[level-membership-for-pathology-category]2
Fetal and neonatal hypoxic–ischemic lesions
 The extraordinarily rapid growth and plasticity of the immature nervous system.
The extraordinarily rapid growth and plasticity of the immature nervous system.
 The major changes in cell and tissue properties that occur during development.
The major changes in cell and tissue properties that occur during development.
 PATHOGENESIS OF HYPOXIC–ISCHEMIC LESIONS
PATHOGENESIS OF HYPOXIC–ISCHEMIC LESIONS
Possible pathophysiologic schema
Principal factors involved in the production of lesions:





Present knowledge of embryology and developmental physiology allows a tentative chronology to be assigned to individual lesions (Fig. 2.1) although this remains relatively imprecise and extreme caution is needed in the forensic arena. Although some pathologic changes closely parallel those found in older individuals, others such as intraventricular hemorrhage, white matter necrosis, and marbling are unique to the perinatal period.
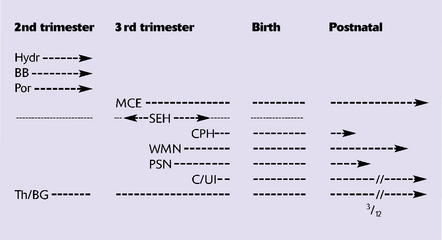
2.1 A timetable for hypoxic–ischemic lesions in early life.
Hydr, hydranencephaly; BB, basket brain; Por, porencephaly; MCE, multicystic encephalopathy; SEH, subependymal hemorrhage; CPH, choroid plexus hemorrhage; WMN (PVL), white matter necrosis (periventricular leukomalacia); PSN, pontosubicular necrosis; C/UI, cortical necrosis/ulegyria; Th/BG, thalamus/basal ganglia damage.
FETAL LESIONS
 CAUSES OF HYPOXIC–ISCHEMIC LESIONS
CAUSES OF HYPOXIC–ISCHEMIC LESIONS













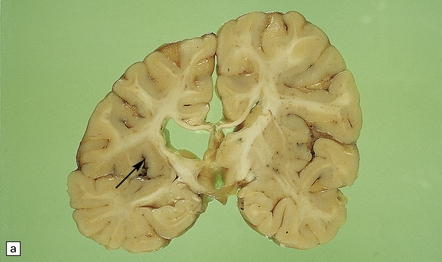
HYDRANENCEPHALY
MACROSCOPIC AND MICROSCOPIC APPEARANCES
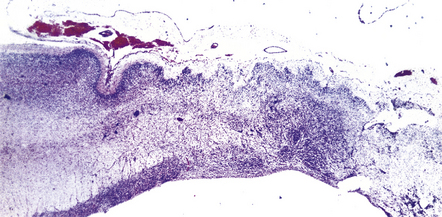
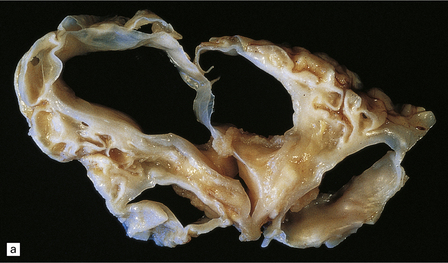
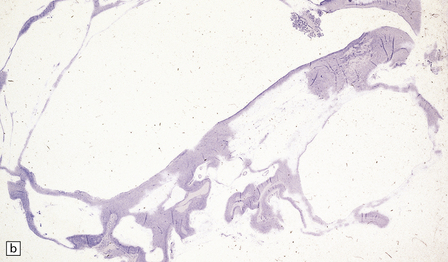
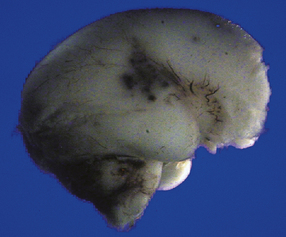
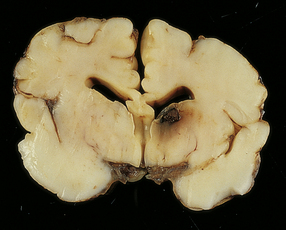
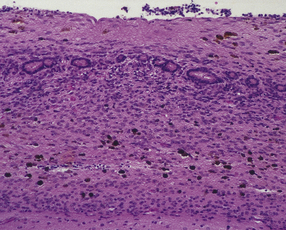
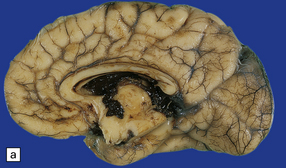
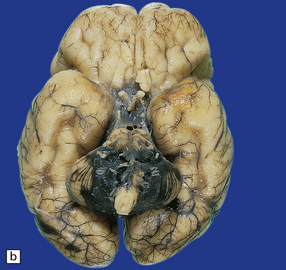
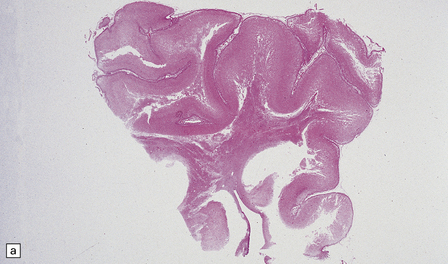
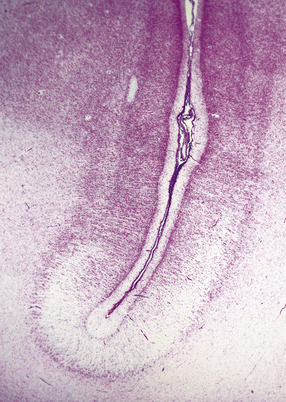
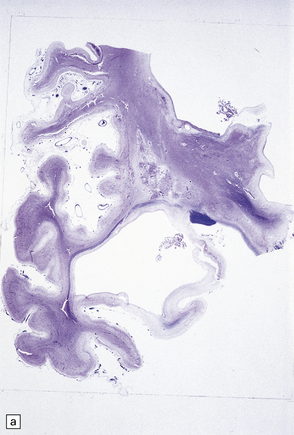
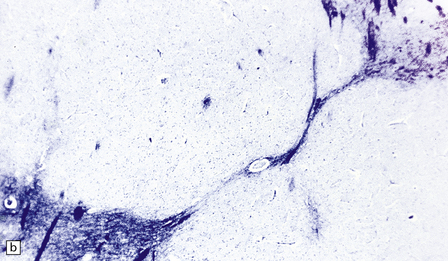
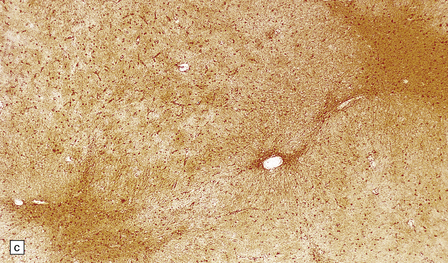
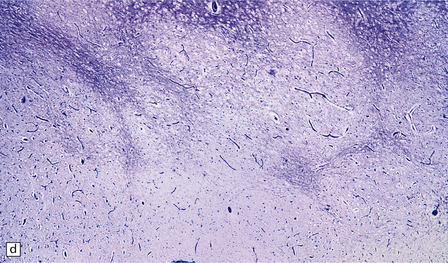
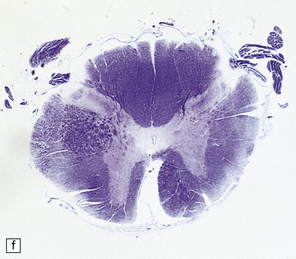
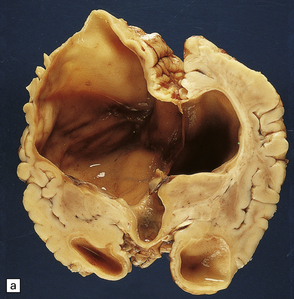
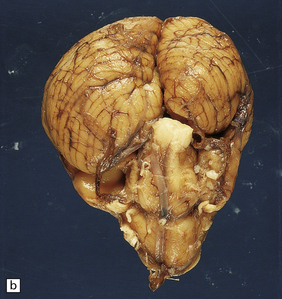
Much of the cerebral mantle is replaced by a thin translucent membrane. The hemispheres are cystic, and lack surface convolutions (Fig. 2.2). Inferior aspects of the temporal, occipital, and frontal lobes are usually spared, but sometimes only the hippocampi remain. The deep gray nuclei may be rotated outwards and the thalami are atrophied. The membranous cerebral mantle comprises an outer connective tissue layer and an inner irregular glial layer which also contains mineralized neurons, debris, and hemosiderin-laden macrophages. The ependyma is usually absent. At the interface with surviving cerebral tissue, the inner glial layer runs into the molecular layer, covering it for a short distance, while adjacent cortex is usually disorganized, often with a pattern of polymicrogyria (Fig. 2.3). This histologic appearance clearly distinguishes hydranencephaly from the macroscopically similar bubble brain in Fowler syndrome (see Fig. 7.2).
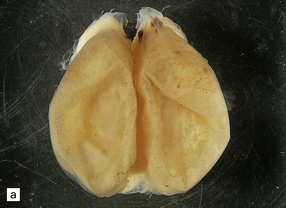
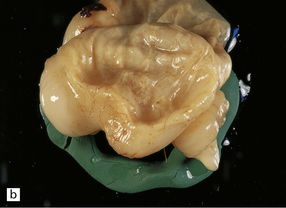
2.2 Hydranencephaly in a twin of 22 weeks’ gestation whose co-twin died at 17 weeks.
(a) The bubble-like hemispheres photographed under water and viewed from above. (b) Removal from water allows the diaphanous membrane of the cyst wall to collapse. Damage is largely restricted to the internal carotid artery territory with sparing of the orbitofrontal cortex and inferior temporal and occipital lobes.
BASKET BRAIN
 ETIOLOGY AND PATHOGENESIS OF FETAL HYPOXIC–ISCHEMIC LESIONS
ETIOLOGY AND PATHOGENESIS OF FETAL HYPOXIC–ISCHEMIC LESIONS








