Causes of Cancer
Epidemiology and Causal Criteria
Known Cancer Risk Factors
Smoking
Table 7-1
Carcinogens in Tobacco Smoke
| Carcinogen Class | No. of Compounds | Example Compound |
| Polycyclic aromatic hydrocarbons | 10 | Benzo[a]pyrene 5-Methylchrysene Dibenz[a,h]anthracene |
| Aza-arenes | 3 | Dibenz[a,h]acridine |
| N-nitrosamines | 7 | 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) N-Nitrosodiethylamine |
| Aromatic amines | 3 | 4-Aminobiphenyl |
| Heterocyclic amines | 8 | 2-Amino-3-methylimidazo[4,5–f]quinoline |
| Aldehydes | 2 | Formaldehyde |
| Miscellaneous organic compounds | 15 | 1,3-Butadiene Ethyl carbamate |
| Inorganic compounds | 7 | Nickel Chromium Cadmium Arsenic |
| Total | 55 |
Adapted from Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 1999;91:1194.
Diet
Occupation
Table 7-2
Environmental Carcinogens Associated with Occupation
| Occupation | Carcinogen Exposure | Associated Cancer Type |
| Iron and steel founding | PAH, chromium, nickel, formaldehyde | Lung |
| Copper mining and smelting | Arsenic | Skin, bronchus, liver |
| Underground mining | Radon (ionizing radiation ) | Lung |
| Aluminum production | PAH | Lung |
| Coke production | PAH | Lung, kidney |
| Painting | Chromium, solvents | Lung |
| Furniture and cabinet making | Wood dust | Nasal sinus |
| Boot and shoe manufacture | Leather dust, benzene | Nasal sinus, leukemia |
| Rubber industry | Aromatic amines, solvents | Bladder, leukemia |
| Nickel refining | Nickel | Nasal sinus, bronchus |
| Vinyl chloride manufacture | Vinyl chloride | Liver |
| Dye and textile production | Benzidine-based dyes | Bladder |
PAH, Polycyclic aromatic hydrocarbons.
Causes of Cancer by Organ Site
Classes and Types of Carcinogens
Carcinogen Evaluation and Classification
Table 7-3
Exposures Associated with Human Cancers, as Identified by the IARC (Partial Listing)
| Cancer Site | Carcinogenic Agents with Sufficient Evidence in Humans | Agents with Limited Evidence in Humans |
| Oral cavity | Alcohol, betel quid, HPV, tobacco smoking, smokeless tobacco | Solar radiation |
| Stomach | Helicobacter pylori, rubber production industry, tobacco smoking, x-rays, gamma radiation | Asbestos, Epstein-Barr virus, lead, nitrate, nitrite, pickled vegetables, salted fish |
| Colon and rectum | Alcohol, tobacco smoking, radiation | Asbestos, Schistosoma japonicum |
| Liver and bile duct | Aflatoxins, alcohol, Clonorchis sinensis, estrogen-progestin contraceptives, HBV, HCV, Opisthorchis viverrini, plutonium, thorium-232, vinyl chloride | Androgenic steroids, arsenic, betel quid, HIV, polychlorinated biphenyls, Schistosoma japonicum, trichloroethylene, x-rays, gamma radiation |
| Pancreas | Tobacco smoking, smokeless tobacco | Alcohol, thorium-232, x-rays, gamma radiation, radioiodines |
| Lung | Tobacco smoking, aluminum production, arsenic, asbestos, beryllium, bis (chloromethyl) ether, chloromethyl methyl ether, cadmium, chromium, coal combustion and coal tar pitch, coke production, hematite mining, iron and steel founding, MOPP, nickel, painting, plutonium, radon, rubber production, silica dust, soot, sulfur mustard, x-rays, gamma radiation | Acid mists, manufacture of glass, indoor emissions from household combustion, carbon electrode manufacture, chlorinated toluenes and benzoyl chloride, cobalt metal with tungsten carbide, creosotes, engine exhaust, insecticides, dioxin, printing processes, welding fumes |
