Chapter 591 Encephalopathies
Encephalopathy is a generalized disorder of cerebral function that may be acute or chronic, progressive or static. The etiologies of the encephalopathies in children include infectious, toxic (carbon monoxide, drugs, lead), metabolic, genetic and ischemic causes. Hypoxic-ischemic encephalopathy is discussed in Chapter 93.5.
591.1 Cerebral Palsy
See Chapters 33 and 91.2.
Clinical Manifestations
CP is generally divided into several major motor syndromes that differ according to the pattern of neurologic involvement, neuropathology, and etiology (Table 591-1). The physiologic classification identifies the major motor abnormality, whereas the topographic taxonomy indicates the involved extremities. CP is also commonly associated with a spectrum of developmental disabilities, including mental retardation, epilepsy, and visual, hearing, speech, cognitive, and behavioral abnormalities. The motor handicap may be the least of the child’s problems.
Table 591-1 CLASSIFICATION OF CEREBRAL PALSY AND MAJOR CAUSES
| MOTOR SYNDROME (APPROX % OF CP) | NEUROPATHOLOGY/MRI | MAJOR CAUSES |
|---|---|---|
| Spastic diplegia (35%) | Periventricular leukomalacia Periventricular cysts or scars in White matter, enlargement of ventricles, squared of posterior ventricles |
Prematurity |
| Ischemia | ||
| Infection | ||
| Endocrine/metabolic (e.g., thyroid) | ||
| Spastic quadriplegia (20%) | Periventricular leukomalacia | Ischemia, infection |
| Multicystic encephalomalacia Cortical malformations |
Endocrine/metabolic, genetic/developmental | |
| Hemiplegia (25%) | Stroke: in utero or neonatal Focal infarct or cortical, subcortical damage Cortical malformations |
Thrombophilic disorders |
| Infection | ||
| Genetic/developmental | ||
| Periventricular hemorrhagic infarction | ||
| Extrapyramidal (athetoid, dyskinetic) (15%) | Asphyxia: symmetric scars in putamen and thalamus Kernicterus: scars in globus pallidus, hippocampus Mitochondrial: scaring globus pallidus, caudate, putamen, brainstem No lesions: ? dopa-responsive dystonia |
Asphyxia |
| Kernicterus | ||
| Mitochondrial | ||
| Genetic/metabolic |
Infants with spastic hemiplegia have decreased spontaneous movements on the affected side and show hand preference at a very early age. The arm is often more involved than the leg and difficulty in hand manipulation is obvious by 1 yr of age. Walking is usually delayed until 18-24 mo, and a circumductive gait is apparent. Examination of the extremities may show growth arrest, particularly in the hand and thumbnail, especially if the contralateral parietal lobe is abnormal, because extremity growth is influenced by this area of the brain. Spasticity refers to the quality of increased muscle tone which increases with the speed of passive muscle stretching and is greatest in antigravity muscles. It is apparent in the affected extremities, particularly at the ankle, causing an equinovarus deformity of the foot. An affected child often walks on tiptoe because of the increased tone in the antigravity gastrocnemius muscles, and the affected upper extremity assumes a flexed posture when the child runs. Ankle clonus and a Babinski sign may be present, the deep tendon reflexes are increased, and weakness of the hand and foot dorsiflexors is evident. About one third of patients with spastic hemiplegia have a seizure disorder that usually develops in the 1st yr or 2; approximately 25% have cognitive abnormalities including mental retardation. MRI is far more sensitive than CT for most lesions seen with CP, although a CT scan may be useful for detecting calcifications associated with congenital infections. In the European CP study, 34% of children with hemiplegia had injury to the white matter that probably dated to the in utero period and 27% had a focal lesion that may have resulted from a stroke. Other children with hemiplegic CP had had malformations from multiple causes including infections (e.g., cytomegalovirus), lissencephaly, polymicrogyria, schizencephaly, or cortical dysplasia. Focal cerebral infarction (stroke) secondary to intrauterine or perinatal thromboembolism related to thrombophilic disorders, like the presence of anticardiolipin antibodies, is an important cause of hemiplegic CP (Chapter 594). Family histories suggestive of thrombosis and inherited clotting disorders, such as factor V Leiden mutation, may be present and evaluation of the mother may provide information valuable for future pregnancies and other family members.
