[level-membership-for-neurology-category]
Chapter 82 Fungal, Rickettsial, and Parasitic Diseases of the Nervous System
Fungal Diseases
The importance of invasive fungal infections, including those of the central nervous system (CNS), has risen as the population of immunologically compromised patients, including preterm infants, has grown. Patients with congenital immunologicial deficiencies, aplastic anemia, or poorly controlled human immunodeficiency virus (HIV) infection are at risk for a wide variety of fungal infections because their illness prevents normal functioning of the immune system. Those patients receiving chemotherapy, hematopoetic stem-cell or solid organ transplants, or medications such as corticosteroids that blunt the normal immune systems, can also be highly susceptible. The normal host can also fall victim to invasive fungal infection, but does so less often and to a smaller spectrum of organisms [Chimelli and Mahler-Araujo, 1997].
Fungal diseases involving the brain are usually spread via the bloodstream after a systemic or pulmonary focus is established. Alternatively, pathogens invade the CNS via direct extension from adjacent sinuses, the cranial vault, orbits, or the spine. With exceptions, such as Candida spp. meningitis in premature neonates and Aspergillus spp. brain abscess in the highly compromised host, fungal infections of the CNS in children are rare. A review from a large university children’s hospital found that, in a 6-year period, only 2 percent of nearly 1500 positive cerebrospinal fluid (CSF) cultures recovered fungal isolates [Arisoy et al., 1994], mostly Candida spp.; nearly half came from low-birth-weight infants. Table 82-1 provides a more extensive list of risk factors, syndromes, and treatments for fungal infections of the nervous system. Since clinical manifestations of CNS fungal infections can be subtle and not specific to individual pathogens, errors and delays in diagnosis occur. Fungi may cause a variety of clinical syndromes, including meningitis, brain abscesses, granulomas, and invasion of the vasculature, further complicating clinical presentation and treatment [Salaki et al., 1984]. Prompt diagnosis and effective treatment often require a medical–surgical team approach.
Cryptococcosis
Epidemiology, Environmental Reservoir, and Pathogenesis
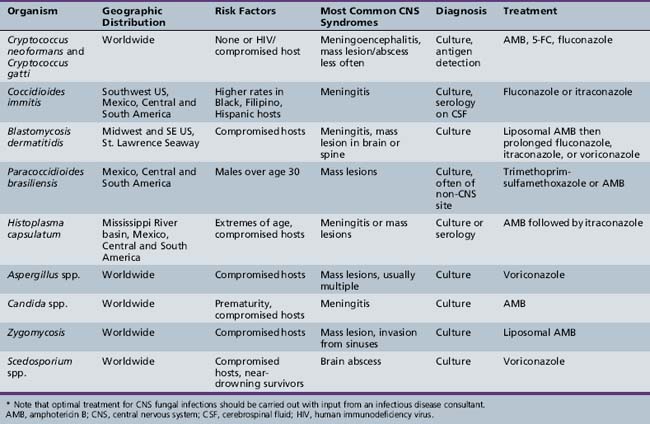
Cryptococcosis is globally distributed and is most commonly seen in patients with acquired immunodeficiency syndrome (AIDS) and in those receiving systemic corticosteroids; infection also occurs in those without apparent predisposition but at a lower frequency [Wendisch et al., 1996]. Although children are less frequently infected than adults, infants have contracted the disease [Hung et al., 1995; Littman and Walter, 1968; Wang et al., 1996], and vertical transmission has been reported from a mother with advanced AIDS [Sirinavin et al., 2004]. The mode of entry in humans is probably through inhalation or ingestion, and dissemination to the nervous system can follow. The most common cause of cryptococcal disease, Cryptococcus neoformans, is an encapsulated yeast found widely in the environment, particularly in bird feces and soil visited by birds (Figure 82-1). Cryptococcus gatti has been the cause of disease in animals and humans in British Columbia and the northwestern United States [Galanis et al., 2009], and appears to have a link to certain tree species in Australia and North America.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Cryptococcus most often causes a meningoencephalitis but can also cause brain abscess and cryptococcomas. Typical meningitis features, such as nausea, vomiting, headache, and fever, are often present. Caution is urged in ruling out the diagnosis based on history and physical examination alone because presenting symptoms may be nonspecific and a substantial number of patients are afebrile and lack meningeal signs [Sabetta and Andriole, 1985]. In AIDS patients, CNS cryptococcosis tends to be acute, with fever and headache in 70 percent; meningismus, photophobia, or mental status changes in 20–25 percent; and focal deficits in a small percentage [Daar and Meyer, 1992]. Despite this, a substantial minority of these patients may have initially normal CSF cell counts, glucose, and protein [Darras-Joly et al., 1996].
When C. neoformans causes mass lesions, it can result in papilledema, cranial neuropathies, and hemiparesis [Hung et al., 1995]. Brain abscess and cyptococcomas may begin with an indolent course, but hydrocephalus occurs and often necessitates shunting; if untreated, the mortality is high [Richardson et al., 1976]. Cryptococcus can be identified by culturing CSF on Sabouraud’s agar, but large-volume culture on more than one occasion may be needed. Latex agglutination detection of cryptococcal antigen is a rapid and useful adjunct test for both serum and CSF specimens. In the CSF, the antigen detection has an estimated sensitivity of 90 percent [Sabetta and Andriole, 1985]; both very high and very low antigen levels can give false-negative results. Since patients may have disseminated disease, C. neoformans can also frequently be cultured from urine or sputum, and the putative cause of CNS disease deduced. Cranial computed tomography (CT) and magnetic resonance imaging (MRI) scans can reveal mass lesions and hydrocephalus.
Management
No pediatric-specific clinical treatment trials have been completed. The Infectious Disease Society of America (IDSA) recently published treatment guidelines and recommends an initial combination of amphotericin B and flucytosine for cryptococcal CNS disease in all age groups, regardless of HIV status [Perfect et al., 2010]. Oral fluconazole is recommended for both consolidation and long-term suppression therapy. All cases of CNS cryptococcal disease should be managed in consultation with an infectious disease specialist.
Coccidioidomycosis
Epidemiology, Microbiology, and Pathology
Coccidioidomycosis is caused by the fungus Coccidioides immitis, a dimorphic fungus that grows as either a mycelium or a unique structure called a spherule. Disease caused by C. immitis most commonly occurs in endemic areas of the southwestern United States, northern Mexico, and portions of Central and South America. The San Joaquin Valley in California is the most notorious endemic zone, giving rise to the term valley fever. Most infections occur during the dry season when spores are borne by dust. Most often, C. immitis infection is unnoticed or causes a self-limited respiratory tract infection; dissemination is uncommon, but the risk is higher in patients of Filipino, black, and Hispanic backgrounds [Einstein and Johnson, 1993], and in the setting of HIV infection, organ transplantation, or prolonged use of corticosteroids. Coccidioidomycosis is acquired by inhalation of spore-laden dust or transcutaneously after skin abrasion; it is not spread from person to person. Because most (60 percent) infections are asymptomatic or mildly symptomatic, reported cases represent only a small fraction of the total burden of disease. CNS infection in children is rarely reported.
The spectrum of neuropathologic change ranges from meningitis to meningoencephalitis and meningomyelitis with extensive parenchymal destruction, sometimes as a result of an associated endarteritis obliterans [Mischel and Vinters, 1995]. Parenchymal brain lesions are rare but may occur in the absence of meningitis [Banuelos et al., 1996].
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Studies done on military recruits in the 1940s, using skin testing to diagnose acute infection, provided detailed data on infection in the normal host. Only 40 percent of these patients experienced acute respiratory symptoms [Smith et al., 1946], typically within 3 weeks after exposure and accompanied by fever, chills, night sweats, cough, anorexia, and weight loss. Dissemination of the infection from the pulmonary focus to distant sites usually occurs 1–6 months after primary infection in approximately 0.5 percent of cases; half of these are meningitis [Banuelos et al., 1996]. Other sites of dissemination include skin, lymph nodes, bones, and joints.
The clinical characteristics of coccidioidal meningitis are nonspecific and prompt diagnosis can be difficult [Caudill et al., 1970]. The most prevalent manifestations are headache, fever, malaise, and weight loss; meningismus may be absent [Saitoh et al., 2000]. Findings can include confusion, personality changes, focal neurological deficits, ataxia, obtundation, and coma. Brain and spinal cord abscesses can occur with or without concurrent meningitis [Banuelos et al., 1996]. Spinal cord symptoms may also occur in conjunction with coccidioidal osteomyelitis of the cervical vertebra [Jackson et al., 1964].
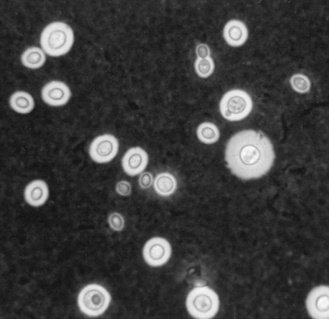
CSF abnormalities include mononuclear pleocytosis, increased protein content, increased chloride concentration, and normal or decreased glucose concentration. Diagnosis can be confirmed by either direct microscopy or culture of the CSF (Figure 82-2). Unfortunately, CSF cultures and microscopy are often negative in C. immitis meningitis, requiring indirect evidence of infection through serologic testing of serum and CSF. Complement fixation and precipitin tests are dependable and provide accurate diagnosis in more than 99 percent of patients with disseminated infection [Pappagianis, 1976]. Any discernible titer of complement fixation antibody in spinal fluid is considered diagnostic [Lyons and Andriole, 1986]. The coccidioidin skin test is not currently used for diagnosis of acute disease.
Imaging by CT or MRI scan may reveal widespread cisternal and cervical subarachnoid meningeal involvement in patients with coccidioidal meningitis [Wrobel et al., 1992], and may demonstrate the need for serial imaging to detect developing hydrocephalus.
Management
The 2008 IDSA guidelines for treatment of coccidioidal meningitis recommend fluconazole or itraconazole by mouth for all patients; intrathecal amphotericin was recommended by some clinicians but was graded C-III (poor evidence) by the IDSA [Ampel et al., 2009]. Fluconazole is well tolerated in both children and adults, and the oral form is highly bioavailable. Patients with C. immitis meningitis must be treated indefinitely with fluconazole to prevent relapse, and should be managed in conjunction with an infectious disease specialist.
North American Blastomycosis
Blastomycosis is caused by inhalation of airborne spores from Blastomycosis dermatitidis, a dimorphic fungus found in soil [Bradsher, 1996]. The endemic area includes the midwest, southeastern, and south central United States, and the Canadian provinces near the Great Lakes and St. Lawrence Seaway. The spectrum of clinical manifestations in adults includes pulmonary disease, acute and chronic skin disease, and disseminated disease involving the bones, the genitourinary tract, or the CNS; disseminated disease, and particularly CNS disease, is more common in those with compromised immune systems. Children are infrequently affected but suffer a similar spectrum of illness as adults [Schutze et al., 1996], and may be at increased risk if they play near beaver dams [Dismukes, 1986; Klein et al., 1986]. The three major CNS clinical manifestations are meningitis, intracranial mass, and spinal mass [Gonyea, 1978]. Patients with advanced HIV disease have accounted for an increase in cases compared with the past and are at greater risk for mortality [Witzig et al., 1994].
Diagnosis
In pulmonary disease, the organism can be identified on microscopy and culture of respiratory tract specimens. For CNS disease, ventricular and cisternal fluid may have a higher yield than lumbar CSF, which is rarely positive. Mass lesions are identified with neuroimaging [Ward et al., 1995]; microscopic study of biopsy material provides positive identification of the fungus. Skin tests and serologic tests are not helpful in the diagnosis of North American blastomycosis.
Management
Lipid formulations of amphotericin B for 4–6 weeks are recommended for moderate to severe disease [Chapman et al., 2008], followed by prolonged treatment with oral fluconazole, itraconazole, or voriconazole. Surgery may be indicated in patients with mass lesions, those with a need for a diagnostic biopsy, or those with osteomyelitis refractory to pharmacotherapy [Ward et al., 1995]. As with other CNS fungal diseases, co-management with an infectious disease specialist is recommended.
South American Blastomycosis
South American blastomycosis is caused by the fungus Paracoccidioides brasiliensis [Lutz, 1908]. The organism is a dimorphic fungal saprophyte found in soil and plants, which grows as a yeast at body temperature. Acquisition of disease is restricted to people living in latitudes between Mexico and Argentina, with most patients reported from Brazil. Most disease occurs in people older than the age of 30 years, with a striking male preponderance; among the rare childhood cases, the gender distribution is equal [Restrepo, 2000].
CNS involvement may follow disseminated infection; one review from Brazil found CNS involvement in 14 percent of symptomatic patients [de Almeida et al., 2004].
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
The most common findings in a series of 24 CNS cases included seizures, hemiparesis, cerebellar signs, and hydrocephalus [de Almeida et al., 2004]. CNS infection usually causes single or multiple mass lesions that appear hypodense and enhance on CT scan. The masses/abscesses may cause focal neurologic signs, increased intracranial pressure, and symptoms of meningeal inflammation.
The diagnosis of South American blastomycosis is suggested by simultaneous involvement of several organs or by characteristic oral and skin lesions in patients who reside in endemic areas. Microscopic study of scrapings from mucous lesions, sputum, gastric contents, lymph nodes, or surgical specimens is most helpful. The etiology of brain lesions must be deduced from culture results outside the CNS or via brain biopsy. CSF findings are typically normal, except for slightly elevated protein; CSF microscopy and culture are typically negative [de Almeida et al., 2004].
Management
Inadequate clinical trials have been completed to allow evidence-based treatment recommendations for CNS paracoccidioidal infection. Brazilian guidelines on treatment [Shikanai et al., 2006] are available in Portuguese and recommend that CNS infection be treated with trimethoprim-sulfamethoxazole at a high dose for at least 2 years; amphotericin B can be used for shorter periods. Voriconazole has been used for mild to moderate paracoccidioidomycosis but its role in CNS disease is unclear [de Almeida et al., 2004; Queiroz-Telles et al., 2007; Araujo et al., 1978].
Histoplasmosis
Epidemiology, Microbiology, and Pathology
Histoplasmosis is globally distributed; the Mississippi River basin, Mexico, and Central and South America are all endemic areas [Loosli, 1957]. In these regions, as many as 80 percent of children manifest positive histoplasmosis skin test results by age 5 years [Rogers, 1967]. In most cases, infection is self-limited, but an estimated 10 percent of those infected with Histoplasma capsulatum develop pulmonary symptoms to a degree prompting medical evaluation; those younger than the age of 2 years, the elderly, and those with immunocompromising conditions are more likely to be symptomatic. Overall, males are more likely to have symptomatic pulmonary disease, whereas the disseminated form of the disease affects males and females equally. Only a small subset of individuals with primary histoplasmosis develop disseminated disease, and an even smaller group develop a CNS infection. A review of 235 cases of histoplasmosis reported between 1952 and 1960 revealed that, of the one-third of the patients who had disseminated disease, one-quarter exhibited CNS impairment [Cooper and Goldstein, 1963]. In contrast, CNS disease was not found in a review of 57 histoplasmosis patients, including 23 with disseminated disease, at a pediatric oncology center in an endemic area [Adderson, 2004].
Histoplasma capsulatum is found in the Americas, and Histoplasma duboisii is found in Africa. This dimorphic fungus grows as a yeast at body temperature, whereas mycelia form at room temperature. The mycelia phase contains macroconidia (8–15 mm) and microconidia (2–5 mm); the latter may be essential for human infectivity because they are small enough to reach the alveoli. The organism is found in soil; dampness and nutrients from bird and bat droppings encourage growth in aviaries, chicken coops, mines, caves, and open fields [Ajello and Zeidberg, 1951]. During dry spells, the organism may become airborne and the spores inhaled. No evidence of person-to-person or animal-to-human transmission exists.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Most children with respiratory infection are asymptomatic or experience little difficulty [Ibach et al., 1954]. Fever, chills, and a productive cough can occur, and initial chest radiographs may document a nodular or diffuse infiltrate. Years later, chest radiographs demonstrate characteristic calcification in the central lymph nodes and peripheral lung fields. If untreated, the infection lasts 2–12 weeks.
Progressive disseminated histoplasmosis is rare in healthy children. Signs and symptoms associated with CNS involvement in children include intracranial hypertension, memory impairment, confusion, seizures, and urinary incontinence [Machado et al., 1993]. Reported CNS complications in children include hydrocephalus [Enarson et al., 1978], meningitis [Couch et al., 1978], ataxia, seizures, and cranial nerve palsy [Rivera et al., 1992]. Approximately 5 percent of adult patients with disseminated histoplasmosis will have CNS involvement [Borges et al., 1997], with a median time to diagnosis of 36 months [Machado et al., 1993].
The histoplasmin skin test converts to positive during the course of the acute infection, but this test is not used diagnostically. A 4-fold rise in complement fixation titers between acute and convalescent specimens or a single elevated titer in the correct clinical setting is diagnostic. A rise in complement fixation titers after treatment suggests relapse. The organism can be directly detected through culture of CSF, blood, and bone marrow, and through biopsy of liver, bone marrow, and lymph nodes. Histoplasma antigen can be detected in serum or urine [Wheat et al., 1986] and in some CSF specimens of patients with chronic Histoplasma meningitis, but its use for CNS disease is unreliable [Wheat et al., 1989].
All patients with disseminated histoplasmosis should be evaluated for adrenal insufficiency. In one series, half of the patients, regardless of treatment, had adrenal insufficiency, which was the most common cause of death [Sarosi et al., 1971]. Because Histoplasma can remain quiescent in the lungs or the adrenal glands for more than 40 years before dissemination, it needs to be considered in unexplained neurologic disease, particularly in people who lived in endemic areas as children [Tan et al., 1992].
Management
CNS infection in children should be treated sequentially with amphotericin B, followed by itraconazole, according to the 2007 IDSA guidelines [Wheat et al., 2007]. The prognosis of Histoplasma CNS infections is guarded: 20–40 percent of patients die, despite treatment, and relapse among survivors is common [Wheat et al., 2000].
Nocardia
Nocardia spp., Gram-positive, branching, filamentous bacteria, are ubiquitous in the environment; Nocardia asteroides, Nocardia farcinica, and Nocardia brasiliensis are the species most often responsible for clinical disease. Although this is a bacterial infection, it is included in this section because diagnosis may need to be sought specifically, and may require techniques different from those used to identify typical causes of bacterial CNS infection [Threlkeld and Hooper, 1997]. Nocardial disease during childhood is relatively uncommon but needs to be considered in immunologically compromised hosts and those with chronic lung disease. CNS infection may be isolated or found at the same time as pulmonary or disseminated infection [Beaman and Beaman, 1994]. In those with disseminated infection, CNS involvement should be viewed with concern because it is associated with mortality rates of 80 percent [Beaman and Beaman, 1994; Threlkeld and Hooper, 1997]. When CNS disease is the only manifestation of nocardiosis, it suggests that an inapparent pulmonary disease has resolved. Risk factors for pediatric CNS infections include malignancy, chronic granulomatous disease, lupus, HIV infection, organ transplantation, and surgery [Marlowe et al., 2000].
Brain abscess is the most frequently reported CNS manifestation of nocardial infection, but meningitis can occur, typically after rupture of an abscess and, more rarely, as the primary presentation [Al Soub et al., 2007]. Nocardia brain abscess presents with signs and symptoms typical for this syndrome, including fever, headache, seizure, and focal neurologic findings. Meningitis is often reported as having a subacute or chronic course [Bross and Gordon, 1991].
Diagnosis of Nocardia brain abscess may require a drainage procedure to obtain an adequate specimen for prolonged laboratory incubation to ensure growth on culture. A nocardial etiology of brain abscess or culture-negative meningitis is strongly suggested by concurrent isolation of this organism from a pulmonary site [Bross and Gordon, 1991].
Treatment of nocardial CNS infection requires multiple agents for prolonged periods. Susceptibility testing done at a reference laboratory can help guide initial therapy and provide options, if intolerance to initial therapy develops. Most Nocardia spp. are susceptible to trimethoprim-sulfamethoxazole, but treatment of CNS infection typically requires parenteral therapy with amikacin plus imipenem or ceftriaxone, with or without trimethoprim-sulfamethoxazole [Gilbert et al., 2004]. A review of antibiotic treatment for nocardial infections has been published [Threlkeld and Hooper, 1997]; most patients require at least 6 months of therapy, and the compromised host requires longer. Treatment composition and duration are based on expert opinion, since no controlled trials have been completed. Serial neuroimaging studies during treatment may be helpful in monitoring resolution of the abscesses [Kirmani et al., 1978]. In selected patients, surgical extirpation of multiple abscesses can be successfully accomplished [Rosenblum and Rosegay, 1979].
Actinomycosis
Epidemiology, Microbiology, and Pathology
Actinomyces spp. are anaerobic or microaerophilic, Gram-positive, non-spore forming, branching bacilli that normally colonize the mouth, intestine, and female genital tract. Invasive disease, usually caused by Actinomyces israelii, is most common in those of ages 10–60 years, with males more frequently affected than females. CNS disease in young children has not been reported [Olah et al., 2004], but risk factors for CNS disease in older children and adults include untreated focal infections, immunosuppression, and congenital heart disease with right-to-left shunts [Olah et al., 2004]. Although CNS disease is rare, it can occur either after direct extension from a contiguous focus or by hematogenous spread from another site.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Actinomycosis is a chronic, slow-growing, inflammatory mass, most frequently affecting the lungs, cervicofacial region, abdomen, and pelvis (in association with intrauterine devices) [Vinard et al., 1992; Winking et al., 1996]. The mass has a tough, fibrous capsule, does not respect tissue planes, and develops sinus tracts. Concurrent chronic infections and other debilitating conditions predispose patients to dissemination.
A review of 181 cases of actinomycosis revealed that less than 2 percent of patients had cerebral involvement, as manifested by abscess (solitary or multiple) and, less commonly, meningitis [Brown, 1973; Fetter et al., 1967; Winking et al., 1996]. Abscesses have been reported in the cerebrum, cerebellum, optic chiasm, parasellar region, and subdural space [Bebrova et al., 1994; Brown, 1973]. Osteomyelitis of the spine may be the origin of spinal cord abscess and compression [Fetter et al., 1967]. Neuroimaging demonstrates abscess easily, but microbiologic diagnosis of CNS disease can be challenging because the organism may be difficult to recover on culture if preceding antibiotics have been given. In addition, infections involving Actinomyces spp. are often polymicrobial, sometimes clouding the role of individual species. The etiology of CNS disease may be inferred if Actinomyces spp. are isolated at another site, particularly the lung.
Management
Recommendations for treatment of actinomycosis at any site are based on experience and in vitro susceptibility testing, rather than clinical trials. Standard recommended treatment for CNS disease is penicillin for at least 6 months. Recently, successful shorter courses of treatment with drugs other than penicillin, such as ceftriaxone and cefotaxime, for CNS disease have been reported [Olah et al., 2004; Sudhakar and Ross, 2004]. Surgical drainage of the abscess frequently is indicated [Jamjoom et al., 1994; Winking et al., 1996] and will speed definitive diagnosis of CNS disease.
Aspergillosis
Epidemiology, Microbiology, and Pathology
Like many fungal pathogens, Aspergillus spp. (Figure 82-3) initially gain entrance to the lung by inhalation. The name of this fungus was established because of the similarity of its fruiting head to the aspergillum, a perforated globe used to sprinkle holy water during religious ceremonies. Disseminated disease and CNS infection are nearly always accompanied by a serious underlying condition.

Aspergillosis of the CNS was rare until the population of immunocompromised hosts increased during the past two decades [Carpentier et al., 1996; Sparano et al., 1992; Torre Cisneros et al., 1993; Wright et al., 1993]. Among compromised hosts, the highest frequency of invasive aspergillosis is in heart and lung transplant recipients (19–26 percent), followed by those with chronic granulomatous disease (24–40 percent), acute leukemia (5–24 percent), bone marrow or hematopoietic stem cell recipients (0.5–9 percent), AIDS (0–12 percent), and solid organ transplant recipients (0.5–10 percent) [Denning, 1998]. CNS disease complicates 10–20 percent of invasive disease and is nearly universally accompanied by concurrent infection at other sites, typically the lung.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Other less common manifestations include meningitis, meningoencephalitis, hemorrhagic or ischemic necrosis, solitary granulomas, and invasive fungal arteritis [Torre Cisneros et al., 1993]. Highly immunocompromised patients may have altered mental status and seizures, whereas more competent hosts may have headache and focal complaints. In one series, the most frequent neurologic symptoms included altered mental status (86 percent), seizures (41 percent), and focal neurologic deficits (32 percent) [Torre Cisneros et al., 1993]. Neuroimaging may reveal sinus invasion, enhancing or nonenhancing hypodense brain lesions indicative of abscess [Sparano et al., 1992], or prominent ischemic lesions in the basal ganglia [Miaux et al., 1995]; concurrent chest radiography or CT scan usually shows multiple nodular lesions.