| Skin—melanoma | Solar radiation, UV-emitting tanning devices | |
| Other skin cancers | Arsenic, azathiopurine, coal tar pitch, coal tar distillation, cyclosporine, methoxsalen plus UVA, mineral oils, shale oils, solar radiation, soot, x-rays, gamma radiation | Creosotes, HIV, HPV, nitrogen mustard, petroleum refining, UV-emitting tanning devices |
| Mesothelioma | Asbestos, erionite, painting | |
| Breast | Alcohol, diethylstilbestrol, estrogen-progesterone contraceptive and menopausal therapy, x-rays, gamma radiation | Estrogen menopausal therapy, ethylene oxide, shift work resulting in circadian disruption, tobacco smoking |
| Uterine cervix | Diethylstilbestrol (exposure in utero), estrogen-progestogen contraception, HIV, HPV, tobacco smoking | Tetrachloroethylene |
| Ovary | Asbestos, estrogen menopausal therapy, tobacco smoking | Talc-based body powder, x-rays, gamma radiation |
| Prostate | Androgenic steroids, arsenic, cadmium, rubber production industry, thorium-232, x-rays, gamma radiation, diethylstilbestrol (exposure in utero) | |
| Kidney | Tobacco smoking, x-rays, gamma radiation | Arsenic, cadmium, printing processes |
| Urinary Bladder | Aluminum production, 4-aminobiphenyl, arsenic, auramine production, benzidine, chlornaphazine, cyclophosphamide, magenta production, 2-naphthylamine, painting, rubber production, Schistosoma haematobium, tobacco smoking, toluidine, x-rays, gamma radiation | Coal tar pitch, coffee, dry cleaning, engine exhaust, printing processes, occupational exposures in hair dressing and barbering, soot, textile manufacturing |
| Brain | X radiation, gamma radiation | |
| Leukemia and/or lymphoma | Azathiopurine, benzene, busulfan, 1,3-butadiene, chlorambucil, cyclophosphamide, cyclosporine, Epstein-Barr virus, etoposide with cisplatin and bleomycin, fission products, formaldehyde, Helicobacter pylori, HCV, HIV, human T-cell lymphotropic virus type 1, Kaposi’s sarcoma herpesvirus, melphalan, MOPP, phosphorus-32, rubber production, semustine, thiotepa, thorium-232, tobacco smoking, treosulfan, X radiation, gamma radiation | Bischloroethyl nitrosourea, chloramphenicol, ethylene oxide, etoposide, HBV, magnetic fields, mitoxantrone, nitrogen mustard, painting, petroleum refining, polychlorophenols, radioiodines, radon-222, styrene, teniposide, tetrachloroethylene, trichloroethylene, dioxin, tobacco smoking (childhood leukemia in smokers’ children) |
HBV, Hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HPV, human papillomavirus; IARC, International Agency for Research on Cancer; MOPP, mustargen-oncovin-procarbazine-prednisone chemotherapy; UVA, ultraviolet A light.
Adapted from Cogliano et al. Preventable exposures associated with human cancers. J Natl Cancer Inst 2011;103:1835.
Table 7-4
IARC Classification of Suspected Carcinogenic Agents
IARC, International Agency for Research on Cancer.
