Fetal Physiology
Mieke Soens MD, Lawrence C. Tsen MD
Chapter Outline
Fetal life in utero differs significantly from postnatal life. The fetus relies completely on the mother and the placenta for basic metabolic needs such as nutrient delivery, gas exchange, acid-base balance, and electrolyte homeostasis. During gestation, the fetus gradually assumes the responsibility for many of the vital physiologic functions that must be assumed after the abrupt transition to physiologic independence at birth. Knowledge of fetal physiology, and the timing associated with these developmental changes, is necessary for the optimal provision of analgesia and anesthesia during pregnancy and childbirth.
Fetal Environment
Amniotic Fluid
The fetus is surrounded by amniotic fluid, a complex fluid that changes as the pregnancy progresses. Amniotic fluid serves a number of vital roles, including the facilitation of fetal growth, the provision of a microgravity environment that cushions the fetus, and the generation of a defense mechanism against invading microbes.1,2 The formation and maintenance of amniotic fluid is an intricate process that depends on fetal maturation and maternal hydration, hormonal status, and uteroplacental perfusion.
Amniotic fluid during early embryogenesis is principally derived from maternal plasma by the passage of water and solutes through fetal membranous and placental layers. Between 10 and 20 weeks’ gestation, the volume of amniotic fluid increases in a predictable and linear manner from approximately 25 mL to 400 mL. During this period, the composition of amniotic fluid is similar to fetal extracellular fluid, owing to the absence of keratin in the fetal skin. After this period, the volume of amniotic fluid is a function of production, from fetal urine (600 to 1200 mL/day near term) and respiratory tract secretions (60 to 100 mL/kg fetal body weight/day), and removal through fetal swallowing (200 to 250 mL/kg fetal body weight/day).3 Amniotic fluid volume is also influenced by intramembranous (between amniotic fluid and fetal blood within the placenta) and transmembranous (between amniotic fluid and maternal blood within the uterus) pathways in both physiologic and pathophysiologic states.4 Finally, the status of maternal hydration and the amount of decidual prolactin may alter the transfer of amniotic fluid through fetal and maternal tissues.
The composition of amniotic fluid undergoes more marked variation than its volume.5,6 Keratinization of the fetal skin is complete by 25 weeks’ gestation and decreases the permeability of fetal tissues to water and solutes. The impact of this process, coupled with the ability of the fetal kidneys to produce urine, results in increased amniotic fluid concentrations of urea and creatinine, decreased concentrations of sodium and chloride, and reduced osmolality. A variety of carbohydrates, proteins, lipids, electrolytes, enzymes, and hormones, which vary in concentration depending on the gestational age, are also present; some of these elements, particularly the amino acids taurine, glutamine, and arginine, serve a nutritive function for mitotic cells involved in trophoblastic growth and placental angiogenesis.1 An abundance of growth factors are found in amniotic fluid, including epidermal growth factor, transforming growth factor-alpha, transforming growth factor-beta 1, insulin-like growth factor-1, erythropoietin, and granulocyte colony-stimulating factor; many of these growth factors play an important role in fetal intestinal development.1,7
Antimicrobial defenses within the amniotic fluid are primarily composed of humoral mediators such as alpha-defensins, which are released from neutrophils, especially in the setting of preterm labor and/or chorioamnionitis. Other humoral mediators include lactoferrin, calprotectin, leukocyte protease inhibitor, and cathelicidin, which have significant activity against bacteria, viruses, and fungi.8–10 Cellular mediators of the immune response are poorly characterized in amniotic fluid, and it remains unclear if the macrophages that are present serve a scavenging or an antimicrobial role. Neutrophils are usually absent from the amniotic fluid of a healthy fetus, and their presence typically signifies an inflammatory or infectious process.1
Biochemical and cellular analyses of amniotic fluid provide valuable information on chromosomal abnormalities, neural tube defects, prenatal infections, and most inborn errors of metabolism.11,12 Several amniotic fluid−based indices, including the lecithin-sphingomyelin ratio, the phosphatidylglycerol level, and the lamellar body count, are commonly used to assess fetal lung maturity.13,14 Bilirubin levels can be determined by measuring the optical density of amniotic fluid, which assists in the monitoring of fetal hemolysis. Estimation of the amniotic fluid levels of S100-β (a protein released from injured astrocytes) and cell-free fetal nucleic acids may serve as early screening tests for perinatal neurologic damage and fetal development, respectively.15,16 Finally, amniotic fluid is a valuable reservoir for cell types of multiple lineages at different maturational ages; approximately 1% of these cells are pluripotent, thereby representing a novel source of stem cells.17,18
Oxygen Supply and Transport
The fetus has almost no oxygen reserve and thus depends on maternal sources of oxygen delivery. Oxygen is an essential substrate for cell survival, because it is the final electron acceptor in the electron transport chain. When oxygen is scarce, the electron transport chain is compromised, resulting in decreased oxidative phosphorylation and adenosine triphosphate (ATP) production.19 Hypoxia ensues when the demand for oxygen exceeds the available supply, and it occurs more frequently in the presence of low oxygen tensions. In adult tissues, hypoxia occurs at oxygen tensions less than 20 mm Hg (normal, 40 mm Hg). By contrast, in fetal tissues, hypoxia occurs at oxygen tensions less than 17 mm Hg (normal, 20 to 25 mm Hg).20,21 This implies that fetal development occurs in an environment that exhibits a smaller margin of safety before reaching a state of oxygen insufficiency and highlights the importance of ensuring fetal oxygen delivery through the maintenance of adequate uteroplacental perfusion and fetal cardiac output. Ultimately, oxygenation of fetal tissues depends principally on the partial pressure of oxygen gradient between maternal and fetal blood as well as on the difference in the types of hemoglobin that exist in maternal and fetal blood.
Placental oxygen concentrations change with gestation. In early pregnancy, the placental intervillous space is free of maternal blood cells, thereby requiring the embryo to rely on endometrial secretions and maternal plasma for its energy requirements.22,23 The first trimester placenta has (1) an oxygen partial pressure (PO2) of approximately 20 mm Hg; (2) only a few capillaries, which are located mainly in the center of the mesenchymal core; and (3) a trophoblastic layer that is twice the thickness of that in the second trimester.24 Moreover, the fetal red blood cells are nucleated and the exocoelomic cavity does not contain an oxygen transport system, but rather it contains antioxidant molecules. These anatomic features, which limit the transfer of oxygen and the creation of free radicals, protect the highly sensitive embryo from the effects of oxidative stress.25 At the end of the first trimester, an exponential increase in fetal growth creates significant demands for oxygen and nutrients (Figure 5-1). In response, cytotrophoblastic cells interact with the smooth muscle of maternal spiral arteries, resulting in vessel dilation (Figure 5-2). This allows oxygen-rich maternal blood to flow to the placenta.26
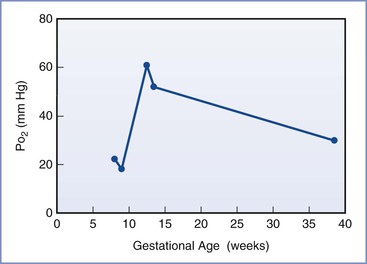
FIGURE 5-1 The mean oxygen partial pressure (PO2) throughout gestation in the human intervillous space. (Data from Jauniaux E, Kiserud T, Ozturk O, et al. Amniotic gas values and acid-base status during acute maternal hyperoxemia and hypoxemia in the early fetal sheep. Am J Obstet Gynecol 2000; 182:661-5; Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol 1992; 80:283-5; and Schaaps JP, Tsatsaris V, Goffin F, et al. Shunting the intervillous space: new concepts in human uteroplacental vascularization. Am J Obstet Gynecol 2005; 192:323-32.)
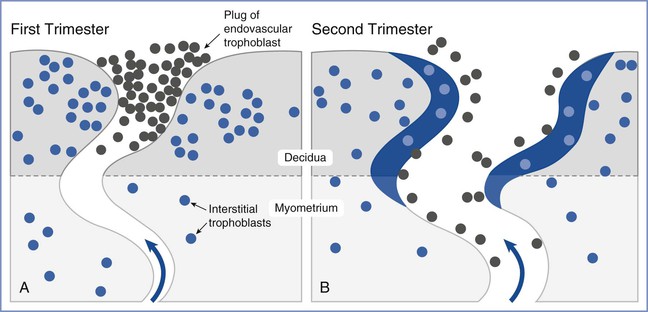
FIGURE 5-2 Invasion and remodeling of the spiral arteries by endovascular and interstitial extravillous trophoblasts. A, In the first trimester, the terminal portion of the spiral artery is blocked by a plug of endovascular trophoblast. Early placental and embryonic development occurs in a state of low oxygen tension, and nutrition at this early stage is derived from secretions from maternal endometrial glands. B, After 10 to 12 weeks’ gestation, the endovascular trophoblast plug dissolves and the endovascular trophoblast migrates into the myometrium, replacing endothelial cells, which undergo apoptosis. Maternal blood is now able to enter the intervillous space, the oxygen tension increases to 60 mm Hg, and nutrition changes from histotrophic to hemotrophic. (Modified from Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 2006; 27:939-58.)
The placenta acts as both a conduit and consumer of oxygen. The placenta is metabolically active and performs important roles in carbohydrate and amino acid metabolism, protein synthesis, and substrate transport. Almost 40% of the oxygen delivered to the pregnant uterus is needed to support the metabolic processes of the placenta.27 During periods of hypoxia, the placenta appears to alter its metabolism to diminish its consumption of oxygen, most likely by increasing glycolysis.28,29 This process can maintain fetal oxygen supply but, if ongoing, may result in fetal growth restriction (also known as intrauterine growth restriction). When the oxygen supply is compromised, the fetus shunts blood flow from peripheral tissues to vital organs (see later discussion), converts to greater use of anaerobic pathways, and undergoes an induction of gene expression that enables improved survival in a low-oxygen environment.19 The presence of fetal hemoglobin (hemoglobin F), with its greater affinity for oxygen than adult hemoglobin (see later discussion), and a hemoglobin concentration higher than that of adults (approximately 18 g/dL) result in a fetal arterial blood oxygen content that is only marginally lower than that in the adult, despite a lower oxygen tension.30
Glucose and Lactate Metabolism
Under normal conditions, gluconeogenesis does not occur to any significant extent in mammalian fetuses; the only source of glucose is that which is transferred across the placenta.31 Fetal glucose concentrations are linearly related to maternal concentrations over a range of 3 to 5 mmol/L (54 to 90 mg/dL; Figure 5-3); studies in isolated placentas suggest that this relationship continues up to a glucose concentration of 20 mmol/L (360 mg/dL).32 The placenta uses the majority of glucose delivered to the uterus for oxidation, glycogen storage, and conversion to lactate, with the remainder being transferred to the umbilical venous blood by facilitated, carrier-mediated diffusion. The amount of glucose supplied to the fetus appears more than adequate during normal conditions; ovine uterine blood flow must be reduced by greater than 50% before a decrease in fetal glucose uptake or fetal arterial glucose concentration is observed.33,34
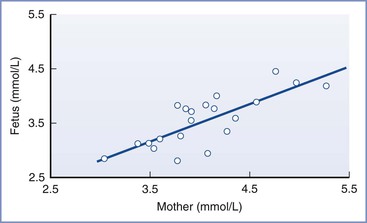
FIGURE 5-3 The linear relationship between maternal and fetal blood glucose concentrations during the third trimester. Fetal blood was obtained by percutaneous umbilical cord blood sampling. (From Kalhan SC. Metabolism of glucose and methods of investigation in the fetus and newborn. In Polin RA, Fox WW, editors. Fetal and Neonatal Physiology. Vol I. Philadelphia, WB Saunders, 1992:477-88.)
