[level-membership-for-neurology-category]
Chapter 55 Epilepsy and Neurodevelopmental Disorders
Introduction
The association of epilepsy with neurodevelopmental disorders is now well established. Children with developmental epilepsies are at increased risk for cognitive [Hermann and Seidenberg, 2007], neurobehavioral [Hermann et al., 2008], and psychiatric disorders [Plioplys et al., 2007]. Specific factors associated with a higher risk of neuropsychological deficits in children with epilepsy include multiple seizures, use of antiepileptic drugs, etiology, and interictal epileptiform discharges (IEDs) on the initial electroencephalogram (EEG) [Fastenau et al., 2009]. In children with epilepsy, younger age at seizure onset, cognitive impairment, temporal or frontal lobe onset of seizures, and intractable epilepsy are associated with an increased likelihood of coexisting social, communication, and behavioral disorders [Hamiwka and Wirrell, 2009]. The multiple complex and confounding risk factors that account for the coexistence of epilepsy and neurodevelopmental disorders include etiology, associated disabilities, seizure type, frequency, duration, control, age of onset, and IEDs, as well as psychosocial factors and medication effects [Austin and Caplan, 2007].
The relationship between epilepsy and neurodevelopmental disorders also reflects common pathologies and mechanisms [Tuchman et al., 2009], and there is accumulating evidence to suggest that cognitive and behavioral impairments may precede the onset of seizures [Austin et al., 2001; Oostrom et al., 2003; Bhise et al., 2009; Fastenau et al., 2009]. On the other hand, there is evidence that recurrent seizures or IEDs can cause acute and long-lasting impairment of brain function and development [Holmes and Lenck-Santini, 2006; Galanopoulou and Moshe, 2009], and significant efforts are being made to determine how risk factors contribute to developmental outcomes in children with epilepsy [Austin and Fastenau, 2009]. The notion that epileptic activity, seizures, or IEDs can lead to cognitive and behavioral impairment above and beyond what might be expected from the underlying pathology is exemplified by the concept of epileptic encephalopathy [Berg et al., 2009]. This idea has important implications for treatment, especially in children with an epileptic encephalopathy, in whom early and successful treatment of seizures and interictal epileptiform activity may be crucial to positive neurodevelopmental outcomes [Freitag and Tuxhorn, 2005; Jonas et al., 2005; Lux et al., 2005; Arts and Geerts, 2009; Bombardieri et al., 2009].
Epilepsy is more frequent in children with intellectual disability [Goulden et al., 1991]; the behavioral, cognitive, and social aspects of epilepsy are multiple and diverse, and are discussed in Chapter 62. This present chapter focuses on the concept of epileptic encephalopathy and the association of epilepsy and IEDs with specific language impairments and autistic spectrum disorders (ASDs). In addition, the common mechanisms of disease and management of children with developmental epilepsies and neurodevelopmental disorders are addressed.
Epileptic Encephalopathies
The epileptic encephalopathies can occur at any age, but are more common and severe in the first decade of life as the brain is developing. Approximately 40 percent of epilepsies occurring during the first 3 years of life are associated with an epileptic encephalopathy [Guerrini, 2006]. Epileptic encephalopathy of childhood includes: early myoclonic encephalopathy (Ohtahara’s or Aicardi’s syndrome), severe myoclonic epilepsy of infancy (Dravet’s syndrome), myoclonic-astatic epilepsy of early childhood (Doose’s syndrome), infantile spasms (West’s syndrome), Lennox–Gastaut syndrome, and Landau–Kleffner syndrome-continuous spike waves during slow-wave sleep [Engel, 2001].
The myoclonic epilepsies and infantile spasms are discussed in Chapter 56 and Lennox–Gastaut syndrome in Chapter 53. In this chapter, Dravet’s syndrome, infantile spasms, Lennox–Gastaut syndrome, Landau–Kleffner syndrome, and continuous spike waves during slow-wave sleep will be discussed from a developmental perspective. Specifically, the impact of seizures and of IEDs on the developmental trajectories of children with these electroclinical syndromes will be emphasized.
Dravet’s Syndrome
Dravet’s syndrome is a genetically determined infantile epileptic encephalopathy, mainly caused by de novo mutations in the SCN1A gene [Scheffer et al., 2009]. Early development is normal, with seizures usually beginning in the first 10 months of life; these become frequent by 2–4 years of age, with focal and generalized myoclonus, atypical absences, and partial complex seizures, as well as seizures characterized by fluctuating alteration of consciousness with reduced postural tone and myoclonic jerks [Millichap et al., 2009]. Despite frequent seizures, the EEG is usually normal in the first 2 years of life and then usually progresses into spike and wave and multifocal discharges, although some children may have persistently normal interictal EEG studies [Korff et al., 2007]. Progressive decline or plateau in development occurs by 1–4 years of age, with intellectual disability and an autism phenotype commonly present, especially in those with more than five seizures per month [Scheffer et al., 2009].
Genes associated with Dravet’s syndrome appear to have relevance to the neurodevelopmental disorders, such as ASDs. Specifically, a susceptibility locus for ASDs has been found on chromosome 2 in the vicinity of the epilepsy-involved genes SCN1A and SCN2A [Weiss et al., 2003], while PCDH10 on chromosome 4 has been linked to ASDs in families with shared ancestry [Morrow et al., 2008]. Recently, a sporadic infantile epileptic encephalopathy that resembles Dravet’s syndrome has been tied to mutations in PCDH19 [Depienne et al., 2009]. These results hint strongly at possible shared molecular underpinnings between ASD and this epileptic encephalopathy.
Infantile Spasms
Infantile spasms constitute an age-specific seizure disorder, with a peak age of presentation between 4 and 8 months of age, a critical time of brain development [Zupanc, 2009]. There is a strong correlation between infantile spasms and intellectual disability [Trevathan et al., 1999], and the majority of children with infantile spasms develop intellectual disability and specific cognitive and behavioral deficits [Riikonen, 2001]. In addition, the association of infantile spasms and ASDs is well recognized [Riikonen and Amnell, 1981]; in children with infantile spasms the prevalence of ASDs is as high as 35 percent, depending on the severity of intellectual disability [Saemundsen et al., 2007], with a heightened risk of ASDs in the presence of identifiable structural lesions of the brain [Saemundsen et al., 2008]. Further, in children with infantile spasms, EEG epileptiform activity – particularly bilateral frontal EEG discharges and persistence of hypsarrhythmia – contributes to the development of the autism phenotype [Kayaalp et al., 2007].
One of the intriguing features of infantile spasms is that the severity and, to a certain extent, the frequency of seizures do not seem to correlate with the degree of cognitive impairment or the occurrence of regression [Riikonen, 1984; Bednarek et al., 1998]. It may be that the interictal EEG pattern of hypsarrhythmia associated with infantile spasms affects cortical and subcortical neuronal networks, resulting in abnormal synaptogenesis and poor developmental outcomes [Zupanc, 2009].
Lennox–Gastaut Syndrome
Lennox–Gastaut syndrome is an age-specific epileptic syndrome that peaks between 3 and 5 years; it is characterized by multiple seizure types that include tonic and atonic seizures, atypical absences, and myoclonic and generalized or focal seizures, in association with a characteristic EEG pattern of slow spike-and-wave complexes, often associated with multifocal epileptiform activity and runs of fast activity [Crumrine, 2002]. Approximately 20 percent of children with this epileptic syndrome have a history of infantile spasms [Markand, 2003]. Cognitive and behavioral abnormalities precede the clinical seizures in approximately 20–60 percent of children with Lennox–Gastaut syndrome [Blume, 2001].
It has been suggested that the epileptic processes associated with this syndrome lead to patterns of abnormal activity and connectivity that compete with normal brain development, thus resulting in subsequent impairment or regression of cognition [Blume, 2004]. It is not known whether it is the underlying brain pathology, the burden of frequent seizures, the persistent epileptiform activity, or all of these factors that is responsible for the cognitive deficits in Lennox–Gastaut syndrome. The age specificity, typically frequent seizures, and unremitting epileptiform activity suggest that the epileptic activity occurring at a critical time in brain development contributes to a progressive disturbance in cerebral function.
Landau–Kleffner Syndrome-Continuous Spike Waves During Slow-Wave Sleep
Landau–Kleffner syndrome (LKS) is an acquired aphasia associated with an epileptiform EEG with spikes, sharp waves, or spike-and-wave discharges that are usually bilateral and occur predominantly over the temporal regions; in approximately 25 percent of children there are no clinical seizures [Landau and Kleffner, 1998]. Continuous spike-wave discharges during slow-wave sleep (CSWS), an epileptic encephalopathy associated with the EEG pattern of electrical status epilepticus during slow-wave sleep, with various seizure types, and with cognitive, motor, and behavioral disturbances, is, along with LKS, considered a sleep-related epileptic encephalopathy [Tassinari et al., 2009].
CSWS and LKS are epileptic encephalopathies with common clinical features, including seizures, regression, and epileptiform abnormalities that are activated by sleep [Nickels and Wirrell, 2008]. In CSWS there is a regression in global skills, while in LKS the primary clinical manifestation is a regression of language. The conceptual thinking is that there is a spectrum of disorders associated with activation of epileptiform activity during slow-wave sleep that includes LKS, CSWS, and “atypical” benign epilepsy with centrotemporal spikes (BECTS) [Fejerman, 2009; Scheltens-de Boer, 2009]. The implications are that the EEG abnormalities lead to regression in language, behavioral, and cognitive manifestations and that treatment requires reversal of the epileptiform pattern on the EEG. However, whether the frequency of the epileptiform discharges, as linked to the appearance or disappearance of electrical status epilepticus during slow-wave sleep, is responsible for the course of language, cognitive, and behavioral deterioration [Tassinari et al., 2000; Seri et al., 2009] remains an unanswered and controversial issue.
Seizures, Interictal Epileptiform Discharges, and Neurodevelopmental Disorders
IEDs, characterized by spikes or sharp waves that appear abruptly from the electrographic background, with or without an associated slow wave, are limited in duration and do not evolve in frequency and distribution over time, but can at times be responsible for disruption of cognitive and behavioral function [Fisch, 2003]. The concept that transitory changes in higher cortical functions can be secondary to EEG discharges not accompanied by seizures was proposed more than 60 years ago [Schwab, 1939]. The term transient cognitive impairment is used to describe individuals with epileptiform EEG discharges in association with a momentary disruption of adaptive cerebral function [Aarts et al., 1984; Binnie, 2003].