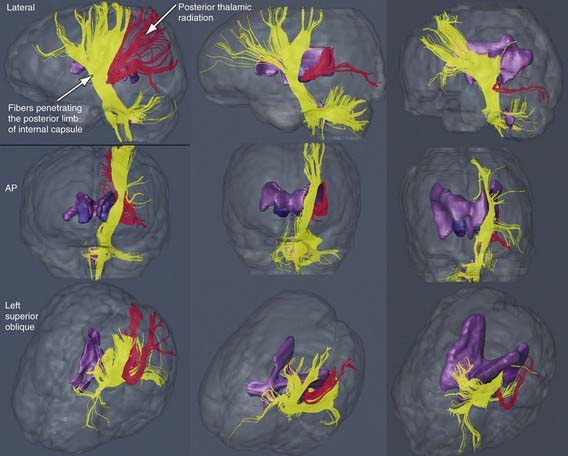
The most common neuropathologic finding in children with spastic diplegia is PVL, which is visualized on MRI in more than 70% of cases. MRI typically shows scarring and shrinkage in the periventricular white matter with compensatory enlargement of the cerebral ventricles. However, neuropathology has also demonstrated a reduction in oligodendroglia in more widespread subcortical regions beyond the periventricular zones, and these subcortical lesions may contribute to the learning problems these patients can have. MRI with diffusion tensor imaging (DTI) is being used to map white matter tracks more precisely in patients with spastic diplegia, and this technique has shown that thalamocortical sensory pathways are often injured as severely as motor corticospinal pathways (Fig 591-1). These observations have led to greater interest in the importance of sensory deficits in these patients, which may be important for designing rehabilitative techniques.
Treatment
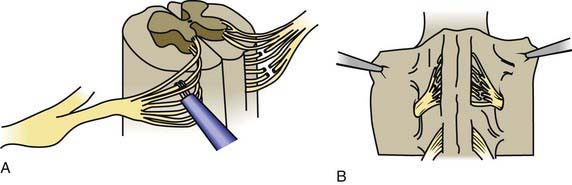
Children with spastic diplegia are treated initially with the assistance of adaptive equipment, such as walkers, poles, and standing frames. If a patient has marked spasticity of the lower extremities or evidence of hip dislocation, consideration should be given to performing surgical soft tissue procedures that reduce muscle spasm around the hip girdle, including an adductor tenotomy or psoas transfer and release. A rhizotomy procedure in which the roots of the spinal nerves are divided produces considerable improvement in selected patients with severe spastic diplegia (Fig. 591-2). A tight heel cord in a child with spastic hemiplegia may be treated surgically by tenotomy of the Achilles tendon. Quadriplegia is managed with motorized wheelchairs, special feeding devices, modified typewriters, and customized seating arrangements. The function of the affected extremities in children with hemiplegic CP can often be improved by therapy in which movement of the good side is constrained with casts while the impaired extremities perform exercises which induce improved hand and arm functioning. This constraint-induced movement therapy is effective in patients of all ages.
Albavera-Hernandez C, Rodriguez JM, Idrovo AJ. Safety of botulinum toxin type A among children with spasticity secondary to cerebral palsy: a systemic review of randomized clinical trials. Clin Rehabil. 2009;23:394-407.
Alberman E, Peckham C. Cerebral palsy and perinatal exposure to neurotropic viruses. BMJ. 2006;332:63-64.
Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy. JAMA. 2006;296:1602-1608. 2006
Brunstrom JE, Bastion AJ, Wong M. Motor benefit from levodopa in spastic quadriplegic cerebral palsy. Ann Neurol. 2000;47:662-665.
Delgado MR, Hirtz D, Aisen M, et al. Practice parameter: pharmacologic treatment of spasticity in children and adolescents with cerebral palsy (an evidence-based review). Neurology. 2010;74:336-343.
Eek MN. Muscle strength training to improve gait function in children with cerebral palsy. Dev Med Child Neurol. 2008;50:759-764.
Fauconnier J, Dickinson HO, Beckung E, et al. Participation in life situations of 8–12 year old children with cerebral palsy: cross sectional European study. BMJ. 2009;338:1116-1121.
Gordon AM, Charles J, Wolf SL. Efficacy of constraint-induced movement therapy on involved upper-extremity use in children with hemiplegic cerebral palsy is not age-dependent. Pediatrics. 2006;117:363-373.
Graham EM, Ruis KA, Hartman AL, et al. A systemic review of the role of intrapartum hypoxia-ischemia in the causation of neonatal encephalopathy. Am J Obstet Gynecol. 2008;199:587-595.