The umbilical cord blood glucose uptake is approximately 5 mg/kg/min at normal maternal arterial plasma glucose concentrations.35 Because the umbilical glucose/oxygen quotient varies from approximately 0.5 in sheep36 to 0.8 in human fetuses during labor,37 it is assumed that substrates other than glucose are used to support fetal oxidative metabolism; it is estimated that lactate and amino acids each provides approximately 25% of the total fetal energy requirements.38,39
Lactate is produced even in well-oxygenated fetal lambs, with total lactate production being approximately 4 mg/kg/min.40 Although the exact origin of fetal lactate is unclear, skeletal muscles and bones have been identified as sources of lactate production under resting conditions. Lactate production increases during episodes of acute hypoxemia, although this response may be blunted in fetuses previously exposed to oxidative stress.41 Lactate consumption occurs in the fetal myocardium and liver.42 Short-term exogenous lactate infusion in fetal lambs (sufficient to lower the pH to 7.20) results in transient fetal bradycardia and increased fetal breathing movements but no other adverse effects.43
Amino Acid and Lipid Metabolism
The fetus uses amino acids for protein synthesis, growth, and oxidation. Most maternal-to-fetal amino acid transfer occurs against a concentration gradient and involves energy-dependent transfer mechanisms. Under conditions in which fetal aerobic metabolism is decreased, amino acid uptake by the placenta and fetus may be reduced because it involves an expenditure of energy. Hypoxia results in a large reduction in nitrogen uptake in fetal lambs.44 During maternal fasting, fetal amino acid uptake does not change; however, enhanced fetal proteolysis may occur, which subsequently results in amino acid oxidation or gluconeogenesis.
Lipid products are transferred from the mother to the fetus. The fetus requires free fatty acids for growth, brain development, and the deposition of body fat for postnatal life. Fatty acids are transferred across the placenta by simple diffusion. Ketones are also transferred by simple diffusion; in humans, the maternal/fetal ketone ratio is approximately 2.0.45 The fetus can use ketones as lipogenic substrates or as energy substrates in the brain, kidney, heart, liver, and placenta.46 Beta-hydroxybutyrate (fatty acid) metabolism can occur in the placenta, brain, and liver during episodes of fetal hypoglycemia that result from maternal fasting.46 Cholesterol synthesis or free cholesterol diffusion does not appear to occur in the placenta.47 However, there is a significant correlation between maternal and fetal concentrations of lipoprotein(a), implying that diffusion of lipoprotein(a) may occur.47
Thermoregulation
Intrauterine fetal temperature largely depends on maternal temperature. However, owing to the high metabolic rate in the fetus, the net flow of heat is from the fetus to the mother. When compared with the mother during the third trimester, the fetus produces approximately twice as much heat (on a weight-adjusted basis) and maintains a temperature 0.5° C higher.48,49 This maternal-fetal difference in temperature remains relatively constant and is referred to as the “heat clamp.”50
The placental circulation is responsible for approximately 85% of the heat exchange between the mother and fetus. The remaining 15% is dissipated through the fetal skin and transferred through the amniotic fluid and the uterine wall to the maternal abdomen.51 As a consequence, fetal temperature may be rapidly affected by changes in umbilical blood flow; fetal temperatures rise quickly on occlusion of umbilical blood flow in both baboons and sheep.52,53 In humans, fetal temperatures increase during uterine contractions, which may be a result of intermittent obstruction of umbilical cord blood flow.54 Whether this rise in fetal temperature contributes to acute hypoxic-ischemic brain damage in the setting of umbilical cord prolapse is currently unknown. However, relatively small increases in temperature increase the sensitivity of the fetal brain to hypoxic injury (see Chapter 10).55
Although the fetus generates heat through high metabolic activity, the ability of the fetus to generate heat through thermogenic mechanisms is not developed until the end of gestation and is largely inactive in utero. Newborns are at high risk for rapid heat loss due to amniotic fluid evaporation and a sudden decrease in ambient temperature.49 They are not capable of significant heat production through shivering owing to their small muscle mass. As a consequence, nonshivering thermogenesis plays an important role in maintaining neonatal temperature. Nonshivering thermogenesis occurs in brown adipose tissue, which is unique from other adipocytes owing to the significant presence of mitochondria, fat vacuoles, sympathetic innervation, and blood vessels. In the mitochondria of brown adipose tissue, ATP production is uncoupled from the oxidative process, resulting in an increase in heat production and oxygen consumption.56 Nonshivering thermogenesis is inhibited in utero, most likely owing to the presence of adenosine and prostaglandin E2, which have strong antilipolytic actions on brown tissue.57–59 Inadequate oxygen levels and low levels of intrauterine catecholamines and thyroid hormones may also inhibit nonshivering thermogenesis. The inhibition of nonshivering thermogenesis is believed to be beneficial to the health of the fetus, in that it allows for conservation of fetal oxygenation and accumulation of brown adipose tissue.50
Fetal Cardiovascular System
The cardiovascular system is one of the first functional organ systems in the developing fetus. The morphologic development of the human heart, from its first appearance as a heart tube to its development as a four-chambered structure, occurs between 20 and 44 days’ gestation. Even before the development of the four-chambered heart, the valveless heart tube generates unidirectional flow, typically around 21 days’ gestation.
Circulatory Pattern
Fetal circulation differs significantly from the postnatal circulation. The fetal cardiovascular system is anatomically arranged in such a way as to allow blood to bypass the lungs and provide maximal perfusion of the placenta, where gas and nutrient exchange occur. The fetal systemic circulation receives cardiac output from both the left and the right ventricle, with the ventricles working in parallel. In contrast, during postnatal life, the left and right circulations are separated and the ventricles work in series.
Fetal blood flow is characterized by three anatomic communications between the left and right circulations: the ductus venosus, the foramen ovale, and the ductus arteriosus (Figure 5-4). Oxygenated blood travels from the placenta through the umbilical vein to the ductus venosus, which connects the umbilical vein with the inferior vena cava, thus bypassing the portal circulation and the liver. At mid gestation, approximately 30% of the umbilical venous blood is shunted through the ductus venosus; from 30 to 40 weeks’ gestation, this fraction decreases to approximately 20%, although a significant increase can occur in response to hypoxia (see later discussion).60 Once in the right atrium, oxygenated blood preferentially flows through the foramen ovale to the left atrium and left ventricle, before entering the aorta and the systemic circulation. This mechanism ensures the delivery of well-oxygenated blood to the brain and the heart, which are the two organs with the highest oxygen requirements. The preferential shunting of ductus venosus blood through the foramen ovale into the left atrium is related to the umbilical venous pressure and the portocaval pressure gradient.
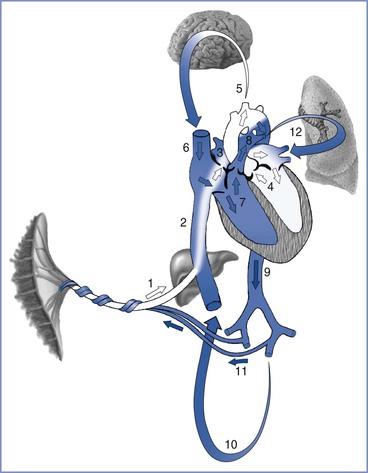
FIGURE 5-4 Oxygenated blood leaves the placenta via the fetal umbilical vein (1), enters the liver where flow divides between the portal sinus and the ductus venosus, and then empties into the inferior vena cava (2). Inside the fetal heart, blood enters the right atrium, where most of the blood is directed through the foramen ovale (3) into the left atrium and ventricle (4), and then enters the aorta. Blood is then sent to the brain (5) and myocardium, ensuring that these cells receive the highest oxygen content available. Deoxygenated blood returning from the lower extremities and the superior vena cava (6) is preferentially directed into the right ventricle (7) and pulmonary trunk. The majority of blood passes through the ductus arteriosus (8) into the descending aorta (9), which in turn supplies the lower extremities (10) and the hypogastric arteries (11). Blood returns to the placenta via the umbilical arteries for gas and nutrient exchange. A small amount of blood from the pulmonary trunk travels through the pulmonary arteries (12) to perfuse the lungs. Arrows in this figure depict the direction and oxygen content [white (oxygenated), blue (deoxygenated)] of the blood in circulation. (Drawing by Naveen Nathan, MD, Northwestern University Feinberg School of Medicine, Chicago, IL.)
Deoxygenated blood from the head and upper extremities enters the right atrium through the superior vena cava and is preferentially directed into the right ventricle and the pulmonary artery. Because fetal pulmonary vascular resistance is higher than systemic vascular resistance, the majority of pulmonary artery blood flow crosses the ductus arteriosus into the descending aorta, which in turn supplies the lower extremities and hypogastric arteries. Deoxygenated blood returns to the placenta via the umbilical arteries for gas and nutrient exchange; only a small percentage travels through the lungs into the left atrium, the left ventricle, and the ascending aorta.
At birth, the fetus undergoes a significant and abrupt transition to a state of physiologic independence (see Chapter 9). Clamping of the umbilical cord results in a sudden increase in systemic vascular resistance, whereas expansion of the lungs and an increased alveolar oxygen tension result in decreased pulmonary vascular resistance. This allows for greater blood flow through the lungs, resulting in a decrease in right atrial pressure and an increase in left atrial pressure, ultimately leading to the functional closure of both the foramen ovale and the ductus arteriosus.
Blood Volume
Human fetal intravascular volume is approximately 110 mL/kg, which is higher than that in postnatal life. However, approximately 25% of this blood volume is contained within the placenta; the blood volume within the fetal body is estimated to be approximately 80 mL/kg.61,62 Fetal intravascular volume is regulated through a complex interplay between the fetal heart, kidneys, and circulation and the placenta.63 The fetus can adapt more quickly to changes in intravascular volume than the adult, owing to higher diffusion rates between fetal compartments.64
Transplacental transfer of water from mother to fetus depends on hydrostatic and osmotic pressures. The hydrostatic pressure is determined by the difference in pressures between the maternal intervillous space or capillaries and the fetal capillaries. The osmotic pressure is mainly determined by the presence of plasma proteins (i.e., colloid osmotic pressure). Transplacental water transfer is further regulated by angiotensin II. Adamson et al.65,66 found that angiotensin II lowered the pressures in fetal placental exchange vessels, thereby promoting fluid transfer from the maternal to the fetal circulation. The production of angiotensin II is under control of the renin-angiotensin-aldosterone system in the fetal kidneys. A reduction in fetal arterial pressure results in an increase in fetal plasma renin activity, which results in subsequent increases in angiotensin I and II. The resulting expansion of intravascular volume augments fetal cardiac output and arterial pressure.