Studies by numerous investigators suggest that transient cognitive impairment is not a consequence of general impairment of attention, but is likely secondary to disruption of functions located in the region or regions of the brain where the epileptiform discharges arise [Kasteleijn-Nolst Trenite, 1995; Aldenkamp and Arends, 2004]. Specific functions, such as perception, reaction time, and scholastic performance, can be disrupted by brief epileptiform discharges in the absence of convulsive seizures [Shewmon and Erwin, 1988; Sengoku et al., 1990]. The spectrum of language and cognitive impairment secondary to an active epileptic focus, even in the absence of clinical seizures, is wide [Deonna and Roulet-Perez, 2005; Roulet-Perez and Deonna, 2006].
Specific Language Impairment
Specific language impairment is a developmental language disorder characterized by varying types and degree of dysfunction in expressive and receptive communication skills (see Chapter 45). Children with specific language impairments differ from children with acquired aphasia characteristic of the epileptic encephalopathies, such as LKS-CSWS, in that there is no regression of language skills. Nevertheless, the impairments of language in epilepsies such as BECTS raises interesting questions regarding the overlap and interactions of epilepsy, IEDs, and specific language impairments [Billard et al., 2009].
An increased association of seizures in children with specific language impairments has been found [Robinson, 1991; Echenne et al., 1992], with prevalence rates from 7–40 percent reflecting the cohort studied and the type and duration of the EEG recording [Parry-Fielder et al., 1997]. In one study that compared the rates of epilepsy in children with autistic spectrum versus developmental language disorders, the prevalence of epilepsy was 8 percent (14 of 168) in the nonautistic, language-impaired children. In addition, when the children were subtyped on the basis of language, the highest risk of epilepsy, regardless of whether the children had ASD or specific language impairment, was seen in those individuals with the most severe receptive language disorder [Tuchman et al., 1991; Klein et al., 2000]. A link between specific language impairment and IEDs during sleep has also been suggested [Parry-Fielder et al., 1997; Ballaban-Gil and Tuchman, 2000; Wheless et al., 2002]. However, the strength of this association has been questioned in a recent study in which 13 percent of children with specific language impairments had IEDs on a sleep EEG recording, which was not significantly different than the non-language-impaired control group [Parry-Fielder et al., 2009].
Autistic Spectrum Disorders
ASD is a broad classification that includes a heterogeneous group of individuals with behaviorally defined impairments in reciprocal social interaction, verbal and nonverbal communication, and restricted and repetitive behaviors (see Chapter 48). The prevalence of epilepsy in children with ASD is highly variable, depending on the study sample, with rates ranging from 5 to 46 percent [Spence and Schneider, 2009]. The major risk factor for epilepsy in children with ASD is moderate to severe intellectual disability [Amiet et al., 2008]. In children with ASD without severe intellectual disability, motor deficit, associated perinatal or medical disorder, or a positive family history of epilepsy, the prevalence of epilepsy is 6 percent, which is not significantly different than in nonautistic children with a specific language impairment [Tuchman et al., 1991]. The prevalence of ASD in individuals with epilepsy has not been investigated with the same intensity as that of epilepsy in ASD, although one study in a tertiary epilepsy clinic found that approximately 30 percent of children with epilepsy screened positive for ASD [Clarke et al., 2005].
The prevalence of IEDs in children with ASD and no clinical history of seizures may be as high as 60 percent [Chez et al., 2006], but varies depending on the cohort studied and the type of EEG performed, with most studies finding prevalence rates ranging from 6 to 31 percent [Kagan-Kushnir et al., 2005]. The higher prevalence of IEDs in children with specific language impairment and in those with ASD compared to the 1.5–4 percent rate of IEDs in the general population [Cavazzuti et al., 1980; Capdevila et al., 2008] has been a source of significant controversy. The significance of these findings remains unclear and is not unique, as IEDs have been reported in 6–30 percent of children with attention-deficit hyperactivity disorders [Hughes et al., 2000; Richer et al., 2002; Kaufmann et al., 2009], which is remarkably similar to the prevalence of IEDs in children with specific language impairment or ASDs without seizures. One suggestion is that the high prevalence of IEDs in all of these disorders is secondary to the neuropathological processes common to both epilepsy and neurodevelopmental disorders. However, the question of whether these IEDs may be a biomarker of prognostic or interventional significance in a subgroup of children with specific language impairment or with ASDs remains to be determined.
Regression
The association of developmental regression in ASD to epilepsy and IEDs became a topic of increased interest after Hagberg and colleagues [Hagberg et al., 1983] published a report of 35 girls with regression in higher brain functions, stereotypical hand movements, and ASD, with a significant proportion of the girls having epilepsy. Childhood disintegrative disorder is characterized by late-onset autistic and cognitive regression that can include motor regression and loss of bowel and bladder function, usually occurring after age 3 [Rapin, 1995]. Childhood disintegrative disorder is based on the description, in the early 1900s, of children with normal development until age 3 or 4 years, who regressed in multiple developmental areas. This entity was first described by Heller in 1908 [Mouridsen, 2003], and differentiation from autism, especially from autistic regression, is still in progress [Kurita et al., 2004]. Disintegrative disorder is very rare, with a prevalence of 2 in 100,000 [Fombonne, 2009].
The prevalence of epilepsy in Rett’s syndrome and in childhood disintegrative disorder is greater than 70 percent, both disorders have EEGs with marked IEDs, and both featuring regression of cognitive, language, and social skills [Mouridsen et al., 1999; Steffenburg et al., 2001]. Some children with childhood disintegrative disorder overlap with those having epilepsy and CSWS [Roulet Perez et al., 1993]. To what extent the high rate of seizures in these groups is secondary to the severe cognitive impairment present in Rett’s syndrome and childhood disintegrative disorder, or what influence other specific variables, such as metabolic or molecular factors (i.e., the role of MECP2), have in the development of seizures remains unknown.
The developmental trajectory in approximately 30 percent of children with ASD is characterized by a regression of the few words acquired and a loss of nonverbal communication skills, usually occurring prior to 24 months of age [Goldberg et al., 2003; Lord et al., 2004]. This regression has been termed autistic regression. The relation of autistic regression to epilepsy or to an epileptiform EEG without seizures remains controversial, with some studies reporting higher rates of epilepsy in children with ASD and regression [Kobayashi and Murata, 1998; Hrdlicka et al., 2004], and others showing no relation between ASD, epilepsy, and regression [Tuchman and Rapin, 1997; Baird et al., 2008]. A recent study found that children with autistic regression had more disrupted sleep, as compared to those with autism without regression, and were more likely to have epilepsy [Giannotti et al., 2008]. In addition, Giannotti and colleagues found that epileptiform activity did not differ among those with and without regression, except that those with autistic regression were more likely than those without regression to have more “frequent epileptiform EEGs.” There is evidence to suggest that, in a subgroup of children with ASD and without convulsive seizures, an epileptiform EEG is significantly more likely to be associated with a history of regression in language [Tuchman and Rapin, 1997]. However, these data must be put into perspective, as they represent a very specific subgroup of children with autism, and because at the present time there are no data regarding the number of children in the general population without seizures and cognitive and behavioral impairments who have interictal epileptiform abnormalities on an overnight EEG study. Others have found no differences in regression in those with epileptiform EEGs and epilepsy and those without seizures and a normal EEG [Canitano et al., 2005]. Children with autistic regression and an epileptiform EEG (AREE) should be differentiated from those with LKS-CSWS, and these differences are highlighted in Table 55-1.
Table 55-1 Landau–Kleffner Syndrome-Continuous Spike Waves During Slow-Wave Sleep Versus Autistic Regression with Epileptiform EEG
| Landau–Kleffner Syndrome-Continuous Spike-Waves During Slow-Wave Sleep (LKS-CSWS) | Autistic Regression with Epileptiform EEG (AREE) | |
|---|---|---|
| Age of regression/(symptoms) | Usually after 3 years. Peak age 3–5 years. In CSWS may be as late as 12 years of age | Usually prior to age 2 years with a mean age of regression of 21 months |
| Seizures | Usually not frequent or intractable. In approximately 25 percent, seizures are not present | Seizures are not part of phenotype. In autistic spectrum disorders, when seizures occur, they are usually not frequent and responded well to antiepileptic medications |
| EEG | Spikes, sharp waves, or spike-and-wave discharges, usually bilateral and occurring predominantly over the temporal regions. They increase during sleep; EEG pattern of electrical status epilepticus during slow-wave sleep is common | Infrequent spikes, usually centrotemporal. Rarely associated with CSWS. No clear correlation with interictal epileptiform discharges and improvement or worsening of underlying language and social dysfunction |
| Treatment | See Box 55-1. Case reports, mostly with use of steroids, suggest improvement in language. Surgical outcomes with multiple subpial transections are variable. No controlled clinical trials | No evidence that present medical interventions (antiepileptic medications) or surgical interventions are indicated |
| Outcome/Comments | Improvement occurs in late childhood/early adolescence. Approximately one-third recover. Prognosis for seizure control is excellent but recovery of language is variable and not as good as for seizures | Improvement seems related to cognitive skills. No data to determine if interictal discharges combined with regression are marker for worse prognosis |
The mean age of onset of language regression in ASDs is 21 months, and over 90 percent of children with autism who undergo a regression do so before age 3 years [Tuchman and Rapin, 1997]. By contrast, in LKS, only 12–14 percent of children experience regression before age 3 years [Bishop, 1985]. The peak age of onset of symptoms in LKS is between 5 and 7 years [Bureau, 1995]. In only 5 percent of individuals does LKS begin after age 9 years and it appears to occur rarely, if ever, after age 12 years [Bureau, 1995]. In children with the CSWS, the first symptoms occur in up to 20 percent of children between 9 and 12 years of age [Bureau, 1995].
Children with ASD are more likely to regress earlier, usually prior to age 2, as contrasted to those with LKS who have a regression in language usually after age 3 years, and seizures are more likely to occur in children who regress in language after age 3 years [Klein et al., 2000; Shinnar et al., 2001; Wilson et al., 2003]. In addition, children with isolated language regression have a higher frequency of epileptiform discharges and seizures than children with both language and autistic (social and behavioral) regression [McVicar et al., 2005]. Furthermore, electrical status epilepticus during slow-wave sleep, the EEG pattern associated with LKS and CSWS, is almost exclusively found in those children with isolated language regression [McVicar et al., 2005], and CSWS with autistic regression is a rare occurrence [Tuchman, 2009]. The age of symptom onset seems to be an important indicator of outcome, at least in LKS. In one study, the prognosis for recovery was worse in children with LKS who lost their language at an early age [Bishop, 1985]. In a series of studies, age of language regression differentiates autistic regression from LKS.