Hagberg H, Mallard C. Effect of inflammation on central nervous system development and vulnerability. Curr Opin Neurol. 2005;18:117-123.
Hansel DE, Hansel CR, Shindle MK, et al. Oral baclofen in cerebral palsy: possible seizure potentiation? Pediatr Neurol. 2003;29:203-206.
Himmelmann K, McManus V, Hagberg G, et al. Dyskinetic cerebral palsy in Europe: trends in prevalence and severity. Arch Dis Child. 2009;94:921-926.
Hoon AHJr, Stashinko EE, Nagae LM, et al. Sensory and motor deficits in children with cerebral palsy born preterm correlate with diffusion tensor imaging abnormalities in thalamocortical pathways. Dev Med Child Neurol. 2009;51:697-704.
Inder TE, Wells SJ, Mogridge NB, et al. Defining the nature of the cerebral abnormalities in the premature infant: a qualitative magnetic resonance imaging study. J Pediatr. 2003;143:171-179.
Jarvis S, Glinianaia SV, Fauconnier AC, et al. SCPE collaboration of European Cerebral Palsy Registers. Arch Dis Child. 2005;90:474-479.
Johnston MV, Hagberg H. Sex and the pathogenesis of cerebral palsy. Dev Med Child Neurol. 2007;49:74-78.
Johnston MV, Hoon AHJr. Cerebral palsy. Neuromolecular Med. 2006;8:435-450.
Krageloh-Mann I, Cans C. Cerebral palsy update. Brain Dev. 2009;31:537-544.
Nagae LM, Hoon AHJr, Stashinko E, et al. Diffusion tensor imaging in children with periventricular leukomalacia: variability of injuries to white matter tracts. AJNR Am J Neuroradiol. 2007;28:1213-1222.
Nelson KB. Causative factors in cerebral palsy. Clin Obstet Gynecol. 2008;51:749-762.
Robertson CM, Watt MJ, Yasul Y. Changes in the prevalence of cerebral palsy for children born very prematurely within a population-based program over 30 years. JAMA. 2007;297:2733-2740.
Robinson MN, Peake LJ, Ditchfield MR, et al. Magnetic resonance imaging findings in a population-based cohort of children with cerebral palsy. Dev Med Child Neurol. 2009;51:39-45.
Rouse DJ, Hirtz DG, Thom E, et al. Eunice Kennedy Shriver NICHD Maternal-Fetal Medicine Units Network: a randomized, controlled trial of magnesium sulfate for the prevention of cerebral palsy. N Engl J Med. 2008;359:895-905.
Sakzewski L, Ziciani J, Boyd R. Systematic review and meta-analysis of therapeutic management of upper-limb dysfunction in children with congenital hemiplegia. Pediatrics. 2009;123:e1111-e1122.
Shevell MI, Dagenais L, Hall N, et al. The relationship of cerebral palsy subtype and functional motor impairment: a population-based study. Dev Med Child Neurol. 2009;51:872-877.
Stearns GE, Burtner P, Keenan KM, et al. Effects of constraint-induced movement therapy on hand skills and muscle recruitment of children with spastic hemiplegic cerebral palsy. NeuroRehabilitation. 2009;24:95-108.
Taylor CL, de Groot J, Blair EM, et al. The risk of cerebral palsy in survivors of multiple pregnancies with co-fetal loss or death. Am J Obstet Gynecol. 2009;201:41-47.
Woodward LJ, Anderson PJ, Austin NC, et al. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N Engl J Med. 2006;355:685-694.
Wu YW, Croen LA, Torres AR, et al. Interleukin-6 and risk for cerebral palsy in term and near-term infants. Ann Neurol. 2009;66:663-670.
Yeargin-Allsopp M, Van Naarden Barun K, Doernberg N, et al. Prevalence of cerebral palsy in 8-year-old children in three areas of the United States in 2002: a multisite collaboration. Pediatrics. 2008;121:547-554.
591.2 Mitochondrial Encephalomyopathies
See Chapters 81.4 and 603.4.