Cardiac Development
During gestation the fetal heart grows quickly and adapts to the continuously changing demands. The fetal myocardium grows primarily through cell division, whereas after delivery, cardiac mass increases as a result of cell enlargement.67 This growth correlates with a pre-birth transition from mononucleated cardiomyocytes, which contribute to heart growth by hyperplasia, to binucleated cardiomyocytes, which contribute to heart growth by hypertrophy.
The number of cardiac myofibrils and the transition in the type of cardiac troponins that are present during prenatal development can alter fetal heart contractility.68 The change from fetal to adult troponin is associated with decreased sensitivity of the contractile apparatus to calcium. A heightened calcium sensitivity is important in the early development of the fetal heart, when the sarcoplasmic reticulum is immature.69 With advancing gestational age, ejection fraction declines but cardiac output (per unit of fetal weight) does not change owing to increasing ventricular volume. The fetal heart rate decreases over the course of gestation from 140 to 150 beats per minute at 18 weeks’ gestation to 120 to 140 beats per minute at term.70,71
Ventricular Responses to Changes in Preload and Afterload
It is unclear whether fetal and adult hearts possess similar responses to preload and afterload. The adult heart responds in accordance to the Frank-Starling curve, which indicates that ventricular distention lengthens the diastolic fibers and results in augmented contractility. A number of studies have indicated that the fetal heart has a limited capacity to increase its stroke volume in response to an increase in preload (e.g., intravenous fluid infusion).72,73 By contrast, other studies have observed that the fetal heart can accommodate increases in preload and afterload in a manner consistent with the Frank-Starling curve.74,75 These seemingly contradictory findings may be partially explained if the fetal heart functions in vivo near the peak of the Frank-Starling curve. However, the left ventricular stroke volume is known to double at birth, which would not be in agreement with this hypothesis. A more plausible explanation is that ventricular constraint, arising from tissues that surround the heart (chest wall, pericardium, and lungs), limits fetal ventricular preload and overall cardiac function in utero. Relief of this constraint at birth, as a result of lung aeration and clearance of liquid from the lungs, may then allow for an increase in left ventricular preload and subsequent stroke volume in the newborn.76
Studies investigating the effects of afterload on fetal ventricular function have observed a significant decrease in right ventricular stroke volume in response to increases in arterial pressure.72 The same phenomenon occurs in the left ventricle, although to a lesser degree. In a study of fetal lambs, in which gradual constriction of the descending aorta was applied, stroke volume was maintained until high mean arterial pressures were achieved, after which decreases were observed. This decrease in stroke volume in the presence of high mean arterial pressure may represent the exhaustion of “preload reserve,” which will typically allow the maintenance of stroke volume in the setting of increased afterload.77
Cardiac Output and Distribution
In postnatal life, the right and left ventricles operate in series and their output is approximately equal; as a consequence, cardiac output is defined through measurements of output from either ventricle. However, in the fetus, the systemic circulation receives blood from both the left and right ventricle in parallel (i.e., the sum of the right and left ventricular outputs, with the exception of a proportion of the right ventricular output that is delivered to the fetal lungs). At mid gestation, the combined ventricular output (CVO) is approximately 210 mL, and it increases to approximately 1900 mL at 38 weeks’ gestation (500 mL/min/kg).73,78,79 During fetal life, the right ventricular volume is greater than the left ventricular volume during both systole and diastole, but stroke volume does not differ significantly between the two ventricles.70
Fetal cardiac output is sensitive to changes in fetal heart rate. As heart rate increases, cardiac output increases. As fetal heart rate decreases, fetal stroke volume increases only slightly, in part because of low fetal myocardial compliance. Although fetal bradycardia results in an extended diastolic filling time, the stiff fetal cardiac ventricles have limited ability to distend. Therefore, fetal bradycardia is associated with a marked drop in fetal cardiac output.
The distribution of the CVO in near-term fetal lambs and resting adult humans is shown in Figure 5-5. The fetal lamb CVO is distributed to the placenta (41%), the bone and skeletal muscle (38%), the gastrointestinal system (6%), the heart (4%), the brain (3%), and the kidneys (2%). In both fetal and adult animals, approximately equal volumes of blood are delivered to oxygen-uptake organs (i.e., the placenta before delivery, the lungs after delivery) and the oxygen-consuming organs.
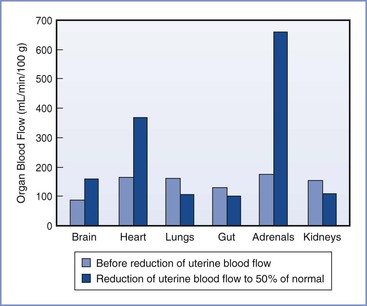
FIGURE 5-5 Redistribution of combined ventricular output in fetal lambs during hypoxemia caused by reduced uterine blood flow. (Modified from Jensen A, Roman C, Rudolph AM. Effects of reducing uterine blood flow on fetal blood flow distribution and oxygen delivery. J Dev Physiol 1991; 15: 309-23.)
The distribution of the CVO changes over the course of gestation and in certain conditions, such as hypoxia and hypovolemia. Interpretation of CVO data should be evaluated with the understanding that significant interspecies differences exist. For example, in humans the fetal lungs receive approximately 20% of CVO, whereas in the fetal lamb the lungs receive 10% or less of CVO. In human fetuses at 10 to 20 weeks’ gestation, the brain receives approximately 15% of CVO,80 but this fraction may be increased during circumstances of decreased placental perfusion, acidosis, and increased PCO2. In the rhesus monkey, the fraction of CVO devoted to cerebral blood flow was observed to increase from 16% to 31% during a hypoxic challenge.81
Fetal Blood Pressure
Fetal blood pressure increases with gestational age. Intracardiac (intraventricular) pressure recordings in the human fetus suggest that systolic pressure increases from 15 to 20 mm Hg at 16 weeks’ gestation to 30 to 40 mm Hg at 28 weeks’ gestation.67 Substantial variation in blood pressure may be observed. The diastolic ventricular pressures undergo similar, albeit slower and smaller increases, from 5 mm Hg or less at 16 to 18 weeks’ gestation to 5 to 15 mm Hg at 19 to 26 weeks’ gestation.67
Regulation of Fetal Circulation
Fetal cardiovascular function continuously adapts to varying metabolic and environmental conditions through regulation by the neurologic and endocrine systems. The predominant form of neuroregulation occurs in response to baroreceptor and chemoreceptor afferent input to the autonomic nervous system and through modulation of myocardial adrenergic receptor activity. Thus, the autonomic nervous system functions to reversibly redirect blood flow and oxygen delivery as required.
Arterial baroreceptor function has been demonstrated in several different fetal animal models. The predominant baroreceptors are located within the vessel walls of the aortic arch and at the bifurcation of the common carotid arteries. These receptors project signals to the vasomotor center in the medulla, from which autonomic responses emanate. The baroreceptors are functional early in fetal development and undergo continuous adaptation to the increases in blood pressure observed over time.82 A sudden increase in fetal mean arterial pressure—as occurs with partial or complete occlusion of the umbilical arteries—results in cholinergic stimulation and subsequent fetal bradycardia.
Peripheral chemoreceptors are present within the vessel walls of the aortic arch and at the bifurcation of the common carotid arteries. In some animal species, peripheral chemoreceptors are transiently present in the adrenal gland but disappear after birth.83 The fetal aortic chemoreceptors are responsive to even small changes in arterial oxygenation,84,85 which contrasts to the less active fetal carotid chemoreceptors. Dawes et al.86 concluded that the carotid chemoreceptors are important for postnatal respiratory control, whereas the aortic chemoreceptors are more involved in the control of cardiovascular responses and the regulation of oxygen delivery. Central chemoreceptors, located in the medullar oblongata, appear to play little if any role in fetal circulatory responses.
The neural control of the fetal circulation is far more dependent on chemoreceptor-mediated responses than neural control of the adult circulation.87 Acute fetal hypotension often stimulates a reflex response, which can include both bradycardia and vasoconstriction. Vasoconstriction is dependent on increases in both sympathetic autonomic activity and the rate of secretion of several vasoactive hormones, including arginine vasopressin, renin, angiotensin, and aldosterone. Fetal bradycardia is most likely caused by activation of peripheral chemoreceptors.87
Autonomic Nervous System
The autonomic nervous system is present early in gestation and plays a critical role in maintaining cardiovascular homeostasis. In the fetal chick heart, evidence of cholinergic innervation occurs as early as 3 days after fertilization (average incubation, 22 days). In the mammalian heart, inotropic and chronotropic responses to adrenergic agents have been measured as early as 4 to 5 weeks’ gestation,88 and the fetal myocardial pacemaker can be inhibited at this time by the cholinergic agonists carbamylcholine and acetylcholine.89
In comparing the parasympathetic cholinergic and sympathetic adrenergic nervous systems during gestation, the majority of studies indicate that the parasympathetic system appears earlier (8 weeks’ gestation versus 9 to 10 weeks’ gestation),88,90,91 becomes more dominant as pregnancy progresses, and is more functionally complete at birth (Figure 5-6). As a result, the baseline fetal heart rate is slower at term than at 26 weeks’ gestation. The administration of atropine can result in fetal tachycardia by 15 to 17 weeks’ gestation, which occurs before fetal bradycardia can be demonstrated with the administration of a beta-adrenergic receptor antagonist.88
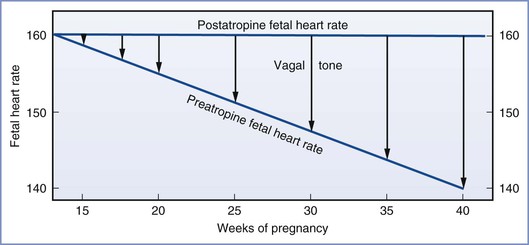
FIGURE 5-6 The growing influence of the parasympathetic nervous system on fetal heart rate as gestation progresses. This parasympathetic activity is reversible with administration of atropine. (From Schifferli P, Caldeyro-Barcia R. Effects of atropine and beta-adrenergic drugs on the heart rate of the human fetus. In Boréus LO, editor. Fetal Pharmacology. New York, Raven Press, 1973:264.)