Definitive diagnosis of invasive Aspergillus is made by smear and culture of biopsy material. A presumptive diagnosis of CNS infection can be made if Aspergillus is identified from another site at the same time that neuroimaging shows multiple mass lesions. Serum galactomannan testing for invasive aspergillosis is available [Herbrecht et al., 2002] and provides a noninvasive alternative to support the diagnosis; however, published cutoffs of sensitivity and specificity are variable [Marr et al., 2004].
Management
Cerebral aspergillosis has a very poor prognosis, particularly in the immunocompromised host. Surgical drainage and removal of abscesses and granulomas are often performed for diagnostic and therapeutic reasons, but may not improve outcome [Casey et al., 1994; Haran and Chandy, 1993]. Case reports and one retrospective review suggest that a subset of patients will respond and some may be cured [Damaj et al., 2004; de Lastours et al., 2003; Schwartz and Thiel, 2004, 2009]. The 2009 IDSA guidelines for Aspergillus spp. infection of the CNS recommend voriconazole as the primary medical therapy, with itraconazole, posaconazole, and liposomal formulations of amphotericin B as alternatives in cases of intolerance. Appropriate surgical debridement of CNS and other sites, and minimization of immunosuppresion are also recommended, while the use of corticosteroids is discouraged.
Candidiasis
Candida spp. are the most common invasive fungal infections in infants and children [Arisoy et al., 1994]. Cerebral candidiasis is a rare sequela of disseminated disease in debilitated patients. Risk factors include prematurity, long-term receipt of broad-spectrum antibiotics, total parenteral nutrition, central venous catheterization, immunosuppression, immunodeficiency (congenital or acquired), chronic renal disease, diabetes, neutropenia, and ventriculoperitoneal shunts [Arisoy et al., 1994; Chiou et al., 1994; McAbee et al., 1995]. In a review from a large pediatric referral hospital, 12 percent of disseminated Candida infections affected the brain [Zaoutis et al., 2004]. Historically, Candida albicans has been the most common species identified in CNS disease, but many others, including Candida tropicalis, Candida glabrata, and Candida parapsilosis, are also frequently reported.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
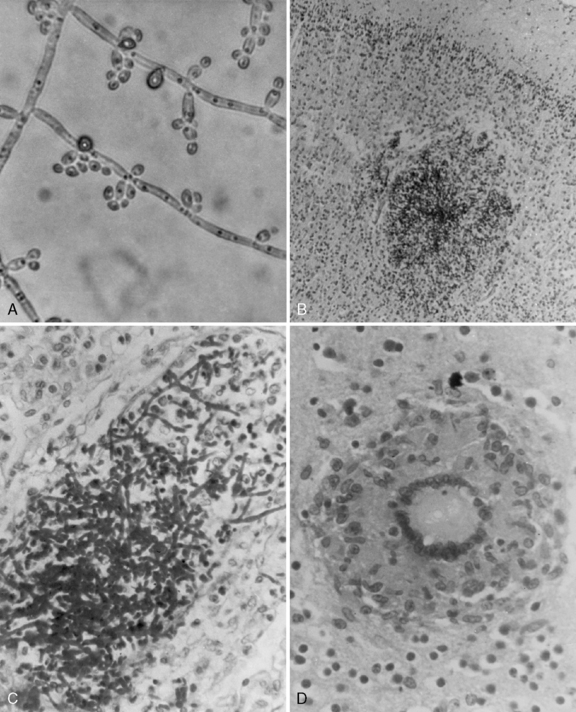
Meningitis is the most common manifestation of candidal CNS disease, but abscesses and granulomas occur independently and in combination with meningitis (Figure 82-4). When brain abscesses occur, they are typically multiple in number, illustrating the consequence of hematogenous dissemination [Incesu et al., 1994]. Intracerebral abscess after Candida septicemia is illustrated in Figure 82-4B. The clinical presentation of meningitis may be subtle in children because many patients have serious underlying illness and may have other concurrent infections. Mass lesions can cause hemiparesis, cranial nerve palsies, or signs of hydrocephalus [Coker and Beltran, 1988]. Fungal arterial and venous involvement can cause serious complications, as can the development of intracranial mycotic aneurysms [Goldman et al., 1979].
Diagnosis can be promptly confirmed by identification of the organism in spinal fluid or infected neural tissue [McGinnis, 1983]. Pleocytosis or reduced CSF glucose occurs in only about half of the patients who are culture-positive [Hughes, 1992].
Management
There are no comparative trials to permit definitive antifungal recommendations. Prompt speciation and susceptibility testing help optimize antifungal therapy. Recent IDSA guidelines for adults for C. albicans infection in the CNS recommend a liposomal formulation of amphotericin, with or without 5-flucytosine followed by fluconazole [Pappas et al., 2009]. Standard amphotericin deoxycholate may be acceptable in some young children, particularly neonates who are not routinely treated with 5-flucytosine. Consultation with an infectious disease specialist, regarding diagnosis, treatment, and follow-up, is recommended.
Zygomycosis
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
This invasive infection rarely, if ever, occurs in the normal host. Medical risk factors include diabetes mellitus, intravenous drug use, solid organ or bone marrow transplantation, and other immunocompromising conditions [Hussain et al., 1995]. Fever, facial and ocular pain, or CNS symptoms may herald the onset, and a black eschar-like plaque may be seen if sought in the oro- or nasopharynx. Etiology of CNS disease seen in MRI or CT imaging can be inferred from biopsy of nasopharyngeal or sinus specimens. Hyphae can be seen with conventional and special stains of biopsy material and are distinguished by their lack of septae, 90-degree branching, and breadth (10–20 μM).
Management
The prognosis for zygomycosis of the CNS is usually poor; however, several case reports describe survival in well-controlled diabetic patients treated with surgical debridement plus local and systemic administration of amphotericin B [Hamilton et al., 2003; Moll et al., 1994; Sandler et al., 1971]. Unfortunately, these organisms tend to infect and obstruct cranial blood vessels, and thus hamper drug delivery to affected tissue. General principles of management include correcting diabetic ketoacidosis if present, minimizing immunosuppressive therapy, and removing infected tissue surgically, often through extensive debridement, in addition to antifungal therapy [Cuadrado et al., 1988; Galetta et al., 1990; Hussain et al., 1995]. The most commonly recommended antifungal agents for zygomycosis are lipid-formulation amphotericin products at high doses. Although voriconazole is not active against zygmycosis, posaconazole is a potential alternative for initial or step-down therapy [van Burik et al., 2006]. Some authors have advocated less radical surgery combined with systemic and local (via Ommaya reservoir) amphotericin, augmented by hyperbaric oxygen therapy [Hamilton et al., 2003]. All cases should be managed by a joint medical-surgical team.
Scedosporium spp. Infection
Two species of Scedosporium environmental molds, prolificans and apiospermum (sexual state known as Pseudallescheria boydii), can cause a wide variety of human disease, including CNS infection [Cortez et al., 2008]. Risk groups include compromised hosts and survivors of near-drowning in polluted water. In a comprehensive review of 38 CNS cases, the youngest patient was 24 months old [Nesky et al., 2000].
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Scedosporium spp. can cause skin, lung, bone, joint, ocular, bloodstream and CNS infections. Brain abscess is the typical CNS manifestation and can be solitary or multiple; meningitis occurs but is less common [Rosen et al., 1965; Bell and Myers, 1978; Cortez et al., 2008]. Definitive diagnosis is through brain biopsy, although culture of the organism from a nonsterile site can support the diagnosis. There are no available noninvasive or antigen detection tests. On microscopy, this organism can be confused with Aspergillus or Fusarium spp., but it is easily distinguished once cultured.
Management
The principles of therapy are similar to that for aspergillosis and zygomycosis, and include surgical resection of accessible brain abscesses, reduction or reversal of immunosuppression, and the use of antifungal agents. Unfortunately, these organisms are notoriously resistant to most agents, and most patients with CNS disease succumb. Recently, however, in vitro data and a small number of clinical reports support the efficacy of voriconazole, possibly in combination with other agents [Cortez et al., 2008; Nesky et al., 2000; Poza et al., 2000; Gosbell et al., 2003; Howden et al., 2003].
Rickettsial Diseases
Rickettsiae are pleomorphic coccobacilli that are obligate intracellular bacteria whose arthropod vectors include ticks, mites, fleas, or lice [Kim and Durack, 1997]. Rickettsial diseases include the spotted fevers, typhus fevers, scrub typhus, Q fever, and ehrlichioses [Marrie and Raoult, 1992; Pai et al., 1997]. Rickettsial infections often have an accompanying skin eruption. Coxiella burnetii, the rickettsia causing Q fever, differs from the other organisms because it does not require a vector and rarely causes a skin rash.
CNS involvement can occur in all forms of rickettsial infections [Marrie and Raoult, 1992]. Pathologic changes may be extensive and, in some infections, systemic and cerebral vasculitis may be prominent. The clinical and pathologic response of the CNS to different infections caused by Rickettsia spp. is generally similar and differs only in severity [Harrell, 1953; Woodward and Osterman, 1984]. Headache is often the earliest and most frequent symptom in all rickettsial infections [Harrell, 1953].
Recommended treatment of rickettsial diseases in almost all clinical situations includes tetracyclines. Most broad-spectrum antibiotics, such as penicillins, cephalosporins, and penems, are ineffective. Doxycycline is considered the drug of choice in almost all clinical situations for both pediatric and adult patients [Dalton et al., 1995; Kirkland et al., 1995]. The dosage is 100 mg twice per day for adults, and the pediatric dose is 4 mg/kg/day, divided twice daily, intravenously or orally, for 10–14 days [Schutze and Jacobs, 1997]. Because tetracyclines are contraindicated during pregnancy, chloramphenicol is used as an alternate drug in this patient population; however, this drug has questionable efficacy for patients with ehrlichiosis and Q fever [Markley et al., 1998]. Empiric treatment with doxycycline should be initiated early in the course if clinical and epidemiologic findings are suggestive of a rickettsial infection because delays in treatment can lead to adverse outcomes. Tetracycline use in pediatric patients is often discouraged because of the potential association with tooth discoloration. For suspected cases of rickettsial illnesses, however, it is important to use a tetracycline. Tetracycline tooth staining is dose-related and studies have found that perceptible tooth staining does not occur until multiple courses have been used [Grossman et al., 1971; Lochary et al., 1998].
Table 82-2 lists rickettsial diseases, organisms, vectors, reservoirs, and geographic distribution. Rocky Mountain spotted fever is the prototypic condition for these infections.
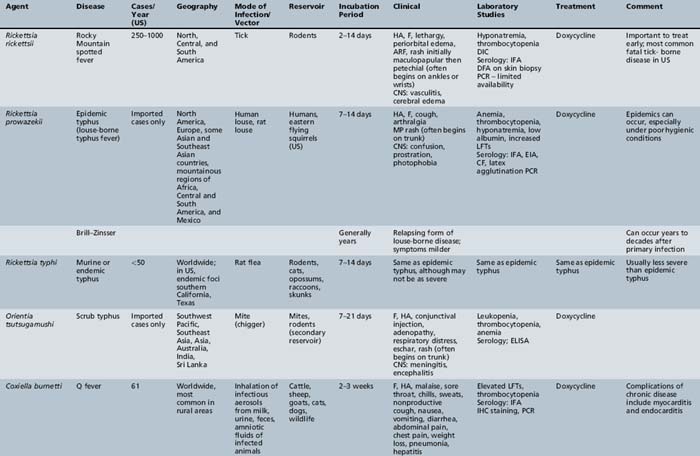
Rocky Mountain Spotted Fever
Rocky Mountain spotted fever (RMSF) is caused by Rickettsia rickettsii and is the most common fatal tick-borne illness in the United States [CDC, 2004]. Early missionaries and settlers discerned the association of tick bites with the disease and called the disease tick fever [Aikawa, 1966]. Howard Ricketts identified the etiologic organism in 1906 and established the tick as the vector in transmitting the organism to humans.
The disease is prevalent in wide geographic areas. RMSF has been reported throughout the United States, with the exception of Hawaii and Vermont [Walker, 1995]. The states with the highest incidence of RMSF are Oklahoma and North Carolina [CDC, 2008a]. In the United States, the number of RMSF cases has increased in recent years, with over 2000 cases a year reported in 2006 and 2007 – the highest recorded levels in more than 80 years [Walker et al., 2008; CDC, 2008b]. Outside the United States, the re-emergence of the disease has been described in several countries of Central and South America [Heldrich, 1987; Hidalgo et al., 2007; Nava et al., 2008]. In the Northern Hemisphere, the incidence of the disease is greatest from April to September, when tick bites are most likely to occur. The American dog tick (Dermacentor variabilis) is the primary vector responsible for the transmission of R. rickettsii along the southern Atlantic coast and the southeastern and south-central United States. In the western region of the United States, the vector is usually the wood tick (Dermacentor andersoni; Figure 82-5). Given the habitat preferences of the vector, rural and suburban populations are more likely to be infected than urban residents.

Children have a higher risk of infection (half of the cases are younger than 19 years of age [Kamper, 1991]) because they are exposed to tick bites during outdoor play. RMSF should be considered in the differential diagnosis of anyone with compatible clinical features and exposure to a grassy or wooded area where ticks may be present [Walker, 1995]. Dogs may carry ticks into households and yards, and are a risk factor for infection [Paddock et al., 2002; Walker, 1995]. The absence of a reported tick bite history should not dissuade a clinician from suspecting RMSF. In a recent study of 92 children with RMSF, only 49 percent recalled a recent tick bite [Buckingham et al., 2007].
The incubation period ranges from 2 to 14 days (mean 7 days) after a tick bite [Thorner et al., 1998]. Transmission of rickettsiae occurs as a tick feeds on the blood of its host. After entering the body, rickettsiae multiply and disrupt the endothelial cells of capillaries and arterioles. Vasculitis is the pathologic hallmark of RMSF, and many of the clinical features are a result of vascular inflammation. In RMSF, the rickettsiae may also infect the smooth muscle cells of the tunica media. Loss of integrity of the small vessels in the brain results in hemorrhage into the subarachnoid space. Subsequent meningeal irritation and an accompanying perivascular mononuclear infiltrate develop. The brain parenchyma is studded with areas of microscopic infarctions. In the healing phase, a proliferative fibroblastic and gliotic response is evident; the ensuing focal nodules develop more often in white matter in spotted fever and more often in gray matter in typhus.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Symptoms of RMSF vary in severity from mild to severe; the course may rapidly lead to death. RMSF is characterized by the classic triad of fever, headache, and rash in the setting of a recent tick bite. Most patients with RMSF, however, do not present with the classic triad. Initial symptoms are nonspecific and include fever, malaise, lethargy, and headache. The headache is usually intense and refractory to most analgesics. Progressive restlessness, irritability, confusion, and delirium may also occur. By day 5 of the illness, 85 percent of patients develop a rash. The rash often begins with a maculopapular appearance and becomes more defined as petechiae as the rash evolves. The rash often begins around the ankles or wrists and then spreads to other parts of the body (Figure 82-6). Acute renal failure, coagulopathy, and cerebral edema are well-recognized complications. Edema, usually periorbital at first and then generalized, is characteristic of and may suggest the diagnosis. Hepatosplenomegaly accompanied by liver dysfunction, including coagulation defects, may confound the diagnostic process. In fulminating disease, shock, coma, and general or focal neurologic impairment, including vertigo, seizures, hemiparesis, and ataxia, may dominate the clinical manifestations [Marrie and Raoult, 1992]. Skin necrosis and gangrene can be a late complication (Figure 82-7). RMSF may manifest as a skeletal muscle disease. Acute temporary hearing impairment has been described, but permanent auditory disruption is uncommon [Kelsey, 1979]. Ophthalmic features in all rickettsial disorders are virtually identical [Raab et al., 1969; Smith and Burton, 1977], and may include photophobia, conjunctivitis, petechiae on the bulbar conjunctiva, exudates and retinal venous engorgement, papilledema, and ocular palsies.
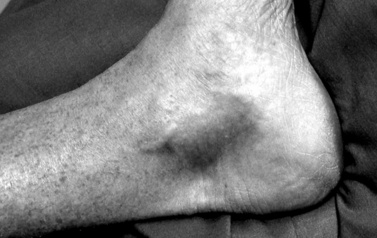
Fig. 82-6 Petechial rash of Rocky Mountain spotted fever involving the ankle.
(Courtesy of Dr. Daniel J Sexton.)
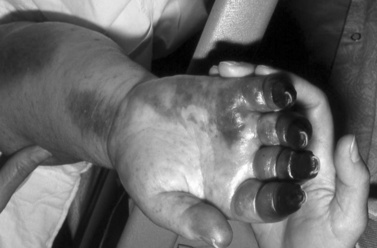
Fig. 82-7 Necrosis and gangrene of digits is a late complication of Rocky Mountain spotted fever.
(Courtesy Dr. Daniel J Sexton.)
The differential diagnosis of RMSF includes meningococcemia, enteroviral infection, scarlet fever, toxic shock syndrome, Kawasaki disease, Epstein–Barr virus, syphilis, typhoid, rat-bite fever, and other tick-borne diseases. Muscle biopsy reveals perivascular infiltrates of chronic inflammatory cells and focal chronic interstitial myositis [Krober, 1978]. Other organs, including kidneys, heart, lungs, spleen, and epididymis, may also be affected [Green et al., 1978]. Muscle weakness may be overwhelming; therefore, Guillain–Barré syndrome, viral myositis, and collagen disease may be erroneously diagnosed (although a case of Guillain–Barré syndrome associated with RMSF has been documented [Toerner et al., 1996]).
Laboratory abnormalities are common in severe disease and often include hyponatremia and thrombocytopenia. Spinal fluid may indicate a lymphocytic pleocytosis and the protein is elevated in about 30–50 percent of patients [Kim and Durack, 1997]. The cellular response in CSF may suggest bacterial meningitis, and the dermal petechiae may mimic those in meningococcemia. MRI of the brain reveals increased signal intensity in the distribution of perivascular spaces, which resolves in relation to clinical improvement [Baganz et al., 1995]. Postmortem examination of RMSF cases has demonstrated that rickettsial vasculitis can result in focal neurologic damage in the brain and spinal cord, as well as myocarditis and vasculitis in the lungs, kidney, and spleen [Walker and Mattern, 1980].
The most widely used and best serologic test for RMSF is the indirect fluorescent antibody assay, which is available through commercial laboratories and most state health departments [Lochary et al., 1998; Sexton and Kaye, 2002]. Immunofluorescent antibody for immunoglobulin M or immunoglobulin G antibodies can be used to make a presumptive diagnosis of acute infection, although these antibodies are often undetectable in the early phase of the illness. Confirmatory indirect fluorescent antibody testing requires demonstration of a 4-fold or greater rise in immunoglobulin G or immunoglobulin M antibody titer between paired serum specimens, and is generally considered as a retrospective diagnosis [Thorner et al., 1998]. Direct immunofluorescence antibody or immunohistochemical staining of skin biopsies is 70–90 percent sensitive for patients with a rash. For patients without a rash, diagnosis by direct immunofluorescence antibody can be problematic. Polymerase chain reaction testing for R. rickettsii DNA can be applied to serum, whole blood, or tissue specimens, but is generally available only at specialized research laboratories, and the sensitivity of the assay is directly related to the severity of the illness [Sexton et al., 1994].
Management
Early therapy with doxycycline for at least 7 days is recommended. The case fatality is 2–10 percent in treated patients and may be as high as 30 percent in untreated patients [Dalton et al., 1995]. Serologic confirmation may be delayed for 2–3 weeks; therefore, therapy should be administered when the diagnosis is suspected, based on clinical and epidemiologic findings. A history of tick bite is a useful epidemiologic clue in making a presumptive diagnosis. With early treatment, the prognosis for full recovery from RMSF is excellent. Neurologic sequelae may occur when therapy is postponed [Miller and Price, 1972]; mild intellectual impairment has been described [Wright, 1972].
Typhus Fever Group
The typhus fever group consists of epidemic (louse-borne typhus), Brill–Zinsser disease, and murine (flea-borne) typhus. Louse-borne typhus fever, caused by Rickettsia prowazekii, is distributed worldwide. Epidemics of typhus fever occur by transmission from human to louse to human, typically after skin is rubbed that is contaminated with infective louse feces. Epidemics of typhus fever are often associated with war, movements of large numbers of people, and squalid living conditions [Zinsser, 1935]. Systemic and neurologic symptoms are similar to RMSF (see above). A severe headache is almost always present. More severe CNS features, such as neck stiffness, seizures, delirium, and coma, can also occur. Treatment (e.g., with doxycycline) is the same as that used for the spotted fever group of infections.
Brill–Zinsser disease is a relapsing form of louse-borne typhus that may occur years to decades after the primary attack [Feigin et al., 1992]. Presumably, the rickettsiae remain dormant in the reticuloendothelial system until a subsequent reactivation occurs. Symptoms are similar to the primary attack, although generally milder, and doxycycline is the recommended therapy.
Murine, or flea-borne typhus, caused by Rickettsia typhi, is one of the most prevalent rickettsial diseases found in humans throughout the world [Azad, 1990]. Before World War II, murine typhus was widespread in the United States, and approximately 42,000 cases were reported between 1931 and 1946 [Azad, 1990; Pratt, 1958]. During the Vietnam War, murine typhus [Miller and Beeson, 1946] was one of the most common causes of acute febrile illness in American military personnel [Berman and Kundin, 1973]. Currently, endemic foci often exist around port cities and adjacent coastal areas [Azad, 1990; Civen and Ngo, 2008]. R. typhi is transmitted from rat to rat by the rat flea. In addition to rats, many animals, such as other rodents, skunks, opossums, shrews, and cats, can serve as hosts for R. typhi and arthropod vectors. Rickettsia felis causes a recently described rickettsial infection with similar clinical manifestations. The life cycle of R. felis is through opossums, cats, and cat fleas; infection has been identified in southern California and Texas [Civen and Ngo, 2008]. Human infection occurs after exposure of abraded or flea-bitten skin to infective flea feces [Feigin et al., 1992]. Unlike many other arthropod-borne infections, murine typhus can be acquired in households because the transmission cycle includes commensal rats and fleas [Azad, 1990]. Symptoms of murine typhus are similar to but generally milder than those occurring in RMSF. R. typhi infections are likely underdiagnosed because the clinical and laboratory features are similar to viral infections. The illness is often characterized by fever (frequently prolonged), headache, myalgia, and sometimes a maculopapular or petechial rash [Gelston and Jones, 1977]. Lymphadenopathy was reported as a common manifestation in a study in children [Bitsori et al., 2002]. When present, the rash often occurs on the trunk. The infection may resemble Kawasaki’s disease [Bitsori et al., 2002]. There is considerable variability in the frequency of reports of neurologic manifestations of murine typhus, ranging from 2 to 20 percent of cases [Allen and Saitz, 1945; Bitsori et al., 2002]. Meningitis and encephalitis are occasionally reported [Bitsori et al., 2002; Galanakis et al., 2002; Gelston and Jones, 1977]. Less commonly, focal symptoms, such as papilledema, focal seizure, and hemiparesis, have also been described [Masalha et al., 1998]. Sensorineural hearing loss has been reported during the convalescent phase of the illness [Tsiachris et al., 2008]. Diagnostic evaluation and treatment are similar to other diseases of the typhus group.
Scrub Typhus
Scrub typhus, caused by Orientia tsutsugamushi, is a systemic illness that causes a generalized vasculitis [Pai et al., 1997]. O. tsutsugamushi was formerly a member of the spotted fever group of Rickettsia spp., but was placed in its own genus based on molecular phylogenetic studies [Tamura et al., 1995]. Trombiculid (chigger) mites serve as reservoirs and vectors of this disease, and human infection occurs after a chigger bite in endemic areas, primarily in the southwest Pacific and Southeast Asia [Feigin et al., 1992]. An eschar, a characteristic feature of the disease, is seen in 60–80 percent of cases [Jelinek and Loscher, 2001] (Figure 82-8). Fever, headache, and rash are also common clinical manifestations. Complications include renal failure, pneumonitis, acute respiratory distress syndrome, and myocarditis. CNS complications include meningitis or meningoencephalitis. Case series have reported varying occurrence/frequencies of meningoencephalitis ranging from 3 to 20 percent [Fang et al., 1997; Pai et al., 1997; Huang et al., 2009]. Other less commonly reported neurologic complications include acute disseminated encephalomyelitis (ADEM), Guillain–Barré syndrome, brachial plexus neuropathy, and acute sensorineural hearing loss [Chen et al., 2006; Lee et al., 2009; Ting et al., 1992; Kang et al., 2009]. Approximately 50 percent of patients will have a mild CSF monocytic pleocytosis or an increase in protein concentration [Pai et al., 1997]. Doxycycline is considered the most effective treatment.
