized seizures in childhood. This listing is based on both efficacy and tolerability, and reflects the author’s personal preferences. AED therapy should always be tailored to the needs of the particular patient.
In addition to treating with AEDs, it is important to counsel the patient and parents about factors that could exacerbate seizures, such as sleep deprivation. Drugs that can lower seizure threshold, such as bupropion, should be avoided [Foley et al., 2006].
Absence Seizures
Clinical Features
Absence seizures, formerly termed petit mal seizures, are characterized by an abrupt cessation of activity, change in facial expression, and impairment of consciousness [Browne et al., 1974; Pearl and Holmes, 2008; Porter et al., 1973]. Absence seizures are not common, accounting for less than 10 percent of all seizure types [Hauser and Hersdorffer, 1990; Hauser and Kurland, 1975; Sofijanov, 1982]. Absence seizures may be the most common seizure type to go undetected. The prevalence of absence seizures is highest during the first 10 years of life and then drops dramatically to a very low level [Hauser and Kurland, 1975; Hertoft, 1963; Sato et al., 1976]. Absence seizures are more common in girls than in boys [Hertoft, 1963; Sato et al., 1976]. Typical absence seizures rarely start before the age of 2 years or after the teenage years [Sato et al., 1983; Sato, 1983]. In a study of 83 patients with absence seizures, Sato and co-workers [Sato et al., 1983] found the average age at onset was 3.8 years.
Absence seizures are classified as typical or atypical in type (Table 53-3). Typical absence seizures are short, rarely lasting over 30 seconds; as with other generalized seizures, they are never associated with an aura or postictal impairment. The sudden onset of impaired consciousness, usually associated with a blank facial appearance without other motor or behavioral phenomena, is characteristic. The degree of impairment of consciousness is variable. Some children remember virtually everything that is said during the seizure, whereas for others, the entire duration of the seizure is “time lost.”
| Clinical Seizure Type | EEG – Interictal | EEG – Ictal |
|---|---|---|
| Typical absence |
Although the absence seizure is commonly thought to consist only of staring, in fact the behavioral changes associated with the seizure type usually are more complex. Most absence seizures are accompanied by motor, behavioral, or autonomic phenomena, and seizures characterized by only staring and altered consciousness are unusual. Penry et al. [1975] reviewed 374 absence seizures recorded on videotape from 48 patients and found simple absences, characterized by staring and cessation of activities, to constitute only 9.4 percent of the seizures. A majority of the patients had other clinical manifestations.
Automatisms, semipurposeful behaviors of which the patient is unaware and subsequently cannot recall, are very common with absence seizures [Holmes et al., 1987; Penry et al., 1975]. They either may be perseverative, reflecting continuation of preictal activities, or may arise de novo. Simple behaviors, such as rubbing the face or hands, licking the lips, chewing, grimacing, scratching, or fumbling with clothes, tend to be de novo automatisms. Complex activities, such as dealing cards, moving a chess piece, or handling a toy, are generally perseverative. Speech, if it occurs during the seizure, usually is perseverative and may be slow and slurred, but also may be totally normal [McKeever et al., 1983]. The longer the absence seizure lasts, the more likely automatisms are to occur [Penry et al., 1975].
Clonic or myoclonic components are common but may be quite subtle, most frequently consisting of blinking. Clonic activity also may be manifested by nystagmus, rapid jerking or trembling of the arms, or head nods. Alterations in muscle tone may lead to stiffening of the trunk or a fall. A study of 426 typical and 500 atypical absence seizures studied in 54 children using simultaneous EEG and video monitoring found myoclonic jerks in 13 percent of typical absences and 12 percent of atypical absences [Holmes et al., 1987]. Children with the syndrome of epilepsy with myoclonic absences have prominent myoclonus during the seizure.
The frequency of absence seizures varies considerably from day to day and even from hour to hour. The number of absence seizures varies significantly with different environmental situations [Borkowski, et al., 1992; Pearl and Holmes, 2008]. Seizures are more likely to occur during periods of inactivity than when the child is busily engaged in a task. Fatigue also may dramatically increase seizure susceptibility.
A majority of children with typical absence seizures have normal findings on neurologic examination [Dalby, 1969; Sato, 1983]. When neurologic abnormalities are found, they usually are mild and nonprogressive. Most children with typical absence seizures have normal or mildly low intelligence [Dalby, 1969; Sato, 1983]. Compared with age-matched controls, children with absence seizures have lower general cognitive function with impaired visual-spatial skills and memory disturbances [Pavone et al., 2001]. Children with absence seizures have higher rates of behavioral problems and psychopathology than in the normal population [Ott et al., 2001].
Typical absence seizures may be associated with generalized tonic-clonic seizures in 40–60 percent of patients [Hertoft, 1963; Loiseau et al., 1983; Sato, 1983]. In most children, the generalized tonic-clonic seizures occur after onset of the absence seizures [Loiseau et al., 1983].
Although atypical absence seizures form a separate category of absences, overlap between the two seizure types is considerable, and they appear to represent a clinical continuum [Holmes et al., 1987]. Diminished postural tone, or tonic or myoclonic activity, is significantly more likely to be the initial clinical feature in atypical than in typical absences. Automatisms are less likely in atypical absences than in typical absences. Like typical absences, atypical absences have a distinct onset and ending, without auras or postictal symptoms. Although atypical absences usually are of longer duration than typical absences, a considerable amount of variability exists.
Epilepsy with Myoclonic Absences
Epilepsy with myoclonic absences consists of typical absence seizures with a sudden onset and offset, in which the child has axial hypertonia with the trunk bent slightly forward and the arms and shoulders raised. In conjuction with the 3-Hz generalized spike and wave on the EEG, there are rhythmic jerks of the arms and shoulders [Genton and Bureau, 2006; Tassinari et al., 1992]. The condition should be differentiated from eyelid myoclonia with absences or Jeavons’ syndrome, which is a generalized epileptic condition characterized by eyelid myoclonia with or without absences [Striano et al., 2009]. Epilepsy with myoclonic absences is often more difficult to treat than typical absence seizures.
Juvenile Myoclonic Epilepsy
Juvenile myoclonic epilepsy is a familial disorder that typically begins in the second decade of life and is characterized by mild myoclonic seizures, generalized tonic-clonic or clonic-tonic-clonic seizures (a variation of generalized tonic-clonic seizures in which an initial clonic phase precedes the usual pattern), and occasionally absence seizures. This syndrome is discussed in Chapter 56.
Differential Diagnosis
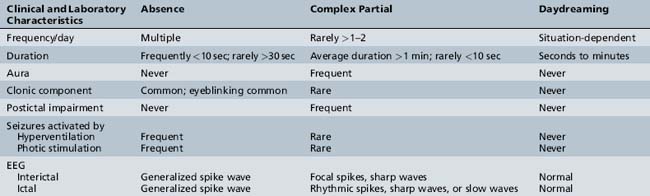
The primary diagnostic considerations in the child referred because of “staring attacks” include absence seizures, complex partial seizures, and daydreaming (Table 53-4). Complex partial seizures are more common than absence seizures and also are manifested by an alteration in consciousness with staring, automatisms, changes in tone, and autonomic symptoms. Complex partial seizures tend to be longer and less frequent, but clinically no absolute distinguishing factor may be present. The presence of an aura or postictal impairment is strongly suggestive of a complex partial seizure. In the child not on antiepileptic medications, 3 minutes of hyperventilation usually precipitates an absence seizure. It is unusual for complex partial seizures to be precipitated by hyperventilation. Abnormalities documented on the EEG constitute the best confirmation of either seizure type.