Both parasympathetic and sympathetic systems undergo significant maturation during postnatal life, and full maturation of the vagal response is not observed until 1 to 2 months after delivery.92–94 Similarly, although the contractile response of the fetal vasculature is less functional than the adult response, the fetal administration of an alpha-adrenergic agonist can result in the redistribution of blood flow away from the kidneys, skin, and splanchnic organs and toward the heart, brain, placenta, and adrenal glands.95 At birth, the autonomic nervous system can mediate a number of hemodynamic adjustments, including changes in heart rate and peripheral vascular resistance, as well as a redistribution of blood flow.88
Fetal Pulmonary System
The lungs begin as small, saccular outgrowths of the ventral wall foregut. Although sacculi with type I and II pneumocytes and ventilatory capacity are present during the last trimester, true alveoli develop at approximately 36 weeks’ gestation. The majority of alveolar development occurs postnatally, within the first 6 to 18 months of life, when further maturation of the microvasculature and the air-blood barrier occurs.96
The pulmonary vasculature develops early in gestation, with continuous circulation being documented at 34 days’ gestation.97,98 The size and number of pulmonary arteries and veins increases over time; however, vessel reactivity to local and hormonal influences is not detectable until after 20 weeks’ gestation.99,100 From 20 to 30 weeks’ gestation, an increase in the size of the pulmonary vascular bed combined with a decrease in the pulmonary vascular resistance results in an increase in pulmonary blood flow (i.e., from 10% to 15% of the CVO to 25% of the CVO). During this time, alterations in maternal oxygenation have no effect on the fetal pulmonary vasculature.80,99 However, after 30 weeks’ gestation, blood flow to the lung decreases slightly owing to a significant increase in pulmonary vascular resistance, diminishing the fraction of CVO to approximately 20%. Contemporaneously, the vasomotor tone and reactivity of the fetal circulation begins to respond to maternal hyperoxygenation with a decrease in pulmonary vascular resistance and an increase in pulmonary blood flow.80,99 A study in near-term fetal lambs observed a 10-fold increase in pulmonary blood flow when fetal oxygen tension was increased from 24 to 46 mm Hg101; this finding emphasizes the importance of ventilation and oxygenation in the newborn to assist in the transition to postnatal circulation.
The reduction in pulmonary vascular resistance at birth is also attributed to a number of mechanical and molecular processes. In utero, the fetal lungs are filled with fluid to maintain an appropriate level of expansion for normal pulmonary development.102 The expulsion of lung liquid, particularly with a vaginal birth, likely decreases extraluminal pressure on the pulmonary vasculature and leads to a decrease in pulmonary vascular resistance.103 Breathing movements, shear stress created by an abrupt surge in pulmonary blood flow, and the creation of alveolar surface tension are other mechanical factors that can decrease pulmonary vascular resistance.104 Finally, the relative predominance of vasodilators (e.g., endothelium-derived nitric oxide, prostacyclin) versus vasoconstrictors (e.g., platelet activating factor) at birth may also significantly decrease pulmonary vascular resistance.105–109
The pulmonary surfactant system is one of the last systems to develop before birth.110 Surfactant is a lipoprotein complex (phospholipoprotein) that reduces and regulates the surface tension at the air-liquid interface to prevent alveolar collapse and reduces the work associated with breathing.111 Pulmonary surfactant is composed predominantly (> 90%) of lipids (i.e., phospholipids and neutral lipids [primarily cholesterol]), with the remaining fraction being proteins.112,113 Surfactant assembly occurs in the endoplasmic reticulum and the Golgi apparatus of the type II alveolar cells, and it is stored in the lamellar bodies. The primary stimuli for surfactant secretion (i.e., exocytosis from the lamellar bodies) are signals from the autonomic nervous system (β2-adrenergic receptor mediated) and mechanical factors (e.g., stretching of the basement membrane of alveolar type II cells with ventilation).114 Dipalmitoylphosphatidylcholine, an important component of surfactant, is present in amniotic fluid and can be found in alveolar lavage samples from human fetuses between 24 and 28 weeks’ gestation.
The amount and composition of surfactant changes over the course of gestation. For example, the ratio of phosphatidylglycerol to phosphatidylinositol, as well as the ratio of lecithin to sphingomyelin, increases with gestation and may be used as markers of fetal lung maturity.115–117 Fetal surfactant production can be accelerated by a number of factors, including glucocorticoids, thyroid hormones, and autonomic neurotransmitters. The maternal administration of glucocorticoids such as betamethasone or dexamethasone has been associated with a 35% to 40% reduction in respiratory distress syndrome in preterm infants.118
Fetal Renal System
Although fluid and electrolyte balance, as well as acid-base homeostasis, are primarily regulated and maintained by the placenta, the fetal kidneys play an important role in fetal development through amniotic fluid production. Fetal glomeruli begin to develop at 8 to 9 weeks’ gestation and start producing urine at 10 weeks’ gestation, which contributes significantly to amniotic fluid production.119,120 By 20 weeks’ gestation, greater than 90% of amniotic fluid is provided by the kidneys. Fetal oliguria and anuria can lead to lung hypoplasia and skeletal and tissue deformities (e.g., Potter sequence).121 Glomerular filtration rate (GFR) increases over the course of gestation but remains low during fetal and early neonatal life. At birth, term newborns have a GFR of approximately 20 mL/min/1.73m2,122,123 which increases to approximately 50 mL/min/1.73 m2 by 1 month of age.123 This early increase in GFR is believed to result from a large increase in the glomerular capillary surface area and the ultrafiltration coefficient, together with a small increase in the filtration pressure.124,125 Thereafter, the GFR undergoes progressive increases and reaches adult levels between 1 and 2 years of age.126
The ability of the fetal kidneys to perform filtration, reabsorption, and secretion (i.e., tubular function) begins by 14 weeks’ gestation and continues to develop postnatally. Immaturity of tubular function in preterm infants can lead to acidosis and salt wasting.127,128 Renal function in utero is regulated by a variety of factors that control renal blood flow, glomerular filtration, and tubular function. The renin-angiotensin system is particularly important for normal fetal renal growth and development129; angiotensin II helps regulate blood pressure as well as the volume of fluid in the extravascular space.130
Fetal Hematologic System
Red blood cells, platelets, neutrophils, monocytes, and macrophages are all derived from a common progenitor cell.131 In the developing embryo, hematopoiesis occurs at several anatomic sites in multiple waves. The first wave occurs in the yolk sac and produces mostly primitive erythroid cells, but also macrophages and megakaryocytes. The second wave also arises in the yolk sac but creates the same cells found in the adult human (i.e., erythroid, megakaryocyte, and several myeloid lineages). The third wave emerges from hematopoietic stem cells located within the major arteries of the embryo, yolk sac, and placenta. Hematopoietic stem cells migrate to the fetal liver and eventually seed the bone marrow. The final wave of hematopoiesis produces all hematopoietic cell lineages, including B- and T-lymphocyte progenitor cells.132,133
Erythroid (red blood cells) are the first blood cells to develop. There are two developmentally and morphologically distinct erythroid lineages: primitive (embryonic) and definitive (adult). Cells of the primitive lineage support the transition from the rapidly growing embryo to fetus; primitive megaloblastic erythrocytes are much larger than definitive erythrocytes, express different globin genes, and differ in their oxygen-carrying capacity and response to low oxygen tension. By contrast, definitive erythrocytes function during the transition from fetal to extrauterine life at birth are produced continuously from hematopoietic stem cells in the bone marrow and participate in a variety of normal physiologic processes throughout postnatal life.131,134
Fetal and adult human erythrocytes can be distinguished by their hemoglobin (hemoglobin F and A, respectively). The tetramer for hemoglobin F consists of two alpha (α) chains and two gamma (γ) chains (α2γ2), whereas the tetramer for hemoglobin A includes two alpha (α) chains and two beta (β) chains (α2β2). The gamma chain and the beta chain contain the same number of amino acids (146), but their sequences differ by a total of 39 amino acids.135 The change in expression from fetal to adult beta-globin genes begins at approximately 32 weeks’ gestation and is completed after birth.136
Hemoglobin F has a greater affinity for oxygen and a lower affinity for 2,3-disphosphoglycerate (DPG) and exhibits a leftward shift in the oxyhemoglobin dissociation curve compared with hemoglobin A (Figure 5-7).137–139 These differences result in greater arterial oxygen saturation in fetal versus maternal blood for any given arterial oxygen pressure. This difference in oxygen affinity can be explained by a decreased interaction between the gamma chains of hemoglobin F and intraerythrocyte 2,3-DPG, which acts to lower oxygen affinity by binding and stabilizing the deoxygenated hemoglobin tetramer. As a consequence, 2,3-DPG decreases the oxygen affinity of hemoglobin F less than that of hemoglobin A.140,141 Although fetuses and adults have similar intraerythrocyte 2,3-DPG concentrations, fetal blood exhibits a lower oxygen tension at which hemoglobin is 50% saturated (P50). Hemoglobin F levels begin to decrease toward the end of pregnancy, resulting in a corresponding increase in the P50. At term, hemoglobin A accounts for approximately 25% of total hemoglobin and the P50 is approximately 19 mm Hg.142,143
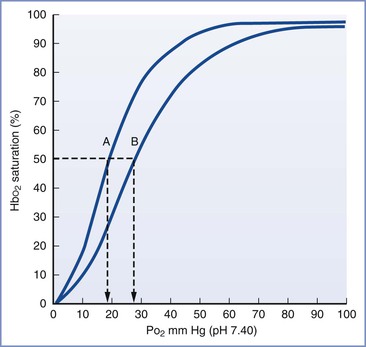
FIGURE 5-7 Oxyhemoglobin saturation curves for fetal (A) and adult (B) human blood. The P50 is indicated by the dashed vertical line. (Modified from Delivoria-Papadopoulos M, DiGiacomo JE. Oxygen transport. In Polin RA, Fox WW, editors. Fetal and Neonatal Physiology. Vol 1. Philadelphia, WB Saunders, 1992:807.)
