[level-membership-for-pediatrics-category]
Chapter 102 Dysmorphology
Dysmorphology is the study of abnormalities of human form and the mechanisms that cause them. It is estimated that 1 in 40, or 2.5% of newborns, have a recognizable malformation or malformations at birth. In about half of these newborns, a single isolated malformation is found, whereas in the other half there are multiple malformations. It is estimated that 10% of pediatric hospital admissions involve known genetic conditions, 18% involve congenital defects of unknown etiology, and 40% of surgical admissions are of patients with congenital malformations. Twenty percent to 30% of infant deaths and 30-50% of deaths after the neonatal period are due to congenital abnormalities (http://www.marchofdimes.com/peristats/). In 2001, birth defects accounted for 1 in 5 infant deaths in the United States, with a rate of 137.6 deaths per 100,000 live births, which is higher than other causes such as preterm/low birthweight (109.5/100,000), sudden infant death syndrome (55.5/100,000), maternal complications of pregnancy (37.3/100,000), and respiratory distress syndrome (25.3/100,000).
Classification of Birth Defects
Congenital birth defects either are isolated, single defects or manifest as multiple anomalies in a single individual. Single primary defects can be classified according to the nature of the presumed cause of the defect as a malformation, dysplasia, deformation, or disruption (Table 102-1). Malformations and dysplasias both affect intrinsic structure. A malformation is a primary structural defect arising from a localized error in morphogenesis and resulting in the abnormal formation of a tissue or organ. Dysplasia refers to an abnormal organization of cells into tissues. The distinction of a malformation from a dysplasia may be helpful, but there is much overlap. Deformations and disruptions are secondary effects that result from forces generated extrinsic to the affected tissue or organ. A deformation is an alteration in shape or structure of a structure or organ that has differentiated normally. A disruption is a structural defect resulting from the destruction of a structure that had formed normally before the insult.
Table 102-1 MECHANISMS, TERMINOLOGY, AND DEFINITIONS OF DYSMORPHOLOGY
| TERMINOLOGY | DEFINITION | EXAMPLE |
|---|---|---|
| Malformation sequence | Single, local tissue morphogenesis abnormality that produces a chain of subsequent defects | DiGeorge sequence of primary fourth brachial arch and 3rd and 4th pharyngeal pouch defects that lead to aplasia or hypoplasia of the thymus and parathyroid glands, aortic arch anomalies, and micrognathia |
| Deformation sequence | Mechanical (uterine) forces that alter structure of intrinsically normal tissue | Oligohydramnios produces deformations by in utero compression of limbs (dislocated hips, equinovarus foot deformity), crumpled ears, dislocated nose, or small thorax |
| Disruption sequence | In utero tissue destruction after a period of normal morphogenesis | Amnionic membrane rupture sequence, leading to amputation of fingers/toes, tissue fibrosis, and destructive tissue bands |
| Dysplasia sequence | Poor organization of cells into tissues or organs | Neurocutaneous melanosis sequence with poor migration of melanocyte precursor cells from the neural crest to the periphery, manifesting as melanocytic hamartosis of skin, meninges, and so forth |
| Malformation syndrome | Appearance of multiple malformations in unrelated tissues without an understandable unifying cause; with enhanced genetic investigation, a single etiology may become identified |
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders.
Malformations and/or Dysplasias
Human malformations and dysplasias are caused by the combined effects of genes and environmental factors (Table 102-2). Some malformations are caused by single gene defects or abnormalities of multiple genes acting in concert, and the environment causes others. In 1996, it was thought that malformations were due to monogenic defects in 7.5% of patients; to chromosomal anomalies in 6%; to multigenic defects in 20%; and to known environmental factors, such as maternal diseases, infections, and teratogens, in 6-7% (Table 102-3); in the remaining 60-70% of patients, malformations were classified as due to unknown etiologies. A decade later, the percentages were somewhat higher for all categories of known causes of malformations, a change resulting from improved cytogenetic methods of detecting small chromosomal abnormalities as well as techniques for mapping and cloning disease genes. Since the previous edition of this book, an additional 10-20% of birth defects result from even smaller chromosomal abnormalities detectable by whole genome arrays using comparative genomic hybridization (array CGH) methods. In spite of these advances, we still do not know the causes or even the diagnosis for 40-50% of birth defects.
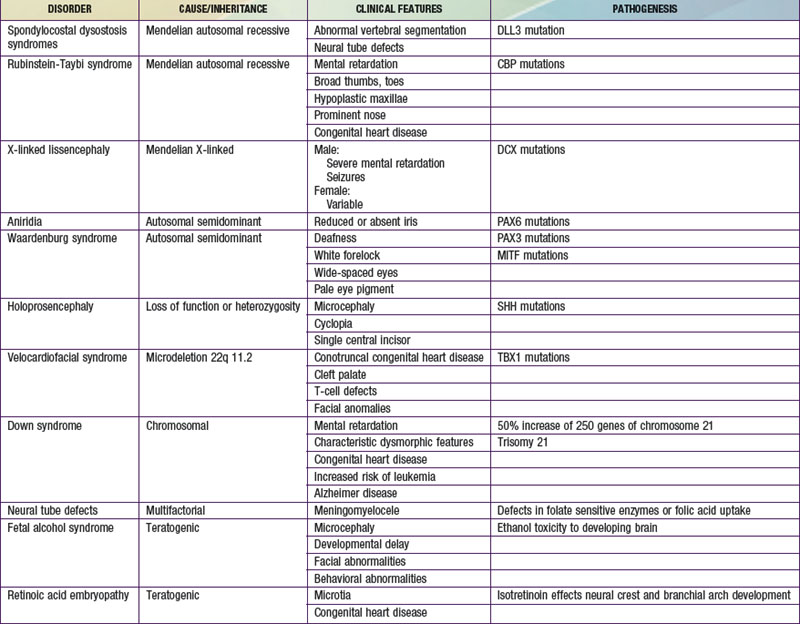
Table 102-3 CAUSES OF CONGENITAL MALFORMATIONS
MONOGENIC (7.5% of serious anomalies)
CHROMOSOMAL (6% of serious anomalies)
MATERNAL INFECTION (2% of serious anomalies)
Intrauterine infections (e.g., herpes simplex virus, cytomegalovirus, varicella-zoster virus, rubella virus, and toxoplasmosis)
MATERNAL ILLNESS (3.5% of serious anomalies)
UTERINE ENVIRONMENT (% unknown)
ENVIRONMENTAL AGENTS (% unknown)
MEDICATIONS (% unknown)
UNKNOWN ETIOLOGIES
SPORADIC SYNDROME COMPLEXES (ANOMALADS)
NUTRITIONAL
Low folic acid–neural tube defects
From Behrman RE, Kliegman RM, editors: Nelson’s essentials of pediatrics, ed 4, Philadelphia, 2002, WB Saunders.