Etiology
Both acquired and inherited factors are implicated in the etiology of absence seizures, reflecting the heterogeneity of the patient population. Genetic factors predominate in children with typical absence seizures, whereas acquired disorders are more common in children with atypical absences. The genetics of absence seizures are discussed in Chapter 52.
Electroencephalographic Findings
The EEG signature of a typical absence seizure is the sudden onset of 3-Hz generalized symmetric spike-and-wave or multiple spike-wave complexes (Figure 53-2). The voltage of the discharges often is maximal in the frontal-central regions. The frequency tends to be faster, about 4 Hz, at the onset and may slow to 2 Hz toward the end of a discharge lasting longer than 10 seconds.
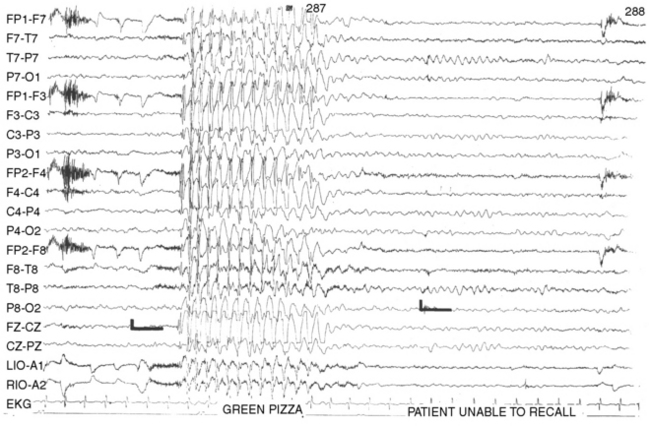
Hyperventilation is a potent activator of typical absence seizures [Adams and Lueders, 1981; Dalby, 1969]. Failure to induce an absence seizure with several trials of hyperventilation of 3–5 minutes’ duration in a child not receiving antiepileptic medication would make the diagnosis of typical absence seizures unlikely. Photic stimulation also may precipitate seizures, although the frequency of activation does not appear as high as with hyperventilation [Newmark and Penry, 1979].
When closely observed, children typically demonstrate behavioral changes with spike-and-wave discharges lasting longer than a few seconds. In studies of auditory reaction times, it has been found that responses are abnormal in 80 percent of the situations in which a stimulus is presented after onset of the spike-and-wave activity [Porter et al., 1973]. In a test of visual motor coordination in children with spike-and-wave activity on the EEG, discharges lasting longer than 3 seconds were associated with impaired function [Opp et al., 1992; Brown et al., 1974; Porter et al., 1973].
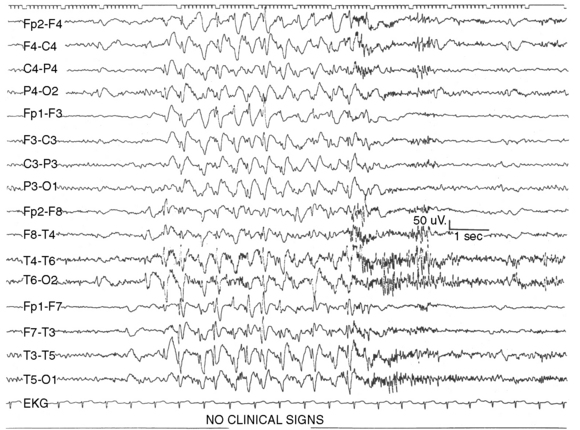
In atypical absences, the ictal EEG is more heterogeneous, showing 1.5- to 2.5-Hz slow spike-and-wave or multiple spike-and-wave discharges that may be irregular or asymmetric (Figure 53-3). Interictal EEG findings usually are abnormal, with background slowing and multifocal epileptiform features. The EEG features of typical and atypical absences are summarized in Table 53-3.
Pathophysiology
A number of studies have demonstrated that the basic underlying mechanism in generalized absence epilepsies involves thalamocortical circuitry and the generation of abnormal oscillatory rhythms in this neuronal network [Coulter and Zhang, 1994; Crunelli and Leresche, 2002; Hosford et al., 1995; Huguenard and Prince, 1994b; Steriade et al., 1993; Steriade and Conteras, 1998]. The neuronal circuits that generate the oscillatory thalamocortical burst firing observed during absence seizures have now been identified. This circuit includes cortical pyramidal neurons, thalamic relay neurons, and the nucleus reticularis thalami (Figure 53-4) [Steriade and Llinas, 1988; Steriade et al., 1993]. The principal synaptic connections of the thalamocortical circuit include glutamatergic fibers between neocortical pyramidal cells and the nucleus reticularis thalami, gamma-aminobutyric acid (GABA)-ergic fibers from nucleus reticularis thalami neurons that activate GABAA and GABAB receptors on thalamic relay neurons, and recurrent collateral GABAergic fibers from nucleus reticularis thalami neurons that activate GABAA receptors on adjacent nucleus reticularis thalami neurons. The nucleus reticularis thalami, therefore, is in a position to influence the flow of information between the thalamus and cerebral cortex [Snead, 1995]. The nucleus reticularis thalami cells undergo rhythmic burst firing during periods of sleep and continuous single-spike firing during wakefulness.
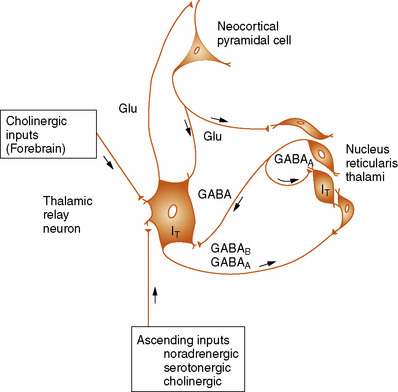
T, T-current; Glu, Glutamatergic transmission.
(From Holmes GL. Basic mechanisms in epilepsy. Int Pediatr 1996;11:343.)
The cellular events that underlie the ability of nucleus reticularis thalami neurons to shift between an oscillatory and a firing mode include activation of the low-threshold, transient Ca2+ channels (T-channels) [Coulter et al., 1989, 1990; Crunelli and Leresche, 2002; Snead, 1995]. These channels appear to be important in the generation of absence seizures [Snead, 1995; Tsakiridou et al., 1995]. Mild depolarization of these neurons is sufficient to activate these T-channels and to allow the influx of extracellular Ca2+. Further depolarization produced by Ca2+ inflow often exceeds the threshold for firing a burst of action potentials. After T-channels are activated, they become inactivated rather quickly – hence the name transient. T-channels require a lengthy, intense hyperpolarization to reverse their inactivation (a process termed deinactivation). The requisite hyperpolarization can be provided by GABAB receptors that are present on thalamic relay neurons. The interplay between GABAB-mediated inhibition and the low-threshold T-type calcium channel therefore plays a critical role in generating the oscillating hyperpolarization/depolarization activity seen in the thalamus. In animal models of absences, GABAB agonists produce an increase in seizure frequency (by facilitating deinactivation of T-channels), whereas GABAB antagonists reduce seizure frequency.