Hemoglobin A levels begin to increase and 2,3-DPG concentrations transiently increase above usual fetal and adult levels during the first few months of life. During this time, the affinity of neonatal blood for oxygen is equivalent to that of the adult despite the persistence of 25% fetal hemoglobin.137,142,143
Fetal Gastrointestinal System
The gastrointestinal tract develops from the primitive digestive tube, which includes the foregut, midgut, and hindgut. The foregut receives its vascular supply from the celiac axis and gives origin to the oral cavity, pharynx, esophagus, stomach, and upper duodenum. The midgut, which receives its vascular supply from the superior mesenteric artery, develops into the distal duodenum, jejunum, ileum, cecum, appendix, and transverse colon. The hindgut receives its vascular supply from the inferior mesenteric artery, and it differentiates into the descending colon, the sigmoid colon, and the upper two thirds of the rectum.144 Intestinal villi appear by 7 weeks’ gestation, and active absorption of glucose and amino acids occurs by 10 and 12 weeks’ gestation, respectively.145 Peristaltic waves and gastrointestinal motility are initiated by 8 weeks’ gestation. Teniae, the three longitudinal ribbons of smooth muscle on the outside of the colon, appear by 12 weeks’ gestation and contract to produce the haustra (bulges) in the colon.146 In the small intestine, Auerbach’s and Meissner’s plexuses of parasympathetic nerves provide motor and secretomotor innervation, respectively; the two plexuses are present as early as 8 weeks’ gestation.145 Aggregations of lymphoid nodules (i.e., Peyer patches) develop by 20 weeks’ gestation in the ileum.147
Swallowing
The fetus starts swallowing at approximately 15 weeks’ gestation, and at term the fetus ingests 500 to 750 mL of amniotic fluid per day.148 Fetal swallowing plays an important role in amniotic fluid homeostasis,148 and the swallowed fluid appears to provide nutritional support for mucosal development within the gastrointestinal tract.149 Avila et al.150 found that surgical obstruction of ingested fluid within the upper gastrointestinal tract resulted in restricted development of the gastrointestinal tract, liver, and pancreas. In addition, the ingestion and intestinal absorption of nutrient-rich amniotic fluid appears to play an important role in general fetal growth and development. In the fetal rabbit model, disorders of the upper gastrointestinal tract (e.g., esophageal obstruction, gastroschisis) lead to decreased intestinal nutrient absorption and decreased birth weight and crown-rump length.151,152 Similar findings have been reported in human neonates with congenital esophageal atresia.153
Meconium
Meconium, which consists of water, intestinal secretions, squamous cells, lanugo hair, bile pigments, and blood, first appears in the fetal intestine between 10 and 12 weeks’ gestation.154 By 16 weeks’ gestation, meconium moves into the colon.155 Between 14 and 22 weeks’ gestation, fetal colonic contents, as indicated by the presence of high levels of intestinal enzymes (disaccharidases, alkaline phosphatase), appear in the amniotic fluid.156 After 22 weeks’ gestation, a subsequent decline in the concentration of these gastrointestinal enzymes within the amniotic fluid is observed, which coincides with the development of anal sphincter tone.156,157
Meconium is continually cleared by fetal swallowing, leading to relatively clear amniotic fluid in the majority of pregnancies. The presence of meconium-stained amniotic fluid may therefore represent either decreased meconium clearance or increased meconium passage, which is observed in the presence of fetomaternal stress factors such as hypoxia and infection, independent of fetal maturation.154 Meconium-stained amniotic fluid occurs more frequently with advanced gestational age and is common in post-term pregnancies.158
Although many fetuses with meconium-stained amniotic fluid are born without adverse sequelae, meconium can have detrimental effects on fetal organs and the placenta. Meconium may cause umbilical cord vessel constriction, vessel necrosis, and the production of thrombi, which can lead to altered coagulation, cerebral palsy, and neonatal seizures.159 In addition, meconium may reduce the antibacterial properties of amniotic fluid by altering zinc levels.154 Fetal aspiration of meconium also may neutralize the action of surfactant, promote lung tissue inflammation through the activation of neutrophils, and possibly result in meconium aspiration syndrome (see Chapter 9). Finally, in the presence of perinatal hypoxia, meconium also may contribute to vascular hypertrophy and possible pulmonary hypertension.154
Fetal Nervous System
Over the course of gestation, the human brain and central nervous system begin to develop from a few embryonic cells to a complex system in which billions of neurons are arranged and interconnected; small, seemingly minor changes may have profound implications. For example, animal studies suggest that intrauterine exposure to a variety of anesthetic agents at specific time intervals appears to result in anatomic, functional, and behavioral changes following birth (see Chapter 10).
Structural and Functional Brain Development
Primary neuromodulation and neural tube formation occur by 4 weeks’ gestation. Between 8 and 12 weeks’ gestation, prosencephalon development is initiated, which is accompanied by neuronal proliferation and migration. Simultaneously, the subplate layer is created to fulfill a critical, albeit transient, role as a location for synapses with cortical and thalamic projections; the subplate layer disintegrates between 24 and 28 weeks’ gestation. A significant increase in cortical development, organization, and synapse formation begins by 20 weeks’ gestation and continues postnatally; during the third trimester alone, the cerebral cortex volume increases fourfold.160–163
The first fetal movements are witnessed near the end of the first trimester. These initial movements have simple patterns and originate from spontaneous discharges within the spine and brainstem. The fetal movements become more organized and complex as the pregnancy progresses, with higher brain centers modulating the activity of the brainstem and spine.
The exact onset of electrocortical activity is unknown, but electroencephalographic (EEG) activity can be recorded in preterm infants as early as 24 weeks’ gestation. Fetal EEG activity differs from that in the adult and is characterized by the presence of intermittent bursts of activity separated by periods of complete suppression. With maturation, these suppressed episodes become shorter and less frequent before completely disappearing in postnatal life. The early electrical activity within the nervous system controls several developmental processes, such as neuronal differentiation, migration, synaptogenesis, and formation of neuronal networks. For example, the initial spontaneous spinal and subcortical discharges are believed necessary for somatosensory development. As they elicit movements in the periphery, afferent signals establish topographic representation on the sensory cortex.164–166
Cerebral Metabolism
The immature brain, similar to the adult brain, relies mostly on oxidative metabolism for the production of energy. However, owing to the limited capacity for mitochondrial oxidative phosphorylation and the lower partial pressures of oxygen observed in utero, anaerobic glycolysis exhibits a greater role during this developmental period than after delivery.167,168 In the presence of aerobic conditions, glucose is converted to pyruvic acid (glycolysis), which enters the Krebs cycle and the mitochondrial cytochrome system to create chemical energy; this process converts 1 mole of glucose into 36 moles of ATP. By contrast, during anaerobic conditions, glycolysis is much less efficient, yielding only 2 moles of ATP for each mole of glucose.169
Although glucose represents the primary and predominant source of cerebral energy, the perinatal brain is uniquely capable of metabolizing other substrates, such as lactic acid and ketone bodies (β-hydroxybutyrate and acetoacetate). Lactic acid concentrations in the peripartum period are significantly elevated and may support over 50% of total cerebral oxidative metabolism in certain conditions such as hypoglycemia and hypoxia.170,171 During hypoxic conditions, the fetal brain will also significantly decrease its energy consumption, as evidenced by fewer fetal movements and a slower EEG wave pattern.172
Cerebral Blood Flow
The development of the neural tube begins with formation of endothelium-lined vascular channels; by 10 weeks’ gestation, an extensive network of leptomeningeal arteries covers the fetal brain, allowing vessels to sprout and penetrate the brain parenchyma. Subsequent vascular growth is most pronounced in rapidly developing areas of the brain.167
The fetal systemic circulation has unique features that ensure optimal oxygen delivery to the brain. Well-oxygenated blood from the umbilical vein and ductus venosus is preferentially shunted through the foramen ovale to the left side of the heart and the ascending aorta to supply the cerebral and coronary circulations. Hypoxia results in acute changes in fetal and placental vascular resistance, which leads to intense peripheral vasoconstriction (likely mediated by stimulation of chemoreceptors) and further shunting of umbilical venous blood through the ductus venosus. The fetal circulatory system is much more sensitive to hypoxemia than that in the adult, which helps maintain oxygen delivery to the developing brain and myocardium (Figure 5-8).173–175
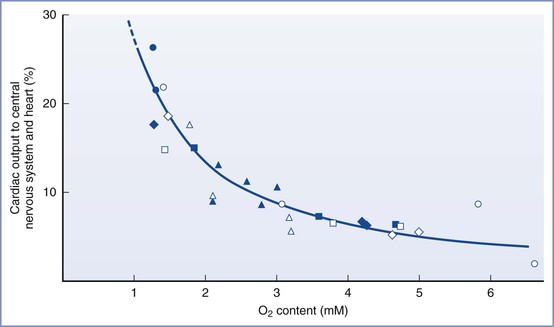
FIGURE 5-8 The redistribution of cardiac output to the heart and central nervous system during hypoxemia in fetal lambs. Each symbol represents a measurement from an individual fetal lamb. (Modified from Sheldon RE, Peeters LLH, Jones MD Jr, et al. Redistribution of cardiac output and oxygen delivery in the hypoxic fetal lamb. Am J Obstet Gynecol 1979; 135:1071-8.)
The redistribution of blood flow to the most actively developing regions of the fetal brain is at least partially the result of an adenosine-mediated mechanism. Adenosine, the breakdown product of ATP, accumulates during failure of ATP resynthesis and causes vasodilation of blood vessels and suppression of neuronal activity.172 Other substances (e.g., nitric oxide, endogenous opioids, adrenomedullin) may also play a role in cerebral blood redistribution, but the exact mechanisms are incompletely understood.176
Nociception
Cutaneous sensory receptors are present in the human fetus at approximately 7 weeks’ gestation, and a widespread network is established by 20 weeks. At term gestation, the density of cutaneous nociceptive receptors in the fetus is comparable to, and may even exceed, that of the adult. Although the development of sensory fiber-to-dorsal horn interneuron synapses has been reported to occur as early as 6 weeks’ gestation,177 differentiation of dorsal horn neurons begins at approximately 13 weeks’ gestation; the laminar arrangement of dorsal horn neurons, replete with synaptic interconnections and neurotransmitter vesicles, is present in some regions of the spinal cord by 30 weeks’ gestation.178 At this time, the A-delta and C fibers make connections at the spinal cord level and with the surrounding dermatomes.
The neurons of the cerebral cortex develop by 20 weeks’ gestation, and synaptogenesis of the thalamocortical connections is established between 20 and 24 weeks’ gestation. Thalamocortical axons reach the somatosensory cortex at 24 to 26 weeks’ gestation. Myelination of the pain pathways of the spinal cord and brainstem is completed during the second and third trimesters of gestation179; however, the process continues postnatally in other areas of the brain and in peripheral nerve fibers. Although optimal pain processing requires myelination of pain pathways, cortical maturation, dendritic arborization, and thalamocortical fiber synaptogenesis, it is unclear when nociception, the capacity to feel pain, develops within the fetus. As early as 18 weeks’ gestation, human fetuses demonstrate pituitary-adrenal, sympathoadrenal, and circulatory stress responses to noxious stimuli.180–182 In studies of intrauterine blood transfusion in the human fetus, surgical needling of the intrahepatic vein (compared with needling of the insensate umbilical cord) is associated with evidence of a stress response, including increases in plasma beta-endorphin and cortisol levels and a diminution in the middle cerebral artery pulsatility index.183 Administration of fentanyl 10 µg/kg blunts this stress response to intrahepatic needling.184
Near-infrared spectroscopy has demonstrated cortical activity in response to noxious stimuli in preterm neonates born and studied as early as 25 weeks’ postmenstrual age.185,186 Similarly, facial responses to painful stimuli (similar to those seen in adults) can be provoked in preterm neonates born and assessed as early as 25 weeks’ gestation, which suggests the development of functional pathways from the spinal cord to the brain.187,188 However, the withdrawal from noxious stimuli or an increased release of stress hormones does not necessarily reflect an awareness of pain, because local spinal reflexes and hormonal release can occur without cortical involvement.189 The experience of pain is a conscious subjective experience with emotional and affective components that requires higher-level cortical processing. Nociceptive processing begins in the peripheral neurons, which relay signals through the spinothalamic tract, the thalamus, and ultimately the cerebral cortex, where conscious perception of pain occurs.190
After birth, neonates appear to be more sensitive to pain, with lower pain thresholds, poor discriminative abilities, and a greater tendency to exhibit central sensitization in response to later noxious stimuli than adults. Early sensory experiences in the neonate can influence the development of nociceptive pathways.191 Neonates and especially preterm infants who undergo numerous procedures in the neonatal intensive care unit and/or surgery have been observed to demonstrate altered pain perceptions later in life.192 In the rodent model, tissue injury in early neonatal life results in an increased magnitude and duration of hyperalgesia after reinjury in later life, compared with those with no early life pain experience.191 Collectively, these observations have prompted some investigators to conclude that noxious events in neonates, when pain pathways are still undergoing a learning or “tuning process,” may result in structural functional and behavioral alterations in adult pain processing. Some of these long-term consequences may be attenuated by preemptive analgesia.193
The foregoing neuroanatomic and neurochemical evidence, in addition to the well-characterized behavioral and physiologic responses to pain, indicate that both the fetus and newborn infant have nociceptive pathways capable of communicating nociceptive stimuli from the periphery to the cerebral cortex and regulating the response via efferent inhibitory pathways. Current evidence suggests that fetal nociception at the level of the cortex occurs after the midpoint of pregnancy (i.e., between 24 and 30 weeks’ gestation). Of note, maternal administration of general anesthesia does not guarantee the presence of fetal anesthesia or analgesia (see Chapter 7). For example, most infants are clearly awake and cry loudly immediately after cesarean delivery during maternal administration of general anesthesia.