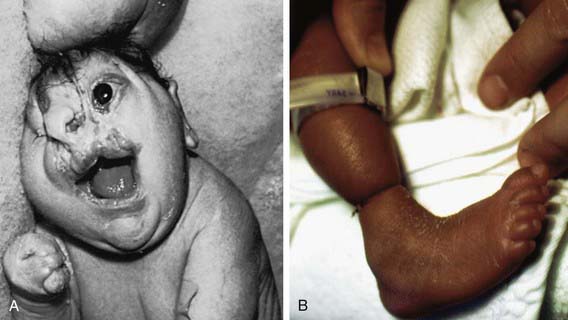
Many developmental abnormalities are caused by mutations in a single gene and display characteristic mendelian patterns of inheritance. The molecular etiology for more than 250 single gene disorders is known. Affected genes are often part of evolutionarily conserved signal transduction pathways, transcription factors, or regulatory proteins required for key developmental events. Some examples are listed in Table 102-2; they include autosomal recessive spondylocostal dysostosis (SCD) syndrome, the autosomal recessive Smith-Lemli-Opitz syndrome (SLOS), the autosomal dominant Rubinstein-Taybi syndrome, and the X-linked lissencephaly (“smooth brain”) syndrome. The SCD syndrome is etiologically heterogeneous and is often caused by mutations in the gene coding for delta-like 3 (DLL3), a ligand of the Notch receptors. The Notch/delta pathway is conserved throughout evolution and regulates a number of developmental events. Patients with SCD display a characteristic pattern of abnormal vertebral segmentation associated with a number of other malformations, such as neural tube defects. SLOS (Fig. 102-1) results from mutations in the sterol delta-7-reductase gene, an enzyme important in cholesterol biosynthesis. Patients with SLOS display syndactyly (fusion of the fingers and toes) often with polydactyly, upturned nose, ptosis, cryptorchidism, central nervous system hypoplasia, and holoprosencephaly. These mutations link cholesterol biosynthesis pathogenetically to the sonic hedgehog (SHH) pathway, because many of the features of the former disorder are related to defects in SHH, which is post-translationally modified by cholesterol (Chapter 80). Rubinstein-Taybi syndrome (see Fig. 102-1) results from heterozygous loss-of-function mutations in the gene coding for a broadly acting transcriptional coactivator called CBP, or CREB-binding protein. The CBP coactivator regulates the transcription of a number of genes, a fact that helps explain why patients with mutations in CBP have a wide-ranging phenotype that includes mental retardation, broad thumbs and toes, and congenital heart disease. One of the transcription factors that binds to CBP is GLI3, a transcription factor that is part of the SHH pathway (see Fig. 102-1). X-linked lissencephaly—a severe neuronal migration defect that in males causes a smooth brain with reduction or absence of gyri and sulci and in females gives rise to a variable pattern of mental retardation and seizures—is caused by mutations in DCX, a protein that regulates the activity of dynein motors and moves the nucleus during neuronal migration.
Other malformation syndromes are caused by chromosomal imbalance, multifactorial inheritance, and teratogens (see Tables 102-2 and 102-3). Down syndrome results from an extra dose of part or all of chromosome 21, a small chromosome that contains ≈ 200 known or predicted genes. It is most commonly caused by trisomy 21, which means that individuals with Down syndrome have an increased dose of as many as 250 genes contained on this chromosome (Chapter 76.1). Neural tube defects (NTDs) are an example of a disorder that displays multifactorial inheritance in the majority of cases. NTDs and a number of other congenital malformations, such as cleft lip and palate, recur in families, but several genes and environmental factors together contribute to the pathogenesis (see Table 102-2). Many of the genes involved in NTDs are unknown, so one cannot predict with certainty a mode of inheritance or a precise recurrence risk. Empiric risks can be provided on the basis of population studies and the presence of single or multiple relatives with the same malformation. However, an important gene/environment interaction has been identified for NTDs (Chapter 585.1). Folic acid status is associated with NTDs and can result from a combination of dietary deficiencies and increased utilization during pregnancy as well as from a common variant in the gene for an enzyme in the folate recycling pathway, 5,10-methylene-tetrahydrofolate reductase, that makes this enzyme less stable. These discoveries led to the recommendation that all women supplement their diets with 400-800 µg of folic acid per day 1 mo before pregnancy and during the 1st 2 mo of pregnancy. This supplementation has resulted in a reduction in the incidence of NTDs by 75%. Several teratogenic causes of birth defects have been described (see Tables 102-2 and 102-3). Ethanol causes a recognizable malformation syndrome called fetal alcohol syndrome (FAS) (Chapter 100.2). Children with FAS display microcephaly, developmental delay, hyperactivity, and facial dysmorphisms. Ethanol, which is toxic to the developing central nervous system, causes cell death of developing neurons.
