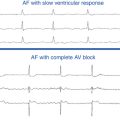
Chapter 3 Electrophysiological Mechanisms of Cardiac Arrhythmias
The mechanisms responsible for cardiac arrhythmias are generally divided into categories of disorders of impulse formation (automaticity or triggered activity), disorders of impulse conduction (reentry), or combinations of both. Automaticity is the property of cardiac cells to initiate an impulse spontaneously, without need for prior stimulation. Triggered activity is impulse initiation in cardiac fibers caused by depolarizing oscillations in membrane voltage (known as afterdepolarizations) that occur consequent to one or more preceding action potentials.1 Reentry occurs when a propagating action potential wave fails to extinguish after initial tissue activation; instead, it blocks in circumscribed areas, circulates around the zones of block, and reenters and reactivates the site of original excitation after it recovers excitability. Reentry is the likely mechanism of most recurrent clinical arrhythmias.
Automaticity
Automaticity, or spontaneous impulse initiation, is the ability of cardiac cells to depolarize spontaneously, reach threshold potential, and initiate a propagated action potential in the absence of external electrical stimulation. Altered automaticity can be caused by enhanced normal automaticity or abnormal automaticity.1
Enhanced Normal Automaticity
Pacemaker Mechanisms
Normal automaticity involves a spontaneous, slow, progressive decline (less negative) in the transmembrane potential during diastole (spontaneous diastolic depolarization or phase 4 depolarization) (see Chap. 1). Once this spontaneous depolarization reaches threshold (approximately −40 mV), a new action potential is generated.2
The ionic mechanisms responsible for normal pacemaker activity in the sinus node are still controversial. The fall in membrane potential during phase 4 seems to arise from a changing balance between positive inward currents, which favor depolarization, and positive outward currents, with a net gain in intracellular positive charges during diastole (i.e., inward depolarizing current; Fig. 3-1).1–6
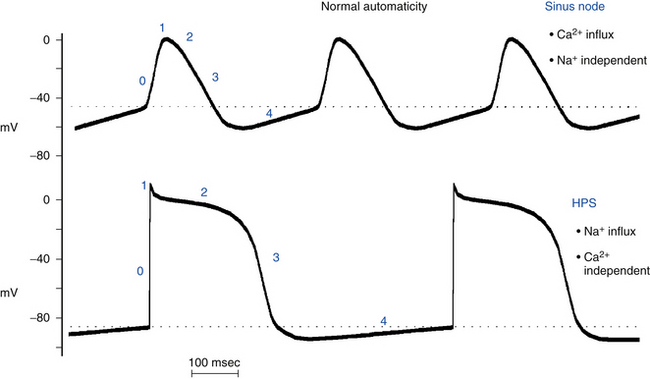
Evidence suggests that If (named the “funny” current because, unlike most voltage-sensitive currents, it is activated by hyperpolarization rather than depolarization) is one of the most important ionic currents involved in the rate regulation of cardiac pacemaker cells, hence its designation as the pacemaker current. If is an inward current carried largely by Na+ and, to a lesser extent, K+ ions. The If channels are deactivated during the action potential upstroke and the initial plateau phase of repolarization. However, they begin to activate at the end of the action potential as repolarization brings the membrane potential to levels more negative than approximately −40 to −50 mV, and If is fully activated at approximately −100 mV. Once activated, If depolarizes the membrane to a level where the Ca2+ current activates to initiate the action potential.7 In its range of activation, which quite properly comprises the voltage range of diastolic depolarization, the current is inward, and its reversal occurs at approximately −10 to −20 mV because of the mixed Na+-K+ permeability of If channels. At the end of the repolarization phase of an action potential, because If activation occurs in the background of a decaying outward (K+ time-dependent) current, current flow quickly shifts from outward to inward, thus giving rise to a sudden reversal of voltage change (from repolarizing to depolarizing) at the maximum diastolic potential. The major role of If has been reinforced by the findings that drugs such as ivabradine targeted to block If slow heart rate and mutations in the If channel are associated with slowed heart rate.2,5,8–10
On the other hand, several studies have shown that If is not the only current that can initiate the diastolic depolarization process in the sinus node. In addition to voltage and time, the electrogenic and regulatory molecules on the surface membrane of sinus node cells are strongly modulated by Ca2+ and phosphorylation, a finding suggesting that intracellular Ca2+ is an important player in controlling pacemaker cell automaticity. Newer evidence points to a substantial impact of another current on the late diastolic depolarization; that is, the Na+-Ca2+ exchanger current activated by submembrane spontaneous rhythmic local Ca2+ releases from the sarcoplasmic reticulum, a major Ca2+ store within sinus node cells, via ryanodine receptors (RyR2). Activation of the local oscillatory Ca2+ releases is independent of membrane depolarization and is driven by a high level of basal state phosphorylation of Ca2+ cycling proteins. Critically timed Ca2+ releases occur during the later phase of diastolic depolarization and activate the forward mode of the Na+-Ca2+ exchanger (one Ca2+ for three Na+). The result is in an inward membrane current that causes the late diastolic depolarization to increase exponentially, thus driving the membrane potential to the threshold to activate a sufficient number of voltage-gated L-type Ca2+ channels and leading to generation of the rapid upstroke of the next action potential (see Fig. 1-3).Although regulated by membrane potential and submembrane Ca2+, the Na+-Ca2+ exchanger does not have time-dependent gating, as do ion channels, but generates an inward current almost instantaneously when submembrane Ca2+ concentration increases.8,9,11
There is some degree of uncertainty about the relative role of If versus that of intracellular Ca2+ cycling in controlling the normal pacemaker cell automaticity and their individual (or mutual) relevance in mediating the positive-negative chronotropic effect of neurotransmitters. Furthermore, the interactions between the membrane ion channel clock and the intracellular Ca2+ clock and the cellular mechanisms underlying this internal Ca2+ clock are not completely elucidated.8,9,11
Hierarchy of Pacemaker Function
Automaticity is not limited to the cells within the sinus node. Under physiological conditions, cells in parts of the atria and within the atrioventricular node (AVN) and the His-Purkinje system (HPS) also possess pacemaking capability. However, the occurrence of spontaneous activity in these cells is prevented by the natural hierarchy of pacemaker function that causes these sites to be latent or subsidiary pacemakers.1 The spontaneous discharge rate of the sinus node normally exceeds that of all other subsidiary pacemakers (see Fig. 3-1). Therefore, the impulse initiated by the sinus node depolarizes subsidiary pacemaker sites and keeps their activity depressed before they can spontaneously reach threshold. However, slowly depolarizing and previously suppressed pacemakers in the atrium, AVN, or ventricle can become active and assume pacemaker control of the cardiac rhythm if the sinus node pacemaker becomes slow or unable to generate an impulse (e.g., secondary to depressed sinus node automaticity) or if impulses generated by the sinus node are unable to activate the subsidiary pacemaker sites (e.g., sinoatrial exit block, or atrioventricular [AV] block). The emergence of subsidiary or latent pacemakers under such circumstances is an appropriate fail-safe mechanism, which ensures that ventricular activation is maintained. Because spontaneous diastolic depolarization is a normal property, the automaticity generated by these cells is classified as normal.
There is also a natural hierarchy of intrinsic rates of subsidiary pacemakers that have normal automaticity, with atrial pacemakers having faster intrinsic rates than AV junctional pacemakers, and AV junctional pacemakers having faster rates than ventricular pacemakers.
Subsidiary Pacemakers
Subsidiary Atrial Pacemakers
Subsidiary atrial pacemakers have been identified in the atrial myocardium, especially in the crista terminalis, at the junction of the inferior right atrium and inferior vena cava, near or on the eustachian ridge, near the coronary sinus ostium, in the atrial muscle that extends into the tricuspid and mitral valves, and in the muscle sleeves that extend into the cardiac veins (venae cavae and pulmonary veins).12
Subsidiary Ventricular Pacemakers
In the ventricles, latent pacemakers are found in the HPS, where Purkinje fibers have the property of spontaneous diastolic depolarization. Isolated cells of the HPS discharge spontaneously at rates of 15 to 60 beats/min, whereas ventricular myocardial cells do not normally exhibit spontaneous diastolic depolarization or automaticity. The relatively slow spontaneous discharge rate of the HPS pacemakers, which further decreases from the His bundle to the distal Purkinje branches, ensures that pacemaker activity in the HPS will be suppressed on a beat-to-beat basis by the more rapid discharge rate of the sinus node and atrial and AV junctional pacemakers. However, enhanced Purkinje fiber automaticity can be induced by certain situations, such as myocardial infarction (MI). In this setting, some Purkinje fibers that survive the infarction develop moderately reduced maximum diastolic membrane potentials and therefore accelerated spontaneous discharge rates.13
Regulation of Pacemaker Function
The intrinsic rate at which the sinus node pacemaker cells generate impulses is determined by the interplay of three factors: the maximum diastolic potential, the threshold potential at which the action potential is initiated, and the rate or slope of phase 4 depolarization (Fig. 3-2). A change in any one of these factors will alter the time required for phase 4 depolarization to carry the membrane potential from its maximum diastolic level to threshold and thus alter the rate of impulse initiation.1
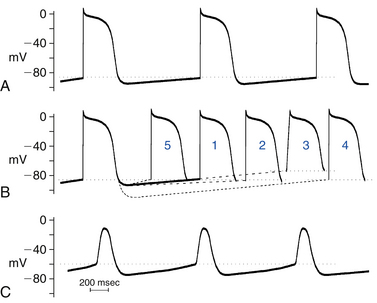
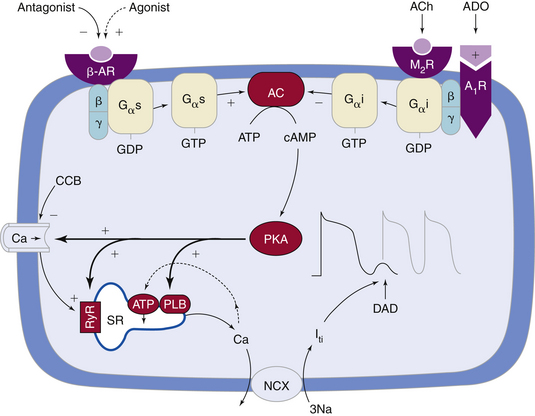
The sinus node is innervated by the parasympathetic and sympathetic nervous systems, and the balance between these systems importantly controls the pacemaker rate. The classic concept has been that of a reciprocal relationship between sympathetic and parasympathetic inputs. More recent investigations, however, stress dynamic, demand-oriented interactions, and the anatomical distribution of fibers that allows both autonomic systems to act quite selectively. Muscarinic cholinergic and beta1-adrenergic receptors are nonuniformly distributed in the sinus node, and they modulate both the rate of depolarization and impulse propagation.1
Parasympathetic Activity
Parasympathetic tone reduces the spontaneous discharge rate of the sinus node, whereas its withdrawal accelerates sinus node automaticity. Acetylcholine, the principal neurotransmitter of the parasympathetic nervous system, inhibits spontaneous impulse generation in the sinus node by increasing K+ conductance. Acetylcholine acts through M2 muscarinic receptors to activate the Gi protein, which subsequently results in activation of IKACh (an acetylcholine-activated subtype of inward rectifying current) in tissues of the sinus node and AVN as well as of the atria, Purkinje fibers, and ventricles. The increased outward repolarizing K+ current (IK) leads to membrane hyperpolarization (i.e., the resting potential and the maximum diastolic potential become more negative). The resulting hyperpolarization of the membrane potential lengthens the time required for the membrane potential to depolarize to threshold and thereby decreases the automaticity of the sinus node (see Fig. 3-2). In addition, activation of the Gi protein results in inhibition of beta-receptor–stimulated adenylate cyclase activity, thus reducing cyclic adenosine monophosphate (cAMP) and inhibiting protein kinase A, with subsequent inhibition of the inward Ca2+ current. This results in reduction of the rate of diastolic depolarization because of less Ca2+ entry and subsequent slowing of the pacemaker activity. Inhibition of beta-receptor–stimulated adenylate cyclase activity can also inhibit the inward If current.
Sympathetic Activity
Increased sympathetic nerve traffic and the adrenomedullary release of catecholamines increase sinus node discharge rate. Stimulation of beta1-receptors by catecholamines enhances the L-type of inward Ca2+ current (ICaL) by increasing cAMP and activating the protein kinase A system; the increment in inward Ca2+ current increases the slope of diastolic depolarization and enhances pacemaker activity (see Fig. 3-2). The redistribution of Ca2+ can also increase the completeness and the rate of deactivation of the rapid (IKr) and slow (IKs) components of the delayed rectifier IK; the ensuing decline in the opposing outward current results in a further net increase in inward current. Catecholamines can also enhance the inward If current by shifting the voltage dependence of If to more positive potentials, thus augmenting the slope of phase 4 and increasing the rate of sinus node firing.10
Atrial, AV junctional, and HPS subsidiary pacemakers are also under similar autonomic control, with the sympathetic nervous system enhancing pacemaker activity through beta1-adrenergic stimulation and the parasympathetic nervous system inhibiting pacemaker activity through muscarinic receptor stimulation.1
Other Influences
Evidence indicates that active and passive changes in the mechanical environment of the heart provide feedback to modify cardiac rate and rhythm and are capable of influencing both the initiation and spread of cardiac excitation. This direction of the crosstalk between cardiac electrical and mechanical activity is referred to as mechanoelectric feedback and is thought to be involved in the adjustment of heart rate to changes in mechanical load, which would help explain the precise beat-to-beat regulation of cardiac performance. Acute mechanical stretch enhances automaticity, reversibly depolarizes the cell membrane, and shortens the action potential duration. Feedback from cardiac mechanics to electrical activity involves mechanosensitive ion channels and ATP-sensitive K+ channels. In addition, Na+ and Ca2+ entering the cells via nonselective ion channels are thought to contribute to the genesis of stretch-induced arrhythmia.14
Abnormal Automaticity
In the normal heart, automaticity is confined to the sinus node and other specialized conducting tissues. Working atrial and ventricular myocardial cells do not normally have spontaneous diastolic depolarization and do not initiate spontaneous impulses, even when they are not excited for long periods of time by propagating impulses. Although these cells do have an If, the range of activation of this current in these cells is much more negative (−120 to −170 mV) than in Purkinje fibers or in the sinus node. As a result, during physiological resting membrane potentials (−85 to −95 mV), the If is not activated, and ventricular cells do not depolarize spontaneously.1 When the resting potentials of these cells are depolarized sufficiently, to approximately −70 to −30 mV, however, spontaneous diastolic depolarization can occur and cause repetitive impulse initiation, a phenomenon called depolarization-induced automaticity or abnormal automaticity (see Fig. 3-2). Similarly, cells in the Purkinje system, which are normally automatic at high levels of membrane potential, show abnormal automaticity when the membrane potential is reduced to approximately −60 mV or less, as can occur in ischemic regions of the heart. When the steady-state membrane potential of Purkinje fibers is reduced to approximately −60 mV or less, the If channels that participate in normal pacemaker activity in Purkinje fibers are closed and nonfunctional, and automaticity is therefore not caused by the normal pacemaker mechanism. It can, however, be caused by an “abnormal” mechanism. In contrast, enhanced automaticity of the sinus node, subsidiary atrial pacemakers, or the AVN caused by a mechanism other than acceleration of normal automaticity has not been demonstrated clinically.15
Several different mechanisms probably cause abnormal pacemaker activity at low membrane potentials, including activation and deactivation of the delayed rectifier IK, intracellular Ca2+ release from the sarcoplasmic reticulum that causes activation of inward Ca2+ currents and the inward INa (through the Na+-Ca2+ exchanger), and a potential contribution by If.16 It has not been determined which of these mechanisms are operative in the different pathological conditions in which abnormal automaticity can occur.1
The upstroke of the spontaneously occurring action potentials generated by abnormal automaticity can be caused by Na+ or Ca2+ inward currents or possibly a combination of the two.1 In the range of diastolic potentials between approximately −70 and −50 mV, repetitive activity is dependent on extracellular Na+ concentration and can be decreased or abolished by Na+ channel blockers. In a diastolic potential range of approximately −50 to −30 mV, Na+ channels are predominantly inactivated; repetitive activity depends on extracellular Ca2+ concentration and is reduced by L-type Ca2+ channel blockers.