 HYDRANENCEPHALY
HYDRANENCEPHALY


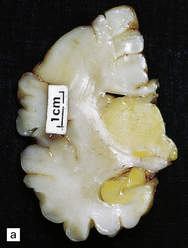
PORENCEPHALY AND SCHIZENCEPHALY
MACROSCOPIC AND MICROSCOPIC APPEARANCES
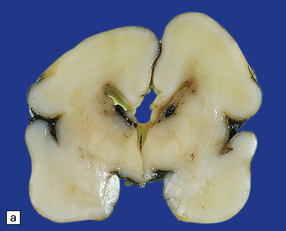
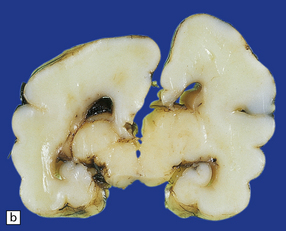
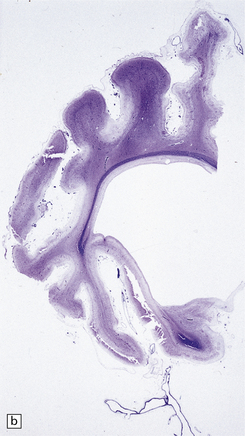
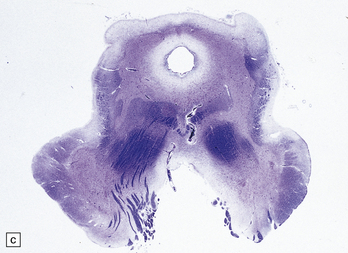
A porus is a smooth-walled defect in the cerebral mantle, usually surrounded by an abnormal gyral pattern. It varies in depth from a slight indentation or fissure to a full-thickness breach of the hemispheric wall connecting subarachnoid space with ventricle. Pori tend to be bilateral, approximately symmetric, and situated over the Sylvian fissures or central sulci. Unilateral defects may be parasagittal, orbital, or occipital, and associated with abnormal convolutions, especially polymicrogyria, which are symmetrically placed in the contralateral hemisphere. Gyri surrounding the defect form irregular or radiating patterns (Fig. 2.4). The cortex around the porus is broken into islands or folded into polymicrogyria, which extends down into the cleft to meet the ventricular wall. The latter is denuded of ependyma, but covered by glial tissue, which extends a short way over the adjacent gray matter (Fig. 2.5). Subependymal nodular heterotopia (Fig. 2.6d) and partial or complete absence of the septum pellucidum are other features.
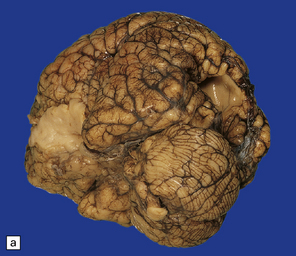
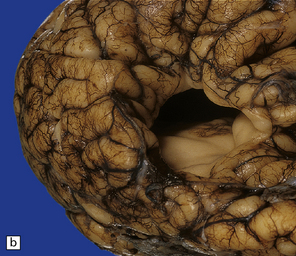
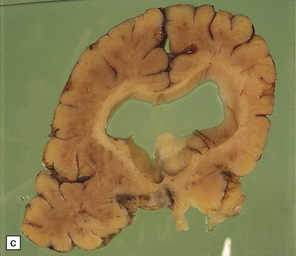
2.4 Porencephaly.
(a) Two unilateral pori are present. The shallow depression in the orbitofrontal cortex contrasts with the wide-mouthed full-thickness defect in the occipital lobe allowing communication between the subarachnoid space and the ventricle. (b) The surrounding convolutions have a radiating pattern. (c) Coronal section through the frontal lobes showing the extent of the anterior defect, which is lined by nodules of gray matter. The septum pellucidum is absent.
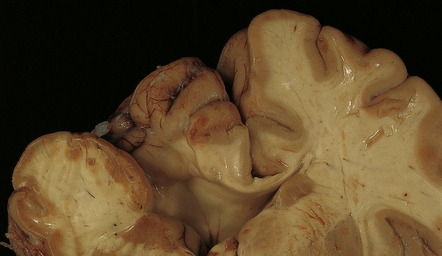
2.5 Porencephaly.
Coronal section through a frontal porus in a 48-year-old man. The cortical gray matter dips down into the cleft to meet the white gliotic ependyma. The cortical ribbon around the edge of the defect is irregularly thickened and disorganized. (Courtesy of Dr T Moss, Frenchay Hospital, Bristol.)
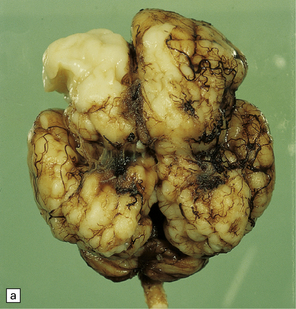
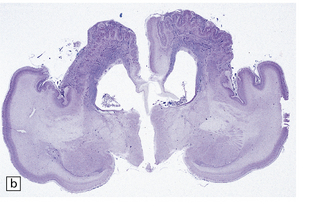
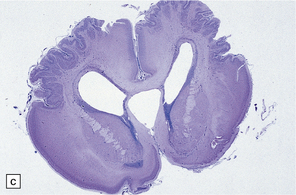
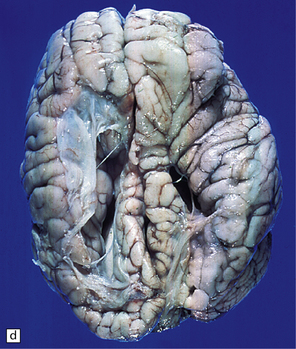
2.6 Schizencephaly.
(a) Bilateral porencephalic clefts centered on the middle cerebral artery territory in a twin of 32 weeks’ gestation whose co-twin died at 19 weeks. The hemispheres are viewed from above. (b) Coronal section at anterior thalamic level. The cortex in the center of the lesion is a completely disorganized mixture of neuroblasts and macrophages surrounded either side by polymicrogyric cortex. (c) Coronal section at striatal level. Polymicrogyric cortex from the anterior border of the cleft. (d) Large bilateral full-thickness defects in a 4-year-old boy with cerebral palsy and epilepsy. The covering leptomeninges ruptured during the dissection, revealing a radial pattern of convolutions around the clefts. (e) Coronal sections through the posterior parts of the hemispheres show thick polymicrogyric cortex curling over the mouths of the clefts and deeply invaginating the ventricle. Note the heterotopic gray nodules in the inferior corners of the ventricles.
Extensive bilateral porencephalic clefts (Fig. 2.6) are sometimes termed schizencephaly, especially in the radiologic literature, although the term schizencephaly refers back to an outmoded concept of circumscribed growth failure of the cerebral wall. The narrowness of the cleft with either closed or open lips was thought to differentiate malformed from acquired lesions, but clinical, morphologic, and experimental evidence favors a destructive origin for most lesions. There is also recent evidence for familial occurrence of schizencephaly in brothers with defects in the homeobox gene EMX2.
 CLINICAL DIAGNOSIS OF PERINATAL ASPHYXIA
CLINICAL DIAGNOSIS OF PERINATAL ASPHYXIA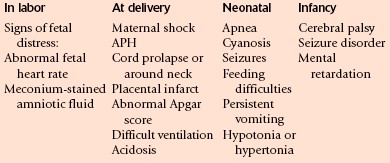
 PORENCEPHALY
PORENCEPHALY


MULTICYSTIC ENCEPHALOPATHY
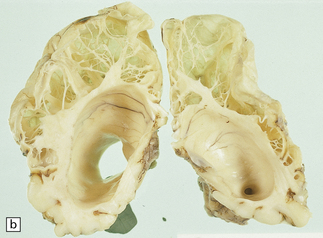
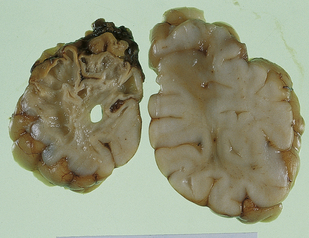
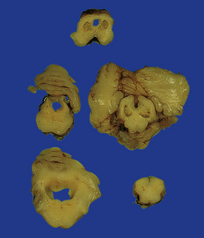
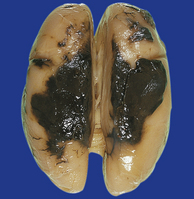
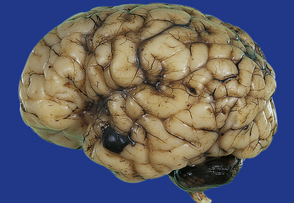
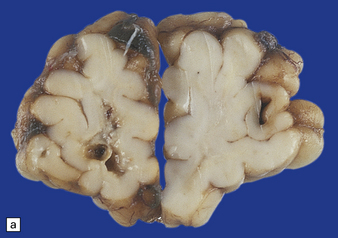
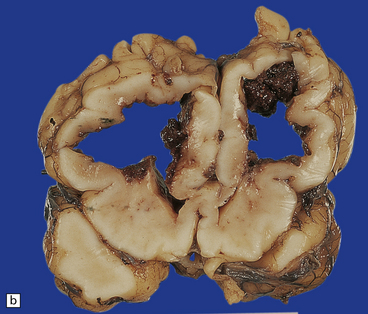
In contrast to the smooth-walled defects of hypoxic–ischemic lesions of early gestation, third trimester insults produce many cysts throughout large areas of cerebral white matter and deeper cortical layers (Figs 2.7, 2.8). Occasional cases are unilateral and circumscribed (Fig. 2.9), but most are extensive and bilateral. Bilateral multicystic encephalopathy is often associated with cystic necrosis in the basal ganglia and brain stem tegmentum (Fig. 2.10).
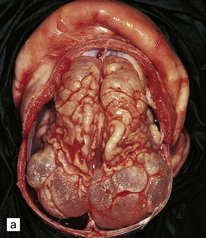
2.7 Multicystic encephalopathy in a smaller first twin at 36 weeks’ gestation.
(a) Superior view of the brain within the skull. The convolutions are either massively cystically dilated or narrow and thin. (b) Coronal section showing dilated ventricles and replacement of much of the cortex and white matter by an irregular firm white cystic meshwork.
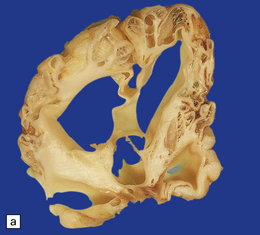
2.8 Multicystic encephalopathy.
(a) Tough gliovascular bands transform much of the deep cortex, white matter, and basal ganglia into a spongy mass. The thalamic nuclei are shrunken and extremely white. (b) Comparable histologic section from another patient showing that not only the cortex and white matter, but also the thalami and basal ganglia have been replaced by large cysts.
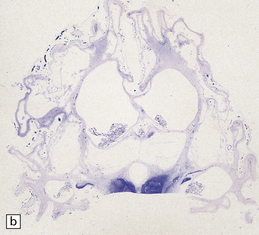
MICROSCOPIC APPEARANCES
A meshwork of thin gliovascular septa form numerous cysts containing lipid-laden macrophages. Global hemispheric necrosis (Fig. 2.11) is the term used to describe morphologically similar but particularly extensive cases following severe birth asphyxia and sudden hyperpyrexia and collapse in the postnatal period.
 MULTICYSTIC ENCEPHALOPATHY
MULTICYSTIC ENCEPHALOPATHY


PERINATAL LESIONS
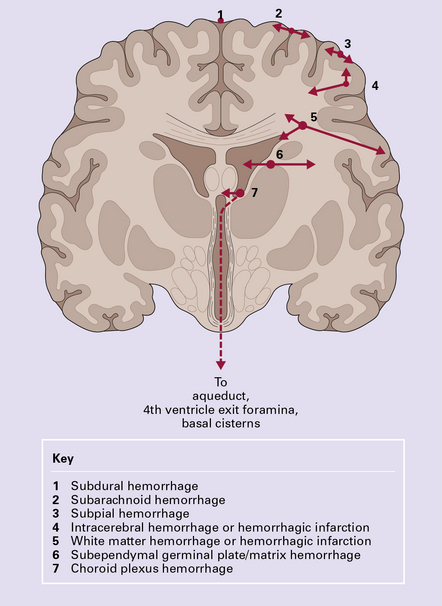
Hemorrhage is the commonest pathology in fetal and neonatal brains. It can start in any intracranial compartment and rapidly spread elsewhere. The pathogenesis is multifactorial. The principal risk factors are immaturity, perinatal distress, and asphyxia, though a similar spatial distribution in still and live births also implicates antenatal factors. The variety of hemorrhages in the neonatal brain is shown in Fig. 2.12.
SUBDURAL HEMORRHAGE (SDH)
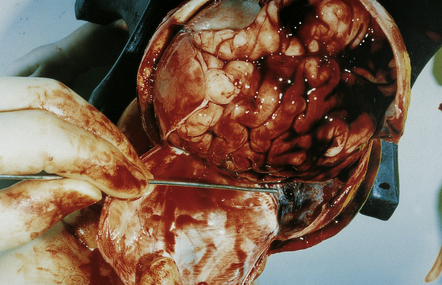
SDH is not related to asphyxia, but is closely related to perinatal distress. Excessive compression and distortion of the head during delivery can disrupt the superior cerebral bridging veins that drain into the sagittal sinus, or the veins to the transverse sinus. More rarely, the falx and sagittal sinus or the tentorium (Fig. 2.13) with the straight sinus or great vein of Galen is torn.
SUBARACHNOID HEMORRHAGE (SAH)
SAH (Figs 2.14–2.16) may manifest as:
2.15 Well-demarcated subarachnoid hematomas localized over parietal cortex.
This was associated with a threatened abortion at 20 weeks’ gestation.
2.16 A small localized SAH on the temporal pole as well as extensive basal SAH secondary to hemorrhagic white matter infarction.
This occurred in a term infant with multiple congenital anomalies who was cyanosed at birth.