Deformations
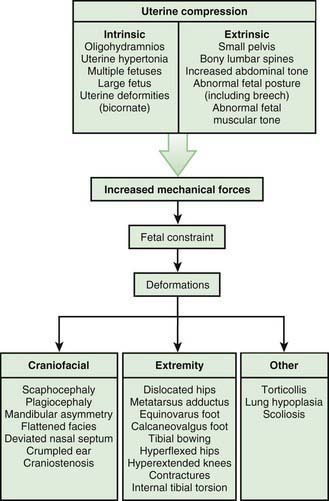
Most deformations involve the musculoskeletal system (Fig. 102-2). Fetal movement is required for the proper development of the normal musculoskeletal system, and anything that restricts fetal movement can cause a musculoskeletal deformation from intrauterine molding. It is important to recognize that deformations can be caused by problems either intrinsic or extrinsic to the developing fetus. Two major intrinsic causes of deformations are primary neuromuscular disorders and oligohydramnios, or decreased amniotic fluid, which is caused by renal defects. The major extrinsic causes of deformation are those that result in fetal crowding to restrict fetal movement. Examples of such extrinsic causes are oligohydramnios from chronic leakage of amniotic fluid, breech presentation (Fig. 102-3), and abnormal shape of the amniotic cavity. When a fetus is in the breech position, the incidence of deformations is increased 10-fold. The shape of the amniotic cavity has a profound effect on the shape of the fetus and is influenced by many factors, including uterine shape; volume of amniotic fluid; size and shape of the fetus; presence of more than one fetus; site of placental implantation; presence of uterine tumors; shape of the abdominal cavity, which is influenced by the pelvis, sacral promontory, and neighboring abdominal organs; and tightness of the abdominal musculature.
Disruption
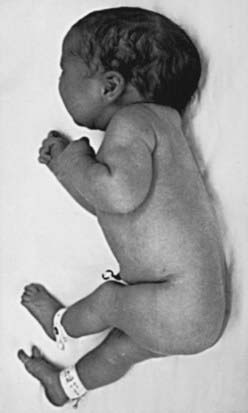
Disruption defects are caused by destruction of a previously normally formed part. At least two basic mechanisms are known to produce disruption. One involves entanglement followed by tearing apart or amputation of a normally developed structure, usually a digit, arm, or leg, by strands of amnion floating within amniotic fluid (amniotic bands) (Fig. 102-4). The second involves interruption of the blood supply to a developing part, which leads to infarction, necrosis, and/or resorption of structures distal to the insult. If interruption of the blood supply occurs early in gestation, the disruptive defect seen at term usually involves atresia, or absence of a particular part. If the infarction occurs later, necrosis is more likely to be present. Examples of disruptive single primary defects for which infarction has been implicated include nonduodenal intestinal atresia, gastroschisis, porencephaly, and terminal transverse limb reduction defects. Genetic factors usually play a minor role in the pathogenesis of disruptions; most are sporadic events in otherwise normal families. The prognosis for a disruptive defect is determined entirely by the extent and location of the tissue loss.
Molecular Mechanisms of Malformations
Inborn Errors of Development
Sonic Hedgehog Pathway as Model
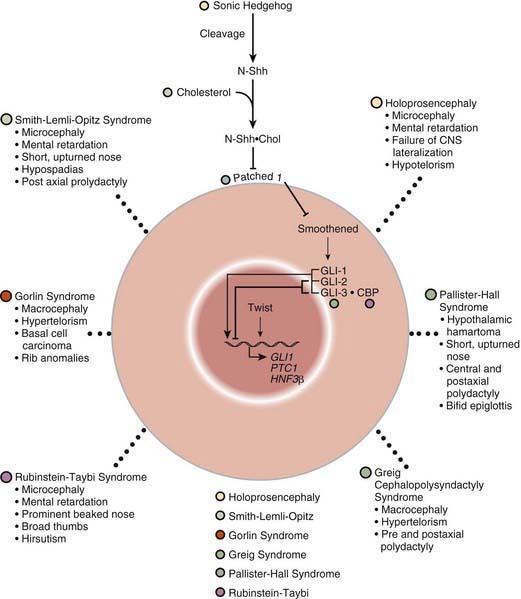
The SHH pathway is developmentally important during embryogenesis to induce controlled proliferation in a tissue-specific manner; disruption of specific steps in this pathway results in a variety of related developmental disorders and malformations (see Fig. 102-1). Activation of this pathway in the adult leads to abnormal proliferation and cancer. The SHH pathway transduces an external signal in the form of a ligand into changes in gene transcription by binding of the ligand to specific cellular receptors. SHH is a ligand expressed in the embryo in a variety of areas important for development of the brain, face, limbs, and the gut. Sporadic and inherited mutations are found to cause holoprosencephaly (see Fig. 102-1), a variably severe midline defect with phenotypes ranging from a single maxillary incisor with hypotelorism to cyclopia. SHH is processed by proteolytic cleavage to an active N-terminal form, which is then further modified by the addition of cholesterol. Defects in cholesterol biosynthesis, in particular the sterol delta-7-reductase gene, result in Smith-Lemli-Opitz syndrome (see Fig. 102-1). SLOS is also associated with holoprosencephaly. The modified and active form of SHH binds to its transmembrane receptor Patched (PTCH); there are two family members, PTCH-1 and PTCH-2. SHH binding to PTCH inhibits the activity of the transmembrane protein Smoothened (SMOH). SMOH act to suppress downstream targets of the SHH pathway, the GLI family of transcription factors, so inhibition of SMOH by PTCH results in activation of GLI1, GLI2, and GLI3, resulting in alteration of transcription of GLI targets. Somatic inactivating mutations in PTCH-1 and PTCH-2 act as tumor suppressors, whereas activating mutations in SMOH function as oncogenes, particularly in basal cell carcinomas and medulloblastomas. Germline inactivating mutations in PTCH-1 result in Gorlin syndrome (see Fig. 102-1), an autosomal dominant disorder characterized by dysmorphic features (short metacarpals, rib defects, broad face, and dental abnormalities), basal cell nevi that undergo malignant transformation, and an increased risk of cancers such as