Abnormal oscillatory rhythms could be caused by abnormalities of the T-channel or enhanced GABAB function [Crunelli and Leresche, 2002; Snead, 1995]. In some animal models of absence seizures, T-channel activation in the nucleus reticularis thalami is significantly different than in control animals [Tsakiridou et al., 1995]. These aberrant T-channels may be one basis for absence seizures. In other animal models, an increase in GABAB receptors in thalamic and neocortical neuronal populations, compared with that in controls, has been described [Hosford et al., 1995]. Because an increase in GABA in the thalamic relay neurons would lead to enhanced hyperpolarization and consequently deinactivation of the T-channels, GABAB agonists produce an increase in seizure frequency, whereas GABAB antagonists reduce seizure frequency in rodent models of absence epilepsy [Hosford et al., 1992; Liu et al., 1992; Marescaux et al., 1992; Tsakiridou et al., 1995].
Supporting these animal findings are the clinical observations that three drugs efficacious in the treatment of absence seizures – valproic acid, ethosuximide, and trimethadione– suppress T-currents [Coulter et al., 1990]. In addition, some clinical evidence indicates that vigabatrin, which increases endogenous GABA levels and thereby increases the activation of GABAB receptors, worsens absence seizures in patients. Clonazepam, which activates GABAA levels in the nucleus reticularis thalami, can be an effective anti-absence drug [Huguenard and Prince, 1994a].
Treatment
In a randomized, controlled clinical trial of 453 children with childhood absence epilepsy who were randomly assigned to one of these three drugs, ethosuximide and valproic acid were more efficacious than lamotrigine whereas there were no significant differences among the three drugs with regard to discontinuation because of adverse events (Glauser et al., 2010). However, ethosuximide had less effect on attention than valproic acid. This study would suggest that when considering both efficacy and side effects ethosuximide is the drug of first choice for childhood absence epilepsy. A single agent should be chosen and, after appropriate laboratory studies, initiated at a low dose and gradually increased. AED levels may be helpful, but dose changes in either direction should follow clinical indications. On dosage modifications, drug levels should be obtained only after sufficient time has elapsed to reach steady-state serum concentrations.
In the United States, many physicians will begin therapy with ethosuximide, primarily because of the rare, but severe, hepatotoxicity and pancreatitis associated with valproic acid [Dreifuss et al., 1987, 1989; Penry and Dean, 1993]. Ethosuximide has proved over the past two decades to be a safe and effective AED used almost exclusively for absence seizures [Schneider, 1993; Perucca, 1990]. Valproic acid is equally effective and generally is considered the drug of choice in the patient who has both absence and generalized tonic-clonic seizures, because ethosuximide is not effective in the treatment of generalized tonic-clonic seizures. Lamotrigine appears to be a promising drug in the treatment of absence seizures [Frank et al., 1999; French et al., 2004; Holmes et al., 2008].
Benzodiazepines, such as clonazepam, may be effective, but they usually are reserved for refractory cases because of the relatively high incidence of drowsiness and behavioral side effects [Pearl and Holmes, 2008]. Topiramate [Cross, 2002; Wheless, 2000], levetiracetam [Cohen, 2003], and acetazolamide [Reiss and Oles, 1996] may be useful. Carbamazepine [Yang et al., 2003], vigabatrin [Yang et al., 2003], and tiagabine [Knake et al., 1999] should be avoided because they may exacerbate absence seizures.
In addition to obtaining an interim history, the clinician should always have the child perform a hyperventilation test at follow-up visits. Eliciting a seizure during hyperventilation strongly indicates that the epilepsy disorder is not totally controlled [Adams and Lueders, 1981].
Prognosis
Approximately two-thirds of children with childhood absence epilepsy can be expected to enter long-term remission [Wirrell, 2003]. Sato et al. [1976] identified favorable prognostic signs in absence seizures as a negative family history of epilepsy, normal EEG background activity, and normal intelligence. Nearly 90 percent of children with these characteristics stopped having absence seizures.
Juvenile absence epilepsy may occasionally persist into adulthood, however, and juvenile myoclonic epilepsy, as discussed later on, does not spontaneously remit [Wirrell et al., 1996]. As a rule, onset of generalized tonic-clonic seizures before absence seizures carries a poorer prognosis than that noted with the reverse order [Sato et al., 1976].
Tonic Seizures
Clinical Features
Tonic seizures are brief seizures (usually lasting less than 60 seconds), consisting of the sudden onset of increased tone in the extensor muscles [Egli et al., 1985; Holmes, 1988]. If standing, the patient typically falls to the ground. Electromyographic activity is dramatically increased in tonic seizures.
Impairment of consciousness during the seizure is characteristic, although when seizures are brief, this change may be difficult to detect. Tonic seizures frequently are seen in patients with Lennox–Gastaut syndrome, a disorder consisting of a mixed seizure disorder, mental retardation, and the EEG findings of a slow spike-and-wave pattern [Aicardi, 1988a, b; Markand, 2003]. The seizures usually occur more frequently at night.
Tonic seizures frequently begin with a tonic contraction of the neck muscles, leading to fixation of the head in an erect position, widely opened eyes, and jaw clenching or mouth opening [Holmes, 1988]. Contraction of the respiratory and abdominal muscles often follows, sometimes leading to a high-pitched cry and brief periods of apnea. The tonic contractions may extend to the proximal musculature of the upper limbs, elevating the shoulders and abducting the arms. Asymmetric tonic seizures vary in severity, ranging from a slight rotation of the head to a tonic contraction of all of the musculature on one side of the body. Eyelid retraction, staring, mydriasis, and apnea also may occur [Aicardi, 1988b]. Occasionally, tonic seizures terminate with a brief clonic phase. Unlike in generalized tonic-clonic seizures, however, the clonic phase is abbreviated. Postictal impairment, with confusion, tiredness, and headache, is common. The degree of postictal impairment usually is related to the duration of the seizure.
Etiology and Prognosis
Tonic seizures usually occur in children with Lennox–Gastaut syndrome, which is discussed later.
Electroencephalographic Findings
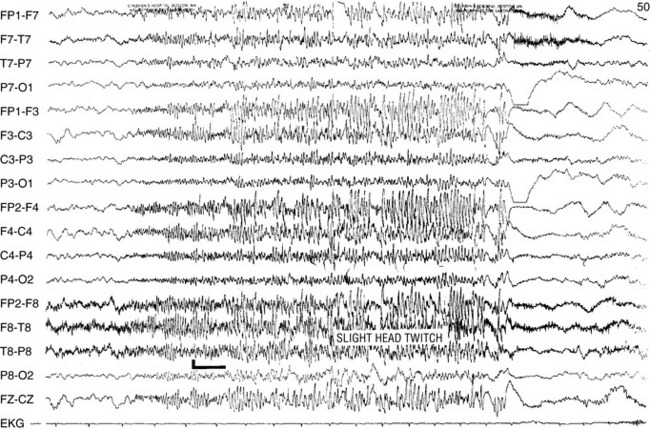
The interictal EEG pattern in patients with tonic seizures usually is quite abnormal, consisting of slowing of the background, with multifocal spikes, sharp waves, and bursts of irregular spike-and-wave activity. The EEG ictal manifestations of tonic seizures usually consist of bilateral synchronous spikes of 10–25 Hz, of medium to high voltage, with a frontal accentuation. Simple flattening or desynchronization may also occur (Figure 53-5). Occasional multiple spike-and-wave or diffuse slow-wave activity may occur during a tonic seizure.
