Chapter 54 Focal and Multifocal Seizures
Introduction
Focal or partial seizures originate in one region of the brain, where they may stay confined or spread to other areas. Multifocal seizures arise from multiple locations and constitute an important type of seizure in infancy and childhood. Both focal and multifocal types have been under-recognized in children, but modern epidemiologic studies show that focal epilepsies account for about 60 percent of all seizure disorders [Berg et al., 1999a,b; Sillanpaa et al., 1999]. The behavioral manifestations of focal seizures relate not only to the region of the brain involved during the ictal discharge, but also to the maturation of the nervous system and the integrity of the pathways necessary for clinical expression.
Focal seizures in the very young are subtler and less declarative than focal seizures seen later in life [Acharya et al., 1997; Hamer et al., 1999; Nordli et al., 1997]. This is particularly true in infants and children with diffuse encephalopathies, in whom brain immaturity, diffuse cerebral dysfunction, or both make manifestations of focal seizures difficult to recognize. Focal seizures also can be mistaken in older children when the presence of secondary convulsive movements prompts casual observers to label the event a “generalized tonic-clonic” seizure. With this misdiagnosis, critical elements of the seizures are overlooked. As described later, careful consideration of the unique features present in pediatric focal seizures can improve diagnostic accuracy.
In a majority of children with focal seizures, no focal structural lesion is present, and the seizures either are the expression of an idiopathic disorder (benign rolandic epilepsy) or are cryptogenic. This finding is in contrast to adults, in whom a focal seizure strongly implies the presence of a focal structural lesion (e.g., stroke, brain tumor). Instead, only 10 percent of children with focal seizures have brain tumors or strokes. In one large epidemiologic study, only 4 of 613 children with epilepsy had a brain tumor [Berg et al., 2000a].
The prognostic value of seizure classification by itself is limited, and a fuller understanding of the patient is achieved by making an epilepsy syndrome diagnosis. As pointed out in Chapter 50, two children with the same seizure type can have markedly different outcomes. Establishing an epilepsy syndrome diagnosis is the best way to determine on management options for different patients. An epilepsy syndrome diagnosis is preferred for assessing prognosis and treatment. Although it may not be possible to diagnose every child immediately on presentation, prospective population-based studies suggest that most children can ultimately be diagnosed with an epilepsy syndrome. Many factors contribute to the diagnosis of a syndrome, but in practice, three are most important:
Children with multifocal seizures (three or more foci, involving both hemispheres) may have unfavorable forms of epilepsy (e.g., migrating partial seizures, Dravet’s syndrome, symptomatic diffuse epileptogenic encephalopathies not otherwise specified). Although these epilepsies manifest with focal seizures, the children usually have evidence of concomitant diffuse cerebral dysfunction on clinical examination, developmental history, and interictal EEG studies. Correct diagnosis is particularly challenging in the group of children with focal seizures and evidence of widespread, diffuse, or multifocal cerebral dysfunction; too often, these patients end up in broad, poorly descriptive “wastebasket” categories, such as Lennox–Gastaut syndrome or generalized symptomatic epilepsy not otherwise specified. Lennox–Gastaut syndrome has particular diagnostic features and is not synonymous with diffuse symptomatic epilepsy, as discussed in Chapter 53. In summary, the importance of correctly recognizing focal seizures cannot be overstated.
Recognition of Focal Seizures in Children
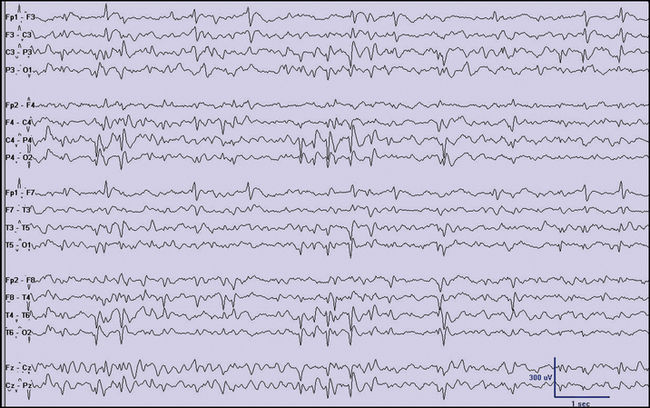
Clinical features alone cannot always allow one to diagnose a focal seizure correctly. Rather, “focal seizure” is actually an electroclinical diagnosis. It usually is made following consideration of multiple factors related to the patient and the clinical event, but may require EEG confirmation, particularly in the very young. Still, a number of important clues can help point to the presence of focal seizures in children (Box 54-1). Experience using video EEG monitoring suggests that certain clinical features tend to have focal ictal EEG correlates [Nordli et al., 2001] (Figure 54-1).








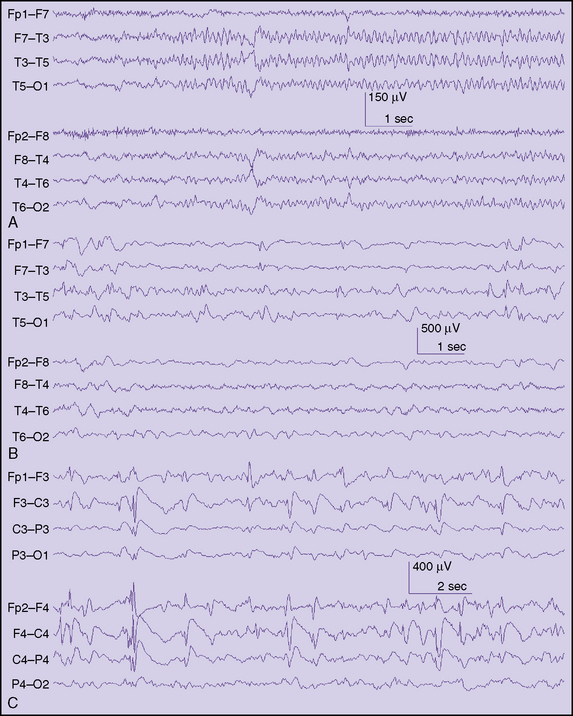
Fig. 54-1 Typical interictal findings on electroencephalogram (EEG) in children with focal seizures.
Behavioral Arrest
In some infants and young children, the most conspicuous feature of a focal seizure may be the sudden, abrupt cessation of on-going activity or a marked change in demeanor, as indicated by subtle but distinct changes in facial expression. Parents easily identify these features because they represent a clear paroxysmal alteration in the child’s behavior. Parents are particularly well attuned to the nature of their child’s habitual behavior, but these behavioral changes may be challenging for a person unacquainted with the child to identify on video tape. In the preverbal child, or in many children with special needs, it is impossible to ascertain alteration of consciousness reliably. Alteration of consciousness cannot be unambiguously inferred from behavior (e.g., daydreaming in school). To assess consciousness accurately, test items must be given and recall tested after the seizure. In children, this often is not possible, so the simple description of a behavioral arrest is more reliably used, rather than trying to infer if a seizure was truly “complex partial.” Behavioral arrest seizures also have been described as hypomotor seizures. This description refers to a sudden reduction in the motor activity of the child. The electrographic ictal accompaniment often emanates from the temporal lobe or posterior quadrant and may be composed of monotonous rhythmic delta, rhythmic theta-alpha patterns with an electrographic “crescendo” appearance, or low-voltage fast discharges that subsequently evolve to other rhythms (Figure 54-2). In children above age 3 years, behavioral arrest may accompany both focal and generalized seizures (absence seizures), so in isolation it is not a reliable indicator of a focal seizure; however, since absence seizures rarely occur in children less than 2.5 years, it is likely to be the correlate of a focal seizure in this age group.
Spasms
Spasms can be recognized by their tendency to recur in clusters, many times in an almost periodic fashion, with a fairly constant interval between some of the individual spasms. Spasms have a quick or myoclonic component at the start, followed by a brief sustained posture (tonic phase), followed in turn by a relaxation. Spasms that are asymmetric, that occur in a child with hemiparesis or other focal pathology, or that are associated with marked interhemispheric asymmetries on EEG are most likely focal seizures. In about 25 percent of patients with spasms, clear electrographic focal seizures can be detected before, during, or after the cluster. The EEG accompaniment of spasms often contains diffuse electrodecrements, even if they are preceded by clear focal seizures [Kubota et al., 1999].