rhabdomyosarcoma and medulloblastoma. GLI1 amplification has been found in several human tumors, including glioblastoma, osteosarcoma, rhabdomyosarcoma, and B cell lymphomas; mutations or alterations in GLI3 have been found in Greig cephalopolysyndactyly syndrome (GCPS), Pallister-Hall syndrome (PHS), and postaxial polydactyly type A (and A/B) and preaxial polydactyly type IV (see Fig. 102-1). GCPS consists of hypertelorism, syndactyly, preaxial polydactyly, and broad thumbs and great toes. PHS is an autosomal dominant disorder characterized by postaxial polydactyly, syndactyly, hypothalamic hamartomas, imperforate anus, and, occasionally, holoprosencephaly. GLI3 binds to CBP, the protein that is haploinsufficient in the Rubinstein-Taybi syndrome. Disorders that are caused by mutations in genes that function together in a genetic developmental pathway commonly have overlapping clinical manifestations. These overlapping manifestations result from the expression domains of SHH important for development of the brain, face, limbs, and gut in the embryo. Brain defects are found in holoprosencephaly, SLOS, and PHS. Facial abnormalities are found in holoprosencephaly, Gorlin syndrome, GCPS, and PHS. Limb defects are found in SLOS, Gorlin syndrome, GCPS, PHS, and the polydactyly syndromes. Overexpression or activating mutations of the SHH pathway results in cancer, including basal cell carcinoma, medulloblastoma, glioblastoma, and rhabdomyosarcoma.
Chromosomal Imbalances
It has been recognized for more than 50 years that genomic imbalances that result from an additional copy of one whole human chromosome can result in a characteristic and recognizable syndrome. As previously discussed, an additional copy of chromosome 21 results in Down syndrome (Chapter 76); loss of one of the X chromosomes results in Turner syndrome (see Chapter 76 for discussion of syndromes with whole chromosomal imbalances). With the advent of higher-resolution cytogenetics techniques and standardization of chromosome identification using chromosomal preparations, it became possible to identify subchromosomal deletions and duplications. A number of recurrent deletions and duplications were identified that resulted in characteristic and recognizable syndrome (Chapter 76, Table 76-12), such as Williams syndrome (deletion of 7q11.23), Miller-Dieker syndrome (deletion of 17p13.3), Smith-Magenis syndrome (deletion of 17p11.2), and velocardiofacial/DiGeorge syndrome (deletion of 22q11.2). Array CGH (or single-nucleotide polymorphism [SNP]–based genotyping with dosage detection) has made it possible to uncover smaller microdeletions and microduplications associated with various birth defects, mental retardation, and neuropsychiatric disorders. The sensitivity and specificity of array CGH is making it the technique of choice for the initial evaluation of a child with multiple congenital anomalies and/or mental retardation, although it is important to note that all individuals carry dozens of small microdeletions and microduplications as normal variants. Therefore, it is important to examine any of these findings in children with birth defects with databases of normal variants detected in individuals without such birth defects. Array CGH is the preferred method for detecting possible genomic abnormalities associated with multiple congenital anomalies and/or mental retardation.
Approach to the Dysmorphic Child
History
The history for a child with birth defects includes a number of elements that are related to etiologic factors. The first is the pedigree or family history that is necessary to assess the inheritance pattern, or lack thereof, of the disorder. For disorders that have simple mendelian inheritance patterns, the recognition of that pattern can be critical to help narrow the differential diagnosis. A number of common birth defects have complex genetic contributions, such as isolated cleft palate and spina bifida. The recognition of a close relative (or the fetus of a close relative) affected with a birth defect that is similar to that of the proband can be quite useful. A three-generation pedigree is sufficient for this purpose (Chapter 75).
The perinatal history is an essential component of the history (Chapter 88.1). It includes the pregnancy history of the mother (useful for recognition of recurrent miscarriages that may be a sign of a familial chromosomal disorder), factors that may relate to deformations or disruptions (oligohydramnios), and maternal exposures to teratogenic drugs or chemicals (methyl mercury, isotretinoin, and ethanol are potential causes of microcephaly). Although recognition of known teratogens is an important part of the history, it is important to know that many more agents are impugned as teratogenic than are confirmed to be so. Physicians are encouraged to consult experts in teratology and expert information sources such as Teris (http://depts.washington.edu/~terisweb/teris) to analyze specific potential teratogens.