Atonic Seizures
Clinical Features
Atonic (astatic) seizures, or “drop attacks,” are characterized by a sudden loss of muscle tone [Oguni et al., 1992, 1997]. They begin suddenly and without warning, and cause the patient, if standing, to fall quickly to the floor. Children with atonic seizures are more likely to fall backward than children with tonic seizures [Oguni et al., 1997]. Because muscle tone may be completely absent, the children have little means by which to protect themselves, and injuries often occur. The attack may be fragmentary and manifest as dropping of the head with slackening of the jaw or dropping of a limb.
At times, it may be difficult to distinguish epileptic from nonepileptic head drops. Children with head drops secondary to seizures often have a change in facial expression and subtle myoclonic extremity movements associated with the head drops. Head drops with a rapid head descent, followed by a slow recovery to the upright position, usually represent seizures [Brunquell et al., 1990].
In atonic seizures, a loss of electromyographic activity is characteristic. Consciousness is impaired during the fall, although the patient may regain alertness immediately on hitting the floor. Atonic attacks are frequently associated with myoclonic jerks either before, during, or after the atonic seizure [Egli et al., 1985; Schneider et al., 1970]. This combination has been described as myoclonic-astatic seizures. Atonic seizures are rare [Egli et al., 1985; Ikeno et al., 1985]; a majority of children with drop attacks have myoclonic or tonic seizures [Ikeno et al., 1985].
Lennox–Gastaut Syndrome
Lennox–Gastaut syndrome is characterized by a mixed seizure disorder, of which tonic seizures are a major component, along with a slow spike-and-wave EEG pattern. The syndrome always begins in childhood and often is accompanied by mental retardation [Markand, 2003].
The child with Lennox–Gastaut syndrome typically has a mixture of seizure types. The most frequently occurring are tonic, tonic-clonic, myoclonic, atypical absences, and “head drops,” which represent a form of atonic, tonic, or myoclonic seizures. Lennox–Gastaut syndrome is characterized by very frequent seizures, usually occurring multiple times per day. Tonic seizures, in particular, are a major problem in this syndrome [Aicardi, 1988b; Chevrie and Aicardi, 1972]. The tonic seizures usually are brief, lasting from a few seconds to a minute, with an average duration of about 10 seconds.
Myoclonic seizures, occurring either in isolation or as a component of an absence seizure, can occur in this disorder. In some patients, the myoclonus may be so prominent that some investigators have described a myoclonic variant of the Lennox–Gastaut syndrome [Aicardi, 1988b]. Atypical absence and generalized tonic-clonic seizures are seen in more than half of the patients with Lennox–Gastaut syndrome [Schneider et al., 1970]. Generalized tonic-clonic seizures usually cause the most concern to parents and often are the seizure type to precipitate hospitalization.
Mental retardation is present before the onset of seizures in 20–60 percent of patients [Aicardi, 1988b]. Some patients, with an idiopathic or cryptogenic etiology of their seizures, have normal intelligence quotient (IQ) scores or developmental histories before the onset of their seizures. The proportion of retarded patients increases with age because of the deterioration that frequently occurs in Lennox–Gastaut syndrome. Fluctuations in cognitive abilities may occur and are correlated to some degree with the intensity of EEG abnormalities. Behavioral problems also are common in Lennox–Gastaut syndrome, ranging from hyperactivity to frank psychotic and autistic behavior. Abnormalities on the neurologic examination have been reported in 30–88 percent of patients with Lennox–Gastaut syndrome [Kurokowa et al., 1980; Markand, 1977; Schneider et al., 1970].
Patients with Lennox–Gastaut syndrome typically have very frequent seizures [Markand, 1977; Papini et al., 1984]. It is not unusual for some children with this syndrome to have hundreds of seizures per day. Seizure frequency may vary during the course of a day, with the highest frequency during drowsiness and inactivity [Papini et al., 1984]. In addition, in many of the patients, a weekly or monthly periodicity in seizure frequency is observed that is unrelated to AED therapy [Aicardi, 1973]. Periods of prolonged repetitive seizures of a mixed type are interspersed with periods of relative freedom from attacks. During times when the child is seizure-free, marked improvements in alertness, motivation, and academic progress can be seen. Unfortunately, these periods usually are short-lived, leading to a great deal of frustration for the child, parent, school personnel, and medical professionals.
Electroencephalographic Findings
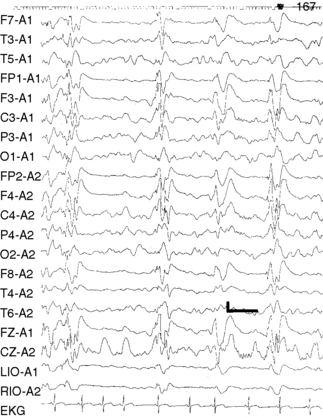
The hallmark of the EEG pattern in Lennox–Gastaut syndrome is the slow spike-and-wave discharge superimposed on an abnormal, slow background. The slow spike-and-wave or sharp-and-slow-wave complexes consist of generalized discharges occurring at a frequency of 1.5–2.5 Hz. The morphology, amplitude, and repetition rate may vary both between bursts and during paroxysmal bursts of spike-and-wave activity, and asymmetries of the discharge are frequent. The area of maximum voltage, although variable, usually is frontal or temporal in location. Often, sleep increases the number of epileptiform discharges, but these discharges may slow in frequency and become even more irregular than during the awake state (Figure 53-6). In REM sleep, the paroxysmal activity decreases markedly. Hyperventilation and photic stimulation rarely activate these discharges.
Etiology
The syndrome may arise de novo in a previously well child or may supervene in an already neurologically or medically handicapped patient. Lennox–Gastaut syndrome has been divided into primary and secondary cases. Primary refers to cases in which the etiology is idiopathic, whereas secondary refers to cases in which the disorder is symptomatic of a definable etiology. Markand [1977] was able to identify an underlying disorder in 64 percent of 83 patients, and Chevrie and Aicardi [1972] were able to do so in 66 percent of 80 patients. Other investigators, however, have reported a smaller incidence of identifiable causes in Lennox–Gastaut syndrome [Kurokowa et al., 1980; Schneider et al., 1970]. The causes of Lennox–Gastaut syndrome are listed in Box 53-1 and are similar to those seen in infantile spasms. In fact, in many patients with infantile spasms, (30–40 percent) the disorder will progress to Lennox–Gastaut syndrome [Kurokawa et al., 1980; Olmos-Garcia de Alba et al., 1984; Riikonen, 1996]. In some children with infantile spasms that evolve into Lennox–Gastaut syndrome the epileptic spasms continue to occur in series and become one of the seizure types (Donat and Wright, 1991).














Although a majority of patients with Lennox–Gastaut syndrome have a static disorder, degenerative disorders may occasionally manifest as this syndrome [Markand, 1977; Pampiglione and Harden, 1977]. Genetic factors do not play a significant role in a majority of individuals with this disorder.
Treatment
In the United States, valproate likely is the most commonly used drug in the treatment of Lennox–Gastaut syndrome. It is a broad-spectrum drug with effectiveness against all of the common seizure types in the syndrome. The lack of sedative side effects and the broad spectrum of action make the drug appealing. The drug has been associated with hepatotoxicity, however, particularly in children younger than 2 years of age. Phenytoin is considered by some investigators to be the drug of choice for tonic seizures. Intravenous phenytoin is used as a first-line drug for status epilepticus in Lennox–Gastaut syndrome (Richard et al., 1993) since benzodiazepines can precipitate tonic status epilepticus in Lennox–Gastaut syndrome [Tassinari et al., 1972].