The intrinsic rate of a focus with abnormal automaticity is a function of the membrane potential. The more positive the membrane potential is, the faster the automatic rate will be (see Fig. 3-2). Abnormal automaticity is less vulnerable to suppression by overdrive pacing (see later). Therefore, even occasional slowing of the sinus node rate can allow an ectopic focus with abnormal automaticity to fire without a preceding long period of quiescence. Catecholamines can increase the rate of discharge caused by abnormal automaticity and therefore can contribute to a shift in the pacemaker site from the sinus node to a region with abnormal automaticity.
Overdrive Suppression of Automatic Rhythms
Suppression of Normal and Abnormal Automatic Subsidiary Pacemakers
The sinus node likely maintains its dominance over subsidiary pacemakers in the AVN and the Purkinje fibers by several mechanisms. During sinus rhythm in a normal heart, the intrinsic automatic rate of the sinus node is faster than that of the other potentially automatic cells. Consequently, the latent pacemakers are excited by propagated impulses from the sinus node before they have a chance to depolarize spontaneously to threshold potential. The higher frequency of sinus node discharge also suppresses the automaticity of other pacemaker sites by a mechanism called overdrive suppression. The diastolic (phase 4) depolarization of the latent pacemaker cells with the property of normal automaticity is actually inhibited because the cells are repeatedly depolarized by the impulses from the sinus node.1 Electrotonic interaction between the pacemaker cells and the nonpacemaker cells in the surrounding myocardium via intercalated discs and gap junctions can also hyperpolarize the latent pacemakers and contribute to their suppression (Fig. 3-3).
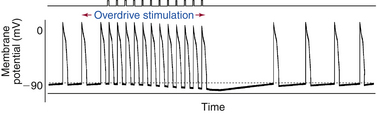
Mechanism of Overdrive Suppression
The mechanism of overdrive suppression is mediated mostly by enhanced activity of the Na+-K+ exchange pump that results from driving a pacemaker cell faster than its intrinsic spontaneous rate. During normal sinus rhythm (NSR), latent pacemakers are depolarized at a higher frequency than their intrinsic rate of automaticity. The increased frequency of depolarizations leads to an increase in intracellular Na+, which enters the cell with every action potential, because more Na+ enters the cell per unit time.1 The increased intracellular Na+ stimulates the Na+-K+ exchange pump. Because the Na+-K+ exchange pump is electrogenic (i.e., moves more Na+ outward than K+ inward), it generates a net outward (hyperpolarizing) current across the cell membrane. This drives the membrane potential more negative, thereby offsetting the depolarizing If being carried into the cell and slowing the rate of phase 4 diastolic depolarization. This effectively prevents the If from depolarizing the cell to its threshold potential and thereby suppresses spontaneous impulse initiation in these cells.
Abnormally automatic cells and tissues at reduced levels of membrane potential are less sensitive to overdrive suppression than cells and tissues that are fully polarized, with enhanced normal automaticity. The amount of overdrive suppression of spontaneous diastolic depolarization that causes abnormal automaticity is directly related to the level of membrane potential at which the automatic rhythm occurs. At low levels of membrane potential, Na+ channels are inactivated, decreasing the fast inward INa; therefore, there are reductions in the amount of Na+ entering the cell during overdrive and the degree of stimulation of the Na+-K+ exchange pump. The more polarized the membrane is during phase 4, the larger the amount will be of Na+ entering the cell with each action potential, and the more overdrive suppression will occur. As a result of the lack of overdrive suppression of abnormally automatic cells, even transient sinus pauses can permit an ectopic focus with a slower rate than the sinus node to capture the heart for one or more beats. However, even in situations in which the cells can be sufficiently depolarized to inactivate the INa and limit intracellular Na+ load, overdrive suppression can still be observed because of increased intracellular Ca2+ loading. Such Ca2+ loading can activate Ca2+-dependent K+ conductance (favoring repolarization) and promote Ca2+ extrusion through the Na+-Ca2+ exchanger and Ca2+ channel phosphorylation, thus increasing Na+ load and thus Na+-K+ exchange pump activity. The increase in intracellular Ca2+ load can also reduce the depolarizing ICaL by promoting Ca2+-induced inactivation of the Ca2+ current.
Arrhythmias Caused by Automaticity
Parasystole
Parasystole is a result of interaction between two fixed rate pacemakers having different discharge rates. Parasystolic pacemakers can exist in either the atrium or the ventricle. The latent pacemaker is protected from being overdriven by the dominant rhythm (usually NSR) by intermittent or constant entrance block (i.e., impulses of sinus origin fail to depolarize the latent pacemaker secondary to block in the tissue surrounding the latent pacemaker focus). Various mechanisms have been postulated to explain the protected zone surrounding the ectopic focus. It is possible that the depolarized level of membrane potential at which abnormal automaticity occurs can cause entrance block, leading to parasystole. This would be an example of an arrhythmia caused by a combination of an abnormality of impulse conduction and impulse initiation. Such block, however, must be unidirectional, so that activity from the ectopic pacemaker can exit and produce depolarization whenever the surrounding myocardium is excitable. The protected pacemaker is said to be a parasystolic focus. In general, under these conditions, a protected focus of automaticity of this type fires at its own intrinsic frequency, and the intervals between the discharges of each pacemaker are multiples of its intrinsic discharge rate (sometimes described as fixed parasystole). Therefore, on the surface ECG, the coupling intervals of the manifest ectopic beats wander through the basic cycle of the sinus rhythm. Accordingly, the traditional ECG criteria used to recognize the fixed form of parasystole are (1) the presence of variable coupling intervals of the manifest ectopic beats, (2) interectopic intervals that are simple multiples of a common denominator, and (3) the presence of fusion beats. Occasionally, the parasystolic focus can exhibit exit block, during which it may fail to depolarize excitable myocardium.17
Although the parasystolic focus is protected, it may not be totally immune to the surrounding electrical activity. The effective electrical communication that permits the emergence of the ectopic discharges can also allow the rhythmic activity of the surrounding tissues to electrotonically influence the periodicity of the pacemaker discharge rate (described as modulated parasystole). Electrotonic influences arriving during the early stage of diastolic depolarization result in a delay in the firing of the parasystolic focus, whereas those arriving late accelerate the discharge of the parasystolic focus. As a consequence, the dominant pacemaker can entrain the partially protected parasystolic focus and force it to discharge at periods that may be faster or slower than its own intrinsic cycle and give rise to premature discharges whose patterns depend on the degree of modulation and the basic heart rate, occasionally mimic reentry, and occur at fixed coupling intervals. Therefore, appropriate diagnosis of modulated parasystole relies on the construction of a phase response curve as theoretical evidence of modulation of the ectopic pacemaker cycle length (CL) by the electrotonic activity generated by the sinus discharges across the area of protection.17
Arrhythmias Caused by Abnormal Automaticity
There appears to be an association between abnormal Purkinje fiber automaticity and the arrhythmias that occur during the acute phase of MI (e.g., an accelerated idioventricular rhythm). However, the role of abnormal automaticity in the development of ventricular arrhythmias associated with chronic ischemic heart disease is less certain. Additionally, isolated myocytes obtained from hypertrophied and failing hearts have been shown to manifest spontaneous diastolic depolarization and enhanced If, findings suggesting that abnormal automaticity can contribute to the occurrence of some arrhythmias in heart failure and left ventricular hypertrophy.
Triggered Activity
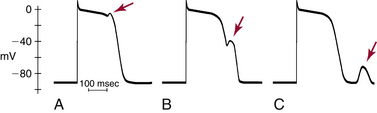
Triggered activity is impulse initiation in cardiac fibers caused by afterdepolarizations that occur consequent to a preceding impulse or series of impulses.1 Afterdepolarizations are depolarizing oscillations in membrane potential that follow the upstroke of a preceding action potential. Afterdepolarizations can occur early during the repolarization phase of the action potential (early afterdepolarization [EAD]) or late, after completion of the repolarization phase (delayed afterdepolarization [DAD]; Fig. 3-4). When either type of afterdepolarization is large enough to reach the threshold potential for activation of a regenerative inward current, a new action potential is generated, which is referred to as triggered.
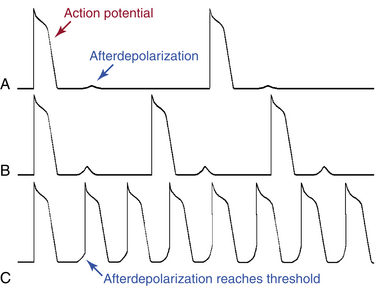
Delayed Afterdepolarizations and Triggered Activity
DADs are oscillations in membrane voltage that occur after completion of repolarization of the action potential (i.e., during phase 4). The transient nature of the DAD distinguishes it from normal spontaneous diastolic (pacemaker) depolarization, during which the membrane potential declines almost monotonically until the next action potential occurs. DADs may or may not reach threshold. Subthreshold DADs do not initiate action potentials or trigger arrhythmias. When a DAD does reach threshold, only one triggered action potential occurs (Fig. 3-5). The triggered action potential can also be followed by a DAD that, again, may or may not reach threshold and may or may not trigger another action potential. The first triggered action potential is often followed by a short or long train of additional triggered action potentials, each arising from the DAD caused by the previous action potential.
Ionic Basis of Delayed Afterdepolarizations
DADs usually occur under a variety of conditions in which Ca2+ overload develops in the cytoplasm and sarcoplasmic reticulum. During the plateau phase of the normal action potential, Ca2+ flows through voltage-dependent L-type Ca2+ channels (ICaL). Although the rise in intracellular Ca2+ is small and not sufficient to induce contraction, the small amount of Ca2+ entering the cell via ICaL triggers a massive release of Ca2+ from the sarcoplasmic reticulum (the major store for Ca2+) into the cytosol by opening the RyR2 channels (present in the membrane of the sarcoplasmic reticulum) in a process known as Ca2+-induced Ca2+ release (CICR).1,6 During repolarization (i.e., diastole), most of the surplus Ca2+ in the cytosol is resequestered into the sarcoplasmic reticulum by the sarcoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA), the activity of which is controlled by the phosphoprotein phospholamban. Additionally, some of the Ca2+ is extruded from the cell by the Na+-Ca2+ exchanger to balance the Ca2+ that enters with Ca2+ current. Recurring Ca2+ release-uptake cycles provide the basis for periodic elevations of the cytosolic Ca2+ concentration and contractions of myocytes, hence for the orderly beating of the heart (Fig. 3-6).18–20
Under various pathological conditions, Ca2+ concentration in the sarcoplasmic reticulum can rise to a critical level during repolarization (i.e., Ca2+ overload), at which time a secondary spontaneous release of Ca2+ from the sarcoplasmic reticulum occurs after the action potential, rather than as a part of excitation-contraction coupling. This secondary release of Ca2+ results in inappropriately timed Ca2+ transients and contractions. Spontaneous Ca2+ waves are arrhythmogenic; they induce Ca2+-dependent depolarizing membrane currents (transient inward current), mainly by activation of the Na+-Ca2+ exchanger, thereby causing oscillations of the membrane potential known as DADs. After one or several DADs, myoplasmic Ca2+ can decrease because the Na+-Ca2+ exchanger extrudes Ca2+ from the cell, and the membrane potential stops oscillating.18–20
When the DADs are of low amplitude, they usually are not apparent or clinically significant. However, during pathological conditions (e.g., myocardial ischemia, acidosis, hypomagnesemia, digitalis toxicity, and increased catecholamines), the amplitude of the Ca2+-mediated oscillations is increased and can reach the stimulation threshold, and an action potential is triggered. If this process continues, sustained tachycardia will develop. Probably the most important influence that causes subthreshold DADs to reach threshold is a decrease in the initiating CL, because that increases both the amplitude and rate of the DADs. Therefore, initiation of arrhythmias triggered by DADs can be facilitated by a spontaneous or pacing-induced increase in the heart rate.
Digitalis causes DAD-dependent triggered arrhythmias by inhibiting the Na+-K+ exchange pump. In toxic amounts, this effect results in the accumulation of intracellular Na+ and, consequentially, an enhancement of the Na+-Ca2+ exchanger in the reverse mode (Na+ removal, Ca2+ entry) and an accumulation of intracellular Ca2+.16 Spontaneously occurring accelerated ventricular arrhythmias that occur during digitalis toxicity are likely to be caused by DADs. Triggered ventricular arrhythmias caused by digitalis also can be initiated by pacing at rapid rates. As toxicity progresses, the duration of the trains of repetitive responses induced by pacing increases.
Catecholamines can cause DADs by increasing intracellular Ca2+ overload secondary to different mechanisms. Catecholamines increase the slow, inward ICaL through stimulation of beta-adrenergic receptors and increasing cAMP, which result in an increase in transsarcolemmal Ca2+ influx and intracellular Ca2+ overload (see Fig. 3-6). Catecholamines can also enhance the activity of the Na+-Ca2+ exchanger, thus increasing the likelihood of DAD-mediated triggered activity. Additionally, catecholamines enhance the uptake of Ca2+ by the sarcoplasmic reticulum and lead to increased Ca2+ stored in the sarcoplasmic reticulum and the subsequent release of an increased amount of Ca2+ from the sarcoplasmic reticulum during contraction.16 Sympathetic stimulation can potentially cause triggered atrial and ventricular arrhythmias, possibly some of the ventricular arrhythmias that accompany exercise and those occurring during ischemia and infarction.