In LKS, improvement occurs usually toward late childhood or early adolescence. Approximately one-third of affected children make a good recovery [Mantovani and Landau, 1980]. The prognosis for seizure control and normalization of the EEG is excellent, but prognosis for recovery of language and cognitive function is variable and in general not as good as it is for the seizures [Smith and Hoeppner, 2003]. In the group of children with regression and global cognitive deficits in the context of electrical status epilepticus during slow-wave sleep (ESES), a significant majority is left with some degree of neurological impairment [Rossi et al., 1999; Robinson et al., 2001]. In childhood disintegrative disorder, the prognosis is generally poor [Gillberg, 1991]. In general, children with autistic regression have significant long-term morbidity [Wilson et al., 2003]. What is still not known is whether children with autistic epileptiform regression have a poorer prognosis than those with regression without an epileptiform EEG.
An emerging concept of epileptic encephalopathy suggests a continuum of disorders in which the interictal epileptiform activity activated by sleep may account for regression in varying neurodevelopmental domains (Figure 55-1). This hypothetical model may provide a framework to understand the impact of seizures and interictal epileptiform activity on the developing brain, as well as to unravel the common complex genetic predisposition of these childhood epilepsies [Rudolf et al., 2009]. It also has important implications for the development of pharmaceutical agents that can target and suppress IEDs.
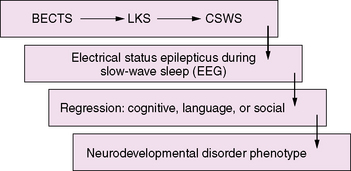
Etiology and Pathophysiology
As a group, the epileptic encephalopathies are associated with regression or slowing of cognitive, language, or behavioral development; the hypothesis is that seizures or the interictal epileptiform activity are responsible for the deterioration [Nabbout and Dulac, 2003]. There are differences between the epileptic encephalopathies in terms of timing, frequency, and severity of the seizures or IEDs, despite the fact that, as a group, they are associated with poor developmental outcomes. In Dravet’s syndrome, the seizures are more predominant than the IEDs, while in infantile spasms the epileptiform activity on the EEG is a more dramatic and likely important determinant of developmental outcome than the seizures. In Lennox–Gastaut syndrome, both the seizures and interictal epileptiform activity seem to play a critical role in terms of neurodevelopmental outcome. To date, studies have failed to dissect the effect of seizures per se from that produced by the underlying disease. A case in point is that individuals with infantile spasms of unknown cause have a better intellectual outcome than those individuals in whom a structural or metabolic etiology is determined.
Numerous etiologies are associated with LKS and CSWS, including neurocysticercosis [Otero et al., 1989], sylvian arachnoid cyst [De Volder et al., 1994], left temporal astrocytoma [Nass et al., 1993; Solomon et al., 1993], and inflammatory demyelinating disease [Perniola et al., 1993], suggesting that this syndrome may result from unilateral brain lesions present during a critical age of development and affecting areas essential for language development, or from multiple and diffuse lesions. In addition, an association between congenital hydrocephalus and CSWS and LKS has been reported [Ben-Zeev et al., 2004], and children with shunted hydrocephalus who develop language or behavioral deterioration may be at high risk for CSWS [Caraballo et al., 2008]. Reports of siblings [Nakano et al., 1989] with LKS have raised the possibility of a genetic basis for this disorder, but other reports of LKS occurring discordantly in monozygotic twins suggest an “environmental trigger” [Feekery et al., 1993]. A variety of different triggers, including an autoimmune process [Nevsimalova et al., 1992], an infectious or inflammatory process [Connolly et al., 1999], and a possible role for an arteritis [Pascual-Castroviejo et al., 1992], have all been proposed as etiologies for LKS. No evidence of encephalitis was found in two patients with LKS who underwent temporal lobectomy [Cole et al., 1988]. LKS, as with other childhood epileptic encephalopathies, has more than one etiology, and what may be important in determining the impact of the epileptic activity on the nervous system and the subsequent neurodevelopmental disorder is the timing of the insult and the extent to which the neural network is affected.
The pathophysiology of epileptic encephalopathies differs and remains poorly understood, but it is likely that the etiology of the epileptic activity, as well as the frequency or location of IEDs alone or in combination, could lead to a specific epileptic encephalopathy. LKS has become the model for understanding the functional mechanisms leading to an epileptic encephalopathy. Findings on functional neuroimaging studies in LKS have been consistent with temporal lobe abnormalities. A study using fluorodeoxyglucose positron emission tomography (PET) during sleep in three males with LKS demonstrated metabolic disturbances that varied among children but predominated over the temporal lobes [Marescaux et al., 1990]. Studies using single photon emission computed tomography imaging (SPECT) have found abnormal perfusion in the left temporal lobe only in LKS [O’Tuama et al., 1992; Guerreiro et al., 1996]. Temporal lobe metabolic abnormalities, as determined with PET scanning in children with LKS, have also been found in association with CSWS [Rintahaka et al., 1995]. A study of four children with LKS demonstrated a reduction in the cortical volume of the superior temporal areas, corresponding to the auditory association cortex with magnetic resonance imaging (MRI) volumetric analysis, with the difference greatest in the two children who had the most frequent temporal lobe epileptiform activity. Although these results do not allow determination of whether the atrophy is the cause of the LKS or the consequence of excitoxicity from the epileptiform discharges, they do support the concept that epileptiform activity may account for language regression in LKS. There is also evidence from magnetoencephalography (MEG) that, in LKS, bilateral discharges are generated in auditory and language areas of the perisylvian cortex, and that 20 percent of children have a unilateral perisylvian pacemaker that triggers bilateral synchrony of the spike discharges [Paetau, 2009].
Two areas of critical importance in understanding the pathophysiology of epileptic encephalopathies are the developmentally related effects of IEDs and seizures, and the common molecular mechanisms that could account for both epilepsy and the neurodevelopmental disorder. Gene defects can affect numerous processes in brain development, such as molecular derangement of ion channels, patterns of cortical neurogenesis leading to malformations of cortical development, and specific protein products, all of which have been implicated in the development of epilepsy and neurodevelopmental disorders [Wisniewski et al., 2001; Crino et al., 2002; Paredes and Baraban, 2002; Noebels, 2003; Muhle et al., 2004]. Genetic abnormalities may be responsible for the EEG pattern and the cognitive dysfunction in children with epilepsy or neurodevelopmental disorders [Roubertie et al., 2003], and the seizures themselves may change neurotransmitter release and gene expression [Kovacs et al., 2003].
Animal model studies have shown that the developmental age at which seizures and IEDs develop is a crucial factor in determining outcome, and that the effects of seizures or IEDs on the developing brain are further modified by age, sex, and underlying pathology [Galanopoulou and Moshe, 2009]. Frequent and prolonged seizures or interictal epileptiform activity are also key factors in determining developmental outcome and likely explain the poor developmental outcomes associated with epileptic encephalopathies [Ben-Ari and Holmes, 2006; Holmes and Lenck-Santini, 2006]. From a treatment perspective, there is animal model evidence that environmental enrichment may ameliorate or reverse the altered gene expression, as well as the cognitive and behavioral aspects caused by the effect of seizures and IEDs on the developing brain [Koh et al., 2005].
Tuberous sclerosis complex (TSC), a neurological disorder commonly associated with both ASD and epilepsy, provides an informative example of common mechanisms that could account for epilepsy and neurodevelopmental disorders [Napolioni et al., 2009]. TSC occurs in about 1 in 6000 live births and is an autosomal-dominant disorder, although up to 60 percent of affected children carry spontaneous mutations of one of two different genes causing the disorder, TSC2 and TSC1 [Curatolo, 2003]. Some 40 percent of individuals with TSC have an IQ below 70 and 30 percent have an IQ below 30; the likelihood of cognitive impairment is associated with a history of seizures and particularly infantile spasms [Joinson et al., 2003]. Even in children with TSC and normal cognitive development, careful neuropsychological testing reveals deficits more commonly than in a control sample of children [Harrison et al., 1999].
Epilepsy occurs in approximately 80–90 percent of children with TSC and is often intractable [Holmes and Stafstrom, 2007]. A retrospective study looking at epileptic risk factors for the development of ASD in TSC found that tubers in the temporal lobes predisposed to ASDs and, more specifically, that it was temporal lobe epileptiform discharges, a history of infantile spasms, and onset of seizures in the first 3 years of life that determined whether or not an individual with TSC developed an ASD [Bolton et al., 2002]. Thus, the relation of epilepsy to neurodevelopmental disorders is complex and determined by underlying pathology, as well as by genetic and epigenetic factors combined with the developmental age, location, frequency, and duration of the epileptic activity.
Management
Management of the child with epilepsy and a neurodevelopmental disorder should be individualized. One needs to weigh the advantages of treating the seizures or IEDs with the potential cognitive and behavioral effects of the antiepileptic medications [French et al., 2004]. This consideration should be balanced with an understanding of the psychotropic mechanisms of action of antiepileptic medications and their role in treating children with epilepsy and coexisting mood and behavioral disorders [Di Martino and Tuchman, 2001; Ettinger, 2006].
Treatment of epileptiform discharges in the absence of clinical seizures remains controversial. The classic teaching is not to treat the EEG but to treat the patient. A degree of skepticism is appropriate when attempting to correlate an epileptiform EEG with specific cognitive, language, or behavioral dysfunction. Evidence from clinical case studies and the occurrence of transient cognitive impairment in some patients suggest that, if appropriate testing is done with concurrent EEG monitoring, a reliable correlation can be made between epileptiform discharges and performance on numerous tasks. The controversy is whether treating this group of children in the absence of seizures is justifiable [Binnie, 2003; Pressler et al., 2005]. Despite the higher prevalence of epileptic activity in neurodevelopmental disorders, such as specific language impairment and ASDs, there is no evidence that treatment of the seizures or the IEDs has a positive impact on language, social, cognitive, or behavioral outcomes [Tharp, 2004; Tuchman, 2004].