Seizure Classification: International League Against Epilepsy
The International League against Epilepsy (ILAE) Commission on Classification proposed a classification of seizures in 1981. This scheme was widely used for almost three decades. Recently, a new ILAE commission on classification proposed a substantial revision [Berg et al., 2010] (Box 54-2). Now, the term “focal” replaces the previous term “partial,” and the obligatory separation of partial seizures into simple, complex, and secondary generalized has been discarded. Focal seizures still may be described further, if desired. One such way is to identify the degree of impairment. These and other descriptive terms are outlined in a previously published glossary [Blume et al., 2001] (Box 54-3). Examples of descriptors listed in the 2010 report include “without impairment of consciousness or awareness,” “with impairment of consciousness or awareness,” and “evolving to bilateral convulsive seizure.” The reader will note that these terms roughly equate to the older terms of simple, complex, and secondary generalized seizures. The important distinction is that these terms are no longer required but may be used by those who wish to maintain continuity with the 1981 classification. This adjustment appears minor, but is very useful because it is often not possible to determine alteration of consciousness reliably in the very young or in those with difficulties with communication. Lüders and colleagues have a logical and simple system that has been used internationally in major epilepsy centers (Lüders et al., 1999]. Others have proposed a much-simplified semiologic classification system for use in the very young [Nordli et al., 1997]. Neither of these schemes has been endorsed by the ILAE [Nordli et al., 1997]. While seizures may sometimes be broadly classified using the most prominent and early feature of the seizure, the various combinations of features, patterns, and time course of the seizure cannot be adequately summarized in a single word or phrase. Nothing can replace a thorough and meticulous description of the seizure. Indeed, the historic narrative of the seizure, as described or observed by parents, is the single most helpful piece of information allowing proper diagnosis of the seizure disorder and should be recorded, as accurately as possible, with few or no editorial comments.






















Epilepsy Syndromes with Focal Seizures
The ILAE classification of epilepsy syndromes is reviewed in Chapter 50. A recent modification of the ILAE classification eliminated the “focal” and “generalized” headings, along with the previous terms “idiopathic” and “symptomatic” (Box 54-4) [Borg et al., 2010]. One way to organize the recognized syndromes is by the specificity of the diagnostic criteria, further organized by age.
Box 54-4 Electroclinical Syndromes Categorized by Age at Onset










































Neonatal Period
Benign Familial Neonatal Epilepsy
Of the three recognized epilepsy syndromes in neonates, only one has prominent focal seizures: benign familial neonatal epilepsy (BFNE). The other two syndromes, early myoclonic encephalopathy (EME) and Ohtahara’s syndrome, may have accompanying focal seizures, but the predominant seizures are myoclonic, tonic, or epileptic spasms. BFNE was first described in 1964 by Rett and Teubel [Rett and Teubel, 1964]. Before the advent of newborn video-EEG recordings, it was thought that the seizures in this syndrome might be generalized, but subsequent recordings showed that the predominant seizures are focal clonic or adversive, even though the accompanying EEG may show diffuse flattening at the onset. Ronen et al., reported the incidence as 14.4 per 100,000 live births [Ronen et al., 1999]. The clinical features have been thoroughly reviewed by Plouin [Plouin, 2008]. Most seizures (80 percent) start on the second or third day of life in term, otherwise healthy, newborns. Clinically, they usually begin with a diffuse tonic component, followed by a variety of motor and autonomic phenomena. Motor manifestations may include prominent oculofacial features, limb clonus, or both. Interictal EEG backgrounds are usually normal or may show a théta pointu alternant pattern, which consists of short bursts of rhythmic theta activity with sharply contoured components. Ictal EEGs have shown initial flattening, which may be focal or diffuse, followed by subsequent ictal rhythms. Family history, by definition, is positive and the inheritance is autosomal-dominant. Mutations in the genes encoding KCNQ2 and KCNQ3 account for the majority of cases. There are no official guidelines for treatment of BFNE and it is uncertain whether treatment is beneficial in the long run. There are clear regional preferences but phenobarbital, sodium valproate, and phenytoin have all been used. The long-term outcome is favorable, although 11 percent of patients may have epilepsy later in life [Plouin, 2008].
Infancy
Benign Nonfamilial Infantile Seizures
Fukuyama in 1963 and later Watanabe were among the first to describe infants with the onset of epilepsy in the first 2 years of life with no known cause and excellent outcome [Fukuyama, 1963; Watanabe and Okumura, 2000]. Fukuyama originally described these seizures as generalized convulsions, but this was before the advent of modern video-EEG recordings and it is likely that these were actually focal seizures with secondary spread. Watanabe and colleagues described a case series in 1987 and noted clear focal features, describing these seizures as “complex partial” [Watanabe and Okumura, 2000]. Whether these are two separate conditions or one syndrome is a matter of some debate. In both cases, there is normal development before the onset of seizures. Imaging studies and metabolic tests are unremarkable. Onset is mostly within the first year of life in both cases. The interictal EEG is normal. In both cases, seizures may manifest with blank staring, and may be followed by secondary generalization in the Fukuyama type. Seizures may occur in clusters in both. The ictal focus is most often in the temporal region in the Watanabe form and in the centroparietal region in the Fukuyama type. Both demonstrate an excellent response to treatment and have normal development. Capovilla and colleagues have described another infantile epilepsy with excellent outcome. These infants have focal seizures and a characteristic interictal EEG finding of a “bell-shaped” discharge, which is maximal at the vertex [Capovilla and Vigevano, 2001]. Infants present between 8 and 30 months with relatively bland seizures characterized by motion arrest, some tonic stiffening, and oxygen desaturation. Seizures are infrequent and usually of short duration. The authors did not recommend treatment with antiepileptic drugs (AEDs) in most cases. A family history of epilepsy is present in half the cases and the outcome is very favorable.
Benign Familial Infantile Seizures
Vigevano and colleagues described cases with similar features but with a positive family history for infantile epilepsy [Vigevano et al., 1992]. In these patients there is a similar age of onset and normal development before the onset of seizures. Seizures usually start between 4 and 8 months of age, occur in clusters, have focal features including behavioral arrest, cyanosis, head/eye version, tonic stiffening of the limbs, and bilateral clonus. The interictal EEG is normal. The ictal EEG usually shows a fast rhythm beginning in the occipitoparietal region. Development is normal and the outcome is excellent. Treatment with antiepileptic medication is not mandatory. Since other family members may have experienced infantile seizures with a very favourable outcome, families may be very amenable to witholding treatment.
Epilepsy of Infancy with Migrating Focal Seizures
This condition was described by Coppola and others in 1995 [Coppola et al., 1995]. It is a severe and devastating epilepsy, with onset within the first 6 months of life in otherwise healthy infants. Seizures are truly multifocal, with a variety of clinical manifestations that shift from side to side. They often involve head/eye version, eyelid twitching, limb jerks, and tonic stiffening of one or both limbs. Initially, seizures may begin with a simple behavioral arrest, subtle eye deviation, and some oral automatisms. The striking feature of this epilepsy is the nearly continuous nature of the seizures. At the start of the disorder, each individual seizure is brief; seizures tend to recur in groups, 5–30 seizures occurring during drowsiness several times per day. Seizures may cluster for days, and the severity increases over the course of days or months to the point where the seizures are more or less continuous. Developmental regression ensues in all affected infants, along with deceleration of head growth. Marsh and colleagues reported a series of infants with slightly better outcome, but even in this series of patients, mental retardation ranged from mild to severe [Marsh et al., 2005]. There is no known cause and treatment has been frustrating. In the original series, two patients benefited from a combination of clonazepam and stiripentol, while in another series two others benefited from treatment with potassium bromide [Okuda et al., 2000].