Elevations in intracellular Ca2+ in the ischemic myocardium are also associated with DADs and triggered arrhythmias. Accumulation of lysophosphoglycerides in the ischemic myocardium, with consequent Na+ and Ca2+ overload, has been suggested as a mechanism for DADs and triggered activity. Cells from damaged areas or surviving the infarction can display spontaneous release of Ca2+ from sarcoplasmic reticulum, which can generate waves of intracellular Ca2+ elevation and arrhythmias.19,20
Abnormal sarcoplasmic reticulum function caused by genetic defects that impair the ability of the sarcoplasmic reticulum to sequester Ca2+ during diastole can lead to DADs and be the cause of certain inherited ventricular tachyarrhythmias. Mutations in the cardiac RyR2, the sarcoplasmic reticulum Ca2+ release channel in the heart, have been identified in kindreds with the syndrome of catecholaminergic polymorphic VT and ventricular fibrillation (VF) with short QT intervals. It seems likely that perturbed intracellular Ca2+, and perhaps also DADs, underlie arrhythmias in this syndrome (see Fig. 3-6).18,21
Several drugs can inhibit DAD-related triggered activity via different mechanisms, including reduction of the inward Ca2+ current and intracellular Ca2+ overload (Ca2+ channel blockers, beta-adrenergic blockers; see Fig. 3-6), reduction of Ca2+ release from the sarcoplasmic reticulum (caffeine, ryanodine, thapsigargin, cyclopiazonic acid), and reduction of the inward INa (tetrodotoxin, lidocaine, phenytoin).
DAD-related triggered activity is thought to be a mechanism for tachyarrhythmia associated with MI, reperfusion injury, some right ventricular outflow tract tachycardia, and some atrial tachyarrhythmias. DADs are more likely to occur with fast spontaneous or paced rates or with increased premature beats.15,18,20,22
Properties of Delayed Afterdepolarizations
The amplitude of DADs and the coupling interval between the first triggered impulse and the last stimulated impulse that induced them are directly related to the drive CL at which triggered impulses are initiated. A decrease in the basic drive CL (even a single drive cycle; i.e., premature impulse), in addition to increasing the DAD amplitude, results in a decrease in the coupling interval between the last drive cycle and the first DAD-triggered impulse, with respect to the last driven action potential, and an increase of the rate of DADs. Triggered activity tends to be induced by a critical decrease in the drive CL, either spontaneous, such as in sinus tachycardia, or pacing induced. The increased time during which the membrane is in the depolarized state at shorter stimulation CLs or after premature impulses increases Ca2+ in the myoplasm and the sarcoplasmic reticulum, thus increasing the transient inward current responsible for the increased afterdepolarization amplitude, causing the current to reach its maximum amplitude more rapidly, and decreasing the coupling interval of triggered impulses. The repetitive depolarizations can increase intracellular Ca2+ because of repeated activation of ICaL. This characteristic property can help distinguish triggered activity from reentrant activity because the relationship for reentry impulses initiated by rapid stimulation is often the opposite; that is, as the drive CL is reduced, the first reentrant impulse occurs later with respect to the last driven action potential because of rate-dependent conduction slowing in the reentrant pathway.
Early Afterdepolarizations and Triggered Activity
Ionic Basis of Early Afterdepolarizations
The plateau of the action potential is a time of high membrane resistance (i.e., membrane conductance to all ions falls to rather low values), when there is little current flow. Consequently, small changes in repolarizing or depolarizing currents can have profound effects on the action potential duration and profile. Normally, during phases 2 and 3, the net membrane current is outward. Any factor that transiently shifts the net current in the inward direction can potentially overcome and reverse repolarization and lead to EADs. Such a shift can arise from blockage of the outward current, carried by Na+ or Ca2+ at that time, or enhancement of the inward current, mostly carried by K+ at that time.1
EADs have been classified as phase 2 (occurring at the plateau level of membrane potential) and phase 3 (occurring during phase 3 of repolarization; see Fig. 3-4). The ionic mechanisms of phase 2 and phase 3 EADs and the upstrokes of the action potentials they elicit can differ.1 At the depolarized membrane voltages of phase 2, Na+ channels are inactivated; hence, the ICaL and Na+-Ca2+ exchanger current are the major currents potentially responsible for EADs. Voltage steady-state activation and inactivation of the L-type Ca2+ channels are sigmoidal, with an activation range over –40 to +10 mV (with a half-activation potential near –15 mV) and a half-inactivation potential near –35 mV. However, a relief of inactivation for voltages positive to 0 mV leads to a U-shaped voltage curve for steady-state inactivation. Overlap of the steady-state voltage-dependent inactivation and activation relations defines a “window” current near the action potential plateau, within which transitions from closed and open states can occur. As the action potential repolarizes into the window region, ICaL increases and can potentially be sufficient to reverse repolarization, thus generating the EAD upstroke (Fig. 3-7).23
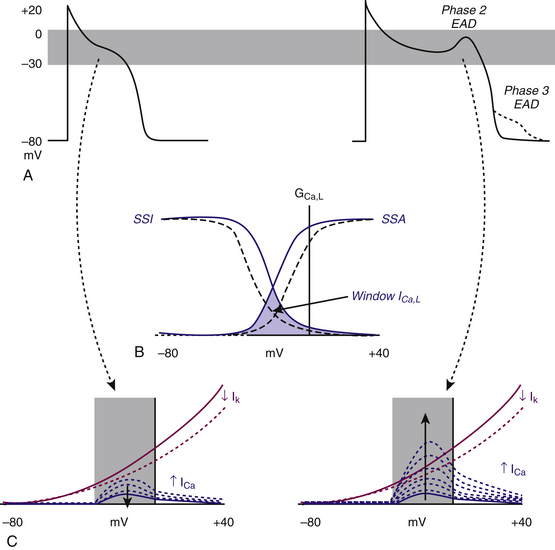
The cardiac Na+-Ca2+ exchanger exchanges three Na+ ions for one Ca2+ ion; the direction is dependent on the Na+ and Ca2+ concentrations on the two sides of the membrane and the transmembrane potential difference. When operating in forward mode, this exchanger generates a net Na+ influx, thereby resisting repolarization. The increase in the window ICaL further increases the Na+-Ca2+ exchanger, thus possibly facilitating EAD formation and increasing the probability of an EAD-triggered action potential.23
EADs occurring late in repolarization develop at membrane potentials more negative than −60 mV in atrial, ventricular, or Purkinje cells that have normal resting potentials. Normally, a net outward membrane current shifts the membrane potential progressively in a negative direction during phase 3 repolarization of the action potential. Despite fewer data, it has been suggested that current through the Na+-Ca2+ exchanger and possibly the INa can participate in the activation of phase 3 EADs. Nevertheless, this concept was questioned by a study suggesting that phase 2 EADs appear to be responsible for inducing phase 3 EADs through electrotonic interactions and that a large voltage gradient related to heterogeneous repolarization is essential for phase 3 EADs.23,24
The upstrokes of the action potentials elicited by phase 2 and phase 3 EADs also differ.1 Phase 2 EAD-triggered action potential upstrokes are exclusively mediated by Ca2+ currents. Even when these triggered action potentials do not propagate, they can substantially exaggerate heterogeneity of the time course of repolarization of the action potential (a key substrate for reentry), because EADs occur more readily in some regions (e.g., Purkinje fibers, mid left ventricular myocardium, right ventricular outflow tract epicardium) than others (e.g., left ventricular epicardium, endocardium). Action potentials triggered by phase 3 EADs arise from more negative membrane voltages. Therefore, the upstrokes can be caused by Na+ and Ca2+ currents and are more likely to propagate.
Under certain conditions, when an EAD is large enough, the decrease in membrane potential leads to an increase in net inward (depolarizing) current, and a second upstroke or an action potential is triggered before complete repolarization of the first. The triggered action potential also can be followed by other action potentials, all occurring at the low level of membrane potential characteristic of the plateau or at the higher level of membrane potential of later phase 3 (Fig. 3-8). The sustained rhythmic activity can continue for a variable number of impulses and terminates when repolarization of the initiating action potential returns membrane potential to a high level. As repolarization occurs, the rate of the triggered rhythm slows because the rate is dependent on the level of membrane potential. Sometimes repolarization to the high level of membrane potential may not occur, and membrane potential can remain at the plateau level or at a level intermediate between the plateau level and the resting potential. The sustained rhythmic activity then can continue at the reduced level of membrane potential and assumes the characteristics of abnormal automaticity. However, in contrast to automatic rhythms, without the initiating action potential, there can be no triggered action potentials.
The ability of the triggered action potentials to propagate is related to the level of membrane potential at which the triggered action potential occurs. The more negative the membrane potential is, the more Na+ channels are available for activation, the greater the influx of Na+ into the cell during phase 0, and the higher the conduction velocity. At more positive membrane potentials of the plateau (phase 2) and early during phase 3, most Na+ channels are still inactivated, and the triggered action potentials most likely have upstrokes caused by the inward ICaL. Therefore, those triggered action potentials have slow upstrokes and are less able to propagate. Increased dispersion of repolarization facilitates the ability of phase 2 EADs to trigger propagating ventricular responses.24
A fundamental condition that underlies the development of EADs is action potential prolongation, which is manifest on the surface ECG by QT prolongation. Hypokalemia, hypomagnesemia, bradycardia, and drugs can predispose to the formation of EADs, invariably in the context of prolonging the action potential duration; drugs are the most common cause. Class IA and III antiarrhythmic agents prolong the action potential duration and the QT interval, effects intended to be therapeutic but frequently causing proarrhythmia. Noncardiac drugs such as some phenothiazines, some nonsedating antihistamines, and some antibiotics can also prolong the action potential duration and predispose to EAD-mediated triggered arrhythmias, particularly when there is associated hypokalemia, bradycardia, or both. Decreased extracellular K+ concentration paradoxically decreases some membrane IK (particularly the IKr) in the ventricular myocyte. This finding explains why hypokalemia causes action potential prolongation and EADs. EAD-mediated triggered activity likely underlies initiation of the characteristic polymorphic VT, torsades de pointes, seen in patients with congenital and acquired forms of long QT syndrome (see Chapter 31). Although the genesis of ventricular arrhythmias in these patients is still unclear, marked transmural dispersion of repolarization can create a vulnerable window for development of reentry. EADs arising from these regions can underlie the premature complexes that initiate or perpetuate the tachycardia.1 Structural heart disease such as cardiac hypertrophy and failure can also delay ventricular repolarization—so-called electrical remodeling—and predispose to arrhythmias related to abnormalities of repolarization. The abnormalities of repolarization in hypertrophy and failure are often magnified by concomitant drug therapy or electrolyte disturbances.
Properties of Early Afterdepolarizations
EAD-triggered arrhythmias exhibit rate dependence. In general, the amplitude of an EAD is augmented at slow rates when action potentials are longer in duration.6 Pacing-induced increases in rate shorten the action potential duration and reduce EAD amplitude. Action potential shortening and suppression of EADs with increased stimulation rate are likely the result of augmentation of delayed rectifier IK and perhaps hastening of Ca2+-induced inactivation of ICaL. Once EADs have achieved a steady-state magnitude at a constant drive CL, any event that shortens the drive CL tends to reduce their amplitude. Hence, the initiation of a single premature depolarization, which is associated with an acceleration of repolarization, will reduce the magnitude of the EADs that accompany the premature action potential; as a result, triggered activity is not expected to follow premature stimulation. The exception is when a long compensatory pause follows a premature ventricular complex. This situation can predispose to the development of an EAD and can be the mechanism of torsades de pointes in some patients with the long QT syndrome. Thus, EADs are more likely to trigger rhythmic activity when the spontaneous heart rate is slow because bradycardia is associated with prolongation of the QT interval and action potential duration (e.g., bradycardia- or pause-induced torsades de pointes). Similarly, catecholamines increase heart rate and decrease action potential duration and EAD amplitude, despite the effect of beta-adrenergic stimulation to increase ICaL.
Reentry
Basic Principles of Reentry
During each normal cardiac cycle, at the completion of normal cardiac excitation, the electrical impulse originating from the sinus node becomes extinct, and the subsequent excitation cycles originate from new pacemaker impulses.6 Physiological excitation waves vanish spontaneously after the entire heart has been activated because of the long duration of refractoriness in the cardiac tissue compared with the duration of the excitation period; therefore, after its first pass, the impulse, having no place to go, expires. Reentry occurs when a propagating impulse fails to die out after normal activation of the heart and persists to reexcite the heart after expiration of the refractory period. In pathological settings, excitation waves can be blocked in circumscribed areas, rotate around these zones of block, and reenter the site of original excitation in repetitive cycles. The wavefront does not extinguish but rather propagates continuously and thus continues to excite the heart because it always encounters excitable tissue.
Reentrant tachycardia, also called reentrant excitation, reciprocating tachycardia, circus movement, or reciprocal or echo beats, is a continuous repetitive propagation of the activation wave in a circular path, returning to its site of origin to reactivate that site.1,25,26 Traditionally, reentry has been divided into two types: (1) anatomical reentry, when there is a distinct relationship of the reentry pathway with the underlying tissue structure; and (2) functional reentry, when reentrant circuits occur at random locations without clearly defined anatomical boundaries (Fig. 3-9). Although this distinction has a historical background and is useful for didactic purposes, both the anatomical and functional forms can coexist in a given pathological setting and share many common basic biophysical mechanisms.

The original 3 criteria for reentry proposed by Mines still hold true: (1) unidirectional block is necessary for initiation; (2) the wave of excitation should travel in a single direction around the pathway, returning to its point of origin and then restarting along the same path; and (3) the tachycardia should terminate when one limb of the pathway is cut or temporarily blocked. The 12 conditions that were proposed to prove or identify the existence of reentrant tachycardia in the EP laboratory are listed in Table 3-1.25,26
Requisites of Reentry
Substrate
The reentrant circuit can be an anatomical structure, such as a loop of fiber bundles in the Purkinje system or accessory pathways, or a functionally defined pathway, with its existence, size, and shape determined by the EP properties of cardiac tissues in which the reentrant wavefront circulates. The circuit can also be an anatomical-functional combination. The cardiac tissue that constitutes the substrate for reentrant excitation can be located almost anywhere in the heart.1 The reentrant circuit can be a variety of sizes and shapes and can include different types of myocardial cells (e.g., atrial, ventricular, nodal, Purkinje; Fig. 3-10).