Q Fever
Q fever, caused by Coxiella burnetii, is a worldwide zoonosis. Infection in humans occurs from inhalation of the organism in areas where infected livestock are kept and, on occasion, from ingestion of contaminated dairy products. Occasional outbreaks in research laboratories that use sheep or cattle have also been reported [Feigin et al., 1992]. Prominent symptoms include fever, headache, and pneumonia. A rash rarely, if ever, occurs. Cases of encephalitis have been reported to accompany Q fever and may occur late in the course [Maurin and Raoult, 1999]. Other reported neurologic manifestations have been described, and these include pseudotumor cerebri syndrome, myelitis, meningitis, hemiparesis, aphasia, ataxia, seizures, tinnitus, cranial nerve palsies, and behavioral abnormalities [Ravid et al., 2008; Bernit et al., 2002; Glaser et al., 2010]. The reported frequencies of neurologic symptoms associated with Q fever vary considerably, ranging from 2 to 22 percent [Clark et al., 1951; Bernit et al., 2002].
Other Rickettsiae
Several new rickettsiae have been described since the early 1990s: Rickettsia japonica in Japan and Korea, Rickettsia honei in Australia and Southeast Asia, Rickettsia africae in Africa and the French West Indies, the Rickettsia mongolotimonae strain in Asia, Europe, and Africa, Rickettsia slovaca in Europe, Rickettsia aeschlimannii in Africa and Europe, Rickettsia heilongjanghensis in Asia, and Rickettsia parkeri in the United States [Raoult, 2004; Walker, 2007]. The extent of neurologic involvement with these emerging infections is unknown.
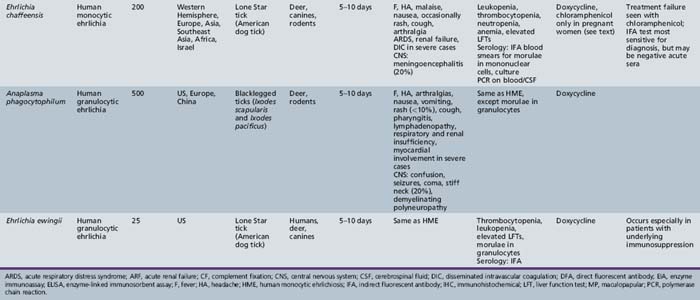
Ehrlichiosis
Emerging infections, akin to those caused by other rickettsial agents, have been increasingly reported in recent years [Walker et al., 1996].
The term ehrlichiosis was initially used to describe veterinary infections with Rickettsia-like bacteria that infected leukocytes. Human infection with Ehrlichia in the United States was first identified in a 51-year-old male with a history of a tick bite, fever, leukopenia, thrombocytopenia, and elevated liver enzymes [Maeda et al., 1987]. The patient was originally diagnosed with RMSF (see above). Cytoplasmic inclusions were observed in peripheral lymphocytes, and the patient was subsequently diagnosed with an Ehrlichia infection (E. chaffeensis) species. Since then, two additional Ehrlichia species have been recognized: Ehrlichia ewingii and Anaplasma (formerly Ehrlichia) phagocytophilum.
Ehrlichia are obligate intracellular bacteria and belong to the family Rickettsiaceae. All Ehrlichia are transmitted by ticks, and the interval from tick exposure to onset of illness is between 2 and 21 days [Harkess, 1991]. E. chaffeensis infects mononuclear cells and causes human monocytic ehrlichiosis. The incidence of human monocytic ehrlichiosis is approximately equal to, and sometimes higher than, that of RMSF [Fishbein et al., 1990]. Anaplasma phagocytophilum causes human anaplasmosis (formerly human granulocytic ehrlichiosis) and infects neutrophils. E. ewingii also infects neutrophils and has serologic cross-reactivity with E. chaffeensis. Rashes are generally less common with the ehrlichioses than with rickettsial infections [CDC, 1988].
Human Monocytic Ehrlichiosis
Human monocyctic ehrlichiosis (HME) generally causes an acute febrile illness occurring after E. chaffeensis is inoculated by the Lone Star tick (Amblyomma americanum). Between 2003 and 2008, 3408 cases of HME were reported in the United States [CDC, 2009]. Fever, headache, and malaise are commonly described. A skin rash, appearing macular or maculopapular, occurs more commonly in children than adults, and is often on the trunk or extremities [Everett et al., 1994; Fishbein et al., 1994; Ratnasamy et al., 1996]. Nausea, vomiting, abdominal pain, and cough are other frequent manifestations. Approximately 20 percent of infections result in meningoencephalitis. Long-term neurologic sequelae have been reported in a limited number of children, including a 7-year-old with reading problems and impairment of fine motor skills, and an 11-year-old with bilateral footdrop and speech impairment [Schutze and Jacobs, 1997].
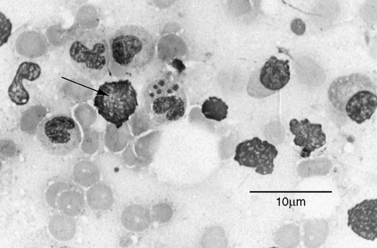
Laboratory testing often reveals leukopenia, thrombocytopenia, anemia, and increases in hepatic transaminases. Peripheral blood examination can reveal intracytoplasmic inclusions (termed morulae) in white blood cells in a small percentage of cases (Figure 82-9). CSF abnormalities often include lymphocytic pleocytosis and high protein [Ratnassamy et al., 1996]. Serologic testing can sometimes be helpful but is often negative during the early stages of the illness. The quickest method of diagnosis is via Giemsa or Wright strain of the buffy coat or peripheral blood to look for intravacuolar bacterial inclusions (morulae) which are stippled and stain dark blue or purple [Thomas et al., 2009]. Morulae identifed in monocytes are indicative of HME. Evaluation of paired serum samples is generally the optimal method of establishing the diagnosis but provides only retrospective confirmation. Molecular testing (e.g., polymerase chain reaction) is available in some settings and is currently the most sensitive modality for diagnosis during the acute phases of infection (particularly before antibiotic use) [Dumler, 2003; Thomas et al., 2009].
Human Anaplasmosis
Human granulocytic anaplasmosis was identified in humans in the 1990s. The organism in humans was initially believed to be the same organism that causes disease in horses (Ehrlichia equi) and ruminants (Ehrlichia phagocytophila) [Chen et al., 1994; Madigan et al., 1995]. Molecular analysis of these organisms resulted in a taxonomic reassignment of human anaplasmosis so that the organism is now considered to belong in the family Anaplasmataceae rather than family Rickettsiaeceae. Current terminology often refers to this disease as human anaplasmosis. Anaplasma phagocytophilum, the organism causing human anaplasmosis, is transmitted by blacklegged ticks (Ixodes scapularis and Ixodes pacificus). Although human granulocytic anaplasmosis was only recently identified, almost 4000 cases were reported in the US from 2003 through 2008 [CDC, 2009].
The clinical features of human anaplasmosis are similar to those of HME, including fever, headache, and myalgias. Gastrointestinal symptoms are seen in about 25 percent and a rash in 10 percent. Unlike HME, however, CNS involvement is uncommon in human granulocytic anaplasmosis; meningoencephalitis is reported in only about 1 percent of cases [Dumler et al., 2007]. Peripheral neuropathies have been described, including cranial nerve palsies and brachial plexopathy [Horowitz et al., 1996]. Severe clinical manifestations include septic shock manifestations, toxic shock syndrome, acute respiratory distress syndrome, myocarditis, and rhabdomyolysis.
Laboratory testing often reveals leukopenia, thrombocytopenia, and mild increases in serum transaminases. Diagnostic testing during the active phase includes checking for morulae in a blood smear (morulae of human granulocytic anaplasmosis found in neutrophils), polymerase chain reaction on blood culture, and possibly serology [Thomas et al., 2009]. Serologic testing with paired sera is helpful for confirmation but not necessarily helpful during the acute stage of infection. When found, morulae are helpful in making a rapid diagnosis, but this method may be insensitive.
Ehrlichia ewingii Ehrlichiosis
In the late 1990s, another Ehrlichia species emerged in the United States: E. ewingii [Buller et al., 1999]. Similar to E. chaffeensis, E. ewingii is transmitted by the Lone Star tick. Of the few cases reported to date, clinical symptoms are similar to HME and human anaplasmosis but are generally milder. Laboratory abnormalities, such as leukopenia, thrombocytopenia, and elevated transaminases, are seen in cases, as in other Ehrlichia infections. Morulae can be identified in neutrophils, yet there can be serologic cross-reactivity to E. chaffeensis.
Parasitic Diseases
Human parasitic infestation occurs worldwide [Bia and Barry, 1986; Markell et al., 1992; Warren and Mahmoud, 1984]. Parasites infect humans in tropical, temperate, and cold climates. Parasitic infections impair the CNS [Brown and Voge, 1982] by a variety of mechanisms, including the following:
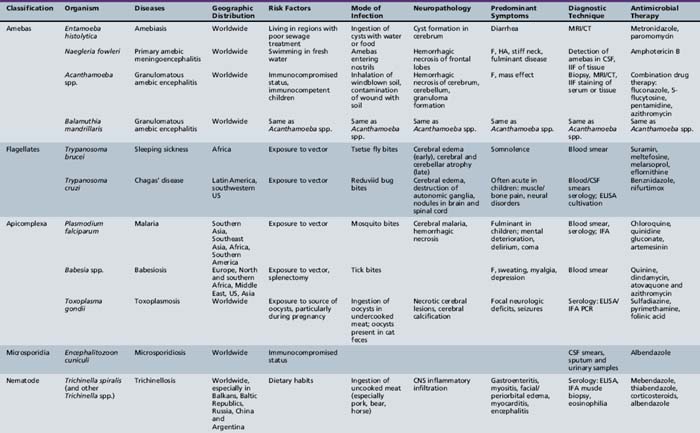
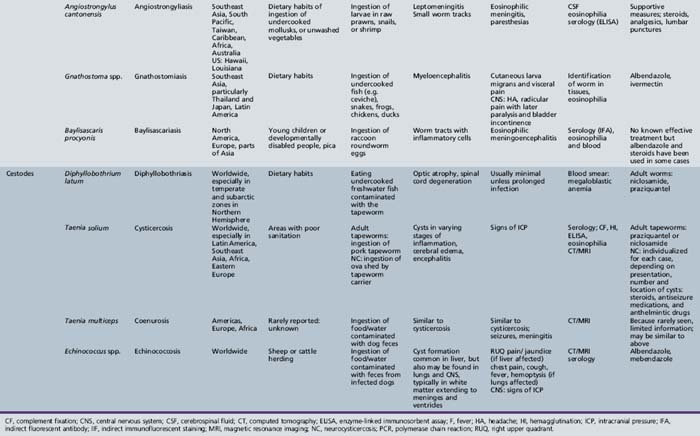
Many different classifications of parasitic disease have been proposed [Garcia, 1992; Markell et al., 1992], but none is completely satisfactory because of the complexity, widespread distribution, and incredible variety of infectious agents that are known. Modified or even new classification systems for parasites are being proposed, based on data from DNA sequencing studies, which have become an important tool in the understanding of phylogeny and taxonomy of these organisms. Table 82-3 lists the most common parasitic diseases that are known to cause serious neurologic disease and are reviewed in this section. A large group of parasites includes the helminths. The helminths (worms) are subdivided into three categories: nematodes, cestodes, and trematodes. Nematodes are cylindrical and nonsegmented worms. Trichinella, Angiostrongylus, and Toxocara are genera in this class that infect the CNS. Cestodes that infect humans belong to the subclass Eucestoda, the true tapeworms. These endoparasites have no epidermis or digestive tract and have elongated segmented bodies. Adults have a multihook organ of attachment at the anterior end called the scolex. Genera of cestodes associated with CNS infection in the larval stage include Dibothriocephalus (Diphyllobothrium), Taenia, Multiceps, and Echinococcus. Trematodes (flukes) are nonsegmented worms that possess a digestive tract. Trematodes infecting the nervous system include the genera Schistosoma and Paragonimus.
Isolation and growth are important for accurate identification of etiologic agents, preparation of antibodies for serology, testing for efficacy of antimicrobial agents, and as a source of DNA for diagnostic and molecular biologic procedures [Visvesvara and Garcia, 2002]. Cultivation of parasitic protozoa is often demanding and may require complex media that are not readily available to most diagnostic laboratories or in the field. For this reason, in vitro cultivation of protozoal parasites is relegated to research or reference laboratories. In the United States, in vitro growth of these parasites is carried out primarily by the Centers for Disease Control and Prevention (CDC), specific (usually academic) research laboratories, or at state public health laboratories [Lowichik and Siegel, 1995]. In such cases, consulting with and sending specimens to these laboratories is usually helpful for early diagnosis and treatment. For the parasitic worms, in vitro cultivation has yet to be realized.
Protozoal Infections of the Central Nervous System
Amebic Infections of the Central Nervous System
Cerebral Amebiasis: Entamoeba histolytica
Epidemiology, microbiology, and pathology
Amebiasis results from gastrointestinal infection by the protozoon E. histolytica. Amebic dysentery was described in ancient times by Galen and Hippocrates, and even earlier descriptions of dysentery-like illness were found in Assyrian and Babylonian writings [Cox, 2002]. The role of E. histolytica in causing pathogenic intestinal and hepatic lesions was described definitively by [Kartulis 1886] and [Councilman and Lafleur 1891]. Its distribution is global, although it is more common in regions that have hot, humid climates. Despite its partiality for tropical climates, the organism was first isolated from a stool sample by Lösch in St. Petersburg, Russia, in 1873, and an outbreak of amebiasis occurred at a Chicago hotel at the time of the 1933 World’s Fair, resulting in about 100 deaths.
Gross postmortem examination of the brain reveals abscesses, petechiae, and suppurative meningitis. Necrotic areas are distributed throughout the brain but are concentrated in the basal ganglia and frontal lobes. Cerebral edema and transtentorial herniation are often seen. Microscopic study demonstrates a zone of disruption; necrotic areas are more common than areas of inflammation. The inflammatory response is attended by mononuclear cells [Lombardo et al., 1964].
Clinical characteristics, clinical laboratory tests, and diagnosis
From the intestine, the liver, lungs, and brain are secondarily affected. Cerebral amebiasis is the most unusual condition and principally occurs in patients with hepatic or pulmonary infections. Most patients with cerebral amebiasis have symptoms that accompany the effects of systemic disease, such as fever, weight loss, and anorexia. Cerebral amebiasis is rare, and many of the patients who have this disorder do not manifest neurologic symptoms. In one series, only 5 of 17 patients had neurologic manifestations, consisting of increased intracranial pressure, hemiplegia, meningeal irritation, nuchal rigidity, confusion, and headache [Lombardoet al., 1964]. The presence of a focal CNS lesion in a febrile patient with hepatic or intestinal disease should suggest amebiasis, particularly if there is a history of travel to an endemic area and if more common causes, such as bacterial brain abscesses or tumors, have been ruled out [Durack, 1997].
The diagnosis of cerebral amebiasis can usually be confirmed by serologic evaluation using an indirect hemagglutination assay or indirect immunofluorescent staining. Commercial kits are available for detection of ameba antigen, and the polymerase chain reaction has been used for detecting Entamoeba DNA [Leber and Novak, 1999; Tanyuksel and Petri, 2003]. CSF evaluation is usually not helpful; cultures are usually sterile, and cytology or changes in the protein or glucose concentrations are nonspecific. The polymerase chain reaction applied to fluid drained from a brain abscess in an adult detected E. histolytica DNA, though intact amebas were not seen [Ohnishi et al., 1994]. In a pediatric case, intact amebas were recovered in fluid aspirated from the thorax but were not detected in the child’s stool [Hughes et al., 1975]. Stool cultures are usually negative when nervous system involvement appears. Neuroimaging may reveal abscess formation with perilesional edema [Tikly et al., 1988]. General testing for amebic colitis and liver abscess is reviewed in Tanyuksel and Petri [2003].
Management
Cerebral amebiasis is rarely treated successfully. Metronidazole is currently the preferred drug (15 mg/kg initially followed by 30 mg/kg/day divided into four doses) [Strickland, 1992], although trinidazole may be more effective than metronidazole [Medical Letter, 2004]. Emetine and chloroquine are alternative medications.
A combination of metronidazole and chloroquine was used successfully in treating a pediatric case of cerebral amebiasis with three abscesses [Hughes et al., 1975]. Amebiasis in an adult was treated with metronidazole and dehydroemetine after drainage of a brain abscess [Ohnishi et al., 1994]; the patient recovered, with minor neurologic impairment. An adult male in a stuporous state, with signs of encephalitis and hepatic abscesses, had a low titer for E. histolytica by indirect hemagglutination, but countercurrent immunoelectrophoresis was positive [Schmutzhard et al., 1986]. CT scans of the brain exhibited approximately 40 small ringlike lesions, mostly in the white matter of both hemispheres. Treatment with metronidazole, chloroquine, and tetracycline effected a partial recovery; further regression of the lesions and recovery occurred after a course of dehydroemetine treatment. Paromomycin has been used for asymptomatic cases [Medical Letter, 2004].
Primary Amebic Meningoencephalitis: Naegleria fowleri
Epidemiology, microbiology, and pathology
Primary amebic meningoencephalitis (PAM) is a fulminant disease occurring in children and young adults. The disease was first reported from Australia and was initially attributed to Acanthamoeba [Fowler and Carter, 1965], which previously had been found to cause death of mice inoculated intranasally with amebas [Culbertson et al., 1958]. The disease was soon reported from Florida and was then characterized as PAM [Butt, 1966]. N. fowleri is a free-living, facultative parasite, with a life cycle that includes an ameboid trophic (feeding) stage, a transitory flagellate stage, and a double-walled cyst able to withstand desiccation. The amebas measure about 15–30 μm, and cysts from 7 to 15 μm in diameter. The ameba is found in water, soil, sewage, or other decaying organic material where there is a bacterial food source [Marshall et al., 1997], and grows optimally at elevated temperatures (37–45°C). Of an estimated 200 cases since the first descriptions of PAM, more than 90 percent of the patients have died. The disease has been reported from Australia, Belgium, Czech Republic, Great Britain, Mexico, India, New Zealand, Nigeria, and the United States. Extended descriptions of the organism and the disease can be found in several review articles [Barnett et al., 1996; Duma, 1984; John, 1993].
In the United States, most of the cases have developed in the southern tier of the country, where warm water conditions are more likely to be encountered. In Florida, where a number of cases have been reported after swimming in lakes, the probability of infection was estimated to be 1 case per 2.6 million exposures [Wellings, 1977]. No cases have resulted from drinking water containing amebas.
Typically, infection with N. fowleri occurs while swimming or washing in warm, fresh water containing the amebas, and early manifestations of the disease ensue. Bodies of water associated with amebic infections have included ponds, man-made lakes, hot springs, wading pools, irrigation canals, thermally polluted streams, and inadequately chlorinated swimming pools. Those affected are children or young adults without underlying disease. Infection has been associated with prolonged immersion in water, diving, underwater swimming, or other activities that allow water to enter the nostrils. Well water that was used to bathe a child was believed to be the source of Naegleria in a 6-month-old child who died with PAM [Hebbar et al., Annls of Trop Peds, 2005]. The onset of PAM is usually within 2–3 days after exposure. Amebas enter the nasal passages and gain access to the nervous system by penetrating the olfactory mucosa, entering the submucosal nervous plexus, migrating along the olfactory nerves, and traversing the cribriform plate. Hemorrhagic destruction of gray matter and devastation of the olfactory bulbs follow sanguinopurulent meningitis and pronounced brain edema [Martinez et al., 1973]. Spinal cord involvement is signaled by similar gray-matter disruption. Perivascular infiltrates include leukocytes and amebas (Figure 82-10). Numerous eosinophils may be present. Simultaneous bronchopneumonia and pulmonary edema, myocarditis, and splenitis may occur.
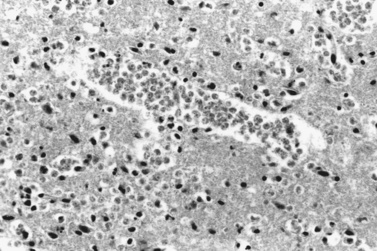
Clinical characteristics, clinical laboratory tests, and diagnosis
Among the most common symptoms of PAM are severe headache, stiff neck, nausea, vomiting, fever (39–40°C), behavioral changes, seizures, diplopia, and coma [Duma, 1984; Martinez, 1985]. One patient complained of a violent headache and foul odors before becoming unconscious [Duma, 1984]. Distortion of taste has also been reported [Martinez, 1993]. CSF is typically cloudy with a slightly decreased glucose concentration, increased protein content (75–970 mg/dL), and innumerable white blood cells (300–24,600/mm3) and erythrocytes [Martinez, 1985]. Polymorphonuclear cells make up 52–99 percent, lymphocytes 1–48 percent, and monocytes 1–2 percent of the white blood cells in the CSF. High opening pressures may be observed.
Differential diagnosis for PAM is bacterial meningitis, but the CSF is sterile in PAM [Martinez, 1993]. A Gram stain of the CSF is not particularly helpful in diagnosis because the amebas may lyse during preparation of the slide. In freshly obtained spinal fluid, motile Naegleria amebas can usually be seen in wet-mount preparations [Lowichik and Ruff, 1995], which are best viewed with a phase-contrast microscope. The CSF should not be refrigerated or frozen before examination. Low-speed centrifugation of the sample may help concentrate the amebas and make them easier to find. In CSF from three PAM patients, amebas numbered from 26 to 118/mm3, as compared with 330 to more than 9000 leukocytes/mm3 [Duma et al., 1971]. The amebas can be recognized by their relatively swift movement with an anterior, eruptive pseudopod (Figure 82-11), contrasting with the sluggish motility of the leukocytes with indistinct pseudopodal activity. As an aid in diagnosis, amebas can be readily cultured on non-nutrient agar plates spread with a suspension of Escherichia coli as a food source [Visvesvara, 1999]. Although not widely available, molecular testing may prove to be a sensitive method for testing even archived samples [Schild et al., 2007]. Immunofluorescent staining for Naegleria antibodies in acute-phase serum is of little diagnostic value because of the fulminant nature of the disease and a delayed humoral response.
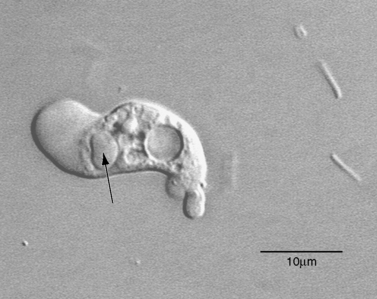
Neuroimaging is not particularly helpful in diagnosis, with results being nonspecific. CT and MRI scans of a 9-year-old child hospitalized with what was diagnosed upon postmortem examination as PAM showed no abnormalities at time of admission [Kidney and Kim, 1998]. Three days later, 1 day before the child died, a CT scan indicated developing hydrocephalus and brain edema but no focal lesions.
Management
The high mortality from PAM may be a consequence of delays in diagnosis and initiation of antimicrobial therapy. Early diagnosis and intensive care may improve the small chance of recovery [Seidel, 1985a]. Differences in virulence of PAM ameba isolates, when tested by mouse inoculations, indicate that this may be a factor affecting survival. Naegleria amebas are sensitive to amphotericin B, the drug of choice in treating PAM. In a well-documented PAM case, the patient, a 9-year-old female, had been swimming in a California hot spring before developing symptoms of meningoencephalitis. She was given intravenous and intrathecal amphotericin B and miconazole, oral rifampin, and sulfisoxazole, and recovered with minimal neurologic sequelae [Seidel et al., 1982]. Other successful treatments have been reported [Apley et al., 1970; Brown, 1991; Jain et al., 2002; Wang et al., 1993]. The macrolide azithromycin has demonstrated amebacidal activity in vitro and in the mouse model and may have potential for treating primary amebic meningoencephalitis [Goswick and Brenner, 2003]. Although azithromycin penetrates the brain, its low concentration in CSF may render it less effective in PAM [Jaruratanasirikul et al., 1996]. Despite a small number of successful treatments, most cases are fatal [Seidel, 1985a]. Various combination therapies have been attempted, including amphotericin B/azithromycin and amphotericin/fluconazole/rifampin, and may improve outcomes [Soltow and Brenner, 2007; Vargas-Zepeda et al., 2005].
Granulomatous Amebic Encephalitis : Acanthamoeb spp. and Balamuthia mandrillaris
Epidemiology, microbiology, and pathology
It was largely through the observations of Culbertson that free-living amebas were recognized as potentially pathogenic organisms. He detected acanthamoebas in monkey kidney cell cultures used for growing poliovirus, and demonstrated their pathogenicity in mice [Culbertson et al., 1958].
Two different, although closely related amebas, cause granulomatous encephalitis: Acanthamoeba spp. and Balamuthia mandrillaris. Acanthamoeba granulomatous encephalitis is an opportunistic, chronic disease that may have a prodromal period of weeks to months, with most patients being immunocompromised [Martinez, 1980]. Predisposing factors to acanthamebiasis include steroid treatments, autoimmune conditions, organ transplants, chemotherapy, radiation therapy, alcoholism, and pregnancy [Martinez, 1980; Cha et al., 2006]. A small number of Acanthamoeba granulomatous encephalitis cases have been described in immunocompetent children.