Types of Carcinogens
Physical Carcinogens
Table 7-5
Selected IARC Known Human Carcinogens
| 4-Aminobiphenyl | Hepatitis B virus |
| Arsenic | Hepatitis C virus |
| Asbestos | Human immunodeficiency virus type 1 |
| Azathioprine | Human papillomavirus |
| Benzene | Human T-cell lymphotropic virus |
| Benzidine | Melphalan |
| Benzo[a]pyrene | 8-Methoxypsoralen |
| Beryllium | Mustard gas |
| N,N-Bis(2-chloroethyl)-2-naphthylamine | 2-Naphthylamine |
| Bis(chloromethyl)ether | Nickel compounds |
| Chloromethyl methyl ether | N′-Nitrosonornicotine (NNN) |
| 1,4-Butanediol dimethanesulfonate | Phosphorus-32 |
| Cadmium | Plutonium-239 |
| Chlorambucil | Radioiodines |
| 1-(2-Chloroethyl)-3-(4-methylcyclohexyl)-1-nitrosourea | Radium-224 |
| Chromium[VI] | Radium-226 |
| Cyclosporine | Radium-228 |
| Cyclophosphamide | Radon-222 |
| Diethylstilbestrol | Silica |
| Epstein-Barr virus | Solar radiation |
| Erionite | Talc-containing asbestiform fibers |
| Estrogen-progestogen menopausal therapy | Tamoxifen |
| Estrogen-progestogen oral contraceptives | 2,3,7,8-Tetrachlorodibenzo-para-dioxin |
| Estrogen therapy | Thiotepa |
| Ethylene oxide | Treosulfan |
| Etoposide | Vinyl chloride |
| Formaldehyde | X- and gamma (γ)-radiation |
| Gallium arsenide | Aflatoxins |
| Helicobacter pylori | Soots
Tobacco Wood dust |
IARC, International Agency for Research on Cancer.
Biologic Carcinogens
Chemical Carcinogens
Organic Carcinogens
Benzene
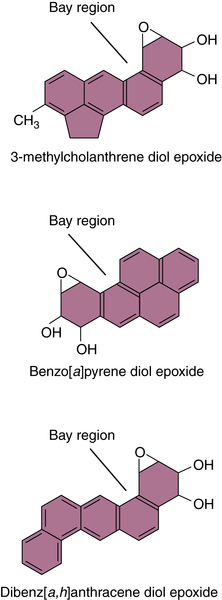
Polycyclic Aromatic Hydrocarbons
Aflatoxin B1
Benzidine
Nitrosamines and Heterocyclic Amines
Inorganic Carcinogens
Beryllium
Cadmium
Arsenic
Chromium
Fibers
Asbestos
Hormones
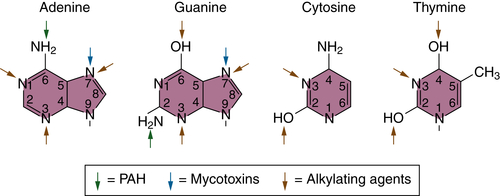
Mechanisms of Chemical Carcinogenesis
Multistage Nature of Carcinogenesis and the Multistage Model of Mouse Skin Carcinogenesis
Initiation and Mutational Theory of Carcinogenesis
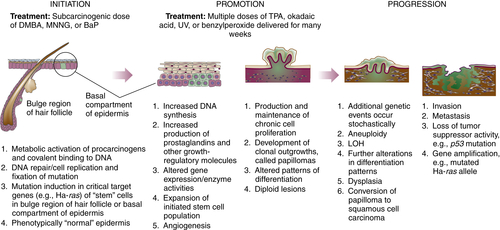
Table 7-6
Selected Proto-oncogenes and Tumor Suppressor Genes and Some Associated Cancers
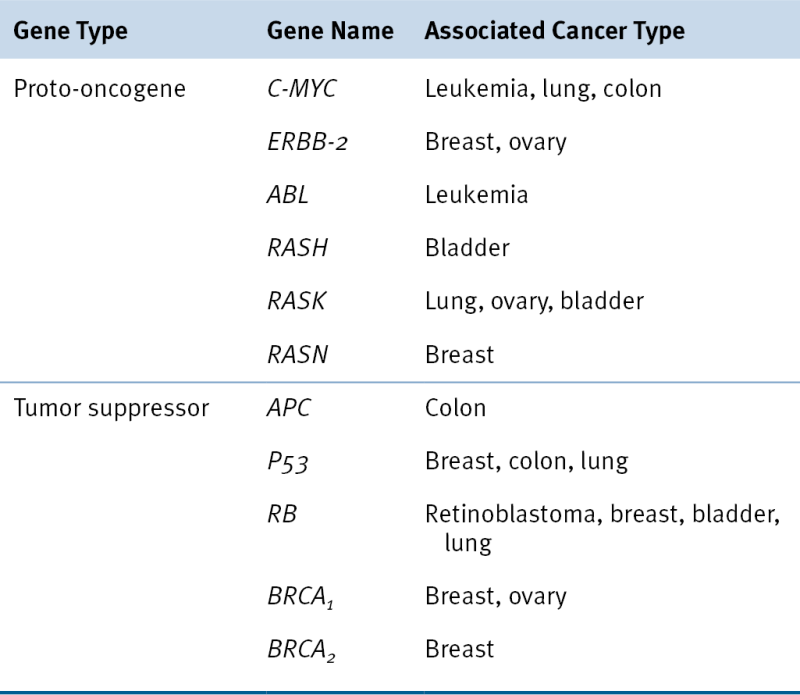
Adapted from Perkins AS, Stern DF: Molecular biology of cancer. Oncogenes. In: DeVita VT, Hellman S, Rosenbert SA, eds. Cancer: Principles & Practice of Oncology. 5th ed. Philadelphia, Pa: Lippincott-Raven Publishers; 1997.