Most clinicians would agree that, in the epileptic encephalopathies, such as Dravet’s syndrome, infantile spasms, Lennox–Gastaut syndrome, and LKS-CSWS, early and aggressive treatment is indicated [Arts and Geerts, 2009]. Interventions commonly used in this group of children include antiepileptic drugs, corticosteroids and adrenocorticotropic hormone (ACTH), immunoglobulins, vagal nerve stimulation, the ketogenic diet, and epilepsy surgery (Box 55-1). In LKS-CSWS, several case reports with poorly defined endpoints suggest that corticosteroid therapy improves language [Hirsch et al., 1990; Lerman et al., 1991; Tsuru et al., 2000; Sinclair and Snyder, 2005; Gallagher et al., 2006]. In addition, a number of case reports using a variety of interventions, including nicardipine [Pascual-Castroviejo et al., 1992], vigabatrin [Appleton et al., 1993; Wakai et al., 1997], sulthiame, clobazam [Gross-Selbeck 1995], levetiracetam [Kossoff et al., 2003], diazepam [Mikati and Shamseddine, 2005] immunoglobulins [Mikati et al., 2002], and ketogenic diet [Kossoff et al., 2009; Nikanorova et al., 2009], have been associated with improvements in behavioral and language functioning in children with LKS-CSWS. There are no controlled clinical trials of interventions for children with LKS-CSWS, limiting the ability to make rational decisions regarding appropriate intervention [Lagae, 2009].












In children with ESES exclusive of LKS, the most effective treatment is corticosteroids, which, in one study, were reported to abate the EEG discharges successfully in 65 percent of children; other treatments, such as intravenous immunoglobulin infusions and administration of antiepileptic drugs, were successful in less than 50 percent of cases [Kramer et al., 2008]. In infantile spasms, one study suggested that the use of vigabatrin in children with infantile spasms and tuberous sclerosis improved developmental outcome [Jambaque et al., 2000]; however, there is no substantial evidence that current intervention strategies for infantile spasms improve developmental outcomes [Mackay et al., 2004]. Etiology may be the most reliable predictor of developmental outcome in children with infantile spasms, regardless of intervention [Lux and Osborne, 2006].
Multiple subpial transection (MST) has been the most common neurosurgical treatment for LKS, with multiple reports suggesting postoperative improvement in language function [Sawhney et al., 1995; Grote et al., 1999; Irwin et al., 2001; Guenot, 2004]. What is less clear is whether successful surgical treatment of seizures or epileptiform discharge abatement improves the developmental trajectories of these children [Caplan et al., 1992; Kanner, 2000]. Intellectual capacity influences response to interventions, at least in children with ASD and epilepsy, and current medical and surgical interventions do not appear to alter intellectual ability dramatically [Danielsson et al., 2009], even when surgical elimination of seizures occurs at an early age.
In the management of the epileptic encephalopathies, there is no consensus on which medication to use, when to start one particular treatment compared to another, and what to use as an endpoint for success [Guerrini, 2006; Arts and Geerts, 2009]. There are no comprehensive studies using a multidisciplinary approach that combines medications or surgery to eliminate the seizures or interictal epileptiform activity, with early intensive educative interventions that target communication, behavioral, and educational deficits. If seizures or interictal epileptiform activity interfering with the normal trajectory of development is eliminated, then intensive, frequent, and structured interventions may allow for the plasticity of the brain, now unburdened of the epileptic activity, to overcome the neurodevelopmental deficits.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Aarts J.H., Binnie C.D., Smit A.M., et al. Selective cognitive impairment during focal and generalized epileptiform EEG activity. Brain. 1984;107(Pt 1):293-308.
Aldenkamp A.P., Arends J. Effects of epileptiform EEG discharges on cognitive function: is the concept of “transient cognitive impairment” still valid? Epilepsy Behav. 2004;5(Suppl 1):S25-S34.
Amiet C., Gourfinkel-An I., Bouzamondo A., et al. Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol Psychiatry. 2008;64(7):577-582.
Appleton R., Hughes A., Beirne M., et al. Vigabatrin in the Landau-Kleffner syndrome. Dev Med Child Neurol. 1993;35(5):457-459.
Arts W.F., Geerts A.T. When to start drug treatment for childhood epilepsy: the clinical-epidemiological evidence. Eur J Paediatr Neurol. 2009;13(2):93-101.
Austin J.K., Caplan R. Behavioral and psychiatric comorbidities in pediatric epilepsy: toward an integrative model. Epilepsia. 2007;48(9):1639-1651.
Austin J.K., Fastenau P.S. Are seizure variables related to cognitive and behavior problems? Dev Med Child Neurol. 2009.
Austin J.K., Harezlak J., Dunn D.W., et al. Behavior problems in children before first recognized seizures. Pediatrics. 2001;107(1):115-122.
Baird G., Charman T., Pickles A., et al. Regression, developmental trajectory and associated problems in disorders in the autism spectrum: the SNAP study. J Autism Dev Disord. 2008;38(10):1827-1836.
Ballaban-Gil K., Tuchman R. Epilepsy and epileptiform EEG: association with autism and language disorders. Ment Retard Dev Disabil Res Rev. 2000;6(4):300-308.
Bednarek N., Motte J., Soufflet C., et al. Evidence of late-onset infantile spasms. Epilepsia. 1998;39(1):55-60.
Ben-Ari Y., Holmes G.L. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5(12):1055-1063.
Ben-Zeev B., Kivity S., Pshitizki Y., et al. Congenital hydrocephalus and continuous spike wave in slow-wave sleep – a common association? J Child Neurol. 2004;19(2):129-134.
Berg A., Berkovic S., Brodie M.J., et al. Revised terminology and concepts for organization of the epilepsies: Report of the Commission on Classification and Terminology. Epilepsia. 2009. Web. August 2009. http://www.ilae-epilepsy.org
Bhise V.V., Burack G.D., Mandelbaum D.E. Baseline cognition, behavior, and motor skills in children with new-onset, idiopathic epilepsy. Dev Med Child Neurol. 2009.
Billard C., Fluss J., Pinton F. Specific language impairment versus Landau-Kleffner syndrome. Epilepsia. 2009;50(Suppl 7):21-24.
Binnie C.D. Cognitive impairment during epileptiform discharges: is it ever justifiable to treat the EEG? Lancet Neurol. 2003;2(12):725-730.
Bishop D.V. Age of onset and outcome in ‘acquired aphasia with convulsive disorder’ (Landau-Kleffner syndrome). Dev Med Child Neurol. 1985;27(6):705-712.
Blume W.T. Lennox-Gastaut syndrome: potential mechanisms of cognitive regression. Ment Retard Dev Disabil Res Rev. 2004;10(2):150-153.
Blume W.T. Pathogenesis of Lennox-Gastaut syndrome: considerations and hypotheses. Epileptic Disord. 2001;3(4):183-196.
Bolton P.F., Park R.J., Higgins J.N., et al. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002;125(Pt 6):1247-1255.
Bombardieri R., Pinci M., Moavero R., et al. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2009.
Bureau M.. Outstanding cases of CSWS and LKS: analysis of data sheets provided by the participants. Beaumanoir A., Bureau M., Deonna T., Mira L., Tassinari C.A., editors. Continuous Spikes and Waves During Slow Sleep, Electrical Status Epilepticus During Slow Sleep, Acquired Epileptic Aphasia and Related Conditions. vol 3. London: John Libbey; 1995:213-216.
Canitano R., Luchetti A., Zappella M. Epilepsy, electroencephalographic abnormalities, and regression in children with autism. J Child Neurol. 2005;20(1):27-31.
Capdevila O.S., Dayyat E., Kheirandish-Gozal L., et al. Prevalence of epileptiform activity in healthy children during sleep. Sleep Med. 2008;9(3):303-309.
Caplan R., Guthrie D., Mundy P., et al. Non-verbal communication skills of surgically treated children with infantile spasms. Dev Med Child Neurol. 1992;34(6):499-506.
Caraballo R.H., Bongiorni L., Cersosimo R., et al. Epileptic encephalopathy with continuous spikes and waves during sleep in children with shunted hydrocephalus: A study of nine cases. Epilepsia. 2008.
Cavazzuti G.B., Cappella L., Nalin A. Longitudinal study of epileptiform EEG patterns in normal children. Epilepsia. 1980;21(1):43-55.
Chez M.G., Chang M., Krasne V., et al. Frequency of epileptiform EEG abnormalities in a sequential screening of autistic patients with no known clinical epilepsy from 1996 to 2005. Epilepsy Behav. 2006;8(1):267-271.
Clarke D.F., Roberts W., Daraksan M., et al. The prevalence of autistic spectrum disorder in children surveyed in a tertiary care epilepsy clinic. Epilepsia. 2005;46(12):1970-1977.
Cole A.J., Andermann F., Taylor L., et al. The Landau-Kleffner syndrome of acquired epileptic aphasia: unusual clinical outcome, surgical experience, and absence of encephalitis. Neurology. 1988;38(1):31-38.
Connolly A.M., Chez M.G., Pestronk A., et al. Serum autoantibodies to brain in Landau-Kleffner variant, autism, and other neurologic disorders. J Pediatr. 1999;134(5):607-613.
Crino P.B., Miyata H., Vinters H.V. Neurodevelopmental disorders as a cause of seizures: neuropathologic, genetic, and mechanistic considerations. Brain Pathol. 2002;12(2):212-233.
Crumrine P.K. Lennox-Gastaut syndrome. J Child Neurol. 2002;17(Suppl 1):S70-S75.
Curatolo P., editor. Tuberous sclerosis Complex: From Basic Science to Clinical Phenotypes. MacKeith Press, 2003.
Danielsson S., Viggedal G., Steffenburg S., et al. Psychopathology, psychosocial functioning, and IQ before and after epilepsy surgery in children with drug-resistant epilepsy. Epilepsy Behav. 2009;14(2):330-337.
Deonna T., Roulet-Perez E. Cognitive and behavioral disorders of epileptic origin in children. London UK: MacKeith Press, Cambridge University Press; 2005.
Depienne C., Bouteiller D., Keren B., et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009;5(2):e1000381.
De Volder A.G., Michel C., Thauvoy C., et al. Brain glucose utilisation in acquired childhood aphasia associated with a sylvian arachnoid cyst: recovery after shunting as demonstrated by PET. J Neurol Neurosurg Psychiatry. 1994;57(3):296-300.
Di Martino A., Tuchman R.F. Antiepileptic drugs: affective use in autism spectrum disorders. Pediatr Neurol. 2001;25(3):199-207.