Physical Examination
The physical examination is essential to the diagnosis of a dysmorphic syndrome. The essential element of the evaluation is objective assessment of the structure of the child. The clinician needs to perform an organized and systematic cataloguing of the size and structure of various body structures. Familiarity with the nomenclature of dysmorphic signs is helpful (Table 102-4). The size and shape of the head is relevant, as many children with Down syndrome have mild microcephaly and brachycephaly (shortened anteroposterior dimension of the skull). Eye position and shape are useful signs for many disorders. There are a number of reference standards with which pediatric physical measurements (interpupillary distance) can be compared. It is also useful to categorize abnormalities as “major” or “minor” birth defects. The former are those that either cause dysfunction (absence of a digit) or require surgical correction (polydactyly), and the latter those that neither cause significant dysfunction nor require surgical correction (mild cutaneous syndactyly) (Table 102-5; Fig. 102-5). By cataloging every available physical parameter, the clinician can recognize the diagnosis or at least have enough information for intelligent discussion of the patient with a consultant.
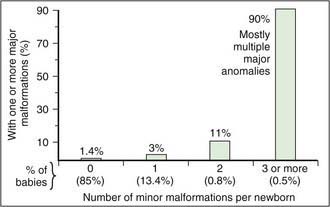
Figure 102-5 Frequency of major malformations in relation to the number of minor anomalies detected in a given newborn baby.
(From Jones KJ: Smith’s recognizable patterns of human malformation, ed 6, Philadelphia, 2006, Saunders.)
Table 102-4 DEFINITIONS OF COMMON CLINICAL SIGNS OF DYSMORPHIC SYNDROMES
| SIGN | DEFINITION |
|---|---|
| Brachycephaly | A condition in which head shape is shortened from front to back along the sagittal plane; the skull is rounder than normal |
| Brachydactyly | A condition of having short digits |
| Brushfield spots | Speckled white rings about two thirds of the distance to the periphery of the iris of the eye |
| Camptodactyly | Permanent flexion of one or more fingers associated with missing inner phalangeal creases indicating lack of finger movement from before 8 wk of gestation |
| Clinodactyly | A medial or lateral curving of the fingers; usually refers to incurving of the 5th finger |
| Hypoplastic nail | An unusually small nail on a digit |
| Low-set ears | This designation is made when the helix meets the cranium at a level below a horizontal plane that is an extension of a line through both inner canthi |
| Melia | A suffix meaning “limb” (e.g., amelia—missing limb; brachymelia—short limb) |
| Ocular hypertelorism | Increased distance between the pupils of the two eyes |
| Plagiocephaly | A condition in which head shape is asymmetric in the sagittal or coronal plane; can result from asymmetry in suture closure or from asymmetry of brain growth |
| Posterior parietal hair whorl | A single whorl occurs to the right or left of midline and within 2 cm anterior to the posterior fontanel in 95% of cases. The whorl represents the focal point from which the posterior scalp skin was under growth tension during brain growth between the 10th and 16th wk of fetal development. Aberrant position of the whorl reflects an early defect in brain development. |
| Postaxial polydactyly | Extra finger or toe present on the lateral side of the hand or foot |
| Preaxial polydactyly | Extra finger or toe present on the medial side of the hand or foot |
| Prominent lateral palatine ridges | Relative overgrowth of the lateral palatine ridges secondary to a deficit of tongue thrust into the hard palate |
| Scaphocephaly | A condition in which the head is elongated from front to back in the sagittal plane; most normal skulls are scaphocephalic |
| Shawl scrotum | The scrotal skin joins around the superior aspect of the penis and represents a mild deficit in full migration of the labial-scrotal folds |
| Short palpebral fissures | Decreased horizontal distance of the eye based on measurement from the inner to the outer canthus |
| Syndactyly | Incomplete separation of the fingers. It most commonly occurs between the 3rd and 4th fingers and between the 2nd and 3rd toes. |
| Synophrys | Eyebrows that meet in the midline |
| Telecanthus | Lateral displacement of the inner canthi. The inner canthal distance is increased, but the inner pupillary distance is normal. |
| Widow’s peak | V-shaped midline, downward projection of the scalp hair in the frontal region. It represents an upper forehead intersection of the bilateral fields of periocular hair growth suppression. It usually occurs because the fields are widely spaced, as in ocular hypertelorism. |
Table 102-5 MINOR ANOMALIES AND PHENOTYPE VARIANTS*
CRANIOFACIAL
EYE
EAR
SKIN
HAND
FOOT
OTHERS
* Approximately 15% of newborns have one minor anomaly, 0.8% have two minor anomalies, and 0.5% have three. If two minor anomalies are present, the probability of an underlying syndrome or a major anomaly (congenital heart disease, renal, central nervous system, limbic) is fivefold that in the general population. If three minor anomalies are present, the probability that there is a major anomaly is 20-30%.
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, Elsevier Saunders, 2004.
Laboratory Studies
The laboratory evaluation of the dysmorphic child is helpful but complex. Cytogenetics with Giemsa-banded peripheral leukocyte karyotype (or chromosome) analysis has been the gold standard and has been performed in most evaluations of the dysmorphic child (Table 102-6). Array CGH and SNP genotyping with copy number variation (dosage detection) are the most sensitive methods for the detection of genomic alterations associated with multiple congenital anomalies. A practical reason for ordering cytogenetic studies early in the diagnostic process is that it typically takes 7-12 days for results. As long as standard karyotype analysis remains the method used in the initial evaluation of birth defects, one must consider other adjunct cytogenetic tests that can be used to evaluate chromosome defects. Because many chromosome abnormalities include derangement of the termini (telomeres) of the chromosomes, assays should detect small (submicroscopic) duplications or deletions of the termini of the chromosomes. These tests are clinically available and may be considered if results of standard chromosome analysis are negative and until it has been supplanted by the more sensitive method of array CGH or SNP-based typing.