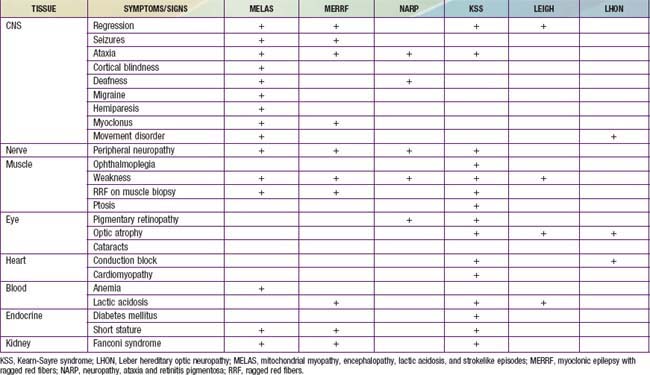
Children with mitochondrial disorders often have multifocal signs that are intermittent or relapsing-remitting, often in association with intercurrent illness. Many of these disorders were described as clinical syndromes before their genetics were understood. Children with mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS) present with developmental delay, weakness and headaches as well as focal signs that suggest a stroke. Brain imaging indicates that injury does not fit within the usual vascular territories. Children with myoclonic epilepsy with ragged red fibers (MERRF) present with myoclonus and myoclonic seizures as well as intermittent muscle weakness. The ragged red fibers referred to in the name of this disorder are clumps of abnormal mitochondria seen within muscle fibers in sections from a muscle biopsy stained with Gomori trichrome stain. NARP syndrome (neuropathy, ataxia and retinitis pigmentosa), Kearn-Sayre syndrome (KSS) (ptosis, ophthalmoplegia, heart block, Leigh disease (subacute necrotizing encephalomyelopathy), and Leber hereditary optic neuropathy (LHON) have also been defined as relatively homogeneous clinical subgroups (Table 591-2). It is important to keep in mind that mitochondrial disorders can be difficult to diagnose. They often present with novel combinations of signs and symptoms due to high mutation rates for mitochondrial DNA (mtDNA), and the severity of disease varies from person to person.
Mitochondrial diseases can be caused by mutations of nuclear DNA (nDNA) or mtDNA (Chapters 75, 80, and 81). Oxidative phosphorylation in the respiratory chain is mediated by 5 intramitochondrial enzyme complexes (complexes I-V) and 2 mobile electron carriers (coenzyme Q and cytochrome c) that are responsible for producing the adenosine triphosphate (ATP) required for normal cellular function. The maintenance of oxidative phosphorylation requires coordinated regulation of nuclear DNA and mitochondrial DNA genes. Human mtDNA is a small (16.6 kb), circular, double-stranded molecule that has been completely sequenced and encodes 13 structural proteins, all of which are subunits of the respiratory chain complexes, as well as 2 ribosomal RNAs and 22 tRNAs needed for translation. The nuclear DNA is responsible for synthesizing approximately 70 subunits, transporting them to the mitochondria via chaperone proteins, ensuring their passage across the inner mitochondrial membrane, and coordinating their correct processing and assembly. Diseases of mitochondrial oxidative phosphorylation can be divided into 3 groups: (1) defects of mtDNA, (2) defects of nDNA, and (3) defects of communication between the nuclear and mitochondrial genome.
Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes (Melas)
Children with MELAS may be normal for the 1st several years, but they gradually display delayed motor and cognitive development and short stature. The clinical syndrome is characterized by (1) recurrent strokelike episodes of hemiparesis or other focal neurologic signs with lesions most commonly seen in the posterior temporal, parietal, and occipital lobes (CT or MRI evidence of focal brain abnormalities); (2) lactic acidosis, ragged red fibers (RRF), or both; and (3) at least 2 of the following: focal or generalized seizures, dementia, recurrent migraine headaches, and vomiting. In 1 series, onset was before age 15 yr in 62% of patients, and hemianopia or cortical blindness was the most common manifestation. Cerebrospinal fluid protein is often increased. The MELAS 3243 mutation on mtDNA can also be associated with different combinations of exercise intolerance, myopathy, ophthalmoplegia, pigmentary retinopathy, hypertrophic or dilated cardiomyopathy, cardiac conduction defects, deafness, endocrinopathy (diabetes mellitus), and proximal renal tubular dysfunction. Two patients have also been described with bilateral rolandic lesions and epilepsia partialis continua associated with mitochondrial DNA mutations at 10158T>C and 10191T>C. MELAS is a progressive disorder that has been reported in siblings. It is punctuated with episodes of stroke leading to dementia (Chapter 603.4).
