Chapter 12 Dystonia
Phenomenology, classification, etiology, pathology, biochemistry, and genetics
Historical highlights
In 1908, Schwalbe published his dissertation on a family with three affected children who are now recognized to have had primary generalized dystonia. An English translation of Schwalbe’s paper is available (Truong and Fahn, 1988). Three years later Oppenheim (1911) described the same disorder in four patients and coined the word “dystonia” to indicate that in this disorder, there would be hypotonia on one occasion and tonic muscle spasms on another, usually but not exclusively elicited on volitional movements. Oppenheim called this syndrome by two different names: “dystonia musculorum deformans” and “dysbasia lordotica progressiva.” The first name relates to the spasms and to the postural deformities that develop in these children; the second name emphasizes the dromedary gait and the progressive nature of the illness. Oppenheim described muscle spasms; bizarre walking with bending and twisting of the torso; rapid, sometimes rhythmic jerking movements; and progression of symptoms leading eventually to sustained fixed postural deformities.
Oppenheim, however, failed to recognize the inherited nature of the disorder, which was emphasized by Flatau and Sterling (1911) later that year; they suggested the name “progressive torsion spasm,” which perhaps would have been the preferred one, according to the full syndrome recognized today. The word “dystonia,” however, was immediately adopted by neurologists and has been used to describe both a distinctive motor phenomenology and a clinical syndrome in which these motoric features are present. Over time, different meanings were used for “dystonia” (for historical details, see Fahn et al., 1987). To clarify the definition, an ad hoc committee of the Dystonia Medical Research Foundation considered the clinical features described by Oppenheim (1911), Flatau and Sterling (1911) and other early observers, and developed the following definition: Dystonia is a syndrome of sustained muscle contractions, frequently causing twisting and repetitive movements, or abnormal postures (Fahn et al., 1987; Fahn, 1988). To emphasize the twisting quality of the abnormal movements and postures, the term “torsion” is often placed in front of the word “dystonia.” Such twisting is one feature that distinguishes dystonic movements from those of other dyskinesias, such as chorea, and distinguishes dystonic postures from other syndromes of increased muscle tone, such as rigidity, stiff-person syndrome, and neuromyotonia (Fahn, 1999). Exceptions to twisting are around joints that do not allow such torsion. Thus, dystonia involving the jaw, focusing on the temporomandibular joint, are jaw-opening and jaw-closing dystonias, rarely lateral jaw dystonia, but not twisting of the jaw.
The early observers described dystonia as a specific disease entity, but by the next decade, dystonia was recognized to be a feature in other neurologic disorders, such as Wilson disease and cerebral palsy, and following encephalitis. Soon, dystonia as a specific entity (today known as primary dystonia) was lost. It was Herz (1944a, 1944b, 1944c) who, in a masterful series of three papers, resurrected torsion dystonia as a specific neurologic entity as well as its presence within other neurologic diseases, who described the motor phenomenology and compared the duration of its contractions with those of chorea and athetosis, who utilized the analysis of cinematography to distinguish these differences in various movement disorders, and who showed the characteristic simultaneous contractions of agonist and antagonist muscles in dystonia. Another major pioneer in dystonia was Zeman (1970), who, along with his colleagues (Zeman et al., 1959, 1960; Zeman and Dyken, 1967), carried out the first epidemiologic study, emphasized the autosomal dominant pattern of inheritance, described the focal dystonias as formes frustes of generalized dystonia, and found the pathologic anatomy to be normal in primary torsion dystonia (PTD). The disparate varieties of focal dystonias were not linked as focal dystonias until Marsden placed them together (1976). Still, patients with dystonia, both generalized and focal, were often considered to be hysterical, i.e., to have a psychogenic disorder and not an organic one, as Lesser and Fahn (1978) pointed out, often with tragic consequences, as described in Cooper’s book (1976). Better awareness of the organic nature of dystonia, both generalized and focal (for torticollis and writer’s cramp were often considered hysterical), began to come about with the holding and publication of the first international symposium on dystonia (Eldridge and Fahn, 1976). The final proof came with the discovery of the gene locus for Oppenheim dystonia (Ozelius et al., 1989) and the discovery that dystonia in the Ashkenazi Jewish population was inherited in an autosomal dominant pattern (Bressman et al., 1989).
Other important events in the development of our understanding of dystonia are the formation of the Dystonia Medical Research Foundation in 1976, the creation of four international symposia on dystonia with their subsequent publications (Eldridge and Fahn, 1976; Fahn et al., 1988, 1998b, 2004), and the investigations of clinical and molecular genetics that are leading to the discovery of the mutated genes and clarifying the classification and clinical features of the dystonias (Bressman et al., 1989, 1994a; Ozelius et al., 1989, 1997; Nygaard et al., 1993; Ichinose et al., 1994). The advances in genetics have led to a better etiologic classification of the dystonias (Fahn et al., 1998a) and to the labeling of many of the dystonias using a “DYT” classification, as displayed in Table 12.1.
AD, autosomal dominant; AR, autosomal recessive.
A number of reviews covering the historical aspects are available to which the reader is referred for more details than are provided in this chapter (Fahn, 1984a; Fahn et al., 1987; Fahn and Marsden, 1987; Rothwell and Obeso, 1987; Fahn et al., 1988; Fahn, 1989a, 1990; Tsui and Calne, 1995; Jankovic and Fahn, 1998; Nemeth, 2002; Stacy, 2007; Breakefield et al., 2008). A collection of historical photographs about dystonia is available for perusal (Goetz et al., 2001).
Impact and overview of genetic discoveries
Perhaps the greatest advance in dystonia in the past 10 years has been the continual unraveling of an increasing number of distinct genetic forms of dystonia based on gene mapping and cloning. In turn, this has led to the beginning of molecular biology research in the dystonias and a more comprehensive classification of the dystonias based on etiology. The first genetic form of dystonia whose gene was mapped was the form described by Oppenheim (1911), which he called dystonia musculorum deformans and which is now called Oppenheim dystonia (Fahn et al., 1998a) and also DYT1 dystonia (because the mapped gene was given that designation). The gene for Oppenheim dystonia has been identified and cloned, but not before the gene responsible for another genetic form of dystonia was identified, namely, dopa-responsive dystonia (DYT5), or DRD, caused by a mutation in the gene for the enzyme guanosine triphosphate (GTP) cyclohydrolase I (GCH1). As genes are being mapped, the clinicians are able to use this information to better define the clinical features of each genetic type. Table 12.1 lists the currently known genetic designations for the dystonias. The last two genes identified in this nomenclature are those of NA-K-ATPase responsible for rapid-onset dystonia-parkinsonism (RDP) (DYT12) (de Carvalho Aguiar et al., 2004) and TAF1 (TATA box-binding protein-associated factor 1) for lubag (DYT3) (Makino et al. 2007). The official nomenclature committee has labeled 20 DYTs, but omitted other genetic forms of dystonia, which are listed in this section after the first 20. Moreover, clinical neurologists usually consider DYT8, DYT9, and DYT10 to be part of the category of movement disorders known as paroxysmal dyskinesias, and not the dystonia category. These are covered in Chapter 22. Also, the designation of DYT14 for a new form of DRD was incorrect. The family with “DYT14” was thought not to have the DYT5 gene deletion in GCH1 (Grotzsch et al., 2002), but subsequently a mutation in GCH1 was found (Wider et al., 2008). DYT14 has been eliminated in Table 12.1.
A number of other dystonias are known to be genetic, but have not received a “DYT” classification. Two new families with suspected DYT2 dystonia were reported (Khan et al., 2003; Moretti et al., 2005). Siblings from two families with consanguinity developed autosomal recessive childhood-onset generalized dystonia. Gene studies for other known genetic forms of dystonia were negative.
The gene causing lubag (DYT3) was reported to involve a multiple transcript system (Nolte et al., 2003). It was not initially possible to say which mutant protein (and there may be up to six from this mutation) is actually responsible for the disease. However, Makino and colleagues (2007) have subsequently found the mutation for lubag to be in the TAF1 gene.
The gene causing DYT6 was reported in 2009 as the THAP1 gene (Fuchs et al., 2009). Its function is unknown. Subsequently a search was made for familial dystonia with young onset and many more mutations were discovered in this gene, and the age at onset expanded to 49 years, with the majority still being young onset (Bressman et al., 2009). Screening for focal dystonia, singleton cases revealed far fewer cases, with a phenotype of young-onset spasmodic dysphonia that later spread to become generalized dystonia (Djarmati et al., 2009). The THAP1 gene was analyzed in a series of 362 British, genetically undetermined, primary dystonia patients, and nine mutations were found (Houlden et al., 2010). The main clinical presentation was early-onset (<30 years) dystonia in the craniocervical region or the limbs (8 of 9 patients); laryngeal or oromandibular dystonia was present in 3 cases. Thus, early-onset dystonia that includes involvement of the larynx or face should arouse suspicion of a THAP1 mutation. A search for THAP1 mutations in 1114 patients with adult-onset primary dystonia revealed different THAP1 sequence variants to be associated with varied anatomical distributions and onset ages of both familial and sporadic primary dystonia (Xiao et al., 2010).
de Carvalho Aguiar and colleagues (2004) reported the identification of missense mutations in the gene for the Na+/K+-ATPase α3 subunit (ATP1A3) as a cause of RDP (DYT12).
A fuller description of the phenotype of DYT13 (craniocervical-brachial dystonia) has been reported in the only known family with this disorder (Bentivoglio et al., 2004). Age at onset ranged from 5 to 43 years. Onset occurred either in the craniocervical region or in the upper limbs. Progression was mild, and the disease course was benign in most affected individuals; generalization occurred in only two cases. There was no anticipation of age at onset or of disease severity through generations. Most subjects presented with jerky, myoclonic-like dystonic movements of the neck or shoulders. DYT13 is an autosomal dominant disease, with incomplete penetrance (58%).
Two independent studies searched for mutations in the ε-sarcoglycan gene in patients with familial and sporadic myoclonus–dystonia and did not find any (Han et al., 2003; Valente et al., 2003). This finding, plus the report of one family having a mutation on a different chromosome, 18p11 (DYT15), provides additional evidence for genetic heterogeneity in myoclonus–dystonia.
Two unrelated consanguineous Brazilian families with early-onset dystonia–parkinsonism were found to have a mutation in the PRKRA gene for protein kinase, interferon-inducible double-stranded RNA-dependent activator and it has been labeled as DYT16 (Camargos et al., 2008).
DYT17 is the designation given to a large consanguineous Lebanese family in which three sisters had primary torsion dystonia beginning with torticollis at ages 14, 17, and 19 years, spreading to segmental and generalized in one of the sisters (Chouery et al., 2008).
DYT18, DYT19, and DYT20 are paroxysmal dyskinesias and are covered in Chapter 22. DYT21 is an autosomal dominant family from northern Sweden of adult onset manifesting combinations of focal, segmental and generalized dystonia (Holmgren et al., 1995; Norgren et al., 2011).
A number of dystonic conditions are not labeled as “DYT,” but dystonia is part of their features (Table 12.2).
Table 12.2 Unlabeled as “DYT” but dystonia is present as well as other neurologic features in some of these disorders
AD, autosomal dominant; AR, autosomal recessive.
The association with a polymorphism in the D5 receptor gene (DRD5) for primary focal dystonias could not be confirmed in German and French patients (Sibbing et al., 2003), but was seen in Italian patients (Brancati et al., 2003).
The autosomal recessive disorder of early-onset cerebellar ataxia with oculomotor apraxia associated with a mutation of the aprataxin gene on chromosome 9p13 can also cause generalized dystonia (Sekijima et al., 2003). Adult-onset craniocervical dystonia preceded ataxia in a case with spinocerebellar ataxia type 1 (SCA1) (Wu et al., 2004). Cervical dystonia was also seen in SCA10 (Gatto et al., 2007), and spasmodic dysphonia has been reported in SCA17 (Hagenah et al., 2004, 2007).
Some patients with adult-onset dystonia have been found to have a missense mutation in the mitochondrial complex I gene (Simon et al., 2003). In addition to dystonia, spasticity and core-type myopathy are present.
A screen for a combination of dystonia with cerebellar atrophy identified eight such families (Le Ber et al. 2006), which raises the possibility that the cerebellum could be involved in dystonia (Jinnah and Hess, 2006), but more likely these families suggest a phenotype of a newly recognized neurodegenerative disease.
Although the GAG mutation in torsinA (TOR1A) is rare and causes young-onset limb-onset dystonia, two polymorphisms in or near the TOR1A gene have been discovered to be common in idiopathic focal dystonia, raising the possible association of a risk factor for this gene and the common adult-onset focal dystonias (Kamm et al., 2006).
Phenomenology of dystonic movements
With few exceptions, the four clinically unifying, consistent, and predominant features of dystonic contractions are (1) their relatively long duration (compared to myoclonus and chorea), although short-duration contractions can occur in dystonia; (2) their simultaneous contractions of agonists and antagonists; (3) their resulting in a twisting of the affected body part; and (4) their continual contractions of the same muscle groups (called patterned movements, see Chapter 1). One exception to the twisting nature of dystonia is that dystonias in facial muscles are rarely twisting; they are patterned and involve sustained contractions of forehead, eyelids, and lower face. As it turns out, twisting facial muscles that move the mouth to one side or the other or back and forth to each side are usually psychogenic (see Chapter 25).
Although the durations of the patterned contractions are usually more sustained than those of chorea, sometimes they can be very short. The range from very short to very prolonged contractions results in the appearance of a wide range in the speed of the patterned dystonic movements from rapid to slow. The movements can be so fast that they have the appearance of repetitive myoclonic jerking. The term “myoclonic-dystonia” has been applied to such dystonia (Davidenkow, 1926; Obeso et al., 1983; Kurlan et al., 1988; Quinn et al., 1988), and the rapid jerks may respond to alcohol (Quinn et al., 1988). In some families, the phenomenology of the combination of myoclonus and dystonia is a major feature, and these families are genetically distinct from those of the originally described PTD (i.e., Oppenheim dystonia) (Wahlström et al., 1994) and have been called by different names, initially dystonia–myoclonus (Fahn et al., 1998a), but now myoclonus–dystonia (Klein et al., 1999; Nygaard et al., 1999). The relationship of familial myoclonus–dystonia and hereditary essential myoclonus, which can also include patients with some dystonic features, is not clear (Fahn and Sjaastad, 1991; Quinn, 1996), but will soon be settled, as the identification of a gene (ε-sarcoglycan) for myoclonus–dystonia has been discovered (Zimprich et al., 2001).