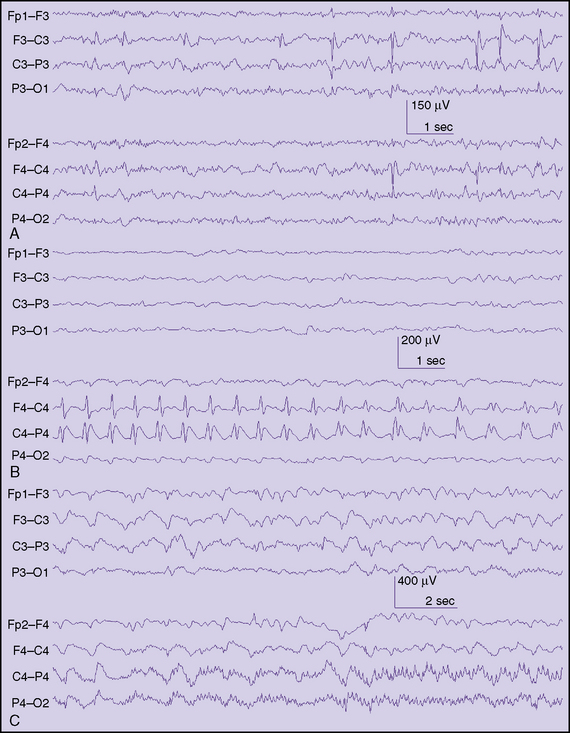
Dravet’s Syndrome
Dravet’s syndrome is another severe form of epilepsy affecting infants usually within the first year of life [Dravet et al., 2005]. Initial seizures are often hemiconvulsive and prolonged, and frequently are found in the setting of fever. Neurological examination and imaging studies are unremarkable. Subsequent seizure types may develop, including myoclonic jerks, and atypical absence and focal seizures. Athough development is usually normal prior to disease onset, children with Dravet’s syndrome may experience a developmental regression that can result in severe global delays. It is not yet known whether modern treatments can prevent, delay, or reduce the severity of this regression. Despite the general trend not to rush to treatment with AEDs in most children with epilepsy, there is a strong suspicion that early and aggressive treatment is warranted in Dravet’s syndrome once the diagnosis is secured [Arts and Geerts, 2009]. The diagnosis can be suspected in an infant with prolonged febrile seizures who later develops prolonged afebrile focal seizures, including hemiclonic convulsions [Millichap et al., 2009]. The majority of cases of Dravet’s syndrome are associated with mutations in the SCN1A gene. In particular, truncating mutations or missense mutations involving the portion of the gene that encodes for the pore of the sodium channel are most common. Initially, EEGs may be normal, or show polyspikes in the posterior quadrant. In time, generalized spike-wave discharges develop, and children may have photoparoxysmal responses (Figure 54-3). Effective treatments include valproate, stiripentol, clobazam, topiramate, ketogenic diet, and bromides [Korff et al., 2007; Wheless 2009; Caraballo and Fejerman, 2006]. Other newer medications may be effective but have not yet been rigorously studied. Since drugs with sodium channel properties, like carbamazepine, phenytoin, and lamotrigine, may exacerbate Dravet’s syndrome, newer medications that have pronounced sodium channel properties should probably be avoided [Guerrini et al., 1998]. Children with Dravet’s syndrome may have gait disorders (walking with a stooped gait), fine motor skill difficulties, and dysarthria so comprehensive that multidisciplinary management of their disorder is warranted. Dravet’s syndrome is also discussed in Chapter 55.
Myoclonic-astatic epilepsy, which was described by Doose, is sometimes confused with Dravet’s syndrome. This is now called epilepsy with myoclonic-atonic seizures or myoclonic-atonic epilepsy (MAE). As in Dravet’s syndrome, children can present with convulsive episodes early in life, but MAE usually begins after the first year of life, whereas almost all children with Dravet’s syndrome start having seizures in the first year of life. The most conspicuous feature of MAE is the presence of myoclonic-atonic seizures, which begin with a sudden jerk and are followed by a drop attack. This sequence can be demonstrated on video-EEG, or with an ordinary EEG using polygraphic techniques. Absence seizures and sometimes tonic seizures may occur, but prolonged focal seizures are very unusual in MAE. Children with Dravet’s syndrome often have myoclonus but it usually does not result in severe drop attacks. Children with MAE do not usually have mutations in the SCN1A gene. MAE is also discussed in Chapter 56.
Childhood
Panayiotopoulos’ Syndrome
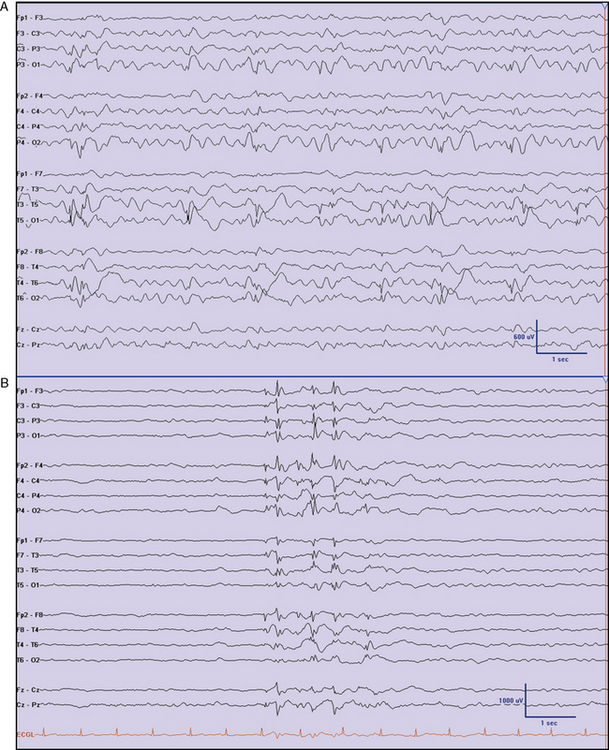
The ILAE recognized early-onset childhood epilepsy with occipital spikes (Panayiotopoulos type) and differentiated it from the later-onset occipital epilepsy, as described by Gastaut [Panayiotopoulos, 1999; Lada et al., 2003]. Panayiotopoulos’ syndrome is about half as frequent as benign focal epilepsy with central-temporal spikes by some estimates, and carries an excellent prognosis. It is characterized by autonomic and behavioral disturbances, with vomiting, deviation of the eyes, and impairment of consciousness that can progress to convulsions. Seizures are long-lasting, often more than 3 minutes. The typical age at onset is 5 years. Occipital spikes may be present, but stereotypic spikes in other locations (cloned spikes) are reported, particularly as the child ages [Ohtsu et al., 2003] (Figure 54-4). Panayiotopoulos has indicated that diagnosis is important for several reasons, including the fact that prophylactic treatment may not be necessary.
Benign Focal Epilepsies of Childhood
Benign focal epilepsy with central-midtemporal spikes (also called central-temporal epilepsy or rolandic epilepsy) is most common in previously healthy children aged 4–13 years. It had previously been classified as an idiopathic localization-related epilepsy by the ILAE, but the widespread nature of the EEG features and the shifting laterality of the clinical expression could equally argue for a more diffuse or multifocal predisposition. Rolandic epilepsy has characteristic EEG and clinical features. In a comprehensive study performed in Connecticut, rolandic epilepsy represented 9.6–10.3 percent of all childhood epilepsies, determined at presentation and 2 years later [Berg et al., 2000]. In its pure form, it is not associated with structural lesions and severe neurocognitive deficits [Lundberg and Eeg-Olofsson, 2003]. When awake, children experience brief focal seizures, with twitching of one side of the face, anarthria, drooling, and paresthesias of the face, gums, tongue, or inner cheeks. These manifestations may be followed by hemiclonic movements or hemitonic posturing. These diurnal seizures are simple; consciousness is preserved. Postictal weakness (Todd’s paralysis) of the involved face and limbs may occur. Most children have purely nocturnal seizures that usually become secondarily generalized. In such cases, the focal onset of the seizure usually is not observed, but parents are alerted by the sounds of the secondarily generalized convulsion. The prognosis is good, and seizures remit by adolescence. The EEG abnormality also resolves, although spikes may persist long after seizures have ceased.
Prior genetic studies indicate that the EEG trait itself is controlled by an autosomal-dominant gene with age-dependent penetrance [Bray and Wiser, 1965; Heijbel et al., 1975]. However, work done from recent multi-institutional twin studies suggests that the genetic contributions to the epilepsy per se appear to play a relatively minor role [Vadlamudi et al., 2006]. In the case of benign focal epilepsy with central-midtemporal spikes, more than half of siblings who have typical central-midtemporal spikes on EEG never have clinical attacks.
EEG findings are distinctive and diagnostic in benign focal epilepsy with central-midtemporal spikes. Focal di- or triphasic sharp waves of almost invariant morphology occur in the central and midtemporal regions (see Figure 54-1). Epileptiform discharges usually are of high voltage (greater than 100 mV), tend to occur in clusters, and activate dramatically during sleep, when they may seem almost continuous. In a single EEG, discharges may be unilateral, but with prolonged or repeated recordings, they are almost always bilateral. Lateralization may switch in serial tracings [Lerman, 1985]. Generalized spikes and spike-and-wave activity occasionally occur, although more often the spikes are maximal in either central region and are simply bisynchronous. “Benign” occipital spikes or multifocal spikes may coexist, especially in younger patients.