SUBPIAL HEMORRHAGE (SPH)
SPH is often confused with SAH. It is a focal hemorrhage, which is usually temporal, parietal, or cerebellar, and occurs with or without other signs of bleeding. It is most common in asphyxiated premature infants, but also occurs in premature or term infants with respiratory distress syndrome or congenital heart disease (Figs 2.17, 2.18).
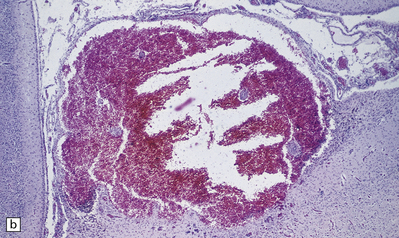
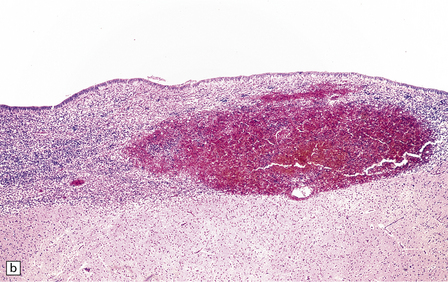
2.17 SPH in a term infant with multiple congenital anomalies including cyanotic congenital heart disease.
(a) Anterior coronal sections in the frontal lobes showing right-sided central white matter softening and several superficial hemorrhages. (b) Microscopy reveals an entirely intracortical bleed covered by pia.
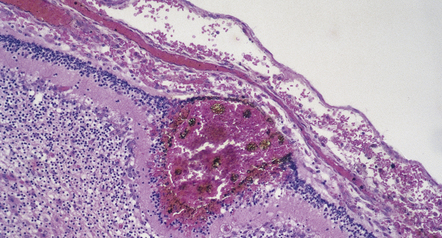
SUBEPENDYMAL GERMINAL PLATE/MATRIX HEMORRHAGE (SEH)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Acute SEH has variable appearances: small, multiple, and bilateral bleeds occur anywhere in periventricular matrix tissue (Fig. 2.19), including the roof of the fourth ventricle. Acute SEH is usually found
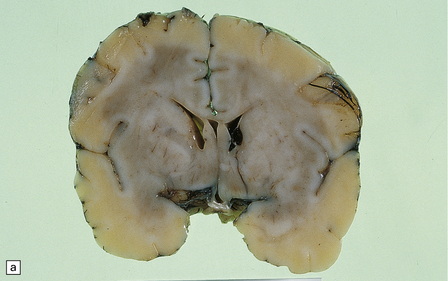
2.19 SEH in an infant with hypoplastic left heart syndrome born by cesarean section at 36 weeks’ gestation who survived 3 days.
(a) Small SEH that has remained within the matrix zone (grade 1) in the most typical position near the terminal vein close to the caudate nucleus and thalamus. (b) The small bleed remains in situ within the cellular matrix tissue. The ependyma is intact. No bleeding point can be ascertained.
 GRADING OF SEH ON CRANIAL ULTRASOUND
GRADING OF SEH ON CRANIAL ULTRASOUND
| Grade | Distribution of hemorrhage |
| 1 | Confined to germinal matrix |
| 2 | Germinal matrix and lateral ventricle, but no ventricular dilatation |
| 3 | Germinal matrix and lateral ventricle, which is acutely distended by hematoma within the ventricle |
| 4 | As above with extension of hemorrhage into adjacent brain parenchyma. (Note: the appearances may resemble those of pure venous infarction without IVH; see Fig. 2.36) |
 SEH
SEH SEH is mainly seen in low birth weight premature infants under 34 weeks’ gestation, but occasionally in term infants and rarely in stillbirths as young as 12 weeks’ gestation.
SEH is mainly seen in low birth weight premature infants under 34 weeks’ gestation, but occasionally in term infants and rarely in stillbirths as young as 12 weeks’ gestation.
 Most intraventricular hemorrhages (IVHs) in neonates are secondary to SEH.
Most intraventricular hemorrhages (IVHs) in neonates are secondary to SEH.

 Hemorrhage begins between 6 hours and 8 days postpartum and in 60% of cases before 48 hours.
Hemorrhage begins between 6 hours and 8 days postpartum and in 60% of cases before 48 hours.

in the germinal zone over the head of the caudate and thalamus, at or just behind the foramen of Monro; hematoma may extend into the centrum semiovale, simulating or accompanying hemorrhagic infarction (Figs 2.20–2.23). Rapidly fatal bleeds burst through the ependymal lining, fill the ventricles, and spill into the basal cisterns and subarachnoid spaces. Ventricular dilatation may result from tamponade or hematoma in the aqueduct, foramen of Monro (Fig. 2.24), or fourth ventricular outlets. Microscopic evidence of ruptured vessels, thrombosis, or infarcts within the densely cellular matrix is rare.
2.20 In situ matrix zone hemorrhage.
This occurred in an infant born by cesarean section for hydrops fetalis at 33 weeks’ gestation who survived 10 days. The clot has retracted and organized.
2.21 SEH 1–2 weeks after the ictus.
Hemosiderin-laden macrophages may be all that remains to indicate previous SEH. Note that the ependyma has been disorganized and glial tissue has overgrown ependymal canaliculi.
2.22 Grade 2 SEH with intraventricular extension.
This occurred in an infant born by cesarean section following antepartum hemorrhage at 26 weeks’ gestation who had respiratory distress syndrome with hyaline membranes and survived 3 days. (a) This shows bilateral bleeds in the typical position. (b) The hemorrhage in the matrix tissue surrounding the right temporal horn has leaked into the ventricle.
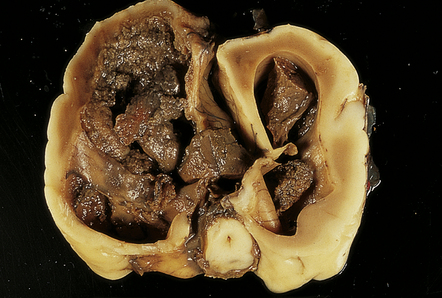
2.23 Grade 3 SEH filling the ventricles.
This occurred in a premature infant born at 30 weeks’ gestation with tracheoesophageal fistula and esophageal atresia, who developed respiratory distress syndrome and survived nine days. (a) Medial surface of the hemisphere. The septum has been removed. The large clot forms a cast of the ventricular system and aqueduct. (b) Blood has flushed through the fourth ventricular outlet foramina to fill the basal cisterns. Note the rusty staining of the temporal lobe due to an earlier SAH.
2.24 Ventricular dilatation resulting from blockage of the foramen of Monro.
This occurred in an infant born at 26 weeks’ gestation with tracheoesophageal fistula and esophageal atresia who survived 3 weeks. The clot has begun to retract and organize.
 PATHOGENESIS OF SEH
PATHOGENESIS OF SEH



macrophages, and granulation tissue. Hydrocephalus often occurs and in some cases is due to occlusion of the aqueduct by hematoma which organizes as a gliotic plug, fibrotic occlusion of the fourth ventricular outlets, or adhesive basal arachnoiditis.
CEREBRAL AND CEREBELLAR HEMORRHAGE
Common causes of cerebral hemorrhage are extension of SEH into periventricular white matter, massive IVH bursting through the ventricular wall (Fig. 2.25), or hemorrhage into areas of periventricular infarction (Fig. 2.26). Other sources include hemorrhagic middle cerebral artery territory infarction, and sinus thrombosis with venous infarction. Cerebellar hemorrhages may be petechial or large hematomas in the cortex or white matter (Figs 2.27–2.29), and are most often seen in low birth weight premature infants.
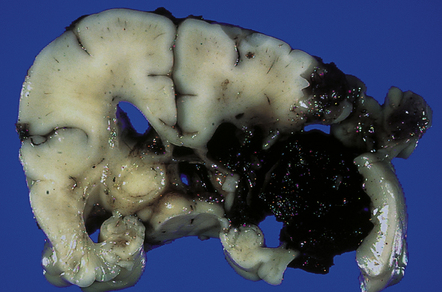
2.25 Grade 4 SEH bursting out into the periventricular tissues.
This occurred in an infant of 26 weeks’ gestation treated with a laparotomy for perforating necrotizing enterocolitis. A grade 4 SEH sometimes reaches the cortical surface as shown here.
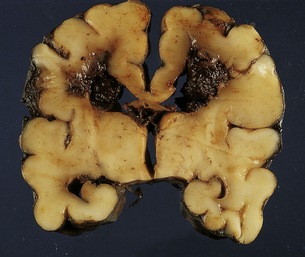
2.26 Coexistent matrix hemorrhage and hemorrhagic WMN.
This occurred in an infant born at 33 weeks’ gestation following pre-eclampsia and fetal distress, postnatal jaundice, hypoglycemia, and disseminated intravascular coagulation. He survived 5 days. Despite its misleading appearance this is not a grade 4 SHE.
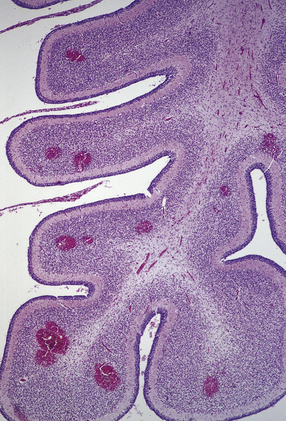
2.27 Cerebellar hemorrhage.
Numerous petechial hemorrhages in cerebellar cortex in a twin with congenital heart disease.
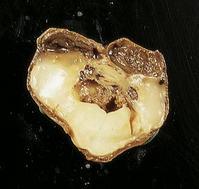
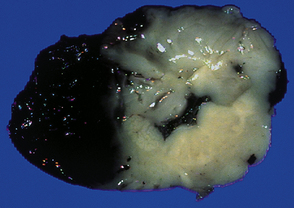
CHOROID PLEXUS HEMORRHAGE (CPH)
CPH is usually a coincidental finding in neonates and is occasionally responsible for massive IVH in term infants (Fig. 2.30).
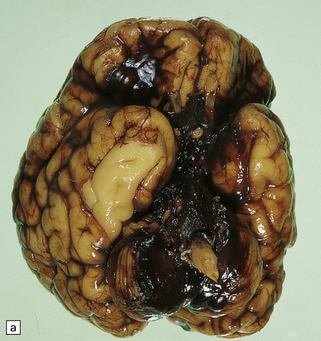
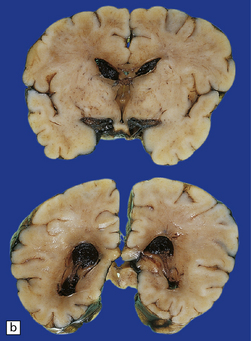
2.30 SAH due to CPH in a second twin born by cesarean section after cord prolapse at 37 weeks’ gestation.
The infant had necrotizing enterocolitis, disseminated intravascular coagulation, a cardiac arrest, and renal failure, and survived 15 days. (a) Massive SAH. (b) The cause is evident as bleeding from the choroid plexus. There is also extensive softening of periventricular white matter, but no trace of SEH.
ACUTE WHITE MATTER LESIONS
 PATHOGENESIS OF WMN
PATHOGENESIS OF WMN The most widely held theory is that WMN results from impaired perfusion at the boundary zone between ventriculopetal and ventriculofugal arteries where the metabolic requirements of myelinating white matter are high (see Fig. 2.41).
The most widely held theory is that WMN results from impaired perfusion at the boundary zone between ventriculopetal and ventriculofugal arteries where the metabolic requirements of myelinating white matter are high (see Fig. 2.41).

 Hemorrhagic lesions may result from venous infarction (see Fig. 2.36).
Hemorrhagic lesions may result from venous infarction (see Fig. 2.36).