Acanthamoeba is perhaps the most common ameba found in soil, which is often the source of infection. It is remarkably tolerant of a range of environmental conditions and can be isolated from seawater and fresh water, and from Arctic soils and beach sands. Acanthamoeba is also found in the home in aquaria, flowerpot soil, humidifiers, sink taps, and drains. It has been isolated from hot tubs, hydrotherapy baths, dental irrigation equipment, and laboratory eye wash stations. The life cycle consists of alternating ameboid and cyst stages. Cysts are particularly resistant to both physical (heat, desiccation, drying) and chemical (chlorine, antimicrobials) agents, and have remained viable in the laboratory in excess of 20 years. Infection may take place through breaks in the skin that are contaminated with soil, or by inhalation of airborne ameba cysts originating from soil. In surveys of healthy groups of individuals (students, military recruits), Acanthamoeba has been cultured from the nasal passages, indicating exposure and infection but not disease [Badenoch et al., 1988; Cerva et al., 1973].
Clinical characteristics, clinical laboratory tests, and diagnosis
In addition to encephalitis, Acanthamoeba can cause nasopharyngeal, cutaneous, and disseminated infections. Nasopharyngeal and cutaneous infections can be precursors to disseminated amebiasis or encephalitis, as amebas spread from their initial portal of entry. The CNS becomes infected by hematogenous transport of amebas, either from the lungs or from cutaneous lesions. A granulomatous reaction occurs, which can be seen in histopathologic slide preparations, but this reaction may be absent in immunocompromised hosts [Martinez, 1980]. Areas of the brain affected are usually the cerebrum, cerebellum, and brainstem, where the amebas produce hemorrhagic necrotic lesions; large numbers of amebas can be found in the perivascular areas of brain tissue. Cysts of Acanthamoeba may be seen in histopathologic sections; Naegleria amebas do not form cysts in tissue. Reports of detecting amebas in CSF are rare. Because of the absence of clear pathognomonic symptoms, Acanthamoeba granulomatous encephalitis is often unrecognized and diagnoses are often made postmortem.
Because the disease is chronic, an antibody response develops over time and the infection can be diagnosed by indirect immunofluorescence staining of the patient’s serum. Surveys of human populations for Acanthamoeba antibodies have found that many healthy people have elevated antibody titers, probably resulting from exposure to the amebas through contact with soil or water [Cerva, 1989; Cursons et al., 1980].
Clinical features are variable but typically have a subacute to chronic presentation, with fever, headache, seizures, personality change, lethargy, or confusion. Cranial nerve palsies, meningeal signs, or hemiparesis may be seen on physical examination [Cha et al., 2006]. Diagnosis of Acanthamoeba granulomatous encephalitis is difficult and the disease mimics other types of encephalitis. Children infected with Acanthamoeba have exhibited headache, stiff neck, vomiting, abnormal behavior, fever, ataxia, and tonic-clonic seizures [Karande et al., 1991; Sharma et al., 1993; Singhal et al., 2001]. Hydrocephalus may also develop [Karande et al., 1991]. Differential diagnoses include neurotuberculosis and neurocysticercosis. Although the amebas are generally not recovered from CSF, several cases were reported in which the amebas were seen in wet-mount preparations of spinal fluid, and/or were cultured from CSF [Callicot et al., 1968; Karande et al., 1991; Kidney and Kim, 1998; Sharma et al., 1993]. In wet-mount slide preparations, trophic Acanthamoeba have radiating fingerlike pseudopodia over the surface and are relatively slow-moving amebas. More often, amebas are cultured from biopsied brain tissue or skin ulcers on to plates of non-nutrient agar coated with E. coli and incubated at 37°C [Visvesvara, 1999]. Recently developed molecular techniques may also be helpful in making the diagnosis [Yagi et al., 2007; Qvarnstrom et al., 2006; Yagi et al., 2008]. CSF protein is elevated (range 31–500 mg/dL), as is the white blood cell count (14–750 cells/μL) and glucose level (17–240 mg/dL); among the white blood cells are lymphocytes (19–100 percent) and polymorphonuclear leukocytes (2–70 percent) [Martinez, 1980, 1985]. The CSF protein in an 8-year-old female with Acanthamoeba meningitis increased from 200 mg/dL at onset to a high of 500 mg/dL, and then decreased to 41 mg/dL after completion of drug therapy 5 months after the patient was first seen [Singhal et al., 2001]. Neuroimaging by MRI of an adult male diagnosed with acanthamebiasis revealed punctate focal lesions, mainly in the cerebellar hemispheres, and fewer lesions in the cerebral hemispheres and the corpus callosum [Kidney and Kim, 1998]. Sequential MRIs often show a rapidly expanding brain mass [Gene et al., 2007].
Unrelated to its role as etiologic agent of Acanthamoeba granulomatous encephalitis, Acanthamoeba can also cause amebic keratitis as a result of corneal trauma or wearing contact lenses. Ophthalmic solutions prepared from nonsterile tap water, and lens cases that harbor bacterial biofilms can support growth of the amebas. Keratitis has not been reported to develop into encephalitis; however, a child diagnosed with uveitis and pharyngitis caused by Acanthamoeba went on to develop fatal encephalitis [Jones et al., 1975].
Management
There are several reported cases of acanthamebiasis in pediatric HIV/AIDS patients. An 8-year-old patient developed cutaneous and sinus lesions, in which Acanthamoeba cysts and trophozoites were seen, along with a weak granulomatous response [Friedland et al., 1992]. The infection progressed after about 3 months to disseminated disease with CNS involvement (multiple lesions within the right basal ganglia by MRI) and ultimately proved fatal, despite treatment with ketoconazole, fluconazole, sulfadiazine, and trimethoprim-sulfamethoxazole. Acanthamebiasis in another pediatric case with congenital HIV/AIDS developed as osteomyelitis and subcutaneous abscesses without neural involvement [Selby et al., 1998]. Initial treatment was with oral fluconazole but was subsequently changed to intravenous pentamidine, oral flucytosine (5-fluorocytosine), and oral itraconazole, and disease progression was halted. In both adult and pediatric acanthamebiasis, death is usually a consequence of multiple AIDS-related infections rather than from the amebic infection per se.
Acanthamoeba infections of the CNS are usually fatal, although some reports of successful antimicrobial treatment are described. Often, however, the same antimicrobial agents used successfully in one case are unsuccessful in other acanthamebiasis cases. Response to therapy depends, to a large extent, on infective dose of amebas, virulence of the infecting strain, the time at which diagnosis is made, the extent of dissemination of the infection, and the immune status of the patient. As yet, the optimal antimicrobial therapy for treatment of Acanthamoeba encephalitis is unclear. Most recoveries have been treated successfully with combinations of antimicrobials: sulfadiazine, pyrimethamine, and fluconazole in an adult HIV/AIDS patient [Martinez et al., 2000], and ketoconazole, rifampin, and trimethoprim-sulfamethoxazole in two pediatric patients [Singhal et al., 2001]. Surgical excision may be warranted in some, if not all, cases [Fung et al., 2008]. In vitro studies suggest that miltefosine and voriconazole have activity against Acanthamoeba [Schuster et al., 2006].
Granulomatous Amebic Encephalitis: Balamuthia mandrillaris
Epidemiology, microbiology, and pathology
Balamuthia granulomatous encephalitis was initially described in immunocompromised humans [Anzil et al., 1991; Visvesvara et al., 1990], but more recent cases have occurred in immunocompetent children [Bakardjiev et al., 2003; Duke et al., 1997; Reed et al., 1997; Rowen et al., 1995]. The importance of Balamuthia as a CNS pathogen is increasingly recognized [Matin et al., 2008]. The life cycle comprises trophic and cystic stages; the cyst is a triple-layered structure without pores. Little is known about the ecology of the ameba, other than that it is found in soil. Like Acanthamoeba, it is likely to enter the body by inhalation of cysts in windblown soil, or through breaks in the skin contaminated by soil, followed by hematogenous spread. Balamuthiasis can develop as cutaneous, nasopharyngeal, or CNS lesions. In a review of 30 cases reported from Peru, all developed initially as cutaneous lesions, mostly on the nose or the central region of the face, and progressed to infections of the CNS [Bravo and Sanchez, 2003]. The disease may be chronic, with a prodrome that can extend as long as 2 years, but it often presents in an acute form [Bakardjiev et al., 2003]. The long incubation period makes it difficult to pinpoint the source and time of infection. A 3-year-old female may have become infected through playing with soil in a flowerpot, from which Balamuthia was subsequently isolated (Figure 82-12) [Schuster et al., 2003]. Evidence of exposure to Balamuthia among healthy individuals is seen in elevated serum antibody titers in studies performed in Australia and the United States [Huang et al., 1999; Schuster et al., 2001]. The relative prevalence of Balamuthia granulomatous encephalitis in Hispanics is high [Schuster et al., 2004]; in the United States, about 50 percent of cases occurred in Hispanic Americans (who comprise approximately 12 percent of the U.S. population), and almost half of the number of cases worldwide have been described from Latin America and Mexico [Bravo and Sanchez, 2003; Galarza et al., 2002; Martinez et al., 1994; Recavarren-Arce et al., 1999; Taratuto et al., 1991; Uribe-Uribe et al., 2001]. The reasons for the high proportion of Hispanics with balamuthiasis could be because of genetic susceptibility, or environmental or sociologic factors, or a combination of these factors.
Clinical characteristics, clinical laboratory tests, and diagnosis
Symptoms of Balamuthia granulomatous encephalitis include fever, headache, vomiting, ataxia, hemiparesis, tonic-clonic seizures, cranial nerve palsies (particularly third and sixth cranial nerves), and diplopia [Bakardjiev et al., 2003; Martinez and Visvesvara, 2001; Schuster et al., 2009]. Areas of the brain that are typically infected are the basal ganglia, midbrain, brainstem, and cerebral hemispheres [Martinez and Visvesvara, 1997]. Otitis media has preceded the onset of Balamuthia granulomatous encephalitis in several pediatric cases [Bakardjiev et al., 2003; Kodet et al., 1998; Rowen et al., 1995]. CSF may have mild to markedly elevated protein and leukocyte levels, but normal or slightly decreased glucose. Hydrocephalus develops in many cases [Duke et al., 1997; Kodet et al., 1998; Schuster et al., 2009]. Because of its vague symptomatology, diagnosis of balamuthiasis requires a high index of suspicion on the part of the physician, and differential diagnoses include bacterial or viral encephalitis, neurotuberculosis, and neurocysticercosis [Bakardjiev et al., 2003]. What was initially identified as brainstem glioma in an infant was, at postmortem examination, recognized as a Balamuthia infection [Lowichik et al., 1995]. Balamuthia granulomatous encephalitis should also be considered in patients with suspected herpes simplex virus encephalitis when testing for the virus yields negative results [Deetz et al., 2003; Reed et al., 1997].
Most diagnoses of the disease are made at postmortem examination. In histopathologic sections of brain tissue, the amebas are found in large numbers within perivascular areas. Because of the chronic nature of the disease, an antibody response to Balamuthia develops over the course of the infection and is the basis for indirect immunofluorescent staining as a diagnostic aid. Likewise, immunohistopathology is useful for visualizing amebas in brain and other tissues. Neuroimaging is helpful in making a diagnosis [Deetz et al., 2003; Healy, 2002]; the presence of single or multiple space-occupying or ring-enhancing lesions may be indicative of balamuthiasis (Figure 82-13). Neuroimaging may also show evidence of enlarged ventricles and atypical demyelinating lesions [Schuster et al., 2009]. In brain and other tissue sections, the ameba is difficult to distinguish from Acanthamoeba because the organisms have similar morphologies. Trophic Acanthamoeba are somewhat smaller (approximately 20 μm) than Balamuthia (approximately 5 μm), though multiple nucleoli are sometimes seen in Balamuthia nuclei. Both amebas form cysts in tissue, but Balamuthia cysts are larger (up to 30 μm) and have a wrinkled wall without pores. Immunofluorescent staining, however, can distinguish between the two amebas. Affected brain tissue often shows areas of necrosis with surrounding areas of ayptical reactive astrocytes and inflammatory cells [Perez and Bush, 2007]. Balamuthia cannot be readily cultured from biopsied tissues because the organism grows slowly and requires tissue culture cells as a feeder layer. Attempts to culture Balamuthia from macerated brain tissue have taken from 10 to about 30 days at 37°C. Polymerase chain reaction technology is available for identification of Balamuthia DNA in tissues and in CSF but is largely limited to research laboratories (e.g., California Department of Health Services, Viral and Rickettsial Disease Laboratory, Richmond; Ohio State University, Department of Molecular Genetics, Columbus) [Schuster et al., 2009; Yagi et al., 2008]. A multiplex, real-time polymerase chain reaction has recently been developed with the ability to detect Balamuthia, Acanthamoeba, and Naegleria simultaneously [Qvarnstrom et al., 2006].
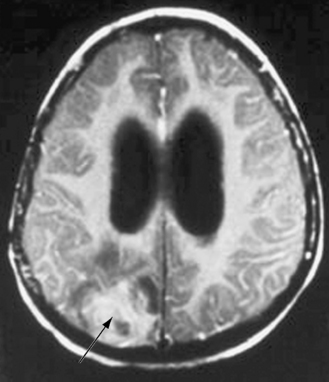
Management
Of the approximately 150 recognized Balamuthia cases, successful outcomes have been reported in three individuals [Deetz et al., 2003; Jung et al., 2004], one of them a 5-year-old female. Therapy included flucytosine, pentamidine isethionate, fluconazole, sulfadiazine, and a macrolide antibiotic (azithromycin or clarithromycin). In vitro testing of antimicrobials on clinical isolates indicates varied sensitivity to some of the drugs. Pentamidine, however, is particularly effective against all isolates in vitro [Schuster and Visvesvara, 2004], but the drug is slow to cross the blood–brain barrier and has marked toxicity [Donnelly et al., 1988].
Other Central Nervous System Amebic Infections
A single case of encephalitis due to Sappinia pedata (formerly known as Sappinia diploidea), previously described in the feces of large herbivores but never associated with disease in humans or animals, has been reported from an immunocompetent adult male [Gelman et al., 2003; Qvarnstrom et al., 2009]. A mass detected by MRI in the left temporal lobe was excised and, upon histopathologic examination, was found to contain an ameba with two closely apposed nuclei characteristic of Sappinia. The patient recovered after treatment with a combination of antimicrobials similar to those used for balamuthiasis. The infection had been preceded by a sinus infection, suggesting that the portal of entry into the body was the respiratory tract, with subsequent transport to the CNS. The recognition of Sappinia as an etiologic agent of human encephalitis suggests that it and other amebas previously regarded as innocuous might also be potential pathogens.
Other amebas have been implicated as etiologic agents of neural disease. The soil ameba, Hartmannella, was isolated from the CSF of a patient with meningoencephalitis, but the ameba was most likely a secondary invader of an existing lesion or possibly a contaminant of clinical specimens [Centeno et al., 1996]. Because of the ubiquity of these free-living amebas, caution is advised when assigning them an etiologic role based on results from cultivation of clinical specimens.
Microsporidial Infections of the Nervous System
The microsporidia are obligate intracellular parasites that cause infections in invertebrates and vertebrates, including humans [Weber et al., 1997]. There are about 150 genera and 1400 species, of which 8 genera and 14 species are human parasites. Host specificity for the group is low, and a number of the species that infect humans also parasitize other mammals. Historic interest in the microsporidia developed in the 19th century largely because of their role in diseases of insects. Pébrine, a disease of silkworms that threatened the industry in France, was studied by Pasteur [Cox, 2002], but the role of microsporidia as etiologic agents of human disease was only recognized in the 20th century, during the HIV/AIDS epidemic.
Microsporidiosis
Epidemiology, microbiology, and pathology
The microsporidia are opportunistic pathogens, and the majority of these infections have been reported from highly immunosuppresssed cases, including individuals with HIV/AIDS and occasionally solid organ transplant patients [Mohindra et al., 2002]. Cases in immunocompetent individuals are rare. Most infections in humans have been attributed to Enterocytozoon bieneusi, which can cause cachexia, chronic diarrhea, and cholangitis. Encephalitozoon spp. infections are less frequently identified. In recent years, three species (Encephalitozoon hellem, Encephalitozoon intestinalis, and Encephalitozoon cuniculi) have been detected, with a range of presentations including hepatitis, peritonitis, keratoconjunctivitis, nephritis, cystitis, bronchiolitis, sinusitis, diarrhea, and disseminated infection. Different species appear to be associated with different clinical syndromes: E. intestinalis can present with chronic diarrhea and cholangitis, E. hellem can often cause conjunctivitis, and E. cuniculi is associated with disseminated infections, neurologic symptoms, or renal failure [Fournier et al., 2000].
The life cycle consists of an infective spore stage, in which the spore may be as small as a bacterium (typically 1–2 μm by 2–5 μm), and an intracellular proliferative stage that ultimately develops into spores. On release from infected cells, spores may be found in urine, sputum, conjunctival fluid, feces, and CSF, depending on the site of infection. Spores discharged in urine can contaminate ground water, which is a possible source of infection. Ingestion of spores is the most common route of infection for all species, although transplacental infection is known for nonhuman mammals, particularly carnivores [Didier and Bessinger, 1999; Weber et al., 1994]. Other routes of infection include zoonotic transmission and aerosol dispersal. Depending on species, spore germination and discharge occur in the presence of enterocytes, respiratory and nasal epithelial cells, corneal epithelial cells or stroma, and macrophages as primary sites of infection. Infection may be secondarily spread to the kidneys and the CNS by macrophages.
Clinical characteristics, clinical laboratory tests, and diagnosis
Human microsporidial infections of the nervous system occur principally in HIV/AIDS adults. Disseminated infection with renal failure in the setting of underlying severe immunosuppression is frequently described. Patients with CNS involvement can present with cognitive dysfunction, visual impairment, and seizures [Fournier et al., 2000; Leder et al., 1998]. Pediatric cases of microsporidiosis in the CNS have been rarely reported . The first recognized case of human microsporidiosis due to E. cuniculi was in a 9-year-old Japanese male with severe headache, vomiting, and convulsions. The child lived on a farm and had contact with a variety of farm animals, and an animal reservoir is likely to have been the source of the infection [Matsubayashi et al., 1959]. In 1984, a second reported pediatric case occurred in a child from Colombia, who was adopted by a Swedish family and developed convulsions [Bergquist et al., 1984]. The child was successfully treated with carbamazepine and an anticonvulsive drug, and, aside from later minor arthritic symptoms that may have been sequelae of the microsporidial infection, remained in good health. The tentative diagnosis in both cases was based on light microscopy detection of Toxoplasma-like bodies, which were actually microsporidial spores, seen in CSF and urine. The infection was transferable to mice by intraperitoneal injection of the patient’s CSF, urine, or blood. Identification of the species at the time remained inconclusive because diagnostic techniques to distinguish among Encephalitozoon-like microsporidia were not yet available. Based on morphologic distinctions from Toxoplasma, the infective agent was tentatively identified as Encephalitozoon spp. [Weber et al., 1997]. In the second case, serologic studies also detected the presence of immunoglobulin G and immunoglobulin M antibodies against E. cuniculi.
A third reported case was in a 9-year-old black female with congenital HIV/AIDS and a CNS tumor, who developed tonic clonic seizures and died after 8 days in the hospital [Yachnis et al., 1996]. Her symptoms before hospitalization included aphasia, hallucinations, inability to walk, and reduced consciousness. A CT scan exhibited multiple lesions in the cortical gray matter and at the gray–white junction. At postmortem examination, necrotic lesions were found in the cerebral cortex, cerebellum, pons, and medulla. Macrophages and astrocytes in histopathologic sections were filled with spores located in vesicles. Free spores released from destroyed cells were seen in the perivascular spaces. Spores were recognized in hematoxylin-eosin sections, in Gram- and Giemsa-stained preparations, and by polarization microscopy, and were later identified as Trachipleistophora anthropophthera.
Clinical suspicion, in most cases, is triggered by exclusion of other more common diagnoses and persistent symptoms suggestive of multi-organ involvement and fever. CT or MRI can reveal multiple ring-enhancing lesions in the brain suggestive of toxoplasmosis, with failure of anti-Toxoplasma therapy to improve these lesions leading to suspicion of microsporidial infection. Detection of the parasite in CSF may be difficult, as the number of spores may be low [Weber et al., 1997].
Diagnosis of infections, in most cases, is by microscopic examination of tissues or body fluids but primarily of urinary sediment, which seems to be almost always positive [Fournier et al., 2000; Weber et al., 1999]. The Gram and Giemsa stains are good for detection of spores, as is hematoxylin-eosin staining. Spores are Gram-positive (purple) or stain blue with Giemsa. The polarization microscope is helpful in visualizing spores because of their birefringence due to the presence of chitin in the spore wall. Body fluids (urine, CSF) containing spores can be inoculated into tissue culture monolayers for cultivation of the parasite [Visvesvara et al., 1999; Weber and Canning, 1999]. Definitive identification of microsporidial species is done by electron microscopy or polymerase chain reaction, or both. MRI reveals multiple ring-enhancing lesions in the brain. A comprehensive review of techniques for identification of microsporidia in clinical specimens is available [Garcia, 2002].
Management
Albendazole and other benzamidazoles, such as thiabendazole, are currently the recommended antimicrobials for treatment of microsporidiosis, although they may not be effective against mature spores [Weber et al., 2000]. The drugs act by binding to tubulin and inhibiting microsporidial divisions. Disseminated infections caused by E. cuniculi are treated in adults with a dose of 400 mg twice a day [Medical Letter, 2004]. While there are no data on drug levels in CSF, the successful use of albendazole in treatment of cerebral cystercercosis suggests diffusion across the blood–brain barrier. In the few patients described, a decrease in the number and size of brain lesions following treatment has been observed, although urinary excretion of spores persisted [Weber et al., 2000].
Apicomplexan Infections of the Central Nervous System
Toxoplasmosis
Epidemiology, microbiology, and pathology
The causal agent of toxoplasmosis was described from a desert rodent, the gondi, in 1908, but it was 1923 before the association with human disease was reported, and 1937 when its role in congenital infections was recognized [Cox, 2002; Remington et al., 2001]. The organism is found worldwide in a variety of mammals and birds. Toxoplasmosis is a major public health problem in both developed and developing countries. Approximately 20–40 percent of the adult population in the United States is seropositive. The cat is the definitive host of the parasite and excretes oocysts in the feces. Ingestion of the oocyst by animals and humans continues the cycle [Frenkel, 1985]. After being ingested by the host, the oocyst releases sporozoites that invade cells, either by active penetration or by phagocytosis, giving rise to rapidly reproducing trophozoites, or tachyzoites. Tachyzoites eventually transform into slower-growing bradyzoites that form intracellular tissue cysts and may remain quiescent for years in the host, particularly in muscle and brain tissues. Cyst formation is triggered by the host’s developing immune response, and the continued release of bradyzoites from tissue cysts stimulates the immune system, resulting in lasting immune protection.
Humans are infected by ingestion of raw or inadequately cooked meat containing tissue or by ingestion of oocysts shed in cat feces. Of the two possible sources of infection, undercooked meat is the more likely vehicle of transmission. Through dissemination of the parasite by tissue cysts in undercooked meats, Toxoplasma has effectively bypassed the need for the cat as an intermediate host, relying entirely upon food as a vehicle for direct transmission; ingestion of undercooked ground beef, rare lamb, and unpasteurized goats’ milk was identified as a risk factor for infection a recent study in the United States [Jones et al., 2009]; [Su et al., 2003]. Risk factors also included the ownership of three or more kittens or having an occupation handling meat products [Jones et al., 2009]. Transfusion of whole blood from asymptomatic carriers can also serve as a mode of transmission. Reactivation of a latent infection can be triggered by the host becoming immunodeficient or immunocompromised.
Intrauterine or congenital toxoplasmosis may develop in the fetus if the mother is infected during pregnancy. The placenta is invaded by parasites from the maternal circulatory system that eventually pass into fetal circulation. Infection may be asymptomatic in the mother; however, the fetus develops a generalized infection involving spleen, liver, eyes, and CNS [Desmonts and Couvreur, 1974; Lowichik and Siegel, 1995]. The incidences of maternal and fetal infection in the United States are 6 and 2 per 1000, respectively [Wilson and Remington, 1992]. The risk of fetal infection increases as the pregnancy progresses: 25 percent in the first trimester, 54 percent in the second trimester, and 65 percent in the third trimester. However, severity of infection is inversely related to when infection occurs during pregnancy. When disseminated, acquired toxoplasmosis produces systemic manifestations consisting of multiple organ infection, including the brain. Calcium is deposited in necrotic cerebral lesions; cerebral calcification is often seen on CT or MRI scans and plain radiographs of neonates (Figure 82-14).