Primary dystonia almost always begins by affecting a single part of the body; this is focal dystonia (Video 12.1). Most patients’ dystonia remains as a focal dystonia without spreading to other parts of the body. However, even within that single body part, multiple muscles can be affected. Thus, in patients with dystonia of the neck (cervical dystonia, or torticollis) a combination of muscles are involved (Video 12.2). Moreover, even if the neck is postured to a stable position, there can be changes in muscle contraction patterns that can be detected with EMG recordings (Munchau et al., 2001a). In a sizeable minority of patients, dystonia that starts in one body part can spread to involve other parts of the body. Most often, the spread is to contiguous body parts; hence, the spread is from focal dystonia to segmental dystonia. The pattern of spread of adult-onset focal dystonias has been analyzed, especially blepharospasm (Fahn, 1985; Weiss et al., 2006), as has the spread of childhood-onset dystonia (Greene et al., 1995). In blepharospasm, spread to other body parts is faster in patients who carry at least one T allele in a polymorphism site on the TOR1A gene (Defazio et al. 2009). As a general rule, the younger the age at onset, the more likely it is that the dystonia will spread; for example, childhood onset with leg involvement usually leads to eventual generalized dystonia (Marsden et al., 1976; Fahn, 1986; Greene et al., 1995). Regardless of genetic etiology, the phenotypes of primary dystonias are affected by age at onset, with a caudal-to-rostral change in the site of onset as a function of age (O’Riordan et al., 2004). The severity of dystonia can be quantified by using clinical rating scales for generalized dystonia and the various focal dystonias (Burke et al., 1985; Fahn, 1989b; Comella et al., 1997, 2003). 
Dystonic movements are almost always aggravated during voluntary movement. The appearance of dystonic movements with voluntary movement is referred to as “action dystonia.” Primary dystonia commonly begins with a specific action dystonia, that is, the abnormal movements appear with a special action (i.e., task-specific action) and are not present at rest, in contrast to secondary dystonias which are more likely to begin with dystonia at rest (Svetel et al., 2004). For example, a child who develops primary dystonia might have the initial symptom in one leg, but only when walking forward. It could be absent when the child runs or walks backward (Video 12.3). Other common examples are the task-specific dystonias that are seen with writing (writer’s cramp) (Video 12.4), playing a musical instrument (musician’s cramp) (Video 12.5), chewing (Video 12.6), and speaking, including auctioneering (Scolding et al., 1995). Often, these task-specific dystonias produce occupational disability; e.g., musicians usually no longer can play their instrument professionally. Robert Schumann’s career as a pianist was impaired, probably because of musician’s cramp (de Yebenes, 1995). Musician’s cramp and other occupational cramps can occur in any part of body that is engaged in repetitive, highly skilled tasks (Altenmüller and Jabusch, 2010). Embouchure (the pattern of lip, jaw, and tongue muscles used to control the flow of air into a mouthpiece) dystonia has been seen in horn and woodwind players (Frucht and Estrin, 2010). Patients with embouchure dystonia can be separated into several groups, including embouchure tremor, involuntary lip movements, and jaw closure (Frucht et al., 2001). The dystonia can spread to other oral tasks, often producing significant disability. Musician’s dystonia, like other focal dystonias (discussed below in pathophysiology of primary dystonia), is associated with increased sensorimotor activation (Haslinger et al., 2010). Focal task-specific tremors might be a form of focal dystonia rather than a manifestation of essential tremor (Soland et al., 1996b). Several reviews of musician’s cramps were presented at an international symposium on dystonia (Brandfonbrener and Robson, 2004; Charness and Schlaug, 2004; Frucht, 2004; Jabusch and Altenmuller, 2004; Pesenti et al., 2004). 
As the dystonic condition progresses, less specific voluntary motor actions of the involved limb can bring out the dystonic movements (see Table 12.3). In the above example, the affected leg might also activate the dystonia when it is tapping the floor. With further evolution, actions in other parts of the body can induce dystonic movements of the involved leg, so-called “overflow.” Talking is the most common mechanism for causing overflow dystonia in other body parts. Such activation of involuntary movements by talking is also particularly common with levodopa-induced dyskinesias and in cerebral palsy. With still further worsening, the affected limb can develop dystonic movements while it is at rest. Eventually, the leg can have sustained posturing. Thus, dystonia at rest is usually a more severe form than pure action dystonia. Whereas primary dystonia often begins as action dystonia and may persist as the kinetic (clonic) form, symptomatic dystonia often begins as sustained postures (tonic form). Sustained postures may appear in specific placements of the body. For example, the trunk may be in normal posture when the patient is lying supine or prone, but develop into kyphosis, scoliosis, or lordosis when sitting or standing. Thus, this dystonia is not a fixed dystonia (non-changing with a change in body posture and unable to be altered by the examiner applying normal strength to move the affected body part). Fixed postures, with or without pain, are often psychogenic in origin (Fahn and Williams, 1988; Lang, 1995; Sa et al., 2003; Schrag et al., 2004), although there may be organic cases. Transcranial magnetic stimulation studies revealed similar physiologic alterations in the cerebral cortex as seen in patients with mobile dystonia (Avanzino et al., 2008), but as Espay and colleagues (2006) have shown, and as discussed in Chapter 25, the changes in the brain can be secondary to the abnormal psychogenic postures or sensory abnormalities.
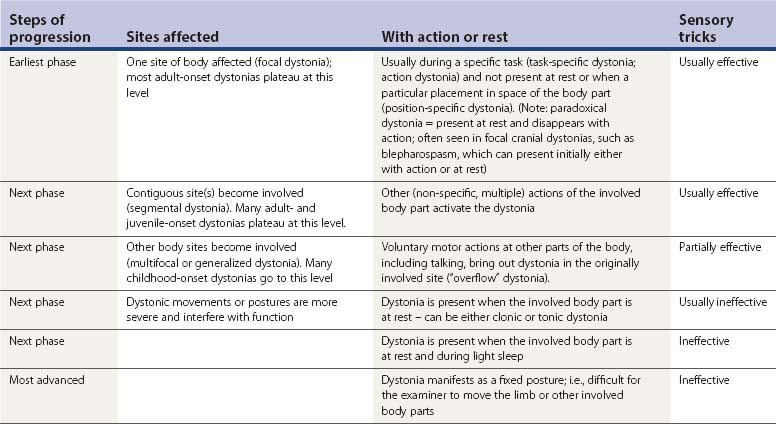
Much less common than action dystonia or overflow dystonia is the reverse phenomenon, i.e., for dystonia at rest to be improved by talking or by other voluntary active movements, so-called paradoxical dystonia (Fahn, 1989c). With paradoxical dystonia the patient is usually observed moving the affected or non-affected body part. The patient does this to obtain relief of the dystonia. When the paradoxical dystonia involves the trunk, the observer can easily mistake the patient’s moving about as being due to restlessness or akathisia, which is the most common differential diagnosis (Video 12.7). The focal dystonia that is most commonly decreased by voluntary motor activity is blepharospasm (Fahn et al., 1985). About 60% of patients with blepharospasm obtain relief when talking; about 40% worsen with talking. 
Dystonia usually is present continually throughout the day whenever the affected body part is in use; and as a sign of more severity, also when the body part is at rest. A notable phenomenologic feature of DRD is that of diurnal variation in many of the patients, with the dystonia being worse at the end of the day, and minimal in the morning hours (Segawa et al., 1976). Dystonic movements tend to increase with fatigue, stress, and emotional states; they tend to be suppressed with relaxation, hypnosis, and sleep (Fish et al., 1991). Dystonia may be precipitated or exacerbated by pregnancy (dystonia gravidarum) (Lim et al., 2006). Unless it is extremely severe, dystonia often disappears with deep sleep. For patients who appear to have persistent postural abnormalities that cannot be overcome by manual manipulation, it might be necessary to put them to sleep with anesthesia to determine whether a contracture is already present (Fahn, 2006). Propofol anesthesia, however, does not entirely suppress dystonia because recurrences occur even under deep propofol anesthesia (Zabani and Vaghadia, 1996).
One of the characteristic and almost unique features of dystonic movements is that they can often be diminished by tactile or proprioceptive “sensory tricks” (geste antagoniste). Thus, touching the involved body part or an adjacent body part can often reduce the muscle contractions (Video 12.8). For example, patients with torticollis will often place a hand on the chin or side of the face to reduce nuchal contractions, and orolingual dystonia is often helped by touching the lips or placing an object in the mouth (Blunt et al., 1994; Lo et al., 2007). In a study of 50 patients with cervical dystonia who were known to have at least one sensory trick, 54% of them had two to five different tricks and 82% had a reduction of head deviation by at least 30%, with a mean of 60% (Muller et al., 2001). In cervical dystonia, applying the trick when the head is in a neutral or even contralateral position was most effective, while no reduction of muscle activity occurs during trick application at the maximum dystonic head position (Schramm et al., 2004). Sometimes a mechanical device can be utilized therapeutically, especially for cervical dystonia (Krack et al., 1998) (Video 12.9). Greene and Bressman (1998) found that in some patients with torsion dystonia, simply thinking about a sensory trick or task affects the dystonia in the same way as actually performing the activity. A positron emission tomography (PET) study has shown that the sensory trick brings about a normalization of the abnormal cortical physiology that is seen in dystonia, and results in increasing activation of the ipsilateral superior and inferior parietal lobule and decreasing activity of the contralateral supplementary motor area and the primary sensorimotor cortex (Naumann et al., 2000). The presence of a sensory trick is not specific for primary dystonias; it can sometimes occur in secondary dystonias, including psychogenic dystonia (Munhoz and Lang, 2004). 
Pain is uncommon in dystonia except in cervical dystonia; 75% of patients with cervical dystonia (spasmodic torticollis) have pain (Chan et al., 1991). The pain perception threshold appears to be lower in patients with primary cervical dystonia (Lobbezoo et al., 1996). Dystonia in most parts of the body rarely is accompanied by pain; when it is, it is not clear whether the pain is due to painful contractions of muscles or some other factor. The high incidence of pain in cervical dystonia appears to be due to muscle contractions because this pain is usually relieved by injections of botulinum toxin (Greene et al., 1990). It is believed that the posterior cervical muscles are rich in pain fibers, and that continual contractions of these muscles results in pain. On the other hand, because no correlation was found between the severity of motor signs and pain, some investigators hypothesize that central mechanisms are also involved (Kutvonen et al., 1997). Quality of life is negatively affected with cervical dystonia (Camfield et al., 2002). It has been difficult to explain fixed painful postural torticollis following trauma, but recent analysis indicates that many of these cases appear to be psychogenic (Sa et al., 2003). Fixed dystonia in other parts of the body is also usually associated with a peripheral injury and overlaps with chronic regional pain syndrome (reflex sympathetic dystrophy); many of these individuals fulfill strict criteria for a somatoform disorder or psychogenic dystonia (Schrag et al., 2004).
Patients with PTD sometimes have rhythmical movements, particularly in the arms (Video 12.10) and neck, manifested as a tremor (Yanagisawa et al., 1972; Jankovic and Fahn, 1980). In one survey, 68% of patients with cervical dystonia had head tremor (Pal et al., 2000). Two basic types of tremors are seen in dystonic patients: (1) an accompanying postural/action tremor of the upper limbs that resembles essential tremor or enhanced physiologic tremor and (2) a tremor that is a rhythmic expression of rapid dystonic movements (Yanagisawa and Goto, 1971); these occur at the site of the tremor, such as arms, neck, or jaw (Schneider and Bhatia, 2007). The latter can usually be distinguished from the former by showing that the tremor appears only when the affected body part is placed in a position of opposition to the major direction of pulling by the abnormal dystonic contractions and disappears when the body part is positioned where the dystonia wants to place it. Dystonic tremor appears to be less regular than essential tremor (Jedynak et al., 1991). Tremor of the hands in patients with cervical dystonia tends to be irregular and therefore dystonic, rather than an accompanying essential tremor (Shaikh et al., 2008). Sometimes, it is very difficult to distinguish between the two types, particularly with writing tremor and cervical tremor. Primary writing tremor can sometimes represent task-specific dystonia or task-specific essential tremor (Cohen et al., 1987; Rosenbaum and Jankovic, 1988; Elble et al., 1990). A family history of tremor (and stuttering) is increased in PTD (Fletcher et al., 1991a). 
Although accompanying essential tremor is recognized in patients with dystonia (Lou and Jankovic, 1991), there is uncertainty as to how common this occurrence is. Tremor of the hands can be seen fairly often in patients with cervical dystonia (spasmodic torticollis) (Couch, 1976). Deuschl and colleagues (1997) analyzed this tremor in 55 patients with cervical dystonia. The mean amplitudes of postural tremor were only slightly higher than those of the controls and much smaller than those found in classic essential tremor; analytic measurements showed evidence of physiologic tremor mechanisms only. In another study, arm tremor in patients with cervical dystonia was found to develop either before or simultaneously with onset of the torticollis; such temporal relationships do not correspond to either dystonic tremor or tremor in the presence of dystonia (Munchau et al., 2001c).
Tics are another type of involuntary movement that appears to occur more commonly in patients with dystonia than in the general population (Shale et al., 1986; Stone and Jankovic, 1991; Damasio et al., 2011).
Although this is rare, some children and adolescents with primary and secondary dystonia can develop a crisis of sudden marked increase in the severity of dystonia, which has been called dystonic storm (Dalvi et al., 1998) and status dystonicus (Manji et al., 1998). It can cause rhabdomyolysis and myoglobinuria, with a threat of death by renal failure (Jankovic and Penn, 1982; Paret et al., 1995). Placing the patient in an intensive care unit and narcotizing him or her with barbiturates is usually necessary to treat this crisis. Intrathecal baclofen might be necessary if the dystonic storm persists and continues to be present when the patient is allowed to awaken (Jankovic and Penn, 1982; Dalvi et al., 1998). More recently, pallidotomy or pallidal stimulation has been utilized in place of intrathecal baclofen.
A case of orthostatic hemidystonia has been reported (Sethi et al., 2002). The patient developed hemidystonia on rising from the sitting position. It was due to poor vascular perfusion in the contralateral frontoparietal cortex and was the result of occlusion of the contralateral internal carotid artery and near-total occlusion of the ipsilateral internal carotid artery.