Echenne B., Cheminal R., Rivier F., et al. Epileptic electroencephalographic abnormalities and developmental dysphasias: a study of 32 patients. Brain Dev. 1992;14(4):216-225.
Engel J.Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42(6):796-803.
Ettinger A.B. Psychotropic effects of antiepileptic drugs. Neurology. 2006;67(11):1916-1925.
Fastenau P.S., Johnson C.S., Perkins S.M., et al. Neuropsychological status at seizure onset in children: risk factors for early cognitive deficits. Neurology. 2009;73(7):526-534.
Feekery C.J., Parry-Fielder B., Hopkins I.J. Landau-Kleffner syndrome: six patients including discordant monozygotic twins. Pediatr Neurol. 1993;9(1):49-53.
Fejerman N. Atypical rolandic epilepsy. Epilepsia. 2009;50(Suppl 7):9-12.
Fisch B.J. Interictal epileptiform activity: diagnostic and behavioral implications: 2002 ACNS presidential address. J Clin Neurophysiol. 2003;20(3):155-162.
Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr Res. 2009.
Freitag H., Tuxhorn I. Cognitive function in preschool children after epilepsy surgery: rationale for early intervention. Epilepsia. 2005;46(4):561-567.
French J.A., Kanner A.M., Bautista J., et al. Efficacy and tolerability of the new antiepileptic drugs II: treatment of refractory epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62(8):1261-1273.
Galanopoulou A.S., Moshe S.L. The epileptic hypothesis: developmentally related arguments based on animal models. Epilepsia. 2009;50(Suppl 7):37-42.
Gallagher S., Weiss S., Oram Cardy J., et al. Efficacy of very high dose steroid treatment in a case of Landau-Kleffner syndrome. Dev Med Child Neurol. 2006;48(9):766-769.
Giannotti F., Cortesi F., Cerquiglini A., et al. An Investigation of Sleep Characteristics, EEG Abnormalities and Epilepsy in Developmentally Regressed and Non-regressed Children with Autism. J Autism Dev Disord. 2008;38(10):1888-1897.
Gillberg C. Outcome in autism and autistic-like conditions. J Am Acad Child Adolesc Psychiatry. 1991;30(3):375-382.
Goldberg W.A., Osann K., Filipek P.A., et al. Language and other regression: assessment and timing. J Autism Dev Disord. 2003;33(6):607-616.
Goulden K.J., Shinnar S., Koller H., et al. Epilepsy in children with mental retardation: a cohort study. Epilepsia. 1991;32(5):690-697.
Gross-Selbeck G. Treatment of “benign” partial epilepsies of childhood, including atypical forms. Neuropediatrics. 1995;26(1):45-50.
Grote C.L., Van Slyke P., Hoeppner J.A. Language outcome following multiple subpial transection for Landau-Kleffner syndrome. Brain. 1999;122(Pt 3):561-566.
Guenot M. Surgical treatment for epilepsy in children: indications and complications. Rev Neurol (Paris). 2004;160(Spec No 1):5S203. 5S209
Guerreiro M.M., Camargo E.E., Kato M., et al. Brain single photon emission computed tomography imaging in Landau-Kleffner syndrome. Epilepsia. 1996;37(1):60-67.
Guerrini R. Epilepsy in children. Lancet. 2006;367(9509):499-524.
Hagberg B., Aicardi J., Dias K., et al. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471-479.
Hamiwka L.D., Wirrell E.C. Comorbidities in pediatric epilepsy: beyond “just” treating the seizures. J Child Neurol. 2009;24(6):734-742.
Harrison J.E., O’Callaghan F.J., Hancock E., et al. Cognitive deficits in normally intelligent patients with tuberous sclerosis. Am J Med Genet. 1999;88(6):642-646.
Hermann B., Seidenberg M. Epilepsy and cognition. Epilepsy Curr. 2007;7(1):1-6.
Hermann B., Seidenberg M., Jones J. The neurobehavioural comorbidities of epilepsy: can a natural history be developed? Lancet Neurol. 2008;7(2):151-160.
Hirsch E., Marescaux C., Maquet P., et al. Landau-Kleffner syndrome: a clinical and EEG study of five cases. Epilepsia. 1990;31(6):756-767.
Holmes G.L., Lenck-Santini P.P. Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 2006;8(3):504-515.
Holmes G.L., Stafstrom C.E. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48(4):617-630.
Hrdlicka M., Komarek V., Propper L., et al. Not EEG abnormalities but epilepsy is associated with autistic regression and mental functioning in childhood autism. Eur Child Adolesc Psychiatry. 2004;13(4):209-213.
Hughes J.R., DeLeo A.J., Melyn M.A. The Electroencephalogram in Attention Deficit-Hyperactivity Disorder: Emphasis on Epileptiform Discharges. Epilepsy Behav. 2000;1(4):271-277.
Irwin K., Birch V., Lees J., et al. Multiple subpial transection in Landau-Kleffner syndrome. Dev Med Child Neurol. 2001;43(4):248-252.
Jambaque I., Chiron C., Dumas C., et al. Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res. 2000;38(2–3):151-160.
Joinson C., O’Callaghan F.J., Osborne J.P., et al. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33(2):335-344.
Jonas R., Asarnow R.F., LoPresti C., et al. Surgery for symptomatic infant-onset epileptic encephalopathy with and without infantile spasms. Neurology. 2005;64(4):746-750.
Kagan-Kushnir T., Roberts S.W., Snead O.C.3rd. Screening electroencephalograms in autism spectrum disorders: evidence-based guideline. J Child Neurol. 2005;20(3):197-206.
Kanner A.M. Commentary: the treatment of seizure disorders and EEG abnormalities in children with autistic spectrum disorders: are we getting ahead of ourselves? J Autism Dev Disord. 2000;30(5):491-495.
Kasteleijn-Nolst Trenite D.G. Transient cognitive impairment during subclinical epileptiform electroencephalographic discharges. Semin Pediatr Neurol. 1995;2(4):246-253.
Kaufmann R., Goldberg-Stern H., Shuper A. Attention-deficit disorders and epilepsy in childhood: incidence, causative relations and treatment possibilities. J Child Neurol. 2009;24(6):727-733.
Kayaalp L., Dervent A., Saltik S., et al. EEG abnormalities in West syndrome: correlation with the emergence of autistic features. Brain Dev. 2007;29(6):336-345.
Klein S.K., Tuchman R.F., Rapin I. The influence of premorbid language skills and behavior on language recovery in children with verbal auditory agnosia. J Child Neurol. 2000;15(1):36-43.
Kobayashi R., Murata T. Setback phenomenon in autism and long-term prognosis. Acta Psychiatr Scand. 1998;98(4):296-303.
Koh S., Chung H., Xia H., et al. Environmental enrichment reverses the impaired exploratory behavior and altered gene expression induced by early-life seizures. J Child Neurol. 2005;20(10):796-802.
Korff C., Laux L., Kelley K., et al. Dravet syndrome (severe myoclonic epilepsy in infancy): a retrospective study of 16 patients. J Child Neurol. 2007;22(2):185-194.
Kossoff E.H., Boatman D., Freeman J.M. Landau-Kleffner syndrome responsive to levetiracetam. Epilepsy Behav. 2003;4(5):571-575.
Kossoff E.H., Zupec-Kania B.A., Rho J.M. Ketogenic diets: an update for child neurologists. J Child Neurol. 2009;24(8):979-988.
Kovacs A., Mihaly A., Komaromi A., et al. Seizure, neurotransmitter release, and gene expression are closely related in the striatum of 4-aminopyridine-treated rats. Epilepsy Res. 2003;55(1–2):117-129.
Kramer U., Sagi L., Goldberg-Stern H., et al. Clinical spectrum and medical treatment of children with electrical status epilepticus in sleep (ESES). Epilepsia. 2008.
Kurita H., Koyama T., Setoya Y., et al. Validity of childhood disintegrative disorder apart from autistic disorder with speech loss. Eur Child Adolesc Psychiatry. 2004;13(4):221-226.
Lagae L. Rational treatment options with AEDs and ketogenic diet in Landau-Kleffner syndrome: still waiting after all these years. Epilepsia. 2009;50(Suppl 7):59-62.
Landau W.M., Kleffner F.R. Syndrome of acquired aphasia with convulsive disorder in children. 1957. Neurology. 1998;51(5):1241. 1248, pages following 1241
Lerman P., Lerman-Sagie T., Kivity S. Effect of early corticosteroid therapy for Landau-Kleffner syndrome. Dev Med Child Neurol. 1991;33(3):257-260.
Lord C., Shulman C., DiLavore P. Regression and word loss in autistic spectrum disorders. J Child Psychol Psychiatry. 2004;45(5):936-955.
Lux A.L., Edwards S.W., Hancock E., et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol. 2005;4(11):712-717.
Lux A.L., Osborne J.P. The influence of etiology upon ictal semiology, treatment decisions and long-term outcomes in infantile spasms and West syndrome. Epilepsy Res. 2006;70(Suppl 1):S77-S86.
Mackay M.T., Weiss S.K., Adams-Webber T., et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004;62(10):1668-1681.
Mantovani J.F., Landau W.M. Acquired aphasia with convulsive disorder: course and prognosis. Neurology. 1980;30(5):524-529.
Marescaux C., Hirsch E., Finck S., et al. Landau-Kleffner syndrome: a pharmacologic study of five cases. Epilepsia. 1990;31(6):768-777.
Markand O.N. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J Clin Neurophysiol. 2003;20(6):426-441.
McVicar K.A., Ballaban-Gil K., Rapin I., et al. Epileptiform EEG abnormalities in children with language regression. Neurology. 2005;65(1):129-131.
Mikati M.A., Saab R., Fayad M.N., et al. Efficacy of intravenous immunoglobulin in Landau-Kleffner syndrome. Pediatr Neurol. 2002;26(4):298-300.
Mikati M.A., Shamseddine A.N. Management of Landau-Kleffner syndrome. Paediatr Drugs. 2005;7(6):377-389.
Millichap J.J., Koh S., Laux L.C., et al. Child Neurology: Dravet syndrome: when to suspect the diagnosis. Neurology. 2009;73(13):e59-e62.
Morrow E.M., Yoo S.Y., Flavell S.W., et al. Identifying autism Loci and genes by tracing recent shared ancestry. Science. 2008;321(5886):218-223.