Central Area of Block
As mentioned earlier, the area of block can be anatomical, functional, or a combination of the two.6,27 Anatomical block is the result of a nonconductive medium in the center of the circuit, such as the tricuspid annulus in typical atrial flutter (AFL). Functional block at the center of a circuit occurs when there is block of impulses in otherwise excitable cardiac muscle. The central area of functional block develops during the initiation of the reentrant circuit by the formation of a line of block that most likely is caused by refractoriness. When the reentrant circuit forms, the line of block then is sustained by centripetal activation from the circulating wavefront that, by repeatedly bombarding the central area of block, maintains the state of refractoriness of this region. A combination of an anatomical and a functional central area of block in the reentrant circuit has been described in some models of AFL such as the orifice of one or both venae cavae and an area of functional block continuous with or adjacent to either or both caval orifice(s). Additionally, it has now been shown that a functional extension of an anatomical line of block can occur such that it plays a role in creating the necessary or critical substrate for reentry. Thus, a surgical incision in the right atrium made to repair a congenital heart lesion can, under certain circumstances, develop a functional extension to one or both of the venae cavae, such that the substrate to create and sustain AFL develops.
Unidirectional Conduction Block
Transient or permanent unidirectional block is usually a result of heterogeneity of EP properties of the myocardium and is essential for the initiation of reentry. The excitation wavefront propagating in the substrate must encounter unidirectional block; otherwise, the excitation wavefronts traveling down both limbs of the reentrant circuit will collide and extinguish each other.27
Area of Slow Conduction
In a successful reentrant circuit, the wavefront of excitation must encounter excitable cells or the tachycardia will terminate. Therefore, a condition necessary for reentry is the maintenance of excitable tissue ahead of the propagating wavefront. In other words, the tissue initially activated by the excitation wavefront should have sufficient time to recover its excitability by the time the reentrant wavefront returns. Thus, conduction of the circulating wavefront must be sufficiently delayed in an alternate pathway to allow for expiration of the refractory period in the tissue proximal to the site of unidirectional block, and there must always be a gap of excitable tissue (fully or partially excitable) ahead of the circulating wavefront (i.e., the length of the reentrant pathway must equal or exceed the reentrant wavelength; see later). This is facilitated by a sufficiently long reentrant pathway (which is especially important when conduction is normal along the reentrant path), sufficiently slow conduction in all or part of the alternative pathway (because sufficiently long pathways are usually not present in the heart), sufficient shortening of the refractory period, or a combination of these factors.1
Initiating Trigger
Another prerequisite for reentrant excitation to occur is often, but not always, the presence of an initiating trigger, which invokes the necessary EP milieu for initiation of reentry.1 Susceptible patients with appropriate underlying substrates usually do not suffer from incessant tachycardia because the different EP mechanisms required for the initiation and maintenance of a reentrant tachycardia are infrequently present at exactly the same time. However, changes in heart rate or autonomic tone, ischemia, electrolyte or pH abnormalities, or the occurrence of a premature depolarization can be sufficient to initiate reentrant tachycardia.
Types of Reentrant Circuits
Anatomical Reentry
A reentrant tachycardia is initiated when an excitation wavefront splits into two limbs after going around the anatomical obstacle and travels down one pathway and not the other, thus creating a circus movement. Tachycardia rates are determined by the wavelength and by the length of the reentrant pathway (the path length). The initiation and maintenance of anatomical reentry depend on conduction velocity and refractory period. Thus, as long as the extension of the refractory zone behind the excitation wave, the so-called wavelength of excitation, is smaller than the entire length of the anatomically defined reentrant pathway, a zone of excitable tissue, the so-called excitable gap, exists between the tail of the preceding wave and the head of the following wave.27 In essence, circus movements containing an excitable gap are stable with respect to their frequency of rotation and can persist at a constant rate for hours. In the setting where the wavelength of excitation exceeds the path length, the excitation wavefront becomes extinct when it encounters the not yet recovered inexcitable tissue. A special case is present in the intermediate situation, when the head of the following wavefront meets the partially refractory tail of the preceding wavefront (i.e., the wavelength approximates the path length). This situation is characterized by unstable reentrant CLs and complex dynamics of the reentrant wavefront. There is often a long excitable gap associated with anatomical reentry.
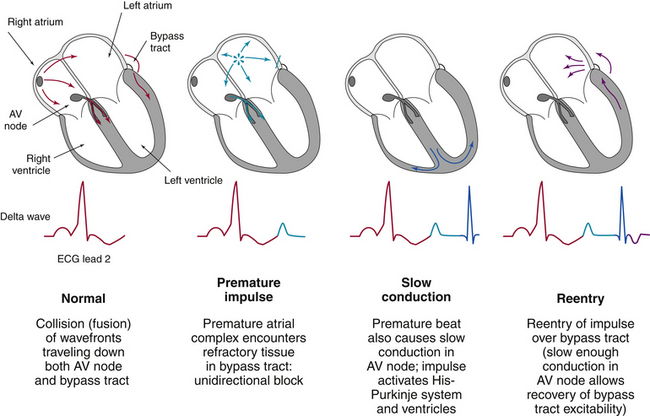
Anatomical circuits therefore are associated with ordered reentry. Examples of this type of reentry are AV reentrant tachycardia associated with an AV bypass tract, AVN reentrant tachycardia, AFL, VT originating within the HPS (bundle branch reentrant VT), and post-MIVT.
Functional Reentry
In functionally determined circuits, the reentrant pathway depends on the intrinsic heterogeneity of the EP properties of the myocardium, not by a predetermined anatomical circuit (i.e., without involvement of an anatomical obstacle or anatomically defined conducting pathway). Such heterogeneity involves dispersion of excitability or refractoriness and conduction velocity, as well as anisotropic conduction properties of the myocardium.28
Functional circuits typically tend to be small and unstable; the reentrant excitation wavefront can fragment, generating other areas of reentry. The location and size of these tachycardias can vary. The circumference of the leading circle around a functional obstacle can be as small as 6 to 8 mm and represents a pathway in which the efficacy of stimulation of the circulating wavefront is just sufficient to excite the tissue ahead, which is still in its relative refractory phase. Therefore, conduction through the functional reentrant circuit is slowed because impulses are propagating in partially refractory tissue. Consequently, this form of functional reentry has a partially excitable gap. The reentry CLs are therefore significantly dependent on the refractory period of the involved tissue.28
Leading Circle Concept
To explain the properties of a single functional reentrant circuit, Allessie and colleagues formulated the leading circle concept (see Fig. 3-9).28 It was postulated that during wavefront rotation in tissue without anatomical inexcitable obstacles, the wavefront impinges on its refractory tail and travels through partially refractory tissue. The interaction between the wavefront and the refractory tail determines the properties of functional reentry. In this model, functional reentry involves the propagation of an impulse around a functionally determined region of inexcitable tissue or a refractory core and among neighboring fibers with different EP properties. The tissue within this core is maintained in a state of refractoriness by constant centripetal bombardment from the circulating wavefront. The premature impulse that initiates reentry blocks in fibers with long refractory periods and conducts in fibers with shorter refractory periods and eventually returns to the initial region of block after excitability has recovered there. The impulse then continues to circulate around a central area that is kept refractory because it is bombarded constantly by wavelets propagating toward it from the circulating wavefront. This central area provides a functional obstacle that prevents excitation from propagating across the fulcrum of the circuit.
The leading circle was defined as “the smallest possible pathway in which the impulse can continue to circulate” and “in which the stimulating efficacy of the wavefront is just enough to excite the tissue ahead which is still in its relative refractory phase.”28 Thus, the “head of the circulating wavefront is continuously biting its tail of refractoriness” and the length of the reentrant pathway equals the wavelength of the impulse; as a result, there is usually no fully excitable gap.28 Because the wavefront propagates through partially refractory tissue, the conduction velocity is reduced.
Anisotropic Reentry
Isotropic conduction is uniform in all directions; anisotropic conduction is not. Anisotropy is a normal feature of heart muscle and is related to the differences in longitudinal and transverse conduction velocities, which are attributable to the lower resistivity of myocardium in the longitudinal (parallel to the long axis of the myocardial fiber bundles) versus the transverse direction (Fig. 3-11). Anisotropy in myocardium composed of tissue with structural features different from those of adjacent tissue results in heterogeneity in conduction velocities and repolarization properties (see later discussion), which can lead to blocked impulses and slowed conduction, thereby setting the stage for reentry (referred to as anisotropic reentry).
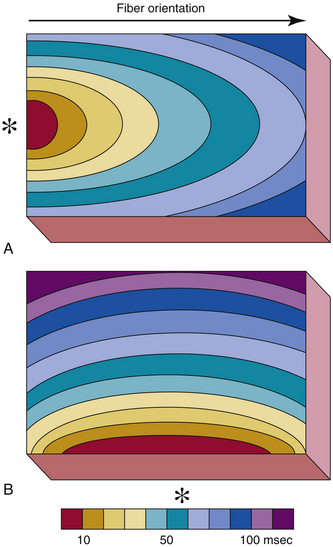
Unlike the functional characteristic that leads to the leading circle type of reentry (differences in refractoriness in adjacent areas caused by local differences in membrane properties), the functional characteristic that is important in functional reentry caused by anisotropy is the difference in effective axial resistance to impulse propagation dependent on fiber direction. In its pure form, the unidirectional conduction block and slow conduction in the reentrant circuit result from anisotropic, discontinuous propagation, and there is no need for variations in membrane properties, such as regional differences in refractoriness or depression of the resting and action potentials.29
Anisotropic reentrant circuits usually remain in a fixed position and cause ordered reentry. The degree of anisotropy (i.e., the ratio of longitudinal to transverse conduction velocity) varies in different regions of the heart, and the circuit can reside only in a region in which the conduction transverse to the longitudinal axis is sufficiently slow to allow reentry. Stability of anisotropic reentrant circuits is also assisted by the presence of an excitable gap, which does not occur in the leading circle functional circuit. The excitable gap is caused by the sudden slowing of conduction velocity and a decrease in the wavelength of excitation as the reentrant impulse turns the corner from the fast longitudinal direction to the slow transverse direction and from the slow transverse direction to the fast longitudinal direction. Anisotropic reentry is typically initiated by a premature stimulus that blocks in the direction of propagation parallel to the long axis of the cells and then propagates slowly in the transverse direction of fiber orientation because of high axial resistance (see later).29
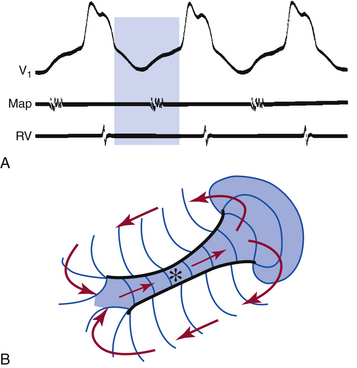
Figure-of-8 Reentry
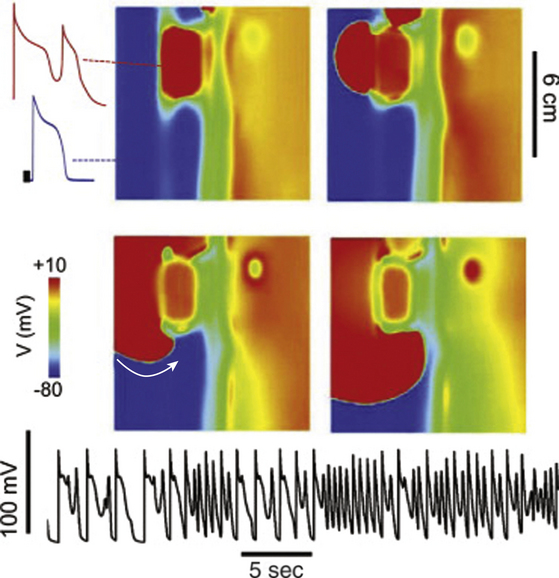
The model of figure-of-8 or double-loop reentry involves two concomitant excitation wavefronts circulating in opposite directions, clockwise and counterclockwise, around a long line of functional conduction block rejoining on the distal side of the block. The wavefront then breaks through the arc of block to reexcite the tissue proximal to the block. The single arc of block is thus divided into two, and reentrant activation continues as two circulating wavefronts that travel clockwise and counterclockwise around the two arcs in a pretzel-like configuration.30 This form of reentry has been shown in atrial and ventricular myocardia  (Fig. 3-12, Video 1).
(Fig. 3-12, Video 1).
Reflection
Reflection is a special subclass of reentry in which the excitation wavefront does not require a circuit but appears to travel back and forth in a linear segment of tissue (e.g., trabecula or Purkinje fiber) containing an area of conduction block (see Fig. 3-9). In such a situation, an action potential propagates toward, but not through, the inexcitable zone. Subsequently, an electrotonic current conducts passively (i.e., without eliciting an action potential) through the inexcitable zone toward the distal portion of the pathway. If the inexcitable zone is sufficiently small and the magnitude of the electrotonic current is sufficiently large, the segment of tissue distal to the blocked area will be excited (i.e., an action potential is elicited) but with a significant delay. The action potential generated in the distal portion of the pathway will then cause electrotonic current to flow back through the inexcitable zone toward the proximal region. Provided the proximal portion of the conduction pathway recovers quickly enough, this current may be sufficient to elicit a second action potential on the proximal side of the inexcitable zone, which propagates in the opposite direction to the first action potential, thus giving the appearance that the inexcitable zone has reflected the initial action potential. Because reflection can occur within areas of tissue as small as 1 to 2 mm2, it is likely to appear of focal origin. Its identification as a mechanism of arrhythmia may be difficult even with very high spatial resolution mapping of the electrical activity of discrete sites.