No correlation has been found between EEG findings and seizure occurrence or frequency. As a rule, EEG abnormalities are much more impressive than clinical seizure activity. Indeed, when central-midtemporal spikes are recorded in children without seizures, in whom EEGs are performed for other reasons, typical seizures eventually develop in only approximately half of the children. Furthermore, in symptomatic children, EEG abnormalities persist long after seizures cease. Thus, EEG does not provide assistance in making decisions about when or how long to treat. When treatment is elected because of recurrent attacks that are interfering with the child’s function or because of parental concern, carbamazepine and gabapentin are options. In some European countries, sulthiame (a drug not readily available in the United States) is used because of its reported beneficial effects of reducing spike frequency and clinical tolerance [Bast et al., 2003; Rating et al., 2000]. In addition, some authors are concerned that certain drugs (carbamazepine and lamotrigine, in particular) may aggravate this form of epilepsy [Cerminara et al., 2004; Parmeggiani et al., 2004], although such exacerbation appears to be rare [Corda et al., 2001].
Benign Focal Epilepsy with Occipital Spikes
Gastaut [1982] described another form of idiopathic localization-related epilepsy in children, in which visual symptoms, either amblyopia or hallucinations, are a common early feature of ictal events. The EEG shows stereotypic, high-voltage (200–300 mV) sharp-wave discharges over one or both occipital regions. Epileptiform activity is attenuated by eye opening and activated by sleep. Discharge morphology resembles that of central-midtemporal spikes. Background activity is normal. This electroclinical entity is more heterogeneous than benign focal epilepsy with central-midtemporal spikes. None the less, in a typical case, outcome usually is excellent, with complete resolution of clinical and EEG abnormalities in most children by 18 years of age. Like central-midtemporal spikes, occipital spikes do not correlate with clinical seizure activity or prognosis, and they often persist after seizures cease. Generalized spike-and-wave discharges and central-midtemporal spikes can coexist.
Acquired Epileptic Aphasia (Landau–Kleffner Syndrome)
Acquired epileptic aphasia, also known as Landau–Kleffner syndrome, is not a primary epileptic disorder, although epileptiform activity is one of the diagnostic criteria (see Chapter 55). Seizures occur in about 70 percent of cases but usually are infrequent. The syndrome is one of typically healthy children who acutely, or sometimes with a fluctuating course, lose previously acquired language skills. The aphasia begins with verbal auditory agnosia, and EEGs show abundant, high-voltage epileptiform activity that can be temporal, bitemporal, or generalized. Considerable slow-wave activity accompanies epileptiform discharges. In the early stages, EEG abnormalities may occur only during sleep, and throughout the illness, slow-wave sleep produces marked activation, sometimes with almost continuous generalized spike-and-wave activity. Review of original and subsequent cases of Landau–Kleffner syndrome does not support any single EEG feature or combination of features as being distinctive of this syndrome [Holmes et al., 1981; Landau and Kleffner, 1957]. AED treatment does not clearly alter the natural evolution of EEG abnormalities, clinical findings, or outcome, but corticosteroids may be of benefit [Lerman, 1991; Marescaux et al., 1990]. Subpial transections or focal resections are sometimes performed to treat acquired epileptic aphasia, but a surgical survey showed that they are rarely carried out and constitute less than 2 percent of all surgical procedures for epilepsy [Harvey et al., 2008].
Autosomal-Dominant Familial Nocturnal Frontal Lobe Epilepsy
Autosomal-dominant familial nocturnal frontal lobe epilepsy is characterized by clusters of brief hypermotor and tonic seizures with a nocturnal predominance. Onset typically is in childhood, and seizures may persist through life, with a wide variation in the severity of presentation. The ictal EEG shows bilateral anterior ictal discharges, but interictal findings usually are normal. Three different mutations in the gene encoding the alpha 4 subunit of a neuronal nicotinic acetylcholine receptor (CHRNA4) on chromosomal region 20q13.2 have been described, and other mutations in the gene encoding the beta 2 subunit of the neuronal nicotinic acetylcholine receptor (CHRNB2) on 1p21 were identified [Phillips et al., 1995, 2001]. Mutations in CHRNA2 have also been noted, and currently, testing is clinically available for all three genes. The topic has been recently updated by Hirose and Kurahashi on GeneReviews. Carbamazepine is effective in 70 percent of individuals; some with mutations in CHRNA4 respond better to zonisamide. At least one-third of patients may be refractory and require trials of multiple different AEDs. The susceptibility to seizures is lifelong, although the severity may lessen as patients reach middle age. The risk to each offspring of inheriting the mutant allele is 50 percent, but penetrance is estimated to be 70 percent and so the risk of inheriting the epilepsy is about 35 percent [Hirose and Kurahashi, 2010].
Adolescence–Adulthood
Autosomal-Dominant Epilepsy with Auditory Features
Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features, a form of familial temporal lobe epilepsy with auditory ictal manifestations. This disorder is characterized by simple partial seizures with auditory symptoms and secondary generalization. Sensory and psychic symptoms also may be present. Typical age at onset is in the first two decades [Ottman et al., 2004]. The most common auditory symptoms are simple, unformed sounds (e.g., buzzing, ringing).
Less Specific Age Relationship
Familial Focal Epilepsy with Variable Foci
Another epilepsy characterized by focal seizures but with a less specific age relationship is familial focal epilepsy with variable foci. Members of affected families have focal seizures with a variety of foci. The onset of seizures typically is in middle childhood, and attacks usually are easy to control. Seizure semiology varies among family members but is stereotypic for each affected person. In some reports, a pattern of nocturnal frontal lobe seizures suggested autosomal-dominant familial nocturnal frontal lobe epilepsy. Linkage to chromosome 22q has been found [Callenbach et al., 2003].
Distinctive Constellations
Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis
In mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE with HS), seizures involving the limbic structures produce characteristic clinical and electrographic features. Patients may complain of a rising epigastric aura or an indescribable sensation. Propagation of the ictal discharges can cause alteration of consciousness. Observers can document alteration of consciousness by asking the child to carry out simple commands such as “point to the door,” and to remember a peculiar test item such as “purple elephant.” Amnesia can be ascertained by asking about this test item after the seizure is finished and the child’s clinical condition has returned to baseline. Other characteristic clinical features that may evolve are ipsilateral picking automatisms and contralateral hand dystonic postures. Seizures may secondarily generalize; this sequence often begins with contralateral head and eye version, followed by contralateral clonus. This sequence represents a mature pattern, usually seen beyond the age of 6 years. In younger children, only fragments of this activity may be seen [Nordli et al., 2001].
Mesial temporal sclerosis (MTS) is a condition characterized by pathologic changes in the medial temporal region (Figure 54-5). It is a common cause of refractory temporal lobe seizures in adults that come to epilepsy surgery, but it is not commonly seen in population-based studies in children. Indeed, Berg et al. [2000a] found that only 3 children of 613 with epilepsy had evidence of hippocampal atrophy on their scans. One of these patients was later found to have a tumor. One possible explanation for this discrepancy is that mesial temporal sclerosis may be an acquired lesion. Indeed, pediatric operations for MTS are relatively rare worldwide [Harvey et al., 2008].
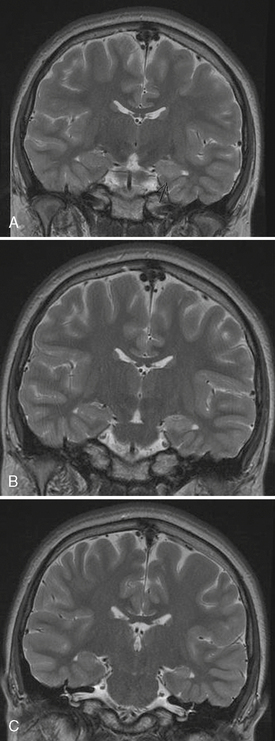
As mentioned, MTS frequently has been found in patients who have undergone temporal lobectomy for uncontrolled complex partial seizures [Corellis and Meldrum, 1976]. Such sclerosis may also affect the amygdala and the parahippocampal gyrus. Patients who have undergone temporal lobectomy with subsequent pathologic documentation of MTS frequently experience prolonged febrile convulsions in infancy and early childhood. Ounsted et al. [1966] stressed that early-onset seizures and severe generalized tonic-clonic seizures, accompanied by a high frequency of temporal lobe attacks, were associated with an adverse prognosis. Meldrum [1978] stressed the metabolic process by which neuronal hippocampal changes could be produced by recurrent seizures. Prospective epidemiologic studies have failed to prove a clear connection between prolonged febrile convulsions and mesial temporal sclerosis, but experience at surgical centers suggests that many patients with MTS have had prolonged febrile convulsions in early life. Magnetic resonance imaging (MRI) studies have shown acute and chronic changes in the hippocampi of children with prolonged febrile seizures [VanLandingham et al., 1998; Provenzale et al., 2008]. In these studies, increased T2 signal in the hippocampus was predictive of development of hippocampal atrophy. A large prospective study of children with febrile status epilepticus (FEBSTAT) is on-going in the US, which will hopefully address this important issue, but an association between prolonged febrile seizures and MTS appears likely from the published literature to date.