Diagnosis
The clinician should now organize the findings by their specificity into potential developmental pathophysiologic processes. The specificity assessment is the simplest. If a child has a patent ductus arteriosus (PDA), mild growth retardation, mild microcephaly, and holoprosencephaly (MRI finding of failure to lateralize the forebrain), micropenis, and ptosis, these findings can be prioritized. The PDA, ptosis, mild growth retardation, and mild microcephaly are nonspecific findings (present in many disorders or often present as isolated features not part of a syndrome), whereas holoprosencephaly and micropenis are present in fewer syndromes and are never normal variants. With this recognition, the clinician can search for disorders that include both holoprosencephaly and micropenis. The search can be performed manually using the features index of a textbook such as Smith’s Recognizable Patterns of Human Malformation or a computerized database such as the Winter-Baraitser Dysmorphology Database (www.lmdatabases.com/about_lmd.html). Searching for disorders with both findings leads quickly to a modest list of only 21 disorders. One of these is SLOS. The identification of this possible diagnosis prompts the physician to return to the bedside, realize that many of the nonspecific features in the child are common in SLOS, and make a tentative diagnosis of this disorder. Although holoprosencephaly is an uncommon manifestation of SLOS, this manifestation makes sense because of the known pathogenetic link between sonic hedgehog and cholesterol biosynthesis. Because this disorder is caused by mutations in the sterol delta-7-reductase gene and is associated with elevated 7-dehydrocholesterol, the pediatrician can initiate a consultation with the clinical geneticist for suspected SLOS. The consultant can then confirm the diagnosis and begin the process of identifying a laboratory to verify the diagnosis.
Management and Counseling
The second major benefit of an accurate diagnosis is that it provides data for appropriate recurrence risk estimates. Genetic disorders may have direct effects on only one member of the family, but the diagnosis of the condition has implications for the entire family. One or both parents may be carriers; siblings may be carriers or may wish to know their at-risk status when they reach their reproductive years. Recurrence risk provision is one facet of genetic counseling, which should be a component of all evaluations for families affected with birth defects or other heritable disorders (Chapter 72).
Ang ESJr, Gluncic V, Duque A, et al. Prenatal exposure to ultrasound waves impacts neuronal migration in mice. Proc Natl Acad Sci U S A. 2006;103:12903-12910.
Berry RJ, Buehler JW, Strauss LT, et al. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep. 1987;102:171-181.
Boyle RJ. Effects of certain prenatal drugs on the fetus and newborn. Pediatr Rev. 2002;23:17-24.
Breen DP, Davenport RJ. Teratogenicity of antiepileptic drugs. BMJ. 2006;333:615-616.
Cassidy SB, Allanson JE. Management of genetic syndromes, Hoboken. NJ. 2005.
Cohen MMJr. An introduction to sonic hedgehog signaling. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Cooper WO, Hernandez-Diaz S, Arbogast PG, et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354:2443-2451.
Epstein CJ. Human malformations and their genetic basis. In: Epstein CJ, Erickson RP, Wynshaw-Boris . Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Epstein CJ. The new dysmorphology: application of insights from basic developmental biology to the understanding of human birth defects. Proc Natl Acad Sci U S A. 1995;92:8566-8573.
Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Gleeson JG. LIS1 and DCX and classical lissencephaly. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Hall JG, Froster-Iskenius UG, Allanson JE. Handbook of normal physical measurements. NewYork: Oxford Medical Publications; 1989.
Hassold TJ, Patterson D. Down syndrome: a promising future together. New York: Wiley-Liss; 1999.
Ho KS, Scott MP. Sonic hedgehog in the nervous system: functions, modifications and mechanisms. Curr Opin Neurobiol. 2002;12:57-63.
Hoekelman RA, Pless IB. Decline in mortality among young Americans during the 20th century: prospects for reaching national mortality reduction goals for 1990. Pediatrics. 1988;82:582-595.
Holmes LB. Inborn errors of morphogenesis: a review of localized hereditary malformations. N Engl J Med. 1974;291:763-773.
Irons M. DHCR7 and the Smith-Lemli-Opitz (RSH) syndrome. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Jones KL. Smith’s recognizable patterns of human malformation, ed 6. Philadelphia: WB Saunders; 2005.
Kingston HM. Genetic assessment and pedigree analysis. In Rimoin DL, Connor JM, Pyeritz RE, et al, editors: Emery and Rimoin’s principles and practice of medical genetics, ed 4, New York: Churchill Livingstone, 2002.
Lu X, Shaw CA, Patel A, et al. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327.
Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417-422.
Mitchell LE, Adzick NS, Melchionne J, et al. Spina bifida. Lancet. 2004;364:1885-1895.
Miyamo A, Weinmaster G. Introduction to notch signaling. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Mulliken JB. The craniofacial surgeon as amateur geneticist. J Craniofac Surg. 2002;13:3-17.
Oskarsdottir S, Persson C, Eriksson BO, et al. Presenting phenotype in 100 children with 22q11 deletion syndrome. Eur J Pediatr. 2005;164:146-153.
Park SM, Mathur R, Smith GCS. Congenital anomalies after treatment for infertility. BMJ. 2006;333:665-666.
Petrij F, Breuning MH, Hennekam RCM, et al. CBP and the Rubinstein-Taybi syndrome. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Plunkett KS, Simpson JL. A general approach to genetic counseling. Obstet Gynecol Clin North Am. 2002;29:265-276.
Quinlan RJ, Tobin JL, Beales PL. Modeling ciliopathies: primary cilia in development and disease. Curr Top Dev Biol. 2008;84:249-310.
Rauch A, Hoyer J, Guth S, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A. 2006;140:2063-2074.