Mouridsen S.E. Childhood disintegrative disorder. Brain Dev. 2003;25(4):225-228.
Mouridsen S.E., Rich B., Isager T. Epilepsy in disintegrative psychosis and infantile autism: a long-term validation study. Dev Med Child Neurol. 1999;41(2):110-114.
Muhle R., Trentacoste S.V., Rapin I. The genetics of autism. Pediatrics. 2004;113(5):e472-e486.
Nabbout R., Dulac O. Epileptic encephalopathies: a brief overview. J Clin Neurophysiol. 2003;20(6):393-397.
Nakano S., Okuno T., Mikawa H. Landau-Kleffner syndrome. EEG topographic studies. Brain Dev. 1989;11(1):43-50.
Napolioni V., Moavero R., Curatolo P. Recent advances in neurobiology of Tuberous Sclerosis Complex. Brain Dev. 2009;31(2):104-113.
Nass R., Heier L., Walker R. Landau-Kleffner syndrome: temporal lobe tumor resection results in good outcome. Pediatr Neurol. 1993;9(4):303-305.
Nickels K., Wirrell E. Electrical status epilepticus in sleep. Semin Pediatr Neurol. 2008;15(2):50-60.
Nikanorova M., Miranda M.J., Atkins M., et al. Ketogenic diet in the treatment of refractory continuous spikes and waves during slow sleep. Epilepsia. 2009.
Noebels J.L. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599-625.
Oostrom K.J., Smeets-Schouten A., Kruitwagen C.L., et al. Not only a matter of epilepsy: early problems of cognition and behavior in children with “epilepsy only” – a prospective, longitudinal, controlled study starting at diagnosis. Pediatrics. 2003;112(6 Pt 1):1338-1344.
Otero E., Cordova S., Diaz F., et al. Acquired epileptic aphasia (the Landau-Kleffner syndrome) due to neurocysticercosis. Epilepsia. 1989;30(5):569-572.
O’Tuama L.A., Urion D.K., Janicek M.J., et al. Regional cerebral perfusion in Landau-Kleffner syndrome and related childhood aphasias. J Nucl Med. 1992;33(10):1758-1765.
Paetau R. Magnetoencephalography in Landau-Kleffner syndrome. Epilepsia. 2009;50(Suppl 7):51-54.
Paredes M.F., Baraban S.C. A review of gene expression patterns in the malformed brain. Mol Neurobiol. 2002;26(1):109-116.
Parry-Fielder B., Collins K., Fisher J., et al. Electroencephalographic abnormalities during sleep in children with developmental speech-language disorders: a case-control study. Dev Med Child Neurol. 2009;51(3):228-234.
Parry-Fielder B., Nolan T.M., Collins K.J., et al. Developmental language disorders and epilepsy. J Paediatr Child Health. 1997;33(4):277-280.
Pascual-Castroviejo I., Lopez Martin V., Martinez Bermejo A., et al. Is cerebral arteritis the cause of the Landau-Kleffner syndrome? Four cases in childhood with angiographic study. Can J Neurol Sci. 1992;19(1):46-52.
Perniola T., Margari L., Buttiglione M., et al. A case of Landau-Kleffner syndrome secondary to inflammatory demyelinating disease. Epilepsia. 1993;34(3):551-556.
Plioplys S., Dunn D.W., Caplan R. 10-year research update review: psychiatric problems in children with epilepsy. J Am Acad Child Adolesc Psychiatry. 2007;46(11):1389-1402.
Pressler R.M., Robinson R.O., Wilson G.A., et al. Treatment of interictal epileptiform discharges can improve behavior in children with behavioral problems and epilepsy. J Pediatr. 2005;146(1):112-117.
Rapin I. Autistic regression and disintegrative disorder: how important the role of epilepsy? Semin Pediatr Neurol. 1995;2(4):278-285.
Richer L.P., Shevell M.I., Rosenblatt B.R. Epileptiform abnormalities in children with attention-deficit-hyperactivity disorder. Pediatr Neurol. 2002;26(2):125-129.
Riikonen R. Infantile spasms: modern practical aspects. Acta Paediatr Scand. 1984;73(1):1-12.
Riikonen R. Long-term outcome of patients with West syndrome. Brain Dev. 2001;23(7):683-687.
Riikonen R., Amnell G. Psychiatric disorders in children with earlier infantile spasms. Dev Med Child Neurol. 1981;23(6):747-760.
Rintahaka P.J., Chugani H.T., Sankar R. Landau-Kleffner syndrome with continuous spikes and waves during slow-wave sleep. J Child Neurol. 1995;10(2):127-133.
Robinson R.J. Causes and associations of severe and persistent specific speech and language disorders in children. Dev Med Child Neurol. 1991;33(11):943-962.
Robinson R.O., Baird G., Robinson G., et al. Landau-Kleffner syndrome: course and correlates with outcome. Dev Med Child Neurol. 2001;43(4):243-247.
Rossi P.G., Parmeggiani A., Posar A., et al. Landau-Kleffner syndrome (LKS): long-term follow-up and links with electrical status epilepticus during sleep (ESES). Brain Dev. 1999;21(2):90-98.
Roubertie A., Humbertclaude V., Rivier F., et al. Interictal paroxysmal epileptic discharges during sleep in childhood: phenotypic variability in a family. Epilepsia. 2003;44(6):864-869.
Roulet Perez E., Davidoff V., Despland P.A., et al. Mental and behavioural deterioration of children with epilepsy and CSWS: acquired epileptic frontal syndrome. Dev Med Child Neurol. 1993;35(8):661-674.
Roulet-Perez E., Deonna T. Autism, Epilepsy, and EEG epileptiform Activity. In: Tuchman R., Rapin I., editors. Autism: A neurological disorder of early brain development. London: MacKeith Press; 2006:174-188.
Rudolf G., Valenti M.P., Hirsch E., et al. From rolandic epilepsy to continuous spike-and-waves during sleep and Landau-Kleffner syndromes: insights into possible genetic factors. Epilepsia. 2009;50(Suppl 7):25-28.
Saemundsen E., Ludvigsson P., Rafnsson V. Autism spectrum disorders in children with a history of infantile spasms: a population-based study. J Child Neurol. 2007;22(9):1102-1107.
Saemundsen E., Ludvigsson P., Rafnsson V. Risk of autism spectrum disorders after infantile spasms: A population-based study nested in a cohort with seizures in the first year of life. Epilepsia. 2008;49(11):1865-1870.
Sawhney I.M., Robertson I.J., Polkey C.E., et al. Multiple subpial transection: a review of 21 cases. J Neurol Neurosurg Psychiatry. 1995;58(3):344-349.
Scheffer I.E., Zhang Y.H., Jansen F.E., et al. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev. 2009;31(5):394-400.
Scheltens-de Boer M. Guidelines for EEG in encephalopathy related to ESES/CSWS in children. Epilepsia. 2009;50(Suppl 7):13-17.
Schwab R. A method of measuring consciousness in petit mal epilepsy. J Nerv Ment Dis. 1939;89:690-691.
Sengoku A., Kanazawa O., Kawai I., et al. Visual cognitive disturbance during spike-wave discharges. Epilepsia. 1990;31(1):47-50.
Seri S., Thai J.N., Brazzo D., et al. Neurophysiology of CSWS-associated cognitive dysfunction. Epilepsia. 2009;50(Suppl 7):33-36.
Shewmon D.A., Erwin R.J. The effect of focal interictal spikes on perception and reaction time. II. Neuroanatomic specificity. Electroencephalogr Clin Neurophysiol. 1988;69(4):338-352.
Shinnar S., Rapin I., Arnold S., et al. Language regression in childhood. Pediatr Neurol. 2001;24(3):183-189.
Sinclair D.B., Snyder T.J. Corticosteroids for the treatment of Landau-Kleffner syndrome and continuous spike-wave discharge during sleep. Pediatr Neurol. 2005;32(5):300-306.
Smith M.C., Hoeppner T.J. Epileptic encephalopathy of late childhood: Landau-Kleffner syndrome and the syndrome of continuous spikes and waves during slow-wave sleep. J Clin Neurophysiol. 2003;20(6):462-472.
Solomon G.E., Carson D., Pavlakis S., et al. Intracranial EEG monitoring in Landau-Kleffner syndrome associated with left temporal lobe astrocytoma. Epilepsia. 1993;34(3):557-560.
Spence S.J., Schneider M.T. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr Res. 2009;65(6):599-606.
Steffenburg U., Hagberg G., Hagberg B. Epilepsy in a representative series of Rett syndrome. Acta Paediatr. 2001;90(1):34-39.
Tassinari C.A., Cantalupo G., Rios-Pohl L., et al. Encephalopathy with status epilepticus during slow sleep: “the Penelope syndrome”. Epilepsia. 2009;50(Suppl 7):4-8.
Tassinari C.A., Rubboli G., Volpi L., et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin Neurophysiol. 2000;111(Suppl 2):S94-S102.
Tharp B.R. Epileptic encephalopathies and their relationship to developmental disorders: do spikes cause autism? Ment Retard Dev Disabil Res Rev. 2004;10(2):132-134.
Trevathan E., Murphy C.C., Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999;40(6):748-751.
Tsuru T., Mori M., Mizuguchi M., et al. Effects of high-dose intravenous corticosteroid therapy in Landau-Kleffner syndrome. Pediatr Neurol. 2000;22(2):145-147.
Tuchman R. AEDs and psychotropic drugs in children with autism and epilepsy. Ment Retard Dev Disabil Res Rev. 2004;10(2):135-138.
Tuchman R. CSWS-related autistic regression versus autistic regression without CSWS. Epilepsia. 2009;50(Suppl 7):18-20.
Tuchman R.F., Rapin I. Regression in pervasive developmental disorders: seizures and epileptiform electroencephalogram correlates. Pediatrics. 1997;99(4):560-566.
Tuchman R., Moshe S.L., Rapin I. Convulsing toward the pathophysiology of autism. Brain Dev. 2009;31(2):95-103.
Tuchman R.F., Rapin I., Shinnar S. Autistic and dysphasic children. II: Epilepsy. Pediatrics. 1991;88(6):1219-1225.
Wakai S., Ito N., Ueda D., et al. Landau-Kleffner syndrome and sulthiame. Neuropediatrics. 1997;28(2):135-136.
Weiss L.A., Escayg A., Kearney J.A., et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;8(2):186-194.