Brain MRI is very helpful in establishing the diagnosis of MTS [Woermann and Vollmar, 2009]. The afflicted hippocampus can be best visualized in coronal sequences obtained orthogonal to the long axis of the temporal lobe. Combinations of high-resolution images, inversion recovery sequences, and thin-cut images are used to compare the morphology, signal characteristics, and size of the hippocampus and adjacent structures. Characteristically, the interictal EEG shows focal pleomorphic interictal epileptiform discharges in the anterior to midtemporal region. Additional electrode placements, including anterior temporal and subtemporal leads, may assist in the detection and characterization of the interictal epileptiform discharges. The ictal EEG may reveal broad, relatively nonlocalizing, delta discharges at onset, but evolution to a rhythmic theta-alpha pattern within the first 10–20 seconds is a supportive feature. Patients with this lesion who have intractable epilepsy usually respond favorably to focal resection of the involved anterior temporal region. A randomized study of patients with temporal lobe epilepsy showed the superiority of surgical over medical management of MTS [Wiebe et al., 2001]. These results argue for early and aggressive surgical management of patients with intractable seizures resulting from MTS.
Epilepsia Partialis Continua and Rasmussen’s Syndrome
Another important epilepsy constellation presenting with focal seizures is epilepsia partialis continua manifesting as Rasmussen’s syndrome. Epilepsia partialis continua may be seen in a wide variety of clinical circumstances related to focal structural and metabolic lesions. In children, it may occur as a progressive entity with concurrent loss of motor skills [Rasmussen and McCann, 1968]. In a majority of patients, onset is before age 10 years. About two-thirds of children or their family members have an infectious or inflammatory illness before the onset of the epilepsia partialis continua. The first seizures are reported as generalized, and the intractable focal nature of the attack becomes apparent only over time. When fully established, the syndrome is characterized by unremitting seizure activity limited to part or one side of the body. Unlike those in more typical simple focal seizures of motor type, muscle movements usually are asynchronous in different muscle groups and seem to ebb and flow in waves, sometimes involving fewer muscles, sometimes more. The clinical course is marked by slow neurologic deterioration with development of hemiparesis, diminished mental capacity, and usually, hemianopia. Although progressive, the disease is only rarely fatal, and a permanent but stable neurologic deficit emerges.
Although childhood epilepsia partialis continua is considered the result of chronic encephalitis, the cause is unknown [Andrews and McNamara, 1996]. Evidence of anti-glutamate receptor (anti-Glu R3) antibodies suggests that an autoimmune process may be involved, but this has not been substantiated. Focal motor seizures often are associated with myoclonus, which may appear early. Progressive motor deficits develop during the illness, as does loss of mental function. The seizures are extremely resistant to treatment, and the condition usually progresses to complete hemiplegia with progressive unilateral brain atrophy [Andermann, 1991].
EEG findings are variable, and their topography often is difficult to characterize precisely [Bancaud, 1985]. Most often, interictal EEGs show excessive dysrhythmic or rhythmic delta activity, which is usually bilateral but accentuated over a large area contralateral to the partial seizures. Epileptiform discharges are rarely well localized and often are sporadic, especially early in the illness. As the disease progresses, more abundant pleomorphic spike and sharp-wave discharges appear over an extensive area or in both hemispheres. With scalp recordings, it is usually difficult to recognize distinct ictal discharges, and correlation of EEG changes with muscle jerking is always poor to nonexistent.
The evaluation of patients with possible Rasmussen’s syndrome is heavily dependent upon brain imaging, with important contributions from the clinical presentation and EEG. Progressive atrophy of one hemisphere, often heralded by a reduced size of the caudate along with a predominance of frontal atrophy, is strongly suggestive of this syndrome. Unilateral EEG findings of slowing, attenuation, and multiple populations of spikes predominantly within one hemisphere support the diagnosis. Patients with anti-GAD (glutamic acid decarboxylase) antibodies have been noted to have epilepsia partialis continua, but this can be easily excluded by a serum test searching for the presence of the antibodies. Children with Alpers’ disease can present with epilepsia partialis continua and recurrent bouts of focal status epilepticus, but brain atrophy and EEG changes will be more widespread. Metabolic acidosis and strokelike episodes (MELAS) can also present with epilepsia partialis continua and may have increased T2 signal, particularly in the posterior regions. Serum lactate measurement will help to exclude this condition. Perhaps the most difficult diagnosis to exclude is cerebral vasculitis. The absence of substantial gadolinium enhancement and calcifications can help to argue against this diagnosis. A European consensus conference concluded that, if the patient has epilepsia partialis continua, the EEG features are unilateral, and the imaging findings show progressive atrophy, then Rasmussen’s syndrome is highly likely and a brain biopsy is not needed [Bien et al., 2005]. In cases where the diagnosis is in doubt, the pathologic findings are microglial nodules and perivascular lymphocytes.
Immunomodulatory treatment with intravenous immunoglobulin or plasmapheresis has been tried and may ameliorate the clinical symptoms, including the epilepsy [Schmalbach and Lang, 2009]. The unanswered question is whether this approach can completely halt progression of the disease and avert the need for surgery. Even if successful, it is conceivable that medical treatment may only delay the inevitable definitive treatment: hemispherectomy. A complete disconnection of the involved hemisphere is necessary for good results. This can be achieved by a variety of surgical approaches, but a common modification of the standard anatomical hemispherectomy involves removing the temporal and inferior frontal lobes, thereby creating a window for the operating microscope. The surgeon can then complete a disconnection of the corpus callosum, as well as the frontal and occipital lobes, without resecting those regions. This modification can reduce the need for ventriculoperitoneal shunting and other complications, but may have a higher failure rate because of the difficulty of completely disconnecting the inferior frontal lobe and insula. Even when successful, surgery may not be able to reverse the cognitive disabilities [Terra-Bustamante et al., 2009].
Epilepsies Attributed to and Organized by Structural Metabolic Causes
The recent ILAE revised terminology suggests this etiological group to replace the former symptomatic epilepsy designation [Berg et al., 2010]. Major causes of epilepsy in this group include cortical malformations, neurocutaneous syndromes, tumors, infections, trauma, vascular lesions, prenatal insults, and others. Epilepsy due to structural or metabolic causes may manifest at any age with focal seizures. The clinical presentation varies widely, depending on the age of the patient, degree of cerebral maturation, location of the epileptogenic focus, and integrity of the underlying nervous system.
Although partial seizures are more likely to be associated with focal hemispheric lesions than are generalized seizures, structural causes are rarely identified. In children, approximately 30–50 percent of these seizures have no determinable etiology, whereas others have vague putative causes, such as a difficult delivery at birth or early childhood head trauma that cannot be substantiated. Moreover, genetic factors evidently determine susceptibility to focal seizures. An important exception is infants, in whom the rate of brain abnormalities may be higher [Hsieh et al., 2010]. In the past, prenatal and perinatal complications were thought to cause pediatric seizure disorders, but more recent epidemiologic evidence suggests that cerebral malformations are the most commonly identified underlying cause. Berg et al. [2000a] found that 16 of 613 children with epilepsy had cortical malformations, whereas 13 had presumed intrauterine insults, 8 had a neurocutaneous syndrome, and 5 had intraventricular hemorrhage. Only 1 had an anoxic encephalopathy, and 3 had defined genetic or chromosomal abnormalities. Four patients had tumors, 3 had vascular lesions, and 3 had clear evidence of an infectious disorder (based on neuroimaging). Only 2 patients had neuroimaging findings indicating previous brain trauma.
Congenital and Perinatal Factors
Chromosomal pathologic conditions may result in malformations. Intrauterine infections, specifically cytomegalic inclusion disease, toxoplasmosis, and rubella, are well known for their ability to cause abnormal brain development. Syphilis, rare in many parts of the world, also may cause intrauterine brain infection, with severe neurologic residua. Maternal exposure to radiation during pregnancy or ingestion by the mother of teratogenic drugs also may lead to cerebral malformations. In addition to other manifestations, these disorders also may result in focal and multifocal seizures. An unusual seizure type, myoclonic spasms, sometimes results from intrauterine infections. Tuberous sclerosis may manifest during the first few months of life and may be accompanied by focal seizures or infantile myoclonic spasms. Other causes of seizures in the perinatal period are discussed in Chapter 16.
