CHAPTER 43 G2 Phase and Control of Entry into Mitosis
The G2 phase was originally defined simply as a gap between the completion of DNA replication and the onset of mitosis. In fact, definition of this phase is not entirely straightforward, as progression from interphase into mitosis is gradual, with early mitotic prophase sharing characteristics of both interphase and mitosis. Here, we define the G2 phase as the period from the end of the S phase until mid-prophase, that is, until activation of the main mitotic kinase, Cdk1–cyclin B1.
Enzymology of the G2/Mitosis Transition
The transition between the G2 phase and mitosis is the most profound morphologic and physiological change that occurs during the life of a growing cell, matched only by the dramatic changes that occur during death by apoptosis (see Chapter 46). Entry into mitosis is controlled by a network of stimulatory and inhibitory protein kinases and phosphatases, presided over by Cdk1–cyclin B1. (Chapter 40 introduced the components involved in the G2/M transition.)
Cdk1, the driving force for entry into mitosis, is present at a constant level throughout the cell cycle. Cdk1 regulation is multifaceted (see Fig. 40-14), including binding of cyclin cofactors, inhibition and activation by phosphorylation, binding of inhibitory molecules, and changes in subcellular localization.
Mammalian cells have at least three B-type cyclins: B1, B2, and B3. Cyclin B1 is essential for triggering the G2/M transition, and disruption of its gene has lethal consequences. Cyclin B1, newly synthesized during the latter part of the cell cycle, binds Cdk1 and shuttles it in and out of the nucleus. Importin b carries the Cdk1–cyclin B1 complex into the nucleus, and then Crm1 rapidly exports it back to the cytoplasm (see Chapter 16). Cdk1–cyclin B2 associates with the Golgi apparatus during interphase and might function in Golgi disassembly during mitosis (see Fig. 44-4). Cyclin B3 appears to function only during meiosis in mammals.
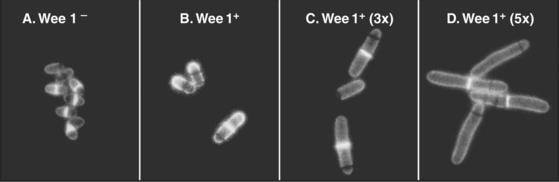
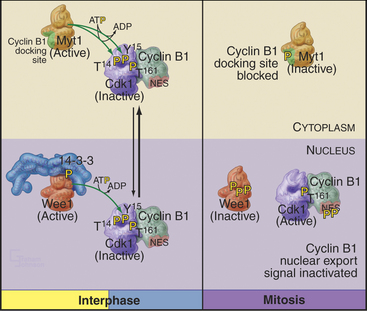
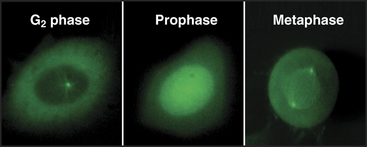
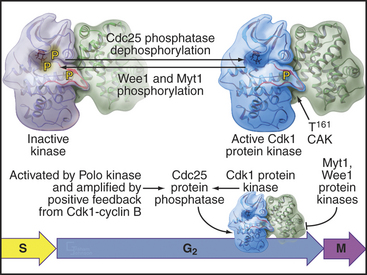
As cells approach the G2/M transition, a combination of one stimulatory and two inhibitory kinases control activation of Cdk1–cyclin B1. The activating kinase, called CAK (Cdk-activating kinase; actually Cdk7–cyclin H), is located in the nucleus. CAK phosphorylates Cdk1 on T161, allowing the refolding of the active site cleft that is required for the enzyme to bind substrates (see Chapter 40). Two other kinases, Wee1 and Myt1, counteract the action of CAK (Fig. 43-1). Wee1 is localized to the nucleus, where it inhibits Cdk1 by phosphoryla-ting Y15 adjacent to the ATP-binding site. The role of Wee1 as a mitotic inhibitor is clearly demonstrated in S. pombe: Overexpression of functional Wee1 delays or prevents the entry of cells into mitosis (Fig. 43-2). Myt1 associated with the Golgi apparatus and endoplasmic reticulum inhibits Cdk1 while it resides in the cytoplasm by phosphorylating T14 and Y15. Together, Wee1 and Myt1 ensure that Cdk1 remains inactive as it shuttles into and out of the nucleus (Fig. 43-3). This combination of stimulatory and inhibitory modifica-tions holds Cdk1–cyclin B1 poised for a burst of activation.