Felbamate, released in the United States in 1993, was the first drug to be demonstrated to be effective in Lennox–Gastaut syndrome [Dodson, 1993a, b; The Felbamate Study Group in Lennox–Gastaut Syndrome, 1993]. After the drug was released, it was found to result in fatal aplastic anemia and hepatic failure. The overall risk of aplastic anemia for patients taking felbamate is estimated to be 1:5000, 100 times greater than the risk in the normal population, and the risk of fatal outcome is approximately 1:10,000 [Kaufman et al., 1997]. Felbamate-associated aplastic anemia has not been reported in children younger than 13 years [Brodie and Pellock, 1995; Pellock and Brodie, 1997]. Risk factors associated with aplastic anemia include a history of blood dyscrasia or of substantial toxicity to other AEDs, serologic or clinical evidence of an autoimmune disorder, and a history of treatment with felbamate for less than 1 year [Pellock, 1999].
Both lamotrigine [Donaldson et al., 1997; Motte et al., 1997] and topiramate [Guerreiro et al., 1997; Ormrod and McClellan, 2001; Sachdeo et al., 1999] have demonstrated efficacy in Lennox–Gastaut syndrome but rarely with total control of the seizures.
In a double-blind, placebo-controlled trial of lamotrigine in 169 patients with Lennox–Gastaut syndrome, either lamotrigine or placebo was added to the maintenance medications. After 16 weeks, 33 percent of children receiving lamotrigine and 16 percent of placebo recipients had more than a 50 percent reduction in seizure frequency [Motte et al., 1997]. In a double-blind, placebo-controlled, multicenter trial of topiramate for adjunctive therapy for Lennox–Gastaut syndrome, the frequency of drop attacks was reduced compared with that in the placebo group [Sachdeo et al., 1999].
The ketogenic diet (see Chapter 60), one of the oldest methods of treating childhood epilepsy, remains a reasonable therapy for children with seizures refractory to standard drug therapy [Vining et al., 1998; Wheless, 2004; Ferrie and Patel, 2009]. Although the diet was considered a therapy of last resort for many years, recent resurgence in interest has occurred, primarily owing to its success in several highly publicized cases. The diet consists of a high proportion of fats and small amounts of carbohydrate and protein, with a fat to carbohydrate and protein ratio of 4:1. Although it is clear that the child must remain in a state of ketosis for the diet to be effective, the basis of the therapeutic effectiveness of the ketogenic diet remains uncertain.
The resurgence in interest in the diet is appropriate because it does provide an attractive alternative in the treatment of childhood epilepsy. The literature supports the view that the ketogenic diet improves seizure control in a significant number of children with medically intractable epilepsy [Prasad et al., 1996; Rubenstein and Vining, 2004]. One-third to one-half of children appear to have an excellent response to the ketogenic diet in terms of a marked or complete cessation of seizures or reduction in seizure severity. In another one-third of the children, a partial reduction in seizure frequency or severity occurs; the remaining children have no appreciable benefit from the diet. Improvement in alertness and behavior often is seen when the child is placed on the diet. It is not clear whether this benefit is secondary to withdrawal of drugs, reduction in seizure frequency, or a direct result of the diet.
Aspects of the surgical evaluation of children with generalized epilepsies, including use of vagal nerve stimulators, are reviewed in Chapter 61.
 The complete list of references for this chapter is available online at
The complete list of references for this chapter is available online at Abou-Khalil B., Ge Q., Desai R., et al. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology. 2001;57:2265-2272.
Adams D.J., Lueders H. Hyperventilation and six-hour EEG recording in evaluation of absence seizures. Neurology. 1981;31:1175-1177.
Aicardi J. The problem of the Lennox syndrome. Dev Med Child Neurol. 1973;15:77-81.
Aicardi J. Epileptic syndromes in childhood. Epilepsia. 1988;29(Suppl 3):S1-S5.
Aicardi J. The Lennox-Gastaut syndrome. Int Pediatr. 1988;3:152-157.
Aldenkamp A.P., Weber B., Overweg-Plandsoen W.C., et al. Educational underachievement in children with epilepsy: a model to predict the effects of epilepsy on educational achievement. J Child Neurol. 2005;20:175-180.
Baulac S., Gourfinkel-An I., Nabbout R., et al. Fever, genes, and epilepsy. Lancet Neurol. 2004;3:421-430.
Beghi M., Beghi E., Cornaggia C.M., et al. Idiopathic generalized epilepsies of adolescence. Epilepsia. 2006;47(Suppl 2):107-110.
Berg A.T., Berkovic S.F., Brodie M.J., et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676-685.
Besag F.M.C., Wallace S.J., Dulac O., et al. Lamotrigine for the treatment of epilepsy in childhood. J Pediatr. 1995;127:991-997.
Betting L.E., Mory S.B., Lopes-Cendes I., et al. EEG features in idiopathic generalized epilepsy: clues to diagnosis. Epilepsia. 2006;47:523-528.
Binnie C.D., Jeavons P.M. Photosensitive epilepsies. In: Roger J., Bureau M., Dravet C., Dreifuss F.E., Perret A., Wolf P., editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey; 1992:299-305.
Biton V., Montouris G.D., Ritter F., et al. A randomized, placebo-controlled study of topiramate in primary generalized tonic-clonic seizures. Topiramate YTC Study Group. Neurology. 1999;52:1330-1337.
Borkowski W.J.Jr, Ellington R.J., Sverdrup E.K. Effect of sleep deprivation on the EEG of learning-impaired children with absence seizures. Clin Electroencephalogr. 1992;23:62-64.
Bourgeois B.F. Pharmacokinetics and pharmacodynamics of topiramate. J Child Neurol. 2000;15(Suppl 1):S27-S30.
Bridge J.A., Iyengar S., Salary C.B., et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA. 2007;297:1683-1696.
Brodie M.J., Pellock J.M. Taming the brain storms: felbamate updated. Lancet. 1995;346:918-919.
Brown T.R., Penry J.K., Porter R.J., et al. Responsiveness before, during and after spike-wave paroxysms. Neurology. 1974;24:659-665.
Brunquell P., McKeever M., Russman B.S. Differentiation of epileptic from nonepileptic head drops in children. Epilepsia. 1990;31:401-405.
Camfield C.S., Camfield P.R. Juvenile myoclonic epilepsy 25 years after seizure onset: a population-based study. Neurology. 2009;73:1041-1045.
Caplan R., Siddarth P., Gurbani S., et al. Depression and anxiety disorders in pediatric epilepsy. Epilepsia. 2005;46:720-730.
Carreno M. Levetiracetam. Drugs Today (Barc). 2007;43:769-794.
Chevrie J.J., Aicardi J. Childhood epileptic encephalopathy with slow spike-wave. A statistical study of 80 cases. Epilepsia. 1972;13:259-271.
Coats D.K. Bilateral angle closure glaucoma in a child receiving oral topiramate. J AAPOS. 2003;7:303.
Cohen J. Levetiracetam monotherapy for primary generalised epilepsy. Seizure. 2003;12:150-153.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:249-260.
Coulter D.A., Huguenard J.R., Prince D.A. Specific petit mal anticonvulsants reduce calcium currents in thalamic neurons. Neurosci Lett. 1989;98:74-78.
Coulter D.A., Huguenard J.R., Prince D.A. Differential effects of petit mal anticonvulsants and convulsants on thalamic neurones: calcium current reduction. Br J Pharmacol. 1990;100:800-805.