Dystonia patients are relatively free of psychopathology, as measured in patients with writer’s cramp (Sheehy and Marsden, 1982) and blepharospasm (Scheidt et al., 1996). However, a recent study reports a prevalence of obsessive-compulsive disorder in 20% of patients with primary focal dystonias (Cavallaro et al., 2002). Attentional-executive cognitive deficits have been found in patients with primary dystonia by using the Cambridge Neuropsychological Test Automated Battery (Scott et al., 2003), but not by using a host of other tests (Jahanshahi et al., 2003). Obsessive-compulsive disorder and alcohol dependence are not uncommon in individuals who carry the DYT11 gene for myoclonus–dystonia (Saunders-Pullman et al., 2002), and the DYT1 gene has been associated with an increase in depression, whether the person is a manifesting or nonmanifesting carrier (Heiman et al., 2004).
Site of body involvement is characteristic for many types of dystonia. Children usually have onset equally in a leg or an arm, at least for Oppenheim dystonia. Adult-onset dystonia is typically in the upper body, cranial, cervical, or brachial. A few adult patients may have focal truncal dystonia. Foot onset in adults is uncommon, but does occur (Schneider et al., 2006b). Isolated foot dystonia following exercise in adults is sometimes seen as the first symptom of Parkinson disease. This has now been verified in a patient with neuroimaging by using dopamine transporter single photon emission computed tomography (SPECT). Childhood-onset dystonia that begins in the cranial region is usually non-DYT1 dystonia (Bressman et al., 2000; Fasano et al., 2006). Dystonia may be the presenting or dominant feature of many parkinsonian disorders besides Parkinson disease (Jankovic, 2005; Ashour and Jankovic, 2006). Sometimes dystonia involving the foot can resemble a Babinski sign. This pseudo-Babinski (striatal toe) can be a dystonic phenomenon and should be differentiated from a true Babinski sign seen with pyramidal tract lesions (Horstink et al., 2007).
Epidemiology
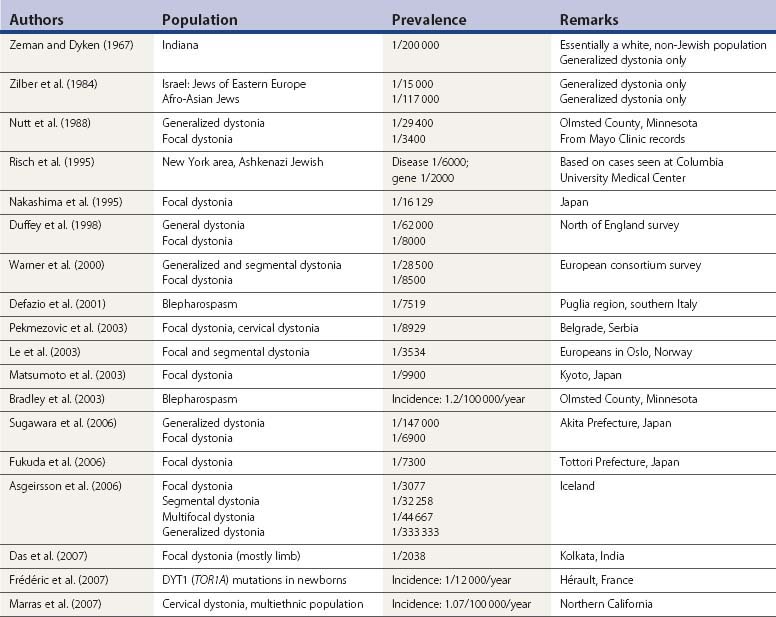
Zeman and his colleagues (Zeman and Dyken, 1967; Zeman et al., 1959, 1960) carried out the first epidemiologic study in dystonia in the population of the state of Indiana and emphasized the autosomal dominant pattern of inheritance in PTD. They considered only generalized dystonia to be PTD, and viewed other types as formes frustes. Today, those other forms are viewed as focal and segmental dystonia, and part of the spectrum of PTD. An epidemiologic study of PTD in the population living in Rochester, Minnesota, found the prevalence of generalized PTD to be 3.4 per 100 000 population, and the prevalence of focal dystonia to be 30 per 100 000 (Nutt et al., 1988). In a study of dystonia in Israel, Zilber and colleagues (1984) estimated the prevalence of generalized dystonia among Jews of Eastern European ancestry to be 1/15 000 or 6.8/100 000, which is double the prevalence in the general population of Rochester. However, the analysis by Risch and colleagues (1995) indicates that the frequency in the Ashkenazim is much higher (between 1/6000 and 1/2000), and they suggest that the Ashkenazi population with PTD descends from a limited group of founders of the DYT1 mutation. These investigators also have traced the origin of the mutation to the northern part of the historic Jewish Pale of settlement (Lithuania and Byelorussia), approximately 350 years ago. In Japan, the DYT1 mutation was looked for in 178 patients with various forms of dystonia and was found in 6 (3.4%) (Matsumoto et al., 2001) and phenotypically resembled Oppenheim dystonia seen in other populations.
In Japan the prevalence rate of focal dystonias was found to be 6.12 (Nakashima et al., 1995), 10.1 (Matsumoto et al., 2003), 13.7 (Fukuda et al., 2006), and 14.4 (Sugawara et al., 2006) per 100 000 population, all of which is considerably lower than the 30/100 000 found by Nutt et al. (1988) in Rochester, Minnesota. The prevalence of focal limb dystonia in India was higher, 49.06/100 000 (Das et al., 2007). In the north of England, the prevalence of focal dystonias was found to be 12 per 100 000, and the prevalence of generalized dystonia was found to be 1.6 per 100 000 (Duffey et al., 1998). A European consortium of investigators published their findings of 11.7 per 100 000 for focal dystonia and 3.5 per 100 000 for segmental and generalized primary dystonias (Warner et al., 2000). A survey of primary blepharospasm in the Puglia region of southern Italy found a prevalence of only 13.3 per 100 000 (Defazio et al., 2001). The incidence rate of primary blepharospasm was found to be 1.2 per 100 000 population per year in Olmsted County, Minnesota (Bradley et al., 2003). Incidence for cervical dystonia was higher in white individuals (1.23/100 000/year) than in persons of other ethnicities (0.15/100 000/year) (Marras et al., 2007).
In Belgrade, the prevalence rate for focal, segmental, and multifocal dystonia was 13.6 per 100 000 population (Pekmezovic et al., 2003). It was almost twice as common (25.4 per 100 000 population) in Oslo (Le et al., 2003). Gender appears to play a role in both the prevalence and the age at onset of focal dystonia, women being more at risk and having an earlier age at onset for writer’s cramp, but men having an earlier age at onset for cervical dystonia, blepharospasm, and laryngeal dystonia (Epidemiologic Study of Dystonia in Europe (ESDE) Collaborative Group, 1999; Marras et al., 2007).
Table 12.4 lists epidemiologic studies of PTD.
Classification of torsion dystonia
Table 12.5 presents the three ways to classify patients with torsion dystonia: by age at onset, by body distribution of abnormal movements, and by etiology. This method allows physicians and health-care providers some understanding of the nature of the dystonia, including prognosis.
| 1. By age at onset |
Classification by age at onset
Classification by age at onset is useful because this is the most important single factor related to prognosis of primary dystonia (Marsden et al., 1976; Fahn, 1986; Greene et al., 1995). Even for secondary dystonias, such as tardive dystonia, age is commonly a factor in the location of the dystonia. As a general rule, the younger the age at onset, the more likely it is that the dystonia will become severe and will spread to involve multiple parts of the body. In contrast, the older the age at onset, the more likely it is that the dystonia will remain focal. Onset of dystonia in a leg is the second most important predictive factor (Greene et al., 1995) (Video 12.11). Because a bimodal age distribution is seen with primary dystonia, the age classification consists of two categories: (1) age 26 years or below and (2) above age 26 (Bressman, 2004). 
Classification by distribution
Since dystonia usually begins by affecting a single part of the body (focal dystonia) and since dystonia can either remain focal or spread to involve other body parts, it is useful to classify dystonia according to its distribution of involvement of the body. Body distribution is one method of defining the severity of dystonia, and knowing the body distribution of dystonia is very important in planning a therapeutic strategy (Fahn, 1995).
Focal dystonia indicates that only a single area of the body is affected. Frequently seen types of focal dystonia tend to have specific labels, such as blepharospasm, torticollis, oromandibular dystonia, spastic dysphonia, writer’s cramp, and occupational cramp. Adult-onset focal dystonias are much more common than generalized dystonias (Fahn, 1986; Marsden, 1986) (Table 12.6). If dystonia spreads, it most commonly does so by next affecting a contiguous body part. When dystonia affects two or more contiguous parts of the body, it is referred to as segmental dystonia. Some of the primary focal dystonias, such as blepharospasm and cervical dystonia, affect females more than males, while the reverse is seen for writer’s cramp (Soland et al., 1996a).
| No. | Percentage | |
|---|---|---|
| Focal | 1230 | 50 |
| Segmental | 837 | 34 |
| Generalized | 383 | 16 |
| 2450 | 100 |
Data from the Center for Parkinson Disease and Other Movement Disorders, Columbia University Medical Center, New York City.
Generalized dystonia is defined as representing a combination of segmental crural dystonia (i.e., both legs or legs plus trunk) plus involvement of any other area of the body (Videos 12.12, 12.13, 12.14, and 12.15). The term multifocal dystonia fills a gap in the above designations. It applies to the involvement of two or more non-contiguous parts of the body. Dystonia affecting one-half of the body is called hemidystonia. Almost always, hemidystonia indicates that the dystonia is symptomatic rather than primary (Narbona et al., 1984; Marsden et al., 1985; Pettigrew and Jankovic, 1985). A review of hemidystonia (Chuang et al., 2002) found that the most common etiologies were stroke, trauma, and perinatal injury; the mean age of onset was between 20 and 26 years; the mean latency from insult to dystonia was between 2.8 and 4.1 years, the longest latencies occurring after perinatal injury; and basal ganglia lesions were identified in 48–60% of the cases, most commonly involving the putamen. When there is a delay by a few years between time of insult and time of onset of dystonia, the condition is named delayed-onset dystonia (Burke et al., 1980). 
Detailed clinical descriptions as to how dystonia manifests itself in the different regions of the body have been summarized by Fahn (1984a). The most common primary focal dystonia seen in a movement disorder clinic is cervical dystonia (torticollis), followed by dystonias affecting cranial musculature, such as blepharospasm and spasmodic dysphonia (Table 12.7). The most common primary segmental dystonia involves the cranial structures, and these are commonly referred to as cranial-cervical dystonia and sometimes as Meige syndrome (Tolosa and Klawans, 1979; Tolosa et al., 1988a) (Table 12.8). Details of cervical dystonia have been reviewed (Dauer et al., 1998). Primary axial dystonia that presents in adulthood is much less common than the cranial-cervical dystonias. In 9 of the 18 patients collected by Bhatia and colleagues (1997), onset was in the back, with the other half spreading to the back from the cranial-cervical region. Probably because of the age at onset, it does not spread to the legs. About one-third of their patients improved with high-dose anticholinergics with or without antidopaminergics.
Table 12.7 Distribution of focal dystonias
| Type of dystonia | No. | Percentage |
|---|---|---|
| Torticollis | 447 | 44.4 |
| Spasmodic dysphonia | 257 | 25.5 |
| Blepharospasm | 140 | 13.9 |
| Right arm | 96 | 9.5 |
| Oromandibular | 31 | 3.1 |
| Left arm | 20 | 2.0 |
| Left leg | 6 | 0.6 |
| Trunk | 5 | 0.5 |
| Right leg | 4 | 0.4 |
| 1006 | 100 |
Data from the Center for Parkinson Disease and Other Movement Disorders, Columbia University Medical Center, New York City.
Table 12.8 Distribution of segmental dystonias
| Type of dystonia | No. | Percentage |
|---|---|---|
| Segmental cranial | 167 | 42.8 |
| Cranial + brachial | 56 | 14.4 |
| Cranial + axial | 14 | 3.6 |
| Segmental brachial | 83 | 21.3 |
| Segmental axial | 31 | 7.9 |
| Segmental crural | 13 | 3.3 |
| Multifocal | 26 | 6.7 |
| 390 | 100 |
Data from the Center for Parkinson Disease and Other Movement Disorders, Columbia University Medical Center, New York City.
Classification by etiology
Awareness of etiology is an ultimate aim in the clinical evaluation of dystonia, not only for treatment and genetic counseling, but also because it should lead to understanding the pathophysiology of the illness and how to prevent dystonia. The etiologic classification (Fahn et al., 1998a) divides the causes of dystonia into five major categories: primary (or idiopathic), secondary (environmental causes) (or symptomatic), dystonia-plus syndromes, heredodegenerative diseases in which dystonia is a prominent feature, and the presence of dystonia as a feature of another neurologic disease (Table 12.9).
Table 12.9 Five categories of the etiologic classification of dystonia
Primary dystonia is characterized as a pure dystonia (with the exception that tremor can be present) and excludes a symptomatic cause. Dystonia-plus syndromes, such as DRD and myoclonus–dystonia, were previously considered to be variants of primary dystonia (Fahn, 1989c). But because symptoms and signs other than dystonia, namely, parkinsonism and myoclonus, respectively, are present, these entities are placed in the dystonia-plus category. Another concept is that dystonia-plus syndromes, like the primary dystonias, are not neurodegenerative disorders, but neurochemical ones (Fahn et al., 1998a). Secondary dystonias are those due to environmental insult, while heredodegenerative dystonias are due to neurodegenerative diseases that are usually inherited.
Primary dystonia
Primary dystonia consists of familial and nonfamilial (sporadic) types. Although most patients with torsion dystonia have a negative family history for this disorder, the presence of other affected family members allows the family to be investigated in terms of localizing the abnormal gene(s) for dystonia. In primary dystonia, the only neurologic abnormality is the presence of dystonic postures and movements, with the exception of tremor that can resemble essential tremor and can even be essential tremor in some individuals. There is no associated loss of postural reflexes, amyotrophy, weakness, spasticity, ataxia, reflex change, abnormality of eye movements, disorder of retina, dementia, or seizures except where they may be the result of a concomitant problem such as a complication from a neurosurgical procedure undertaken to correct the dystonia, or the presence of some other incidental neurologic disease. Since many of the secondary dystonias have these neurologic findings, the presence of any of these findings in a patient with dystonia immediately suggests that one is dealing with a secondary dystonia, a dystonia-plus syndrome, or a heredodegenerative disorder (Table 12.9). However, the absence of such neurologic findings does not necessarily exclude the possibility of a secondary dystonia, which may rarely present as a pure dystonia.