Brain Tumors
Tumors are an uncommon cause of seizures during childhood. Tumors occur less frequently in children than in adults, and are more commonly found in regions of the brain that are not predisposed to developing epileptogenic activity (e.g., thalamus, cerebellum, brainstem). None the less, focal seizures in children can be caused by tumors [Backus and Millichap, 1962]. In a study of 100 patients with seizures of focal origin, 4 had associated hemispheric gliomas and 2 had arteriovenous malformations. During the subsequent 10 years, temporal lobe tumors were discovered in 3 additional patients. A more recent study using a community-based approach and modern imaging techniques revealed brain tumors in only 4 of 613 children with epilepsy. One case each of ganglioglioma, astrocytoma grade II, dysembryoblastic neuroepithelial tumor, and oligodendroglioma was reported [Berg et al., 2000a].
Postnatal Infectious Diseases
Focal or multifocal seizures may be associated with viral encephalitis. Many patients with viral encephalitis have only minor neurologic impairment, and their seizures may be transient. Conversely, severe encephalitis, resulting from herpes simplex virus or Epstein–Barr virus, both of which preferentially affect the temporal lobes, may result in severe sequelae with intractable seizures and permanent intellectual and memory dysfunction. Subacute sclerosing panencephalitis is associated with rubeola infection. This condition often is heralded by multifocal myoclonic seizures and focal and multifocal seizures. Diphtheria-pertussis-tetanus immunization may be followed by focal or generalized seizures. It generally is believed that these seizures are fever-induced and that pertussis encephalopathy is exceedingly rare. Parasitic infestation (see Chapter 82) may result in focal seizures. In particular, cysticercosis is accompanied by circumscribed cortical cysts and acute obstructive hydrocephalus. Echinococcosis is another parasitic infection that results in focal brain lesions. Tuberculosis with tuberculoma formation in the brain continues to be a problem in certain areas of the world.
Trauma
One of the most important consequences of traumatic brain injury is the development of convulsions, although the incidence depends on many factors, including injury severity. Epilepsy occurs more frequently after penetrating wounds [Raymont et al., 2010]. In closed head injuries, which constitute most of the wounds suffered by the general population, the incidence of traumatic epilepsy is relatively low. Cerebral damage during or near the time of birth may be manifested by early or late seizures. The effect of closed head injuries depends on the mechanical factors involved. In children, the most frequent traumatic causes of epilepsy are linear or depressed skull fractures. Except for some instances of early traumatic epilepsy after relatively trivial injury, the incidence of epilepsy after head trauma is generally proportional to the duration of post-traumatic amnesia.
Although epilepsy develops in nearly 50 percent of patients with penetrating head wounds and dural tears, seizures develop in only about 5 percent of patients after closed head injury [Jennett, 1975]. In a community-based study of 613 children with epilepsy in Connecticut, Berg and co-investigators found that only 2 children had neuroimaging findings consistent with trauma [Berg et al., 2000a].
Cerebrovascular Disease
Focal seizures in children rarely result from cerebrovascular disease [Chadehumbe et al., 2009]. Sturge–Weber syndrome (i.e., encephalofacial angiomatosis) usually manifests with a port-wine nevus in the distribution of one or more divisions of cranial nerve V. The associated angiomatosis is found over the ipsilateral cortex in the pia-arachnoid. The associated gyri are atrophied, and linear calcifications may be present, most often in the occipital lobes. Other features may include brain hypoplasia and focal seizures; hemiparesis may compromise the contralateral extremities [Thomas-Sohl et al., 2004]. Hemispherectomy often is indicated and likely should be undertaken early in life when seizures are intractable, and particularly when learning is impaired and a hemiplegia exists [Vining et al., 1997]. As a cautionary note, Kosoff and colleagues did not find a clear improvement in development in those children who underwent surgical treatment for their disease [Kossoff et al., 2002].
Management
Evaluation
Epilepsy syndromes can be identified by considering the age, functional developmental level of the child, clinical features of the seizures, and the interictal EEG findings. Those with favorable prognoses often occur against the backdrop of a normally developing child with either a normal interictal EEG or an EEG that shows highly stereotyped spikes. If the clinical features and EEG findings are not consistent with an epilepsy with a favorable prognosis, then MRI scanning is indicated. In the experience of Berg and colleagues, 177 of 613 children had symptomatic localization-related epilepsy, and findings on MRI were abnormal in 28.3 percent of these children [Berg et al., 2000a]. Any focal features on the neurologic examination or concerning interictal EEG characteristics (focal slowing, focal attenuation, focal polymorphic spikes) require follow-up imaging, and the location of the same findings can help to guide the MRI so that a specific target can be carefully examined (see Figure 54-1). Infants are a special category. While there are favorable forms of epilepsy in infancy, these are relatively uncommon. Indeed, recent imaging studies have suggested that all infants with recurrent seizures should have an MRI because of the low rate of generalized seizures and high rates of cortical malformations [Hsieh et al., 2010].
Berg et al. [2000a] studied 613 children with epilepsy. Neuroimaging was performed in a majority of these children (78.7 percent), but there was a very low rate of abnormalities in those with clear idiopathic localization-related epilepsy. Interictal EEG findings that suggest an idiopathic form of localization-related epilepsy include the presence of a normal background, lack of focal slowing, and stereotypic spikes in which each interictal epileptiform discharge closely resembles the other (see Figure 54-1). Guidelines for the evaluation of the first afebrile seizure have been published, and the recommendations concur that neuroimaging is not necessary in all cases [Hirtz et al., 2000].
Treatment
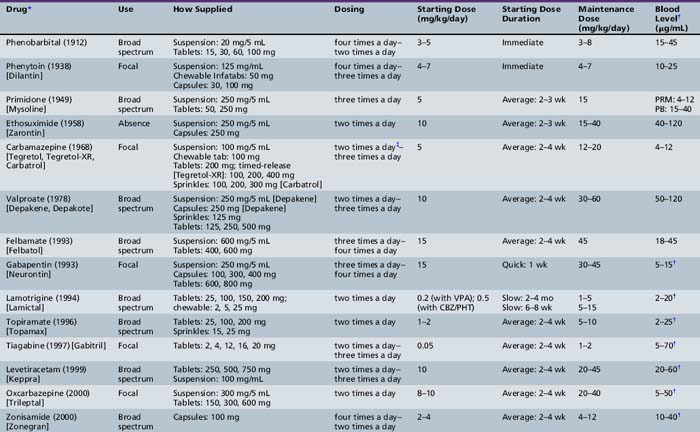
If the child is determined to have a favorable form of epilepsy presenting with focal seizures, then therapy should not be automatic, even if several seizures have occurred. It can be particularly useful to discuss precipitating factors with the parents and to give appropriate anticipatory guidance and counseling. A useful point to highlight is the importance of good sleep hygiene, keeping the same bedtime within an hour or so for weekdays and weekends. If therapy is warranted, carbamazepine, which is widely used in the United States, can be given, although many European practitioners avoid this drug because of concerns of aggravating the epilepsy. In Europe, valproate is still one of the most widely used medications [van de Vrie-Hoekstra et al., 2008]. A task force from the American Academy of Neurology and American Epilepsy Society recently reviewed the literature on AEDs for treatment of new-onset seizures and concluded that treatment could begin with any of the classic medications: carbamazepine, phenobarbital, phenytoin, or valproate. In addition, the task-force findings supported use of gabapentin, lamotrigine, oxcarbazepine, or topiramate as an alternative [French et al., 2004a]. A careful consideration of the side-effect profile of the various drugs in pediatrics is important before a specific AED is selected.
Four drugs, all released before 1978, have been most frequently used in pediatrics for focal seizures: carbamazepine, phenobarbital, phenytoin, and valproate. None is clearly superior to the others [Mattson et al., 1985], although randomized studies have found a disproportionately high incidence of side effects with phenobarbital [de Silva et al., 1996]. For this reason, chronic use of phenobarbital should be avoided, even in infants, unless special circumstances prevail. Most clinicians use carbamazepine as a first-line drug for management of epilepsies with focal seizures. Phenytoin was a favorite choice in the past, but cosmetic side effects and complicated kinetics make it less attractive than other agents. With valproate, a concern is the possibility of idiosyncratic, potentially fatal hepatotoxicity, particularly in children younger than 2 years of age who are receiving two or more antiepileptic agents [Dreifuss and Santilli, 1986].