Wheless J.W., Simos P.G., Butler I.J. Language dysfunction in epileptic conditions. Semin Pediatr Neurol. 2002;9(3):218-228.
Wilson S., Djukic A., Shinnar S., et al. Clinical characteristics of language regression in children. Dev Med Child Neurol. 2003;45(8):508-514.
Wisniewski K.E., Zhong N., Philippart M. Pheno/genotypic correlations of neuronal ceroid lipofuscinoses. Neurology. 2001;57(4):576-581.
Zupanc M.L. Clinical evaluation and diagnosis of severe epilepsy syndromes of early childhood. J Child Neurol. 2009;24(8 Suppl):6S-14S.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 55 Epilepsy and Neurodevelopmental Disorders
Introduction
The association of epilepsy with neurodevelopmental disorders is now well established. Children with developmental epilepsies are at increased risk for cognitive [Hermann and Seidenberg, 2007], neurobehavioral [Hermann et al., 2008], and psychiatric disorders [Plioplys et al., 2007]. Specific factors associated with a higher risk of neuropsychological deficits in children with epilepsy include multiple seizures, use of antiepileptic drugs, etiology, and interictal epileptiform discharges (IEDs) on the initial electroencephalogram (EEG) [Fastenau et al., 2009]. In children with epilepsy, younger age at seizure onset, cognitive impairment, temporal or frontal lobe onset of seizures, and intractable epilepsy are associated with an increased likelihood of coexisting social, communication, and behavioral disorders [Hamiwka and Wirrell, 2009]. The multiple complex and confounding risk factors that account for the coexistence of epilepsy and neurodevelopmental disorders include etiology, associated disabilities, seizure type, frequency, duration, control, age of onset, and IEDs, as well as psychosocial factors and medication effects [Austin and Caplan, 2007].
The relationship between epilepsy and neurodevelopmental disorders also reflects common pathologies and mechanisms [Tuchman et al., 2009], and there is accumulating evidence to suggest that cognitive and behavioral impairments may precede the onset of seizures [Austin et al., 2001; Oostrom et al., 2003; Bhise et al., 2009; Fastenau et al., 2009]. On the other hand, there is evidence that recurrent seizures or IEDs can cause acute and long-lasting impairment of brain function and development [Holmes and Lenck-Santini, 2006; Galanopoulou and Moshe, 2009], and significant efforts are being made to determine how risk factors contribute to developmental outcomes in children with epilepsy [Austin and Fastenau, 2009]. The notion that epileptic activity, seizures, or IEDs can lead to cognitive and behavioral impairment above and beyond what might be expected from the underlying pathology is exemplified by the concept of epileptic encephalopathy [Berg et al., 2009]. This idea has important implications for treatment, especially in children with an epileptic encephalopathy, in whom early and successful treatment of seizures and interictal epileptiform activity may be crucial to positive neurodevelopmental outcomes [Freitag and Tuxhorn, 2005; Jonas et al., 2005; Lux et al., 2005; Arts and Geerts, 2009; Bombardieri et al., 2009].
Epilepsy is more frequent in children with intellectual disability [Goulden et al., 1991]; the behavioral, cognitive, and social aspects of epilepsy are multiple and diverse, and are discussed in Chapter 62. This present chapter focuses on the concept of epileptic encephalopathy and the association of epilepsy and IEDs with specific language impairments and autistic spectrum disorders (ASDs). In addition, the common mechanisms of disease and management of children with developmental epilepsies and neurodevelopmental disorders are addressed.
Epileptic Encephalopathies
The epileptic encephalopathies can occur at any age, but are more common and severe in the first decade of life as the brain is developing. Approximately 40 percent of epilepsies occurring during the first 3 years of life are associated with an epileptic encephalopathy [Guerrini, 2006]. Epileptic encephalopathy of childhood includes: early myoclonic encephalopathy (Ohtahara’s or Aicardi’s syndrome), severe myoclonic epilepsy of infancy (Dravet’s syndrome), myoclonic-astatic epilepsy of early childhood (Doose’s syndrome), infantile spasms (West’s syndrome), Lennox–Gastaut syndrome, and Landau–Kleffner syndrome-continuous spike waves during slow-wave sleep [Engel, 2001].
The myoclonic epilepsies and infantile spasms are discussed in Chapter 56 and Lennox–Gastaut syndrome in Chapter 53. In this chapter, Dravet’s syndrome, infantile spasms, Lennox–Gastaut syndrome, Landau–Kleffner syndrome, and continuous spike waves during slow-wave sleep will be discussed from a developmental perspective. Specifically, the impact of seizures and of IEDs on the developmental trajectories of children with these electroclinical syndromes will be emphasized.
Dravet’s Syndrome
Dravet’s syndrome is a genetically determined infantile epileptic encephalopathy, mainly caused by de novo mutations in the SCN1A gene [Scheffer et al., 2009]. Early development is normal, with seizures usually beginning in the first 10 months of life; these become frequent by 2–4 years of age, with focal and generalized myoclonus, atypical absences, and partial complex seizures, as well as seizures characterized by fluctuating alteration of consciousness with reduced postural tone and myoclonic jerks [Millichap et al., 2009]. Despite frequent seizures, the EEG is usually normal in the first 2 years of life and then usually progresses into spike and wave and multifocal discharges, although some children may have persistently normal interictal EEG studies [Korff et al., 2007]. Progressive decline or plateau in development occurs by 1–4 years of age, with intellectual disability and an autism phenotype commonly present, especially in those with more than five seizures per month [Scheffer et al., 2009].
Genes associated with Dravet’s syndrome appear to have relevance to the neurodevelopmental disorders, such as ASDs. Specifically, a susceptibility locus for ASDs has been found on chromosome 2 in the vicinity of the epilepsy-involved genes SCN1A and SCN2A [Weiss et al., 2003], while PCDH10 on chromosome 4 has been linked to ASDs in families with shared ancestry [Morrow et al., 2008]. Recently, a sporadic infantile epileptic encephalopathy that resembles Dravet’s syndrome has been tied to mutations in PCDH19 [Depienne et al., 2009]. These results hint strongly at possible shared molecular underpinnings between ASD and this epileptic encephalopathy.
Infantile Spasms
Infantile spasms constitute an age-specific seizure disorder, with a peak age of presentation between 4 and 8 months of age, a critical time of brain development [Zupanc, 2009]. There is a strong correlation between infantile spasms and intellectual disability [Trevathan et al., 1999], and the majority of children with infantile spasms develop intellectual disability and specific cognitive and behavioral deficits [Riikonen, 2001]. In addition, the association of infantile spasms and ASDs is well recognized [Riikonen and Amnell, 1981]; in children with infantile spasms the prevalence of ASDs is as high as 35 percent, depending on the severity of intellectual disability [Saemundsen et al., 2007], with a heightened risk of ASDs in the presence of identifiable structural lesions of the brain [Saemundsen et al., 2008]. Further, in children with infantile spasms, EEG epileptiform activity – particularly bilateral frontal EEG discharges and persistence of hypsarrhythmia – contributes to the development of the autism phenotype [Kayaalp et al., 2007].
One of the intriguing features of infantile spasms is that the severity and, to a certain extent, the frequency of seizures do not seem to correlate with the degree of cognitive impairment or the occurrence of regression [Riikonen, 1984; Bednarek et al., 1998]. It may be that the interictal EEG pattern of hypsarrhythmia associated with infantile spasms affects cortical and subcortical neuronal networks, resulting in abnormal synaptogenesis and poor developmental outcomes [Zupanc, 2009].
Lennox–Gastaut Syndrome
Lennox–Gastaut syndrome is an age-specific epileptic syndrome that peaks between 3 and 5 years; it is characterized by multiple seizure types that include tonic and atonic seizures, atypical absences, and myoclonic and generalized or focal seizures, in association with a characteristic EEG pattern of slow spike-and-wave complexes, often associated with multifocal epileptiform activity and runs of fast activity [Crumrine, 2002]. Approximately 20 percent of children with this epileptic syndrome have a history of infantile spasms [Markand, 2003]. Cognitive and behavioral abnormalities precede the clinical seizures in approximately 20–60 percent of children with Lennox–Gastaut syndrome [Blume, 2001].
It has been suggested that the epileptic processes associated with this syndrome lead to patterns of abnormal activity and connectivity that compete with normal brain development, thus resulting in subsequent impairment or regression of cognition [Blume, 2004]. It is not known whether it is the underlying brain pathology, the burden of frequent seizures, the persistent epileptiform activity, or all of these factors that is responsible for the cognitive deficits in Lennox–Gastaut syndrome. The age specificity, typically frequent seizures, and unremitting epileptiform activity suggest that the epileptic activity occurring at a critical time in brain development contributes to a progressive disturbance in cerebral function.
Landau–Kleffner Syndrome-Continuous Spike Waves During Slow-Wave Sleep
Landau–Kleffner syndrome (LKS) is an acquired aphasia associated with an epileptiform EEG with spikes, sharp waves, or spike-and-wave discharges that are usually bilateral and occur predominantly over the temporal regions; in approximately 25 percent of children there are no clinical seizures [Landau and Kleffner, 1998]. Continuous spike-wave discharges during slow-wave sleep (CSWS), an epileptic encephalopathy associated with the EEG pattern of electrical status epilepticus during slow-wave sleep, with various seizure types, and with cognitive, motor, and behavioral disturbances, is, along with LKS, considered a sleep-related epileptic encephalopathy [Tassinari et al., 2009].
CSWS and LKS are epileptic encephalopathies with common clinical features, including seizures, regression, and epileptiform abnormalities that are activated by sleep [Nickels and Wirrell, 2008]. In CSWS there is a regression in global skills, while in LKS the primary clinical manifestation is a regression of language. The conceptual thinking is that there is a spectrum of disorders associated with activation of epileptiform activity during slow-wave sleep that includes LKS, CSWS, and “atypical” benign epilepsy with centrotemporal spikes (BECTS) [Fejerman, 2009; Scheltens-de Boer, 2009]. The implications are that the EEG abnormalities lead to regression in language, behavioral, and cognitive manifestations and that treatment requires reversal of the epileptiform pattern on the EEG. However, whether the frequency of the epileptiform discharges, as linked to the appearance or disappearance of electrical status epilepticus during slow-wave sleep, is responsible for the course of language, cognitive, and behavioral deterioration [Tassinari et al., 2000; Seri et al., 2009] remains an unanswered and controversial issue.