References
1. Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: not just fetal urine anymore. J Perinatol. 2005;25:341–348.
2. Ross MG, Brace RA. National Institute of Child Health and Development Conference summary: amniotic fluid biology—basic and clinical aspects. J Matern Fetal Med. 2001;10:2–19.
3. Brace RA, Wolf EJ. Normal amniotic fluid volume changes throughout pregnancy. Am J Obstet Gynecol. 1989;161:382–388.
4. Gilbert WM, Brace RA. Amniotic fluid volume and normal flows to and from the amniotic cavity. Semin Perinatol. 1993;17:150–157.
5. Lind T, Billewicz WZ, Cheyne GA. Composition of amniotic fluid and maternal blood through pregnancy. J Obstet Gynaecol Br Commonw. 1971;78:505–512.
6. Gillibrand PN. Changes in the electrolytes, urea and osmolality of the amniotic fluid with advancing pregnancy. J Obstet Gynaecol Br Commonw. 1969;76:898–905.
7. Hirai C, Ichiba H, Saito M, et al. Trophic effect of multiple growth factors in amniotic fluid or human milk on cultured human fetal small intestinal cells. J Pediatr Gastroenterol Nutr. 2002;34:524–528.
8. Akinbi HT, Narendran V, Pass AK, et al. Host defense proteins in vernix caseosa and amniotic fluid. Am J Obstet Gynecol. 2004;191:2090–2096.
9. Espinoza J, Chaiworapongsa T, Romero R, et al. Antimicrobial peptides in amniotic fluid: defensins, calprotectin and bacterial/permeability-increasing protein in patients with microbial invasion of the amniotic cavity, intra-amniotic inflammation, preterm labor and premature rupture of membranes. J Matern Fetal Neonatal Med. 2003;13:2–21.
10. Yoshio H, Tollin M, Gudmundsson GH, et al. Antimicrobial polypeptides of human vernix caseosa and amniotic fluid: implications for newborn innate defense. Pediatr Res. 2003;53:211–216.
11. Kramer K, Cohen HJ. Intrauterine fetal diagnosis for hematologic and other congenital disorders. Clin Lab Med. 1999;19:239–253.
12. Wilson RD. Amniocentesis and chorionic villus sampling. Curr Opin Obstet Gynecol. 2000;12:81–86.
13. Lewis PS, Lauria MR, Dzieczkowski J, et al. Amniotic fluid lamellar body count: cost-effective screening for fetal lung maturity. Obstet Gynecol. 1999;93:387–391.
14. Dubin SB. Assessment of fetal lung maturity. Practice parameter. Am J Clin Pathol. 1998;110:723–732.
15. Michetti F, Gazzolo D. S100B protein in biological fluids: a tool for perinatal medicine. Clin Chem. 2002;48:2097–2104.
16. Hui L, Bianchi DW. Cell-free fetal nucleic acids in amniotic fluid. Hum Reprod Update. 2011;17:362–371.
18. Shaw SW, David AL, De Coppi P. Clinical applications of prenatal and postnatal therapy using stem cells retrieved from amniotic fluid. Curr Opin Obstet Gynecol. 2011;23:109–116.
19. Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med. 2010;10:653–666.
20. Gross MW, Karbach U, Groebe K, et al. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer. 1995;61:567–573.
21. Raleigh JA, Calkins-Adams DP, Rinker LH, et al. Hypoxia and vascular endothelial growth factor expression in human squamous cell carcinomas using pimonidazole as a hypoxia marker. Cancer Res. 1998;58:3765–3768.
22. Burton GJ, Watson AL, Hempstock J, et al. Uterine glands provide histotrophic nutrition for the human fetus during the first trimester of pregnancy. J Clin Endocrinol Metab. 2002;87:2954–2959.
23. Foidart JM, Hustin J, Dubois M, Schaaps JP. The human placenta becomes haemochorial at the 13th week of pregnancy. Int J Dev Biol. 1992;36:451–453.
24. Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80:283–285.
25. Schneider H. Oxygenation of the placental-fetal unit in humans. Respir Physiol Neurobiol. 2011;178:51–58.
26. Jauniaux E, Watson A, Burton G. Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks’ gestation. Am J Obstet Gynecol. 2001;184:998–1003.
27. Carter AM. Placental oxygen consumption. Part I: in vivo studies—a review. Placenta. 2000;21(Suppl A):S31–S37.
28. van Patot MC, Ebensperger G, Gassmann M, Llanos AJ. The hypoxic placenta. High Alt Med Biol. 2012;13:176–184.
29. Kay HH, Zhu S, Tsoi S. Hypoxia and lactate production in trophoblast cells. Placenta. 2007;28:854–860.
30. Mollison PL, Veall N, Cutbush M. Red cell and plasma volume in newborn infants. Arch Dis Child. 1950;25:242–253.
31. Kalhan SC, D’Angelo LJ, Savin SM, Adam PA. Glucose production in pregnant women at term gestation: sources of glucose for human fetus. J Clin Invest. 1979;63:388–394.
32. Hauguel S, Desmaizieres V, Challier JC. Glucose uptake, utilization, and transfer by the human placenta as functions of maternal glucose concentration. Pediatr Res. 1986;20:269–273.
33. Oh W, Omori K, Hobel CJ, et al. Umbilical blood flow and glucose uptake in lamb fetus following single umbilical artery ligation. Biol Neonate. 1975;26:291–299.
34. Wilkening RB, Battaglia FC, Meschia G. The relationship of umbilical glucose uptake to uterine blood flow. J Dev Physiol. 1985;7:313–319.
35. Hay WW Jr, Sparks JW, Wilkening RB, et al. Fetal glucose uptake and utilization as functions of maternal glucose concentration. Am J Physiol. 1984;246:E237–E242.
36. Boyd RD, Morriss FH Jr, Meschia G, et al. Growth of glucose and oxygen uptakes by fetuses of fed and starved ewes. Am J Physiol. 1973;225:897–902.
37. Morriss FH Jr, Makowski EL, Meschia G, Battaglia FC. The glucose/oxygen quotient of the term human fetus. Biol Neonate. 1974;25:44–52.
38. Gresham EL, James EJ, Raye JR, et al. Production and excretion of urea by the fetal lamb. Pediatrics. 1972;50:372–379.
39. Burd LI, Jones MD Jr, Simmons MA, et al. Placental production and foetal utilisation of lactate and pyruvate. Nature. 1975;254:710–711.
40. Battaglia FC, Meschia G. An Introduction to Fetal Physiology. Academic Press: Orlando, FL; 1986.
41. Gardner DS, Giussani DA, Fowden AL. Hindlimb glucose and lactate metabolism during umbilical cord compression and acute hypoxemia in the late-gestation ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2003;284:R954–R964.
42. Sparks JW, Hay WW Jr, Bonds D, et al. Simultaneous measurements of lactate turnover rate and umbilical lactate uptake in the fetal lamb. J Clin Invest. 1982;70:179–192.
43. Bocking AD, Challis JR, White SE. Effect of acutely-induced lactic acidemia on fetal breathing movements, heart rate, blood pressure, ACTH and cortisol in sheep. J Dev Physiol. 1991;16:45–50.
44. Milley JR. Protein synthesis during hypoxia in fetal lambs. Am J Physiol. 1987;252:E519–E524.
45. Palacin M, Lasuncion MA, Herrera E. Lactate production and absence of gluconeogenesis from placental transferred substrates in fetuses from fed and 48-H starved rats. Pediatr Res. 1987;22:6–10.
46. Shambaugh GE, Mrozak SC, Freinkel N. Fetal fuels. I. Utilization of ketones by isolated tissues at various stages of maturation and maternal nutrition during late gestation. Metabolism. 1977;26:623–635.
47. Neary RH, Kilby MD, Kumpatula P, et al. Fetal and maternal lipoprotein metabolism in human pregnancy. Clin Sci (Lond). 1995;88:311–318.
48. Power GG, Schroder H, Gilbert RD. Measurement of fetal heat production using differential calorimetry. J Appl Physiol. 1984;57:“917–22.
49. Power GG, Blood AB. Perinatal thermal physiology. Polin RA, Fox MD, Abman SH. Fetal and Neonatal Physiology. 4th edition. Saunders: Philadelphia; 2011:615–624.
50. Asakura H. Fetal and neonatal thermoregulation. J Nippon Med Sch. 2004;71:360–370.
51. Gilbert RD, Schroder H, Kawamura T, et al. Heat transfer pathways between fetal lamb and ewe. J Appl Physiol. 1985;59:634–638.
52. Morishima HO, Yeh MN, Niemann WH, James LS. Temperature gradient between fetus and mother as an index for assessing intrauterine fetal condition. Am J Obstet Gynecol. 1977;129:443–448.
53. Kubonoya K, Yoneyama Y, Sawa R, et al. Brain temperature and metabolic responses during umbilical cord occlusion in fetal sheep. Pflugers Arch. 1998;436:667–672.
54. Rooth G, Huch A, Huch R, Peltonen R. Fetal-maternal temperature differences during labour. Contrib Gynecol Obstet. 1977;3:54–62.
55. Suzuki S, Murata T, Jiang L, Power GG. Hyperthermia prevents metabolic and cerebral flow responses to hypoxia in the fetal sheep. J Soc Gynecol Investig. 2000;7:45–50.
56. Heim T, Hull D. The blood flow and oxygen consumption of brown adipose tissue in the new-born rabbit. J Physiol. 1966;186:42–55.
57. Ball KT, Takeuchi M, Yoneyama Y, Power GG. Role of prostaglandin I2 and prostaglandin E2 in the initiation of nonshivering thermogenesis during the simulation of birth in utero. Reprod Fertil Dev. 1995;7:399–403.
58. Sawa R, Asakura H, Power GG. Changes in plasma adenosine during simulated birth of fetal sheep. J Appl Physiol. 1991;70:1524–1528.
59. Takeuchi M, Yoneyama Y, Power GG. Role of prostaglandin E2 and prostacyclin in nonshivering thermogenesis during simulated birth in utero. Prostaglandins Leukot Essent Fatty Acids. 1994;51:“373–80.
60. Kiserud T, Rasmussen S, Skulstad S. Blood flow and the degree of shunting through the ductus venosus in the human fetus. Am J Obstet Gynecol. 2000;182:147–153.