Clinical characteristics, clinical laboratory tests, and diagnosis
Clinical manifestations of congenital infection are extremely variable. The well-known clinical constellation of hydrocephalus, cerebral calfications, and chorioretinitis is often not present in affected infants [Petersen, 2007]. The approximate incidence of abnormalities in 180 cases of intrauterine toxoplasmosis included microcephaly (20 percent), hydrocephalus (25 percent), microphthalmia (35 percent), seizures (40 percent), psychomotor retardation (45 percent), cerebral calcification (60 percent), and chorioretinitis (95 percent) [Feldman, 1969]. Other studies have reported similar percentages [Lowichik and Siegel, 1995; Wilson and Remington, 1992]. Approximately 80–90 percent of survivors of overt congenital toxoplasmosis have mental retardation, cerebral palsy, epilepsy, or visual impairment [Stern et al., 1969]. Although the manifestations of some cases have been similar to those of congenital cytomegalic inclusion disease, CNS involvement is usually more common and extensive in toxoplasmosis. Hydrocephalus with a tense or bulging fontanel, vomiting, a high-pitched cry, and opisthotonic posturing may dominate the neonatal period (Figure 82-15). Skull radiographs and ultrasound demonstrate punctate, scattered calcifications and separated cranial sutures. Hydranencephaly also has been reported [Altshuler, 1973], as has infantile diabetes insipidus [Silver and Dixon, 1954] and growth hormone deficiency [Massa et al., 1989]. Delayed onset of seizures, chorioretinitis, and mental retardation can also occur [Miller et al., 1971]. Up to 85 percent of children with congenital toxoplasmosis will develop retinal and choroidal involvement; lesions initially are edematous with hemorrhage, and a yellowish exudate gives rise to areas of peripheral irregular hyperpigmentation with a pale atrophic center [Wilson et al., 1980]. Even in those infants with toxoplasmosis who are asymptomatic at birth, up to 85 percent may develop chorioretinitis or serious neurologic disability by 8.3 years [Wilson et al., 1980]. Toxoplasmosis in the fetus and newborn is reviewed by Remington and colleagues [2001], and methods for diagnosis of the disease are discussed by Wilson and McAuley [1999] and by Wilson and associates [2002].

Outside the newborn period, the infection is often asymptomatic in the immunocompetent host. Approximately 10 percent of individuals with acute toxoplasmosis will experience a self-limited flulike illness [Montoya and Liesenfeld, 2004]. In contrast, immunosuppressed individuals can develop a life-threatening illness with either acute infection or reactivation. The acquired form of toxoplasmosis causes meningoencephalitis, and long-term sequelae are common [Townsend et al., 1975]. There have been reports of disseminated generalized toxoplasmosis after immunosuppressive therapy in patients receiving transplantation [Ghatak et al., 1970; Khan and Correa, 1997; Seong et al., 1993] and, to a lesser extent, in those with malignancy [Israelski and Remington, 1993]. Immunodeficiency can lead to reactivation of dormant tissue cysts and the release of bradyzoites. Meningoencephalitis, often fatal, is a major complication. Optic neuritis has also been reported in children [Roach et al., 1985]. Although cerebral toxoplasmosis is an important complication of AIDS in adults [Bertoli et al., 1995], it is not common in neonates or older children [Lowichik and Ruff, 1995; Miller and Remington, 1990]. When it is present, children appear to have similar clinical and neuroimaging findings (although less severe) than adults. Maternal HIV infection appears to reactivate a latent toxoplasmosis, which can be transmitted to the fetus. These infants with both HIV and toxoplasmosis usually present at 3–4 months of age with similar symptoms to those seen in infants with severe intrauterine toxoplasmosis infection [Lowichik and Ruff, 1995]. Neuroimaging studies in infants with both infections are similar to those in infants with just toxoplasmosis. However, in older patients, it is frequently difficult to correlate neuroimaging abnormalities in gray–white matter regions or basal ganglia with either infection or the presence of lymphoma [Ciricillo and Rosenblum, 1990]. This subject has recently been reviewed, and there are recommended treatment protocols for children infected with HIV and CNS toxoplasmosis [Khan and Correa, 1997].
Immunologic diagnosis of toxoplasmosis relies on the Sabin–Feldman dye test (which detects live parasites), the direct hemagglutination test, the double-sandwich enzyme-linked immunosorbent assay, the immunosorbent agglutination assay, the complement fixation test, the evaluation of immunoglobulin M levels, and the indirect fluorescent antibody test. Comparative information on commercially available kits for detecting immunoglobulin M in serum samples is available [Wilson et al., 1997]. The antibody test may be the most dependable, but the hemagglutination test is used most often (see Table 82-2). Diagnosis of congenital toxoplasmosis can be especially challenging, and consultation and testing with a laboratory experienced with toxoplasmosis diagnostics is warranted when toxoplasmosis is suspected [Montoya and Rosso, 2005]. Prenatal diagnosis of congenital toxoplasmosis with a polymerase chain reaction test on amniotic fluid is possible and cost-effective [Hohlfeld et al., 1994; Sahai and Onyett, 1996]. Testing of placental testing has a sensitivity of only 25 percent [Filisetti et al., 2010].
Management
The value of therapy for congenital toxoplasmosis has been demonstrated [McAuley et al., 1994]. Early treatment with spiramycin for the mother and sulfadiazine, pyrimethamine, and folinic acid for the infant is beneficial and 70 percent effective [Lowichik and Siegel, 1995; Roizen et al., 1995; Wilson and Remington, 1992; Montoya and Remington, 2008]. Outside the newborn period, treatment is usually not required, except in immunocompromised hosts.
Malaria
Epidemiology, microbiology, and pathology
Malaria is caused by protozoal parasites of the genus Plasmodium, with life cycle stages alternating between the mosquito and a variety of mammals and birds. Several species of Plasmodium cause malaria in humans – Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, and the recently discovered Plasmodium knowlesi – but P. falciparum is the only member of the genus that has been well documented as affecting the host’s nervous system. Awareness of the disease dates back some 5000 years, with the description of malaria-like symptoms in writings from China, and later in texts from India, Sumer, Egypt, and Ancient Greece [Carter and Mendis, 2002]. Hippocrates (ca. 400 bc) recognized the regular occurrence of febrile episodes of the disease, and used the terms “quotidian” (daily), “tertian” (every 3 days), and “quartan” (every 4 days) to characterize them. The disease derives its name from the misplaced belief that it was caused by miasmas, or “bad air.” The recent sequencing of the P. falciparum genome presents opportunities for development of drugs and vaccines targeted to specific gene products of the parasite [Gardner et al., 2002]. Malaria persists as a major public health problem throughout the world, primarily in developing tropical and subtropical regions. Poverty, poor medical and public health infrastructure, emerging resistance to antimalarial drugs, insecticide-resistant species of mosquitoes, ecologic and practical constraints on drainage of swamps, and limits on widespread use of insecticides make it likely that malaria will persist as an important public health problem for many years to come.
From 200 to 500 million people are affected annually. Malaria’s biggest impact is in sub-Saharan Africa, where it is responsible for at least 20 percent of mortality in young children, or approximately 3000 deaths per day; children younger than 5 years of age account for about 90 percent of the more than 2 million deaths that occur each year. Severe malaria in Africa in children has also been associated with HIV/AIDS, with some studies showing increased incidence, parasite density, and severe clinical illness [Summer et al., 2005].
In endemic areas, such as sub-Saharan Africa, neonates are protected from infection by maternal (transplacental) immunoglobulin G and, in part, from the vestigial presence of fetal hemoglobin. Studies using transgenic mice indicate that the parasite cannot readily break down fetal hemoglobin, thus impairing intraerythrocytic development and replication [Shear et al., 1998]. This protection lasts for about 1 year, after which the infant becomes susceptible to disease. The child of 1–4 years of age is the most vulnerable to infection and the development of severe malaria, with a mortality of about 5 percent. Once this period has passed, the host and parasite exist in a state of détente, with the host exhibiting chronic asymptomatic parasitemia [Kyes et al., 2001]. Additional infections due to continued exposure to parasites from mosquito bites result either in death or in the development of clinical immunity. In endemic areas, a large percentage of adults and children will be infected, but only a small number of these will go on to develop clinical disease [Kyes et al., 2001].
Malaria acquired by recipients of blood transfusions from infected but asymptomatic carriers is also a major problem in endemic regions of the world. Because of more frequent international travel, increasing numbers of cases in adults and children are being recognized in the United States [Emanuel et al., 1993]. A recent report of 30 GeoSentinel sites involving 17,553 travelers observed that malaria was one of the three most frequent causes of systemic febrile illness [Freedman et al., 2006]. Close to 100 cases have appeared in individuals living in the vicinity of or working at airports, probably as the result of inadvertent import of mosquitoes from malarial areas of the world in airplane cabins or luggage [Lusina et al., 2000; Thang et al., 2002]. Of note, the major determinant of outcome in resource-rich areas where malaria is seen infrequently is misdiagnosis, which can occur in 40–50 percent of cases [Summer et al., 2005]. Further, the CDC has estimated that over 80 percent of deaths from malaria in the United States in the last several decades were preventable and were caused by lack of or improper malaria chemoprophylaxis in travelers, delay in establishing diagnosis or misdiagnosis, and/or inappropriate therapy [Summer et al., 2005]. The malaria literature is voluminous and is frequently reviewed, reflecting the importance of the disease [Birbeck, 2004; Maitland and Marsh, 2004; Newton and Krishna, 1998; Newton et al., 2000; Satpathy et al., 2004].
The most serious consequence of falciparum malaria for the CNS develops when the parasitized erythrocytes lodge in the brain vasculature. Cerebral involvement follows binding of infected red blood cells to the microvascular endothelium. Sequestration and hemolysis follow and cause obstruction of blood vessels [Sein et al., 1993]. Parasite-induced knobby projections on the surface of infected erythrocytes bind to the endothelial cells in brain, heart, liver, and lungs, causing capillary congestion. By attaching to the endothelial lining of blood vessels, the parasitized erythrocytes effectively avoid destruction that might otherwise occur upon passage through the spleen. This is accompanied by a change in the erythrocyte from a readily pliant state to one of rigidity, which further contributes to blood vessel occlusion. Additionally, rosette formation, resulting from clustering of parasitized and nonparasitized blood cells, may facilitate transfer of parasites from hemolyzed cells to uninfected red blood cells. Local hypoxia, microinfarction, necrosis, hemorrhage, and inflammation subsequently occur. The sequestration and resulting occlusion may deplete the oxygen level in cerebral vasculature, providing a more optimal environment for metabolism and replication of the parasites [Chen et al., 2000]. Thrombi may form and be associated with clotting aberrations, including disseminated intravascular coagulation [Butler et al., 1973]. The blood–brain barrier is disrupted by changes in endothelial cell junctional permeability leading to cerebral edema, more commonly encountered in children than adults [Adams et al., 2002].
A number of theories have been proposed to explain the pathology of cerebral malaria. Hypoxia is one such theory, based on the impedance of blood flow to the brain and the consequent oxygen deficiency. On recovery, however, there is no evidence of the neuronal damage that might be expected to occur as a result of oxygen deprivation. Another explanation is that the surge of mediators that occurs in the CNS may be responsible for the pathology of cerebral malaria [Medana et al., 2001]. Release of proinflammatory cytokines and interleukins (tumor necrosis factor-α, interleukin-1, interleukin-6, nitric oxide lymphotoxin) results in endothelial damage and mimics the clinical condition of sepsis [Clark et al., 2004; Maitland and Marsh, 2004]. Presence of malarial antigens stimulates release of tumor necrosis factor-α, which, in turn, upregulates endothelial adhesion molecules and promotes attachment of parasitized erythrocytes to the vascular lining [Gimenez et al., 2003]. Once parasites are cleared from the blood, the effects of cerebral malaria are reversible and long-term sequelae generally do not develop [Adams et al., 2002]. The role of proinflammatory cytokines in malarial pathology has been reviewed by Clark and associates [2004] in the context of other infectious diseases and noninfectious conditions, and the molecular aspects of cerebral malaria have been reviewed by [Chen et al., 2000].
Postmortem studies of gross specimens demonstrate fulminating cerebral edema and accompanying transtentorial herniation (Figure 82-16). Congestion of arachnoid vessels causes the brain to appear pink or bright cherry-red. Petechiae are evident on the cut surface. Microscopic examination reveals that small arterioles and capillaries are obstructed with parasitized erythrocytes. Mononuclear cells are readily discernible in meningeal and perivascular infiltrates. Microinfarction and ensuing multifocal necrosis and demyelination are widespread.
Clinical characteristics, clinical laboratory tests, and diagnosis
The clinical features of malaria are notoriously nonspecific, especially in children. Initial symptoms can include influenza-like illness with fever, chills, rigors, headache, and myalgias. These vague prodromal symptoms precede the development of acute paroxysms of high fever and chills, with the classically described periodic fever patterns observed after several days of untreated illness. Gastrointestinal and repiratory symptoms are also common; a study in Nigeria found that vomiting and abdominal pain were the most common gastrointestinal complaints in children infected with P. falciparum, followed by decreased appetite and diarrhea [Summer et al., 2005]. Lactic acidosis due to metabolic impairment is responsible for respiratory distress [Birbeck, 2004]. Severe hypoglycemia is also common, and is postulated to be due to increased metabolic rate and direct inhibition of gluconeogenesis by the parasite [Summer et al., 2005]; it may be exacerbated further by the hyperinsulinemic effect of the antimalarial treatments, quinidine and quinine.
Cerebral malaria is defined as coma without apparent etiology, inability to localize painful stimuli, and falciparum parasitemia, as demonstrated by blood smears [Birbeck, 2004; Newton and Krishna, 1998; Warrell et al., 1982]. Coma is assessed on the basis of the Blantyre or Glasgow coma scales, taking into account eye movements and motor and verbal abilities. The scales may be of greater value in predicting mortality rather than as a diagnostic tool [Newton and Krishna, 1998].
Cerebral malaria is most often heralded by mental deterioration, particularly confusion or delirium, followed by progression to stupor and coma that transpires during the next 1–3 days. In children, cerebral involvement can be more fulminant, with most patients becoming comatose within 2 days [Molyneux et al., 1989]. New-onset seizures may be confused with febrile seizures in young children [Osuntokun, 1977]. Extensor plantar responses, cerebellar impairment, abnormal movements, and psychosis may also occur [Daroff et al., 1967]. Children are more likely to have decreased tone compared with adults, and impaired brainstem cranial nerve responses and abnormal respiratory function [Molyneux et al., 1989; Newton et al., 1991]. Children with severe illness exhibit evidence of transtentorial herniation [Newton et al., 1991]. Papilledema and extramacular retinal edema are markers of a poor prognosis [Lewallen et al., 1993]. The presence of impaired consciousness, prolonged seizure activity, hypoglycemia, or respiratory distress identifies those at high risk for death or long-term sequelae [Marsh et al., 1995; Steele and Baffoe-Bonnie, 1995]. In one study of 308 children with cerebral malaria, the mortality rate was 14 percent [Bruster et al., 1990].
Severe cerebral malaria can be indistinguishable from other common illnesses, including pneumonia, meningitis, and sepsis, with several case series documenting concurrent bacteremia or meningitis in children. While some studies have not found an association between presumptive antibiotic treatment and improved survival, others have observed that bacteremia is significantly associated with an increased risk of death despite routine use of antibiotics [Gwer et al., 2007].
Neurologic sequelae are not uncommon in children recovering from cerebral malaria. In studies combining data on African children, sequelae develop in about 10–30 percent of patients [Birbeck, 2004; Newton and Krishna, 1998; Bruster et al., 1990]. The depth and duration of coma and multiple convulsions were found to be three independent risk factors for neurologic sequelae in one large prospective study in Gambia, West Africa; the most common neurologic abnormalities were ataxia and paresis, which had resolved in the majority of patients (>95 percent) by 6 months post infection. Other sequelae observed include epilepsy, hemiparesis, cortical blindness, impaired hearing, behavioral abnormalities, and cognitive deficits (impaired learning, reasoning, dexterity, memory, perception, etc.). Some sequelae, such as epilepsy, may persist long-term [Summer et al., 2005].
In cases with impaired consciousness, a lumbar puncture should be performed to assess for concurrent meningitis. Caution, however, should be exercised due to the elevated intracranial pressure. CSF in cerebral malaria is clear, with protein ranging up to a high of 0.4 g/L (400 mg/dL); septic CSF is cloudy, with neutrophils comprising about 99 percent of leukocytes and protein ranging from approximately 1 to 3 g/L (1000–3000 mg/dL). Blood cultures to exclude bacteremia are indicated. The electroencephalogram (EEG) in children with cerebral malaria reveals nonspecific generalized slowing [Molyneux et al., 1989] and may be helpful in detection of subclinical seizures that might otherwise pass unnoticed [Maitland and Marsh, 2004]. Neuroimaging may also be abnormal. Acutely, MRI may demonstrate multifocal T2-enhancing lesions indicative of capillary occlusion and cerebral edema [Chia et al., 1992]. CT scan has been reported to demonstrate generalized and sulcal atrophy as a sequela of disease [Newton et al., 1994].
Close scrutiny is required to ensure accuracy in ascertaining the presence of small, ring-form trophozoites and crescent-shaped gametocyte [Coatney et al., 1971; Lowichik and Ruff, 1995]. In the hands of a skilled technician in field conditions, thick films can detect 1 infected cell in 1000 red blood cells (0.001 percent), or 50–100 parasites/μL of blood [Moody, 2002]. Erythrocytes containing Plasmodium ring stages are more likely to be found in peripheral blood because the mature stages undergoing intraerythrocytic replication become sequestered in brain and other deep areas of the body. Blood smears should be prepared at 8- to 12-hour intervals, until diagnosis can be made [Birbeck, 2004]. If antimalarial therapy was started before preparation of blood smears, the smears may be negative.
While examination of Giemsa-stained blood films remains the gold standard, the introduction of rapid diagnostic tests is rapidly changing recommended algorithms for diagnosis and treatment of malaria. These assays detect malaria antigen in a small (5–15 μL) amount of blood by immunochromatographic methodology with monoclonal antibodies directed against the target parasite antigen and impregnated on a test strip. They generally detect histidine-rich protein 2 (HRP-2) and lactate dehydrogenase isoenzymes, with the HRP-2 being specific for P. falciparum [Summer et al., 2005]. The result, usually a colored test line, is obtained in 5–20 minutes. These tests require no capital investment or electricity, and are simple to perform and easy to interpret. Most commonly used rapid diagnostic tests only detect P. falciparum, although some can also distinguish P. falciparum from other non-falciparum species. Depending on the parasite load, most rapid diagnostic tests have achieved greater than 95 percent sensitivity for P. falciparum, with less sensitivity for non-falciparum species [Wongsrichanalai et al., 2007]. Immunochromatography in the form of a dipstick kit has also been employed for Plasmodium detection [Moody, 2002]. Sensitive indirect fluorescent antibody and hemagglutination tests provide further diagnostic capability [Wilson et al., 1975]. [Moody, 2002] has reviewed testing for malarial parasites, with emphasis on methods capable of giving results in 1 hour or less.
In 2010, the World Health Organization published a revised version of evidence-based guidelines for the treatment of malaria [WHO, 2010]. The new guidelines emphasize the importance of parasitological confirmation of diagnosis in all patients suspected of having malaria before treating. Prompted in part by the increasing availability of rapid diagnostic tests, the move towards universal diagnostic testing of malaria is a critical step forward in the fight against malaria, as it will allow for the targeted use of artemesinin-based combination therapy for those who actually have malaria. This will help to reduce the emergence and spread of drug resistance. It will also help identify patients who do not have malaria, so that alternative diagnoses can be made and appropriate treatment provided [WHO, 2010].
Management
As early as 1640, long before the etiologic agent was demonstrated, the therapeutic attributes of cinchona (quinine, quinidine) for treatment of malaria were recognized. In the 1950s, chloroquine became the mainstay of therapy, but efficacy was compromised in many parts of the world by the appearance of chloroquine-resistant strains of the parasite in Southeast Asia, Africa, and South America. Quinine returned to widespread use in the last decades of the 20th century. More recently, the development of artemisinin derivatives, the most rapidly acting of all antimalarial drugs, has transformed current treatment strategies. This Chinese medicinal herb, qinghao (Artemisia annua), also known as annual or sweet wormwood, has been used since antiquity to treat malaria. Recent efforts to understand its chemistry and pharmacology have been very positive. The plant yields a sesquiterpene compound called qinghaosu or artemisinin, which is active against Plasmodium. Artemisinin and its derivatives have been found to cure malaria with much less toxicity than quinoline antimalarials [Hien and White, 1993], and resistance to artemisinin has not yet been encountered. Artemisinin is also effective against the gametocyte stage in the bloodstream (the stage taken up by the mosquito), thereby reducing disease transmission in malaria-endemic areas. To lessen the chance of emergence of drug resistance further, artemisinin has been coupled with other differently acting drugs in artemisinin combination therapy [Adjuik et al., 2004]. Artemether (arthemeter), artesunate, and arteether are artemisinin derivatives that have been effective when used in combination with a second drug (e.g., artesunate-mefloquine) [Ashley, 2004; Olliaro and Taylor, 2004].
Randomized trials comparing artesunate and quinine from Southeast Asia show clear evidence of benefit with artesunate. In the largest multicenter trial, which enrolled 1461 patients (including 202 children below 15 years of age), mortality was reduced by 34.7 percent in the artesunate group when compared to the quinine group. The results of this and smaller trials are consistent and suggest that artesunate is the treatment of choice for adults with severe malaria. There are, however, still insufficient data for children, particularly from high-transmission settings, to enable us to draw the same conclusion. An individual patient data meta-analysis of trials comparing artemether and quinine showed no difference in mortality in African children. There is currently an on-going multicenter study comparing artesunate and quinine in the management of severe malaria in children in several African countries [Day and Dondorp, 2007].
The current World Health Organization treatment recommendations for severe (cerebral) malaria in children include rapid initiation of artesunate or quinine, given intravenously or intramuscularly. Oral artemisinin-based monotherapy is strongly discouraged because of concern about the development of resistant parasites. Parenteral antimalarials should be given for a minimum of 24 hours. Once the patient can tolerate oral therapy, it is essential to continue and complete treatment with an effective oral antimalarial using a full course of an effective ACT (artesunate plus amodiaquine or artemether plus lumefantrine or dihydroartemisinin plus piperaquine) or artesunate (plus clindamycin or doxycycline) or quinine (plus clindamycin or doxycycline). Where available, clindamycin should be substituted for doxycycline in children under the age of 8 years, given concerns in this age group for permanent yellowing or graying of the teeth and impairment of growth. Regimens containing mefloquine should be avoided in patients presenting initially with impaired consciousness because of an increased incidence of neuropsychiatric complications associated with mefloquine following cerebral malaria. The full World Health Organization recommendations are available at http://www.who.int/malaria/publications/atoz/9789241547925/en/index.html.
Supportive care
In cerebral malaria, the focus shifts from treating an infectious disease to dealing with a medical emergency. Fluid and electrolyte balance has to be restored or managed. This includes hypovolemia (remedied by hydration with saline), hypoglycemia (50 percent dextrose intravenously), hyponatremia and hypokalemia (hydration with saline), and renal failure (hemodialysis). Metabolic acidosis is a common complicaton of cerebral malaria and is strongly associated with a fatal outcome. Anemia occurs prominently in children. Hemodynamic shock and acute respiratory distress syndrome are also poor prognostic signs [Day and Dondorp, 2007]. Routine use of parental antibiotics is warranted because of the association between invasive bacterial infections and increased mortality.
In developed country settings, mechanical ventilation is often used in the unconscious cerebral malaria patient to protect the airway, although efficacy in prevention of mortality and sequelae has not been proven. Mannitol or glycerol can reduce intracranial pressure by creating an osmotic gradient across the blood–brain barrier and drawing fluid into the vascular system. In a small study of Kenyan children with cerebral malaria, mannitol was successful in reducing intracranial pressure for short periods, but no current data exist to support its routine use. Seizures can be treated with rectal diazepam, intravenous lorazepam, or other standard anticonvulsants, although prophylactic anticonvulsive therapy is not recommended because of concern for respiratory depression. Dexamethasone, used to relieve intracranial pressure, was found to be no better than a placebo in Thai patients (6–70 years); it prolongs coma, leads to complications, and is usually contraindicated [Warrell et al., 1982]. A meta-analysis combining results of the Thai study with a second malaria steroid trial in Indonesia concluded there was insufficient power to exclude a mortality effect either way. Exchange transfusion is also a popular adjunctive therapy utilized in well-resourced settings, with the goals of removal of parasitized erythrocytes, as well as cytokines and other soluble toxins, although no clear benefit has been proven in small studies. Nevertheless, some experts have suggested that exchange transfusion should be performed in any severely ill patient with a parasitemia that exceeds 15 percent or in any patient with parasitemia in the range of 5–15 percent with signs of poor prognosis [Summer et al., 2005]. Neither heparin nor dextran has been shown to be beneficial in reducing intracranial pressure by lowering blood viscosity [Newton and Krishna, 1998]. Similarly, studies of anti-tumor necrosis factor and iron chelators have not demonstrated clear benefit [Day and Dondorp, 2007].