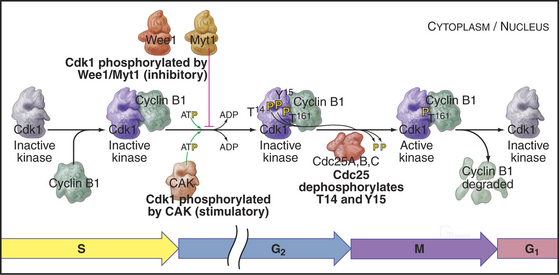
Figure 43-1 Regulation of Cdk1 by cyclin binding and protein phosphorylation from the late S phase through mid-mitosis.
(Structure for Wee1 kinase provided by E. N. Baker prior to re-lease to the Protein Data Bank; based on Squire CJ, Dickson JM, Ivanovic I, Baker EN: Structure of human Wee1A kinase: Kinase domain complexed with inhibitor PD0407824. Structure 13:541–550, 2005. PDB file: 1X8B.)
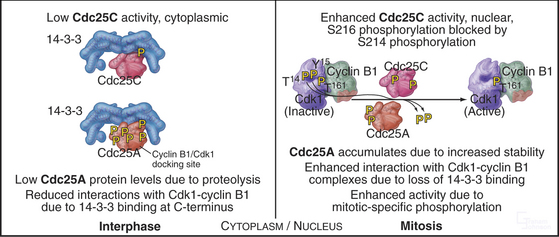
The three Cdc25 protein phosphatases remove the inhibitory phosphates from Cdk1–cyclin B1 and trigger the G2/M transition. Cdc25s are dual-specificity protein phosphatases (see Fig. 25-5) that remove phosphates from serine (S), threonine (T), and tyrosine (Y) residues, including the inhibitory phosphates from T14 and Y15 of Cdk1. Cdc25s are regulated by stimulatory and inhibitory phosphorylation, by alterations in their subcellular localization, and by ubiquitin-mediated proteolysis. Cdc25A, the only one of the three to be indispensable for life, functions at both the G1/S and G2/M transitions, whereas Cdc25B and Cdc25C have roles in the G2/M transition. The preferred targets of the individual Cdc25 isoforms are not known.
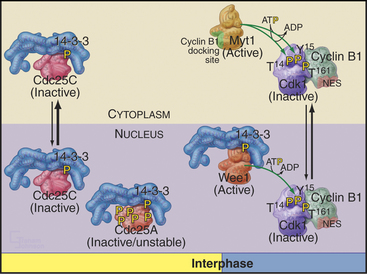
Cdc25A and Cdc25C are relatively inactive during interphase for three reasons (Fig. 43-4). First, phosphorylation on a serine residue creates a binding site for a member of the 14-3-3 group of adapter proteins (see Fig. 25-10). These proteins bind sites on target proteins containing serines flanked by several other characteristic amino acids, but only when the critical serine is phosphorylated. This is an example of the general mechanism whereby phosphorylation regu-lates interactions between proteins in response to physiological signals (see Chapter 25). Association of 14-3-3 with Cdc25A interferes with its binding to Cdk1–cyclin B1 and inhibits its import into the nucleus. Because Cdc25C has an intrinsic nuclear export sequence, the 14-3-3-bound form of Cdc25C is primarily cytoplasmic.
Changes in Subcellular Localization at the G2/M Transition
Cyclin B1 (with associated Cdk1) and Cdc25C have both nuclear import and nuclear export signals, so they shuttle in and out of the nucleus throughout interphase (Fig. 43-5). Cdc25A resides in the nucleus. During interphase, Cdk1–cyclin B1 and Cdc25C spend most of their time in the cytoplasm. Inhibitory phosphorylation of Cdk1 and Cdc25C keeps Cdk1 activity low.
Early in prophase, phosphorylation of cyclin B1 inactivates its nuclear export signal and promotes its nuclear import (see Chapter 16), allowing Cdk1–cyclin B1 to accumulate rapidly in the nucleus within five minutes (Figs. 43-3 and 43-6). Cdc25C also stops shuttling at the G2/M transition, probably as a result of phosphorylation by Polo kinase. The best evidence seems to indicate that the Cdk1–cyclin B that accumulates in the nucleus is already active, but restriction of Cdk–cyclin B complexes to the nucleus together with Cdc25A and Cdc25C significantly increases their local concentration (the volume of the nucleus is much less than the volume of the cytoplasm) and might contribute to the final burst of Cdk1–cyclin B1 activation.