Phase 2 Reentry
As discussed in Chapter 1, substantial differences in the expression levels of ion channels underlie the substantial heterogeneity in action potential duration and configuration between cardiomyocytes across the ventricular wall. Heterogeneity in the distribution of the transient outward IK (Ito) channels across the myocardial wall, being more prominent in ventricular epicardium than endocardium, results in the shorter duration and the prominent phase 1 notch and the “spike and dome” morphology of the epicardial action potential as compared with the endocardium. The resultant transmural voltage gradient during the early phases (phases 1 and 2) of the action potential is thought to be responsible for the inscription of the J wave on the surface ECG (see Fig. 31-6). A significant outward shift of currents, secondary to either a decrease in the inward currents (INa and ICaL) or an increase in the outward IK (Ito, IKr, IKs, IKACH, IKATP), or both, can cause partial or complete loss of the dome of the action potential in the epicardium that leads to exaggeration of transmural voltage gradient and dispersion of repolarization between the epicardium and endocardium. The type of the ion current affected and its regional distribution in the ventricles determine the particular phenotype (including the Brugada syndrome, early repolarization syndrome, hypothermia-induced ST segment elevation, and infarction-induced ST segment elevation).31
In this setting, the phase 2 dome (plateau) of the action potential can potentially propagate from regions where it is preserved (midmyocardium and endocardium) to regions where it is abolished (epicardium), thus causing local reexcitation (phase 2 reentry) and the generation of a closely coupled extrasystole that, in turn, can initiate VT or VF (see Fig. 31-7).32
Spiral Wave (Rotor) Activity
The leading circle concept was based on properties of impulse propagation in a one-dimensional tissue that forms a closed pathway (e.g., a ring). The concept was a major breakthrough in the understanding of the mechanisms of reentrant excitation. However, it became evident that these considerations alone do not fully describe wave rotation in two- and three-dimensional cardiac tissue.33
Spiral waves typically describe reentry in two dimensions. The term rotor initially described the rotating source, and the spiral wave defined the shape (i.e., curvature) of the wave emerging from the rotating source. In many publications, this difference has been blurred, and terms used in the literature include rotors, vortices, and reverberators.27 The center of the spiral wave is called the core, and the distribution of the core in three dimensions is referred to as the filament. The three-dimensional form of the spiral wave is called a scroll wave.30,33
Under appropriate circumstances, a pulse in two-dimensional, homogeneous, excitable media can be made to circulate as a rotor. When heterogeneities in recovery exist, the application of a second stimulus over a large geometric area to initiate a second excitation wave only excites a region in which there has been sufficient time for recovery from the previous excitation, not regions that have not yet recovered. An excitation wave is elicited at the excitable site in the form of a rotor because the wave cannot move in the direction of the wake of the previous wave but only in the opposite direction, thus moving into adjacent regions as they in turn recover. The inner tip of the wavefront circulates around an organizing center or core, which includes cells with transmembrane potentials that have a reduced amplitude, duration, and rate of depolarization (i.e., slow upstroke velocity of phase 0); these cells are potentially excitable, but they remain unexcited, instead of a region of conduction block. In the center of the rotating wave, the tip of the wave moves along a complex trajectory and radiates waves into the surrounding medium. In addition, the spiral waves can give rise to daughter spirals that can result in disorganized electrical activity.34
The curvature of the spiral wave is the key to the formation of the core and the functional region of block.27,30 Propagation of two- and three-dimensional waves also depends on wavefront curvature, a property that is not present in one-dimensional preparations.27 Because the maximal velocity of a convex rotating wavefront can never exceed the velocity of a flat front and the period of rotation remains constant in a stable rotating wave, the velocity has to decrease from the periphery (where the highest value corresponds to linear velocity) to the center of a rotating wave. As a consequence, any freely rotating wave in an excitation-diffusion system has to assume a spiral shape. A prominent curvature of the spiral wave is generally encountered following a wave break, a situation in which a planar wave encounters an obstacle and breaks up into two or more daughter waves. Because it has the greatest curvature, the broken end of the wave moves most slowly. As curvature decreases along the more distal parts of the spiral, propagation speed increases.30 The rotor, by definition, has a marked curvature, and this curvature slows down its propagation. Slow conduction results from an increased electrical load; that is, not only must a curved wavefront depolarize cells ahead of it in the direction of propagation, but also current flows to cells on its sides.
The location of the rotor can occur wherever the second stimulated excitation encounters the wake of the first excitation with the appropriate characteristics. Spirals can be stationary, continuously drift or migrate away from their origin, or anchored, initially drifting and then becoming stationary by anchoring to a small obstacle.34,35
In the heart, spiral waves have been implicated in the generation of cardiac arrhythmias for a long time. Both two-dimensional spiral waves and three-dimensional scroll waves have been implicated in the mechanisms of reentry in atrial and ventricular tachycardia and fibrillation.27 Monomorphic VT results when the spiral wave is anchored and cannot drift within the ventricular myocardium away from its origin. In contrast, a polymorphic VT, such as the torsades de pointes encountered with long QT syndromes, is thought to be caused by a meandering or drifting spiral wave. VF seems to be the most complex representation of rotating spiral waves in the heart. VF develops when the single spiral wave responsible for VT breaks up, leading to the development of multiple spirals that are continuously extinguished and recreated.30,34,35
Excitable Gaps in Reentrant Circuits
Wavelength Concept
The wavelength is defined as the product of the conduction velocity of the circulating excitation wavefront and the effective refractory period of the tissue in which the excitation wavefront is propagating.6 The wavelength quantifies how far the impulse travels relative to the duration of the refractory period. The wavelength of the reentrant excitation wavefront must be shorter than the length of the pathway of the potential reentrant circuit for reentrant excitation to occur; that is, the impulse must travel a distance during the refractory period that is less than the complete reentrant path length to give myocardium ahead of it sufficient time to recover excitability. Slowing of impulse conduction or shortening of refractoriness shortens the wavelength and increases the excitable gap.
Excitable Gaps
The excitable gap in a reentrant circuit is the region of excitable myocardium that exists between the head of the reentrant wavefront and the tail of the preceding wavefront and, at any given time, is no longer refractory (i.e., is capable of being excited) if the excitation wavelength is shorter than the length of the reentrant circuit (Fig. 3-13).
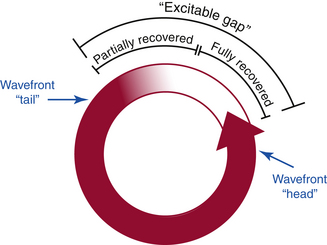
The occurrence of an excitable gap is dependent on the recovery of excitability of the myocardium from its previous excitation by the reentrant wavefront. A fully excitable gap is defined as the segment of the reentrant circuit in which the tail of the preceding wavefront does not affect the head and velocity of the following wavefront (absence of head-tail interaction). A partially excitable gap is defined as the zone where the rotating wave can be captured by local stimulation in the presence of head-tail interaction. Whereas the excitable gap denotes a length of a segment within the reentrant circuit, the fully or partially excitable period denotes the time period during which a segment within the reentrant circuit is fully or partially excitable, respectively (see Fig. 3-13).
The characteristics of the excitable gap can be different in different types of reentrant circuits. Many anatomically determined reentrant circuits have large excitable gaps with a fully excitable component, although, even in anatomically determined circuits, the gap can sometimes be only partially excitable. On the other hand, functional reentrant circuits caused by the leading circle mechanism have very small gaps that are only partially excitable, although parts of some functionally determined reentrant circuits (anisotropic reentrant circuits) can have fully excitable gaps. An excitable gap has been shown to occur during AF, VF, and AFL; these are examples of arrhythmias caused by functional reentrant mechanisms, possibly including spiral waves. The relationship between the excitable gap and the excitable period can be complex if the velocity of propagation changes within the reentry circuit.27
The existence and the extent of an excitable gap in a reentrant circuit have important implications.27 The presence of an excitable gap enables modulation of the frequency of a reentrant tachycardia by a locally applied stimulus or by field stimulation; the longer the excitable gap is, the more likely it will be for an extrastimulus to be able to enter the reentrant circuit and initiate or terminate a reentrant arrhythmia. In addition, resetting and entrainment are more likely to occur when the excitable gap is longer. The excitable gap can be exploited to terminate a reentrant tachycardia. The presence of a significant temporal and spatial excitable gap in some reentrant circuits enables reentry to be terminated by a single premature stimulus or by overdrive stimulation. Termination of arrhythmias by stimulation would be expected to be much more difficult when the reentrant circuit has only a small partially excitable gap. Additionally, the excitable gap can influence the effects of drugs on the reentrant circuit, so that reentry with a partially excitable gap and mainly functional components may respond more readily to drugs that prolong repolarization, without slowing conduction, whereas fixed anatomical reentry with a large excitable gap responds to drugs that decrease conduction velocity, preferentially at pivot points.
The properties of the excitable gap influence the characteristics of arrhythmias caused by reentry. Arrhythmias caused by leading circle reentry, in which the wavefront propagates in the just-recovered myocardium of the refractory tail and in which there is only a small partially excitable gap, are inherently unstable and often terminate after a short period or go on to fibrillation. On the other hand, the reentrant wavefront in anatomical and nonuniform anisotropic reentrant circuits, in general, is not propagating in myocardium that has just recovered excitability, and the excitable gap can be large. This property can contribute to the stability of these reentrant circuits.27
The shape of the anatomical obstacle determines the path of a reentrant wave in fixed anatomical reentry. Therefore, instability of anatomical reentry is confined to variations of the rotating interval and wavelength of excitation. This instability is characterized by the wavefront’s invading the repolarizing phase of the preceding wave, with resulting oscillations of the rotation period.27
Resetting Reentrant Tachycardias
Definition
Resetting is the advancement (acceleration) of a tachycardia impulse by timed premature electrical stimuli. The extrastimulus is followed by a pause that is less than fully compensatory before resumption of the original rhythm. The tachycardia complexes that return first should have the same morphology and CL as the tachycardia before the extrastimulus, regardless of whether single or multiple extrastimuli are used.36–38
The introduction of a single extrastimulus (S2) during a tachycardia yields a return cycle (S2X3) if the tachycardia is not terminated (Fig. 3-14). If S2 does not affect the arrhythmogenic focus, the coupling interval (X1S2) plus the return cycle (S2X3) will be equal to twice the tachycardia cycle (2 × [X1X1]); that is, a fully compensatory pause will occur. Resetting of the tachycardia occurs when a less than fully compensatory pause occurs. In this situation, X1S2 + S2X3 will be less than 2 × (X1X1), as measured from the surface ECG. Tachycardia CL stability should be taken into account when the return cycle is measured. To account for any tachycardia CL instability, at least a 20-millisecond shortening of the return cycle is required to demonstrate resetting.39
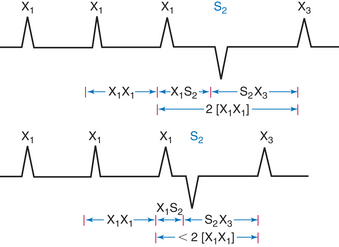
When more than a single extrastimulus is used, the relative prematurity should be corrected by subtracting the coupling interval or intervals from the spontaneous tachycardia cycles when the extrastimuli are delivered.40
Reentrant Tachycardia Resetting
To reset reentrant tachycardia, the stimulated wavefront must reach the reentrant circuit, encounter excitable tissue within the circuit (i.e., enter the excitable gap of the reentrant circuit), collide in the antidromic (retrograde) direction with the previous tachycardia impulse, and continue in the orthodromic (anterograde) direction to exit at an earlier than expected time and perpetuate the tachycardia (Fig. 3-15).36–38 If the extrastimulus encounters fully excitable tissue, which commonly occurs in reentrant tachycardias with large excitable gaps, the tachycardia will be advanced by the extent that the stimulated wavefront arrives at the entrance site prematurely. If the tissue is partially excitable, which can occur in reentrant tachycardias with small or partially excitable gaps or even in circuits with large excitable gaps when the extrastimulus is very premature, the stimulated wavefront will encounter some conduction delay in the orthodromic direction within the circuit (see Fig. 3-15). Therefore, the degree of advancement of the next tachycardia beat depends on both the degree of prematurity of the extrastimulus and the degree of slowing of its conduction within the circuit. The reset tachycardia beat consequently can be early, on time, or delayed.
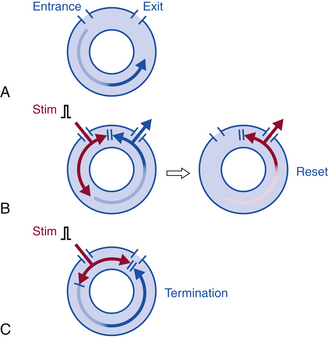
Termination of the tachycardia occurs when the extrastimulus collides with the preceding tachycardia impulse antidromically and blocks in the reentrant circuit orthodromically (see Fig. 3-15). This occurs when the premature impulse enters the reentrant circuit early in the relative refractory period; it fails to propagate in the anterograde direction because it encounters absolutely refractory tissue. In the retrograde direction, it encounters increasingly recovered tissue and can propagate until it encroaches on the circulating wavefront and terminates the arrhythmia.39,40
Resetting does not require that the pacing site be located in the reentrant circuit. The closer the pacing site is to the circuit, however, the less premature a single stimulus can be and reach the circuit without being extinguished by collision with a wave emerging from the circuit. The longest coupling interval for an extrastimulus to be able to reset a reentrant tachycardia depends on the tachycardia CL, the duration of the excitable gap of the tachycardia, refractoriness at the pacing site, and the conduction time from the pacing site to the reentrant circuit.39
Resetting Zone and Excitable Gap
For an extrastimulus to be able to reset the reentrant circuit, it has to penetrate the circuit during its excitable gap. The difference between the longest and shortest coupling intervals resulting in resetting is defined as the resetting interval or resetting zone.37,39 Thus, the coupling intervals over which resetting occurs, the resetting zone, can be considered a measure of the duration of the temporal excitable gap existing in the reentrant circuit. Therefore, the entire extent of the fully excitable gap would be the zone of coupling intervals from the onset of tachycardia resetting until tachycardia termination. The excitable gap, however, can be underestimated by using only a single extrastimulus or by using single or double extrastimuli in the absence of tachycardia termination by the extrastimuli.
Return Cycle
The return cycle is the time interval from the resetting stimulus to the next excitation of the pacing site by the new orthodromic wavefront. This corresponds to the time required for the stimulated impulse to reach the reentrant circuit, conduct through the circuit, exit the circuit, and travel back to the pacing site.37,39 The noncompensatory pause following the extrastimulus and the return cycle are typically measured at the pacing site; however, they may also be measured to the onset of the tachycardia complex on the surface ECG.
Orthodromic and Antidromic Resetting
Antidromic resetting occurs when intracardiac sites are directly captured by the premature stimulus without traversing the reentrant circuit and the zone of slow conduction.37 Therefore, antidromic resetting of intracardiac sites occurs with a conduction interval from the pacing stimulus to the captured electrogram that is less than the tachycardia CL and with differing morphology of the captured as compared with the spontaneous electrogram. Although demonstration of an antidromic resetting response can indicate a tachycardia mechanism other than reentry, an antidromic resetting pattern can also be observed during reentry with an excitable gap if the pacing site is located distal to a region of slow conduction in the reentry circuit. Conversely, if the recording sites are located in regions activated proximal to a region of slow conduction, an antidromic resetting response will be observed.