As discussed above in the phenomenology section, tremor in the primary dystonias may be due to a dystonic tremor (Fahn, 1984b) that results from rhythmic group action potentials that occur in dystonia (Yanagisawa and Goto, 1971). It remains to be elucidated whether tremor that mimics essential tremor and enhanced physiologic tremor that is seen in many patients with dystonia as well as in members of their families (Zeman et al., 1960; Yanagisawa et al., 1972; Couch, 1976; Deuschl et al., 1997) is actually a component of idiopathic dystonia (i.e., dystonic tremor). Hand tremor in patients with cervical dystonia more closely resembles enhanced physiologic tremor than dystonic tremor or essential tremor (Deuschl et al., 1997).
Within the primary dystonias are a variety of genetic disorders, some of which have had their genes already mapped to DYT1, DYT2, DYT4, DYT6, DYT7, and DYT13. Other familial primary dystonias have not yet been mapped. But most primary dystonias are sporadic and with an onset during adulthood, usually presenting as one of a variety of focal and segmental dystonias (Tables 12.7 and 12.8); these are discussed below. DYT1 (Oppenheim dystonia) is discussed in more detail later in the chapter. Here we comment on some of the other primary familial dystonias listed in Table 12.10.
Table 12.10 Detailed etiologic classification of the dystonias
Mutations in Aristaless related homeobox gene, ARX (Stromme et al., 2002)
Adapted from Fahn S, Bressman SB, Marsden CD. Classification of dystonia. In: Fahn S, Marsden CD, DeLong MR, eds. Dystonia 3. Advances in Neurology vol. 78. Lippincott-Raven, Philadelphia, 1998, pp. 1–10, with more recent additions.
In the Mennonite and Amish populations, a mixed type of autosomal dominant dystonia has been seen in which onset can be either in childhood or adulthood, with involvement of limbs and the cervical and cranial regions and designated DYT6. Dysphonia and dysarthria are often the most disabling features, and when present with segmental or generalized dystonia, one should suspect the possibility that the disorder may be DYT6. The mutation was found on the THAP1 gene on 8p11.21 (Fuchs et al., 2009). Since this gene discovery, the THAP1 mutation has been found in populations other than the Mennonite/Amish sect.
Several families with adult-onset familial torticollis have been reported, with one of them (Family K in northwest Germany) mapped to chromosome 18p, this locus being designated DYT7 (Leube et al., 1996). Investigation of more families and of apparently sporadic cases of torticollis from this region showed that most have inherited the same mutation as Family K from a common ancestor and, in fact, owe their disease to autosomal dominant inheritance at low penetrance (Leube et al., 1997a, 1997b). However, subsequent information from these authors now questions whether their findings are incorrect (Leube and Auburger, 1998; Klein et al., 1998b). Other families with torticollis have been excluded from the chromosome 18p region and from DYT1 (Bressman et al., 1996; Cassetta et al., 1999; Jarman et al., 1999).
Cervical-cranial predominant dystonia is another form of autosomal dominant primary dystonia; it has been seen in non-Jewish families that do not link to DYT1 (Bressman et al., 1994b, 1996; Bentivoglio et al., 1997). The site of onset is usually in the neck, which continues to dominate, but dystonia often spreads to involve the cranial structures as well, and occasionally the arm. Onset may be in childhood (Bentivoglio et al., 1997) or adulthood (Bressman et al., 1994b). An autosomal dominant Italian family described by Bentivoglio and colleagues (1997, 2004) with predominantly early onset with cervical-cranial or brachial dystonia has been mapped to chromosome1p36.13–p36.32 and has been categorized as DYT13 (Valente et al., 2001).
Two unrelated consanguineous Brazilian families with early-onset dystonia–parkinsonism were found to have a mutation in the PRKRA gene for protein kinase, interferon-inducible double-stranded RNA-dependent activator (Camargos et al., 2008). Onset began with abnormal gait and leg pain around age 12 years. Later the upper body became involved with dysphagia, spasmodic dysphonia, torticollis, upper limb dystonia, and opisthotonus. Orofacial dystonia and facial grimacing were prominent features. Some patients also had bradykinesia, one with tremor. Subsequently, a deletion in this gene was discovered in a German boy, whose dystonia began in the legs and became generalized (Seibler et al., 2008).
Adult-onset focal dystonias
These are the most common forms of dystonias. Their appearance varies depending on which body part is affected. Although most cases remain focal, there can be contiguous spread to a neighboring segment, thus becoming segmental myoclonus. Seemingly distinct from each other (Table 12.11), the focal dystonias do have some overlap in that when they spread, they affect a contiguous body part and therefore exist in the same patient. This observation and some shared physiological changes have suggested to Defazio and his colleagues (2007) that the different focal dystonias probably share common genetic factors in a multifactorial disease.
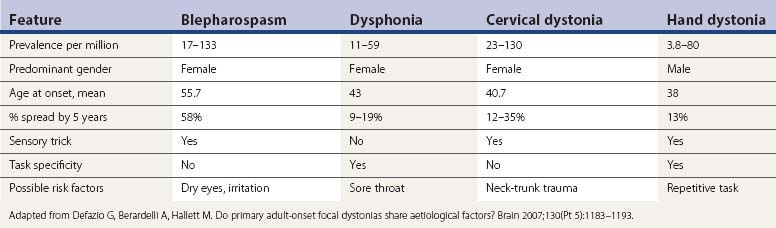
The most common focal dystonia involves the neck musculature, known as spasmodic torticollis or cervical dystonia (Table 12.7). The head can turn (rotational torticollis), tilt, or shift to one side, or bend forward (antecollis) or backwards (retrocollis). Any combination of head positions can be found in individual patients. About 10% have a remission within a year, but relapses usually occur, even many years later (Friedman and Fahn, 1986; Lowenstein and Aminoff, 1988; Jahanshahi et al., 1990; Chan et al., 1991). The average age at onset is between 20 and 50 years. The muscles involved are innervated by cranial nerve (CN) XI and the upper cervical nerve roots. Some cervical dystonias are manifested as a static pulling of the head into one direction, but most have a jerky, irregular rhythmic feature. Some patients try to fight the pulling of the neck muscles and the physician can be misled by seeing or feeling contracted muscles, thinking that these are the dystonic muscles, when in fact they could be the compensatory muscles contracting. To distinguish between involuntary and compensatory/voluntary contractions, the patient should be told to let the movements occur without trying to overcome them. Then the true direction of which muscles are involved by the dystonia is revealed. This is especially important when deciding which muscles to inject with botulinum toxin. Hypermetabolism of dystonic muscles was detected by fluorodeoxyglucose (FDG) PET (Sung et al., 2007), but it is not certain that a compensating muscle would not show the same pattern. Common sensory tricks to reduce dystonia are touching the face or the back of the head. The variety of sensory tricks have been enumerated (Ochudło et al., 2007). Mechanical devices to place cutaneous pressure on the occiput can sometimes be used to advantage. Pain in the neck muscles is common in cervical dystonia (Chan et al., 1991).
Blepharospasm usually occurs in older individuals, women more than men. It begins as excessive blinking, and many patients complain of eye irritation or dryness, although dry eyes from Sjögren disease are usually ruled out. This blinking phase then leads to some longer closing of the eyes, even to very long durations, which has been mistaken for weakness of the levator palpebrae (ptosis) instead of contraction of the orbicularis oculi. Usually there is a combination of eyelid closing and blinking. The muscles involved are the orbicularis oculi innervated by CN VII, and the contractions are symmetrical in the two eyes, quite distinct from hemifacial spasm, which is unilateral. Another interesting difference is that usually in hemifacial spasm the eyebrow may elevate due to simultaneous contraction of the frontalis muscles (known as “the other Babinski sign” – Devoize, 2001; Stamey and Jankovic, 2007), whereas in blepharospasm the eyebrows come down. Dystonia can spread from the upper face causing blepharospasm, to the lower face with movements around the mouth. Common sensory tricks to reduce blepharospasm are touching the corner of the eye, coughing, and talking. Bright light notoriously aggravates blepharospasm and patients have difficulty being in sunlight or bright light and they often wear sunglasses most of the time. Driving at night is very difficult because of oncoming headlights. Blepharospasm tends to spread from upper face to involve lower face, and may spread to involve muscles innervated by CN V, tongue, and neck (Fahn, 1985). Spread of blepharospasm is more common than with other adult-onset dystonias (Svetel et al., 2007; Abbruzzese et al., 2008). Blepharospasm needs to be differentiated from so-called apraxia of eyelid opening in which the lids fail to open due to either “freezing,” levator inhibition, or dystonia, and is often seen in atypical parkinsonian disorders, such as progressive supranuclear palsy. Injection of botulinum toxin into the Riolan muscle can be effective in treating lid “apraxia,” believed to be analogous to a sensory trick (Inoue and Rogers, 2007). About 4% of patients with blepharospasm also have eyelid-opening apraxia (Peckham et al., 2011).
Ormandibular dystonia (OMD) (jaw muscles innervated by CN V) is often associated with lingual dystonia (tongue muscles by CN XII), and is less frequent than blepharospasm. Jaw-opening dystonia is where the jaw is pulled down by the pterygoids; in jaw-clenching dystonia the masseters and temporalis muscles are the prime movers. The jaw can also be moved laterally. It is important to distinguish the latter from facial muscle pulling the mouth to one side, which is often due to psychogenic etiology. OMD can markedly affect chewing and swallowing. Some OMDs appear only with action and are not present at rest. Such actions can involve talking or chewing. Often a patient attempts to overcome jaw-opening and jaw-closing dystonias by purposefully moving the jaw in the opposite direction. This maneuvering has often led to misdiagnosis of tardive dyskinesia because the movements superficially appear rhythmic. To distinguish between OMD and tardive dyskinesia, the patient should be told to let the movements occur without trying to overcome them. In this manner, the true direction of where the dystonia wants to take the jaw is revealed, and the rhythmic movements stop in OMD. Sensory tricks that have been useful are the placing of objects in the mouth or biting down on an object, such as a tongue blade or pencil. Dental implants have sometimes helped by the physical application of a continual sensory trick. Pure lingual dystonia, with protrusion of the tongue, can affect speech and sometimes swallowing and breathing (Schneider et al., 2006a).
Dystonia-plus syndromes
A disorder that is analogous to DRD is dopamine agonist-responsive dystonia, which is considered here briefly. It was first described by Hyland and colleagues (1992) (Video 12.16) and a second family was described by Maller and colleagues (1997). It is due to an autosomal recessive disorder beginning in the first few months of life with hypotonia, hypokinesia, and developmental delay. Eventually, the patient experiences autonomic dysfunction (hyperhidrosis, miosis, and ptosis), dystonia–parkinsonism, episodes of oculogyria, and other paroxysmal movements and bouts of deep sleep (Hyland et al., 1992; Pons et al., 2004). It is the result of reduced activity of aromatic L-amino acid decarboxylase (AADC), so there is reduced metabolism of dopa to dopamine and 5-hydroxytryptophan to serotonin (Hyland et al., 1992). There is reduced concentration of the metabolites of dopamine and serotonin in urine, namely, homovanillic acid and 5-hydroxyindoleacetic acid, respectively. AADC deficiency is one of the pediatric CSF neurotransmitter disorders (Patterson, 2010). The symptoms respond partially to dopamine agonists plus a monoamine oxidase inhibitor. Mild cases do occur. Two siblings presenting with fatigability, hypersomnolence, and dystonia had an excellent response to treatment with dopamine agonist and MAO inhibitor (Tay et al., 2007). The variability of response from excellent to almost none (Pons et al., 2004) is unexplained. The diagnosis depends on reduced CSF biogenic amines, metabolites and plasma AADC levels. CSF 3-O-methyldopa is elevated (Tay et al., 2007). Direct sequencing of the AADC gene should be performed. 
In a series of 78 patients with AADC deficiency (46 reported previously), symptoms (hypotonia 95%, oculogyric crises 86%), and developmental delay (63%) became clinically evident during infancy or childhood (Brun et al., 2010). A total of 24 mutations in the AADC gene were detected in that series. No autopsies have been performed, so there remains uncertainty whether this is a neurochemical rather than a neurodegenerative disease; but without an autopsy to determine this, it is placed now in the dystonia-plus category.
A similar clinical presentation occurs with several pterin disorders (Hyland et al., 1998). The autosomal recessive biopterin deficiency states that are listed with the dopa-responsive dystonias in Table 12.10 should be mentioned here because they also manifest features of decreased norepinephrine and serotonin in addition to dystonia and parkinsonism. In this way they more closely resemble the phenotype of aromatic amino acid decarboxylase deficiency. Their clinical features include miosis, oculogyria, rigidity, hypokinesia, chorea, myoclonus, seizures, temperature disturbance, and hypersalivation. The clinical syndrome can present at any age with generalized dystonia–parkinsonism and with marked diurnal fluctuation (Hanihara et al., 1997). These pterin enzyme deficiencies cause hyperphenylalaninemia, and they may respond partially to levodopa and 5-hydroxytryptophan. There are no neuropathologic observations, and they have arbitrarily been listed in the dystonia-plus syndrome category rather than as a neurodegeneration. But intellectual disability or delay is common. Dihydropteridine reductase deficiency can respond partially to levodopa without inducing dyskinesias despite long-term therapy (Sedel et al., 2006). Indicating this is a neurochemical disorder rather than a neurodegenerative one is the lack of dopaminergic cell loss as suggested by normal PET imaging of the dopamine transporter (Sedel et al., 2006). Sepiapterin reductase deficiency can be associated with hypersomnolence (Friedman et al., 2006), as seen also in aromatic decarboxylase deficiency syndrome.
In DYT16, onset begins with abnormal gait and leg pain around age 12 years (Camargos et al., 2008). Later the upper body becomes involved with dysphagia, spasmodic dysphonia torticollis, upper limb dystonia, and opisthotonus. Orofacial dystonia and facial grimacing are prominent features. Some patients also had bradykinesia, one with tremor. Subsequently, a deletion in this gene was discovered in a German boy, whose dystonia began in the legs and became generalized (Seibler et al., 2008).
Secondary dystonia and heredodegenerative diseases
Secondary dystonias
The secondary dystonias are subdivided into several categories. Those that are due to environmental causes are mainly due to lesions causing structural brain damage, and many result in hemidystonia. See the discussion of hemidystonia (almost always secondary dystonia) in the section “Classification by distribution.” But the secondary dystonias also include psychogenic dystonia (Fahn et al., 1987; Calne and Lang, 1988; Fahn and Williams, 1988; Table 12.10). The heredodegenerative disorders are those conditions associated with various hereditary neurologic disorders, and those in which neuronal degeneration is present. Most secondary dystonias due to structural lesions in the brain are due to insults to the basal ganglia, particularly the putamen, or its afferent and efferent connections, but brainstem lesions can also result in dystonia (Loher and Krauss, 2009).