Cdk1–Cyclin A and the Initiation of Prophase
As is noted in Chapter 42, Cdk2–cyclin A plays a critical role during the S phase. Several lines of evidence have revealed that a Cdk–cyclin A complex also helps to trigger the G2/M transition. First, inactivation of cyclin A, either by mutation in Drosophila or by injection of anti–cyclin A antibodies into cultured cells, arrests the cell cycle in G2. Second, Cdk–cyclin A activity peaks at G2/M, before the peak of Cdk1–cyclin B1 activity. Finally, if activated Cdk2–cyclin A complexes are injected into cells just after completion of the S phase, cells enter mitosis prematurely.
Cdk–cyclin A kinase is most likely to regulate several events at the transition from the G2 phase to prophase, including changes in microtubule behavior and chromosome condensation. Late in G2, the half-life of microtubules drops dramatically from about 10 minutes to about 30 seconds (see Table 44-1). This, coupled with centrosomes’ enhanced ability to initiate microtubule polymerization, completely transforms the organization of the microtubule cytoskeleton. Centrosomes take on the appearance of spindle poles and migrate apart over the surface of the nucleus. At the same time, chromatin begins to condense in the nucleus. The mechanism of chromosome condensation is not known, but a protein complex called condensin is required for this condensation to begin during prophase (see Fig. 13-19). Condensin does not associate tightly with chromosomes during interphase; association with chromosomes requires phosphorylation of two of its subunits by a Cdk. This occurs at G2/M. As chromosomes condense, several mitosis-specific kinetochore proteins also move into the nucleus and associate with kinetochores (see Chapter 13).
These events occur while most Cdk1–cyclin B1 is in the cytoplasm. It is most likely, therefore, that Cdk1–cyclin A triggers at least the nuclear events of prophase (Fig. 43-7). In fact, microinjection of a specific inhibitor of Cdk1–cyclin A causes prophase cells to return rapidly to interphase; chromosomes decondense, rounded prophase cells flatten, and the interphase microtubule network returns. Commitment to mitosis appears to be irreversible only after Cdk1–cyclin B1 enters the nucleus.
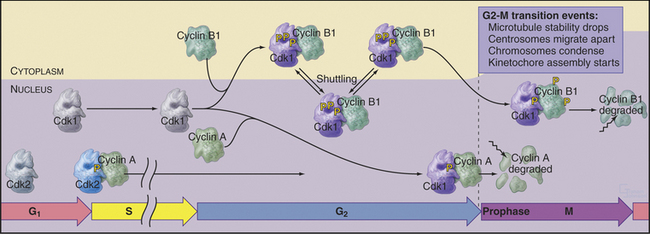
Summary of the Main Events of the G2/M Transition
Synthesis of cyclin B1 in the latter portion of the S and G2 phases leads to assembly of Cdk1–cyclin B heterodimers that shuttle into and out of the nucleus, spending most of their time in the cytoplasm associated with microtubules. In the late S and G2 phases, activation of Cdk1–cyclin A initiates mitotic prophase, beginning with changes in microtubule dynamics and chromosome condensation. Several events trigger entry into the active phase of mitosis. Cdc25A becomes stabilized and no longer binds 14-3-3 proteins. This results in the accumulation of Cdc25A and allows more efficient interactions between Cdc25A and Cdk1–cyclin B. The phosphate-binding site for 14-3-3 proteins on Cdc25C becomes dephosphorylated, allowing it to accumulate in the nucleus. In addition, phosphorylation of cyclin B1 blocks its export from the nucleus and promotes its import, thus causing Cdk1–cyclin B1 to accumulate rapidly in the nucleus. The inhibitory kinase Wee1 is also dephosphorylated, and its activity drops. Cdc25A and Cdc25C activate Cdk1–cyclin B1 by removing inhibitory phosphates on T14 and Y15. This starts in the cytoplasm and then may be stimulated as the proteins concentrate in the nucleus. There, the action of Cdk1–cyclin B1 on the nuclear lamina triggers nuclear envelope breakdown and drives the cell into mitosis. Fig. 43-8 summarizes the network regulating Cdk1–cyclin B activation.