Orthodromic resetting occurs when the premature stimulus traverses the reentrant circuit, including the zone of slow conduction, in the same direction as the spontaneous tachycardia impulse and with an identical exit site.37 Intracardiac areas that are orthodromically reset are advanced by the premature extrastimulus but retain the same morphology because they are activated from the impulse emerging from the same reentrant circuit exit site. The conduction interval from the pacing stimulus to the orthodromically captured electrogram exceeds the tachycardia CL by the time required for the extrastimulus to travel from the pacing site to the reentrant circuit. Thus, an orthodromic resetting response implies that the pacing site is located proximal to a region of slow conduction in the reentry circuit and that the recording site is located distal to this region. The ability to demonstrate orthodromic resetting is critically dependent on the location of pacing and recording electrodes relative to the region of slow conduction in the circuit. Therefore, failure to demonstrate an orthodromic resetting response does not exclude reentry with an excitable gap as a possible tachycardia mechanism.
Resetting Response Curves
Response patterns during resetting are characterized by plotting the coupling interval of the extrastimulus producing resetting versus the return cycle measured at the pacing site. Alternatively, the return cycle is measured to the onset of the first tachycardia complex following stimulation on the surface ECG; then, qualitatively similar but quantitatively different response curves are obtained.39 Demonstration of a noncompensatory pause following the extrastimulus is required, and the interval encompassing the extrastimulus should be 20 or more milliseconds earlier than the expected compensatory pause following a single extrastimulus and 20 or more milliseconds less than three tachycardia CLs when double extrastimuli are used.39 As always, it is important to establish the stability of tachycardia CL before assessing any perturbation in tachycardia presumed to be caused by resetting.
Four resetting response patterns are possible (Fig. 3-16)39:
1. During the flat response pattern, the return cycle is constant (less than a 10-millisecond difference) over a 30-millisecond range of coupling intervals.
2. With the increasing response pattern, the return cycle increases as the coupling interval increases.
3. With a decreasing response pattern, the return cycle decreases as the coupling interval increases.
4. A mixed response pattern meets the criteria for a flat response at long coupling intervals and for an increasing response at shorter coupling intervals.
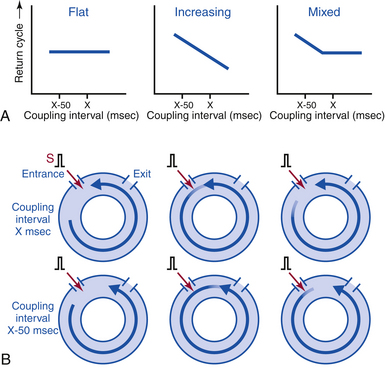
The types of resetting curves can vary depending on the site of stimulation. Extrastimuli from different pacing sites likely engage different sites in the reentrant circuit that are in different states of excitability or refractoriness and therefore result in different conduction velocities and resetting patterns.39
Flat Response Curves in Reentrant Rhythms
A flat resetting curve implies the presence of a fully excitable gap within the reentrant circuit over a range of coupling intervals. The total duration of the excitable gap should exceed the range of coupling intervals that produce resetting with a flat response. Large excitable gaps are more likely to result in flat response curves, because the increasingly premature extrastimuli are less likely to encroach on the trailing edge of refractoriness and encounter decremental conduction (see Fig. 3-16). The flat return cycle also suggests the presence of fixed sites of entrance and exit from the circuit and fixed conduction time from the stimulation site through the reentrant circuit over a wide range of coupling intervals.
If a single extrastimulus produced resetting with a flat response, the response to double extrastimuli would also be flat. However, because the use of double extrastimuli allows engagement of the reentrant circuit at relatively long coupling intervals with greater prematurity, resetting will begin at longer coupling intervals and will continue over a greater range of coupling intervals than observed with a single extrastimulus. Therefore, double extrastimuli can produce a flat and then increasing response curve.
Increasing Response Curves in Reentrant Rhythms
Increasing resetting curves result from progressively longer return cycles in response to increasingly premature extrastimuli and indicate a zone of decremental slow conduction, usually located within the reentrant circuit. The most probable mechanism underlying the decremental conduction is encroachment of the advancing wavefront from the premature extrastimuli on an increasingly more refractory tissue within the reentrant circuit, most likely within the zone of slow conduction (see Fig. 3-16).39 This response pattern is possible only for reentrant arrhythmias and is not observed in triggered or automatic rhythms.
Mixed Response Curves in Reentrant Rhythms
In a mixed response curve, the initial coupling intervals demonstrate a flat portion of the curve of variable duration (but less than 30 milliseconds), followed by a zone during which the return cycle increases (see Fig. 3-16). Occasionally, a flat curve is seen with a single extrastimulus; it is only by using double extrastimuli that an increasing response can be observed.
Resetting with Fusion
Fusion of the stimulated impulse can be observed on surface ECG or intracardiac recordings if the stimulated impulse is intermediate in morphology between a fully paced complex and the tachycardia complex. The ability to recognize ECG fusion requires a significant mass of myocardium to be depolarized by both the extrastimulus and the tachycardia.37,39,41 With early extrastimuli, the paced antidromic wavefront captures all or most of the myocardium prior to the orthodromic wavefront of the tachycardia impulse exiting from the reentrant circuit. Thus, no ECG fusion is present, although resetting can occur. With later coupled extrastimuli, the orthodromic wavefront exits from the reentrant circuit, thus capturing a certain portion of myocardium before colliding with the paced antidromic wavefront. In this situation, ECG fusion occurs. Resetting with ECG fusion requires wide separation of the entrance and exit of the reentrant circuit, with the stimulus wavefront preferentially engaging the entrance.
If presystolic activity in the reentrant circuit is recorded before delivery of the extrastimulus that resets the tachycardia, one must consider this to represent local fusion (Fig. 3-17).39 Thus, an extrastimulus that is delivered after the onset of the tachycardia complex and enters and resets the circuit always demonstrates local fusion. Resetting with local fusion and a totally paced complex morphology provides evidence that the reentrant circuit is electrocardiographically small.
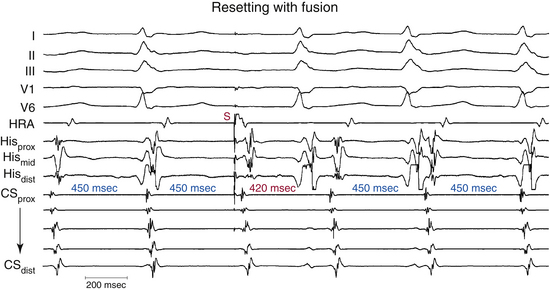
Resetting of Tachycardias with Diverse Mechanisms
Resetting can be demonstrated for tachycardias based on different mechanisms, including reentry, normal or abnormal automaticity, and triggered activity. Although the ability to reset an arrhythmia is not helpful in distinguishing the underlying mechanism, certain features of the resetting response can be useful for the differential diagnosis.39
Resetting with Fusion
The ability to reset tachycardia after it has begun activating the myocardium (i.e., resetting with fusion) is diagnostic of reentry and excludes automatic and triggered mechanisms.37,39,41
In automaticity or triggered activity, resetting of the arrhythmia by an extrastimulus requires depolarization of the site of origin by the paced wavefront. Because the entrance and exit sites of focal rhythms (automatic or triggered) are not separate, a tachycardia wavefront cannot exit the focus once the exit or entrance site has already been depolarized and rendered refractory by the paced wavefront.
Entrainment of Reentrant Tachycardias
Basic Principles of Entrainment
Entrainment of reentrant tachycardias by external stimuli was originally defined in the clinical setting as “an increase in the rate of a tachycardia to a faster pacing rate, with resumption of the intrinsic rate of the tachycardia upon either abrupt cessation of pacing or slowing of pacing beyond the intrinsic rate of the tachycardia” and taken to indicate an underlying reentrant mechanism. The ability to entrain a tachycardia also establishes that the reentrant circuit contains an excitable gap.27,39,42,43
Orthodromic resetting and transient entrainment are manifestations of the same phenomenon (i.e., premature penetration of a tachycardia circuit by a paced wavefront), and the ability to demonstrate resetting is a strong indication that entrainment can occur from that specific pacing site. Entrainment is the continuous resetting of a reentrant circuit by a train of capturing stimuli. However, following the first stimulus of the pacing train that penetrates and resets the reentrant circuit, the subsequent stimuli interact with the reset circuit, which has an abbreviated excitable gap.27
During entrainment, each pacing stimulus creates two activation wavefronts, one in the orthodromic direction and the other in the antidromic direction. The wavefront in the antidromic direction collides with the existing tachycardia wavefront. The wavefront that enters the reentrant circuit in the orthodromic direction (i.e., the same direction as the spontaneous tachycardia wavefront) conducts through the reentrant pathway, resets the tachycardia, and emerges through the exit site to activate the myocardium and collide with the antidromically paced wavefront from the next paced stimulus. This sequence continues until cessation of pacing or block somewhere within the reentrant circuit develops.37 The first entrained stimulus results in retrograde collision between the stimulated and tachycardia wavefronts, whereas for all subsequent stimuli, the collision occurs between the currently stimulated wavefront and the one stimulated previously. Depending on the degree to which the excitable gap is preexcited (and abbreviated) by the first resetting stimulus, subsequent stimuli fall on fully or partially excitable tissue. Entrainment is said to be present when two consecutive extrastimuli conduct orthodromically through the circuit with the same conduction time while colliding antidromically with the preceding paced wavefront. Because all pacing impulses enter the tachycardia circuit during the excitable gap, each paced wavefront advances and resets the tachycardia. Thus, when pacing is terminated, the last paced impulse will continue to activate the entire tachycardia reentrant circuit orthodromically at the pacing CL and also will activate the entire myocardium orthodromically on exiting the reentrant circuit.42,43
Overdrive pacing at long CLs (approximately 10 to 20 milliseconds shorter than the tachycardia CL) can almost always entrain reentrant circuits with large flat resetting curves and a post-pacing interval (PPI) equal to the return cycle observed during the flat part of the resetting curve.39 During overdrive pacing, once the nth pacing stimulus resets the circuit, the following pacing stimulus (n + 1)th will reach the circuit more prematurely. Depending on how premature it is, this (n + 1)th extrastimulus may produce no change in the return cycle (compared with that in response to the nth extrastimulus), encounter progressive conduction delay (until a fixed, longer return cycle is reached), or terminate the tachycardia. The larger the flat curve observed during resetting or the longer the pacing CL, or both, the more likely the return cycle of the nth and (n + 1)th extrastimuli will be the same. In this case, no matter how many subsequent extrastimuli are delivered, the return cycle will be the same and equal to that observed during the flat portion of the resetting curve. However, if the flat portion of the resetting curve is small, the pacing CL is short, or both, the (n + 1)th extrastimulus will fall on partially refractory tissue and the return cycle will increase. Continued pacing at the same CL will result in a stable but longer return cycle than the nth extrastimulus or termination of the tachycardia. Consequently, circuits with large fully excitable gaps (i.e., large flat resetting curves) can demonstrate prolonged return cycles or even termination at pacing CLs equal to coupling intervals of a single extrastimulus demonstrating a fully excitable gap.
Entrainment Response Curves
During entrainment, the orthodromic wavefront of the last extrastimulus propagates around the circuit to become the first complex of the resumed tachycardia. The conduction time of this impulse to the exit site of the circuit is termed the last entrained interval, and it characterizes the properties of the reset circuit during entrainment.37,39 Measurement of the interval between the last paced extrastimulus to the first nonpaced tachycardia complex (on the surface ECG or presystolic electrogram) during entrainment at progressively shorter pacing CLs characterizes an entrainment response curve, analogous but not identical to resetting response curves with single extrastimuli. In this case, the return cycle depends critically on the number of extrastimuli delivered that reset the circuit before the return cycle is measured, because following the first extrastimulus producing resetting (the nth extrastimulus), subsequent extrastimuli are relatively more premature and can lead to a different return cycle.42,43
During entrainment, the return cycle measured at an orthodromically captured presystolic electrogram should equal the pacing CL regardless of the site of pacing, as long as the presystolic electrogram is orthodromically activated at a fixed stimulus-to-electrogram interval (i.e., fixed orthodromic conduction time). This would not be observed if the electrogram were captured antidromically. If the time from the orthodromically activated electrogram to the onset of the return cycle on the surface ECG remains constant, which is a requirement to prove that the electrogram is within or attached to the reentrant circuit proximal to the exit site, then the interval from the stimulus to the surface ECG complex will remain constant. The same is true for the electrogram measured at the stimulation site. In the absence of recording of a presystolic electrogram, other measurements may be used to characterize the last entrained interval at any pacing CL. Therefore, during entrainment, curves relating the pacing CL to the last entrained interval can be measured from the stimulus to the orthodromic presystolic electrogram, to the onset of the surface ECG of the first tachycardia (nonpaced) complex, or to the local activation time at the pacing site of the first tachycardia (nonpaced) complex. These measurements will be qualitatively identical but have different absolute values. As always, it is important to establish the stability of tachycardia CL and document the presence of entrainment before assessing the return cycle and PPI.42,43
Termination is the usual response to overdrive pacing of circuits that demonstrate an increasing curve in response to resetting by a single extrastimulus. However, if the number of extrastimuli following the nth extrastimulus is limited to one or two (especially at long pacing CLs), termination may not occur, although the return cycle will be progressively longer following each extrastimulus. Entrainment is not present until two consecutive PPIs are identical. In such cases, when termination does not occur, the return cycle will be longer than that observed if pacing were discontinued following the nth extrastimulus at that pacing CL.37,39 Thus, if only the return cycle following entrainment is used to analyze the excitable gap, an increasing curve suggesting decremental conduction can result, even though a flat curve is observed with single or double extrastimuli, or both. Therefore, only resetting phenomena describe the characteristics of the reentrant circuit. Entrainment analyzes a reset circuit that has a shorter excitable gap. Flat, mixed (flat and increasing), and increasing curves can be seen during entrainment of macroreentrant circuits. Increasing curves are almost always observed during entrainment of small or microreentrant circuits.