MACROSCOPIC APPEARANCES
Typical lesions manifest as yellow–white spots or cavities with a chalky white border. These measure up to several millimeters in diameter and may be single or multiple, and unilateral or bilateral. They are situated in periventricular white matter anterior to the frontal horns, adjoining the lateral angles of the lateral ventricles, and in the temporal acoustic and optic radiations. WMN may also be invisible, or evident as a diffuse gray area with soft consistency. It occasionally presents as hemorrhagic softening in the subcortical, callosal, and capsular regions, which may show extensive cavitation (Figs 2.31–2.37).
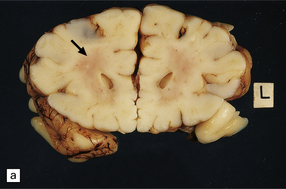
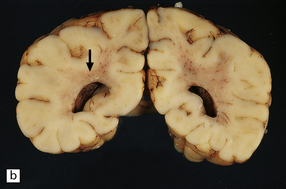
2.31 WMN.
An infant born by cesarean section for fulminating pre-eclampsia at 31 weeks’ gestation who suffered bronchial stenosis and pulmonary collapse and survived 2 months. Small white spots and linear streaks (arrows) in the centrum semiovale indicate focal necrosis. The lesions stand out against the rather congested white matter. (a) Frontal lobes. (b) Parietal lobes.
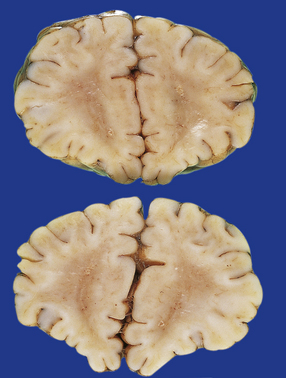
2.32 WMN.
Tiny necrotic foci in the frontal white matter have a central cavity surrounded by a chalky white rim. This is the same case as shown in Fig. 2.28.
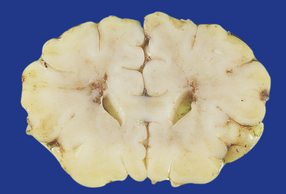
2.33 WMN resulting in bilateral periventricular softening around the angles of the frontal horns.
This occurred in an infant delivered at term who had congenital heart disease and survived 10 days.
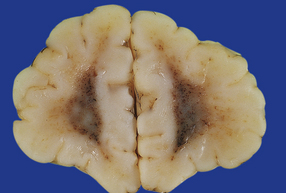
2.34 WMN producing only a diffuse duskiness of the white matter.
There are no focal lesions in this case, which is the same as that shown in Fig. 2.16.
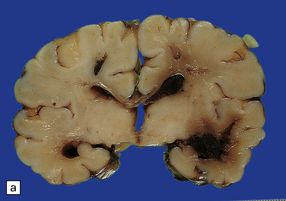
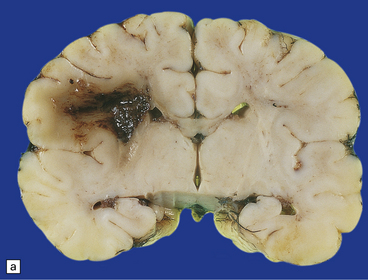
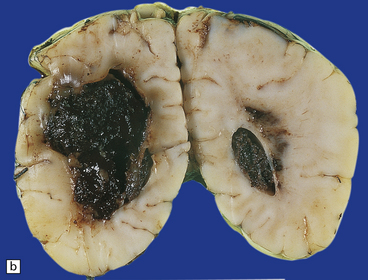
2.35 WMN.
This occurred in an infant born at 37 weeks’ gestation with hypoglycemia and hyperinsulinemia due to nesidioblastosis who survived 10 days. Irregular linear cavities fan out from the lateral angle of the ventricle into frontal and parietal white matter. There is also considerable IVH due to hemorrhagic softening of the temporal white matter. (a) Coronal section at mid-thalamic level. (b) Coronal section at anterior thalamic level.
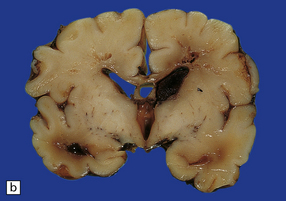
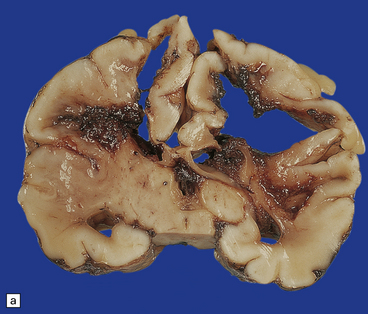
2.36 WMN.
Markedly asymmetric hemorrhagic necrosis dorsal and lateral to the angles of the lateral ventricles. This may mimic grade 4 IVH on ultrasound examination. (a) Coronal section at mid-thalamic level. (b) Coronal section through occipital lobes. Obstruction to the terminal vein is thought responsible for venous infarction.
MICROSCOPIC APPEARANCES
Soon after the insult, irregular zones of coagulative necrosis are surrounded by rings of intense eosinophilia. At a later stage, fragmented and swollen axons are seen at the periphery of the lesion, with microglia, reactive astrocytes, and macrophages. Alternative appearances include cavities surrounded by gliosis and mineralized axons and vessels; a gliotic and microcystic parenchyma with clusters of foamy macrophages; and (rarely) massive infarcts consisting largely of macrophages (Figs 2.38–2.43).
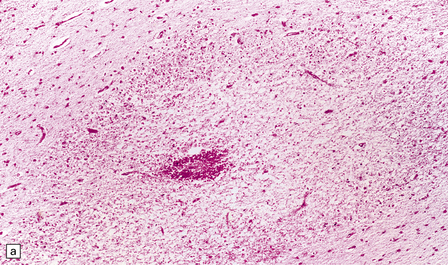
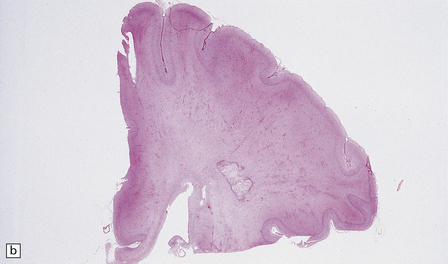
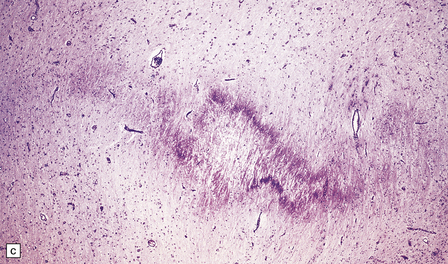
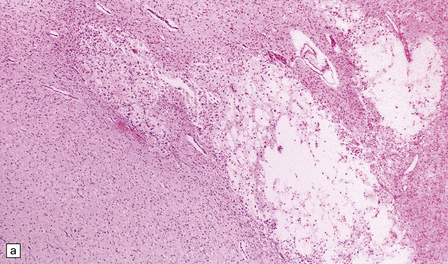
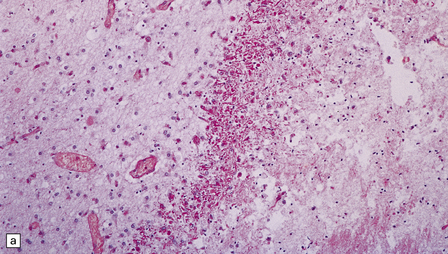
2.38 Microscopic features of white matter necrosis.
(a) With hematoxylin and eosin, the acute lesion (a focus of coagulative necrosis) is indicated only by a zone of pallor. In this case there is also a central petechial hemorrhage. (b) Periodic acid–Schiff (PAS) often outlines acute focal lesions more clearly, as shown here where it reveals a typical focus in the periventricular white matter of the frontal lobe. There was a coexisting germinal matrix hemorrhage and polymicrogyria. This occurred in a twin born at 36 weeks’ gestation who survived 12 days. (c) Luxol fast blue outlines acute focal lesions. Here there is central pallor surrounded by an irregular rim of accentuated staining. This occurred in an infant born at 33 weeks’ gestation with multiple congenital abnormalities including congenital heart disease, who survived 13 days. (d) Occasionally, acute lesions are evident as ill-defined patches of increased PAS staining.
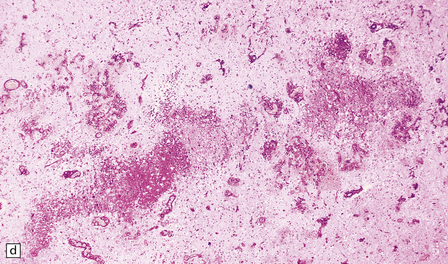
2.39 Acute lesions of WMN.
(a) Multiple foci of necrosis have coalesced within the central white matter (in the same case as shown in Fig. 2.37). (b) Focal necrosis within the corpus callosum is evident as a central cavity and macrophages surrounded by astrocytosis.
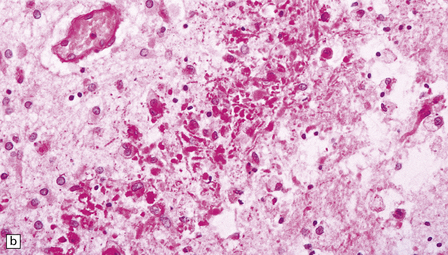
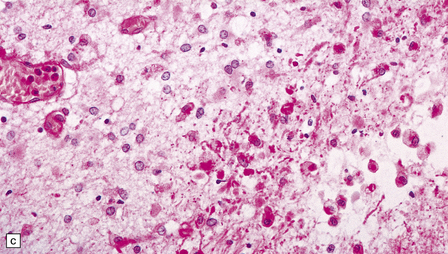
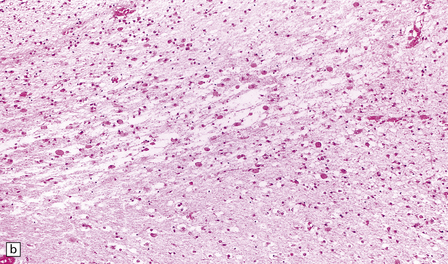
2.40 WMN; early course of the lesion.
Within a few days there is marked cellular infiltration by foamy macrophages and reactive astrocytes around the necrotic focus that contains stick-like debris residual to either necrotic axons or vessels. (a) Low power view of a necrotic focus surrounded by tissue debris and cellular infiltrate. (b) Debris and reactive astrocytes at the edge of the lesion. (c) Infiltrate of reactive astrocytes and foamy macrophages neighboring the necrotic zone.
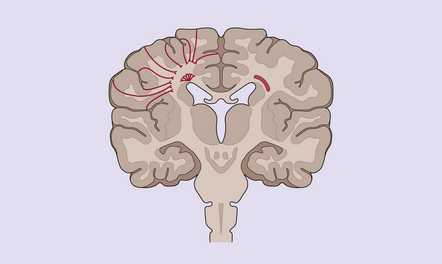
2.41 WMN.
Indicated is the most widely held view of the pathogenesis of the focal softenings of WMN. Note the correspondence between the most common position of these lesions in periventricular regions (shown in the right hemisphere) and the putative boundary zone between ventriculopetal deep branches of superficially derived cerebral arteries and ventriculofugal arteries derived from lenticulostriate and choroidal arteries.
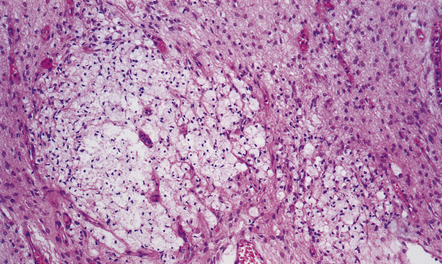
2.42 WMN showing further organization of the lesion into nodular collections of lipid phagocytes associated with capillary proliferation.
This occurred in an infant who survived 3 weeks.
2.43 Extensive WMN.
If white matter destruction is very extensive, soft friable tissue may appear autolyzed at brain cut. This occurred in a term infant who was delivered by forceps after fetal distress in the second stage and who then developed seizures from 6 hours of age and survived 1 week. (a) Frontal lobe section. (b) Microscopy of this lesion. The white matter has been replaced by sheets of macrophages, some in mitosis.
 WMN
WMN