Promotion
Growth Factor Receptor Signaling Pathway Engagement
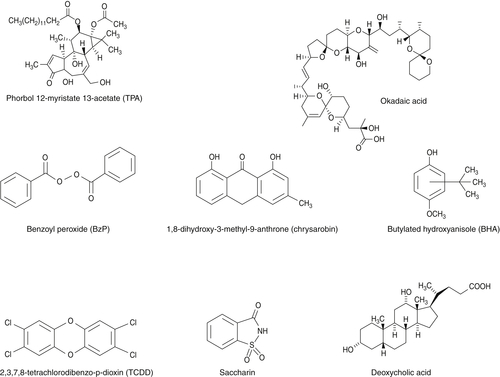
Examples of Tumor Promoters
Progression
Multistage Environmental Carcinogenesis in Humans
Endogenous Defense Systems against Chemical Carcinogenesis
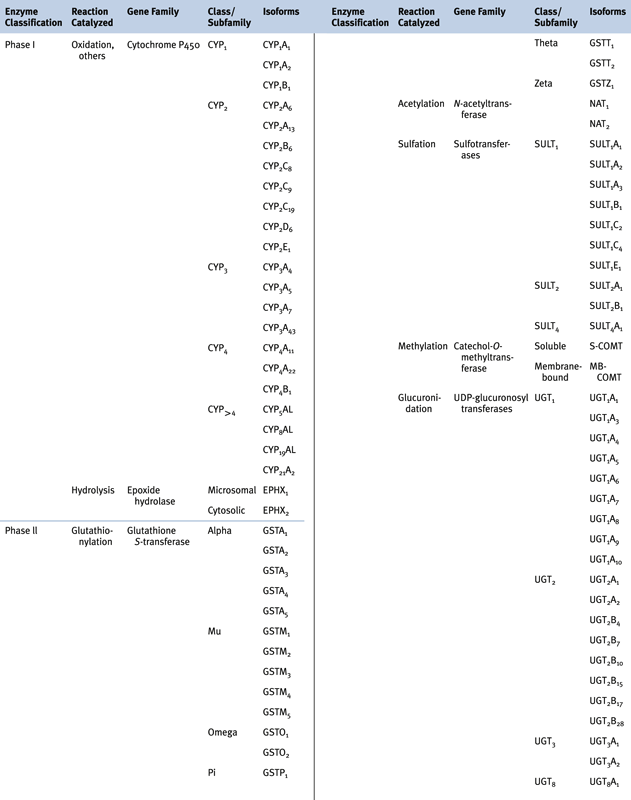
Carcinogen Metabolism
Phase I and Phase II Biotransformation Reactions
PAH Biotransformation
Aflatoxin Biotransformation
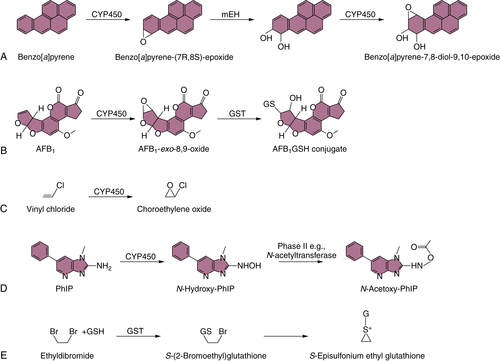
Vinyl Chloride Biotransformation
Benzidine Biotransformation
Heterocyclic Amine Biotransformation
Ethylene Dibromide (1,2-Dibromoethane) Biotransformation
DNA Repair
Defense against Oxidative Stress
Host Susceptibility to Environmental Carcinogenesis
Genetic Susceptibility
Biotransformation Enzyme Polymorphisms and Cancer Risk
GWAS and Cancer Risk
Clinical Susceptibility
Cancer Prevention
Hazard Identification
Assays
Ames Assay
HPRT Assay
Additional In Vitro Carcinogen Identification Assays
In Vivo Assays
Non-genotoxic Carcinogens
Risk Assessment and Regulation of Carcinogen Exposure
Prevention Strategies
Vaccination
Chemoprevention
Summary and Conclusions
1. Nature and nurture—lessons from chemical carcinogenesis . Nat Rev Cancer . 2005 ; 5 : 113 – 125 .
2. Historical overview of chemical carcinogenesis . In: Penning T.M. , ed. Chemical Carcinogenesis . New York, NY : Springer ; 2011 : 1 – 26 .
3. The identification of a carcinogenic compound in coal-tar . Br Med J . 1955 ; 2 ( 4942 ) : 749 – 752 .
4. Global patterns of cancer incidence and mortality rates and trends . Cancer Epidemiol Biomarkers Prev . 2010 ; 19 ( 8 ) : 1893 – 1907 .