Clinicians have recognized that dystonia could become worse if an affected limb is casted and immobilized. Okun and colleagues (2002) reported on four patients who developed segmental dystonia following removal of a cast, which the authors attribute to the trauma of prolonged immobilization (Okun et al., 2002).
Dystonia is perhaps the most common movement disorder (often with hypokinesia seen with disorders causing striatal necrosis). Some of these causes are Wilson disease, toxins, metabolic acidosis (such as from 3-oxothiolase deficiency, propionic acidemia, methylmalonic acidemia, isovaleric acidemia, and glutaric adiduria type 1), HIV and other infections, Leigh disease and other mitochondrial encephalopathies, anoxia, wasp sting encephalopathy (Leopold et al., 1999), hemolytic uremia, vascular disease, and head trauma.
A major portion of the clinical investigation of dystonia (Fahn et al., 1987) concerns the tests that are required to uncover the etiology of secondary and heredodegenerative dystonias. Almost yearly, new etiologies of these types of dystonia are reported. These include mildewed sugar cane ingestion containing the mitochondrial toxin 3-nitroproprionic acid (Ludolph et al., 1992), toxoplasmosis (in AIDS) (Tolge and Factor, 1991), disulfiram intoxication (Krauss et al., 1991), Creutzfeldt–Jakob disease (Sethi and Hess, 1991; Hellmann and Melamed, 2002), primary antiphospholipid syndrome (Angelini et al., 1993), spinal cord lesions (Uncini et al., 1994; Madhusudanan et al., 1995), lumbar canal stenosis resulting in foot dystonia on standing or walking (Blunt et al., 1996), ataxia telangiectasia (Koepp et al., 1994), alternating hemiplegia of childhood (Andermann et al., 1994), organophosphorus insecticide poisoning (Senanayake and Sanmuganathan, 1995), pure thalamic degeneration (Yamamoto and Yamashita, 1995), midbrain hemorrhage (Munoz et al., 1996), bilateral lesions of the mesencephalon and vermis (Rousseaux et al., 1996), posterior fossa tumors (Krauss et al., 1997), electrical injury (Adler and Caviness, 1997), intracerebral arteritis from herpes zoster ophthalmicus (Burbaud et al., 1997), childhood-onset (Uc et al., 2000) or adult-onset Niemann–Pick type C disease (Lossos et al., 1997), adult GM1 gangliosidosis (Kobayashi and Suzuki, 1981; Hirayama et al., 1997; Campdelacreu et al., 2002), optic glioma (Vandertop et al., 1997), neuroferritinopathy (Curtis et al., 2001; Mir et al., 2005; Chinnery et al., 2007), dystonia in spinocerebellar ataxia type 1 (Wu et al., 2004) and in type 6 (Sethi and Jankovic, 2002), familial dystonia associated with cerebellar atrophy (Le Ber et al., 2006), hand dystonia due to neurofibromatosis type 1 (Di Capua et al., 2001), striatal necrosis-induced dystonia with acidosis from 3-oxothiolase deficiency (Yalcinkaya et al., 2001), and striatal necrosis-induced dystonia from hereditary biotin deficiency (Debs et al., 2010). From HIV, there can be dystonia from striatal necrosis (Abbruzzese et al., 1990) or from secondary infections, such as toxoplasmosis (Tolge and Factor, 1991). Homocystinuria can result in recurrent dystonia (Sinclair et al., 2006). It is due to cystathionine β-synthase deficiency. Two biochemical markers for homocystinuria, homocystine and methionine, were markedly elevated during periods when dystonia is manifested. In Lesch–Nyhan disease, although originally described as a choreathetotic disorder, when more carefully analyzed, the major movement disorder is dystonia (Jinnah et al., 2006).
Onset of dystonia and chorea of the limbs in adolescence and associated with alopecia, hypogonadism, diabetes mellitus, intellectual disability and sensory neural deafness is known as the Woodhouse–Sakati syndrome (Schneider and Bhatia, 2008). It is an autosomal recessive disorder.
Dystonia can present in patients with pyruvate dehydrogenase deficiency (Head et al., 2004). The main clue to the biochemical diagnosis is a raised concentration of lactate in the cerebrospinal fluid. These patients can respond to levodopa.
If dystonia occurs in the first year of life, the leading cause is cerebral palsy or a metabolic error, such as glutaric aciduria (Kyllerman et al., 1994). A number of cases of symptomatic dystonias, so-called delayed-onset dystonia, appear months to years after the cerebral insult (Burke et al., 1980). Often such a delayed onset is seen with perinatal or early childhood asphyxia (Saint-Hilaire et al., 1991). In a series of long-term follow-up of patients with perinatal asphyxia, only about 1% develop delayed-onset dystonia (Cerovac et al., 2007). Delayed onset of dystonia can be seen also with central pontine myelinolysis (Tison et al., 1991; Maraganore et al., 1992), cyanide intoxication (Valenzuela et al., 1992), head trauma (Lee et al., 1994), and a variety of other static brain lesions (Scott and Jankovic, 1996). Ingestion causes striatal necrosis and coma, and the dystonia evolves as the patient comes out of the coma (He et al., 1995). Dystonia from severe head trauma is usually delayed. Of 221 patients who survived severe head trauma with a Glasgow Coma Score of ≤8, 4% later developed dystonia (Krauss et al., 1996). The dystonia appeared with a latency of 2 months to 2 years. Delayed-onset generalized dystonia due to cerebral anoxia can worsen over time, and show a delay in magnetic resonance imaging (MRI) changes in the globus pallidi (Kuoppamaki et al., 2002). The delayed-onset dystonia from the ingestion of mildewed sugar cane containing the Arthrinium-produced mycotoxin 3-nitroproprionic acid (3-NP) is not the same phenomenon. 3-NP is a mitochondrial toxin that irreversibly inhibits complex II (Beal, 1995).
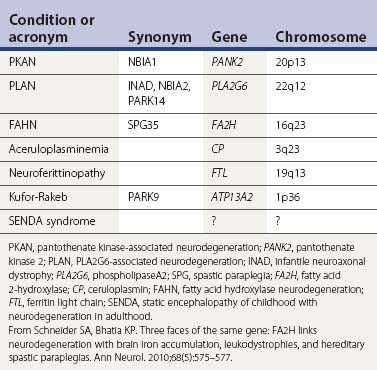
Neurodegenerations with brain iron accumulation (Table 12.12)
The gene for autosomal recessive neurodegeneration with brain iron accumulation type 1 (NBIA) (formerly known as Hallervorden–Spatz syndrome) has been identified as pantothenate kinase (PANK2) (Zhou et al., 2001; Hartig et al., 2006; Valentino et al., 2006). In 49 families with typical phenotype and MRI for NBIA, Hayflick and colleagues (2003) found that all had the PANK2 deficiency; in another 49 families with an atypical phenotype, only 17 had the enzyme deficiency. In another study, looking at 10 families with MRI positive for iron accumulation, only four were found to have a mutation on PANK2 (Thomas et al., 2004). The presence of a mutation in the PANK2 gene was associated with younger age at onset and a higher frequency of dystonia, dysarthria, intellectual impairment, and gait disturbance. Parkinsonism was seen predominantly in adult-onset patients whereas dystonia seemed to be more frequent in the earlier-onset cases. In the most comprehensive study, involving 72 patients with NBIA, 48 (67%) were found to have a PANK2 mutation (Hartig et al., 2006). No strict correlation between the “eye-of-the-tiger” sign and PANK2 mutations was found. Not all patients with PANK2 deficiency have the eye-of-the-tiger sign on MRI; there is a report of one child whose eye-of-the-tiger sign disappeared on the MRI scan over time (Baumeister et al., 2005). Although the “eye-of-the-tiger”sign is very characteristic of PANK2 mutation, it may also be seen in patients with other NBIA syndromes, such as neuroferritinopathy and aceruloplasminemia, and in corticobasal degeneration and progressive supranuclear palsy (Kumar et al., 2006).
Another iron-accumulating dystonia presenting as infantile neuroaxonal dystrophy (NBIA2) can also present in older individuals, including adult-onset dystonia–parkinsonism with pyramidal tract signs and ataxia. When it presents in adults, it can be without iron accumulation (Kurian et al., 2008; Paisan-Ruiz et al., 2009, 2010). It is due to mutations in a phospholipase gene, PLA2G6, and has been labeled PARK14 and as NBIA2, and also called PLAN. Its pathology can show widespread Lewy bodies and hyperphosphorylated tau (Paisan-Ruiz et al., 2010).
Neurodegeneration with brain iron accumulation type 3 is also called neuroferritinopathy. Chinnery and colleagues (2007) studied a large pedigree. Symptoms began in adulthood, and manifested first as chorea in 50%, lower limb dystonia in 42.5%, and parkinsonism in 7.5%. Serum ferritin levels were low in the majority of males and postmenopausal females, but within normal limits for premenopausal females. MR brain imaging was abnormal on all affected individuals and one presymptomatic carrier. A gradient echo brain MRI identified all symptomatic cases.
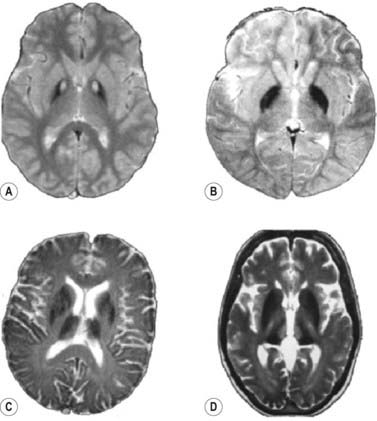
A fourth neurodegeneration with brain iron accumulation (NBIA4) is aceruloplasminemia, an autosomal recessive disorder affecting the ceruloplasmin gene. NBIA is also seen with a mutation in the fatty acid hydroxylase gene (FA2H), manifested with ataxia, spasticity, and dystonia and referred to as FAHN (Kruer et al., 2010). It was originally referred to as a leukodystrophy and as part of the hereditary spastic paraparesis syndromes (Edvardson et al., 2008; Dick et al., 2010). McNeill and colleagues (2008) evaluated gradient and fast spin echo MRI scans and showed that the pattern of hypointensities from accumulated iron and hyperintensities differ in the four subtypes of hereditary neurodegenerations with brain iron accumulation, namely pantothenate kinase-associated neurodegeneration (PKAN), neuroferritinopathy, infantile neuroaxonal dystrophy (INAD), and aceruloplasminemia (Table 12.13 and Fig. 12.1).

Other movement disorders in which dystonia can be present
There are some movement disorders, including Parkinson disease and Parkinson-plus syndromes, dystonic tics, and paroxysmal dyskinesias, in which dystonia is present, but that are not typically classified as dystonia (Table 12.14). Hypnogenic dystonia is commonly a manifestation of frontal lobe epilepsy (Sellal et al., 1991; Meierkord et al., 1992; Montagna, 1992) (see Chapter 22).
Table 12.14 Other movement disorders in which dystonia may be present
Pseudodystonia
There are other neurologic syndromes in which sustained abnormal postures may be present, but that are not considered true dystonias, hence are called pseudodystonia (Table 12.15). These include stiff-person syndrome, Isaacs syndrome, Satoyoshi syndrome (Merello et al., 1994), chronic inflammatory myopathy with involuntary complex repetitive discharges of muscle (Preston et al., 1996), and many others (Table 12.15). It has been found that congenital torticollis not only might be due to thickening and tightness of the sternomastoid muscle (labeled congenital muscular torticollis in Table 12.15), but is even more commonly associated with a palpable sternomastoid tumor (Cheng et al., 2001) and may also be due to other causes, including ocular problems. Most commonly, this is due to weakness of the superior oblique muscle, but it can also be due to paresis of the lateral rectus muscle or nystagmus (Williams et al., 1996). Non-dystonic torticollis may be due to inflammation of joints (Uziel et al., 1998), soft tissue (Shale et al., 1988), and arteriovenous fistula at the craniocervical junction (Bayrakci et al., 1999). Treatment of congenital muscular torticollis by manual stretching is usually safe and effective; surgical treatment is necessary if this noninvasive treatment fails (Cheng et al., 2001).
| (These are not classified as dystonia, but can be mistaken for dystonia because of sustained postures) |
Following are discussions of a few of the specific entities listed in Table 12.10, in the order presented in the table.
Oppenheim dystonia (DYT1)
Clinical
The phenotypes of Oppenheim dystonia (also known as DYT1 dystonia) was characterized in the Ashkenazi Jewish population when detection of individuals with the DYT1 mutation became possible because of the identification of the special genetic haplotype around the DYT1 gene in this population (Bressman et al., 1994a). The mean (± SD) age at onset of symptoms is 12.5 ± 8.2 years. In 94% of patients, symptoms begin in a limb (arm or leg equally) (Videos 12.17 and 12.18); rarely the disorder starts in the neck (3.3%) or larynx (2.2%). Even in the non-Jewish population the same gene is responsible for most cases of early-onset and limb-onset PTD (Kramer et al., 1994). Over time, as diagnostic laboratory examinations for DYT1 have become available, some variations of the phenotype have been observed (Edwards et al., 2003). The phenotype varies from generalized to focal dystonia even in the same family as has been reported in two large families with proven DYT1 gene mutation (Opal et al., 2002; Kostić et al., 2006). The proband of this family died with a dystonic storm, while other family members carrying the same mutation either were asymptomatic or displayed dystonia that was focal, segmental, multifocal, or generalized in distribution. One family member had onset of her dystonia at age 64 years. 
The phenotypes in families with non-DYT1 dystonia overlap with each other but differ from those with DYT1 dystonia. In the majority of non-DYT1 families, the dystonia most commonly begins in the cranial-cervical region (Bentivoglio et al., 1997), whereas this site of onset is rare in DYT1 dystonia (Bressman et al., 1994a). Only in DYT6 dystonia in the Mennonite population is there some clinical phenotypic overlap with Oppenheim dystonia (Almasy et al., 1997).
Sequence learning of motor tasks is reduced in manifesting and nonmanifesting gene carriers (Ghilardi et al., 2003). PET imaging obtained during motor task testing showed increased activation in the left premotor cortex and right supplementary motor area, with concomitant reduction in the posterior medial cerebellum. During motor sequence learning, nonmanifesting DYT1 carriers overactivated the lateral cerebellum and the right inferotemporal cortex, and had relative deficits in the dorsolateral prefrontal cortex bilaterally, left anterior cingulate, and the dorsal premotor cortex (Carbon et al., 2008a). These findings suggest that abnormalities in motor behavior and brain function exist in clinically nonmanifesting DYT1 carriers. Similarly, depression has been found in both manifesting and nonmanifesting DYT1 carriers (Heiman et al., 2004). However, evaluation of manifesting and nonmanifesting DYT1 carried out with a comprehensive neuropsychological test battery found no differences from controls on cognitive tests evaluating verbal and nonverbal abstract abilities, attention, information processing speed, and spatial organization (Balas et al., 2006).