Seizures, Interictal Epileptiform Discharges, and Neurodevelopmental Disorders
IEDs, characterized by spikes or sharp waves that appear abruptly from the electrographic background, with or without an associated slow wave, are limited in duration and do not evolve in frequency and distribution over time, but can at times be responsible for disruption of cognitive and behavioral function [Fisch, 2003]. The concept that transitory changes in higher cortical functions can be secondary to EEG discharges not accompanied by seizures was proposed more than 60 years ago [Schwab, 1939]. The term transient cognitive impairment is used to describe individuals with epileptiform EEG discharges in association with a momentary disruption of adaptive cerebral function [Aarts et al., 1984; Binnie, 2003].
Studies by numerous investigators suggest that transient cognitive impairment is not a consequence of general impairment of attention, but is likely secondary to disruption of functions located in the region or regions of the brain where the epileptiform discharges arise [Kasteleijn-Nolst Trenite, 1995; Aldenkamp and Arends, 2004]. Specific functions, such as perception, reaction time, and scholastic performance, can be disrupted by brief epileptiform discharges in the absence of convulsive seizures [Shewmon and Erwin, 1988; Sengoku et al., 1990]. The spectrum of language and cognitive impairment secondary to an active epileptic focus, even in the absence of clinical seizures, is wide [Deonna and Roulet-Perez, 2005; Roulet-Perez and Deonna, 2006].
Specific Language Impairment
Specific language impairment is a developmental language disorder characterized by varying types and degree of dysfunction in expressive and receptive communication skills (see Chapter 45). Children with specific language impairments differ from children with acquired aphasia characteristic of the epileptic encephalopathies, such as LKS-CSWS, in that there is no regression of language skills. Nevertheless, the impairments of language in epilepsies such as BECTS raises interesting questions regarding the overlap and interactions of epilepsy, IEDs, and specific language impairments [Billard et al., 2009].
An increased association of seizures in children with specific language impairments has been found [Robinson, 1991; Echenne et al., 1992], with prevalence rates from 7–40 percent reflecting the cohort studied and the type and duration of the EEG recording [Parry-Fielder et al., 1997]. In one study that compared the rates of epilepsy in children with autistic spectrum versus developmental language disorders, the prevalence of epilepsy was 8 percent (14 of 168) in the nonautistic, language-impaired children. In addition, when the children were subtyped on the basis of language, the highest risk of epilepsy, regardless of whether the children had ASD or specific language impairment, was seen in those individuals with the most severe receptive language disorder [Tuchman et al., 1991; Klein et al., 2000]. A link between specific language impairment and IEDs during sleep has also been suggested [Parry-Fielder et al., 1997; Ballaban-Gil and Tuchman, 2000; Wheless et al., 2002]. However, the strength of this association has been questioned in a recent study in which 13 percent of children with specific language impairments had IEDs on a sleep EEG recording, which was not significantly different than the non-language-impaired control group [Parry-Fielder et al., 2009].
Autistic Spectrum Disorders
ASD is a broad classification that includes a heterogeneous group of individuals with behaviorally defined impairments in reciprocal social interaction, verbal and nonverbal communication, and restricted and repetitive behaviors (see Chapter 48). The prevalence of epilepsy in children with ASD is highly variable, depending on the study sample, with rates ranging from 5 to 46 percent [Spence and Schneider, 2009]. The major risk factor for epilepsy in children with ASD is moderate to severe intellectual disability [Amiet et al., 2008]. In children with ASD without severe intellectual disability, motor deficit, associated perinatal or medical disorder, or a positive family history of epilepsy, the prevalence of epilepsy is 6 percent, which is not significantly different than in nonautistic children with a specific language impairment [Tuchman et al., 1991]. The prevalence of ASD in individuals with epilepsy has not been investigated with the same intensity as that of epilepsy in ASD, although one study in a tertiary epilepsy clinic found that approximately 30 percent of children with epilepsy screened positive for ASD [Clarke et al., 2005].
The prevalence of IEDs in children with ASD and no clinical history of seizures may be as high as 60 percent [Chez et al., 2006], but varies depending on the cohort studied and the type of EEG performed, with most studies finding prevalence rates ranging from 6 to 31 percent [Kagan-Kushnir et al., 2005]. The higher prevalence of IEDs in children with specific language impairment and in those with ASD compared to the 1.5–4 percent rate of IEDs in the general population [Cavazzuti et al., 1980; Capdevila et al., 2008] has been a source of significant controversy. The significance of these findings remains unclear and is not unique, as IEDs have been reported in 6–30 percent of children with attention-deficit hyperactivity disorders [Hughes et al., 2000; Richer et al., 2002; Kaufmann et al., 2009], which is remarkably similar to the prevalence of IEDs in children with specific language impairment or ASDs without seizures. One suggestion is that the high prevalence of IEDs in all of these disorders is secondary to the neuropathological processes common to both epilepsy and neurodevelopmental disorders. However, the question of whether these IEDs may be a biomarker of prognostic or interventional significance in a subgroup of children with specific language impairment or with ASDs remains to be determined.
Regression
The association of developmental regression in ASD to epilepsy and IEDs became a topic of increased interest after Hagberg and colleagues [Hagberg et al., 1983] published a report of 35 girls with regression in higher brain functions, stereotypical hand movements, and ASD, with a significant proportion of the girls having epilepsy. Childhood disintegrative disorder is characterized by late-onset autistic and cognitive regression that can include motor regression and loss of bowel and bladder function, usually occurring after age 3 [Rapin, 1995]. Childhood disintegrative disorder is based on the description, in the early 1900s, of children with normal development until age 3 or 4 years, who regressed in multiple developmental areas. This entity was first described by Heller in 1908 [Mouridsen, 2003], and differentiation from autism, especially from autistic regression, is still in progress [Kurita et al., 2004]. Disintegrative disorder is very rare, with a prevalence of 2 in 100,000 [Fombonne, 2009].
The prevalence of epilepsy in Rett’s syndrome and in childhood disintegrative disorder is greater than 70 percent, both disorders have EEGs with marked IEDs, and both featuring regression of cognitive, language, and social skills [Mouridsen et al., 1999; Steffenburg et al., 2001]. Some children with childhood disintegrative disorder overlap with those having epilepsy and CSWS [Roulet Perez et al., 1993]. To what extent the high rate of seizures in these groups is secondary to the severe cognitive impairment present in Rett’s syndrome and childhood disintegrative disorder, or what influence other specific variables, such as metabolic or molecular factors (i.e., the role of MECP2), have in the development of seizures remains unknown.
The developmental trajectory in approximately 30 percent of children with ASD is characterized by a regression of the few words acquired and a loss of nonverbal communication skills, usually occurring prior to 24 months of age [Goldberg et al., 2003; Lord et al., 2004]. This regression has been termed autistic regression. The relation of autistic regression to epilepsy or to an epileptiform EEG without seizures remains controversial, with some studies reporting higher rates of epilepsy in children with ASD and regression [Kobayashi and Murata, 1998; Hrdlicka et al., 2004], and others showing no relation between ASD, epilepsy, and regression [Tuchman and Rapin, 1997; Baird et al., 2008]. A recent study found that children with autistic regression had more disrupted sleep, as compared to those with autism without regression, and were more likely to have epilepsy [Giannotti et al., 2008]. In addition, Giannotti and colleagues found that epileptiform activity did not differ among those with and without regression, except that those with autistic regression were more likely than those without regression to have more “frequent epileptiform EEGs.” There is evidence to suggest that, in a subgroup of children with ASD and without convulsive seizures, an epileptiform EEG is significantly more likely to be associated with a history of regression in language [Tuchman and Rapin, 1997]. However, these data must be put into perspective, as they represent a very specific subgroup of children with autism, and because at the present time there are no data regarding the number of children in the general population without seizures and cognitive and behavioral impairments who have interictal epileptiform abnormalities on an overnight EEG study. Others have found no differences in regression in those with epileptiform EEGs and epilepsy and those without seizures and a normal EEG [Canitano et al., 2005]. Children with autistic regression and an epileptiform EEG (AREE) should be differentiated from those with LKS-CSWS, and these differences are highlighted in Table 55-1.
Table 55-1 Landau–Kleffner Syndrome-Continuous Spike Waves During Slow-Wave Sleep Versus Autistic Regression with Epileptiform EEG
| Landau–Kleffner Syndrome-Continuous Spike-Waves During Slow-Wave Sleep (LKS-CSWS) | Autistic Regression with Epileptiform EEG (AREE) | |
|---|---|---|
| Age of regression/(symptoms) | Usually after 3 years. Peak age 3–5 years. In CSWS may be as late as 12 years of age | Usually prior to age 2 years with a mean age of regression of 21 months |
| Seizures | Usually not frequent or intractable. In approximately 25 percent, seizures are not present | Seizures are not part of phenotype. In autistic spectrum disorders, when seizures occur, they are usually not frequent and responded well to antiepileptic medications |
| EEG | Spikes, sharp waves, or spike-and-wave discharges, usually bilateral and occurring predominantly over the temporal regions. They increase during sleep; EEG pattern of electrical status epilepticus during slow-wave sleep is common | Infrequent spikes, usually centrotemporal. Rarely associated with CSWS. No clear correlation with interictal epileptiform discharges and improvement or worsening of underlying language and social dysfunction |
| Treatment | See Box 55-1. Case reports, mostly with use of steroids, suggest improvement in language. Surgical outcomes with multiple subpial transections are variable. No controlled clinical trials | No evidence that present medical interventions (antiepileptic medications) or surgical interventions are indicated |
| Outcome/Comments | Improvement occurs in late childhood/early adolescence. Approximately one-third recover. Prognosis for seizure control is excellent but recovery of language is variable and not as good as for seizures | Improvement seems related to cognitive skills. No data to determine if interictal discharges combined with regression are marker for worse prognosis |
The mean age of onset of language regression in ASDs is 21 months, and over 90 percent of children with autism who undergo a regression do so before age 3 years [Tuchman and Rapin, 1997]. By contrast, in LKS, only 12–14 percent of children experience regression before age 3 years [Bishop, 1985]. The peak age of onset of symptoms in LKS is between 5 and 7 years [Bureau, 1995]. In only 5 percent of individuals does LKS begin after age 9 years and it appears to occur rarely, if ever, after age 12 years [Bureau, 1995]. In children with the CSWS, the first symptoms occur in up to 20 percent of children between 9 and 12 years of age [Bureau, 1995].
[/not-level-membership-for-neurology-category]