Relationship of Pacing Site and Cycle Length to Entrainment
Overdrive pacing at long CLs (approximately 10 to 20 milliseconds shorter than the tachycardia CL) can almost always entrain reentrant tachycardias. However, the number of pacing stimuli required to entrain the reentrant circuit depends on the tachycardia CL, the duration of the excitable gap of the tachycardia, refractoriness at the pacing site, and the conduction time from the stimulation site to the reentrant circuit.39,42–45
Diagnostic Criteria of Entrainment
Entrainment is the continuous resetting of a tachycardia circuit. Therefore, during constant rate pacing, entrainment of tachycardia results in the activation of all myocardial tissue responsible for maintaining the tachycardia at the pacing CL, with the resumption of the intrinsic tachycardia morphology and rate after cessation of pacing. Unfortunately, it is almost impossible to document the acceleration of all tissues responsible for maintaining the reentrant circuit to the pacing CL. Therefore, certain surface ECG criteria have been proposed for establishing the presence of entrainment: (1) fixed fusion of the paced complexes at a constant pacing rate; (2) progressive fusion or different degrees of fusion at different pacing rates (i.e., the surface ECG and intracardiac morphology progressively look more like the purely paced configuration and less like the pure tachycardia complex in the course of pacing at progressively shorter pacing CLs); and (3) resumption of the same tachycardia morphology following cessation of pacing, with the first PPI displaying no fusion but occurring at a return cycle equal to the pacing CL.46 These criteria are discussed in more detail in Chapter 5.42,43,47
Mechanism of Slow Conduction in the Reentrant Circuit
As mentioned earlier, a condition necessary for reentry is that the impulse be delayed sufficiently in the alternative pathway or pathways to allow tissues proximal to the site of unidirectional block to recover excitability.27 All types of reentrant arrhythmias have a basic feature in common—the wavefront must encounter a zone of tissue where local electrical inhomogeneity is present. This inhomogeneity can be related to (1) electrical properties of the individual cardiac myocyte that generates the action potential (inhomogeneity in electrical excitability or refractoriness, or both), (2) passive properties governing the flow of current among cardiac cells (cell-to-cell coupling and tissue geometry), or (3) combinations of those conditions.48 Such changes can be permanent (e.g., in remodeling after ventricular hypertrophy or infarction), or they can be purely functional (e.g., inhomogeneity of refractoriness in acutely ischemic tissue). Additionally, some of those changes are needed only to set the initial condition for the deviation of the impulse, the so-called unidirectional conduction block. Once the disturbance is initiated, an arrhythmia can develop in a perfectly homogeneous electrical medium.48
Slow conduction within the reentrant circuit is also an important determinant of the size of the reentrant circuit. A decrease in conduction velocity results in a reduction in the wavelength and a lessening of the amount of tissue needed to sustain reentry. In contrast, an increase in conduction velocity prolongs the wavelength of excitation, and, hence, a larger anatomical circuit is necessary to sustain reentry. If a larger circuit is not possible, initiation or maintenance of tachycardia cannot occur.27
In some cardiac tissues (e.g., the AVN), slow conduction is normal physiology. Slow conduction also can be secondary to pathophysiological settings (e.g., MI) or caused by functional properties that can develop as a result of premature stimulation or evolve during a rapid transitional rhythm.27
As discussed in Chapter 1, action potential propagation and conduction velocity in cardiac tissue are determined by source-sink relationships, which reflect the interplay between the active membrane properties of cardiac cells (the source generating the action potential) and the passive properties determined by architectural features of the myocardium (sink), as well as the coupling resistance between source and sink. The current provided by the source must reach the sink. The pathway between the source and sink includes intracellular resistance (provided by the cytoplasm) and intercellular resistance (provided by the gap junctions). Extracellular resistance plays a role, but it can often be neglected. The coupling resistance is mainly determined by resistance of the gap junctions. Therefore, the number and distribution of gap junctions, as well as the conductance of the gap junction proteins (connexins), are important factors for conduction of the action potential.
The safety factor for conduction predicts the success of action potential propagation and is defined as the ratio of the current generated by the depolarizing ion channels of a cell (source) to the current consumed during the excitation cycle of a single cell in the tissue (sink).39 Thus, the safety factor for propagation is proportional to the excess of source current over the sink needs. By this definition, conduction fails when the safety factor drops to less than 1 and becomes increasingly stable as it rises to more than 1.49 This concept of propagation safety provides information about the dependence of propagation velocity on the state of the ion channels, cell-to-cell coupling, and tissue geometry. In essence, local source-sink relationships determine the formation of conduction heterogeneities and provide conditions for the development of slow conduction, unidirectional block, and reentry.27,50,51
Reduced Membrane Excitability
Excitability of a cardiac cell describes the ease with which the cell responds to a stimulus with a regenerative action potential, and it depends on the passive and active properties of the cell membrane.7 The passive properties include the membrane resistance and capacitance and the intercellular resistance. The more negative the membrane potential is, the more Na+ channels are available for activation, the greater the influx of Na+ into the cell during phase 0, and the greater the conduction velocity.
In contrast, membrane depolarization to levels of −60 to −70 mV can inactivate half the Na+ channels, and depolarization to −50 mV or less can inactivate all the Na+ channels. Therefore, when stimulation occurs during phase 3 (e.g., during premature stimulation during the relative refractory period), before full recovery and at less negative potentials of the cell membrane, a portion of Na+ channels will still be refractory and unavailable for activation. As a result, INa and phase 0 of the next action potential are reduced, and conduction of the premature stimulus is slowed, facilitating reentry.6,27
Reduced membrane excitability occurs in numerous physiological and pathophysiological conditions. Genetic mutations that result in loss of Na+ channel function, as occurs in the Brugada syndrome, can cause reduced membrane excitability. Reduced membrane excitability is also present in cardiac cells with persistently low levels of resting potential caused by disease (e.g., during acute ischemia, tachycardia, certain electrical remodeling processes, and treatment with class I antiarrhythmic agents).27,50 At low resting potentials, the availability of excitable Na+ channels is reduced because of inactivation of a significant proportion of the Na+ channels and prolonged recovery of Na+ channels from inactivation. With progressive reduction of excitability, less Na+ source current is generated, and conduction velocity and the safety factor decrease monotonically. When the safety factor falls to less than 1, conduction can no longer be sustained, and failure (conduction block) occurs. Action potentials with reduced upstroke velocity resulting from partial inactivation of Na+ channels are called depressed fast responses. These action potential changes are likely to be heterogeneous, with unequal degrees of Na+ inactivation that create areas with minimally reduced velocity, more severely depressed zones, and areas of complete block. In addition, refractoriness in cells with reduced membrane potentials can outlast voltage recovery of the action potential; that is, the cell can still be refractory or partially refractory after the resting membrane potential returns to its most negative value.50
Reduced Cellular Coupling
Intercellular communication is maintained by gap junctional channels that connect neighboring cells and allow biochemical and low-resistance electrical coupling.52 Although the resistivity of the gap junctional membrane for the passage of ions and small molecules and for electrical propagation is several orders of magnitude higher than the cytoplasmic intracellular resistivity, gap junction coupling provides a resistance pathway that is several orders of magnitude lower compared with uncoupled cell membranes.53
In normal adult working myocardium, a given cardiomyocyte is electrically coupled to an average of approximately 11 adjacent cells, with gap junctions being predominantly localized at the intercalated discs at the ends of the rod-shaped cells. Lateral (side-to-side) gap junctions in nondisc lateral membranes of cardiomyocytes are much less abundant and occur more often in atrial than ventricular tissues.27,53–55 This particular subcellular distribution of gap junctions is a main determinant of anisotropic conduction in the heart; a wavefront must traverse more cells in the transverse direction than over an equivalent distance in the longitudinal direction, because cell diameter is much smaller than cell length. Additionally, less intercellular gap junctional coupling occurs and, hence, greater resistance and slower conduction transversely than longitudinally.27,52,55
Modification of intercellular coupling occurs in numerous physiological and pathophysiological conditions.27 Physiologically, cell-to-cell coupling is reduced in atrial and ventricular myocardium in the transverse direction to the main fiber axis relative to the longitudinal direction. Reduced intercellular coupling also likely contributes to slow impulse conduction in the AVN. In pathophysiological settings (e.g., myocardial ischemia, ventricular hypertrophy, cardiomyopathy),56 modification of intercellular coupling can occur as a consequence of acute changes in the average conductance of gap junctions secondary to ischemia, hypoxia, acidification, or increase in intracellular Ca2+, or it can be produced by changes in expression or cellular distribution patterns of gap junctions.57,58 Changes in the distribution and number of gap junctions (gap junction remodeling) have been reported in almost all cardiac diseases predisposing to arrhythmias. Remodeling includes a decrease in some gap junction channels resulting from the interruption of communication between cells by fibrosis and downregulation of connexin-43 (Cx43) formation or trafficking to the intercalated disc. Additionally, gap junctions can become more prominent along lateral membranes of myocytes (so-called structural remodeling).54,59
A reduction in the gap junctional conductance increases the intercellular resistance, thus leading to conduction delay or block. Similar to its behavior during reduced membrane excitability, conduction velocity decreases monotonically with reduction in intercellular coupling.52 However, the resulting reduction of conduction velocities is more than an order of magnitude larger than that observed during a reduction of excitability.27 Importantly, the changes in the safety factor with uncoupling are opposite to those observed with a reduction in membrane excitability. As cells become less coupled, there is greater confinement of depolarizing current to the depolarizing cell, with less electrotonic load and axial flow of charge to the downstream cells. As a result, individual cells depolarize with a high margin of safety, but conduction proceeds with long intercellular delays. At such low levels of coupling, conduction is very slow but, paradoxically, very robust.27,57,58
Tissue Structure and Geometry
Tissue geometry can influence action potential propagation and conduction velocity. In contrast to an uncoupled cell strand, in which the high resistance junctions alternate with the low cytoplasmic resistance of the cells, a high degree of discontinuity can be produced by large tissue segments (consisting of a segment with side branches) alternating with small tissue segments having a small tissue mass (connecting segments without branches).27
When a small mass of cells has to excite the large mass (e.g., an impulse passing abruptly from a fiber of small diameter to one of large diameter or propagating into a region where there is an abrupt increase in branching of the myocardium), transient slowing of the conduction velocity can be observed at the junction. This occurs because of sink-source mismatch, during which the current provided by the excitation wavefront (source) is insufficient to charge the capacity and thus excite the much larger volume of tissue ahead (sink).27,49,60,61
Anisotropy and Reentry
The anisotropic cellular structure of the myocardium is important for the understanding of normal propagation and arrhythmogenesis. Structural anisotropy can relate to cell shape and to the cellular distribution pattern of proteins involved in impulse conduction, such as gap junction connexins and membrane ion channels. The anisotropic architecture of most myocardial regions, consisting of elongated cells forming strands and layers of tissue, leads to a dependence of propagation velocity on the direction of impulse spread.27,54 In normal ventricular myocardium, conduction in the direction parallel to the long axis of the myocardial fiber bundles is approximately three to five times more rapid than that in the transverse direction. This is attributable principally to the lower resistivity of myocardium in the longitudinal versus the transverse direction; hence, intercellular current flow occurs primarily at the cell termini (although propagation also occurs transversely). As discussed previously, the gap junctions of the intercalated discs form a major source of intercellular resistance to current flow between fiber bundles. Therefore, the structure of the myocardium that governs the extent and distribution of these gap junctions has a profound influence on axial resistance and conduction.55
Cellular Coupling: Gap Junctional Organization
Alterations in distribution and function of cardiac gap junctions are associated with conduction delay or block. Inactivation of gap junctions decreases transverse conduction velocity to a greater degree than longitudinal conduction, thereby resulting in exaggeration of anisotropy and providing a substrate for reentrant activity and increased susceptibility to arrhythmias.54,59
Myocyte Packing and Tissue Geometry
Discontinuities in myocardial architecture exist at several levels. In addition to discontinuities imposed by cell borders, microvessels and connective tissue sheets separating bundles of excitable myocytes can act as resistive barriers. A propagating impulse is expected to collide with such barriers and travels around them wherever it encounters excitable tissue.48
In some regions of the myocardium (e.g., the papillary muscles), connective tissue septa subdivide the myocardium into unit bundles composed of 2 to 30 cells surrounded by a connective tissue sheath.6 Within a unit bundle, cells are tightly connected or coupled to each other longitudinally and transversely through intercalated discs that contain the gap junctions and are activated uniformly and synchronously as an impulse propagates along the bundle. Adjacent unit bundles also are connected to each other. Unit bundles are coupled better in the direction of the long axis of its cells and bundles, because of the high frequency of the gap junctions within a unit bundle, than in the direction transverse to the long axis, because of the low frequency of interconnections between the unit bundles. This is reflected as a lower axial resistivity in the longitudinal direction than in the transverse direction in cardiac tissues composed of many unit bundles. Additionally, anisotropy on a macroscopic scale can influence conduction at sites at which a bundle of cardiac fibers branches or separate bundles will coalesce. Marked slowing can occur when there is a sudden change in the fiber direction, causing an abrupt increase in the effective axial resistivity. Conduction block, which sometimes can be unidirectional, can occur at such junction sites, particularly when membrane excitability is reduced.
Uniform Versus Nonuniform Anisotropy
Uniform anisotropy is characterized by smooth wavefront propagation in all directions and measured conduction velocity changes monotonically on moving from fast (longitudinal) to slow (transverse) axes, thus indicating relatively tight coupling between groups of fibers in all directions. However, this definition is based on the characteristics of activation at a macroscopic level, where the spatial resolution encompasses numerous myocardial cells and bundles, and therefore it describes the behavior of the myocardial syncytium. In contrast, when the three-dimensional network of cells is broken down into linear single-cell chains, gap junctions can be shown to limit axial current flow and induce saltatory conduction because of the recurrent increases in axial resistance at the sites of gap junctional coupling; that is, conduction is composed of rapid excitation of individual cells followed by a transjunctional conduction delay. In two- and three-dimensional tissue, these discontinuities disappear because of lateral gap junctional coupling, which serves to average local small differences in activation times of individual cardiomyocytes at the excitation wavefront.49,55
In multicellular tissue, saltatory conduction reappears only under conditions of critical gap junctional uncoupling, in which it leads to a functional unmasking of the cellular structure and induces ultraslow and meandering conduction, well known to be a key ingredient in arrhythmogenesis.49 Change in the characteristics of anisotropic propagation at the macroscopic scale from uniform to nonuniform strongly predisposes to reentrant arrhythmias. Nonuniform anisotropy has been defined as tight electrical coupling between cells in the longitudinal direction but uncoupling to the lateral gap junctional connections. Therefore, there is disruption of the smooth transverse pattern of conduction characteristic of uniform anisotropy that results in a markedly irregular sequence or zigzag conduction, producing the fractionated extracellular electrograms characteristic of nonuniform anisotropic conduction (Fig. 3-18). In nonuniformly anisotropic muscle, there also can be an abrupt transition in conduction velocity from the fast longitudinal direction to the slow transverse direction, unlike the case with uniform anisotropic muscle, in which intermediate velocities occur between the two directions.