Genetics
Previously, Eldridge (1970) proposed that dystonia in the Ashkenazi Jewish population was inherited as an autosomal recessive disorder, while non-Jews inherited dystonia as an autosomal dominant disorder. Reanalysis of Eldridge’s data by segregation analysis has shown that dystonia in the Jewish population was also inherited as autosomal dominant (Pauls and Korczyn, 1990). A detailed analysis of the clinical course of dystonia was compared in the Jewish and non-Jewish populations with inherited dystonia, and no major difference was found (Burke et al., 1986). The prevalence of dystonia among Jews of Eastern European ancestry has been estimated to be 1 per 15 000 (Zilber et al., 1984), while for non-Jews the prevalence is 1 per 200 000 (Zeman and Dyken, 1967).
In both Jewish (Bressman et al., 1989) and non-Jewish groups (Zeman and Dyken, 1967), Oppenheim dystonia is inherited as an autosomal dominant disorder, and the gene has been localized to the long arm of chromosome 9 (9q34.1) (Ozelius et al., 1989; Kramer et al., 1990). This abnormal gene has been given the name DYT1, but has been officially renamed TOR1A. The mutation that causes the disease has been identified as a deletion of one of a pair of GAG triplets (codes for glutamic acid) in exon 5 near the carboxy terminal in a previously unknown protein, designated torsinA (Ozelius et al., 1997). The GAG deletion is the only sequence change that has been found thus far to be associated uniquely with the disease status, regardless of ethnic origin. Mutations causing this deletion are uncommon, but a few have been encountered (Klein et al., 1998a; Valente et al., 1999), and the unique haplotype found in North American and Russian Ashkenazi Jews with DYT1 has also been seen in Great Britain as well (Valente et al., 1999).
Not every non-Jewish family with dystonia has the DYT1 gene, so dystonia is genetically heterogeneous (Bressman et al., 1994b, 1994c). But all Ashkenazi Jewish families with dystonia with limb onset and onset below the age of 49 in at least one member have so far been found to have the DYT1 gene. Also most cases of non-Jewish individuals with limb-onset and childhood-onset have DYT1 dystonia (Kramer et al., 1994). Intrafamilial correlation for age at onset of dystonia is low (Fletcher et al., 1991b). There is some evidence that dystonia in the Ashkenazi Jewish population tends to begin on the dominant side (Inzelberg et al., 1993). The penetrance rate of gene expression in the Ashkenazi Jewish population is approximately 30% (Bressman et al., 1989; Risch et al., 1990). Variations in the TOR1A gene are at least one factor associated with the susceptibility in expressing the disease (Risch et al., 2007). THAP1 encodes a transcription factor that regulates the expression of TOR1A. Wild-type THAP1 represses the expression of TOR1A, whereas DYT6-associated mutant THAP1 results in decreased repression of TOR1A (Gavarini et al., 2010; Kaiser et al., 2010). These observations suggest that transcriptional dysregulation may be a cause of dystonia.
With the availability of direct testing for the DYT1 mutation, Bressman and her colleagues (2000) have developed an algorithm as to which individuals who have the clinical diagnosis of PTD should be tested. They suggest testing in conjunction with genetic counseling for patients with PTD with onset before age 26 years, as this single criterion detected 100% of clinically ascertained carriers, with specificities of 43–63%. Testing patients with onset after age 26 years also may be warranted in those having an affected relative with early onset. The DYT1 gene rarely is found in patients with musician’s cramp (Friedman et al., 2000) or in patients with sporadic or familial writer’s cramp (Kamm et al., 2000).
The relationship between essential tremor and dystonia has been debated (Lou and Jankovic, 1991; Lang et al., 1992). The finding that families with essential tremor do not have the DYT1 gene by linkage analysis (Conway et al., 1993; Durr et al., 1993) indicates that the two disorders are not genetically identical.
Brain networks and pathways
Neuroimaging studies on Oppenheim dystonia are discussed in the section “Biochemistry and neuroimaging.” Based on FDG PET scans and on physiologic monitoring during pallidal surgery in patients with dystonia, there appears to be a disruption of activity within the globus pallidus interna (GPi), suggesting excessive inhibitory input from the striatum. As measured by diffusion tensor imaging, all mutation carriers had reduced connectivity in the cerebellothalamic pathways, with connectivity reduced by approximately 50% compared to normal controls (Argyelan et al., 2009). The reduction was localized to the cerebellar outflow pathway in the white matter of lobule VI, adjacent to the dentate nucleus. Both manifesting and nonmanifesting carriers had reduced connectivity in the proximal segment of the cerebellothalamocortical pathway (i.e., the cerebellothalamic segment), but the nonmanifesting carriers had an additional disruption in the distal, thalamocortical, segment of the pathway. The investigators suggested that perhaps the benefit from pallidal surgery is by affecting the distal connections, bringing the patients to a state resembling nonmanifesting carriers.
Molecular biology
TorsinA is related to homologous proteins throughout the multicellular animal kingdom (Breakefield et al., 2001). It is a 332-amino acid protein and has a molecular weight of 37 813. The torsin family is a member of the AAA+ family of proteins. Their structures indicate that they are heat-shock and ATP-binding proteins. The DYT1 mutation is the first example of a role for any heat-shock protein in a human disease. It suggests a susceptibility to a “second hit.” This might explain the low penetrance rate of 30–40% in gene carriers and why many patients with Oppenheim dystonia experience a stress situation prior to the development or exacerbation of the dystonia, e.g., influenza, trauma, surgery, or casting of a body part. Normal torsinA appears to protect against oxidative stress (Kuner et al., 2003), and torsinA is increased following exposure to the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Kuner et al., 2004). A malfunctioning torsinA would have difficulty carrying out its chaperoning of damaged proteins that had been altered by a variety of stress factors.
TorsinA mRNA has been mapped in normal human brain and found to be localized in specific regions (Augood et al., 1998, 1999), which are listed in Table 12.16. A similar localization was found by using immunostaining for an antibody raised against torsinA (Augood et al., 2003). Although its location within dopamine neurons, among other types, has suggested to some investigators that dopamine is implicated in Oppenheim dystonia, clinical pharmacology has not yet seen a relationship, and biochemically, dopamine concentration is normal except for a 50% reduction in the rostral portions of the putamen and caudate nucleus in a single case (Furukawa et al., 2000) and a possible increase in striatal dopamine turnover in three cases (Augood et al., 2002). A morphometric analysis of the pigmented substantia nigra pars compacta (SNc) neurons found no difference in neuron number, but the average size of neuronal cell bodies was increased, and dopaminergic SNc neurons were arranged in much closer apposition to each other than was observed in control tissue (Rostasy et al., 2003).
| Intense expression |
Data from Augood SJ, Martin DM, Ozelius LJ, et al. Distribution of the mRNAs encoding torsinA and torsinB in the normal adult human brain. Ann Neurol 1999;46(5):761–769.
Antibodies to torsinA have been prepared in several laboratories. The normal protein is widely distributed in human, rat, and mouse brain (Shashidharan et al., 2000a; Konakova et al., 2001; Konakova and Pulst, 2001; Walker et al., 2001) and is found in endoplasmic reticulum (ER) (Hewett et al., 2000; Kustedjo et al., 2000). TorsinA immunohistochemistry investigation of a brain from a patient with Oppenheim dystonia failed to show any difference from control brains (Walker et al., 2002; Rostasy et al., 2003). The cholinergic neurons in rat striatum have a high content of torsinA during development, but then this concentration fades as the animal gets older (Oberlin et al., 2004). This temporal and anatomical pattern could fit with some clinical features of Oppenheim dystonia: young onset with usual sparing after age 28 years, and response to high-dosage antimuscarinic agents.
The AAA+ family of proteins is believed to serve as a chaperone in the processing and repair of damaged proteins. Six torsinA proteins are thought to join together to form a ring, with each of these six proteins bound to its neighbor by ATP. This structure is believed to function to repair other proteins that have been damaged. If the repair is unsuccessful, the damaged proteins can aggregate or die by apoptosis. In neural cultures (Hewett et al., 2000), the normal protein is found throughout the cytoplasm and neurites with a high degree of co-localization with the ER. In contrast, overexpression the mutant protein in transgenic mice showed an accumulation of the protein in multiple, large inclusions in the cytoplasm around the nucleus. These inclusions were composed of membrane whorls, apparently derived from the ER. If disrupted processing of the mutant protein leads to its accumulation in multilayer membranous structures in vivo, these may interfere with membrane trafficking in neurons.
Immunohistochemical studies showed that torsinA is present in Lewy bodies (Shashidharan et al., 2000b; Sharma et al., 2001). A role of torsinA in α-synuclein, a major component of the Lewy body, was subsequently sought. Overexpression of wild-type torsinA dramatically reduced the number of transfected cells containing α-synuclein aggregates, whereas mutant torsinA failed to suppress α-synuclein aggregation (McLean et al., 2002).
TorsinA immunohistochemistry investigation of brains from patients with DYT1 dystonia failed to show any difference from control brains (Walker et al., 2002; Rostasy et al., 2003). A morphometric analysis of the pigmented SNc neurons found no difference in neuron number, but the average size of neuronal cell bodies was increased, and dopaminergic SNc neurons were arranged in much closer apposition to each other than was observed in control tissue (Rostasy et al., 2003). Other evidence for a role in preventing protein aggregation comes from studies of the torsinA homolog in Caenorhabditis elegans, TOR-2 (Caldwell et al., 2003). TOR-2 also appears to be an ER protein. Overexpression of either TOR-2 or human wild-type torsinA significantly suppressed the aggregation of a polyglutamine repeat protein, whereas a mutant form of TOR-2 failed to reduce protein aggregation (Caldwell et al., 2003).
Wild-type torsinA is distributed throughout the cell, and is particularly enriched in the lumen of the ER (Hewett et al., 2000; Kustedjo et al., 2000). Mutant torsinA concentrates in large clumps that are largely or completely segregated from the ER. The clumps of mutant torsinA immunostaining are not insoluble aggregates (Kustedjo et al., 2000), and they appear to be composed of whorled double-membrane structures, apparently derived from the ER (Hewett et al., 2000). This DeltaE302/303 mutation appears to be a stable protein, and not toxic. The DeltaE302/3 mutation causes a striking redistribution of torsinA from the ER to the nuclear envelope (Goodchild and Dauer, 2004; Gonzalez-Alegre and Paulson, 2004; Naismith et al., 2004; Hewett et al., 2004). Oppenheim dystonia, therefore, appears to be a previously uncharacterized nuclear envelope disease and the first to selectively affect CNS function. Goodchild and Dauer (2005) found that normal torsinA binds a substrate in the lumen of the nuclear envelope (NE), and the DeltaE mutation enhances this interaction. They identified lamina-associated polypeptide 1 (LAP1) as a torsinA-interacting protein. Goodchild and her colleagues (2005) went on to show that genetic animal models without torsinA or with abnormal torsinA have severely abnormal nuclear membranes in neurons, whereas non-neuronal cell types appear normal. These observations demonstrate that neurons have a unique requirement for nuclear envelope localized torsinA function and suggest that loss of this activity is a key molecular event in the pathogenesis of DYT1 dystonia.
The mutant torsinA has also been shown to have a reduction of its normal ATPase activity (Konakova and Pulst, 2005). Membranous inclusions, which were found to occur in cells transfected with the mutant torsinA (Hewett et al., 2000), have now also been found in patients with Oppenheim dystonia (McNaught et al., 2004). They are present in the perinuclear region within brainstem cholinergic and other neurons in the pedunculopontine nucleus, cuneiform nucleus, and griseum centrale mesencephali. The inclusions stain positively for ubiquitin, torsinA, and the nuclear envelope protein lamin A/C. No inclusions were detected in the SNc, striatum, hippocampus, or selected regions of the cerebral cortex. McNaught and colleagues (2004) also noted tau/ubiquitin-immunoreactive aggregates in pigmented neurons of the SNc and locus coeruleus.
A novel protein, printor, interacts and co-distributes with torsinA in multiple brain regions and co-localizes with torsinA in the endoplasmic reticulum. The interaction of printor with torsinA is abolished by the DYT1 mutant torsinA (Giles et al., 2009).
Genetic models
Homozygous DYT1 knock-in and knock-out mouse models point to a loss-of-function mutation as animals die shortly after birth, suggesting that torsinA is indispensable for specific developmental processes in mammalian brain (Goodchild et al., 2005). Transgenic mice overexpressing either the wild type or the mutant develop neurologic dysfunction (Grundmann et al., 2007). In the former mice, both dopamine and serotonin are reduced in the striatum, whereas the mutant transgenic mice have reduced serotonin in the brainstem. In other transgenic mutant mice studies, most studies on dopamine function in the striatum were normal, including concentration of dopamine and its metabolites, function of the dopamine transporter and the vesicular monoamine transporter, and the D1 and D2 receptors (Balcioglu et al., 2007). However, a reduction of amphetamine-induced release of dopamine was impaired.
Dopa-responsive dystonia (DRD)
It should be mentioned, that in addition to the genetic types described in this section, some patients with focal dystonias, including familial cases, might respond to levodopa therapy. A report of two families with a total of four members with childhood-onset cervical dystonia, all of whom who had an excellent response to levodopa, illustrates this point (Schneider et al., 2006c). Genetic testing on these patients showed that they did not have any of the genetic types discussed in this section. It is possible that these cases may represent new forms of dopa-responsive dystonia.
Classic dopa-responsive dystonia (DRD)
Segawa and his colleagues (1976) described a syndrome of dystonia in children that has a diurnal pattern. The children may be relatively free of dystonic movements and postures in the morning and be severely afflicted in the late afternoon, evening, and night (Videos 12.19, 12.20, and 12.21). Segawa and colleagues (1976) called this disorder hereditary progressive dystonia with diurnal variation; they also mentioned that it responds to levodopa. Independently, Allen and Knopp (1976) described a form of childhood hereditary dystonia that had features of parkinsonism that showed sustained control by levodopa and anticholinergic medication. This was the same disorder. 