Coulter D.A., Zhang Y. Thalamocortical rhythm generation in vitro: physiological mechanisms, pharmacological control, and relevance to generalized absence epilepsy. In: Malafosse A., Genton P., Hirsch E., Marescaux C., Broglin D., Bernasconi R., editors. Idiopathic Generalized Epilepsies: Clinical, Experimental, and Genetic Aspects. London: John Libbey; 1994:123-131.
Cross J.H. Topiramate monotherapy for childhood absence seizures: an open label pilot study. Seizure. 2002;11:406-410.
Crunelli V., Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371-382.
Dalby M.A. Epilepsy and 3 per second spike and wave rhythm. A clinical electroencephalographic and prognostic analysis of 346 patients. Acta Neurol Scand. 1969;45(Suppl 40):1-183.
Davis R., Peters D.H., McTavish D. Valproic acid. A reappraisal of its pharmacological properties and clinical efficacy in epilepsy. Drugs. 1994;47:332-372.
Dodson W.E. Felbamate in the treatment of Lennox-Gastaut syndrome: 12 months follow-on experience after a randomized controlled trial of felbamate versus placebo. Epilepsia. 1993;34(Suppl 7):S17-S23.
Dodson W.E. Felbamate in the treatment of Lennox-Gastaut syndrome: results of a 12-month open-label study following a randomized clinical trial. Epilepsia. 1993;34(Suppl 7):S18-S24.
Donaldson J.A., Glauser T.A., Olberding L.S. Lamotrigine adjunctive therapy in childhood epileptic encephalopathy (the Lennox-Gastaut syndrome). Epilepsia. 1997;38:68-73.
Donat J.F., Wright F.S. Seizures in series: similarities between seizures of the West and Lennox-Gastaut syndromes. Epilepsia. 1991;32:504-509.
Donner E.J., Smith C.R., Snead O.C.III. Sudden unexplained death in children with epilepsy. Neurology. 2001;57:430-434.
Dreifuss F.E., Langer D.H., Moline K.A., et al. Valproic acid hepatic fatalities: US experience since 1984. Neurology. 1989;39:201-207.
Dreifuss F.E., Santilli N., Langer D.H., et al. Valproic acid hepatic fatalities: a retrospective review. Neurology. 1987;37:379-385.
Duchowny M., Gilman J., Messenheimer J., et al. Long-term tolerability and efficacy of lamotrigine in pediatric patients with epilepsy. J Child Neurol. 2002;17:278-285.
Dunn D.W., Austin J.K., Huster G.A. Symptoms of depression in adolescents with epilepsy. J Am Acad Child Adolesc Psychiatry. 1999;38:1132-1138.
Dunn D.W., Austin J.K., Perkins S.M. Prevalence of psychopathology in childhood epilepsy: categorical and dimensional measures. Dev Med Child Neurol. 2009;51:364-372.
Dunn D.W., Kronenberger W.G. Childhood epilepsy, attention problems, and ADHD: review and practical considerations. Semin Pediatr Neurol. 2005;12:222-228.
Egli M., Mothersill I., O’Kane M., et al. The axial spasm – the predominant type of drop seizure in patients with secondary generalized epilepsy. Epilepsia. 1985;26:401-415.
Eriksson A.S., Nergardh A., Hoppu K. The efficacy of lamotrigine in children and adolescents with refractory generalized epilepsy: a randomized, double-blind, crossover study. Epilepsia. 1998;39:495-501.
Fastenau P.S., Shen J., Dunn D.W., et al. Neuropsychological predictors of academic underachievement in pediatric epilepsy: moderating roles of demographic, seizure, and psychosocial variables. Epilepsia. 2004;45:1261-1272.
Faught E. Clinical trials for treatment of primary generalized epilepsies. Epilepsia. 2003;44(Suppl 7):44-50.
Ferrie C.D., Patel A. Treatment of Lennox-Gastaut Syndrome (LGS). Eur J Paediatr Neurol. 2009;13:493-504.
Foley K.F., DeSanty K.P., Kast R.E. Bupropion: pharmacology and therapeutic applications. Expert Rev Neurother. 2006;6:1249-1265.
Frank L.M., Enlow T., Holmes G.L., et al. Lamictal (lamotrigine) monotherapy for typical absence seizures in children. Epilepsia. 1999;40:973-979.
French J.A., et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new onset epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
Gastaut H., Broughton R. Epileptic Seizures; Clinical and Electrographic Features, Diagnosis and Treatment. Springfield: Charles C. Thomas; 1972.
Genton P., Bureau M. Epilepsy with myoclonic absences. CNS Drugs. 2006;20:911-916.
Gilliam F.G., Chiappa K.H. Significance of spontaneous epileptiform abnormalities associated with a photoparoxysmal response. Neurology. 1995;45:453-456.
Glauser T.A., Cnaan A., Shinnar S., et al. Childhood Absence Epilepsy Study Group. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. Mar 4 2010;362(9):790-799.
Glauser T.A., Pellock J.M., Bebin E.M., et al. Efficacy and safety of levetiracetam in children with partial seizures: an open-label trial. Epilepsia. 2002;43:518-524.
Guerreiro M.M., Manreza M.L., Guerreiro C.A., et al. A pilot study of topiramate in children with Lennox-Gastaut syndrome. Neurology. 1997;48:A39.
Guerrini R., Belmonte A., Genton P. Antiepileptic drug-induced worsening of seizures in children. Epilepsia. 1998;39(Suppl 3):S2-S10.
Hauser W.A., Hersdorffer D.C. Epilepsy: Frequency, Causes and Consequences. New York: Demos; 1990.
Hauser W.A., Kurland L.T. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia. 1975;16:1-66.
Hertoft P. The clinical, electroencephalographic and social prognosis in petit mal epilepsy. Epilepsia. 1963;4:298-314.
Hirsch L.J., Weintraub D.B., Buchsbaum R., et al. Predictors of Lamotrigine-associated rash. Epilepsia. 2006;47:318-322.
Holmes G.L. Myoclonic, tonic, and atonic seizures in children. J Epilepsy. 1988;1:173-195.
Holmes G.L. New drugs in the treatment of epilepsy. Ann Nestlé. 2003;61:107-117.
Holmes G.L., Frank L.M., Sheth R.D., et al. Lamotrigine monotherapy for newly diagnosed typical absence seizures in children. Epilepsy Res. 2008.
Holmes G.L., McKeever M., Adamson M. Absence seizures in children: clinical and electroencephalographic features. Ann Neurol. 1987;21:268-273.
Hosford D.A., Clark S., Cao Z., et al. The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science. 1992;257:398-401.
Hosford D.A., Lin F.H., Kraemer D.L., et al. Neural network of structures in which GABAB receptors regulate absence seizures in the lethargic (lh/lh) mouse model. J Neurosci. 1995;15:7367-7376.
Huguenard J.R., Prince D.A. Clonazepam suppresses GABAB-mediated inhibition in thalamic relay neurons through effects in nucleus reticularis. J Neurophysiol. 1994;71:2576-2581.
Huguenard J.R., Prince D.A. Intrathalamic rhythmicity studied in vitro: nominal T current modulation causes robust anti-oscillatory effects. J Neurosci. 1994;14:5485-5502.
Ikeno T., Shigematsu H., Miyakoshi M., et al. An analytic study of epileptic falls. Epilepsia. 1985;26:612-621.