Reye Syndrome
This encephalopathy, which has become uncommon, is associated with pathologic features characterized by fatty degeneration of the viscera (microvesicular steatosis) and mitochondrial abnormalities and biochemical features consistent with a disturbance of mitochondrial metabolism (Chapter 353).
Bianchi MC, Sgandurra G, Tosetti M, et al. Brain magnetic resonance in the diagnostic evaluation of mitochondrial encephalopathies. Biosci Rep. 2007;27:69-95.
Crest C, Dupont S, Leguern E, et al. Levetiracetam in progressive myoclonic epilepsy: an exploratory study. Neurology. 2004;62:640-643.
Davidzon G, Mancuso M, Ferraris S, et al. POLG mutations and Alpers syndrome. Ann Neurol. 2005;57:921-923.
DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91-123.
Donato S. Multisystem manifestations of mitochondrial disorders. J Neurol. 2009;256:693-710.
Horvath R, Kemp JP, Tuppen HAL, et al. Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain. 2009;132:3165-3174.
Hudson G, Chinnery PF. Mitochondrial DNA polymerase-γ and human disease. Human Mol Genet. 2006;15:R244-R252.
Oldfors A, Tulinius M. Mitochondrial encephalopathies. J Neuropathol Exp Neurol. 2003;62:217-227.
Valente L, Piga D, Lamantea E, et al. Identification of novel mutations in five patients with mitochondrial encephalopathy. Biochim Biophys Acta. 2009;1787:491-501.
Wani AA, Ahanger SH, Bapat SA, et al. Analysis of mitochondrial DNA sequences in childhood encephalomyopathies reveals new disease-associated variants. PLoS One. 2007;2:e942.
Werner KG, Morel CF, Benseler SM, et al. Rolandic mitochondrial encephalomyelopathy and MT-ND3 mutations. Pediatr Neurol. 2009;41:27-33.
591.3 Other Encephalopathies
HIV Encephalopathy
Encephalopathy is an unfortunate and common manifestation in infants and children with HIV infection (Chapter 268). Neurologic signs in congenitally infected patients may appear during early infancy or may be delayed to as late as 5 yr of age. The primary features of HIV encephalopathy include an arrest in brain growth, evidence of developmental delay, and the evolution of neurologic signs including weakness with pyramidal tract signs, ataxia, myoclonus, pseudobulbar palsy, and seizures. However, the introduction of highly active antiretroviral therapy (HAART) and CNS-penetrating antiretroviral regimens for perinatally infected children has been associated with a 10-fold decrease in the incidence of HIV encephalopathy starting in 1996. Introduction of HAART for children has also resulted in an increase in CD4 T-cell count and a reduction in opportunistic infections and organ-specific diseases including wasting syndrome, thrombocytopenia, cardiomyopathy, and lymphoid interstitial pneumonia. High CNS-penetrating regimens are associated with 74% reduction in the risk of death in children with a diagnosis of HIV encephalopathy compared to low CNS-penetrating drugs.
Burn Encephalopathy
An encephalopathy develops in about 5% of children with significant burns in the 1st several weeks of hospitalization (Chapter 68). There is no single cause of burn encephalopathy but rather a combination of factors that include anoxia (smoke inhalation, carbon monoxide poisoning, laryngospasm), electrolyte abnormalities, bacteremia and sepsis, cortical vein thrombosis, a concomitant head injury, cerebral edema, drug reactions, and emotional distress. Seizures are the most common clinical manifestation of burn encephalopathy, but altered states of consciousness, hallucinations, and coma may also occur. Management of burn encephalopathy is directed to a search for the underlying cause and treatment of hypoxemia, seizures, specific electrolyte abnormalities, or cerebral edema. The prognosis for complete neurologic recovery is generally excellent, particularly if seizures are the primary abnormality.
Hypertensive Encephalopathy
Hypertensive encephalopathy is most commonly associated with renal disease in children, including acute glomerulonephritis, chronic pyelonephritis, and end-stage renal disease (Chapters 439 and 529). In some cases, hypertensive encephalopathy is the initial manifestation of underlying renal disease. Marked systemic hypertension produces vasoconstriction of the cerebral vessels, which leads to vascular permeability, causing areas of focal cerebral edema and hemorrhage. The onset may be acute, with seizures and coma, or more indolent, with headache, drowsiness and lethargy, nausea and vomiting, blurred vision, transient cortical blindness, and hemiparesis. Examination of the eyegrounds may be nondiagnostic in children, but papilledema and retinal hemorrhages may occur. MRI often shows increased signal intensity in the occipital lobes on T2 weighted images, which is known as posterior reversible leukoencephalopathy (PRES) and may be confused with cerebral infarctions. These high signal areas may appear in other regions of the brain as well. Treatment is directed at restoration of a normotensive state and control of seizures with appropriate anticonvulsants.