OTHER PATTERNS OF WHITE MATTER DAMAGE
 Subcortical leukomalacia, which is focal necrosis preferentially sited beneath the deep sulci.
Subcortical leukomalacia, which is focal necrosis preferentially sited beneath the deep sulci.
 Telencephalic leukoencephalopathy (Fig. 2.44), which is characterized by diffuse depletion of myelination glia, astrocytic proliferation and hypertrophy, karyorrhectic glial nuclei, and amphophilic globules. The hypertrophic reactive glia should not be confused with normal myelination glia, that have a smaller, more hyperchromatic nucleus, and a more fusiform cytoplasmic profile (Fig. 2.45).
Telencephalic leukoencephalopathy (Fig. 2.44), which is characterized by diffuse depletion of myelination glia, astrocytic proliferation and hypertrophy, karyorrhectic glial nuclei, and amphophilic globules. The hypertrophic reactive glia should not be confused with normal myelination glia, that have a smaller, more hyperchromatic nucleus, and a more fusiform cytoplasmic profile (Fig. 2.45).
2.44 Telencephalic leukoencephalopathy.
This is characterized by poorly cellular white matter, loss of myelination glia, diffuse gliosis, karyorrhexis in glial nuclei, and amphophilic globules. Histology from the corpus callosum of a 1-month-old infant with severe congenital heart disease, cleft lip, and pituitary and adrenal hypoplasia. Typical features are demonstrated here. (a) The white matter is poorly cellular, with loss of myelination glia, and diffuse gliosis. Some of the glial nuclei are karyorrhectic. (b) Amphophilic globules are also observed within the gliotic white matter.
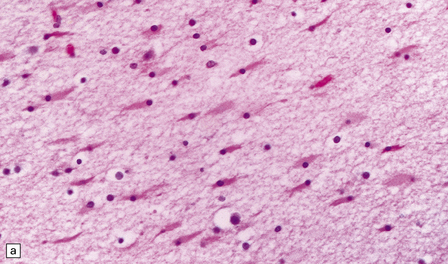
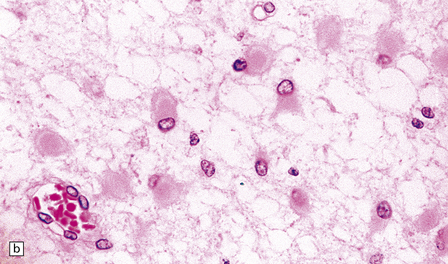
2.45 Myelination glia and reactive gliosis in the neonatal brain.
(a) Myelination glia have small round hyperchromatic nuclei eccentrically set in oval or elongated basophilic cytoplasm. (b) Astrocytic hypertrophy is less pronounced than in older subjects. Reactive astrocytes have an eccentrically placed nucleus and plump cytoplasm, often containing a central region of strong eosinophilia surrounded by an ill-defined, less intensely stained halo.
ACUTE GRAY MATTER LESIONS
MACROSCOPIC APPEARANCES
Diffuse cerebral edema may lead to flattened gyri and obliterated sulci, and occasionally there will be massive swelling with softening of the entire brain. A white or hemorrhagic ribbon of cortex contrasts with dusky congested white matter (Figs 2.46, 2.47). Smaller lesions may be scattered, restricted to an arterial territory, or localized to watershed regions. The depths of sulci are preferentially involved (Fig. 2.48).
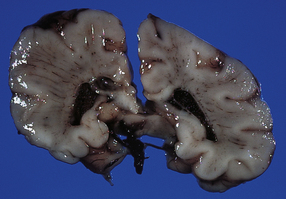
2.46 Cerebral necrosis: the ribbon effect.
Produced by severe hypoxia affecting both cortex and white matter, the paradoxically pale cerebral cortex stands out against the gray duskiness of the white matter. This occurred in an infant delivered by emergency cesarean section following a flat fetal tocograph trace at 38 weeks’ gestation whose Apgar score was 1 at 10 min. Autopsy 1 day later revealed recent colonic perforation with peritonitis.
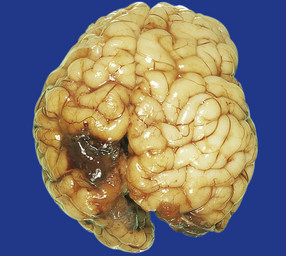
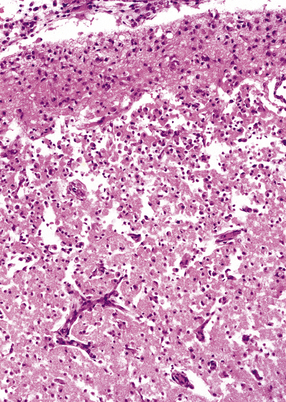
MICROSCOPIC APPEARANCES
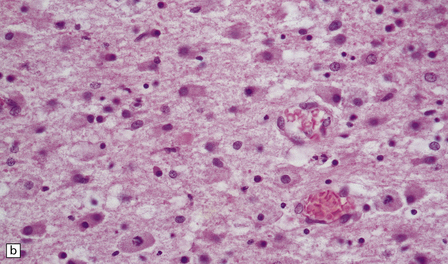
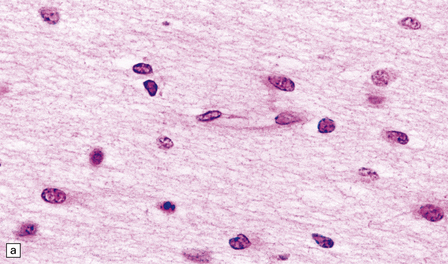
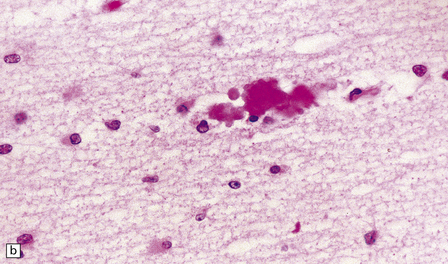
Very early lesions may exhibit only focal pallor of staining in the cortical gray matter. Hypoxic–ischemic changes in immature neurons are characterized by nuclear pyknosis and karyorrhexis (see Figs 2.48, 2.54). Laminar or full-thickness necrosis may also be observed (Fig. 2.49). Activated microglia, astrocytosis, foamy macrophages, and capillary proliferation are seen in lesions after survival for several days (Fig. 2.50). Mineralized neurons are seen when survival exceeds 2 weeks (Fig. 2.51).
2.49 Organizing cortical necrosis with a pseudolaminar pattern.
Astrocytic hyperplasia and hypertrophy in the superficial layers overlie a zone of lipid phagocytes and capillary proliferation.
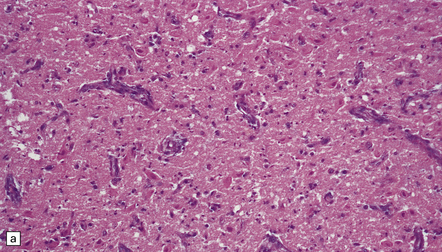
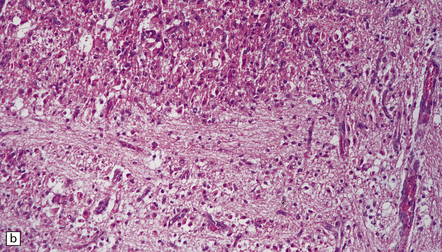
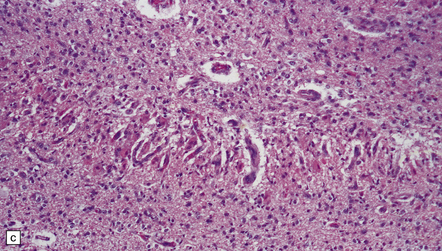
2.50 Severe birth asphyxia resulting in extensive gray matter damage.
This occurred in a 6-day-old neonate who had no registrable heart rate on the monitor during delivery, an Apgar score of 4 at 10 min, no spontaneous respiration, and later seizures and disseminated intravascular coagulation. There was diffuse cortical necrosis and widespread involvement of the basal ganglia, brain stem, and cerebellum. Subacute lesions show marked cellularity, prominent capillaries, neuronal loss, and the characteristic changes of hypoxia in mature neurons (i.e. cytoplasmic eosinophilia) in the remaining cells. (a) Subacute lesions in the substantia nigra. (b) Subacute lesions in the dentate nucleus. (c) Subacute lesions in pontine gray matter.
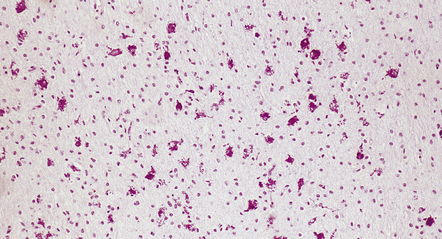
PONTOSUBICULAR NECROSIS
This term describes the occasional association between hypoxic death of neurons in the ventral pontine nuclei and the subiculum in neonates; despite the terminology, the mode of neuronal death in the pontine and subicular neurons is predominantly apoptotic. Pontosubicular ‘necrosis’ rarely occurs in isolation from other hypoxic lesions (Fig. 2.52).
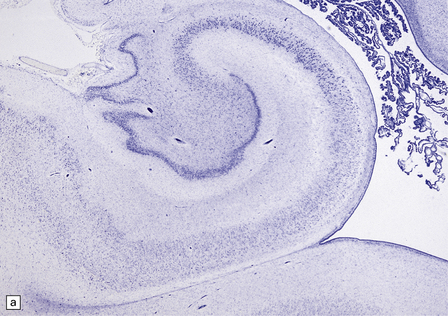
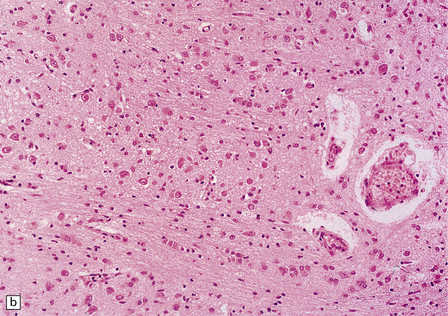
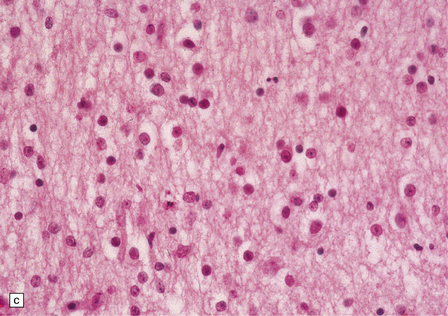
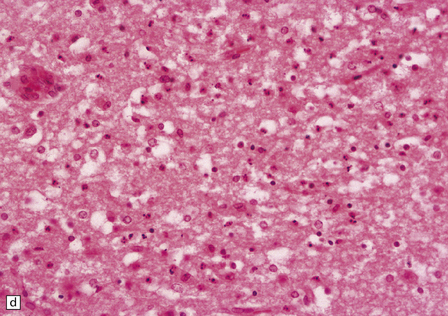
2.52 Pontosubicular necrosis.
This is the association between ischemic destruction of the subiculum and pontine nuclei. It is uncommon, and rarely occurs in isolation from more widespread hypoxic damage. (a) Low power view of Ammon’s horn showing neuronal loss from Sommer’s sector and subiculum. (b) Severe neuronal depletion and astrocytosis in the basis pontis. (c) Karyorrhectic nuclei indicate ischemic neuronal damage to immature neurons. (d) Karyorrhectic neuronal nuclei in hippocampal pyramidal sector.
 PATHOGENESIS OF GRAY MATTER LESIONS
PATHOGENESIS OF GRAY MATTER LESIONS
 As in older subjects, the pathology of HIE is complex and multifactorial in the immature brain.
As in older subjects, the pathology of HIE is complex and multifactorial in the immature brain.


• glutamate-mediated excessive stimulation of NMDA, kainate and AMPA receptors (excitotoxicity)
• increased intracellular uptake of sodium ions and water, causing swelling and lysis of organelles
• increased intracellular uptake of calcium ions, causing activation of nitric oxide synthases and calcium-dependent proteases, and delayed cell death.
 Excess production of free radicals (NO, OH,
Excess production of free radicals (NO, OH, 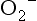 ) enhances tissue damage.
) enhances tissue damage.