Over time, several clinical features have been distinguished as highlighting this disorder: (1) Many patients with onset of dystonia in childhood have features of parkinsonism, including rigidity, bradykinesia, flexed posture, and loss of postural reflexes. (2) The disorder usually, but not exclusively, begins in childhood with a presentation of a peculiar gait, namely a tendency to walk on the toes. (3) The disease can begin in infancy, thereby resembling cerebral palsy (Nygaard et al., 1994) (Videos 12.22 and 12.23), and there has now been a report of a neonatal onset of rigidity, tremor, and dystonia (Nardocci et al., 2003) (Video 12.24). (4) When the disorder begins in adulthood, it can present as a focal dystonia of arm, neck, or cranium, or present as parkinsonism, mimicking Parkinson disease (Nygaard et al., 1992). (5) The patients respond remarkably well to low-dosage levodopa. (6) Not all patients with childhood-onset dystonia have the diurnal fluctuation pattern. (7) Writer’s cramp can be brought out with continued writing (Deonna et al., 1997). (8) Cerebellar signs have also been seen, with response to levodopa (Chaila et al., 2006). The term dopa-responsive dystonia (DRD) appears to be the preferred label for this disorder (Nygaard, 1989; Nygaard et al., 1990), because response to levodopa is the consistent finding, and the term also highlights the appropriate treatment. Moreover, the term hereditary progressive dystonia could apply to many genetic forms of dystonia, and certainly to the more virulent Oppenheim dystonia. 
If levodopa therapy is delayed for a great many years, patients still respond to low doses of levodopa (Harwood et al., 1994) (Video 12.25). Even if the onset is in adulthood presenting as parkinsonism, and resembling Parkinson disease, it responds to low doses of levodopa and without adverse effects of response fluctuations (Nygaard et al., 1992; Harwood et al., 1994). Long-term treatment with levodopa does not cause the wearing-off phenomenon. Worsening of dystonia does not occur until 29 hours or more after levodopa withdrawal; the subjective feeling of wearing-off that is experienced by patients with DRD might be from one of the nonmotor effects of levodopa, such as mood elevation (Dewey et al., 1998). 
DRD is inherited in an autosomal dominant pattern and needs to be recognized because it is so easily treated. The gene causing DRD has been mapped to chromosome 14q by Nygaard et al. (1993) and identified by Ichinose et al. (1994) as the gene for the enzyme GTP cyclohydrolase I (GCH), the rate-limiting enzyme in the biosynthesis of tetrahydrobiopterin (BH4) (Fig. 12.2), which is the cofactor of monoamine-synthesizing enzymes TH for dopamine and norepinephrine and tryptophan hydroxylase for serotonin (and also phenylalanine hydroxylase). The genetic designation DYT5 has been assigned to this form of dystonia (Table 12.1). Clinically, only dystonia and parkinsonism are manifested. Approximately 40–50% of patients with DRD have no known mutations (Furukawa and Kish, 1999). GCH1 point mutations occur in approximately 54% of DRD patients, and deletions in 8% (Zirn et al., 2008). Different families have different mutations on the same GCH gene (Ichinose et al., 1995; Hirano et al., 1995; Bandmann et al., 1996; Furukawa et al., 1996; Ichinose and Nagatsu, 1997; Jarman et al., 1997; Thony and Blau, 1997; Furukawa et al. 1998b; Steinberger et al., 1998; Tamaru et al., 1998), and spontaneous mutations have been identified. The multiple mutations are scattered over the entire coding region for the six exon-containing GCH gene.
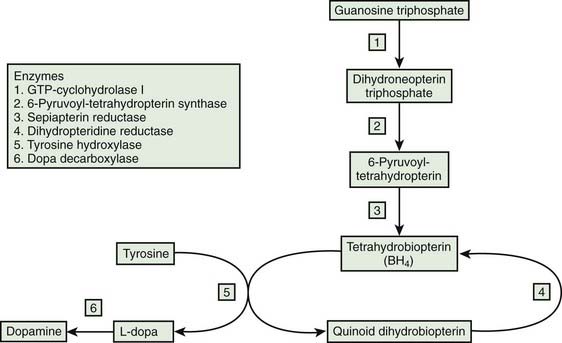
Phenylalanine hydroxylase also requires the cofactor BH4 for its activity, and homozygous mutations and compound heterozygous mutations have been found in the GCH gene to cause the rare autosomal recessively inherited form of hyperphenylalaninemia (Thony and Blau, 1997; Furukawa et al., 1998a). Such mutations are therefore clinically similar to the compound heterozygous or homozygous mutations spread over all six exons encoding 6-pyruvoyl-tetrahydropterin synthase, which manifests as an autosomal recessively inherited variant of hyperphenylalaninemia, along with a deficiency of dopamine and serotonin. Hyperphenylalaninemia may be mild and go undetected neonatally; the child can develop dystonia later in childhood, along with developmental delay and seizures. The condition is treatable with levodopa, 5-hydroxytryptophan, and tetrahydrobiopterin (Demos et al., 2005). The other autosomal recessive pterin deficiency disorders also respond to levodopa, and they often have developmental delay, oculogyria, and dysautonomic features. Sepiapterin reductase deficiency appears to be fairly common in Malta (Neville et al., 2005). Although dystonia can respond to levodopa, cognitive impairment does not.
In some pedigrees with DRD a mutation in the GCH gene has not been found. This fact, plus the multiple mutations in the GCH gene that have been discovered so far, indicated that genetic testing will not be an easy method to determine the presence of the molecular defect. Ichinose and Nagatsu (1997) found that GCH activity in lymphocytes was decreased to less that 20% of the mean value of healthy controls, and in unaffected carriers was 37%. The enzyme had normal activity in juvenile parkinsonism. These authors suggest that assay of GCH in blood cells might become a useful biochemical marker for the gene defect. A deficiency of neopterin is found in CSF, but this is reduced in juvenile parkinsonism also.
PET scans for FDOPA uptake and dopamine transporter are normal, whereas those for the D2 receptor show increased binding, reflecting supersensitivity (Rinne et al., 2004). Network analysis, using FDG PET, shows that DRD has a unique metabolic architecture that differs from other inherited forms of dystonia (Asanuma et al., 2005a). The characteristic features are increases in metabolism in the dorsal midbrain, cerebellum, and supplementary motor area and reductions in motor and lateral premotor cortex and in the basal ganglia.
There has been a report of a patient with DRD who had a remission (Di Capua and Bertini, 1995), which perhaps should not come as a surprise because not every carrier of the mutation has symptoms. In fact, affected females outnumber affected males by a ratio of approximately 4 : 1 (Ichinose and Nagatsu, 1997). This ratio has been explained by the observation that males have a higher GCH activity normally, so missing one of the two genes by a mutation may still leave sufficient enzyme intact to avoid symptoms. There is also a report that SSRI antidepressants reversed the benefit from levodopa therapy (Mathen et al., 1999).
The phenotype of DRD is not always easily distinguished from juvenile parkinsonism, but features that help in the differential diagnosis are presented in Table 12.17. Fluorodopa PET scanning reveals a normal or modest reduction of dopa uptake (Sawle et al., 1991; Nygaard et al., 1992; Snow et al., 1993; Turjanski et al., 1993), in contrast to the marked reduction in juvenile parkinsonism. A similar result is seen with β-CIT SPECT (Naumann et al., 1997; Jeon, 1997) or other dopamine transporter ligands (Huang et al., 2002). Another important difference is that long-term treatment with levodopa in DRD is not associated with the motor complications that are seen with levodopa therapy in juvenile (and adult) Parkinson disease (Nygaard et al., 1991, 1992; Harwood et al., 1994).
Table 12.17 Differential features between juvenile parkinsonism, dopa-responsive dystonia, and primary torsion dystonia
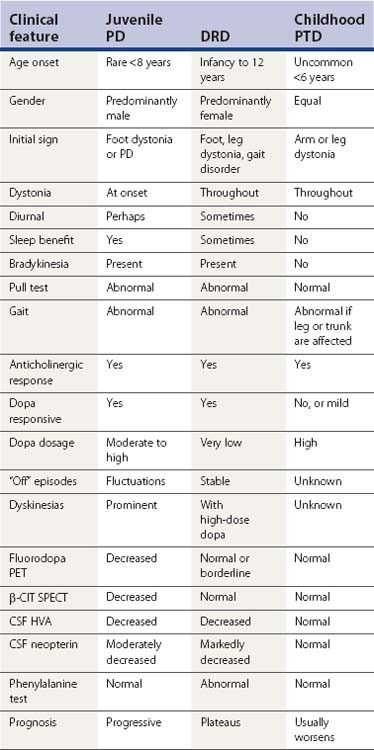
An early sign of dopa-responsive dystonia in neonates may be difficulty with feeding, including vomiting, rigidity, and opisthotonus (Chieng et al., 2007). One unusual phenotypic expression is myoclonus, which occurred in one kindred and preceded the dystonia and bradykinesia (Leuzzi et al., 2002). Molecular genetics investigation found a missense mutation in exon 6 of the GCH1 gene. Another phenotype of DRD is adult-onset oromandibular dystonia and no obvious family history of dystonia, which was found in one individual who had a mutation of GCH and who responded positively to treatment with levodopa (Steinberger et al., 1999). An adult with exercise-induced limb and trunk dystonia and with diurnal variation was found to respond to levodopa (Regula et al., 2007).
The first report of the pathology of a case of DRD-GCH type revealed decreased neuromelanin in the substantia nigra, but otherwise normal nigral cell counts and morphology (Rajput et al., 1994). Biochemically, this patient had reduced concentrations of dopamine and its metabolite, homovanillic acid, in both the caudate and putamen. Furukawa and his colleagues (1999) carried out more extensive postmortem biochemistry in two patients with typical DRD. One had two GCH mutations, but the other had no mutation in the coding region of this gene. Striatal biopterin and neopterin levels were markedly reduced. Both had severely reduced (<3%) TH protein levels and normal concentrations of dopa decarboxylase protein, dopamine transporter, and vesicular monoamine transporters. The authors suggested that the reduction of TH protein might be explained by reduced enzyme stability/expression consequent to congenital BH4 deficiency.
A second autopsy report of DRD was on a patient with a presumed different genetic locus, chromosome 14q13, and called DYT14 by Grotzsch and colleagues (2002). An autopsy in the elderly proband revealed a normal number of cells in the substantia nigra and locus coeruleus, which is typical of DRD and not Parkinson disease, and there was a decrease in neuromelanin. No Lewy bodies were seen. These pathologic features are typical for DRD. Subsequently more careful and thorough genetic analysis revealed that this family had a deletion in GCH1, so was just another pedigree with DYT5 dystonia (Wider et al., 2008). Deletions are harder to detect and care must be made to search for them (Gasser, 2008).
Intracortical inhibition of the primary motor cortex was studied in DRD patients and found to be normal, in contrast to adult-onset focal dystonia (see below in the pathophysiology section) (Hanajima et al., 2007). It is difficult to explain why loss of striatal dopamine in PD produces parkinsonian features while such a loss in DRD produces predominantly dystonic features. Using a transgenic mouse model for DRD, Sato and colleagues (2008) showed that dopamine loss is mainly anteriorly in the putamen affecting the striosomes. They propose that in PD, the major loss of dopamine is in the posterior putamen affecting mainly the matrix, and that this differential loss results in the different phenomenology between the two disorders (Fig. 12.3).
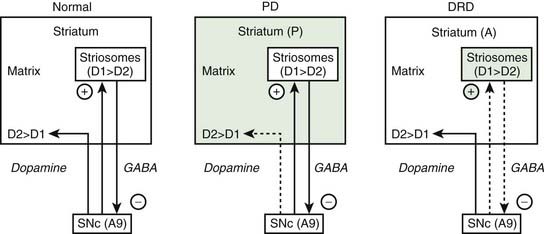
Tyrosine hydroxylase (TH) deficiency
At least 10 different point mutations have now been discovered in the human TH gene, and more are reported regularly (Knappskog et al., 1995; Lüdecke et al., 1995, 1996; van den Heuvel et al., 1998; Swaans et al., 2000; de Rijk-van Andel et al., 2000; Janssen et al., 2000; Furukawa et al., 2001; Giovanniello et al., 2007). Many of the reports are in Dutch families. The clinical features range from a mild syndrome of juvenile DRD to a more severe parkinsonism, dystonia, and oculogyric crises (all three representing dopamine deficiency) and also ptosis, miosis, oropharyngeal secretions, and postural hypotension (all representing norepinephrine deficiency). In the most severe form, the infant was virtually immobile, rigid, drooling saliva, with a tremor in tongue and hands. She responded dramatically to levodopa. The severity appears to be associated with the amount of loss of TH activity. Genetically, there can be homozygous mutations to heterozygous mutations. Compound heterozygotes have presented as spastic paraparesis, with a complete or very good response to levodopa (Furukawa et al., 2001; Giovanniello et al., 2007). Some patients with a mild form of the disorder live a normal life with a response to low-dose levodopa, into adulthood. One reported problem is hypersensitivity to levodopa (Grattan-Smith et al., 2002). Hoffmann and colleagues (2003) reported four patients with TH deficiency and emphasized that most patients with this disorder have an encephalopathy that is not predominantly dystonia, but a progressive, often lethal encephalopathic disorder, which can be improved but not cured by levodopa. The DRD of TH deficiency can be as responsive to levodopa as is DYT5 with GCH involvement. Schiller and colleagues (2004) report sustained benefit, and excellent response to levodopa even when treatment was delayed for 20 years. Most patients with tyrosine hydroxylase deficiency can be successfully treated with levodopa.
A review of the literature and a report of 36 cases of TH deficiency divides the disorder into two phenotypes: an infantile-onset, progressive, hypokinetic-rigid syndrome with dystonia (type A), and a complex encephalopathy with neonatal onset (type B) (Willemsen et al., 2010). The homovanillic acid concentrations and homovanillic acid/5-hydroxyindoleacetic acid ratio in cerebrospinal fluid correlate with the severity of the phenotype.
A phenylalanine-loading test has been recommended as a means to distinguish between DRD with GCH deficiency and that with TH deficiency (Hyland et al., 1997). In the former, phenylalanine is not metabolized rapidly and the blood levels remain elevated for a prolonged period. In TH deficiency, this test would be normal. One other potential method of differentiating GCH-deficient DRD from other DRDs or juvenile parkinsonism is the exquisite sensitivity to centrally acting anticholinergics. Some of the trihexyphenidyl responders reported by Fahn (1983) had responded to small doses, such as 6 mg/day. These patients were subsequently determined to have DRD. When trihexyphenidyl was first reported for the treatment of Parkinson disease, some papers pointed out that small doses could have a dramatic benefit in some children with dystonia (Corner, 1952; Burns, 1959). These children had diurnal fluctuations of their dystonia. Jarman and colleagues (1997) screened some of their patients with responsiveness to anticholinergics and found that some had a GCH mutation.