5. Migration patterns and breast cancer risk in Asian-American women . J Natl Cancer Inst . 1993 ; 85 ( 22 ) : 1819 – 1827 .
6. Worldwide variations in colorectal cancer . CA Cancer J Clin . 2009 ; 59 ( 6 ) : 366 – 378 .
7. Cancer epidemiology . In: Pollock R.E. , ed. UICC Manual of Clinical Oncology . New York, NY : John Wiley & Sons, Inc ; 2004 : 91 – 120 .
8. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today . J Natl Cancer Inst . 1981 ; 66 ( 6 ) : 1191 – 1308 .
9. Progress and challenges in selected areas of tobacco carcinogenesis . Chem Res Toxicol . 2008 ; 21 ( 1 ) : 160 – 171 .
10. Tobacco smoke carcinogens and lung cancer . In: Penning T.M. , ed. Chemical Carcinogenesis . New York, NY : Springer ; 2011 : 53 – 74 .
11. Molecular mechanisms of cancer development in obesity . Nat Rev Cancer . 2011 ; 11 ( 12 ) : 886 – 895 .
12. Obesity, energy balance, and cancer: new opportunities for prevention . Cancer Prev Res (Phila) . 2012 ; 5 ( 11 ) : 1260 – 1272 .
13. Environmental causes of human cancers . Eur J Cancer . 2001 ; 37 ( Suppl 8 ) : S67 – S87 .
14. Cancer chemoprevention through dietary antioxidants: progress and promise . Antioxid Redox Signal . 2008 ; 10 ( 3 ) : 475 – 510 .
15. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi) . N Engl J Med . 2000 ; 342 ( 23 ) : 1686 – 1692 .
16. Ionizing radiation as a carcinogen . In: Sipes G. , McQueen C.A. , Gandolfi A.J. , eds. Comprehensive Toxicology . New York, NY : Elsevier Science, Ltd ; 1997 : 281 – 302 .
17. Carcinogenic aromatic amines and amides . In: Sipes G. , McQueen C.A. , Gandolfi A.J. , eds. Comprehensive Toxicology . New York, NY : Elsevier Science, Ltd ; 1997 : 141 – 170 .