The G2 Checkpoint
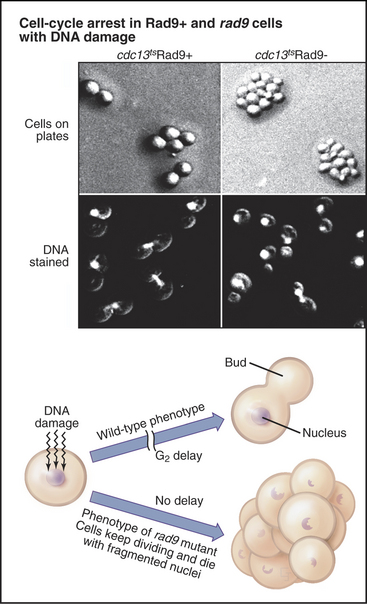
Exposure of cells to agents that damage DNA, including certain chemicals or ionizing radiation, halts the cell cycle temporarily in the G2 phase. This G2 delay gives cells an opportunity to repair damaged DNA before entering mitosis. Studies of radiation induced G2 delay in budding yeast identified a major cell-cycle checkpoint in G2 sensitive to the status of the cellular DNA. Cells that are defective in this checkpoint are much more sensitive to radiation injury than are wild-type cells because they continue to divide, despite the presence of broken or otherwise damaged chromosomes (Fig. 43-9). This continued division in the face of DNA damage leads to cell death, presumably due to accumulated chromosomal defects, as well as chromosome loss.
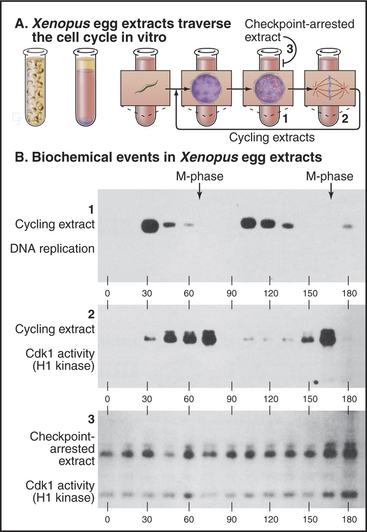
The G2 checkpoint also monitors the completion of DNA replication. Remarkably, this aspect of the checkpoint even works in vitro in cell-free extracts. As is described in Box 40-3, highly concentrated extracts made from Xenopus eggs can be induced to undergo a cyclic alteration in cell-cycle phases, even in the absence of added nuclei. Passage through the different phases can be followed by monitoring the activity of Cdk1–cyclin B kinase, which is high in mitosis and low in interphase. In the absence of nuclei, an inhibitor of DNA polymerases (such as the fungal toxin aphidicolin) has no effect on such extracts, which continue to cycle unabated between the S and M phases. On the other hand, if the extract contains more than 400 nuclei/mL undergoing synchronous cycles of DNA replication and mitosis, aphidicolin brings the cycling to a halt before M phase. This experiment appears to reconstitute the G2 checkpoint in vitro (Fig. 43-10). Furthermore, it reveals that partly replicated nuclei release an inhibitory signal that stops the cycle.
The G2 Checkpoint and Cancer
Defects in the G2 checkpoint are associated with cancer, since failure to correct damage to tumor suppressor genes can compromise the G1 checkpoint in subsequent cell cycles. For example, suppose that radiation damages a nucleotide base in the gene for the retinoblastoma susceptibility protein (Rb; see Fig. 41-10). If this happens in the S or G2 phase, informa-tion in the undamaged copy of the gene can be used to repair the damage. Delaying the cell cycle increases the chance of successful repair. On the other hand, if the checkpoint fails and the cell enters mitosis, sister chromatids separate from one another, and the damage is never repaired. As a consequence, one daughter cell from that division has a defective pRb gene, impairing its ability to regulate its growth at the restriction point in the next G1 phase. Loss of tumor suppressor genes such as pRb and p53 is a major cause of cancer (see Chapter 41).
How the G2 Checkpoint Works
The minimal machinery of a DNA damage checkpoint (see Fig. 40-4) involves sensors that detect DNA damage, transducers (usually protein kinases) that produce a biochemical signal as a result of the detected damage, and effectors (both protein kinases and transcriptional activators) that either directly or indirectly block cell-cycle progression. The sensors are not yet as well characterized as are the transducers and effectors. This section discusses this machinery as a prelude to consideration in Box 43-1 of the mechanisms that repair damaged DNA.
BOX 43-1 DNA Repair in Vertebrates
Every human cell experiences about 105 DNA damage events each day. Cell division must not occur with inaccurately replicated or damaged genomes, as this may cause cell death or heritable mutation. A number of systems have evolved to repair damaged DNA (Fig. 43-13). Their activities depend on the particular form of DNA damage sustained by the cell. These repair mechanisms also act in concert with the apoptotic machinery to ensure that if the DNA damage cannot be repaired, the cell will die (see Chapter 46). DNA damage checkpoints are a critical component of the cellular response to DNA damage (see Fig. 40-4), as they impose a delay in the cell cycle during which cells have a chance to repair their genomes.