61. Brace RA. Fetal blood volume responses to intravenous saline solution and dextran. Am J Obstet Gynecol. 1983;147:777–781.
62. Brace RA. Regulation of blood volume in utero. Hanson MA SJ, Rodeck CH. Fetus and Neonate Physiology and Clinical Application. Cambridge University Press: Cambridge; 1993:75–99.
63. Faber JJ, Anderson DF. The placenta in the integrated physiology of fetal volume control. Int J Dev Biol. 2010;54:391–396.
64. Kiserud T. Physiology of the fetal circulation. Semin Fetal Neonatal Med. 2005;10:493–503.
65. Adamson SL, Morrow RJ, Bull SB, Langille BL. Vasomotor responses of the umbilical circulation in fetal sheep. Am J Physiol. 1989;256:R1056–R1062.
66. Adamson SL, Whiteley KJ, Langille BL. Pulsatile pressure-flow relations and pulse-wave propagation in the umbilical circulation of fetal sheep. Circ Res. 1992;70:761–772.
67. Kiserud T, Acharya G. The fetal circulation. Prenat Diagn. 2004;24:1049–1059.
68. Posterino GS, Dunn SL, Botting KJ, et al. Changes in cardiac troponins with gestational age explain changes in cardiac muscle contractility in the sheep fetus. J Appl Physiol. 2011;111:236–243.
70. Hamill N, Yeo L, Romero R, et al. Fetal cardiac ventricular volume, cardiac output, and ejection fraction determined with 4-dimensional ultrasound using spatiotemporal image correlation and virtual organ computer-aided analysis. Am J Obstet Gynecol. 2011;205:76 e1–10.
71. Elmstedt N, Ferm-Widlund K, Lind B, et al. Fetal cardiac muscle contractility decreases with gestational age: a color-coded tissue velocity imaging study. Cardiovasc Ultrasound. 2012;10:19.
72. Thornburg KL, Morton MJ. Filling and arterial pressures as determinants of RV stroke volume in the sheep fetus. Am J Physiol. 1983;244:H656–H663.
73. Gilbert RD. Control of fetal cardiac output during changes in blood volume. Am J Physiol. 1980;238:H80–H86.
74. Weil SR, Russo PA, Heckman JL, et al. Pressure-volume relationship of the fetal lamb heart. Ann Thorac Surg. 1993;55:470–475.
75. Kirkpatrick SE, Pitlick PT, Naliboff J, Friedman WF. Frank-Starling relationship as an important determinant of fetal cardiac output. Am J Physiol. 1976;231:495–500.
76. Grant DA, Fauchere JC, Eede KJ, et al. Left ventricular stroke volume in the fetal sheep is limited by extracardiac constraint and arterial pressure. J Physiol. 2001;535:231–239.
77. Hawkins J, Van Hare GF, Schmidt KG, Rudolph AM. Effects of increasing afterload on left ventricular output in fetal lambs. Circ Res. 1989;65:127–134.
78. Anderson DF, Bissonnette JM, Faber JJ, Thornburg KL. Central shunt flows and pressures in the mature fetal lamb. Am J Physiol. 1981;241:H60–H66.
79. Rudolph AM, Heymann MA. Circulatory changes during growth in the fetal lamb. Circ Res. 1970;26:289–299.
80. Rasanen J, Wood DC, Weiner S, et al. Role of the pulmonary circulation in the distribution of human fetal cardiac output during the second half of pregnancy. Circulation. 1996;94:1068–1073.
81. Behrman RE, Lees MH, Peterson EN, et al. Distribution of the circulation in the normal and asphyxiated fetal primate. Am J Obstet Gynecol. 1970;108:956–969.
82. Blanco CE, Dawes GS, Hanson MA, McCooke HB. Studies of carotid baroreceptor afferents in fetal and newborn lambs. Jones CT, Nathanielsz PW. The Physiological Development of the Fetus and Newborn. Academic Press: Orlando, FL; 1985.
83. Long WA. Developmental pulmonary circulatory physiology. Long WA. Fetal and Neonatal Cardiology. WB Saunders: Philadelphia; 1990.
84. Boekkooi PF, Baan J Jr, Teitel D, Rudolph AM. Chemoreceptor responsiveness in fetal sheep. Am J Physiol. 1992;263:H162–H167.
85. Walker AM. Physiological control of the fetal cardiovascular system. Beard RW, Nathanielsz PW. Fetal Physiology and Medicine. Marcel Dekker: New York; 1984.
86. Dawes GS, Duncan SL, Lewis BV, et al. Cyanide stimulation of the systemic arterial chemoreceptors in foetal lambs. J Physiol. 1969;201:117–128.
87. Wood CE, Tong H. Central nervous system regulation of reflex responses to hypotension during fetal life. Am J Physiol. 1999;277:R1541–R1552.
88. Papp JG. Autonomic responses and neurohumoral control in the human early antenatal heart. Basic Res Cardiol. 1988;83:2–9.
89. Long WA, Henry GW. Autonomic and central neuroregulation of fetal cardiovascular function. Polin RA, Fox WW. Fetal and Neonatal Physiology. WB Saunders: Philadelphia; 1992.
90. Taylor IM, Smith RB. Cholinesterase activity in the human fetal heart between the 35- and 160-millimeter crown-rump length stages. J Histochem Cytochem. 1971;19:498–503.
91. Smith RB. The development of the intrinsic innervation of the human heart between the 10 and 70 mm stages. J Anat. 1970;107:271–279.
92. Hata T, Matsuura H, Miyata M, et al. Autonomic modulation of sinus and atrioventricular nodes in premature low-birth-weight infants. Pacing Clin Electrophysiol. 2005;28(Suppl 1):S288–S291.
93. Schifferli P, Caldeyro-Barcia R. Effect of atropine and beta-adrenergic drugs on the heart rate of the human fetus. Boréus LO. Fetal Pharmacology. Raven Press: New York; 1973:259–279.
94. Pickoff AS, Rios R, Stolfi A, Wang SN. Postnatal maturation of the response of the canine sinus node to critically timed, brief vagal stimulation. Pediatr Res. 1994;35:55–61.
95. Lorijn RH, Longo LD. Norepinephrine elevation in the fetal lamb: oxygen consumption and cardiac output. Am J Physiol. 1980;239:R115–R122.
96. Langston C, Kida K, Reed M, Thurlbeck WM. Human lung growth in late gestation and in the neonate. Am Rev Respir Dis. 1984;129:607–613.
97. Hall SM, Hislop AA, Haworth SG. Origin, differentiation, and maturation of human pulmonary veins. Am J Respir Cell Mol Biol. 2002;26:333–340.
98. Hall SM, Hislop AA, Pierce CM, Haworth SG. Prenatal origins of human intrapulmonary arteries: formation and smooth muscle maturation. Am J Respir Cell Mol Biol. 2000;23:194–203.
99. Rasanen J, Wood DC, Debbs RH, et al. Reactivity of the human fetal pulmonary circulation to maternal hyperoxygenation increases during the second half of pregnancy: a randomized study. Circulation. 1998;97:257–262.
100. Lewis AB, Heymann MA, Rudolph AM. Gestational changes in pulmonary vascular responses in fetal lambs in utero. Circ Res. 1976;39:536–541.
101. Morin FC 3rd, Egan EA. Pulmonary hemodynamics in fetal lambs during development at normal and increased oxygen tension. J Appl Physiol. 1992;73:213–218.
102. Hooper SB, Wallace MJ. Role of the physicochemical environment in lung development. Clin Exp Pharmacol Physiol. 2006;33:273–279.
103. Polglase GR, Wallace MJ, Morgan DL, Hooper SB. Increases in lung expansion alter pulmonary hemodynamics in fetal sheep. J Appl Physiol. 2006;101:273–282.
104. Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev. 2010;90:1291–1335.
105. Ibe BO, Hillyard RM, Raj JU. Heterogeneity in prostacyclin and thromboxane synthesis in ovine pulmonary vascular tree: effect of age and oxygen tension. Exp Lung Res. 1996;22:351–374.
106. Haworth SG, Hislop AA. Effect of hypoxia on adaptation of the pulmonary circulation to extra-uterine life in the pig. Cardiovasc Res. 1982;16:293–303.
107. Ibe BO, Hibler S, Raj JU. Platelet-activating factor modulates pulmonary vasomotor tone in the perinatal lamb. J Appl Physiol. 1998;85:1079–1085.
108. Ibe BO, Portugal AM, Chaturvedi S, Raj JU. Oxygen-dependent PAF receptor binding and intracellular signaling in ovine fetal pulmonary vascular smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288:L879–L886.
109. Iwamoto HS, Teitel D, Rudolph AM. Effects of birth-related events on blood flow distribution. Pediatr Res. 1987;22:634–640.
110. Hallman M, Glumoff V, Ramet M. Surfactant in respiratory distress syndrome and lung injury. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:287–294.
111. Goerke J, Clements J. Alveolar surface tension and lung surfactant. Macklem P, Mead J. Handbook of Physiology: The Respiratory System. Vol. III, Part I. American Physiological Society: Bethesda, MD; 1986:247–260.
112. Haagsman HP, Diemel RV. Surfactant-associated proteins: functions and structural variation. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:91–108.
113. Veldhuizen R, Nag K, Orgeig S, Possmayer F. The role of lipids in pulmonary surfactant. Biochim Biophys Acta. 1998;1408:90–108.
114. Mason RJ, Voelker DR. Regulatory mechanisms of surfactant secretion. Biochim Biophys Acta. 1998;1408:226–240.
115. Torday JS, Nielsen HC. Surfactant phospholipid ontogeny in fetal rabbit lung lavage and amniotic fluid. Biol Neonate. 1981;39:266–271.
116. Orgeig S, Morrison JL, Daniels CB. Prenatal development of the pulmonary surfactant system and the influence of hypoxia. Respir Physiol Neurobiol. 2011;178:129–145.
117. Gluck L, Kulovich MV. Lecithin-sphingomyelin ratios in amniotic fluid in normal and abnormal pregnancy. Am J Obstet Gynecol. 1973;115:539–546.
118. Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2006;(3).
120. Moritz KM, Macris M, Talbo G, Wintour EM. Foetal fluid balance and hormone status following nephrectomy in the foetal sheep. Clin Exp Pharmacol Physiol. 1999;26:857–864.
121. Cuckow PM, Nyirady P, Winyard PJ. Normal and abnormal development of the urogenital tract. Prenat Diagn. 2001;21:908–916.
122. Vogt BA, Dell KM. The kidney and the urinary tract. Martin RJ, Fanaroff AA, Walsh ML. Fanaroff and Martin’s Neonatal Perinatal Medicine. 9th edition. Elsevier: Philadelphia; 2008:1681–1704.
123. Hunley TE, Kon V, Ichiwaka J. Glomerular circulation and function. Avner ED, Harmon WE, Niaudet P, Yoshiwaka N. Pediatric Nephrology. 6th edition. Springer: Philadelphia; 2009:31–64.
124. Turner AJ, Brown RD, Carlstrom M, et al. Mechanisms of neonatal increase in glomerular filtration rate. Am J Physiol Regul Integr Comp Physiol. 2008;295:R916–R921.