Rapid-onset dystonia–parkinsonism (RDP)
RDP is an autosomal dominant movement disorder characterized by sudden onset of persistent dystonia and parkinsonism, generally during adolescence or early adulthood (Dobyns et al., 1993). Symptoms evolve over hours or days and generally stabilize within a few weeks, with slow or no progression. Members of a family who are wide apart in age may develop symptoms around the same time. Further follow-up of the original family revealed two members to have a more gradual progression of their disorder over 6–18 months. One of them experienced a rapid progression of symptoms 2 years after an initial stabilization of his condition. The phenotype therefore shows considerable variability (Brashear et al., 1996). Two other, seemingly unrelated, kindreds were subsequently reported (Brashear et al., 1997; Pittock et al., 2000). Variability occurs within a kindred, with some members in a kindred becoming stable after their worsening and others having fluctuating worsening or even improving over time (McKeon et al., 2007).
Now that the gene responsible for RDP has been discovered (de Carvalho Aguiar et al., 2004), it is possible to test a wide clinical spectrum of dystonia and determine the expanded phenotype (Brashear et al., 2007). The mutation is in the ATP1A3 gene responsible for the α3 subunit of Na-K-ATPase. The phenotype of 36 affected individuals from 10 families included abrupt onset of dystonia with features of parkinsonism, a rostrocaudal gradient, and prominent bulbar findings. Other features found in some mutation carriers included common reports of triggers, minimal or no tremor at onset, occasional mild limb dystonia before the primary onset, lack of response to dopaminergic medications, rare abrupt worsening of symptoms later in life, stabilization of symptoms within a month, and minimal improvement overall. Tremor at onset of symptoms, a reversed rostrocaudal gradient, and significant limb pain excluded a diagnosis of RDP. A positive family history is not required.
Although there is little or no response to levodopa or dopamine agonists, a low concentration of homovanillic acid in the CSF in patients has been reported (Brashear et al., 1998). PET studies indicate no loss of dopaminergic nerve terminals in RDP (Brashear et al., 1999), suggesting that this disorder results from a functional deficit rather than being a neurodegenerative disease. An autopsied case revealed no neurodegeneration (Pittock et al., 2000). These findings suggest RDP is a neurochemical rather than a neurodegenerative disorder. Deep brain stimulation has not been helpful (Kamm et al., 2008).
The ATP1A3 gene for RDP is on chromosome 19q13 (Kramer et al., 1999). Functional studies and structural analysis of the protein suggest that these mutations impair enzyme activity or stability. This finding implicates the Na+/K+ pump, a crucial protein that is responsible for the electrochemical gradient across the cell membrane. The authors suggest that the rapid onset that is seen in the disorder is related to an inability to “keep up with a high demand for ion transport activity” in response to stressful situations. However, there is genetic heterogeneity because one large family with the RDP phenotype was found not to have a mutation in the ATP1A3 gene (Kabakci et al., 2005). Although most patients have been of European descent, at least one Asian (Korean) has been reported, and with the typical phenotype (Lee et al., 2007).
Now that at least one gene for RDP has been found, genetic analysis can be carried out in dopa-nonresponsive parkinsonism. After being stable for 2.5 years, one patient thought to have Parkinson disease developed overnight oromandibular dystonia and more severe parkinsonian symptoms; he was found to have a missense mutation in the ATP1A3 gene (Kamphuis et al., 2006).
Myoclonus–dystonia
Myoclonic movements were mentioned early in the description of the phenomenology of dystonia. Although lightning-like movements can occur in Oppenheim dystonia, they can also be seen in a distinct autosomal dominant disorder. Both conditions have unfortunately been called myoclonic dystonia, and in both, the myoclonic jerks can respond to alcohol. It has been recommended that when myoclonic jerks are part of Oppenheim dystonia, it be referred to as dystonia with lightning-like jerks or as myoclonic dystonia (Quinn et al., 1988). The autosomal dominant myoclonus–dystonia that is a distinct entity has been mapped to chromosome 7q21and the mutated gene has been identified as the gene for ε-sarcoglycan (Zimprich et al., 2001; Asmus et al., 2002). The report of a mutation in the gene for the dopamine D2 receptor in a single family (Klein et al., 1999) has now been recognized to most likely represent a polymorphism (Klein et al., 2000a). This family has since been found to have genetic linkage to 7q21–q31 like the other families (Klein et al., 2000b). Whether myoclonus–dystonia is an entity separate from hereditary essential myoclonus (Fahn and Sjaastad, 1991; Quinn, 1996) remains to be determined through genetic studies. At least one family originally published as familial essential myoclonus has been reported to have a mutation in the ε-sarcoglycan gene and thus has myoclonus–dystonia (Korten et al., 1974; Foncke et al., 2006).
The onset can be in childhood or in adulthood. Age at onset is highly associated with gender; the median age onset for girls is 5 years versus 8 years for boys (Raymond et al., 2008). The myoclonus and dystonia are located predominantly in the arms and neck (Videos 12.26 and 12.27), and the symptoms tend to plateau after a period of progression (Kyllerman et al., 1990). There is clinical heterogeneity, and leg involvement can occur (Nardocci et al., 2008). Obsessive-compulsive disorder (OCD) and alcohol dependence are not uncommon in individuals who carry the gene (Saunders-Pullman et al., 2002; Hess et al., 2007). Psychiatric problems also include substance abuse, anxiety/panic/phobic disorders, and psychosis (Doheny et al., 2002). Cognitive testing showed impaired verbal learning and memory in one family, impaired memory in the second family, and no cognitive deficits in the third family. In a large Dutch family, there was no cognitive deficit, nor OCD, but there was depression and anxiety with panic attacks (Foncke et al., 2009). 
In SGCE mutation carriers, there is hyperresponsiveness in contralateral inferior parietal cortical areas, ipsilateral premotor and primary somatosensory cortex, and ipsilateral cerebellum during the motor task compared with healthy control subjects (Beukers et al., 2010). This is similar to that in DYT1 and DYT6 dystonia and points to a disorganized sensorimotor integration.
There is markedly reduced penetrance if the parent carrying the mutation is the mother (penetrance about 5–10%), whereas if the father carries the mutation, the penetrance is about 90%. This pattern of phenotypic expression of myoclonus–dystonia is called maternal imprinting. As Asmus and Gasser (2004) explained, if the mutated allele is inherited from the father, inactivation of the maternal allele due to imprinting leads to complete ε-sarcoglycan deficiency, and hence to the development of clinical symptoms. If, on the other hand, the mutated allele is inherited from the mother, the intact paternal allele is sufficient to sustain ε-sarcoglycan function. Making the inheritance pattern more complicated is one report of reduced penetrance in which the mutated gene was inherited from the father (Muller et al., 2002). Deletions in the maternal allele, as well as point mutations, of the SCGE gene occur, and these can cause phenotypic variations, depending on the size of the deletion, with the largest deletion causing a split-hand/split-foot malformation and sensorineural hearing loss (Asmus et al., 2007).
Another gene locus has been found for one family with myoclonus–dystonia on chromosome 18p11 (Grimes et al., 2002). In a study of Dutch families with myoclonus–dystonia, only 7 of 31 patients had carried a mutation in the ε-sarcoglycan gene (Gerrits et al., 2006). In one large Dutch family, originally published as essential myoclonus (Korten et al., 1974), the onset of myoclonus can be delayed as late as 75 years (Foncke et al., 2006), and although emphasis was made that the myoclonus was mainly distal in the arms in contrast to an assumption that myoclonus is usually proximal in this disorder, our personal experience is that myoclonus is commonly distal.
A phenotype similar to myoclonus–dystonia has been encountered with the SCA14 mutation (Foncke et al., 2010).
Secondary dystonias
In a movement disorders center, primary dystonias are much more common than secondary dystonias. A screen for the DYT1 gene in Ashkenazi Jewish patients with secondary dystonia failed to find any evidence that the DYT1 mutation contributes to secondary dystonia (Bressman et al., 1997), including tardive dystonia and other environmental insults. The most common secondary dystonias seen at the Dystonia Clinical Research at Columbia University Medical Center are presented in Table 12.18.
| Cause | No. |
|---|---|
| Primary | 1762 |
| Tardive dystonia | 184 |
| Birth injury | 83 |
| Psychogenic | 64 |
| Peripheral trauma | 51 |
| Head injury | 39 |
| Stroke | 27 |
| Encephalitis | 24 |
| Miscellaneous | 164 |
| Total | 2398 |
Data from the Center for Parkinson Disease and Other Movement Disorders, Columbia University Medical Center, New York City.
Tardive dystonia
Tardive dystonia is the most common form of symptomatic dystonia seen at the Movement Disorders Center at Columbia University Medical Center (Table 12.18). It can manifest exactly like PTD, but there is often accompanying classic tardive dyskinesia or tardive akathisia, which establishes the diagnosis. One form of clinical presentation seems more common in tardive dystonia. Many patients have retrocollis and extension of the elbows with internal rotation of the shoulders and flexion of the wrists. Tardive dystonia is a persistent dystonia as a result of a complication of drugs that block dopamine receptors. Clinical features of tardive dystonia are discussed in detail in Chapter 19.
Acute dystonic reactions
Acute dystonic reactions can occur from drugs that block dopamine receptors; these reactions are widely recognized and easily treated with antihistaminics and anticholinergics. Acute dystonia has also been reported with exposure to domperidone (Bonuccelli et al., 1991), amitriptyline (Ornadel et al., 1992), fluoxetine (Dave, 1994), clozapine (Kastrup et al., 1994; Thomas et al., 1994), and dextromethorphan (Graudins and Fern, 1996). Since domperidone is a peripherally-acting dopamine receptor blocker, the acute dystonic reaction suggests that some of the drug entered the CNS, at least in the affected patient, who had polycystic ovary syndrome. Amitriptyline would ordinarily not be expected to produce such a reaction; the clinical description is that typical for acute dystonia after a neuroleptic, but no such drug was known to have been taken by the patient.
The so-called atypical antipsychotics drugs have been shown to have less risk for causing acute dystonic reactions. In one study evaluating a population of patients consecutively admitted to a psychiatric intensive care unit, 1337 cases were treated with antipsychotics (Raja and Azzoni, 2001). The authors observed 41 cases (3.1%) of acute dystonic reactions. Four occurred with risperidone monotherapy, one with olanzapine, and one with quetiapine. The remaining cases were with medications labeled as typical antipsychotics.
Serotonergic agents have also been reported to induce acute dystonic reactions (Lopez-Alemany et al., 1997; Madhusoodanan and Brenner, 1997; Olivera, 1997). Serotonergic agents, such as fluoxetine, can inhibit dopaminergic neurons in the substantia nigra, worsening parkinsonism (Baldessarini and Marsh, 1992). This might somehow be related to the development of the dystonic reaction. How clozapine induces an acute dystonic reaction is unknown; it could relate to binding to the D2 receptor, the D4 receptor, or the 5-HT2 receptor.
Other secondary dystonias
A large number of injuries to the nervous system can result in secondary dystonia (Table 12.10). Head trauma and peripheral trauma, including dental procedures (Schrag et al., 1999), can induce generalized and segmental dystonia and focal dystonia, respectively. Neck trauma can result in cervical dystonia, and in one child, also camptocormia (Shuper et al., 2007). Retrocollis is more commonly associated with tardive dystonia, but can also be the result of head or neck trauma (Papapetropoulos et al., 2007a). Delayed-onset focal dystonia can follow electrical injury (Lim and Seet, 2007). Trauma may provoke the onset of PTD in an individual who carries the gene for this disorder (Fletcher et al., 1991c). Segmental axial dystonia was described with a closed head injury with small areas of encephalomalacia, including the caudate nucleus (Jabbari et al., 1992). Cervical dystonia can sometimes be secondary to lesions in the CNS, such as lacunar infarction in the putamen (Molho and Factor, 1993), and to lesions in the upper cervical spinal cord (Klostermann et al., 1993; Cammarota et al., 1995), including syringomyelia (Hill et al., 1999). Torticollis has also been reported to occur after electrocution (Colosimo et al., 1993) and in Moya-Moya disease (Yasutomo et al., 1993). Strokes can occasionally lead to delayed-onset hemidystonia. Perhaps in this category is the primary antiphospholipid syndrome (PAPS), an immune-mediated vascular disorder that has been found to result in hemidystonia in children (Angelini et al., 1993). Sjögren syndrome, also considered an autoimmune disorder, has been associated with dystonia (van den Berg, et al., 1999). X-linked agammaglobulinemia can result in a fatal dementing, dystonia–parkinsonism syndrome (Papapetropoulos et al., 2007b).
Encephalitis, usually from severe equine encephalitis, results in permanent dystonia. The common Asian encephalitis known as Japanese encephalitis can cause severe dystonia (Kalita and Misra, 2000; Murgod et al., 2001). Occasionally, dystonia can be psychogenic in origin (see Chapter 25), and it is reasonable to consider psychogenic dystonia as one of the many etiologies of secondary dystonia. Sometimes psychogenic dystonia occurs following trauma. When associated with pain, it may be diagnosed as reflex sympathetic dystrophy or complex regional pain syndrome, but many of the patients have a psychogenic mechanism (Ochoa, 1999); this syndrome has also been called causalgia–dystonia (Bhatia et al., 1993a).
Clues that dystonia might be secondary
In examining patients with dystonia, there are some clues in the history and neurologic examination that would suggest to the clinician that the patient’s dystonia is secondary rather than primary (Table 12.19).
Perhaps the most difficult form of dystonia to diagnose is psychogenic dystonia, which can occur in up to 5% of children presenting with what otherwise appears to be primary dystonia. Clues suggesting a psychogenic etiology are false (give-way) weakness, false sensory findings, inconsistent movements with changing patterns of involvement, incongruent movements not fitting with typical organic dystonia, self-inflicted injuries, deliberate slowness of movement, and multiple types of abnormal dyskinesias that do not fit into a single organic etiology (Fahn and Williams, 1988). Table 12.20 lists the clinical situations that provide clues that the clinician might be encountering a psychogenic movement disorder.
Table 12.20 Clues suggesting psychogenic dystonia (see also Chapter 25)
| A. Clues relating to the movements |
|
3 Incongruous movements and postures (movements don’t fit with recognized patterns or with normal physiological patterns)
|