Base Excision Repair
Bases in DNA can become oxidized, reduced, alkylated, or deaminated owing to endogenous activities or environmental stress. Damaged bases are cut away from the DNA sugar-phosphate backbone by a damage-recognizing glycosylase, leaving an abasic site (Fig. 43-14A). Abasic sites, which also can be generated directly by DNA damage, are then removed by cleavage of the sugar-phosphate backbone mediated by certain glycosylases and endonucleases. The missing sequence is then reconstructed from its complementary strand by DNA polymerase β, with DNA ligase III-Xrcc1 completing the repair by sealing the gaps in the backbone.
Nucleotide Excision Repair
Bulky DNA adducts caused by chemical agents or environmental stress (particularly ultraviolet radiation from sunlight) are excised by a complex, though well-understood, reaction (Fig. 43-14B). Defects in nucleotide excision repair genes cause the human genetic disease xeroderma pigmentosum (XP), which is characterized by hypersensitivity to sunlight and predisposition to skin cancer. Eight proteins encoded by genes mutated in xeroderma pigmentosum (Table 43-1) take part in nucleotide excision repair, providing one of the best examples in which human genetics has helped to unravel a complicated biological process. Recognition of the DNA lesion involves the heterotrimeric replication protein RPA, XPA, and XPC and the nine-subunit transcription factor TFIIH, which contains XPB and XPD. ATP-dependent unwinding of the DNA by XPB and XPD forms a preincision complex. XPG, which replaces XPC in the complex, makes an incision six to nine bases 3′ of the damaged base, and XPF-Ercc1 cuts 20 to 25 bases 5′ of the damage site. This releases a short single-stranded DNA fragment containing the damaged DNA. After excision, DNA polymerases d or e fills in the gap by copying the undamaged strand. Prokaryotes have a similar system of adduct recognition, removal, and repair involving the UvrA, UvrB, and UvrC proteins; however, the enzymes that are involved are not conserved between kingdoms, an unusual occurrence for DNA repair systems.
Mismatch Repair
Errors in DNA replication that have not been detected by the proofreading activity of the DNA polymerase are recognized by a dimer consisting of the MSH2/MSH6 proteins. When a mismatch is detected, this heterodimer undergoes an ATP-dependent transition to a sliding clamp and recruits a second heterodimer, consisting of MLH1 and PMS2 (Fig. 43-14C). To distinguish between the original (“correct”) sequence and the newly synthesized DNA strand, this sliding clamp complex can then translocate along the DNA until a break is reached, such as that found between Okazaki fragments. The broken strand is therefore identified as the newly synthesized DNA strand. The mismatch repair complex then recruits the exonuclease EXO1 and degrades the newly synthesized DNA strand all the way back to the misincorporated base. The resultant long single-stranded region is stabilized by binding of RPA and eventually filled in by the replicative DNA polymerases d and/or e.
Double-Strand Break Repair
DNA double-strand breaks are particularly hazardous forms of damage, as they carry the risk of losing chromosomal material or, if misrepaired, causing chromosomal translocations. Two major pathways are responsible for dealing with these lesions, which can be caused by ionizing radiation or radiomimetic drugs or arise spontaneously after replication. Homologous recombinational repair uses undamaged DNA sequence as a template for the accurate repair of double-strand breaks, this sequence usually being derived from the sister chromatid after replication. Nonhomologous end-joining repairs double-strand breaks with no requirement for homology and therefore carries a much higher risk of introducing mutations. Nonhomologous end-joining is the predominant activity that repairs double-strand breaks in the G1 and early S phase, while homologous recombination becomes more important in the late S and G2 phase. Both pathways require the activity of the MRN protein complex (Mre11/RAD50/Nbs1), which localizes to DNA double-strand breaks and is also found at telomeres (see Fig. 12-13). The exonuclease activity of this com-plex resects (chews back) broken DNA ends to provide single-stranded DNA substrates for the repair systems.
The key protein that is required for homologous recombinational repair in mammalian cells is Rad51, the eukaryotic homologue of E. coli RecA (Fig. 43-15A). Rad51 forms an extended nucleoprotein filament on single-stranded DNA and catalyses the search for homologous sequences, strand pairing, and strand exchange. Also involved in this process is Rad54, a helicase that is believed to facilitate strand invasion when the single-stranded region forces its way into the complementary DNA duplex on the undamaged sister chromatid. Following invasion of the recombining DNA strands, polymerase activity extends the DNA beyond the site of the double-strand break and forms a Holliday junction (Fig. 43-15B). Resolution of the Holliday junction and filling-in of the repaired DNA sequences results in complete repair of the lesion.