Izzedine H., Launay-Vacher V., Deray G. Topiramate-induced renal tubular acidosis. Am J Med. 2004;116:281-282.
Jarrar R.G., Buchhalter J.R. Therapeutics in pediatric epilepsy, Part 1: The new antiepileptic drugs and the ketogenic diet. Mayo Clin Proc. 2003;78:359-370.
Jayakar P., Chiappa K.H. Clinical correlations of photoparoxysmal responses. Electroencephalogr Clin Neurophysiol. 1990;75:251-254.
Jeavons P.M. Photosensitive epilepsy. In: Laidlaw J., Richens A., editors. A Textbook of Epilepsy. Edinburgh: Churchill Livingstone; 1982:195-211.
Jobst B.C., Williamson P.D., Neuschwander T.B., et al. Secondarily generalized seizures in mesial temporal epilepsy: clinical characteristics, lateralizing signs, and association with sleep-wake cycle. Epilepsia. 2001;42:1279-1287.
Juul-Jensen P., Foldspang A. Natural history of epileptic seizures. Epilepsia. 1983;24:297-312.
Kaufman D.W., Kelly J.P., Anderson T., et al. Evaluation of case reports of aplastic anemia among patients treated with felbamate. Epilepsia. 1997;38:1265-1269.
Khurana D.S., Kothare S.V., Valencia I., et al. Levetiracetam monotherapy in children with epilepsy. Pediatr Neurol. 2007;36:227-230.
Knake S., Hamer H.M., Schomburg U., et al. Tiagabine-induced absence status in idiopathic generalized epilepsy. Seizure. 1999;8:314-317.
Kossoff E.H., Bergey G.K., Freeman J.M., et al. Levetiracetam psychosis in children with epilepsy. Epilepsia. 2001;42:1611-1613.
Kuo R.L., Moran M.E., Kim D.H., et al. Topiramate-induced nephrolithiasis. J Endourol. 2002;16:229-231.
Kurokawa T., Goya N., Fukuyama Y., et al. West syndrome and Lennox-Gastaut syndrome: a survey of natural history. Pediatrics. 1980;65:81-88.
Lamb E.J., Stevens P.E., Nashef L. Topiramate increases biochemical risk of nephrolithiasis. Ann Clin Biochem. 2004;41:166-169.
Levy R.H., Shen D.D., Abbott F.S., et al. Valproic Acid. Chemistry, Biotransformation, and Pharmacokinetics. In: Levey R.H., Mattson R.H., Meldrum B.S., Perucca E., editors. Antiepileptic Drugs. ed 5. Philadelphia: Lippincott Williams & Wilkins; 2002:780-800.
Liu Z., Vernes M., Depaulis A., et al. Involvement of intrathalamic GABAB neurotransmission in the control of abscence seizures in the rat. Neuroscience. 1992;48:87-93.
Loiseau P., Pestre M., Dartigues J.F., et al. Long-term prognosis in two forms of childhood epilepsy: Typical absence seizures and epilepsy with rolandic (centrotemporal) EEG foci. Ann Neurol. 1983;13:642-648.
Marescaux C., Vergnes M., Bernasconi R. GABAB receptor antagonists: potential new anti-absence drugs. J Neural Transm. 1992;35:179-188.
Markand O.N. Slow spike-wave activity in EEG and associated clinical features: Often called ‘Lennox’ or ‘Lennox-Gastaut’ syndrome. Neurology. 1977;27:746-757.
Markand O.N. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J Clin Neurophysiol. 2003;20:426-441.
Marson A.G., et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1016-1026.
McGregor A., Wheless J. Pediatric experience with sudden unexplained death in epilepsy at a tertiary epilepsy center. J Child Neurol. 2006;21:782-787.
McKeever M., Holmes G.L., Russman B.S. Speech abnormalities in seizures: a comparison of absence and partial complex seizures. Brain Lang. 1983;19:25-32.
Messenheimer J.A. Rash in adult and pediatric patients treated with lamotrigine. Can J Neurol Sci. 1998;25:S14-S18.
Mikati M.A., Holmes G.L. Lamotrigine in absence and primary generalized epilepsies. J Child Neurol. 1997;12(Suppl 1):S29-S37.
Motte J., Trevathan E., Arvidsson J.F.V., et alLamictal Lennox-Gastaut Study Group. Lamotrigine for generalized seizures associated with the Lennox-Gastaut Syndrome. N Engl J Med. 1997;337:1807-1812.
Murphy K., Delanty N. Primary Generalized Epilepsies. Curr Treat Options Neurol. 2000;2:527-542.
Newmark M.E., Penry J.K. Photosensitivity and Epilepsy: A Review. New York: Raven Press; 1979.
Niedermeyer E. Immediate transition from a petit mal absence into a grand mal seizure; case report. Eur Neurol. 1976;14:11-16.
Noachtar S., Andermann E., Meyvisch P., et al. Levetiracetam for the treatment of idiopathic generalized epilepsy with myoclonic seizures. Neurology. 2008;70:607-616.
Oguni H., Fukuyama Y., Imaizumi Y., et al. Video-EEG analysis of drop seizures in myoclonic astatic epilepsy of early childhood (Doose syndrome). Epilepsia. 1992;33:805-813.
Oguni H., Uehara T., Imai K., et al. Atonic epileptic drop attacks associated with generalized spike-and-slow complexes: video-polygraphic study of two patients. Epilepsia. 1997;38:813-818.
Olmos-Garcia de Alba G., Valdez J.M., Crespo F.V. West syndrome evolving into the Lennox-Gastaut syndrome. Clin Electroencephalogr. 1984;15:61-68.
Opp J., Wenzel D., Brandl U. Visuomotor coordination during focal and generalized EEG discharges. Epilepsia. 1992;33:836-840.
Ormrod D., McClellan K. Topiramate: a review of its use in childhood epilepsy. Paediatr Drugs. 2001;3:293-319.
Ott D., Caplan R., Guthrie D., et al. Measures of psychopathology in children with complex partial seizures and primary generalized epilepsy with absence. J Am Acad Child Adolesc Psychiatry. 2001;40:907-914.
Pampiglione E., Harden A. So-called neuronal ceroid lipofuscinosis: Neurophysiological studies in 60 children. J Neurol Neurosurg Psychiatry. 1977;40:323-330.
Papini M., Pasquinelli A., Armellini M., et al. Alertness and incidence of seizures in patients with Lennox-Gastaut syndrome. Epilepsia. 1984;25:161-167.
Pavone P., Bianchini R., Trifiletti R.R., et al. Neuropsychological assessment in children with absence epilepsy. Neurology. 2001;56:1047-1051.
Pearl P.L., Holmes G.L. Childhood Absence Epilepsies. In: Pellock J.M., Bourgeois B.F.D., Dodson W.E., editors. Pediatric Epilepsy. New York: Demos; 2008:323-334.
Pellock J.M. Felbamate in epilepsy therapy: evaluating the risks. Drug Saf. 1999;21:225-239.
Pellock J.M. Understanding co-morbidities affecting children with epilepsy. Neurology. 2004;62:S17-S23.
Pellock J.M., Brodie M.J. Felbamate: 1997 update. Epilepsia. 1997;38:1261-1264.
Penry J.K., Dean J.C. Valproate. In: Dodson W.E., Pellock J.M., editors. Pediatric Epilepsy: Diagnosis and Therapy. New York: Demos Publications; 1993:315-324.