Rimoin DL, Connor JM, Pyeritz RE, et al, editors. Emery and Rimoin’s principles and practice of medical genetics, ed 4, New York: Churchill Livingstone, 2002.
Salman M, Jhanwar SC, Ostrer H. Will the new cytogenetics replace the old cytogenetics? Clin Genet. 2004;66:265-275.
Sharp AJ, Locke DP, McGrath SD, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78-88.
Turnpenny PD, Kusumi K. DLL3 and spondylocostal dysostosis. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford University Press, 2004.
Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-GLI pathway in human development and disease. Am J Hum Genet. 2000;67:1047-1054.
Wagenstaller J, Spranger S, Lorenz-Depiereux B, et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet. 2007;81:768-779.
Wilson GN. Genomics of human dysmorphogenesis. Am J Med Genet. 1992;42:187-196.
Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366-1373.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 102 Dysmorphology
Dysmorphology is the study of abnormalities of human form and the mechanisms that cause them. It is estimated that 1 in 40, or 2.5% of newborns, have a recognizable malformation or malformations at birth. In about half of these newborns, a single isolated malformation is found, whereas in the other half there are multiple malformations. It is estimated that 10% of pediatric hospital admissions involve known genetic conditions, 18% involve congenital defects of unknown etiology, and 40% of surgical admissions are of patients with congenital malformations. Twenty percent to 30% of infant deaths and 30-50% of deaths after the neonatal period are due to congenital abnormalities (http://www.marchofdimes.com/peristats/). In 2001, birth defects accounted for 1 in 5 infant deaths in the United States, with a rate of 137.6 deaths per 100,000 live births, which is higher than other causes such as preterm/low birthweight (109.5/100,000), sudden infant death syndrome (55.5/100,000), maternal complications of pregnancy (37.3/100,000), and respiratory distress syndrome (25.3/100,000).
Classification of Birth Defects
Congenital birth defects either are isolated, single defects or manifest as multiple anomalies in a single individual. Single primary defects can be classified according to the nature of the presumed cause of the defect as a malformation, dysplasia, deformation, or disruption (Table 102-1). Malformations and dysplasias both affect intrinsic structure. A malformation is a primary structural defect arising from a localized error in morphogenesis and resulting in the abnormal formation of a tissue or organ. Dysplasia refers to an abnormal organization of cells into tissues. The distinction of a malformation from a dysplasia may be helpful, but there is much overlap. Deformations and disruptions are secondary effects that result from forces generated extrinsic to the affected tissue or organ. A deformation is an alteration in shape or structure of a structure or organ that has differentiated normally. A disruption is a structural defect resulting from the destruction of a structure that had formed normally before the insult.
Table 102-1 MECHANISMS, TERMINOLOGY, AND DEFINITIONS OF DYSMORPHOLOGY
| TERMINOLOGY | DEFINITION | EXAMPLE |
|---|---|---|
| Malformation sequence | Single, local tissue morphogenesis abnormality that produces a chain of subsequent defects | DiGeorge sequence of primary fourth brachial arch and 3rd and 4th pharyngeal pouch defects that lead to aplasia or hypoplasia of the thymus and parathyroid glands, aortic arch anomalies, and micrognathia |
| Deformation sequence | Mechanical (uterine) forces that alter structure of intrinsically normal tissue | Oligohydramnios produces deformations by in utero compression of limbs (dislocated hips, equinovarus foot deformity), crumpled ears, dislocated nose, or small thorax |
| Disruption sequence | In utero tissue destruction after a period of normal morphogenesis | Amnionic membrane rupture sequence, leading to amputation of fingers/toes, tissue fibrosis, and destructive tissue bands |
| Dysplasia sequence | Poor organization of cells into tissues or organs | Neurocutaneous melanosis sequence with poor migration of melanocyte precursor cells from the neural crest to the periphery, manifesting as melanocytic hamartosis of skin, meninges, and so forth |
| Malformation syndrome | Appearance of multiple malformations in unrelated tissues without an understandable unifying cause; with enhanced genetic investigation, a single etiology may become identified |
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders.
Malformations and/or Dysplasias
Human malformations and dysplasias are caused by the combined effects of genes and environmental factors (Table 102-2). Some malformations are caused by single gene defects or abnormalities of multiple genes acting in concert, and the environment causes others. In 1996, it was thought that malformations were due to monogenic defects in 7.5% of patients; to chromosomal anomalies in 6%; to multigenic defects in 20%; and to known environmental factors, such as maternal diseases, infections, and teratogens, in 6-7% (Table 102-3); in the remaining 60-70% of patients, malformations were classified as due to unknown etiologies. A decade later, the percentages were somewhat higher for all categories of known causes of malformations, a change resulting from improved cytogenetic methods of detecting small chromosomal abnormalities as well as techniques for mapping and cloning disease genes. Since the previous edition of this book, an additional 10-20% of birth defects result from even smaller chromosomal abnormalities detectable by whole genome arrays using comparative genomic hybridization (array CGH) methods. In spite of these advances, we still do not know the causes or even the diagnosis for 40-50% of birth defects.
Table 102-3 CAUSES OF CONGENITAL MALFORMATIONS
MONOGENIC (7.5% of serious anomalies)
CHROMOSOMAL (6% of serious anomalies)
MATERNAL INFECTION (2% of serious anomalies)
Intrauterine infections (e.g., herpes simplex virus, cytomegalovirus, varicella-zoster virus, rubella virus, and toxoplasmosis)
MATERNAL ILLNESS (3.5% of serious anomalies)
UTERINE ENVIRONMENT (% unknown)
ENVIRONMENTAL AGENTS (% unknown)
MEDICATIONS (% unknown)
UNKNOWN ETIOLOGIES
SPORADIC SYNDROME COMPLEXES (ANOMALADS)
NUTRITIONAL
Low folic acid–neural tube defects
From Behrman RE, Kliegman RM, editors: Nelson’s essentials of pediatrics, ed 4, Philadelphia, 2002, WB Saunders.