Prevention
Prevention of malaria is challenging in resource-poor settings. Early efforts at vector control methods, with widespread use of insecticides and reduction of vector breeding sites (e.g., swamp drainage and environmental control with engineering methods for water source protection), were unsuccessful largely due to lack of resources, poor medical and public health infrastructure, and the emergence of resistance to insecticides. Since the 1990s, control efforts have focused more on prevention of mosquito bites through the use of insecticide-treated nets and on prompt recognition and effective treatment of malarial disease. Insecticide-treated nets have been shown to reduce child mortality in several large randomized controlled trials, with up to 33 percent reduction in mortality and 50 percent reduction in incidence of infections. Elimination of standing water near the household (e.g., in puddles, potted plant trays, old tires, urns, etc.) is also encouraged [Summer et al., 2005].
A vaccine for the prevention of malaria has yet to be realized, although several promising clinical trials are being conducted [Moorthy et al., 2004; Webster and Hill, 2003]. Development of an effective vaccine has been hampered by the presence of different stage-specific antigens displayed during the complex life cycle of the parasite. Of the 5300 genes present in the P. falciparum genome, approximately 4 percent are involved in immune evasion, including antigenic variation [Gardner et al., 2002]. Thus, it is likely that an effective vaccine will have to contain a combination of antigens able to protect against the infective sporozoite stage, the erythrocytic stage, and the transmissive gametocyte (sexual) stage. The role of antigenic variation in pathology of falciparum malaria has been reviewed by Kyes and associates [2001].
Babesiosis
Babesia is the etiologic agent of babesiosis, a malaria-like tick-borne disease occurring in many species of wild and domestic animals, and occasionally humans. The organism was named for Viktor Babès, who first recognized it in blood cells of cattle in the 19th century, but it was not until 1957 that the first human infection was recognized. Babesiosis is a zoonotic disease transmitted to humans from rodents and other mammals. The parasites are contained within erythrocytes and are transmitted by ixodid (hard-bodied) ticks. Human-to-human transmission can occur through blood transfusion [Gerber et al., 1994]. Unlike Plasmodium spp., Babesia does not have an exoerythrocytic stage, divides in the erythrocyte by binary fission rather than schizogony, develops within the cytoplasm of the host cell and not in a parasitophorous vacuole, and does not form hemozoin pigment from hemoglobin, as do the malaria parasites. Two species of Babesia are important in human disease: Babesia microti from rodents and Babesia divergens from bovines. B. microti is the species present in the United States, whereas B. divergens is found in Europe. Of the two species, B. divergens is the more virulent, with 42 percent mortality, compared with 5 percent mortality for B. microti [Gorenflot et al., 1998]. In the northeastern and upper midwestern United States, the transmission cycle of B. microti overlaps that of another well-known zoonotic agent, Borrelia burgdorferi, the causative agent of borreliosis, or Lyme disease [Persing and Conrad, 1995], which is reviewed in Chapter 80. Most cases of human infections have involved elderly adults [Meldrum et al., 1992]. Asplenic individuals are also at higher risk for infection; splenectomy predisposed four adult males to infection (two died) in a mini-epidemic in northern California [Persing et al., 1995]. Human infections on the west coast of the United States have been due to Babesia-like parasites (the WA-1 strain, after a strain first detected in Washington State) that defy classification as either B. microti or B. divergens, and may be related to Babesia spp. from canines and rodents [Quick et al., 1993]. Other unusual strains have been isolated in northern California, Missouri, and Wisconsin.
Clinical characteristics, clinical laboratory tests, and diagnosis
Clinical manifestations consist of a persistent fever, sweating, chills, myalgia, and mildly to moderately severe hemolytic anemia. Emotional depression has been reported, but severe neurologic symptoms are rare. Babesia in bovines can cause encephalitis, as does P. falciparum in humans. Parasitized red blood cells adhere to the endothelial lining of brain capillaries, thus evading destruction in the spleen [O’Connor et al., 1999; Wright et al., 1989].
Babesiosis should be suspected in patients who have spent time in tick-infested areas, had recent blood transfusions, or have undergone splenectomy [Homer et al., 2000]. The diagnosis is made by identification of the organism on thick and thin blood smears, but only if the parasitemia is high. In blood smears, the organism can be mistaken for the malaria parasite, but infected erythrocytes lack the malaria-characteristic hemozoin pigment (Figure 82-17). Inoculation of hamsters with suspect blood samples is useful in cases with low parasitemia but may require weeks for the disease to develop. However, the development of indirect fluorescent antibody assays and the polymerase chain reaction provide more sensitive detection of the parasitemia associated with babesiosis [Homer et al., 2000; Pruthi et al., 1995]. Serologic confirmation of infection has been determined in asymptomatic children [Ruebush et al., 1977]. Cross-reactivity with Plasmodium surface antigens has been reported [Gorenflot et al., 1998].
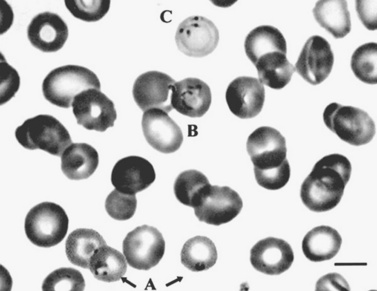
Fig. 82-17 Blood film showing human erythrocytes, some of which are infected with Babesia.
(Courtesy of Dr. Anne Kjemptrup. From Schuster FL. Cultivation of Babesia and Babesia-like blood parasites: agents of an emerging zoonotic disease. Clin Microbiol Rev 2002;15:365–373. Copyright © 2002, American Society of Microbiology.)
Management
Babesiosis was described in a neonate whose mother had been bitten by a tick 1 week before giving birth [Esernio-Jenssen et al., 1987]. Treatment with clindamycin and quinine was successful in effecting recovery. Two other cases of babesiosis in pregnancy were reported; in both, the pregnancies went to term and the infants were unaffected [Feder et al., 2003; Raucher et al., 1984].
Atovaquone (for pediatric patients 20 mg/kg twice a day for 7–10 days) plus azithromycin (12 mg/kg daily for 7–10 days) are the drugs of choice in treating babesiosis [Medical Letter, 2004], with clindamycin (20–40 mg/kg/day orally in three doses for 7 days) plus quinine (25 mg/kg/day orally in three doses for 7 days) as alternatives [Medical Letter, 2004]. Chloroquine therapy has been ineffective [Miller et al., 1978]. In seriously ill patients, exchange transfusion in combination with the above medications was reported to be efficacious, presumably by reducing the acute parasitic load [Doan-Wiggins, 1991]. [Homer et al., 2000] have prepared a comprehensive review of the organism and of the disease.
Trypanosomal Infections of the Nervous System
Chagas’ Disease
Epidemiology, microbiology, pathology
Chagas’ disease (American trypanosomiasis) is an important public health problem, affecting 15–16 million people in Latin America and extending from Argentina to the Mexican border with the United States [Pittella, 2009]. It is a disease of poverty, as the vector triatomine bug thrives in poor housing conditions (e.g., mud walls, thatched roofs), so in endemic countries, people living in rural areas are at greatest risk for acquiring infection.
The disease can also be passed by transplacental infection or through breastfeeding, though data to distinguish this route from intrauterine transmission are not available. Blood transfusions are another source of infection in endemic areas where the blood supply is not monitored for trypanosomes. The disease has also crossed into the United States, mostly in the southern tier, because of immigration from Latin America, where 8 percent of the population is seropositive. As a result, more than 50,000–100,000 people living in the United States have been infected [Kirchoff, 1993].
The vector, the triatomine bug or “kissing bug” (Triatoma spp.), lives in thatched walls and roofs of houses. Humans, cats, dogs, and other mammals serve as reservoirs from which the vector acquires the parasite. The parasite is found in the midgut and rectum of the insect. At the time the triatomine bug bites a human, it defecates on the skin surface, leaving a drop containing parasites. The parasite penetrates the broken skin when the presumptive host scratches or rubs the deposited fecal matter into the site of the insect bite, or when fecal matter is transferred by fingers on to the corneal surface of the eye or mucous membranes. Inside the host, the trypomastigotes invade cells near the site of inoculation, where they differentiate into intracellular amastigotes. The amastigotes multiply by binary fission and differentiate into trypomastigotes, and then are released into the circulation as bloodstream trypomastigotes. Trypomastigotes infect cells from a variety of tissues and transform into intracellular amastigotes in new infection sites, including in the CNS. Clinical manifestations, such as meningoencephalitis, can result from this infective cycle. The bloodstream trypomastigotes do not replicate (different from the African trypanosomes). Replication resumes only when the parasites enter another cell or are ingested by another vector. The “kissing bug” becomes infected by feeding on human or animal blood that contains circulating parasites. The ingested trypomastigotes transform into epimastigotes in the vector’s midgut. The parasites multiply and differentiate in the midgut and differentiate into infective metacyclic trypomastigotes in the hindgut [CDC website http://www.cdc.gov/chagas/epi.html].
Following remission of the acute symptoms, untreated T. cruzi infection can develop into a chronic form of the disease, which can manifest after an asymptomatic latent period. Without treatment, the chronic phase persists for a lifetime, and most remain asymptomatic. However, 15–30 percent of cases will develop symptomatic chronic disease, usually between the second and fourth decades of life. The symptoms of chronic Chagas’ disease result from long-term denervation of the autonomic nervous system, mostly affecting the cardiac and gastrointestinal systems [Pittella, 2009].
Clinical characteristics, clinical laboratory tests, and diagnosis
In endemic areas, T. cruzi infection is often acquired in childhood. Chagas’ disease in childhood can arise either as a result of congenital infection or after being bitten by the insect vector. Intrauterine Chagas’ disease occurs in 2–4 percent of infants born to mothers who have acute or chronic infection with T. cruzi [Lowichik and Ruff, 1995]. Such infants are usually born prematurely and are small for gestational age. Systemic symptoms are similar to other intrauterine infections and include jaundice, hepatosplenomegaly, edema, petechiae, and dysrhythmias. When meningoencephalitis occurs, seizures, hypotonia, and a weak suck are characteristically present [Bittencourt, 1976]. Almost half the infants who acquire infection in utero die by age 4 months.
When Chagas’ disease is acquired from an insect vector, it can manifest in two phases: acute and chronic. The acute phase begins about a week after infection, and is often asymptomatic, subclinical, and undiagnosed in 66–99 percent of cases, most of whom are babies or young children [Pittella, 2009]. The acute phase lasts a few weeks and is characterized by mild, nonspecific symptoms and signs (e.g., fever, vomiting, diarrhea, and hepatosplenomegaly). Inflammation develops at the portal of entry of the trypanosomes, producing a nodule (chagoma), and causing swelling of proximal lymph nodes. Romaña’s sign (unilateral palpebral and periocular swelling) may appear as a result of conjunctival contamination with the vector’s feces, and is a hallmark of the disease. A small percentage of acutely infected patients develop cardiac symptoms suggestive of myocarditis (e.g., dysrythmias) or signs of meningoencephalitis (e.g., generalized encephalopathy and seizures) [Pittella, 2009].
The neurologic manifestations mostly likely to be observed in children are a consequence of acute meningoencephalitis. The symptoms and signs are usually mild, occasionally with stupor or irritability, but can also present with seizures and focal neurologic signs. Almost all cases with CNS involvement will also have concurrent chagasic myocarditis. If it is untreated, the mortality rate following acute Chagas’ infection is approximately 5–10 percent and is usually related to heart failure and/or encephalitis [Pittella, 2009].
Following the latent asymptomatic phase of 10–25 years, chronic Chagas’ disease may re-present in young adulthood. Most symptoms are due to severe cardiac (cardiac arrhythmias, congestive heart failure, apical aneurysms, thromboembolisms) [Spina-Franca, 1996] or gastrointestinal (dysphagia and severe constipation leading to megaesophagus and megacolon) involvement. Reactivated chronic Chagas’ disease is also observed in immunosuppressed patients, most commonly those with AIDS (75–80 percent of cases), and less frequently in patients following organ transplant or with chronic use of immunosuppressive medications [Pittella, 2009; Cordova et al., 2008; Diazgranados et al., 2009].
In the child with acute meningoencephalitis due to Chagas’ disease, CSF reveals a mild to moderate pleocytosis (<100 cells/mm3), predominantly of lymphocytes, and protein elevation. Neuroimaging studies may reveal nonspecific ring-enhancing lesions seen with many CNS infections [Leiguarda et al., 1990]. No EEG abnormalities were seen in children with Chagas’ disease, although temporary irregularities appeared [Moya, 1994]. Sequence data on the T. cruzi genome are now available, which may lead to future attempts at intervention at specific genic targets [El-Sayed et al., 2005b].
The diagnosis of CNS Chagas’ disease is often made by direct examination of the CSF, which will reveal T. cruzi trypomastigotes. T. cruzi can be isolated from CSF in up to 75 percent of adult Chagas’ patients, but there is no evidence of overt CNS clinical symptoms [Cegielski and Durack, 1997]. Rarely, a fulminant meningoencephalitis will occur and be fatal. In a series of 11 children with CNS infection from Chagas’ disease, T. cruzi was isolated from the spinal fluid in 8 [Hoff et al., 1978]. Culture, direct microscopy, or histologic detection of T. cruzi in the blood or tissue obtained from brain biopsy are also likely to be positive. Serology is useful for diagnosis of neonatal Chagas’ disease but can also be misleading due to the presence of maternal antibodies in circulation. Positive serology in the absence of parasitemia and symptoms is probably due to antibody cross-reactivity [Pittella, 2009].
Brain biospy or autopsy will show edema, congestion, and scattered petechial hemorrhages. The fundamental histopathologic finding in encephalitis is multiple foci throughout the CNS, characterized by nodular arrangement of microglia, macrophages, neutrophils, and astrocytes, and mild perivascular lymphocytic infiltrate. T. cruzi are present in the form of amastigotes. Lymphocytic leptomeningitis is invariably present, and likely represents extension of the subadjacent cerebral inflammatory lesions [Pittella, 2009].
Management
Nifurtimox (for 1–10 years: 15–20 mg/kg/day in four doses for 90 days) is used for the treatment of Chagas’ disease [Medical Letter, 2004]. Benznidazole (for ages up to 12 years, 10 mg/kg/day in two doses for 30–90 days) is an alternative drug [Medical Letter, 2004]. Nifurtimox has been associated with a higher risk of chromosomal abnormalities, suggesting that alternative medications might be better. Allopurinol has been found to be as effective as nifurtimox or benznidazole in treating T. cruzi infection at a dose of 300 mg two or three times a day for 60 days [Gallerano et al., 1990].
The intracellular location of the parasite protects it from most drug therapies. Resistance to the basic drug therapy is a concern, and there is interest in finding or developing new drugs that are effective in treatment of Chagas’ disease. Itraconazole [McCabe et al., 1986] and ketoconazole [McCabe et al., 1987] were effective against T. cruzi in a mouse model of disease, but no trials have been conducted in humans. Ketoconazole, combined with benznidazole, demonstrated synergism when tested in the mouse model, but different strains of parasites exhibited variability in response to the drugs [Araújo et al., 2000]. Miltefosine (hexadecylphosphocholine), a phospholipid analog and anticancer drug that has been used with success in treatment of visceral leishmaniasis [Sundar et al., 2002], has been tested in vitro against intracellular stages of T. cruzi and has promise as an antimicrobial [Croft et al., 2003; de Castro et al., 2004]. Alkylphosphocholines, including miltefosine, in combination with ketoconazole, had in vitro activity against T. cruzi [Lira et al., 2001].
African Sleeping Sickness
Human African trypanosomiasis, or sleeping sickness, is prevalent in 36 sub-Saharan countries situated between latitudes 14° North and 29° South. The disease results from infection by the complex Trypanosoma brucei, which is transmitted by the bite of an insect vector, the tsetse fly. Only two subspecies are infectious in man: Trypanosoma brucei gambiense in West and Central Africa, and Trypanosoma brucei rhodesiense in east and southern Africa (Figure 82-18). Three major epidemics have occurred in the past century. The first, occurring between 1896 and 1906 in mostly equatorial Africa, killed an estimated 800,000 people. A second major epidemic in the 1920–1940s prompted establishment of vector control and surveillance programs that kept the disease in check. However, the past several decades in Africa have witnessed civil strife, population shifts, development of disease-resistant trypanosomes, and inadequate funding of programs to control trypanosomiasis, with a resultant resurgence of the disease. Large areas of Africa have been rendered unsuitable for raising and grazing cattle because of the risk of sleeping sickness in domestic stock [Brun et al., 2010].
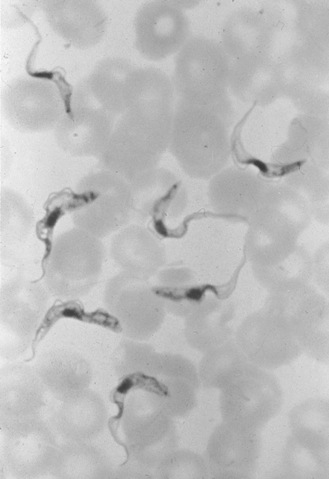
Epidemiology, microbiology, and pathology
Approximately 60 million people are at risk for infection [Kennedy, 2004]; most live in poverty and reside in remote rural regions [Rodgers, 2009]. Disease transmission occurs during activities such as farming, hunting, fishing, or washing clothes. The World Health Organization estimates that about 20–30 cases of Gambian sleeping sickness are diagnosed annually outside of Africa [Lejon et al., 2003a]. With increasing tourism to trypanosome-endemic regions of Africa, there is a greater likelihood that physicians outside of these areas will be seeing cases of sleeping sickness. Of the two types of sleeping sickness, the Gambian form is more likely to be encountered than the fulminant Rhodesian form because it can develop as a chronic disease [Lejon et al., 2003a]. Endemic geographic areas of the diseases do not overlap, which is helpful in distinguishing between the two types of trypanosomiasis. Cattle are an important reservoir for T. b. rhodesiense, although most wild animal species in game parks can harbor human pathogenic trypanosomes. For T. b. gambiense, humans are the main reservoir, with pigs and some wild animals possibly serving as a secondary reservoir [Brun et al., 2010].
At postmortem examination, dural thickening and adhesion to surrounding structures are often present. The perivascular infiltrate in the gray and white matter is delineated so clearly that it has been called a gray sleeve. A mononuclear infiltrate is seen in the meninges and Virchow–Robin spaces during microscopic examination. Lymphocytes are more numerous than plasma cells. Endothelial proliferation of the capillaries may be present. The characteristic morular cell, a plasma cell with immunoglobulin M-containing inclusions (Russell bodies), was first reported by Mott [1906]. Differentiating the neuropathologic changes in African sleeping sickness from Chagas’ disease is practically impossible.
Tryptophol, the 3-indole ethanol formed by the parasite in trypanosomal sleeping sickness, produces the characteristic sleep [Cornford et al., 1979; Feldstein, 1973; Stibbs and Seed, 1973a; 1973b]. Because tryptophol also gains access to lymphoid tissues, such as spleen, thymus, and mesenteric lymph nodes, it may foster immunosuppression in sleeping sickness [Ackerman and Seed, 1976]. The trypanosome induces formation of antibodies to antigens on its surface called variable surface glycoproteins. The variable surface glycoproteins are under genic control and generate antigenic variation by activation and expression of a succession of variable surface glycoprotein genes – of which there are about 1000 in a genome containing 10,000 genes [Donelson, 2003]. Thus, host antibodies aimed at one set of surface antigens are soon rendered ineffectual by selection of bloodstream forms exhibiting newly synthesized surface glycoproteins. The recurring appearance in the bloodstream of trypanosomes with new surface antigens gives rise to periodic fluctuations in parasitemia. Development of a mechanism to modify or terminate this process appears necessary before control or eradication of this disease is feasible [Donelson and Turner, 1985]. Given the immunologic evasion conferred by antigenic switching, the availability of a vaccine in the near future appears unlikely. The trypanosome genome and, in particular, the phenomenon of antigenic variation has been reviewed by Donelson [2003]. Comparative information on trypanosomatid genomes is now available [El-Sayed et al., 2005a], as are sequence data on the T. brucei genome [Berriman et al., 2005].
Clinical characteristics, clinical laboratory tests, and diagnosis
The clinical presentation of trypanosomiasis can mimic malaria, syphilis, tuberculosis, toxoplasmosis, HIV, typhoid fever, borreliosis with neural involvement, mumps, and meningoencephalitis [Lejon et al., 2003b]. The typical symptoms and signs can be divided into two stages, although with T. b. rhodesiense infection there may be no clear distinction between the first and second stages.
Stage 2: Encephalitic Stage
Criteria for diagnosis of trypanosomiasis include three steps. The first step is serologic testing. The card agglutination trypanosomiasis test (CATT) detects the presence of immunoglobulin M and is a widely used screening test, with a reported sensitivity of 87–98 percent and specificity of 93–95 percent. [Lejon et al., 2003b; Truc et al., 2002]. Other highly sensitive serologic tests used in nonendemic countries for screening include immunofluorescence or enzyme-linked immunosorbent assays.
Lumbar puncture is the third step needed for staging. The presence of flagellates in the CSF, elevated CSF leukocyte levels (5–20/μL), and the increase of intrathecal immunoglobulin M (as much as a 40-fold rise in late-stage trypanosomiasis) are suggestive of late-stage disease [Lejon et al., 2003a, 2003b]. Increase in CSF protein (≥750 mg/L [≥75 mg/dL]) is a reflection of the elevated immunoglobulin levels [Lejon et al., 2003b]. Supporting evidence of infection includes irregular EEG patterns, and nonspecific CNS abnormalities seen on MRI and CT scans [Kennedy, 2004]. Polymerase chain reaction has been used to detect trypanosomal DNA in CSF.
African sleeping sickness resulting from infection by T. b. gambiense is a less severe but more chronic illness than rhodesiense sleeping sickness. A furuncle or chancre may appear at the site of the fly bite, followed by fever of about 1 week’s duration and a rash [Lejon et al., 2003a]. Regional lymphadenitis follows the bite and is the dominant feature for approximately 14 days. Fever, malaise, headache, arthralgia, erythematous and edematous cutaneous eruption, and generalized lymphadenopathy, particularly in the posterior cervical nodes (Winterbottom’s sign), compose the next stage, which persists for a week to a month. In the CNS, the disease progresses insidiously from meningitis to encephalopathy through sequential stages, including inflammation and cellular infiltration, alteration of EEG patterns, edema, breakdown of the blood–brain barrier, and stimulation of cytokine production [Pentreath, 1995]. Disseminated intravascular coagulation may be a consequence of complement activation by immune complexes. Production of interferon-γ and tumor necrosis factor-α contributes to the overall inflammatory response in the CNS. Autoimmunity may develop, more likely as a consequence of late-stage neuropathology. Although acute rheumatoid arthritis and rheumatic fever are included in the differential diagnosis, they are readily excluded after appearance of manifestations of nervous system impairment, including confusion, somnolence, memory loss, ataxia, and loss of sphincter control.
Criteria for diagnosis of trypanosomiasis are the presence of flagellates in the CSF, elevated CSF leukocyte levels (5–20/μL), and the presence of intrathecal immunoglobulin M, as shown by the card agglutination trypanosomiasis test [Lejon et al., 2003b; Truc et al., 2002]. The elevated leukocyte count and the increase of intrathecal immunoglobulin M (as much as a 40-fold rise in late-stage trypanosomiasis) are taken as evidence of an intrathecal inflammatory process [Lejon et al., 2003a; 2003b]. Increase in CSF protein (≥750 mg/L [≥75 mg/dL]) is a reflection of the elevated immunoglobulin levels [Lejon et al., 2003b].
A sensitive and specific study results from inoculation of the patient’s blood into a mouse, with subsequent demonstration of the organisms in the mouse’s blood. Blood stages of the trypanosomes can be cultured in a biphasic medium (blood-nutrient agar slant with an overlay of glucose-containing Locke’s solution) [Tobie et al., 1950], but this is best carried out in a reference laboratory.
Management
Suramin is recommended for patients who present with T. b. rhodesiense disease before the development of CNS disease. The drug is avoided in western and central Africa because, where Onchocerca spp. are also present, its high activity against these parasites can cause severe allergic reactions [Brun et al., 2010]. Pentamidine is the treatment of choice for first-stage disease caused by T. b. gambiense. In contrast, pentamidine treatment in intermediate-stage disease (defined as 10–20 white blood cells per μL CSF) does not appear to be effective, and is not generally recommended [Brun et al., 2010]. Pentamidine is reportedly less effective in treating rhodesiense than gambiense disease [Fairlamb, 2003]. Both drugs penetrate poorly across the blood–brain barrier into the CNS, making them ineffective in late-stage disease, and both have serious side effects. The following drugs and dosages are recommended for pediatric patients: for suramin, 20 mg/kg on days 1, 3, 7, 14, and 21; for pentamidine, 4 mg/kg/day intramuscularly for 10 days [Medical Letter, 2004].