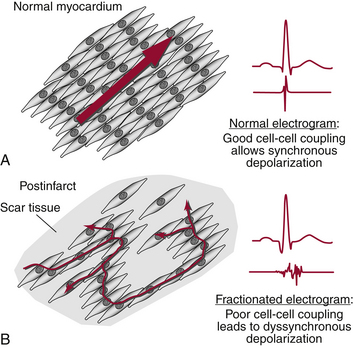
Nonuniform anisotropic properties can exist in normal cardiac tissues secondary to separation of the fascicles of muscle bundles in the transverse direction by fibrous tissue that proliferates with aging to form longitudinally oriented insulating boundaries (e.g., crista terminalis, interatrial band in adult atria, or ventricular papillary muscle). Similar connective tissue septa cause nonuniform anisotropy in pathological situations such as chronic ischemia or a healing MI, in which fibrosis in the myocardium occurs (see Fig. 3-18).6,55
Mechanism of Unidirectional Block in the Reentrant Circuit
Unidirectional block occurs when an impulse cannot conduct in one direction along a bundle of cardiac fibers but can conduct in the opposite direction. As mentioned earlier, this condition is necessary for the occurrence of classic reentrant rhythms. Several mechanisms, involving active and passive electrical properties of cardiac cells, can cause unidirectional block.29
Inhomogeneity of Membrane Excitability and Refractoriness
Unidirectional block develops when the activation wavefront interacts with the repolarization phase (tail) of a preceding excitation wave. There is a critical or vulnerable window during the relative refractory period of a propagating action potential within which unidirectional block occurs. When a premature stimulus is delivered outside this window, the induced action potential propagates or blocks in both directions; specifically, a stimulus delivered too early fails to induce a propagating action potential in either direction (bidirectional block), whereas a late stimulus results in bidirectional conduction (no block). In contrast, when a stimulus is applied within the vulnerable window, the induced action potential propagates incrementally in the retrograde direction because the tissue is progressively more recovered as the distance from the window increases in this direction, but it blocks in the anterograde direction following a short distance of decremental conduction because the tissue is progressively less excitable as the distance from the window increases in this direction.27
Unidirectional conduction block in a reentrant circuit also can be persistent and independent of premature activation, in which case it often occurs in a region of depressed and heterogeneous excitability (as occurs in acute ischemia); this leads to a widening of the vulnerable window. Asymmetry in excitability, which can occur because of asymmetrical distribution of a pathological event, can lead to an abrupt rise in the threshold for excitation in one direction and to a more gradual rise in the other. Conduction fails when the wavefront encounters the least depressed site first and is successful in the direction in which it encounters the most depressed site first. Additionally, impulses are conducted more easily from a rapidly conducting tissue to a slowly conducting tissue than in the opposite direction.29
Local dispersion of refractory periods is a normal feature of ventricular myocardium. Critical increases in the dispersion of refractoriness, the difference between the shortest and longest refractory periods, can result in the local widening of the vulnerability window and an increased probability for the generation of unidirectional block and reentry.6,27 Increased heterogeneity of repolarization and dispersion of refractoriness can be caused by acute or prolonged ischemia, the long QT syndrome, or electrical remodeling in the setting of ventricular hypertrophy and failure and in the setting of MI. When differences in the duration of the refractory periods occur in adjacent areas, conduction of an appropriately timed premature impulse can be blocked in the region with the longest refractory period, which then becomes a site of unidirectional block, whereas conduction continues through regions with a shorter refractory period.6
Anisotropy and Unidirectional Block
The anisotropic properties of cardiac muscle can contribute to the occurrence of unidirectional block.6 As mentioned earlier, in the anisotropic muscle, the safety factor for conduction is lower in the longitudinal direction of rapid than in the transverse direction of slow conduction. The low safety factor longitudinally is a result of a large current load on the membrane associated with the low axial resistivity and a large membrane capacitance in the longitudinal direction. This low safety factor can result in a preferential conduction block of premature impulses in the longitudinal direction while conduction in the transverse direction continues. The site of block in the longitudinal direction can become a site of unidirectional block that leads to reentry.6 In contrast to the propensity of premature impulses to block in the longitudinal direction in nonuniformly anisotropic myocardium because of the decreased depolarizing current and low safety factor, when coupling resistance between cells is increased, conduction of all impulses will block first in the transverse direction. Preferential block in this direction occurs because an increase in coupling resistance will reduce the safety factor below the critical level needed to maintain transverse conduction before the safety factor for longitudinal conduction is reduced to this critical level.6,55
Discontinuities in Tissue Structure and Geometry
Geometric factors related to tissue architecture also can influence impulse conduction and, under certain conditions, lead to unidirectional block.27 Structural discontinuities exist in the normal heart in the form of trabeculations of the atrial and ventricular walls, sheets interconnected by small trabeculae, or myocardial fibers with different diameters packed in a connective tissue matrix. Structural discontinuities can also be secondary to pathophysiological settings, such as the connective tissue septa characteristic of aging, infarcted, hypertrophic, and failing myocardium. The propagating excitation wave is expected to interact with these normal and abnormal structural discontinuities. These structural features influence conduction by affecting the axial currents that flow ahead of the propagating wavefront.27 Therefore, an impulse conducting in one direction can encounter a different sequence of changes in fiber diameter, branching, and frequency and distribution of gap junctions than it does when traveling in the opposite direction. The configuration of pathways in each direction is not the same.
1. Peters N.S., Cabo C., Wit A.L. Arrhythmogenic mechanisms: automaticity, triggered activity, and reentry. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 3. Philadelphia: Saunders; 2000:345-355.
2. Mangoni M.E., Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev. 2008;88:919-982.
3. Dobrzynski H., Boyett M.R., Anderson R.H. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation. 2007;115:1921-1932.
4. Couette B., Marger L., Nargeot J., Mangoni M.E. Physiological and pharmacological insights into the role of ionic channels in cardiac pacemaker activity. Cardiovasc Hematol Disord Drug Targets. 2006;6:169-190.
5. Barbuti A., Baruscotti M., DiFrancesco D. The pacemaker current: from basics to the clinics. J Cardiovasc Electrophysiol. 2007;18:342-347.
6. Waldo A.L., Wit A.L. Mechanisms of cardiac arrhythmias and conduction disturbances. In: Fuster V., Alexander R.W., O’Rourke R.A., editors. Hurst’s the heart. ed 11. Columbus, Ohio: McGraw-Hill; 2004:787-816.
7. Grant A.O. Cardiac ion channels. Circ Arrhythm Electrophysiol. 2009;2:185-194.
8. Lakatta E.G. A paradigm shift for the heart’s pacemaker. Heart Rhythm. 2010;7:559-564.
9. Venetucci L.A., Trafford A.W., O’Neill S.C., Eisner D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285-292.
10. DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434-446.
11. Maltsev V.A., Lakatta E.G. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res. 2008;77:274-284.
12. Haissaguerre M., Jais P., Shah D.C., et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659-666.
13. Boyden P.A., Hirose M., Dun W. Cardiac Purkinje cells. Heart Rhythm. 2010;7:127-135.
14. Kohl P., Bollensdorff C., Garny A. Effects of mechanosensitive ion channels on ventricular electrophysiology: experimental and theoretical models. Exp Physiol. 2006;91:307-321.
15. Dun W., Boyden P.A. The Purkinje cell: 2008 style. J Mol Cell Cardiol. 2008;45:617-624.
16. Venetucci L.A., Trafford A.W., O’Neill S.C., Eisner D.A. Na/Ca exchange: regulator of intracellular calcium and source of arrhythmias in the heart. Ann N Y Acad Sci. 2007;1099:315-325.
17. Kalbfleisch S., Hart D., Weiss R. Two ventricular tachycardias with cycle length and QRS alternans: insights into the mechanism from mapping and ablation of the tachycardias. Pacing Clin Electrophysiol. 2009;32:e31-e35.
18. Gyorke S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2009;6:123-129.
19. Laurita K.R., Rosenbaum D.S. Mechanisms and potential therapeutic targets for ventricular arrhythmias associated with impaired cardiac calcium cycling. J Mol Cell Cardiol. 2008;44:31-43.
20. Ter Keurs H.E., Boyden P.A. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457-506.
21. Kaufman E.S. Mechanisms and clinical management of inherited channelopathies: long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm. 2009;6(Suppl):S51-S55.
22. Scheinman M.M. Role of the His-Purkinje system in the genesis of cardiac arrhythmia. Heart Rhythm. 2009;6:1050-1058.
23. Weiss J.N., Garfinkel A., Karagueuzian H.S., et al. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891-1899.
24. Maruyama M., Lin S.F., Xie Y., et al. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol. 2011;4:103-111.
25. Mines G.R. On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;4:43.
26. Mines G.R. On dynamic equilibrium in the heart. J Physiol. 1913;46:349-383.
27. Kleber A.G., Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431-488.
28. Allessie M.A., Bonke F.I., Schopman F.J. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 1977;41:9-18.
29. Segal O.R., Chow A.W., Peters N.S., Davies D.W. Mechanisms that initiate ventricular tachycardia in the infarcted human heart. Heart Rhythm. 2010;7:57-64.
30. Antzelevitch C. Basic mechanisms of reentrant arrhythmias. Curr Opin Cardiol. 2001;16:1-7.
31. Benito B., Guasch E., Rivard L., Nattel S. Clinical and mechanistic issues in early repolarization of normal variants and lethal arrhythmia syndromes. J Am Coll Cardiol. 2010;56:1177-1186.
32. Antzelevitch C., Yan G.X. J wave syndromes. Heart Rhythm. 2010;7:549-558.
33. Lim Z.Y., Maskara B., Aguel F., et al. Spiral wave attachment to millimeter-sized obstacles. Circulation. 2006;114:2113-2121.
34. Tung L., Zhang Y. Optical imaging of arrhythmias in tissue culture. J Electrocardiol. 2006;39(Suppl):S2-S6.
35. Chang M.G., Zhang Y., Chang C.Y., et al. Spiral waves and reentry dynamics in an in vitro model of the healed infarct border zone. Circ Res. 2009;105:1062-1071.
36. Almendral J.M., Rosenthal M.E., Stamato N.J., et al. Analysis of the resetting phenomenon in sustained uniform ventricular tachycardia: incidence and relation to termination. J Am Coll Cardiol. 1986;8:294-300.
37. Kay G.N., Epstein A.E., Plumb V.J. Resetting of ventricular tachycardia by single extrastimuli: relation to slow conduction within the reentrant circuit. Circulation. 1990;81:1507-1519.
38. Stevenson W.G., Weiss J.N., Wiener I., et al. Resetting of ventricular tachycardia: implications for localizing the area of slow conduction. J Am Coll Cardiol. 1988;11:522-529.
39. Josephson M.E. Recurrent ventricular tachycardia. In: Josephson M., editor. Clinical cardiac electrophysiology. ed 4. Philadelphia: Lippincott Williams & Wilkins; 2002:425-610.
40. Rosman J.Z., John R.M., Stevenson W.G., et al. Resetting criteria during ventricular overdrive pacing successfully differentiate orthodromic reentrant tachycardia from atrioventricular nodal reentrant tachycardia despite interobserver disagreement concerning QRS fusion. Heart Rhythm. 2011;8:2-7.
41. Rosenthal M.E., Stamato N.J., Almendral J.M., et al. Resetting of ventricular tachycardia with electrocardiographic fusion: incidence and significance. Circulation. 1988;77:581-588.
42. Deo R., Berger R. The clinical utility of entrainment pacing. J Cardiovasc Electrophysiol. 2009;20:466-470.
43. Waldo A.L. From bedside to bench: entrainment and other stories. Heart Rhythm. 2004;1:94-106.
44. Tritto M., De P.R., Zardini M., et al. Comparison of single premature versus continuous overdrive stimulation for identification of a protected isthmus in macro-reentrant atrial tachycardia circuits. Am J Cardiol. 2003;91:1485-1489.
45. Morton J.B., Sanders P., Deen V., et al. Sensitivity and specificity of concealed entrainment for the identification of a critical isthmus in the atrium: relationship to rate, anatomic location and antidromic penetration. J Am Coll Cardiol. 2002;39:896-906.
46. Almendral J.M., Gottlieb C.D., Rosenthal M.E., et al. Entrainment of ventricular tachycardia: explanation for surface electrocardiographic phenomena by analysis of electrograms recorded within the tachycardia circuit. Circulation. 1988;77:569-580.
47. Das M.K., Scott L.R., Miller J.M. Focal mechanism of ventricular tachycardia in coronary artery disease. Heart Rhythm. 2010;7:305-311.
48. Kleber A.G., Fast V. Molecular and cellular aspects of re-entrant arrhythmias. Basic Res Cardiol. 1997;92(Suppl 1):111-119.
49. Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004;62:309-322.
50. Antzelevitch C. Cellular basis and mechanism underlying normal and abnormal myocardial repolarization and arrhythmogenesis. Ann Med. 2004;36(Suppl 1):5-14.
51. Santana L.F., Nunez-Duran H., Dilly K.W., Lederer W.J. Sodium current and arrhythmogenesis in heart failure. Heart Fail Clin. 2005;1:193-205.
52. Sohl G., Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62:228-232.
53. Noorman M., van der Heyden M.A., van Veen T.A., et al. Cardiac cell-cell junctions in health and disease: electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23-31.
54. Saffitz J.E., Lerner D.L., Yamada K.A. Gap junction distribution and regulation in the heart. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 4. Philadelphia: Saunders; 2004:181-191.
55. Valderrabano M. Influence of anisotropic conduction properties in the propagation of the cardiac action potential. Prog Biophys Mol Biol. 2007;94:144-168.
56. Cascio W.E., Yang H., Muller-Borer B.J., Johnson T.A. Ischemia-induced arrhythmia: the role of connexins, gap junctions, and attendant changes in impulse propagation. J Electrocardiol. 2005;38(Suppl):55-59.
57. van Rijen H.V., van Veen T.A., Gros D., et al. Connexins and cardiac arrhythmias. Adv Cardiol. 2006;42:150-160.
58. Lee P.J., Pogwizd S.M. Micropatterns of propagation. Adv Cardiol. 2006;42:86-106.
59. Dhein S. Cardiac ischemia and uncoupling: gap junctions in ischemia and infarction. Adv Cardiol. 2006;42:198-212.
60. Fast V.G., Kleber A.G. Cardiac tissue geometry as a determinant of unidirectional conduction block: assessment of microscopic excitation spread by optical mapping in patterned cell cultures and in a computer model. Cardiovasc Res. 1995;29:697-707.
61. Wang Y., Rudy Y. Action potential propagation in inhomogeneous cardiac tissue: safety factor considerations and ionic mechanism. Am J Physiol Heart Circ Physiol. 2000;278:H1019-H1029.