 CEREBRAL NECROSIS
CEREBRAL NECROSIS


BASAL GANGLIA AND THALAMIC LESIONS
Hypoxic–ischemic damage in these regions varies from focal neuronal necrosis to large areas of infarction. The latter usually occur in conjunction with widespread CNS involvement (Fig. 2.53). The initiating injury may be postnatal, perinatal, or intrauterine. Damage to these regions also forms part of an extensive destruction of brain stem reticular formation and central gray nuclei which follows perinatal cardiac arrest (Fig. 2.54).
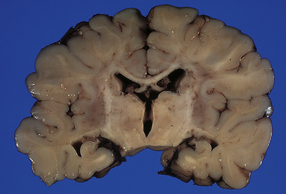
2.53 Prominent hypoxic–ischemic damage in the dusky putamina and shrunken mottled thalami.
The grayish periventricular tissue indicates that white matter is also involved. This pathology was found in a 6-month-old infant whose mother had hypertension in the third trimester and a spontaneous delivery at 36 weeks’ gestation.
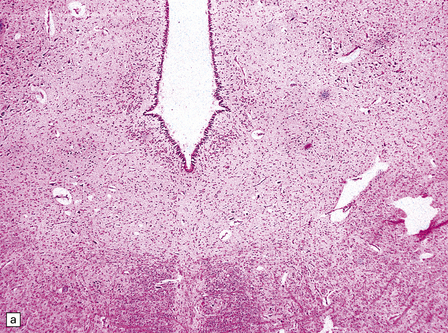
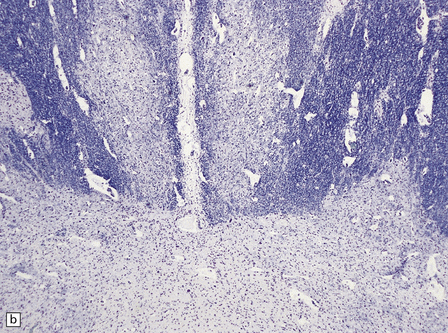
2.54 Reticular core necrosis.
Destruction was confined to the thalamic and brain stem nuclei in this 7-week-old infant born at term with a true knot in the cord resulting in apnea and hypotonia, an Apgar score of 2 at 10 min, seizures from 30 min, and later hypotonia and striking cranial nerve palsies. (a) There is marked neuronal loss and astrocytosis in the XII nucleus. (b) Marked neuronal loss and astrocytosis is also evident in the gracile and cuneate nuclei in the lower medulla.
CEREBELLAR LESIONS
Although all cortical layers and dentate neurons (Fig. 2.50b) may be affected, the immaturity of Purkinje cells before 37 weeks’ gestation means that they are less vulnerable to hypoxia than are the internal granular layer cells. Various patterns of involvement are seen: diffuse, focal damage at the SCA/PICA watershed, or infarction of the deeper parts of the folia (Fig. 2.55). In premature neonates, hypoxia may cause apoptosis of internal granule cells.
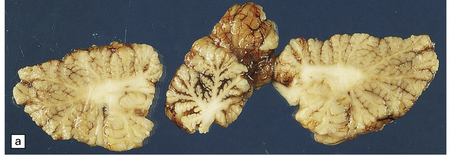
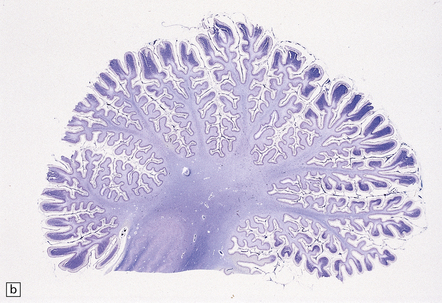
2.55 Necrosis of the deep cerebellar cortex with sparing of only the more superficial parts of the folia.
This is one particular pattern of selective vulnerability to hypoxia in neonates. (a) Cerebellum from a 5-month-old infant delivered by cesarean section for fetal distress who had an Apgar score of 5 at 10 min. (b) Cerebellum from a 6-month-old infant delivered by emergency cesarean section for cord prolapse and multiple abnormalities including congenital heart disease.


CHRONIC LESIONS
PERIVENTRICULAR CYSTS
Small bleeds within the germinal matrix become transformed by macrophage infiltration into unilocular or honeycomb subependymal cysts (Fig. 2.56). Longitudinal imaging studies have shown rapid resolution without neurologic deficit. Similar cysts have also been related to congenital infection.
POST-WHITE MATTER NECROSIS
The hallmarks of birth injury comprise two overlapping patterns (Figs 2.57–2.59):
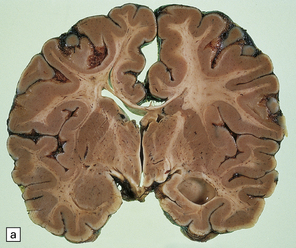
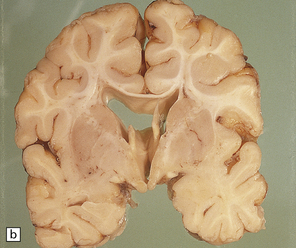
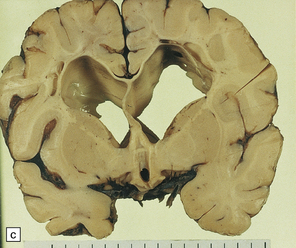
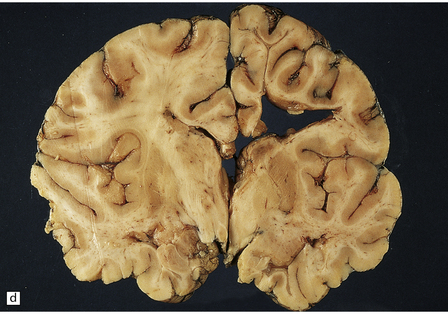
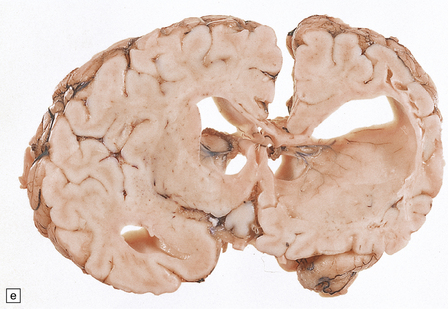
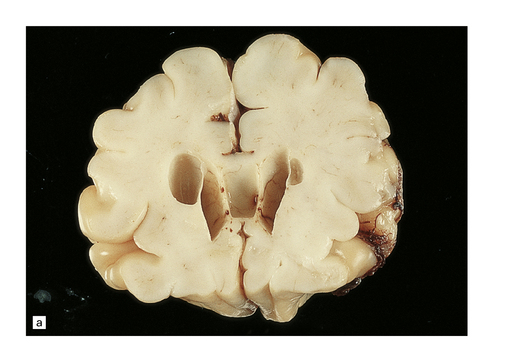
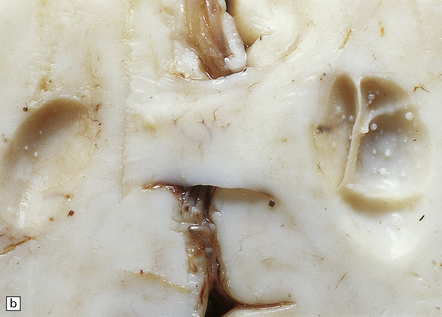
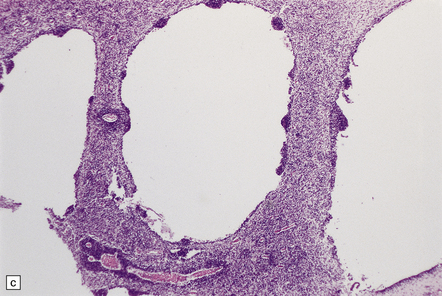
2.57 Chronic lesions in white matter.
These so-called hallmarks of birth injury have widely varying appearances. Both unilateral and bilateral lesions are usually accompanied by ventricular dilatation. (a) A partly cystic linear gliotic scar in the left frontal white matter above the dilated left frontal horn. (b) Bilateral gliotic white matter scars and small periventricular cysts. There is also marked ventriculomegaly. (c) Prominent cyst formation following the contours of the dilated frontal horns. (d) Extensive unilateral cystic destruction of the central and subcortical white matter in the frontal lobe. (e) Unilateral white matter atrophy and ventricular dilatation. Here the overlying cortex is also involved and shows ulegyria.
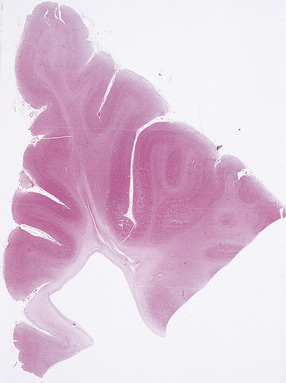
2.58 Chronic slit-like defect in the frontal white matter.
Ultrasound scanning in the perinatal period had shown a considerably larger area of cystic necrosis, which had dramatically reduced in size over several months.
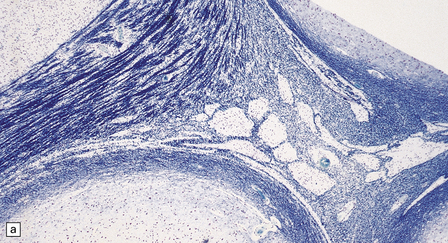
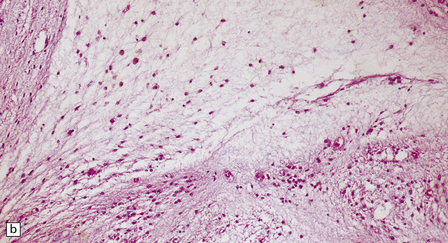
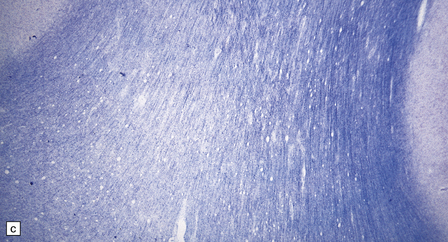
2.59 Chronic longstanding white matter lesions.
(a) Cyst formation in the central white matter of an 8-year-old girl. (b) Dense gliosis surrounding a paucicellular area containing foamy macrophages in a 17-year-old man. (c) Poor myelination, which is the likely sequel to telencephalic leukoencephalopathy.
 Sclerotic atrophy characterized by myelin loss, gliotic scars, macrophage clusters, and mineralized debris in the centrum semiovale and callosum, accompanied by ventricular dilatation.
Sclerotic atrophy characterized by myelin loss, gliotic scars, macrophage clusters, and mineralized debris in the centrum semiovale and callosum, accompanied by ventricular dilatation.

In practice there is often a combination of white and gray matter scars.
POST-GRAY MATTER NECROSIS
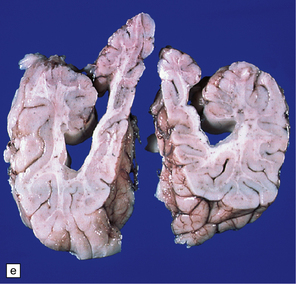
ULEGYRIA AND CORTICAL MARBLING
Many factors influence the final pathology of gray matter necrosis including:



Extreme cases appear severely microcephalic, with cysts and a thin, firm, white cortex (Fig. 2.60). The sparing of gyral crowns results in a mushroom-like appearance called ulegyria (Figs 2.60d, 2.61). Cortical scarring may be full-thickness or laminar. The glial scar is sometimes associated with hypermyelination (état fibromyélinique) (Fig. 2.60c); mineralized neurons and lipid-laden macrophages can remain in situ for many years.
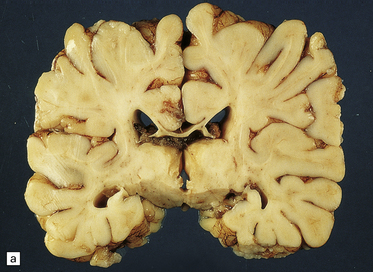
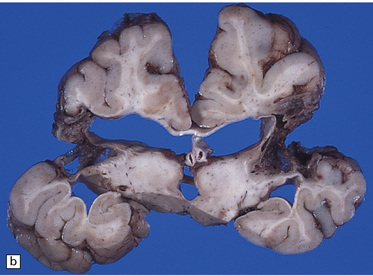
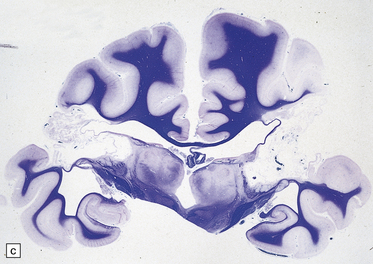
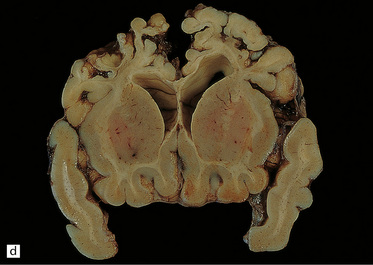
2.60 Various patterns of chronic gray matter damage.
(a) Global atrophy following diffuse cortical involvement. (b) Bilateral symmetric scars due to perinatal cortical infarctions restricted to the territory of the middle cerebral arteries in a 21-year-old man. (c) Stained section from (b) showing irregular myelination (marbling) in the thalami and left superior temporal gyrus (état fibromyélinique). (d) Severe cerebral atrophy with sparing of the crests of the convolutions producing mushroom-shaped gyri, termed ulegyria (compare with Fig. 2.61).
2.61 Chronic gray matter damage.
Severe and extensive gyral scarring or ulegyria, hippocampal sclerosis, sclerotic atrophy of the thalamus, and cystic destruction of the lentiform nucleus and internal capsule are seen in a 6-month-old infant, who was delivered by emergency cesarean section for cord prolapse and had multiple congenital abnormalities including tracheoesophageal fistula, congenital heart disease, and a rudimentary limb. (a) Low power coronal section at anterior thalamic level. (b) Low power coronal section of temporal lobe. (c) Horizontal section of the midbrain showing secondary atrophy of the cerebral peduncles.
STATUS MARMORATUS
Chronic lesions of the basal ganglia and thalamus are gliotic and/or cystic (Fig. 2.61); mineralized neurons are common, but marbling is rare. Marbling appears as irregular white mottling and shrinkage of the deep gray matter (Fig. 2.62) and is due to abnormal hypermyelination in association with glial scars. There is evidence that the hypermyelination is, in part, due to the formation of myelin sheaths around astrocyte processes. Status marmoratus occurs after both prenatal and postnatal hypoxia, provided that the damage takes place before the onset of myelination at about six months postpartum.
2.62 Status marmoratus.
(a) Coronal section at the level of the corpus striatum. Both striatal nuclei are shrunken, their dorsal surfaces abutting the ventricle are corrugated, while their cut surfaces show an irregular white mottling characteristic of marbling. (b) Microscopic appearance of marbling in the basal ganglia showing an excessive and abnormally situated myelination. (c) An adjacent microscopic section to (b) showing the close association between the hypermyelination and gliosis as demonstrated by GFAP immunoreactivity. (d) Similar aberrant myelination in the surviving cortex neighboring an infarcted area (état fibromyélinique).
UNILATERAL HYPERTROPHY OF THE PYRAMIDAL TRACT
An uncommon consequence of midgestational unilateral lesions involving sensorimotor cortex or internal capsule is hypotrophy of the ipsilateral corticospinal pathway and enlargement (reflecting excess fibers) of the contralateral tract (Fig. 2.63).
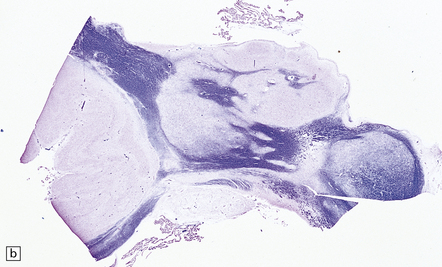
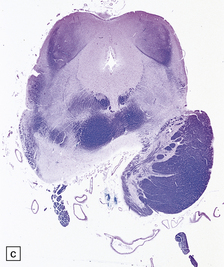
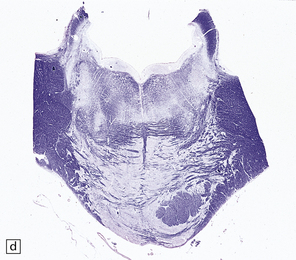
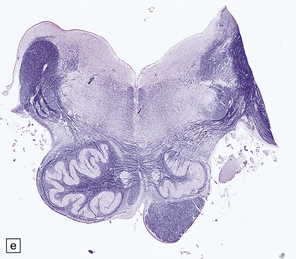
2.63 Unilateral hypertrophy of the pyramidal tract in an 11-month-old girl.
This infant had a gestational history of threatened miscarriage at eight weeks followed by recurrent maternal ill health. (a) Coronal section at the anterior thalamic level shows left-sided ventricular dilatation, atrophy of the thalamus, internal capsule, and lentiform nucleus, and polymicrogyria of the insula (arrow). (b) At a slightly more posterior level on the left side there is shrinkage of the internal capsule and atrophy with marbling of the left thalamus. (c)–(e) Horizontal sections through the brain stem showing ipsilateral (left) hypotrophy and contralateral (right) enlargement of the pyramidal tract. (c) Midbrain. (d) Mid-pons. (e) Medulla. (f) In the cervical spinal cord the contrast between the pyramidal tracts is maintained in both crossed and uncrossed pathways.
CROSSED CEREBELLAR ATROPHY
Rarely, extensive unilateral cerebral damage resulting from an intrauterine or postnatal insult results in atrophy of the contralateral cerebellar hemisphere (Fig. 2.64). The histologic findings vary, and with them the suggested pathogenesis. Some cases show a simple reduction in hemispheric size following transneuronal atrophy of the ipsilateral pontine nuclei. A few show additional granule cell degeneration suggesting tertiary anterograde transneuronal degeneration.
2.64 Crossed cerebellar atrophy.
Presentation in a five-month-old twin with ventriculomegaly detected ultrasonically at 32 weeks’ gestation. (a) Coronal view of the hemispheres showing marked left ventricular dilatation, longstanding destruction of much of the hemispheric cortex and white matter, and thalamic atrophy. (b) There is obvious asymmetry of the hindbrain when viewed from below with ipsilateral (to the supratentorial damage) atrophy of the left pontine base and contralateral atrophy of the right cerebellar hemisphere.
KERNICTERUS
Kernicterus is selective yellow staining of deep gray matter and brain stem nuclei occurring in bilirubin encephalopathy (Fig. 2.65).
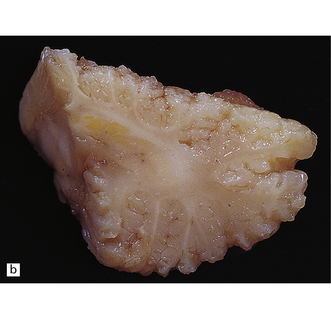
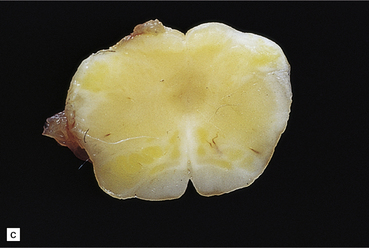
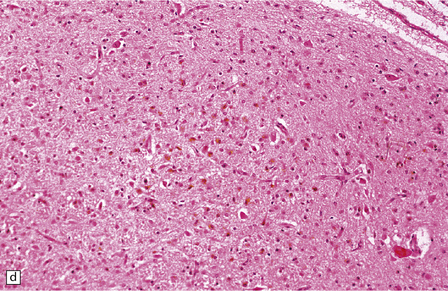
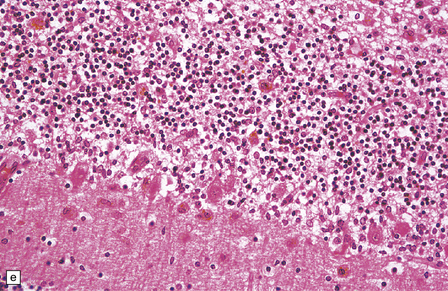
2.65 Kernicterus.
This is characterized by bright yellow staining of affected gray matter structures. (a) Hippocampus and subthalamic nucleus. (b) Dentate nucleus. (c) Inferior olives. (d) Histologically, there is intracellular yellow pigment (bilirubin) in the gracile nucleus. (e) Intracellular bilirubin in the cerebellar cortex.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The subthalamic nucleus, globus pallidus, and lateral thalamus are characteristically affected. Other affected areas are the hippocampus CA2 field, lateral geniculate nucleus, the colliculi, the substantia nigra pars reticularis, the brain stem reticular formation, the cranial nerve nuclei, Purkinje cells, and the dentate and olivary nuclei (Fig. 2.66). The staining is best appreciated in unstained frozen sections, but remains for prolonged periods after formalin fixation. Yellow pigment is also visible in paraffin sections within the vacuoles of swollen cells and in glia (Fig. 2.65). The accumulation of bilirubin causes neuronal necrosis and later mineralization and gliosis.
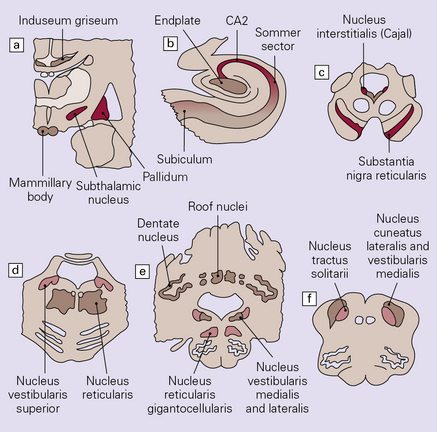
2.66 Diagrammatic representation of the regional distribution of lesions in kernicterus.
(a) Coronal section at the level of the mamillary bodies. (b) Hippocampus. (c) Mid-brain. (d) Pons. (e) Cerebellum and upper medulla. (f) Lower medulla. Key: red, most vulnerable; pink, moderate vulnerability; beige, slight vulnerability.
 PATHOGENESIS OF KERNICTERUS
PATHOGENESIS OF KERNICTERUS