18. Vinyl chloride and the liver . J Hepatol . 2009 ; 51 ( 6 ) : 1074 – 1081 .
19. . Environmental Factors in Cancer . Indianapolis, Ind. Bethesda, Md : Dept. of Health & Human Services National Institutes of Health, National Cancer Institute ; October 21, 2008 .
20. Chemical carcinogenesis . In: Klaassen C.D. , ed. Casarett & Doull’s Toxicology, The Basic Science of Poisons . New York, NY : McGraw-Hill ; 1996 : 201 – 267 .
21. Comprehensive toxicology . In: Bowden G.T. , Fischer S.M. , eds. Chemical Carcinogens and Anticarcinogens . Vol 12 . New York, NY : Elsevier Science Inc ; 1997 .
22. Ultraviolet radiation and skin cancer . Int J Dermatol . 2010 ; 49 ( 9 ) : 978 – 986 .
23. Cell cycle kinetics following UVA irradiation in comparison to UVB and UVC irradiation . Photochem Photobiol . 1996 ; 63 ( 4 ) : 492 – 497 .
24. The optics of human skin . J Invest Dermatol . 1981 ; 77 ( 1 ) : 13 – 19 .
25. Early events in UV carcinogenesis—DNA damage, target cells and mutant p53 foci . Photochem Photobiol . 2008 ; 84 ( 2 ) : 382 – 387 .
26. From genes to drugs: targeted strategies for melanoma . Nat Rev Cancer . 2012 ; 12 ( 5 ) : 349 – 361 .
27. The cancer epidemiology of radiation . Oncogene . 2004 ; 23 ( 38 ) : 6404 – 6428 .
28. Radiation carcinogenesis . Carcinogenesis . 2000 ; 21 ( 3 ) : 397 – 404 .
29. What are the risks of low-level exposure to alpha radiation from radon? Proc Natl Acad Sci U S A . 1997 ; 94 ( 12 ) : 5996 – 5997 .
30. Polycyclic aromatic hydrocarbon carcinogenesis . In: Harvey R.G. , ed. Polycyclic Hydrocarbons and Carcinogenesis. ACS Symposium Series 283 . Philadelphia, Pa : American Chemical Society ; 1985 : 1 – 17 .
31. Benzene in the environment: an assessment of the potential risks to the health of the population . Occup Environ Med . 2001 ; 58 ( 1 ) : 2 – 13 .
32. Current understanding of the mechanism of benzene-induced leukemia in humans: implications for risk assessment . Carcinogenesis . 2012 ; 33 ( 2 ) : 240 – 252 .
33. Covalent polycyclic aromatic hydrocarbon-DNA adducts: carcinogenicity, structure and function . In: Penning T.M. , ed. Chemical Carcinogenesis . New York, NY : Springer ; 2011 : 181 – 208 .
34. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates . Carcinogenesis . 2001 ; 22 ( 12 ) : 1903 – 1930 .
35. Aflatoxin: a 50-year odyssey of mechanistic and translational toxicology . Toxicol Sci . 2011 ; 120 ( Suppl 1 ) : S28 – S48 .
36. Aflatoxin and hepatocellular carcinoma . In: Penning T.M. , ed. Chemical Carcinogenesis . New York, NY : Springer ; 2011 : 113 – 134 .
37. Carcinogenicity of azo colorants: influence of solubility and bioavailability . Toxicol Lett . 2004 ; 151 ( 1 ) : 203 – 210 .
38. Metabolism of N-acetylbenzidine and initiation of bladder cancer . Mutat Res . 2002 ; 506-507 : 29 – 40 .
39. Benzidine mechanistic data and risk assessment: species- and organ-specific metabolic activation . Pharmacol Ther . 1996 ; 71 ( 1-2 ) : 107 – 126 .
40. Risk of colorectal and other gastro-intestinal cancers after exposure to nitrate, nitrite and N-nitroso compounds: a follow-up study . Int J Cancer . 1999 ; 80 ( 6 ) : 852 – 856 .
41. Formation and human risk of carcinogenic heterocyclic amines formed from natural precursors in meat . Nutr Rev . 2005 ; 63 ( 5 ) : 158 – 165 .