Penry J.K., Porter R.J., Dreifuss F.E. Simultaneous recording of absence sizures with videotape and electroencephalography. Brain. 1975;98:427-440.
Perucca E. Succinimides. In: Dam M., Gram L., editors. Comprehensive Epileptology. New York: Raven Press; 1990:603-611.
Perucca E., Gram L., Avanzini G., et al. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5-17.
Porter R.J., Penry J.K., Dreifuss F.E. Responsiveness at the onset of spike-wave bursts. Electroencephalogr Clin Neurophysiol. 1973;34:239-245.
Prasad A.N., Stafstrom C.F., Holmes G.L. Alternative epilepsy therapies: the ketogenic diet, immunoglobulins, and steroids. Epilepsia. 1996;37(Suppl 1):S81-S95.
Ramsay R.E., DeToledo J. Tonic-clonic seizures: a systematic review of antiepilepsy drug efficacy and safety. Clin Ther. 1997;19:433-446.
Reilly E.L., Peters J.F. Relationship of some varieties of electroencephalographic photosensitivity to clinical convulsive disorders. Neurology. 1973;23:1050-1057.
Reiss W.G., Oles K.S. Acetazolamide in the treatment of seizures. Ann Pharmacother. 1996;30:514-519.
Richard M.O., Chiron C., d’Athis P., et al. Phenytoin monitoring in status epilepticus in infants and children. Epilepsia. 1993:144-150.
Riikonen R. Long-term outcome of West syndrome: a study of adults with a history of infantile spasms. Epilepsia. 1996;37:367-372.
Rosenfeld W.E., Benbadis S., Edrich P., et al. Levetiracetam as add-on therapy for idiopathic generalized epilepsy syndromes with onset during adolescence: analysis of two randomized, double-blind, placebo-controlled studies. Epilepsy Res. 2009;85:72-80.
Rubenstein J.E., Vining E.P.G. Efficacy of the Ketogenic Diet. In: Stafstrom E.E., Rho J.M., editors. Epilepsy and the Ketogenic Diet. Totowa, New Jersey: Humana Press; 2004:95-102.
Sachdeo R.C., Glauser T.A., Ritter F., et al. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Topiramate YL Study Group. Neurology. 1999;52:1882-1887.
Sato S. Generalized seizures: Absence. In: Dreifuss F.E., editor. Pediatric Epileptology. Littleton, Mass: John Wright; 1983:65-91.
Sato S., Dreifuss F.E., Penry J.K. Prognostic factors in absence seizures. Neurology. 1976;26:788-796.
Sato S., Dreifuss F.E., Penry J.K., et al. Long-term follow-up of absence seizures. Neurology. 1983;33:1590-1595.
Scheffer I.E., Berkovic S.F. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479-490.
Scheyer R.D. Valproic Acid. Drug Interactions. In: Levey R.H., Mattson R.H., Meldrum B.S., Perucca E., editors. Antiepileptic Drugs. ed 5. Philadelphia: Lippincott Williams & Wilkins; 2002:801-807.
Schmidt D., Tsai J.J., Janz D. Generalized tonic-clonic seizures in patients with complex partial seizures. Natural history and prognosis relevance. Epilepsia. 1983;24:43-48.
Schneider H., Vassella F., Karbowski K. The Lennox syndrome. A clinical study of 40 children. Eur Neurol. 1970;4:289-300.
Schneider S. Clinical utilization of ethosuximide, methsuximide, and trimethadione in children. In: Dodson W.E., Pellock J.M., editors. Pediatric Epilepsy: Diagnosis and Therapy. New York: Demos Publications; 1993:325-330.
Snead O.C.III. Basic mechanisms of generalized absence seizures. Ann Neurol. 1995;37:146-157.
Sofijanov N.G. Clinical evolution and prognosis of childhood epilepsies. Epilepsia. 1982;23:61-69.
Steriade M., Conteras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80:1439-1455.
Steriade M., Llinas R.R. The functional states of the thalamus and the associated neuronal interplay. Physiol Rev. 1988;68:649-742.
Steriade M., McCormick D.A., Sejnowski T.J. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679-685.
Striano S., Capovilla G., Sofia V., et al. Eyelid myoclonia with absences (Jeavons syndrome): a well-defined idiopathic generalized epilepsy syndrome or a spectrum of photosensitive conditions? Epilepsia. 2009;50(Suppl 5):15-19.
Takeoka M., Holmes G.L., Thiele E., et al. Topiramate and metabolic acidosis in pediatric epilepsy. Epilepsia. 2001;42:387-392.
Tassinari C.A., Bureau M., Thomas P. Epilepsy with myoclonic absences. In: Roger J., Bureau M., Dravet C., Dreifuss F.E., Perret A., Wolf P., editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey; 1992:151-160.
Tassinari C.A., Dravet C., Roger J., et al. Tonic status epilepticus precipitated by intravenous benzodiazepine in five patients with Lennox-Gastaut syndrome. Epilepsia. 1972;13:421-435.
Taylor S., Tudur S.C., Williamson P.R., et al. Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalized onset tonic-clonic seizures. Cochrane Database Syst Rev. 2001. CD002217
Tellez-Zenteno J.F., Patten S.B., Jette N., et al. Psychiatric comorbidity in epilepsy: a population-based analysis. Epilepsia. 2007;48:2336-2344.
The Felbamate Study Group in Lennox-Gastaut Syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut Syndrome). N Engl J Med. 1993;328:29-33.
Todt H. The late prognosis of epilepsy in childhood: results of a prospective follow-up study. Epilepsia. 1984;25:137-144.
Trevathan E., Kerls S.P., Hammer A.E., et al. Lamotrigine adjunctive therapy among children and adolescents with primary generalized tonic-clonic seizures. Pediatrics. 2006;118:e371-e378.
Tsakiridou E., Bertollini L., de Curtis M., et al. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110-3117.
Unterberger I., Trinka E., Luef G., et al. Idiopathic generalized epilepsies with pure grand mal: clinical data and genetics. Epilepsy Res. 2001;44:19-25.
Vining E.P., Freeman J.M., Ballaban-Gil K., et al. A multicenter study of the efficacy of the ketogenic diet. Arch Neurol. 1998;55:1433-1437.
Wallace R.H., Scheffer I.E., Barnett S., et al. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68:859-865.
Wallace R.H., Scheffer I.E., Parasivam G., et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology. 2002;58:1426-1429.
Wheless J.W. Use of topiramate in childhood generalized seizure disorders. J Child Neurol. 2000;15(Suppl 1):S7-S13.
Wheless J.W. Nonpharmacologic treatment of the catastrophic epilepsies of childhood. Epilepsia. 2004;45(Suppl 5):17-22.
Wheless J.W. Levetiracetam in the treatment of childhood epilepsy. Neuropsychiatr Dis Treat. 2007;3:409-421.
Wheless J.W., Ng Y.T. Levetiracetam in refractory pediatric epilepsy. J Child Neurol. 2002;17:413-415.
Wirrell E.C. Natural history of absence epilepsy in children. Can J Neurol Sci. 2003;30:184-188.
Wirrell E.C., Camfield C.S., Camfield P.R., et al. Long-term prognosis of typical childhood absence epilepsy: remission or progression to juvenile myoclonic epilepsy. Neurology. 1996;47:912-918.
Yang M.T., Lee W.T., Chu L.W., et al. Anti-epileptic drugs-induced de novo absence seizures. Brain Dev. 2003;25:51-56.