Nonhomologous end-joining is initiated at a DNA double-strand break by binding of the Ku70 and Ku80 heterodimer as a ring to which the DNA-dependent protein kinase catalytic subunit binds, stimulating other repair factors and aligning the broken ends of the DNA (Fig. 43-15C). The ends are finally sealed by DNA ligase IV, which exists in complex with XRCC4. Nonhomologous end-joining is also necessary for V(D)J recombination and therefore for the development of the immune system (see Fig. 28-10).
Given the importance of accurate transmission of the genetic material, it is not surprising that a number of diseases, key among which is cancer, are associated with deficiencies in DNA repair. Table 43-1 summarizes the impact that mutations in the key DNA repair genes can have in disease. Note that many DNA repair activities are essential for life and therefore have not been described in any human diseases.
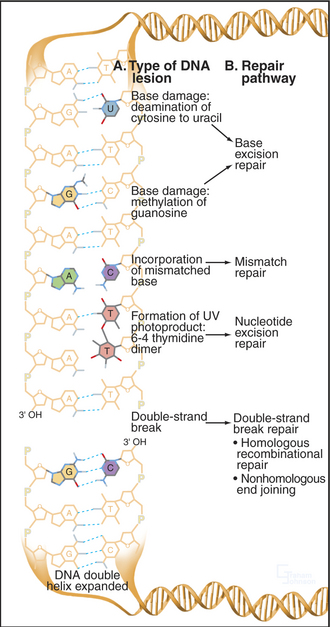
Figure 43-13 EXAMPLES OF DNA DAMAGE AND THE REPAIR PATHWAYS THAT RESPOND TO DIFFERENT TYPES OF LESION.
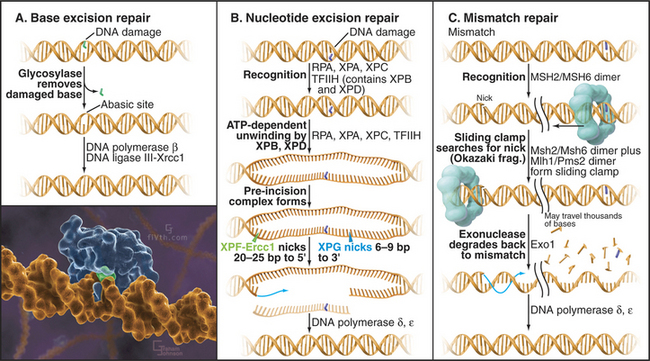
(Inset illustration by Graham Johnson (www.fivth.com) for the Howard Hughes Medical Institute, copyright 2004, all rights reserved. Reference: Lau AY, Scharer OD, Samson L, et al: Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: Mechanisms for nucleotide flipping and base excision. Cell 95:249–258, 1998.)
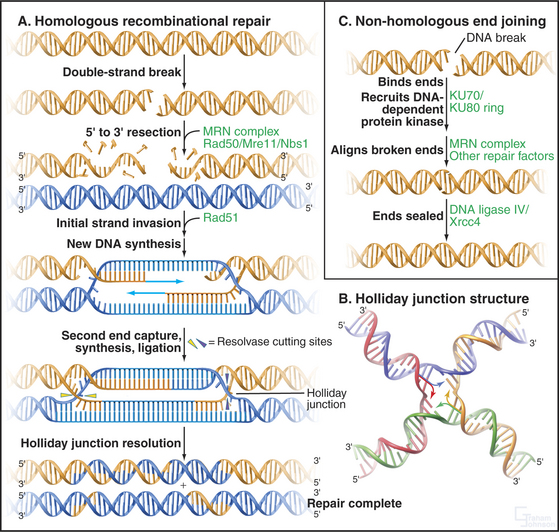
Figure 43-15 pathways for the repair of dna double-strand breaks. A double-strand break is recognized by the PI3-kinase family members ATM or ATR and a cell-cycle delay ensues (see Fig. 40-4). The break is then bound by repair factors in the homologous recombination or the nonhomologous end-joining pathway of DNA repair. It is not known what factors control the “choice” between these pathways. A, Homologous recombination pathway of DNA repair. The MRN complex chews back the DNA at a break, leaving a single-stranded overhang that is stabilized by RPA (not shown). It is believed that the MRN complex also plays a role in keeping the broken ends in proximity to one another. Next, RAD51 forms a nucleoprotein filament on the single-stranded DNA, displacing the RPA. The RAD51 nucleoprotein filament then initiates homology searching and repairs the DNA break by inserting the extended single-stranded DNA into homologous sequences (usually on the sister chromatid [blue]) and allowing homologous recombination and DNA repair/resynthesis to occur. Capture of the second single-stranded DNA end allows the formation of a joint molecule with a double Holliday junction. Resolution of this Holliday junction structure results in accurate, templated repair of the double-strand break. B, A Holliday junction formed by four complementary oligonucleotides complexed to the enzyme Cre (not shown). The Holliday junction is a dynamic structure (arrows) that can migrate along the DNA. (PDB file: 3CRX.) C, Nonhomologous end-joining pathway of DNA repair. Nonhomologous end-joining is initiated by break recognition by the Ku70/Ku80 heterodimer, which recruits DNA-PK and tethers the broken ends. The breaks are then processed in a reaction involving the MRN complex and other repair factors. DNA-PK’s precise role is not yet entirely clear. Next, DNA ligase IV/XRCC4 is recruited to the processed double-strand break, which is ligated back together.