42. Meat-related mutagens/carcinogens in the etiology of colorectal cancer . Environ Mol Mutagen . 2004 ; 44 ( 1 ) : 44 – 55 .
43. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines . Chem Res Toxicol . 1998 ; 11 ( 6 ) : 559 – 603 .
44. Beryllium: genotoxicity and carcinogenicity . Mutat Res . 2003 ; 533 ( 1-2 ) : 99 – 105 .
45. Occupational exposure to beryllium and cancer risk: a review of the epidemiologic evidence . Crit Rev Toxicol . 2012 ; 42 ( 2 ) : 107 – 118 .
46. Beryllium and lung cancer: a weight of evidence evaluation of the toxicological and epidemiological literature . Crit Rev Toxicol . 2009 ; 39 ( Suppl 1 ) : 1 – 32 .
47. Mechanisms of cadmium carcinogenesis . Toxicol Appl Pharmacol . 2009 ; 238 ( 3 ) : 272 – 279 .
48. Arsenic in the aetiology of cancer . Mutat Res . 2006 ; 612 ( 3 ) : 215 – 246 .
49. Some drinking-water disinfectants and contaminants, including arsenic . IARC Monogr Eval Carcinog Risks Hum . 2004 ; 84 : 1 – 477 .
50. Molecular mechanisms of arsenic carcinogenesis . Mol Cell Biochem . 2004 ; 255 ( 1-2 ) : 57 – 66 .
51. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis . Environ Health Perspect . 2011 ; 119 ( 1 ) : 11 – 19 .
52. Chromium in drinking water: sources, metabolism, and cancer risks . Chem Res Toxicol . 2011 ; 24 ( 10 ) : 1617 – 1629 .
53. Molecular mechanisms of Cr(VI)-induced carcinogenesis . Mol Cell Biochem . 2002 ; 234-235 ( 1-2 ) : 293 – 300 .
54. Malignant mesothelioma . Lancet . 2005 ; 366 ( 9483 ) : 397 – 408 .
55. Molecular pathways: targeting mechanisms of asbestos and erionite carcinogenesis in mesothelioma . Clin Cancer Res . 2012 ; 18 ( 3 ) : 598 – 604 .
56. Case-control study of lung cancer among sugar cane farmers in India . Occup Environ Med . 1999 ; 56 ( 8 ) : 548 – 552 .
57. Prevention of hormone-related cancers: breast cancer . J Clin Oncol . 2005 ; 23 ( 2 ) : 357 – 367 .
58. Prevention of hormone-related cancers: prostate cancer . J Clin Oncol . 2005 ; 23 ( 2 ) : 368 – 377 .
59. Multistage carcinogenesis . In: Penning T.M. , ed. Chemical Carcinogenesis . New York, NY : Springer ; 2011 : 27 – 52 .
60. Hallmarks of cancer: the next generation . Cell . 2011 ; 144 ( 5 ) : 646 – 674 .
61. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications . Nat Protoc . 2009 ; 4 ( 9 ) : 1350 – 1362 .
62. Evidence for the binding of polynuclear aromatic hydrocarbons to the nucleic acids of mouse skin: relation between carcinogenic power of hydrocarbons and their binding to deoxyribonucleic acid . Nature . 1964 ; 202 : 781 – 784 .
63. Growth factor signaling pathways as targets for prevention of epithelial carcinogenesis . Mol Carcinog . 2011 ; 50 ( 4 ) : 264 – 279 .
64. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis . J Clin Invest . 2004 ; 114 ( 5 ) : 720 – 728 .
65. Role of PI3K/Akt signaling in insulin-like growth factor-1 (IGF-1) skin tumor promotion . Mol Carcinog . 2005 ; 44 ( 2 ) : 137 – 145 .
66. Mechanisms of tumor promotion . In: Slaga T.J. , ed. Tumor Promotion in Internal Organs . Vol. 1 . Boca Raton, Fla : CRC Press, Inc ; 1983 .