Many developmental abnormalities are caused by mutations in a single gene and display characteristic mendelian patterns of inheritance. The molecular etiology for more than 250 single gene disorders is known. Affected genes are often part of evolutionarily conserved signal transduction pathways, transcription factors, or regulatory proteins required for key developmental events. Some examples are listed in Table 102-2; they include autosomal recessive spondylocostal dysostosis (SCD) syndrome, the autosomal recessive Smith-Lemli-Opitz syndrome (SLOS), the autosomal dominant Rubinstein-Taybi syndrome, and the X-linked lissencephaly (“smooth brain”) syndrome. The SCD syndrome is etiologically heterogeneous and is often caused by mutations in the gene coding for delta-like 3 (DLL3), a ligand of the Notch receptors. The Notch/delta pathway is conserved throughout evolution and regulates a number of developmental events. Patients with SCD display a characteristic pattern of abnormal vertebral segmentation associated with a number of other malformations, such as neural tube defects. SLOS (Fig. 102-1) results from mutations in the sterol delta-7-reductase gene, an enzyme important in cholesterol biosynthesis. Patients with SLOS display syndactyly (fusion of the fingers and toes) often with polydactyly, upturned nose, ptosis, cryptorchidism, central nervous system hypoplasia, and holoprosencephaly. These mutations link cholesterol biosynthesis pathogenetically to the sonic hedgehog (SHH) pathway, because many of the features of the former disorder are related to defects in SHH, which is post-translationally modified by cholesterol (Chapter 80). Rubinstein-Taybi syndrome (see Fig. 102-1) results from heterozygous loss-of-function mutations in the gene coding for a broadly acting transcriptional coactivator called CBP, or CREB-binding protein. The CBP coactivator regulates the transcription of a number of genes, a fact that helps explain why patients with mutations in CBP have a wide-ranging phenotype that includes mental retardation, broad thumbs and toes, and congenital heart disease. One of the transcription factors that binds to CBP is GLI3, a transcription factor that is part of the SHH pathway (see Fig. 102-1). X-linked lissencephaly—a severe neuronal migration defect that in males causes a smooth brain with reduction or absence of gyri and sulci and in females gives rise to a variable pattern of mental retardation and seizures—is caused by mutations in DCX, a protein that regulates the activity of dynein motors and moves the nucleus during neuronal migration.
Other malformation syndromes are caused by chromosomal imbalance, multifactorial inheritance, and teratogens (see Tables 102-2 and 102-3). Down syndrome results from an extra dose of part or all of chromosome 21, a small chromosome that contains ≈ 200 known or predicted genes. It is most commonly caused by trisomy 21, which means that individuals with Down syndrome have an increased dose of as many as 250 genes contained on this chromosome (Chapter 76.1). Neural tube defects (NTDs) are an example of a disorder that displays multifactorial inheritance in the majority of cases. NTDs and a number of other congenital malformations, such as cleft lip and palate, recur in families, but several genes and environmental factors together contribute to the pathogenesis (see Table 102-2). Many of the genes involved in NTDs are unknown, so one cannot predict with certainty a mode of inheritance or a precise recurrence risk. Empiric risks can be provided on the basis of population studies and the presence of single or multiple relatives with the same malformation. However, an important gene/environment interaction has been identified for NTDs (Chapter 585.1). Folic acid status is associated with NTDs and can result from a combination of dietary deficiencies and increased utilization during pregnancy as well as from a common variant in the gene for an enzyme in the folate recycling pathway, 5,10-methylene-tetrahydrofolate reductase, that makes this enzyme less stable. These discoveries led to the recommendation that all women supplement their diets with 400-800 µg of folic acid per day 1 mo before pregnancy and during the 1st 2 mo of pregnancy. This supplementation has resulted in a reduction in the incidence of NTDs by 75%. Several teratogenic causes of birth defects have been described (see Tables 102-2 and 102-3). Ethanol causes a recognizable malformation syndrome called fetal alcohol syndrome (FAS) (Chapter 100.2). Children with FAS display microcephaly, developmental delay, hyperactivity, and facial dysmorphisms. Ethanol, which is toxic to the developing central nervous system, causes cell death of developing neurons.
Deformations
Most deformations involve the musculoskeletal system (Fig. 102-2). Fetal movement is required for the proper development of the normal musculoskeletal system, and anything that restricts fetal movement can cause a musculoskeletal deformation from intrauterine molding. It is important to recognize that deformations can be caused by problems either intrinsic or extrinsic to the developing fetus. Two major intrinsic causes of deformations are primary neuromuscular disorders and oligohydramnios, or decreased amniotic fluid, which is caused by renal defects. The major extrinsic causes of deformation are those that result in fetal crowding to restrict fetal movement. Examples of such extrinsic causes are oligohydramnios from chronic leakage of amniotic fluid, breech presentation (Fig. 102-3), and abnormal shape of the amniotic cavity. When a fetus is in the breech position, the incidence of deformations is increased 10-fold. The shape of the amniotic cavity has a profound effect on the shape of the fetus and is influenced by many factors, including uterine shape; volume of amniotic fluid; size and shape of the fetus; presence of more than one fetus; site of placental implantation; presence of uterine tumors; shape of the abdominal cavity, which is influenced by the pelvis, sacral promontory, and neighboring abdominal organs; and tightness of the abdominal musculature.