125. Guignard JP, Gouyon JB. Glomerular filtration rate in neonates. Polin RA. Nephrology and Fluid Electrolyte Physiology. Saunders: Philadelphia; 2008:1681–1704.
126. Hoseini R, Otukesh H, Rahimzadeh N, Hoseini S. Glomerular function in neonates. Iran J Kidney Dis. 2012;6:166–172.
127. Haycock GB, Aperia A. Salt and the newborn kidney. Pediatr Nephrol. 1991;5:65–70.
128. Baum M, Quigley R. Ontogeny of proximal tubule acidification. Kidney Int. 1995;48:1697–1704.
129. Gomez RA, Norwood VF. Developmental consequences of the renin-angiotensin system. Am J Kidney Dis. 1995;26:409–431.
130. Iwamoto HS, Rudolph AM. Effects of endogenous angiotensin II on the fetal circulation. J Dev Physiol. 1979;1:283–293.
131. Baron MH, Isern J, Fraser ST. The embryonic origins of erythropoiesis in mammals. Blood. 2012;119:4828–4837.
132. Tavian M, Peault B. Embryonic development of the human hematopoietic system. Int J Dev Biol. 2005;49:243–250.
133. Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. 2008;9:129–136.
134. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644.
135. Perutz MF. The hemoglobin molecule. Sci Am. 1964;211:64–76.
136. Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149:181–194.
137. Delivoria-Papadopoulos MMJ. Oxygen transport and delivery. Polin RA, Fox WW, Abman SH. Fetal and Neonatal Physiology. 4th edition. Elsevier Saunders: Philadelphia; 2011:970–980.
138. Beer R, Doll E, Wenner J. Shift in oxygen dissociation curve of the blood of infants in the first month of life. Pflugers Arch. 1958;265:526.
139. Allen DW, Wyman J Jr, Smith CA. The oxygen equilibrium of fetal and adult human hemoglobin. J Biol Chem. 1953;203:81–87.
140. Arnone A. X-ray diffraction study of binding of 2,3-diphosphoglycerate to human deoxyhaemoglobin. Nature. 1972;237:146–149.
141. Lorkin PA. Fetal and embryonic haemoglobins. J Med Genet. 1973;10:50–64.
142. Delivoria-Papadopoulos M, Morrow G 3rd, Oski FA. Exchange transfusion in the newborn infant with fresh and “old” blood: the role of storage on 2,3-diphosphoglycerate, hemoglobin-oxygen affinity, and oxygen release. J Pediatr. 1971;79:898–903.
143. Oski FA, Delivoria-Papadopoulos M. The red cell, 2,3-diphosphoglycerate, and tissue oxygen release. J Pediatr. 1970;77:941–956.
144. Ross AI. Organogenesis of the gastrointestinal tract. Polin R, Fox W, Abman S. Fetal and Neonatal Physiology. 4th edition. Elsevier Saunders: Philadelphia; 2011:1187–1197.
145. Grand RJ, Watkins JB, Torti FM. Development of the human gastrointestinal tract: a review. Gastroenterology. 1976;70:790–810.
146. Pace JL. The age of appearance of the haustra of the human colon. J Anat. 1971;109:75–80.
147. Cornes JS. Peyer’s patches in the human gut. Proc R Soc Med. 1965;58:716.
148. Pritchard JA. Fetal swallowing and amniotic fluid volume. Obstet Gynecol. 1966;28:606–610.
149. Trahair JF, Harding R. Ultrastructural anomalies in the fetal small intestine indicate that fetal swallowing is important for normal development: an experimental study. Virchows Arch A Pathol Anat Histopathol. 1992;420:305–312.
150. Avila CG, Harding R. The development of the gastrointestinal system in fetal sheep in the absence of ingested fluid. J Pediatr Gastroenterol Nutr. 1991;12:96–104.
151. Mulvihill SJ, Stone MM, Debas HT, Fonkalsrud EW. The role of amniotic fluid in fetal nutrition. J Pediatr Surg. 1985;20:668–672.
152. Shaw K, Buchmiller TL, Curr M, et al. Impairment of nutrient uptake in a rabbit model of gastroschisis. J Pediatr Surg. 1994;29:376–378.
153. Jacobs DG, Wesson DE, Mago-Cao H, et al. Effect of esophageal ligation on the growth of fetal rabbits. J Pediatr Gastroenterol Nutr. 1989;8:245–251.
154. Ahanya SN, Lakshmanan J, Morgan BL, Ross MG. Meconium passage in utero: mechanisms, consequences, and management. 2005:45–56. Obstet Gynecol Surv. 60.
155. Shwachman H, Antonowicz I. Studies on meconium. Lebenthal E. Textbook of Gastroenterology and Nutrition in Infancy. Raven Press: New York; 1981:81–93.
156. Potier M, Melancon SB, Dallaire L. Developmental patterns of intestinal disaccharidases in human amniotic fluid. Am J Obstet Gynecol. 1978;131:73–76.
157. Mulivor RA, Mennuti MT, Harris H. Origin of the alkaline phosphatases in amniotic fluid. Am J Obstet Gynecol. 1979;135:77–81.
158. Usher RH, Boyd ME, McLean FH, Kramer MS. Assessment of fetal risk in postdate pregnancies. Am J Obstet Gynecol. 1988;158:259–264.
159. Altshuler G, Arizawa M, Molnar-Nadasdy G. Meconium-induced umbilical cord vascular necrosis and ulceration: a potential link between the placenta and poor pregnancy outcome. Obstet Gynecol. 1992;79:760–766.
160. Kostovic I, Judas M. Transient patterns of cortical lamination during prenatal life: do they have implications for treatment? Neurosci Biobehav Rev. 2007;31:1157–1168.
161. Kostovic I, Judas M, Rados M, Hrabac P. Laminar organization of the human fetal cerebrum revealed by histochemical markers and magnetic resonance imaging. Cereb Cortex. 2002;12:536–544.
162. Kostovic I, Jovanov-Milosevic N. The development of cerebral connections during the first 20-45 weeks’ gestation. Semin Fetal Neonatal Med. 2006;11:415–422.
163. Kostovic I. Structural and histochemical reorganization of the human prefrontal cortex during perinatal and postnatal life. Prog Brain Res. 1990;85:223–239.
164. Khazipov R, Sirota A, Leinekugel X, et al. Early motor activity drives spindle bursts in the developing somatosensory cortex. Nature. 2004;432:758–761.
165. Khazipov R, Luhmann HJ. Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 2006;29:414–418.
166. Hellstrom-Westas L, Rosen I, Svenningsen NW. Cerebral function monitoring during the first week of life in extremely small low birthweight (ESLBW) infants. Neuropediatrics. 1991;22:27–32.
167. du Plessis AJ. Cerebral blood flow and metabolism in the developing fetus. Clin Perinatol. 2009;36:531–548.
168. Murthy MR, Rappoport DA. Biochemistry of the developing rat brain. II. Neonatal mitochondrial oxidations. Biochim Biophys Acta. 1963;74:51–59.
169. Vannucci S, Vannucci R. Perinatal brain metabolism. Polin R, Fox W, Abman S. Fetal and Neonatal Physiology. 4th edition. Elsevier Saunders: Philadelphia; 2011.
170. Hernandez MJ, Vannucci RC, Salcedo A, Brennan RW. Cerebral blood flow and metabolism during hypoglycemia in newborn dogs. J Neurochem. 1980;35:622–628.
171. Hellmann J, Vannucci RC, Nardis EE. Blood-brain barrier permeability to lactic acid in the newborn dog: lactate as a cerebral metabolic fuel. Pediatr Res. 1982;16:40–44.
172. Lutz PL. Mechanisms for anoxic survival in the vertebrate brain. Annu Rev Physiol. 1992;54:601–618.
173. Peeters LL, Sheldon RE, Jones MD Jr, et al. Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol. 1979;135:637–646.
174. Jensen A, Roman C, Rudolph AM. Effects of reducing uterine blood flow on fetal blood flow distribution and oxygen delivery. J Dev Physiol. 1991;15:309–323.
176. Pearce W. Hypoxic regulation of the fetal cerebral circulation. J Appl Physiol. 2006;100:731–738.
177. Okado N. Onset of synapse formation in the human spinal cord. J Comp Neurol. 1981;201:211–219.
178. Bijlani V, Rizvi TA, Wadhwa S. Development of spinal substrate for nociception in man. NIDA Res Monogr. 1988;87:167–179.
179. Gilles FJ, Shankle W, Dooling EC. Myelinated tracts: growth patterns. Gilles FJ, Leviton A, Dooling EC. The Developing Human Brain. Wright & Co: Boston; 1983:117.
180. Giannakoulopoulos X, Teixeira J, Fisk N, Glover V. Human fetal and maternal noradrenaline responses to invasive procedures. Pediatr Res. 1999;45:494–499.
181. Gitau R, Fisk NM, Teixeira JM, et al. Fetal hypothalamic-pituitary-adrenal stress responses to invasive procedures are independent of maternal responses. J Clin Endocrinol Metab. 2001;86:104–109.
182. Teixeira J, Fogliani R, Giannakoulopoulos X, et al. Fetal haemodynamic stress response to invasive procedures. Lancet. 1996;347:624.
183. Giannakoulopoulos X, Sepulveda W, Kourtis P, et al. Fetal plasma cortisol and beta-endorphin response to intrauterine needling. Lancet. 1994;344:77–81.
184. Fisk NM, Gitau R, Teixeira JM, et al. Effect of direct fetal opioid analgesia on fetal hormonal and hemodynamic stress response to intrauterine needling. Anesthesiology. 2001;95:828–835.
185. Slater R, Cantarella A, Gallella S, et al. Cortical pain responses in human infants. J Neurosci. 2006;26:3662–3666.
186. Bartocci M, Bergqvist LL, Lagercrantz H, Anand KJ. Pain activates cortical areas in the preterm newborn brain. Pain. 2006;122:109–117.
187. Whit Hall R, Anand KJS. Physiology of pain and stress in the newborn. NeoReviews. 2005;6:e61–e67.
188. Boyle EM, Freer Y, Wong CM, et al. Assessment of persistent pain or distress and adequacy of analgesia in preterm ventilated infants. Pain. 2006;124:87–91.
189. Benatar D, Benatar M. A pain in the fetus: toward ending confusion about fetal pain. Bioethics. 2001;15:57–76.
190. Fitzgerald MHR. The neurobiologic basis of pediatric pain. Schechter NL, Berde CB, Yaster M. Pain in Infants, Children and Adolescents. 2nd edition. Lippincott Williams and Wilkins: Philadelphia; 2003.
191. Beggs S, Currie G, Salter MW, et al. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain. 2012;135:404–417.
192. Derbyshire SW, Fitzgerald M. The painful consequences of neonatal nociceptive input. Pain. 2010;150:220–221.
193. Laprairie JL, Johns ME, Murphy AZ. Preemptive morphine analgesia attenuates the long-term consequences of neonatal inflammation in male and female rats. Pediatr Res. 2008;64:625–630.