(B, Reference: Gopaul DN, Guo F, Van Duyne GD: Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J 17:4175–4187, 1998.)
The sensors that detect DNA damage have not yet been entirely characterized (Fig. 43-11). The two apical kinases ATM and ATR (see Fig. 40-4) are mobilized quickly to the sites of DNA damage. However, ATM is activated before it gets to the DNA, and the identity of the earliest sensor of DNA damage remains unclear. The 9-1-1 protein complex (described later) and the enzymes that are involved in processing damaged DNA are also possible sensors of the DNA damage.
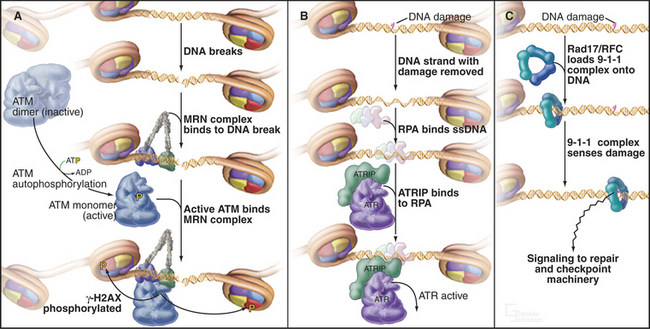
Two kinases, ATM and ATR (see Fig. 40-4), are key transducers of the DNA damage response. ATM responds to DNA damage any time during the cell cycle, but ATR appears to respond only if DNA is damaged prior to the completion of replication. Thus, ATM is essential for the G2 DNA damage checkpoint to arrest the cell cycle when DNA damage occurs after replication is complete.
The activation of ATM involves autophosphorylation, which causes inactive dimers to dissociate into active monomers. Subsequently the Nbs1 subunit of the MRN complex (Fig. 43-11A; see also Fig. 12-13) recruits ATM monomers to the DNA break and initiates the full-blown checkpoint response. The MRN complex has a key role in repair of double-strand breaks in DNA. The Nbs1 gene is mutated in humans with Nijmegen breakage syndrome (Table 43-1).
Table 43-1 KEY DNA REPAIR GENE DEFECTS ASSOCIATED WITH HUMAN DISEASE
| Human Disease | Pathway | Genes Defective* |
|---|---|---|
| Ataxia-telangiectasia (AT) | Checkpoint | ATM |
| Seckel syndrome | Checkpoint | ATR |
| Xeroderma pigmentosum | NER | XP-A, XP-B, XP-C, XP-D, DDB-2, XP-F, XP-G, POLH |
| Cockayne syndrome | NER | XP-B, XP-D, CSA, CSB |
| Trichothiodystrophy | NER | XP-D |
| Hereditary nonpolyposis colon cancer | MMR | MSH2, PMS2, MLH1 |
| AT-like disorder | DSB repair | MRE11 |
| Nijmegen breakage syndrome | DSB repair | NBS1 |
| Breast cancer predisposition | HR | BRCA1, BRCA2 |
| LIG4 syndrome | NHEJ | LIGIV |
| Severe combined immune deficiency | NHEJ | ARTEMIS |
NER, nucleotide excision repair; MMR, mismatch repair; DSB, double-strand break; HR, homologous recombination; NHEJ, nonhomologous end joining.