At least ten new drugs have become widely available since 1993: felbamate, gabapentin, lacosamide, lamotrigine, levetiracetam, oxcarbazepine, rufinamide, topiramate, vigabatrin, and zonisamide. A comprehensive review of eight of these demonstrated that there was a general lack of well-designed randomized controlled trials in children and virtually no comparative efficacy trials [Hwang and Kim, 2008]. It might be tempting to rely on recommendations from experts but this information could be wrong and certainly is not a substitute for more definitive trials. A detailed understanding of the side effects and pharmacology of these medications are helpful. This has been recently reviewed by Sarco and Bourgeois, and medications are discussed in detail in Chapter 59 of this book [Sarco and Bourgeois, 2010].
In those epilepsies with multifocal seizures or focal seizures in combination with generalized seizures, drugs with a broad spectrum of action are usually preferred to those with action only against focal seizures. No published guidelines or practice parameters that apply to these patients are available. The literature on the subject is limited, so only anecdotal advice can be provided. These epilepsies appear to respond better to an agent with a wider spectrum of action, such as valproate. Newer agents offer the hope of better side-effect profiles with equivalent effectiveness (Table 54-1), but comparative data in children are not sufficient to permit a strong recommendation for their use as first-line treatment. The classic broad-spectrum agent is valproate, but newer agents with broad profiles include felbamate, lamotrigine, levetiracetam, topiramate, and zonisamide.

Refractory Focal Seizures
Evidence to guide the order of selection of AEDs for children with refractory focal seizures rationally is scarce. According to the most recently published practice parameters, gabapentin, lamotrigine, oxcarbazepine, and topiramate are useful adjunctive agents in the treatment of refractory pediatric focal seizures [French et al., 2004b]. Evidence was insufficient to make any recommendations concerning the use of levetiracetam, tiagabine, or zonisamide for the treatment of refractory focal seizures. Updated guidelines are probably in order.
Long-Term Outcome and Multidisciplinary Issues
Pediatric epilepsy is more than just two afebrile seizures. Environmental and genetic factors that cause susceptibility to recurrent seizures also may increase the risk for other manifestations of central nervous system dysfunction, including psychopathology, attention difficulties, behavioral disorders, and socialization problems (see Chapter 62). Like the seizures themselves, these clinical features may be expressed only during special developmental windows and, if not promptly recognized and treated, can have long-lasting insidious effects. Sillanpaa et al. [1998] have found that children with epilepsy are at increased risk of failing to match the social, educational, and vocational achievement of their peers. A multidisciplinary team approach to epilepsy can address these concerns by effectively screening at-risk patients and referring affected children to health-care professionals with special expertise. This process moves in parallel with the medical evaluation of the seizures, but careful case management by experienced personnel also is required. Resources vary in different regions and facilities, but intermittent multidisciplinary meetings can facilitate optimal care. Specialized epilepsy centers often are staffed with such teams, and children with refractory epilepsies may benefit from these services. The ultimate goal of treatment is not just seizure control but also maximization of the long-term potential of the child with epilepsy.
Acknowledgments
I would like to recognize the contribution of the late Dr. Fritz Dreifuss to this chapter.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Acharya J.N., Wyllie E., Luders H.O., et al. Seizure symptomatology in infants with localization-related epilepsy. Neurology. 1997;48:189.
Andermann F. Chronic encephalitis and epilepsy: Rasmussen’s syndrome. Stoneham, Mass: Butterworth-Heinemann; 1991.
Andrews P.I., McNamara J.O. Rasmussen’s encephalitis: An autoimmune disorder? Curr Opin Neurol. 1996;9:141.
Arts W.F., Geerts A.T. When to start drug treatment for childhood epilepsy: the clinical-epidemiological evidence. Eur J Paediatr Neurol. 2009;13(2):93-101.
Backus R.E., Millichap J.G. The seizure as a manifestation of intracranial tumor in children. Pediatrics. 1962;29:978.
Bancaud J. Kojewnikow’s syndrome (epilepsia partialis continua) in children. In: Roger J., Dravet C., Bureau M., et al, editors. Epileptic syndromes in infancy, childhood, and adolescence. London: John Libbey, 1985.
Bast T., Volp A., Wolf C., et al. Sulthiame Study Group. The influence of sulthiame on EEG in children with benign childhood epilepsy with centrotemporal spikes (BECTS). Epilepsia. 2003;44:215.
Berg A.T., Berkovic S.F., Brodie M.J., et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51(4):676-685.
Berg A.T., Levy S.R., Testa F.M., et al. Classification of childhood epilepsy syndromes in newly diagnosed epilepsy: Interrater agreement and reasons for disagreement. Epilepsia. 1999;40:439.
Berg A.T., Shinnar S., Levy S.R., et al. Newly diagnosed epilepsy in children: Presentation at diagnosis. Epilepsia. 1999;40:445.
Berg A.T., Shinnar S., et al. How well can epilepsy syndromes be identified at diagnosis? A reassessment 2 years after initial diagnosis. Epilepsia. 2000;41(10):1269-1275.
Berg A.T., Testa F.M., Levy S.R., et al. Neuroimaging in children with newly diagnosed epilepsy: A community-based study. Pediatrics. 2000;106:527.
Bien C.G., Granata T., et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. 2005;128(Pt 3):454-471.
Blume W.T., Luders H.O., Mizrahi E., et al. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia. 2001;42(9):1212-1218.
Bray P.F., Wiser W.C. The relation of focal to diffuse epileptiform EEG discharges in genetic epilepsy. Arch Neurol. 1965;13:223.
Callenbach P.M., van den Maagdenberg A.M., Hottenga J.J., et al. Familial partial epilepsy with variable foci in a Dutch family: Clinical characteristics and confirmation of linkage to chromosome 22q. Epilepsia. 2003;44:1298.
Capovilla G., Vigevano F. Benign idiopathic partial epilepsies in infancy. J Child Neurol. 2001;16:874.
Caraballo R.H., Fejerman N. Dravet syndrome: a study of 53 patients. Epilepsy Res. 2006;70(Suppl 1):S231-S238.
Cerminara C., Montanaro M.L., Curatolo P., et al. Lamotrigine-induced seizure aggravation and negative myoclonus in idiopathic rolandic epilepsy. Neurology. 2004;63:373.
Chadehumbe M.A., Khatri P., et al. Seizures are common in the acute setting of childhood stroke: a population-based study. J Child Neurol. 2009;24(1):9-12.
Coppola G., Plouin P., Chiron C., et al. Migrating partial seizures in infancy: A malignant disorder with developmental arrest. Epilepsia. 1995;36:1017.
Corda D., Gelisse P., Genton P., et al. Incidence of drug-induced aggravation in benign epilepsy with centrotemporal spikes. Epilepsia. 2001;42:754.
Corellis J.A.N., Meldrum B.S. Epilepsy. In Blackwood W., Corsellis J.A.N., editors: Greenfield’s neuropathology, ed 3, London: Edward Arnold, 1976.
de Silva M., MacArdle B., McGowan M., et al. Randomised comparative monotherapy trial of phenobarbitone, phenytoin, carbamazepine, or sodium valproate for newly diagnosed childhood epilepsy. Lancet. 1996;347:709.
Dravet C., Bureau M., et al. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol. 2005;95:71-102.
Dreifuss F.E., Santilli N. Fatal hepatotoxicity with valproate: Analysis of 37 cases. Neurology. 1986;36:175.
French J., Kanner A.M., Bautista J., et al. Efficacy and tolerability of the new antiepileptic drugs, I: Treatment of new-onset epilepsy: Report of the TTA and QSS subcommittees of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 2004;45:401.
French J.A., Kanner A.M., Bautista J., et al. Efficacy and tolerability of the new antiepileptic drugs, II: Treatment of refractory epilepsy: Report of the TTA and QSS subcommittees of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 2004;45:410.
Fukuyama Y. Borderland of epilepsy with special reference to febrile convulsions and so-called infantile convulsions. Seishin-Igaku (Clin Psychiatry). 1963;5:211-223.
Gastaut H. A new type of epilepsy: Benign partial epilepsy of childhood with occipital spike-waves. Clin Electroencephalogr. 1982;13:13.
Guerrini R., Dravet C., et al. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39(5):508-512.
Hamer H.M., Wyllie E., Luders H.O., et al. Symptomatology of epileptic seizures in the first three years of life. Epilepsia. 1999;40:837.