Radiation Encephalopathy
Acute radiation encephalopathy is most likely to develop in young patients who have received large daily doses of radiation. Excessive radiation injures vessel endothelium, resulting in enhanced vascular permeability, cerebral edema, and numerous hemorrhages. The child may suddenly become irritable and lethargic, complain of headache, or present with focal neurologic signs and seizures. Patients occasionally develop hemiparesis due to an infarct secondary to vascular occlusion of the cerebral vessels. Steroids are often beneficial in reducing the cerebral edema and reversing the neurologic signs. Late radiation encephalopathy is characterized by headaches and slowly progressive focal neurologic signs, including hemiparesis and seizures. Exposure of the brain to radiation for treatment of childhood cancer increases the risk of later cerebrovascular disease, including stroke, moyamoya disease, aneurysm, vascular malformations, mineralizing microangiopathy and strokelike migraines. Some children with acute lymphocytic leukemia treated with a combination of intrathecal methotrexate and cranial irradiation develop neurologic signs months or years later; signs consist of increasing lethargy, loss of cognitive abilities, dementia, and focal neurologic signs and seizures (Chapter 488). The CT scan shows calcifications in the white matter, and the postmortem examination demonstrates a necrotizing encephalopathy. This devastating complication of the treatment of leukemia has prompted re-evaluation and reduction in the use of cranial radiation in the treatment of these children.
Zellweger Syndrome (Cerebrohepatorenal Syndrome [CHRS])
This rare, lethal disorder is inherited as an autosomal recessive trait. It represents the prototype of a group of peroxisomal disorders that have overlapping symptoms, signs, and biochemical abnormalities (Chapter 80.2). The cause of the severe neurologic abnormalities is related to an arrest of migrating neuroblasts during early development, resulting in cerebral pachygyria with neuronal heterotopia (Chapter 585.7).
Florance NR, Davis RL, Lam C, et al. Anti–N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66:11-18.
Gaynon PS, Angiolillo AL, Carroll WL, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: A Children’s Oncology Group Report. Leukemia. 2010;24:285-297.
Guillén S, García San Miguel L, Resino S, et al. Opportunistic infections and organ-specific diseases in HIV-1-infected children: a cohort study (1990–2006). HIV Med. 2010;11:245-252.
Gupta S, Shah I. Neurological disorders in HIV-infected children in India. Ann Trop Paediatr. 2009;29:177-181.
Haberlandt E, Bast T, Ebner A, et al. Limbic encephalitis in children and adolescents. Arch Dis Child. 2010;96:186-191.
Lee VH, Wijidicks EFM, Manno EM, et al. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65:205-210.
Leroux G, Sellam J, Costedoat-Chalumeau N, et al. Posterior reversible encephalopathy syndrome during systemic lupus erythematosus: four new cases and review of the literature. Lupus. 2008;17:139-147.
Morris B, Partap S, Yeom K, et al. Cerebrovascular disease in childhood cancer survivors: a Children’s Oncology Group Report. Neurology. 2009;73:1906-1913.
Morris EB, Laninigham FH, Sandlund JT, et al. Posterior reversible encephalopathy syndrome in children with cancer. Pediatr Blood Cancer. 2007;48:152-159.
Onder AM, Lopez R, Teomete U, et al. Posterior reversible encephalopathy syndrome in the pediatric renal population. Pediatr Nephrol. 2007;22:1921-1929.
Patel K, Ming X, Williams PL, et al. Impact of HAART and CNS-penetrating antiretroviral regimens on HIV encephalopathy among perinatally infected children and adolescents. AIDS. 2009;23:1893-1901.
Pavlakis SG, Frank Y, Chusid R. Hypertensive encephalopathy, reversible occipitoparietal encephalopathy, or reversible posterior leukoencephalopathy: three names for an old syndrome. J Child Neurol. 1999;14:277-281.
Prasad N, Gulati S, Gupta RK, et al. Spectrum of radiological changes in hypertensive children with reversible posterior leukoencephalopathy. Br J Radiol. 2007;80:422-429.