* This list is an outline. For an updated list of the approximately 130 human DNA repair genes known to date, the reader is referred to http://www.cgal.icnet.uk/DNA_Repair_Genes.html#refs.
ATR exists in cells in a constitutive complex with a second subunit, ATR-interacting protein (ATRIP). ATRIP binds to the single-strand DNA binding protein RPA, an essential component of the DNA replication fork (Fig. 43-11B). DNA damage often results directly or indirectly in the presence of single-stranded DNA, which can be produced by the damaging event itself or as a by-product of the DNA repair process. This single-stranded DNA binds RPA and then, through the action of ATRIP, recruits and activates ATR.
ATR depends on two other protein complexes to mount a checkpoint response. One is the trimeric 9-1-1 complex, which gets its name from its subunits Rad9, Hus1, and Rad1 (Fig. 43-11C). The 9-1-1 complex resembles proliferating cell nuclear antigen (PCNA), the doughnut-shaped processivity factor that is indispensable during DNA replication (see Fig. 42-11). PCNA is loaded onto DNA by the pentameric replication factor C (RFC) ATPase during DNA replication and anchors DNA polymerases and other factors to DNA. A similar enzyme composed of one special subunit, Rad17, plus the four small subunits of RFC is believed to load the 9-1-1 complex onto DNA at or near sites of damage. This speculation is supported by the observation that mutants in four of the RFC subunits are defective in G2 checkpoint control in yeasts and Drosophila.
When DNA damage activates ATM and ATR, they phosphorylate several important substrates, including the tumor suppressor protein p53, and two protein checkpoint kinases called Chk1 and Chk2. When activated by phosphorylation, Chk1 phosphorylates the Cdc25A protein phosphatase (Fig. 43-12). This has at least two consequences. First, it produces binding sites for a 14-3-3 protein, which blocks Cdc25A from activating Cdk1–cyclin B. Second, phosphorylation by Chk1 targets Cdc25A for ubiquitin-mediated proteoly-sis. This ensures that levels of Cdc25A remain low. In addition, activation of the G2 checkpoint maintains Cdc25C in a 14-3-3-bound form. This prevents Cdc25C from accumulating in the nucleus and from activat-ing Cdk1–cyclin B as it shuttles in and out of the nucleus.
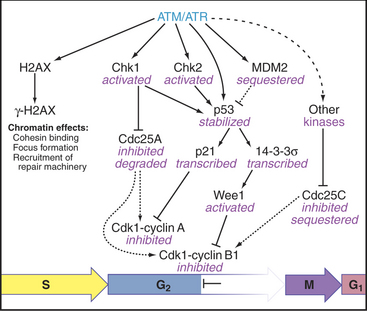
Another substrate of ATM and ATR is the specialized histone isoform H2AX. H2AX phosphorylated by ATM and/or ATR is known as γ-H2AX. γ-H2AX spreads rapidly across sites of DNA damage, called “foci.” These g-H2AX foci recruit key proteins for signaling, DNA repair machinery, and the cohesin complex, which holds replicated sister chromatids together (see Fig. 13-19). In the case of double-strand DNA breaks, a particularly dangerous form of DNA damage, the unbroken sister chromatid provides a template to ensure that the break is repaired correctly.
Ideally, checkpoint activation has one of two outcomes. If DNA damage is so extensive that it cannot be repaired, the cell commits suicide by a pathway called apoptosis (see Chapter 46). Less serious damage can be repaired by one of the systems described in Box 43-1.
Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177-2196.
Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14:237-245.
Morgan DO. Cyclin-dependent kinases: Engines, clocks and microprocessors. Annu Rev Cell Biol. 1997;13:261-291.
Nigg EA. Cell division: Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21-32.
O’Connell MJ, Walworth NC, Carr AM. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 2000;10:296-303.
Ohi R, Gould KL. Regulating the onset of mitosis. Curr Opin Cell Biol. 1999;11:267-273.
Petrini JHJ, Stracker TH. The cellular response to DNA double-strand breaks: Defining the sensors and mediators. Trends Cell Biol. 2003;13:458-462.
Pines J. Four-dimensional control of the cell cycle. Nat Cell Biol. 1999;1:E73-E79.
Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39-85.
Shiloh Y, editor. Bridge over broken ends: The cellular response to DNA breaks in health and disease. Special issue of DNA Repair. 2004;3:779-1251.
Smits VA, Medema RH. Checking out the G(2)/M transition. Biochim Biophys Acta. 2001;1519:1-12.
Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284-1289.