A more recent alternative that is apparently less toxic and perhaps more effective is difluoromethylornithine (eflornithine) for both early- and late-stage disease [Doua and Boa Yapo, 1994; Fairlamb, 2003]. Difluoromethylornithine is not available in the United States but can be obtained through the World Health Organization [Medical Letter, 2004]. Recommended dosage is 400 mg/kg/day intravenously in four divided doses for 14 days [Fairlamb, 2003; Medical Letter, 2004]. Difluoromethylornithine is effective against the gambiense but not the rhodesiense form of the disease. Synergy has been found in combining suramin with eflornithine [Fairlamb, 2003].
In late stages of the disease, melarsoprol is recommended and is administered intravenously (for pediatric patients, the initial dose is 0.36 mg/kg, increasing to a maximum of 3.6 mg/kg at intervals of 1–5 days for a total of 9 or 10 doses, for a total of 18–25 mg/kg over 1 month) [Medical Letter, 2004]. The drug can cross the blood–brain barrier. It is effective against both forms of trypanosomiasis. However, adverse reactions are frequent and can be life-threatening. The most important reaction is an encephalopathic syndrome associated with increased intracranial pressure, which occurs in an average of 4.7 percent of T. b. gambiense and 8 percent of T. b. rhodesiense patients, with a case fatality rate of 44 percent and 57 percent respectively. Administration of corticosteroids (prednisolone) and diazepam can lessen the likelihood of post-treatment sequelae and prevent arsenical encephalopathy [Pepin et al., 1995]. Other serious side effects include seizures, psychosis and coma, skin reactions, and peripheral neuropathies. The clinician must be aware of the side effects of these drugs before therapy is initiated, and gradual increase in dosage is advised [Medical Letter, 2004; Seidel, 1985b]. Nifurtimox, used in treating Chagas’ disease, has also been employed in late-stage disease, when other late-stage antimicrobials are ineffective. After successful treatment, all patients need to be followed up with blood and CSF analysis at 6-month intervals for 2 years, after which the patient is considered cured. Detection of intrathecal IgM suggests treatment failure [Kennedy, 2008].
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 82 Fungal, Rickettsial, and Parasitic Diseases of the Nervous System
Fungal Diseases
The importance of invasive fungal infections, including those of the central nervous system (CNS), has risen as the population of immunologically compromised patients, including preterm infants, has grown. Patients with congenital immunologicial deficiencies, aplastic anemia, or poorly controlled human immunodeficiency virus (HIV) infection are at risk for a wide variety of fungal infections because their illness prevents normal functioning of the immune system. Those patients receiving chemotherapy, hematopoetic stem-cell or solid organ transplants, or medications such as corticosteroids that blunt the normal immune systems, can also be highly susceptible. The normal host can also fall victim to invasive fungal infection, but does so less often and to a smaller spectrum of organisms [Chimelli and Mahler-Araujo, 1997].
Fungal diseases involving the brain are usually spread via the bloodstream after a systemic or pulmonary focus is established. Alternatively, pathogens invade the CNS via direct extension from adjacent sinuses, the cranial vault, orbits, or the spine. With exceptions, such as Candida spp. meningitis in premature neonates and Aspergillus spp. brain abscess in the highly compromised host, fungal infections of the CNS in children are rare. A review from a large university children’s hospital found that, in a 6-year period, only 2 percent of nearly 1500 positive cerebrospinal fluid (CSF) cultures recovered fungal isolates [Arisoy et al., 1994], mostly Candida spp.; nearly half came from low-birth-weight infants. Table 82-1 provides a more extensive list of risk factors, syndromes, and treatments for fungal infections of the nervous system. Since clinical manifestations of CNS fungal infections can be subtle and not specific to individual pathogens, errors and delays in diagnosis occur. Fungi may cause a variety of clinical syndromes, including meningitis, brain abscesses, granulomas, and invasion of the vasculature, further complicating clinical presentation and treatment [Salaki et al., 1984]. Prompt diagnosis and effective treatment often require a medical–surgical team approach.
Cryptococcosis
Epidemiology, Environmental Reservoir, and Pathogenesis
Cryptococcosis is globally distributed and is most commonly seen in patients with acquired immunodeficiency syndrome (AIDS) and in those receiving systemic corticosteroids; infection also occurs in those without apparent predisposition but at a lower frequency [Wendisch et al., 1996]. Although children are less frequently infected than adults, infants have contracted the disease [Hung et al., 1995; Littman and Walter, 1968; Wang et al., 1996], and vertical transmission has been reported from a mother with advanced AIDS [Sirinavin et al., 2004]. The mode of entry in humans is probably through inhalation or ingestion, and dissemination to the nervous system can follow. The most common cause of cryptococcal disease, Cryptococcus neoformans, is an encapsulated yeast found widely in the environment, particularly in bird feces and soil visited by birds (Figure 82-1). Cryptococcus gatti has been the cause of disease in animals and humans in British Columbia and the northwestern United States [Galanis et al., 2009], and appears to have a link to certain tree species in Australia and North America.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Cryptococcus most often causes a meningoencephalitis but can also cause brain abscess and cryptococcomas. Typical meningitis features, such as nausea, vomiting, headache, and fever, are often present. Caution is urged in ruling out the diagnosis based on history and physical examination alone because presenting symptoms may be nonspecific and a substantial number of patients are afebrile and lack meningeal signs [Sabetta and Andriole, 1985]. In AIDS patients, CNS cryptococcosis tends to be acute, with fever and headache in 70 percent; meningismus, photophobia, or mental status changes in 20–25 percent; and focal deficits in a small percentage [Daar and Meyer, 1992]. Despite this, a substantial minority of these patients may have initially normal CSF cell counts, glucose, and protein [Darras-Joly et al., 1996].
When C. neoformans causes mass lesions, it can result in papilledema, cranial neuropathies, and hemiparesis [Hung et al., 1995]. Brain abscess and cyptococcomas may begin with an indolent course, but hydrocephalus occurs and often necessitates shunting; if untreated, the mortality is high [Richardson et al., 1976]. Cryptococcus can be identified by culturing CSF on Sabouraud’s agar, but large-volume culture on more than one occasion may be needed. Latex agglutination detection of cryptococcal antigen is a rapid and useful adjunct test for both serum and CSF specimens. In the CSF, the antigen detection has an estimated sensitivity of 90 percent [Sabetta and Andriole, 1985]; both very high and very low antigen levels can give false-negative results. Since patients may have disseminated disease, C. neoformans can also frequently be cultured from urine or sputum, and the putative cause of CNS disease deduced. Cranial computed tomography (CT) and magnetic resonance imaging (MRI) scans can reveal mass lesions and hydrocephalus.
Management
No pediatric-specific clinical treatment trials have been completed. The Infectious Disease Society of America (IDSA) recently published treatment guidelines and recommends an initial combination of amphotericin B and flucytosine for cryptococcal CNS disease in all age groups, regardless of HIV status [Perfect et al., 2010]. Oral fluconazole is recommended for both consolidation and long-term suppression therapy. All cases of CNS cryptococcal disease should be managed in consultation with an infectious disease specialist.
Coccidioidomycosis
Epidemiology, Microbiology, and Pathology
Coccidioidomycosis is caused by the fungus Coccidioides immitis, a dimorphic fungus that grows as either a mycelium or a unique structure called a spherule. Disease caused by C. immitis most commonly occurs in endemic areas of the southwestern United States, northern Mexico, and portions of Central and South America. The San Joaquin Valley in California is the most notorious endemic zone, giving rise to the term valley fever. Most infections occur during the dry season when spores are borne by dust. Most often, C. immitis infection is unnoticed or causes a self-limited respiratory tract infection; dissemination is uncommon, but the risk is higher in patients of Filipino, black, and Hispanic backgrounds [Einstein and Johnson, 1993], and in the setting of HIV infection, organ transplantation, or prolonged use of corticosteroids. Coccidioidomycosis is acquired by inhalation of spore-laden dust or transcutaneously after skin abrasion; it is not spread from person to person. Because most (60 percent) infections are asymptomatic or mildly symptomatic, reported cases represent only a small fraction of the total burden of disease. CNS infection in children is rarely reported.
The spectrum of neuropathologic change ranges from meningitis to meningoencephalitis and meningomyelitis with extensive parenchymal destruction, sometimes as a result of an associated endarteritis obliterans [Mischel and Vinters, 1995]. Parenchymal brain lesions are rare but may occur in the absence of meningitis [Banuelos et al., 1996].
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Studies done on military recruits in the 1940s, using skin testing to diagnose acute infection, provided detailed data on infection in the normal host. Only 40 percent of these patients experienced acute respiratory symptoms [Smith et al., 1946], typically within 3 weeks after exposure and accompanied by fever, chills, night sweats, cough, anorexia, and weight loss. Dissemination of the infection from the pulmonary focus to distant sites usually occurs 1–6 months after primary infection in approximately 0.5 percent of cases; half of these are meningitis [Banuelos et al., 1996]. Other sites of dissemination include skin, lymph nodes, bones, and joints.
The clinical characteristics of coccidioidal meningitis are nonspecific and prompt diagnosis can be difficult [Caudill et al., 1970]. The most prevalent manifestations are headache, fever, malaise, and weight loss; meningismus may be absent [Saitoh et al., 2000]. Findings can include confusion, personality changes, focal neurological deficits, ataxia, obtundation, and coma. Brain and spinal cord abscesses can occur with or without concurrent meningitis [Banuelos et al., 1996]. Spinal cord symptoms may also occur in conjunction with coccidioidal osteomyelitis of the cervical vertebra [Jackson et al., 1964].
CSF abnormalities include mononuclear pleocytosis, increased protein content, increased chloride concentration, and normal or decreased glucose concentration. Diagnosis can be confirmed by either direct microscopy or culture of the CSF (Figure 82-2). Unfortunately, CSF cultures and microscopy are often negative in C. immitis meningitis, requiring indirect evidence of infection through serologic testing of serum and CSF. Complement fixation and precipitin tests are dependable and provide accurate diagnosis in more than 99 percent of patients with disseminated infection [Pappagianis, 1976]. Any discernible titer of complement fixation antibody in spinal fluid is considered diagnostic [Lyons and Andriole, 1986]. The coccidioidin skin test is not currently used for diagnosis of acute disease.
Imaging by CT or MRI scan may reveal widespread cisternal and cervical subarachnoid meningeal involvement in patients with coccidioidal meningitis [Wrobel et al., 1992], and may demonstrate the need for serial imaging to detect developing hydrocephalus.
Management
The 2008 IDSA guidelines for treatment of coccidioidal meningitis recommend fluconazole or itraconazole by mouth for all patients; intrathecal amphotericin was recommended by some clinicians but was graded C-III (poor evidence) by the IDSA [Ampel et al., 2009]. Fluconazole is well tolerated in both children and adults, and the oral form is highly bioavailable. Patients with C. immitis meningitis must be treated indefinitely with fluconazole to prevent relapse, and should be managed in conjunction with an infectious disease specialist.
North American Blastomycosis
Blastomycosis is caused by inhalation of airborne spores from Blastomycosis dermatitidis, a dimorphic fungus found in soil [Bradsher, 1996]. The endemic area includes the midwest, southeastern, and south central United States, and the Canadian provinces near the Great Lakes and St. Lawrence Seaway. The spectrum of clinical manifestations in adults includes pulmonary disease, acute and chronic skin disease, and disseminated disease involving the bones, the genitourinary tract, or the CNS; disseminated disease, and particularly CNS disease, is more common in those with compromised immune systems. Children are infrequently affected but suffer a similar spectrum of illness as adults [Schutze et al., 1996], and may be at increased risk if they play near beaver dams [Dismukes, 1986; Klein et al., 1986]. The three major CNS clinical manifestations are meningitis, intracranial mass, and spinal mass [Gonyea, 1978]. Patients with advanced HIV disease have accounted for an increase in cases compared with the past and are at greater risk for mortality [Witzig et al., 1994].
Diagnosis
In pulmonary disease, the organism can be identified on microscopy and culture of respiratory tract specimens. For CNS disease, ventricular and cisternal fluid may have a higher yield than lumbar CSF, which is rarely positive. Mass lesions are identified with neuroimaging [Ward et al., 1995]; microscopic study of biopsy material provides positive identification of the fungus. Skin tests and serologic tests are not helpful in the diagnosis of North American blastomycosis.
Management
Lipid formulations of amphotericin B for 4–6 weeks are recommended for moderate to severe disease [Chapman et al., 2008], followed by prolonged treatment with oral fluconazole, itraconazole, or voriconazole. Surgery may be indicated in patients with mass lesions, those with a need for a diagnostic biopsy, or those with osteomyelitis refractory to pharmacotherapy [Ward et al., 1995]. As with other CNS fungal diseases, co-management with an infectious disease specialist is recommended.
South American Blastomycosis
South American blastomycosis is caused by the fungus Paracoccidioides brasiliensis [Lutz, 1908]. The organism is a dimorphic fungal saprophyte found in soil and plants, which grows as a yeast at body temperature. Acquisition of disease is restricted to people living in latitudes between Mexico and Argentina, with most patients reported from Brazil. Most disease occurs in people older than the age of 30 years, with a striking male preponderance; among the rare childhood cases, the gender distribution is equal [Restrepo, 2000].
CNS involvement may follow disseminated infection; one review from Brazil found CNS involvement in 14 percent of symptomatic patients [de Almeida et al., 2004].
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
The most common findings in a series of 24 CNS cases included seizures, hemiparesis, cerebellar signs, and hydrocephalus [de Almeida et al., 2004]. CNS infection usually causes single or multiple mass lesions that appear hypodense and enhance on CT scan. The masses/abscesses may cause focal neurologic signs, increased intracranial pressure, and symptoms of meningeal inflammation.
The diagnosis of South American blastomycosis is suggested by simultaneous involvement of several organs or by characteristic oral and skin lesions in patients who reside in endemic areas. Microscopic study of scrapings from mucous lesions, sputum, gastric contents, lymph nodes, or surgical specimens is most helpful. The etiology of brain lesions must be deduced from culture results outside the CNS or via brain biopsy. CSF findings are typically normal, except for slightly elevated protein; CSF microscopy and culture are typically negative [de Almeida et al., 2004].
Management
Inadequate clinical trials have been completed to allow evidence-based treatment recommendations for CNS paracoccidioidal infection. Brazilian guidelines on treatment [Shikanai et al., 2006] are available in Portuguese and recommend that CNS infection be treated with trimethoprim-sulfamethoxazole at a high dose for at least 2 years; amphotericin B can be used for shorter periods. Voriconazole has been used for mild to moderate paracoccidioidomycosis but its role in CNS disease is unclear [de Almeida et al., 2004; Queiroz-Telles et al., 2007; Araujo et al., 1978].
Histoplasmosis
Epidemiology, Microbiology, and Pathology
Histoplasmosis is globally distributed; the Mississippi River basin, Mexico, and Central and South America are all endemic areas [Loosli, 1957]. In these regions, as many as 80 percent of children manifest positive histoplasmosis skin test results by age 5 years [Rogers, 1967]. In most cases, infection is self-limited, but an estimated 10 percent of those infected with Histoplasma capsulatum develop pulmonary symptoms to a degree prompting medical evaluation; those younger than the age of 2 years, the elderly, and those with immunocompromising conditions are more likely to be symptomatic. Overall, males are more likely to have symptomatic pulmonary disease, whereas the disseminated form of the disease affects males and females equally. Only a small subset of individuals with primary histoplasmosis develop disseminated disease, and an even smaller group develop a CNS infection. A review of 235 cases of histoplasmosis reported between 1952 and 1960 revealed that, of the one-third of the patients who had disseminated disease, one-quarter exhibited CNS impairment [Cooper and Goldstein, 1963]. In contrast, CNS disease was not found in a review of 57 histoplasmosis patients, including 23 with disseminated disease, at a pediatric oncology center in an endemic area [Adderson, 2004].
Histoplasma capsulatum is found in the Americas, and Histoplasma duboisii is found in Africa. This dimorphic fungus grows as a yeast at body temperature, whereas mycelia form at room temperature. The mycelia phase contains macroconidia (8–15 mm) and microconidia (2–5 mm); the latter may be essential for human infectivity because they are small enough to reach the alveoli. The organism is found in soil; dampness and nutrients from bird and bat droppings encourage growth in aviaries, chicken coops, mines, caves, and open fields [Ajello and Zeidberg, 1951]. During dry spells, the organism may become airborne and the spores inhaled. No evidence of person-to-person or animal-to-human transmission exists.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Most children with respiratory infection are asymptomatic or experience little difficulty [Ibach et al., 1954]. Fever, chills, and a productive cough can occur, and initial chest radiographs may document a nodular or diffuse infiltrate. Years later, chest radiographs demonstrate characteristic calcification in the central lymph nodes and peripheral lung fields. If untreated, the infection lasts 2–12 weeks.
Progressive disseminated histoplasmosis is rare in healthy children. Signs and symptoms associated with CNS involvement in children include intracranial hypertension, memory impairment, confusion, seizures, and urinary incontinence [Machado et al., 1993]. Reported CNS complications in children include hydrocephalus [Enarson et al., 1978], meningitis [Couch et al., 1978], ataxia, seizures, and cranial nerve palsy [Rivera et al., 1992]. Approximately 5 percent of adult patients with disseminated histoplasmosis will have CNS involvement [Borges et al., 1997], with a median time to diagnosis of 36 months [Machado et al., 1993].
The histoplasmin skin test converts to positive during the course of the acute infection, but this test is not used diagnostically. A 4-fold rise in complement fixation titers between acute and convalescent specimens or a single elevated titer in the correct clinical setting is diagnostic. A rise in complement fixation titers after treatment suggests relapse. The organism can be directly detected through culture of CSF, blood, and bone marrow, and through biopsy of liver, bone marrow, and lymph nodes. Histoplasma antigen can be detected in serum or urine [Wheat et al., 1986] and in some CSF specimens of patients with chronic Histoplasma meningitis, but its use for CNS disease is unreliable [Wheat et al., 1989].
All patients with disseminated histoplasmosis should be evaluated for adrenal insufficiency. In one series, half of the patients, regardless of treatment, had adrenal insufficiency, which was the most common cause of death [Sarosi et al., 1971]. Because Histoplasma can remain quiescent in the lungs or the adrenal glands for more than 40 years before dissemination, it needs to be considered in unexplained neurologic disease, particularly in people who lived in endemic areas as children [Tan et al., 1992].
Management
CNS infection in children should be treated sequentially with amphotericin B, followed by itraconazole, according to the 2007 IDSA guidelines [Wheat et al., 2007]. The prognosis of Histoplasma CNS infections is guarded: 20–40 percent of patients die, despite treatment, and relapse among survivors is common [Wheat et al., 2000].
Nocardia
Nocardia spp., Gram-positive, branching, filamentous bacteria, are ubiquitous in the environment; Nocardia asteroides, Nocardia farcinica, and Nocardia brasiliensis are the species most often responsible for clinical disease. Although this is a bacterial infection, it is included in this section because diagnosis may need to be sought specifically, and may require techniques different from those used to identify typical causes of bacterial CNS infection [Threlkeld and Hooper, 1997]. Nocardial disease during childhood is relatively uncommon but needs to be considered in immunologically compromised hosts and those with chronic lung disease. CNS infection may be isolated or found at the same time as pulmonary or disseminated infection [Beaman and Beaman, 1994]. In those with disseminated infection, CNS involvement should be viewed with concern because it is associated with mortality rates of 80 percent [Beaman and Beaman, 1994; Threlkeld and Hooper, 1997]. When CNS disease is the only manifestation of nocardiosis, it suggests that an inapparent pulmonary disease has resolved. Risk factors for pediatric CNS infections include malignancy, chronic granulomatous disease, lupus, HIV infection, organ transplantation, and surgery [Marlowe et al., 2000].
Brain abscess is the most frequently reported CNS manifestation of nocardial infection, but meningitis can occur, typically after rupture of an abscess and, more rarely, as the primary presentation [Al Soub et al., 2007]. Nocardia brain abscess presents with signs and symptoms typical for this syndrome, including fever, headache, seizure, and focal neurologic findings. Meningitis is often reported as having a subacute or chronic course [Bross and Gordon, 1991].
Diagnosis of Nocardia brain abscess may require a drainage procedure to obtain an adequate specimen for prolonged laboratory incubation to ensure growth on culture. A nocardial etiology of brain abscess or culture-negative meningitis is strongly suggested by concurrent isolation of this organism from a pulmonary site [Bross and Gordon, 1991].
Treatment of nocardial CNS infection requires multiple agents for prolonged periods. Susceptibility testing done at a reference laboratory can help guide initial therapy and provide options, if intolerance to initial therapy develops. Most Nocardia spp. are susceptible to trimethoprim-sulfamethoxazole, but treatment of CNS infection typically requires parenteral therapy with amikacin plus imipenem or ceftriaxone, with or without trimethoprim-sulfamethoxazole [Gilbert et al., 2004]. A review of antibiotic treatment for nocardial infections has been published [Threlkeld and Hooper, 1997]; most patients require at least 6 months of therapy, and the compromised host requires longer. Treatment composition and duration are based on expert opinion, since no controlled trials have been completed. Serial neuroimaging studies during treatment may be helpful in monitoring resolution of the abscesses [Kirmani et al., 1978]. In selected patients, surgical extirpation of multiple abscesses can be successfully accomplished [Rosenblum and Rosegay, 1979].
Actinomycosis
Epidemiology, Microbiology, and Pathology
Actinomyces spp. are anaerobic or microaerophilic, Gram-positive, non-spore forming, branching bacilli that normally colonize the mouth, intestine, and female genital tract. Invasive disease, usually caused by Actinomyces israelii, is most common in those of ages 10–60 years, with males more frequently affected than females. CNS disease in young children has not been reported [Olah et al., 2004], but risk factors for CNS disease in older children and adults include untreated focal infections, immunosuppression, and congenital heart disease with right-to-left shunts [Olah et al., 2004]. Although CNS disease is rare, it can occur either after direct extension from a contiguous focus or by hematogenous spread from another site.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Actinomycosis is a chronic, slow-growing, inflammatory mass, most frequently affecting the lungs, cervicofacial region, abdomen, and pelvis (in association with intrauterine devices) [Vinard et al., 1992; Winking et al., 1996]. The mass has a tough, fibrous capsule, does not respect tissue planes, and develops sinus tracts. Concurrent chronic infections and other debilitating conditions predispose patients to dissemination.
A review of 181 cases of actinomycosis revealed that less than 2 percent of patients had cerebral involvement, as manifested by abscess (solitary or multiple) and, less commonly, meningitis [Brown, 1973; Fetter et al., 1967; Winking et al., 1996]. Abscesses have been reported in the cerebrum, cerebellum, optic chiasm, parasellar region, and subdural space [Bebrova et al., 1994; Brown, 1973]. Osteomyelitis of the spine may be the origin of spinal cord abscess and compression [Fetter et al., 1967]. Neuroimaging demonstrates abscess easily, but microbiologic diagnosis of CNS disease can be challenging because the organism may be difficult to recover on culture if preceding antibiotics have been given. In addition, infections involving Actinomyces spp. are often polymicrobial, sometimes clouding the role of individual species. The etiology of CNS disease may be inferred if Actinomyces spp. are isolated at another site, particularly the lung.
Management
Recommendations for treatment of actinomycosis at any site are based on experience and in vitro susceptibility testing, rather than clinical trials. Standard recommended treatment for CNS disease is penicillin for at least 6 months. Recently, successful shorter courses of treatment with drugs other than penicillin, such as ceftriaxone and cefotaxime, for CNS disease have been reported [Olah et al., 2004; Sudhakar and Ross, 2004]. Surgical drainage of the abscess frequently is indicated [Jamjoom et al., 1994; Winking et al., 1996] and will speed definitive diagnosis of CNS disease.
Aspergillosis
Epidemiology, Microbiology, and Pathology
Like many fungal pathogens, Aspergillus spp. (Figure 82-3) initially gain entrance to the lung by inhalation. The name of this fungus was established because of the similarity of its fruiting head to the aspergillum, a perforated globe used to sprinkle holy water during religious ceremonies. Disseminated disease and CNS infection are nearly always accompanied by a serious underlying condition.
Aspergillosis of the CNS was rare until the population of immunocompromised hosts increased during the past two decades [Carpentier et al., 1996; Sparano et al., 1992; Torre Cisneros et al., 1993; Wright et al., 1993]. Among compromised hosts, the highest frequency of invasive aspergillosis is in heart and lung transplant recipients (19–26 percent), followed by those with chronic granulomatous disease (24–40 percent), acute leukemia (5–24 percent), bone marrow or hematopoietic stem cell recipients (0.5–9 percent), AIDS (0–12 percent), and solid organ transplant recipients (0.5–10 percent) [Denning, 1998]. CNS disease complicates 10–20 percent of invasive disease and is nearly universally accompanied by concurrent infection at other sites, typically the lung.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Other less common manifestations include meningitis, meningoencephalitis, hemorrhagic or ischemic necrosis, solitary granulomas, and invasive fungal arteritis [Torre Cisneros et al., 1993]. Highly immunocompromised patients may have altered mental status and seizures, whereas more competent hosts may have headache and focal complaints. In one series, the most frequent neurologic symptoms included altered mental status (86 percent), seizures (41 percent), and focal neurologic deficits (32 percent) [Torre Cisneros et al., 1993]. Neuroimaging may reveal sinus invasion, enhancing or nonenhancing hypodense brain lesions indicative of abscess [Sparano et al., 1992], or prominent ischemic lesions in the basal ganglia [Miaux et al., 1995]; concurrent chest radiography or CT scan usually shows multiple nodular lesions.