67. Models of carcinogenesis: an overview . Carcinogenesis . 2010 ; 31 ( 10 ) : 1703 – 1709 .
68. The role of epigenetics in environmental and occupational carcinogenesis . Chem Biol Interact . 2010 ; 188 ( 2 ) : 340 – 349 .
69. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors . Mutagenesis . 2007 ; 22 ( 2 ) : 91 – 103 .
70. Modelling the molecular circuitry of cancer . Nat Rev Cancer . 2002 ; 2 ( 5 ) : 331 – 341 .
71. Lessons from hereditary colorectal cancer . Cell . 1996 ; 87 ( 2 ) : 159 – 170 .
72. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma . Oncogene . 1999 ; 18 ( 3 ) : 643 – 650 .
73. TP53 Mutations in human cancers: origins, consequences and clinical use . In: Levine A. , Lane D. , eds. Cold Spring Harbor Perspectives in Biology . Cold Spring Harbor, NY : Cold Spring Harbor Laboratory Press ; 2012 : 1 – 17 .
74. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53 . Science . 1996 ; 274 ( 5286 ) : 430 – 432 .
75. Detoxification of chemical carcinogens and chemoprevention . In: Penning T.M. , ed. Chemical Carcinogenesis . New York, NY : Springer ; 2011 : 159 – 180 .
76. Role of cytochrome p4501 family members in the metabolic activation of polycyclic aromatic hydrocarbons in mouse epidermis . Chem Res Toxicol . 2004 ; 17 ( 12 ) : 1667 – 1674 .
77. Concise review of the glutathione S-transferases and their significance to toxicology . Toxicol Sci . 1999 ; 49 ( 2 ) : 156 – 164 .
78. Identification of amino acid residues essential for high aflatoxin B1-8,9-epoxide conjugation activity in alpha class glutathione S-transferases through site-directed mutagenesis . Toxicol Appl Pharmacol . 1998 ; 152 ( 1 ) : 166 – 174 .
79. Evidence for formation of an S-[2-(N7-guanyl)ethyl]glutathione adduct in glutathione-mediated binding of the carcinogen 1,2-dibromoethane to DNA . Proc Natl Acad Sci U S A . 1983 ; 80 ( 17 ) : 5266 – 5270 .
80. DNA polymerases and cancer . Nat Rev Cancer . 2011 ; 11 ( 2 ) : 96 – 110 .
81. Glutathione S-transferase polymorphisms: cancer incidence and therapy . Oncogene . 2006 ; 25 ( 11 ) : 1639 – 1648 .
82. Role of xenobiotic metabolic enzymes in cancer epidemiology . Methods Mol Biol . 2009 ; 472 : 243 – 264 .
83. Current status of genome-wide association studies in cancer . Hum Genet . 2011 ; 130 ( 1 ) : 59 – 78 .
84. Developmental reprogramming of cancer susceptibility . Nat Rev Cancer . 2012 ; 12 ( 7 ) : 479 – 486 .
85. The Ames Salmonella/microsome mutagenicity assay . Mutat Res . 2000 ; 455 ( 1-2 ) : 29 – 60 .
86. Characterizing and predicting carcinogenicity and mode of action using conventional and toxicogenomics methods . Mutat Res . 2010 ; 705 ( 3 ) : 184 – 200 .
87. Mechanisms of nongenotoxic carcinogenesis and assessment of the human hazard . Regul Toxicol Pharmacol . 2000 ; 32 ( 2 ) : 135 – 143 .
88. Cancer prevention: from 1727 to milestones of the past 100 years . Cancer Res . 2009 ; 69 ( 13 ) : 5269 – 5284 .
89. Vaccines for tumour prevention . Nat Rev Cancer . 2006 ; 6 ( 3 ) : 204 – 216 .
90. Modulation of xenobiotic metabolising enzymes by anticarcinogens—focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colorectal carcinogenesis . Mutat Res . 2005 ; 591 ( 1-2 ) : 74 – 92 .
91. An update on chemoprevention strategies in prostate cancer for 2006 . Curr Opin Urol . 2006 ; 16 ( 3 ) : 132 – 137 .