REFERENCES
Ahbdab-Barmada, M., Moossy, J., Painter, M. Pontosubicular necrosis and hyperoxemia. Pediatrics. 1980;66:840–847.
Ahbdab-Barmada, M., Moossy, J. The neuropathology of kernicterus in the premature neonate; diagnostic problems. J Neuropathol Exp Neurol.. 1984;43:45–56.
Armstrong, D.D. Neonatal encephalopathies. In: Duckett S., Malver P.A., eds. Pediatric neuropathology. Baltimore: Williams & Wilkins; 1995:334–351.
Armstrong, D.L., Sauls, C.D., Goddard-Finegold, J. Neuropathologic findings in short-term survivors of intraventricular hemorrhage. Am J Dis Child.. 1987;141:617–621.
Back, S.A., Luo, N.L., Borenstein, N.S., et al. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci.. 2001;21:1302–1312.
Bignami, A., Ralsron, H.J. Myelination of fibrillary astroglial processes in long term Wallerian degeneration. The possible relationship to ‘status marmoratus’. Brain Res.. 1968;11:710–713.
DeReuck, J., Chattha, A.S., Richardson, E.P., Jr. Pathogenesis and evolution of periventricular leukomalacia in infancy. Arch Neurol.. 1972;27:229–236.
De Vries, M.S., Groenendaal, F., Eken, P., et al. Infarcts in the vascular distribution of the middle cerebral artery in preterm and fullterm infants. Neuropediatrics.. 1997;28:88–96.
Dubowitz, L.M., Bydder, G.M., Mushin, J. Developmental sequence of periventricular leukomalacia. Correlation of ultrasound, clinical, and nuclear magnetic resonance functions. Arch Dis Child.. 1985;60:349–355.
Edwards, A.D., Yue, X., Cox, P., et al. Apoptosis in the brains of infants suffering intrauterine cerebral injury. Pediatr Res.. 1997;42:684–689.
Folkerth, R.D., Kinney, H.C. Disorders of the perinatal period. In Love S., Louis D.N., Ellison D.W., eds.: Greenfield’s Neuropathology, 8th ed., London: Edward Arnold, 2008.
Friede, R.L. Ponti-subicular lesions in perinatal anoxia. Arch Pathol.. 1972;94:343–354.
Friede, R.L., Schachenmayr, W. Early stages of status marmoratus. Acta Neuropathol.. 1977;38:123–127.
Ghazi-Birry, H.S., Brown, W.R., Moody, D.M., et al. Human germinal matrix: venous origin of hemorrhage and vascular characteristics. Am J Neuroradiol.. 1997;18:219–229.
Gilles, F.H., Price, R.A., Kevy, S.V., et al. Fibrinolytic activity in the ganglionic eminence of the premature human brain. Biol Neonate.. 1971;18:426–432.
Goddard-Finegold, J. Periventricular intraventricular hemorrhages in the premature newborn. Update on pathologic features, pathogenesis, and possible means of prevention. Arch Neurol.. 1984;41:766–771.
Gourley, G.R. Bilirubin metabolism and kernicterus. Adv Pediatr.. 1987;44:173–229.
Larroche, J.C., Encha-Razavi, F. The central nervous system. In: Wigglesworth J.S., Singer D.B., eds. Textbook of fetal and perinatal pathology. Oxford: Blackwell Scientific; 1991:778–842.
Leech, R.W., Alvord, E.C., Jr. Morphologic variations in perventricular leukomalacia. Am J Pathol.. 1974;74:591–602.
Leviton, A., Gilles, F.H. Acquired perinatal leukoencephalopathy. Ann Neurol.. 1984;16:1–8.
Marin-Padilla, M. Developmental neuropathology and impact of perinatal brain damage. I: Hemorrhagic lesions of neocortex. J Neuropathol Exp Neurol.. 1996;55:758–773.
Marin-Padilla, M. Developmental neuropathology and impact of perinatal brain damage. II: White matter lesions of the neocortex. J Neuropathol Exp Neurol.. 1997;56:219–235.
Marin-Padilla, M. Developmental neuropathology and impact of perinatal brain damage. III: Grey matter lesions of the neocortex. J Neuropathol Exp Neurol.. 1999;58:407–429.
Mito, T., Kamei, A., Takashima, S., et al. Clinicopathological study of pontosubicular necrosis. Neuropediatrics.. 1993;24:204–207.
Nakamura, Y, Okudera, T., Hashimoto, T. Vascular architecture in white matter of neonates: its relationship to periventricular leukomalacia. J Neuropathol Exp Neurol.. 1994;53:582–589.
Papile, L.A., Burnstein, J., Burnstein, R., et al. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500gm. J Pediatr.. 1978;92:529–534.
Portera-Cailliau, C., Price, D.L., Martin, L.J. Excitotoxic neuronal death in the immature brain is an apoptosis–necrosis morphological continuum. J Comp Neurol.. 1997;378:70–87.
Rorke, L.B. Pathology of perinatal birth injury. New York: Raven Press; 1982.
Sherwood, A.J., Smith, J.F. Bilirubin encephalopathy. Neuropathol Appl Neurobiol.. 1983;9:271–285.
Volpe, J.J. Neurology of the newborn, 3 rd ed. Philadelphia: Saunders; 1995.
Volpe, J.J. Subplate neurons – missing link in brain injury of the premature infant? Pediatrics.. 1996;97:112–113.
Wilson, E.R., Mirra, S.S., Schwartz, J.F. Congenital diencephalic and brain stem damage; neuropathologic study of three cases. Acta Neuropathol.. 1982;57:70–74.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]2
Fetal and neonatal hypoxic–ischemic lesions
The extraordinarily rapid growth and plasticity of the immature nervous system.
The major changes in cell and tissue properties that occur during development.
PATHOGENESIS OF HYPOXIC–ISCHEMIC LESIONS
Possible pathophysiologic schema
Principal factors involved in the production of lesions:
Present knowledge of embryology and developmental physiology allows a tentative chronology to be assigned to individual lesions (Fig. 2.1) although this remains relatively imprecise and extreme caution is needed in the forensic arena. Although some pathologic changes closely parallel those found in older individuals, others such as intraventricular hemorrhage, white matter necrosis, and marbling are unique to the perinatal period.
2.1 A timetable for hypoxic–ischemic lesions in early life.
Hydr, hydranencephaly; BB, basket brain; Por, porencephaly; MCE, multicystic encephalopathy; SEH, subependymal hemorrhage; CPH, choroid plexus hemorrhage; WMN (PVL), white matter necrosis (periventricular leukomalacia); PSN, pontosubicular necrosis; C/UI, cortical necrosis/ulegyria; Th/BG, thalamus/basal ganglia damage.
FETAL LESIONS
HYDRANENCEPHALY
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Much of the cerebral mantle is replaced by a thin translucent membrane. The hemispheres are cystic, and lack surface convolutions (Fig. 2.2). Inferior aspects of the temporal, occipital, and frontal lobes are usually spared, but sometimes only the hippocampi remain. The deep gray nuclei may be rotated outwards and the thalami are atrophied. The membranous cerebral mantle comprises an outer connective tissue layer and an inner irregular glial layer which also contains mineralized neurons, debris, and hemosiderin-laden macrophages. The ependyma is usually absent. At the interface with surviving cerebral tissue, the inner glial layer runs into the molecular layer, covering it for a short distance, while adjacent cortex is usually disorganized, often with a pattern of polymicrogyria (Fig. 2.3). This histologic appearance clearly distinguishes hydranencephaly from the macroscopically similar bubble brain in Fowler syndrome (see Fig. 7.2).
2.2 Hydranencephaly in a twin of 22 weeks’ gestation whose co-twin died at 17 weeks.
(a) The bubble-like hemispheres photographed under water and viewed from above. (b) Removal from water allows the diaphanous membrane of the cyst wall to collapse. Damage is largely restricted to the internal carotid artery territory with sparing of the orbitofrontal cortex and inferior temporal and occipital lobes.
BASKET BRAIN
PORENCEPHALY AND SCHIZENCEPHALY
MACROSCOPIC AND MICROSCOPIC APPEARANCES
A porus is a smooth-walled defect in the cerebral mantle, usually surrounded by an abnormal gyral pattern. It varies in depth from a slight indentation or fissure to a full-thickness breach of the hemispheric wall connecting subarachnoid space with ventricle. Pori tend to be bilateral, approximately symmetric, and situated over the Sylvian fissures or central sulci. Unilateral defects may be parasagittal, orbital, or occipital, and associated with abnormal convolutions, especially polymicrogyria, which are symmetrically placed in the contralateral hemisphere. Gyri surrounding the defect form irregular or radiating patterns (Fig. 2.4). The cortex around the porus is broken into islands or folded into polymicrogyria, which extends down into the cleft to meet the ventricular wall. The latter is denuded of ependyma, but covered by glial tissue, which extends a short way over the adjacent gray matter (Fig. 2.5). Subependymal nodular heterotopia (Fig. 2.6d) and partial or complete absence of the septum pellucidum are other features.
2.4 Porencephaly.
(a) Two unilateral pori are present. The shallow depression in the orbitofrontal cortex contrasts with the wide-mouthed full-thickness defect in the occipital lobe allowing communication between the subarachnoid space and the ventricle. (b) The surrounding convolutions have a radiating pattern. (c) Coronal section through the frontal lobes showing the extent of the anterior defect, which is lined by nodules of gray matter. The septum pellucidum is absent.
2.5 Porencephaly.
Coronal section through a frontal porus in a 48-year-old man. The cortical gray matter dips down into the cleft to meet the white gliotic ependyma. The cortical ribbon around the edge of the defect is irregularly thickened and disorganized. (Courtesy of Dr T Moss, Frenchay Hospital, Bristol.)
2.6 Schizencephaly.
(a) Bilateral porencephalic clefts centered on the middle cerebral artery territory in a twin of 32 weeks’ gestation whose co-twin died at 19 weeks. The hemispheres are viewed from above. (b) Coronal section at anterior thalamic level. The cortex in the center of the lesion is a completely disorganized mixture of neuroblasts and macrophages surrounded either side by polymicrogyric cortex. (c) Coronal section at striatal level. Polymicrogyric cortex from the anterior border of the cleft. (d) Large bilateral full-thickness defects in a 4-year-old boy with cerebral palsy and epilepsy. The covering leptomeninges ruptured during the dissection, revealing a radial pattern of convolutions around the clefts. (e) Coronal sections through the posterior parts of the hemispheres show thick polymicrogyric cortex curling over the mouths of the clefts and deeply invaginating the ventricle. Note the heterotopic gray nodules in the inferior corners of the ventricles.
Extensive bilateral porencephalic clefts (Fig. 2.6) are sometimes termed schizencephaly, especially in the radiologic literature, although the term schizencephaly refers back to an outmoded concept of circumscribed growth failure of the cerebral wall. The narrowness of the cleft with either closed or open lips was thought to differentiate malformed from acquired lesions, but clinical, morphologic, and experimental evidence favors a destructive origin for most lesions. There is also recent evidence for familial occurrence of schizencephaly in brothers with defects in the homeobox gene EMX2.
MULTICYSTIC ENCEPHALOPATHY
In contrast to the smooth-walled defects of hypoxic–ischemic lesions of early gestation, third trimester insults produce many cysts throughout large areas of cerebral white matter and deeper cortical layers (Figs 2.7, 2.8). Occasional cases are unilateral and circumscribed (Fig. 2.9), but most are extensive and bilateral. Bilateral multicystic encephalopathy is often associated with cystic necrosis in the basal ganglia and brain stem tegmentum (Fig. 2.10).
2.7 Multicystic encephalopathy in a smaller first twin at 36 weeks’ gestation.
(a) Superior view of the brain within the skull. The convolutions are either massively cystically dilated or narrow and thin. (b) Coronal section showing dilated ventricles and replacement of much of the cortex and white matter by an irregular firm white cystic meshwork.
2.8 Multicystic encephalopathy.
(a) Tough gliovascular bands transform much of the deep cortex, white matter, and basal ganglia into a spongy mass. The thalamic nuclei are shrunken and extremely white. (b) Comparable histologic section from another patient showing that not only the cortex and white matter, but also the thalami and basal ganglia have been replaced by large cysts.
MICROSCOPIC APPEARANCES
A meshwork of thin gliovascular septa form numerous cysts containing lipid-laden macrophages. Global hemispheric necrosis (Fig. 2.11) is the term used to describe morphologically similar but particularly extensive cases following severe birth asphyxia and sudden hyperpyrexia and collapse in the postnatal period.
PERINATAL LESIONS
Hemorrhage is the commonest pathology in fetal and neonatal brains. It can start in any intracranial compartment and rapidly spread elsewhere. The pathogenesis is multifactorial. The principal risk factors are immaturity, perinatal distress, and asphyxia, though a similar spatial distribution in still and live births also implicates antenatal factors. The variety of hemorrhages in the neonatal brain is shown in Fig. 2.12.
SUBDURAL HEMORRHAGE (SDH)
SDH is not related to asphyxia, but is closely related to perinatal distress. Excessive compression and distortion of the head during delivery can disrupt the superior cerebral bridging veins that drain into the sagittal sinus, or the veins to the transverse sinus. More rarely, the falx and sagittal sinus or the tentorium (Fig. 2.13) with the straight sinus or great vein of Galen is torn.
SUBARACHNOID HEMORRHAGE (SAH)
SAH (Figs 2.14–2.16) may manifest as:
2.15 Well-demarcated subarachnoid hematomas localized over parietal cortex.
This was associated with a threatened abortion at 20 weeks’ gestation.
2.16 A small localized SAH on the temporal pole as well as extensive basal SAH secondary to hemorrhagic white matter infarction.
This occurred in a term infant with multiple congenital anomalies who was cyanosed at birth.
SUBPIAL HEMORRHAGE (SPH)
SPH is often confused with SAH. It is a focal hemorrhage, which is usually temporal, parietal, or cerebellar, and occurs with or without other signs of bleeding. It is most common in asphyxiated premature infants, but also occurs in premature or term infants with respiratory distress syndrome or congenital heart disease (Figs 2.17, 2.18).
2.17 SPH in a term infant with multiple congenital anomalies including cyanotic congenital heart disease.
(a) Anterior coronal sections in the frontal lobes showing right-sided central white matter softening and several superficial hemorrhages. (b) Microscopy reveals an entirely intracortical bleed covered by pia.
SUBEPENDYMAL GERMINAL PLATE/MATRIX HEMORRHAGE (SEH)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Acute SEH has variable appearances: small, multiple, and bilateral bleeds occur anywhere in periventricular matrix tissue (Fig. 2.19), including the roof of the fourth ventricle. Acute SEH is usually found
2.19 SEH in an infant with hypoplastic left heart syndrome born by cesarean section at 36 weeks’ gestation who survived 3 days.
(a) Small SEH that has remained within the matrix zone (grade 1) in the most typical position near the terminal vein close to the caudate nucleus and thalamus. (b) The small bleed remains in situ within the cellular matrix tissue. The ependyma is intact. No bleeding point can be ascertained.