Definitive diagnosis of invasive Aspergillus is made by smear and culture of biopsy material. A presumptive diagnosis of CNS infection can be made if Aspergillus is identified from another site at the same time that neuroimaging shows multiple mass lesions. Serum galactomannan testing for invasive aspergillosis is available [Herbrecht et al., 2002] and provides a noninvasive alternative to support the diagnosis; however, published cutoffs of sensitivity and specificity are variable [Marr et al., 2004].
Management
Cerebral aspergillosis has a very poor prognosis, particularly in the immunocompromised host. Surgical drainage and removal of abscesses and granulomas are often performed for diagnostic and therapeutic reasons, but may not improve outcome [Casey et al., 1994; Haran and Chandy, 1993]. Case reports and one retrospective review suggest that a subset of patients will respond and some may be cured [Damaj et al., 2004; de Lastours et al., 2003; Schwartz and Thiel, 2004, 2009]. The 2009 IDSA guidelines for Aspergillus spp. infection of the CNS recommend voriconazole as the primary medical therapy, with itraconazole, posaconazole, and liposomal formulations of amphotericin B as alternatives in cases of intolerance. Appropriate surgical debridement of CNS and other sites, and minimization of immunosuppresion are also recommended, while the use of corticosteroids is discouraged.
Candidiasis
Candida spp. are the most common invasive fungal infections in infants and children [Arisoy et al., 1994]. Cerebral candidiasis is a rare sequela of disseminated disease in debilitated patients. Risk factors include prematurity, long-term receipt of broad-spectrum antibiotics, total parenteral nutrition, central venous catheterization, immunosuppression, immunodeficiency (congenital or acquired), chronic renal disease, diabetes, neutropenia, and ventriculoperitoneal shunts [Arisoy et al., 1994; Chiou et al., 1994; McAbee et al., 1995]. In a review from a large pediatric referral hospital, 12 percent of disseminated Candida infections affected the brain [Zaoutis et al., 2004]. Historically, Candida albicans has been the most common species identified in CNS disease, but many others, including Candida tropicalis, Candida glabrata, and Candida parapsilosis, are also frequently reported.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Meningitis is the most common manifestation of candidal CNS disease, but abscesses and granulomas occur independently and in combination with meningitis (Figure 82-4). When brain abscesses occur, they are typically multiple in number, illustrating the consequence of hematogenous dissemination [Incesu et al., 1994]. Intracerebral abscess after Candida septicemia is illustrated in Figure 82-4B. The clinical presentation of meningitis may be subtle in children because many patients have serious underlying illness and may have other concurrent infections. Mass lesions can cause hemiparesis, cranial nerve palsies, or signs of hydrocephalus [Coker and Beltran, 1988]. Fungal arterial and venous involvement can cause serious complications, as can the development of intracranial mycotic aneurysms [Goldman et al., 1979].
Diagnosis can be promptly confirmed by identification of the organism in spinal fluid or infected neural tissue [McGinnis, 1983]. Pleocytosis or reduced CSF glucose occurs in only about half of the patients who are culture-positive [Hughes, 1992].
Management
There are no comparative trials to permit definitive antifungal recommendations. Prompt speciation and susceptibility testing help optimize antifungal therapy. Recent IDSA guidelines for adults for C. albicans infection in the CNS recommend a liposomal formulation of amphotericin, with or without 5-flucytosine followed by fluconazole [Pappas et al., 2009]. Standard amphotericin deoxycholate may be acceptable in some young children, particularly neonates who are not routinely treated with 5-flucytosine. Consultation with an infectious disease specialist, regarding diagnosis, treatment, and follow-up, is recommended.
Zygomycosis
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
This invasive infection rarely, if ever, occurs in the normal host. Medical risk factors include diabetes mellitus, intravenous drug use, solid organ or bone marrow transplantation, and other immunocompromising conditions [Hussain et al., 1995]. Fever, facial and ocular pain, or CNS symptoms may herald the onset, and a black eschar-like plaque may be seen if sought in the oro- or nasopharynx. Etiology of CNS disease seen in MRI or CT imaging can be inferred from biopsy of nasopharyngeal or sinus specimens. Hyphae can be seen with conventional and special stains of biopsy material and are distinguished by their lack of septae, 90-degree branching, and breadth (10–20 μM).
Management
The prognosis for zygomycosis of the CNS is usually poor; however, several case reports describe survival in well-controlled diabetic patients treated with surgical debridement plus local and systemic administration of amphotericin B [Hamilton et al., 2003; Moll et al., 1994; Sandler et al., 1971]. Unfortunately, these organisms tend to infect and obstruct cranial blood vessels, and thus hamper drug delivery to affected tissue. General principles of management include correcting diabetic ketoacidosis if present, minimizing immunosuppressive therapy, and removing infected tissue surgically, often through extensive debridement, in addition to antifungal therapy [Cuadrado et al., 1988; Galetta et al., 1990; Hussain et al., 1995]. The most commonly recommended antifungal agents for zygomycosis are lipid-formulation amphotericin products at high doses. Although voriconazole is not active against zygmycosis, posaconazole is a potential alternative for initial or step-down therapy [van Burik et al., 2006]. Some authors have advocated less radical surgery combined with systemic and local (via Ommaya reservoir) amphotericin, augmented by hyperbaric oxygen therapy [Hamilton et al., 2003]. All cases should be managed by a joint medical-surgical team.
Scedosporium spp. Infection
Two species of Scedosporium environmental molds, prolificans and apiospermum (sexual state known as Pseudallescheria boydii), can cause a wide variety of human disease, including CNS infection [Cortez et al., 2008]. Risk groups include compromised hosts and survivors of near-drowning in polluted water. In a comprehensive review of 38 CNS cases, the youngest patient was 24 months old [Nesky et al., 2000].
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Scedosporium spp. can cause skin, lung, bone, joint, ocular, bloodstream and CNS infections. Brain abscess is the typical CNS manifestation and can be solitary or multiple; meningitis occurs but is less common [Rosen et al., 1965; Bell and Myers, 1978; Cortez et al., 2008]. Definitive diagnosis is through brain biopsy, although culture of the organism from a nonsterile site can support the diagnosis. There are no available noninvasive or antigen detection tests. On microscopy, this organism can be confused with Aspergillus or Fusarium spp., but it is easily distinguished once cultured.
Management
The principles of therapy are similar to that for aspergillosis and zygomycosis, and include surgical resection of accessible brain abscesses, reduction or reversal of immunosuppression, and the use of antifungal agents. Unfortunately, these organisms are notoriously resistant to most agents, and most patients with CNS disease succumb. Recently, however, in vitro data and a small number of clinical reports support the efficacy of voriconazole, possibly in combination with other agents [Cortez et al., 2008; Nesky et al., 2000; Poza et al., 2000; Gosbell et al., 2003; Howden et al., 2003].
Rickettsial Diseases
Rickettsiae are pleomorphic coccobacilli that are obligate intracellular bacteria whose arthropod vectors include ticks, mites, fleas, or lice [Kim and Durack, 1997]. Rickettsial diseases include the spotted fevers, typhus fevers, scrub typhus, Q fever, and ehrlichioses [Marrie and Raoult, 1992; Pai et al., 1997]. Rickettsial infections often have an accompanying skin eruption. Coxiella burnetii, the rickettsia causing Q fever, differs from the other organisms because it does not require a vector and rarely causes a skin rash.
CNS involvement can occur in all forms of rickettsial infections [Marrie and Raoult, 1992]. Pathologic changes may be extensive and, in some infections, systemic and cerebral vasculitis may be prominent. The clinical and pathologic response of the CNS to different infections caused by Rickettsia spp. is generally similar and differs only in severity [Harrell, 1953; Woodward and Osterman, 1984]. Headache is often the earliest and most frequent symptom in all rickettsial infections [Harrell, 1953].
Recommended treatment of rickettsial diseases in almost all clinical situations includes tetracyclines. Most broad-spectrum antibiotics, such as penicillins, cephalosporins, and penems, are ineffective. Doxycycline is considered the drug of choice in almost all clinical situations for both pediatric and adult patients [Dalton et al., 1995; Kirkland et al., 1995]. The dosage is 100 mg twice per day for adults, and the pediatric dose is 4 mg/kg/day, divided twice daily, intravenously or orally, for 10–14 days [Schutze and Jacobs, 1997]. Because tetracyclines are contraindicated during pregnancy, chloramphenicol is used as an alternate drug in this patient population; however, this drug has questionable efficacy for patients with ehrlichiosis and Q fever [Markley et al., 1998]. Empiric treatment with doxycycline should be initiated early in the course if clinical and epidemiologic findings are suggestive of a rickettsial infection because delays in treatment can lead to adverse outcomes. Tetracycline use in pediatric patients is often discouraged because of the potential association with tooth discoloration. For suspected cases of rickettsial illnesses, however, it is important to use a tetracycline. Tetracycline tooth staining is dose-related and studies have found that perceptible tooth staining does not occur until multiple courses have been used [Grossman et al., 1971; Lochary et al., 1998].
Table 82-2 lists rickettsial diseases, organisms, vectors, reservoirs, and geographic distribution. Rocky Mountain spotted fever is the prototypic condition for these infections.
Rocky Mountain Spotted Fever
Rocky Mountain spotted fever (RMSF) is caused by Rickettsia rickettsii and is the most common fatal tick-borne illness in the United States [CDC, 2004]. Early missionaries and settlers discerned the association of tick bites with the disease and called the disease tick fever [Aikawa, 1966]. Howard Ricketts identified the etiologic organism in 1906 and established the tick as the vector in transmitting the organism to humans.
The disease is prevalent in wide geographic areas. RMSF has been reported throughout the United States, with the exception of Hawaii and Vermont [Walker, 1995]. The states with the highest incidence of RMSF are Oklahoma and North Carolina [CDC, 2008a]. In the United States, the number of RMSF cases has increased in recent years, with over 2000 cases a year reported in 2006 and 2007 – the highest recorded levels in more than 80 years [Walker et al., 2008; CDC, 2008b]. Outside the United States, the re-emergence of the disease has been described in several countries of Central and South America [Heldrich, 1987; Hidalgo et al., 2007; Nava et al., 2008]. In the Northern Hemisphere, the incidence of the disease is greatest from April to September, when tick bites are most likely to occur. The American dog tick (Dermacentor variabilis) is the primary vector responsible for the transmission of R. rickettsii along the southern Atlantic coast and the southeastern and south-central United States. In the western region of the United States, the vector is usually the wood tick (Dermacentor andersoni; Figure 82-5). Given the habitat preferences of the vector, rural and suburban populations are more likely to be infected than urban residents.
Children have a higher risk of infection (half of the cases are younger than 19 years of age [Kamper, 1991]) because they are exposed to tick bites during outdoor play. RMSF should be considered in the differential diagnosis of anyone with compatible clinical features and exposure to a grassy or wooded area where ticks may be present [Walker, 1995]. Dogs may carry ticks into households and yards, and are a risk factor for infection [Paddock et al., 2002; Walker, 1995]. The absence of a reported tick bite history should not dissuade a clinician from suspecting RMSF. In a recent study of 92 children with RMSF, only 49 percent recalled a recent tick bite [Buckingham et al., 2007].
The incubation period ranges from 2 to 14 days (mean 7 days) after a tick bite [Thorner et al., 1998]. Transmission of rickettsiae occurs as a tick feeds on the blood of its host. After entering the body, rickettsiae multiply and disrupt the endothelial cells of capillaries and arterioles. Vasculitis is the pathologic hallmark of RMSF, and many of the clinical features are a result of vascular inflammation. In RMSF, the rickettsiae may also infect the smooth muscle cells of the tunica media. Loss of integrity of the small vessels in the brain results in hemorrhage into the subarachnoid space. Subsequent meningeal irritation and an accompanying perivascular mononuclear infiltrate develop. The brain parenchyma is studded with areas of microscopic infarctions. In the healing phase, a proliferative fibroblastic and gliotic response is evident; the ensuing focal nodules develop more often in white matter in spotted fever and more often in gray matter in typhus.
Clinical Characteristics, Clinical Laboratory Tests, and Diagnosis
Symptoms of RMSF vary in severity from mild to severe; the course may rapidly lead to death. RMSF is characterized by the classic triad of fever, headache, and rash in the setting of a recent tick bite. Most patients with RMSF, however, do not present with the classic triad. Initial symptoms are nonspecific and include fever, malaise, lethargy, and headache. The headache is usually intense and refractory to most analgesics. Progressive restlessness, irritability, confusion, and delirium may also occur. By day 5 of the illness, 85 percent of patients develop a rash. The rash often begins with a maculopapular appearance and becomes more defined as petechiae as the rash evolves. The rash often begins around the ankles or wrists and then spreads to other parts of the body (Figure 82-6). Acute renal failure, coagulopathy, and cerebral edema are well-recognized complications. Edema, usually periorbital at first and then generalized, is characteristic of and may suggest the diagnosis. Hepatosplenomegaly accompanied by liver dysfunction, including coagulation defects, may confound the diagnostic process. In fulminating disease, shock, coma, and general or focal neurologic impairment, including vertigo, seizures, hemiparesis, and ataxia, may dominate the clinical manifestations [Marrie and Raoult, 1992]. Skin necrosis and gangrene can be a late complication (Figure 82-7). RMSF may manifest as a skeletal muscle disease. Acute temporary hearing impairment has been described, but permanent auditory disruption is uncommon [Kelsey, 1979]. Ophthalmic features in all rickettsial disorders are virtually identical [Raab et al., 1969; Smith and Burton, 1977], and may include photophobia, conjunctivitis, petechiae on the bulbar conjunctiva, exudates and retinal venous engorgement, papilledema, and ocular palsies.
Fig. 82-6 Petechial rash of Rocky Mountain spotted fever involving the ankle.
(Courtesy of Dr. Daniel J Sexton.)
Fig. 82-7 Necrosis and gangrene of digits is a late complication of Rocky Mountain spotted fever.
(Courtesy Dr. Daniel J Sexton.)
The differential diagnosis of RMSF includes meningococcemia, enteroviral infection, scarlet fever, toxic shock syndrome, Kawasaki disease, Epstein–Barr virus, syphilis, typhoid, rat-bite fever, and other tick-borne diseases. Muscle biopsy reveals perivascular infiltrates of chronic inflammatory cells and focal chronic interstitial myositis [Krober, 1978]. Other organs, including kidneys, heart, lungs, spleen, and epididymis, may also be affected [Green et al., 1978]. Muscle weakness may be overwhelming; therefore, Guillain–Barré syndrome, viral myositis, and collagen disease may be erroneously diagnosed (although a case of Guillain–Barré syndrome associated with RMSF has been documented [Toerner et al., 1996]).
Laboratory abnormalities are common in severe disease and often include hyponatremia and thrombocytopenia. Spinal fluid may indicate a lymphocytic pleocytosis and the protein is elevated in about 30–50 percent of patients [Kim and Durack, 1997]. The cellular response in CSF may suggest bacterial meningitis, and the dermal petechiae may mimic those in meningococcemia. MRI of the brain reveals increased signal intensity in the distribution of perivascular spaces, which resolves in relation to clinical improvement [Baganz et al., 1995]. Postmortem examination of RMSF cases has demonstrated that rickettsial vasculitis can result in focal neurologic damage in the brain and spinal cord, as well as myocarditis and vasculitis in the lungs, kidney, and spleen [Walker and Mattern, 1980].
The most widely used and best serologic test for RMSF is the indirect fluorescent antibody assay, which is available through commercial laboratories and most state health departments [Lochary et al., 1998; Sexton and Kaye, 2002]. Immunofluorescent antibody for immunoglobulin M or immunoglobulin G antibodies can be used to make a presumptive diagnosis of acute infection, although these antibodies are often undetectable in the early phase of the illness. Confirmatory indirect fluorescent antibody testing requires demonstration of a 4-fold or greater rise in immunoglobulin G or immunoglobulin M antibody titer between paired serum specimens, and is generally considered as a retrospective diagnosis [Thorner et al., 1998]. Direct immunofluorescence antibody or immunohistochemical staining of skin biopsies is 70–90 percent sensitive for patients with a rash. For patients without a rash, diagnosis by direct immunofluorescence antibody can be problematic. Polymerase chain reaction testing for R. rickettsii DNA can be applied to serum, whole blood, or tissue specimens, but is generally available only at specialized research laboratories, and the sensitivity of the assay is directly related to the severity of the illness [Sexton et al., 1994].
Management
Early therapy with doxycycline for at least 7 days is recommended. The case fatality is 2–10 percent in treated patients and may be as high as 30 percent in untreated patients [Dalton et al., 1995]. Serologic confirmation may be delayed for 2–3 weeks; therefore, therapy should be administered when the diagnosis is suspected, based on clinical and epidemiologic findings. A history of tick bite is a useful epidemiologic clue in making a presumptive diagnosis. With early treatment, the prognosis for full recovery from RMSF is excellent. Neurologic sequelae may occur when therapy is postponed [Miller and Price, 1972]; mild intellectual impairment has been described [Wright, 1972].
Typhus Fever Group
The typhus fever group consists of epidemic (louse-borne typhus), Brill–Zinsser disease, and murine (flea-borne) typhus. Louse-borne typhus fever, caused by Rickettsia prowazekii, is distributed worldwide. Epidemics of typhus fever occur by transmission from human to louse to human, typically after skin is rubbed that is contaminated with infective louse feces. Epidemics of typhus fever are often associated with war, movements of large numbers of people, and squalid living conditions [Zinsser, 1935]. Systemic and neurologic symptoms are similar to RMSF (see above). A severe headache is almost always present. More severe CNS features, such as neck stiffness, seizures, delirium, and coma, can also occur. Treatment (e.g., with doxycycline) is the same as that used for the spotted fever group of infections.
Brill–Zinsser disease is a relapsing form of louse-borne typhus that may occur years to decades after the primary attack [Feigin et al., 1992]. Presumably, the rickettsiae remain dormant in the reticuloendothelial system until a subsequent reactivation occurs. Symptoms are similar to the primary attack, although generally milder, and doxycycline is the recommended therapy.
Murine, or flea-borne typhus, caused by Rickettsia typhi, is one of the most prevalent rickettsial diseases found in humans throughout the world [Azad, 1990]. Before World War II, murine typhus was widespread in the United States, and approximately 42,000 cases were reported between 1931 and 1946 [Azad, 1990; Pratt, 1958]. During the Vietnam War, murine typhus [Miller and Beeson, 1946] was one of the most common causes of acute febrile illness in American military personnel [Berman and Kundin, 1973]. Currently, endemic foci often exist around port cities and adjacent coastal areas [Azad, 1990; Civen and Ngo, 2008]. R. typhi is transmitted from rat to rat by the rat flea. In addition to rats, many animals, such as other rodents, skunks, opossums, shrews, and cats, can serve as hosts for R. typhi and arthropod vectors. Rickettsia felis causes a recently described rickettsial infection with similar clinical manifestations. The life cycle of R. felis is through opossums, cats, and cat fleas; infection has been identified in southern California and Texas [Civen and Ngo, 2008]. Human infection occurs after exposure of abraded or flea-bitten skin to infective flea feces [Feigin et al., 1992]. Unlike many other arthropod-borne infections, murine typhus can be acquired in households because the transmission cycle includes commensal rats and fleas [Azad, 1990]. Symptoms of murine typhus are similar to but generally milder than those occurring in RMSF. R. typhi infections are likely underdiagnosed because the clinical and laboratory features are similar to viral infections. The illness is often characterized by fever (frequently prolonged), headache, myalgia, and sometimes a maculopapular or petechial rash [Gelston and Jones, 1977]. Lymphadenopathy was reported as a common manifestation in a study in children [Bitsori et al., 2002]. When present, the rash often occurs on the trunk. The infection may resemble Kawasaki’s disease [Bitsori et al., 2002]. There is considerable variability in the frequency of reports of neurologic manifestations of murine typhus, ranging from 2 to 20 percent of cases [Allen and Saitz, 1945; Bitsori et al., 2002]. Meningitis and encephalitis are occasionally reported [Bitsori et al., 2002; Galanakis et al., 2002; Gelston and Jones, 1977]. Less commonly, focal symptoms, such as papilledema, focal seizure, and hemiparesis, have also been described [Masalha et al., 1998]. Sensorineural hearing loss has been reported during the convalescent phase of the illness [Tsiachris et al., 2008]. Diagnostic evaluation and treatment are similar to other diseases of the typhus group.
Scrub Typhus
Scrub typhus, caused by Orientia tsutsugamushi, is a systemic illness that causes a generalized vasculitis [Pai et al., 1997]. O. tsutsugamushi was formerly a member of the spotted fever group of Rickettsia spp., but was placed in its own genus based on molecular phylogenetic studies [Tamura et al., 1995]. Trombiculid (chigger) mites serve as reservoirs and vectors of this disease, and human infection occurs after a chigger bite in endemic areas, primarily in the southwest Pacific and Southeast Asia [Feigin et al., 1992]. An eschar, a characteristic feature of the disease, is seen in 60–80 percent of cases [Jelinek and Loscher, 2001] (Figure 82-8). Fever, headache, and rash are also common clinical manifestations. Complications include renal failure, pneumonitis, acute respiratory distress syndrome, and myocarditis. CNS complications include meningitis or meningoencephalitis. Case series have reported varying occurrence/frequencies of meningoencephalitis ranging from 3 to 20 percent [Fang et al., 1997; Pai et al., 1997; Huang et al., 2009]. Other less commonly reported neurologic complications include acute disseminated encephalomyelitis (ADEM), Guillain–Barré syndrome, brachial plexus neuropathy, and acute sensorineural hearing loss [Chen et al., 2006; Lee et al., 2009; Ting et al., 1992; Kang et al., 2009]. Approximately 50 percent of patients will have a mild CSF monocytic pleocytosis or an increase in protein concentration [Pai et al., 1997]. Doxycycline is considered the most effective treatment.
Q Fever
Q fever, caused by Coxiella burnetii, is a worldwide zoonosis. Infection in humans occurs from inhalation of the organism in areas where infected livestock are kept and, on occasion, from ingestion of contaminated dairy products. Occasional outbreaks in research laboratories that use sheep or cattle have also been reported [Feigin et al., 1992]. Prominent symptoms include fever, headache, and pneumonia. A rash rarely, if ever, occurs. Cases of encephalitis have been reported to accompany Q fever and may occur late in the course [Maurin and Raoult, 1999]. Other reported neurologic manifestations have been described, and these include pseudotumor cerebri syndrome, myelitis, meningitis, hemiparesis, aphasia, ataxia, seizures, tinnitus, cranial nerve palsies, and behavioral abnormalities [Ravid et al., 2008; Bernit et al., 2002; Glaser et al., 2010]. The reported frequencies of neurologic symptoms associated with Q fever vary considerably, ranging from 2 to 22 percent [Clark et al., 1951; Bernit et al., 2002].
Other Rickettsiae
Several new rickettsiae have been described since the early 1990s: Rickettsia japonica in Japan and Korea, Rickettsia honei in Australia and Southeast Asia, Rickettsia africae in Africa and the French West Indies, the Rickettsia mongolotimonae strain in Asia, Europe, and Africa, Rickettsia slovaca in Europe, Rickettsia aeschlimannii in Africa and Europe, Rickettsia heilongjanghensis in Asia, and Rickettsia parkeri in the United States [Raoult, 2004; Walker, 2007]. The extent of neurologic involvement with these emerging infections is unknown.
Ehrlichiosis
Emerging infections, akin to those caused by other rickettsial agents, have been increasingly reported in recent years [Walker et al., 1996].
The term ehrlichiosis was initially used to describe veterinary infections with Rickettsia-like bacteria that infected leukocytes. Human infection with Ehrlichia in the United States was first identified in a 51-year-old male with a history of a tick bite, fever, leukopenia, thrombocytopenia, and elevated liver enzymes [Maeda et al., 1987]. The patient was originally diagnosed with RMSF (see above). Cytoplasmic inclusions were observed in peripheral lymphocytes, and the patient was subsequently diagnosed with an Ehrlichia infection (E. chaffeensis) species. Since then, two additional Ehrlichia species have been recognized: Ehrlichia ewingii and Anaplasma (formerly Ehrlichia) phagocytophilum.
Ehrlichia are obligate intracellular bacteria and belong to the family Rickettsiaceae. All Ehrlichia are transmitted by ticks, and the interval from tick exposure to onset of illness is between 2 and 21 days [Harkess, 1991]. E. chaffeensis
[/not-level-membership-for-neurology-category]