Harvey A.S., Cross J.H., et al. Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia. 2008;49(1):146-155.
Heijbel J., Blom S., Rasmuson M. Benign epilepsy of childhood with centrotemporal EEG foci: A genetic study. Epilepsia. 1975;16:285.
Hirose S., Kurahashi H. Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. GeneReviews. 2010. Retrieved May 16, 2002, 2002
Hirtz D., Ashwal S., Berg A., et al. Practice parameter: Evaluating a first nonfebrile seizure in children. Neurology. 2000;55:616.
Holmes G.L., McKeever M., Saunders Z. Epileptiform activity in aphasia of childhood: An epiphenomenon? Epilepsia. 1981;22:631.
Hsieh D.T., Chang T., Tsuchida T.N., et al. New-onset afebrile seizures in infants: role of neuroimaging. Neurology. 2010;74(2):150-156.
Hwang H., Kim K.J. New antiepileptic drugs in pediatric epilepsy. Brain Dev. 2008;30(9):549-555.
Jennett B. Epilepsy after non-missile head injuries, ed 2. London: William Heinemann Medical Books; 1975.
Korff C., Laux L., et al. Dravet syndrome (severe myoclonic epilepsy in infancy): a retrospective study of 16 patients. J Child Neurol. 2007;22(2):185-194. t
Kossoff E.H., Buck C., Freeman J.M. Outcomes of 32 hemispherectomies for Sturge-Weber syndrome worldwide. Neurology. 2002;59:1735.
Kubota T., Aso K., Negoro T., et al. Epileptic spasms preceded by partial seizures with a close temporal association. Epilepsia. 1999;40:1572.
Lada C., Skiadas K., Theodorou V., et al. A study of 43 patients with Panayiotopoulos syndrome, a common and benign childhood seizure susceptibility. Epilepsia. 2003;44:81.
Landau W.M., Kleffner F.R. Syndrome of acquired aphasia with convulsive disorder in children. Neurology. 1957;7:523.
Lerman P. Benign partial epilepsy with centro-temporal spikes. In: Roger J., Dravet C., Bureau M., et al, editors. Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey, 1985.
Lerman P. Effects of corticosteroid therapy for Landau-Kleffner syndrome. Develop Med Child Neurol. 1991;33:257.
Luders H., Acharya J., Baumgartner C., et al. A new epileptic seizure classification based exclusively on ictal semiology. Acta Neurol Scand. 1999;99(3):137-141.
Lundberg S., Eeg-Olofsson O. Rolandic epilepsy: a challenge in terminology and classification. Eur J Paediatr Neurol. 2003;7(5):239-241.
Marescaux C., Hirsch E., Finck P., et al. Landau-Kleffner syndrome: A pharmacologic study of five cases. Epilepsia. 1990;31:768.
Marsh E., Melamed S.E., Barron T., et al. Migrating partial seizures in infancy: expanding the phenotype of a rare seizure syndrome. Epilepsia. 2005;46(4):568-572.
Mattson R.H., Cramer J.A., Collins J.F., et al. Comparison of carbamazepine, phenobarbital, phenytoin and primidone in partial and secondary generalized tonic-clonic seizures. N Engl J Med. 1985;313:145.
Meldrum B.S. Physiological changes during prolonged seizures and epileptic brain damage. Neuropaediatrie. 1978;9:203.
Millichap J.J., Koh S., et al. Child Neurology: Dravet syndrome: when to suspect the diagnosis. Neurology. 2009;73(13):e59-e62.
Nordli D.R.Jr, Bazil C.W., Scheuer M.L., et al. Recognition and classification of seizures in infants. Epilepsia. 1997;38(5):553.
Nordli D.R., Kuroda M.M., Hirsch L.J. The ontogeny of partial seizures in infants and young children. Epilepsia. 2001;42:986.
Ohtsu M., Oguni H., Hayashi K., et al. EEG in children with early-onset benign occipital seizure susceptibility syndrome: Panayiotopoulos syndrome. Epilepsia. 2003;44:435.
Okuda K., Yasuhara A., Kamei A., et al. Successful control with bromide of two patients with malignant migrating partial seizures in infancy. Brain Dev. 2000;22(1):56-59.
Ottman R., Winawer M.R., Kalachikov S., et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62:1120.
Ounsted C., Lindsey J., Norman R. Biological factors in temporal lobe epilepsy. London: William Heinemann Medical Books; 1966.
Panayiotopoulos C.P. Early-onset benign childhood occipital seizure susceptibility syndrome: A syndrome to recognize. Epilepsia. 1999;40:621.
Parmeggiani L., Seri S., Bonanni P., et al. Electrophysiological characterization of spontaneous and carbamazepine-induced epileptic negative myoclonus in benign childhood epilepsy with centro-temporal spikes. Clin Neurophysiol. 2004;115:50.
Phillips H.A., Favre I., Kirkpatrick M., et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225.
Phillips H.A., Scheffer I.E., Berkovic S.F., et al. Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q13.2. Nat Genet. 1995;10:117.
Plouin P. Benign familial neonatal seizures and benign idiopathic neonatal seizures. In: Engel J., Pedley T.A., editors. Epilepsy: a comprehensive textbook. ed 2. Philadelphia: Lippincott Williams &Wilkins; 2008:2287-2295.
Provenzale J.M., Barboriak D.P., et al. Hippocampal MRI signal hyperintensity after febrile status epilepticus is predictive of subsequent mesial temporal sclerosis. Am J Roentgenol. 2008;190(4):976-983.
Rasmussen T., McCann W. Clinical studies of patients with focal epilepsy due to “chronic encephalitis”. Trans Am Neurol Assoc. 1968;93:89.
Rating D., Wolf C., Bast T. Sulthiame as monotherapy in children with benign childhood epilepsy with centrotemporal spikes: A 6-month randomized, double-blind, placebo-controlled study. Sulthiame Study Group. Epilepsia. 2000;41:1284.
Raymont V., Salazar A.M., et al. Correlates of posttraumatic epilepsy 35 years following combat brain injury. Neurology. 2010;75(3):224-229.
Rett A., Teubel R. Neugeborenen Krampfe im Rahmen einer epileptisch belasten Familie. Wien Klin Wochenschr. 1964;76:609-613.
Ronen G.M., Penney S., Andrews W. The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr. 1999;134(1):71-75.
Sarco D.P., Bourgeois B.F. The safety and tolerability of newer antiepileptic drugs in children and adolescents. CNS Drugs. 2010;24(5):399-430.
Schmalbach B., Lang N. New hope for Rasmussen encephalitis? Discov Med. 2009;8(42):130-132.
Sillanpaa M., Jalava M., Kaleva O., et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715.
Sillanpaa M., Jalava M., Shinnar S. Epilepsy syndromes in patients with childhood-onset epilepsy. Pediatr Neurol. 1999;21:533.
Terra-Bustamante V.C., Machado H.R., et al. Rasmussen encephalitis: long-term outcome after surgery. Childs Nerv Syst. 2009;25(5):583-589.
Thomas-Sohl K.A., Vaslow D.F., Maria B.L. Sturge-Weber syndrome: A review. Pediatr Neurol. 2004;30:303.
Vadlamudi L., Kjeldsen M.J., Corey L.A., et al. Analyzing the etiology of benign rolandic epilepsy: a multicenter twin collaboration. Epilepsia. 2006;47(3):550-555.
van de Vrie-Hoekstra N.W., de Vries T.W., et al. Antiepileptic drug utilization in children from 1997–2005–a study from the Netherlands. Eur J Clin Pharmacol. 2008;64(10):1013-1020.
VanLandingham K.E., Heinz E.R., et al. Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol. 1998;43(4):413-426.
Vigevano F., Fusco L., Di Capua M., et al. Benign infantile familial convulsions. Eur J Pediatr. 1992;151(8):608-612.
Vining E.P., Freeman J.M., Pillas D.J., et al. Why would you remove half a brain? The outcome of 58 children after hemispherectomy – the Johns Hopkins experience: 1968 to 1996. Pediatrics. 1997;100:163.
Watanabe K., Okumura A. Benign partial epilepsies in infancy. Brain Dev. 2000;22:296.
Wheless J.W. Managing severe epilepsy syndromes of early childhood. J Child Neurol. 2009;24(8 Suppl):24S-32S. quiz 33S-6S
Wiebe S., Blume W.T., Girvin J.P., et al. Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med. 2001;345:311.
Woermann F.G., Vollmar C. Clinical MRI in children and adults with focal epilepsy: a critical review. Epilepsy Behav. 2009;15(1):40-49.