CHAPTER 13 DNA Packaging in Chromatin and Chromosomes
Chromosomal DNA molecules of eukaryotes are thousands of times longer than the diameter of the nucleus and must therefore be highly compacted throughout the cell cycle. This folding is accomplished by combining the DNA with structural proteins to make chromatin. A hierarchy of levels of chromatin folding compacts the DNA but permits transcriptional machinery access to those regions of the chromosome required for gene expression.
The First Level of Chromosomal DNA Packaging: The Nucleosome
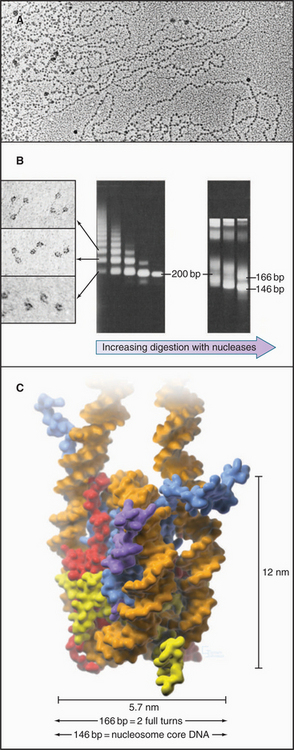
The continuous DNA fiber of each chromosome links hundreds of thousands of nucleosomes in series. Individual nucleosomes can be isolated following cleavage of DNA between neighboring particles. Random digestion of chromatin by DNA-cutting enzymes called nucleases initially yields a mixture of particles consisting of one or more nucleosomes containing multiples of about 200 base pairs of DNA (Fig. 13-1). Continued nuclease cleavage yields a stable particle with 146 base pairs of DNA (1.75 turns of the DNA around the protein core). This is called a nucleosome core particle.
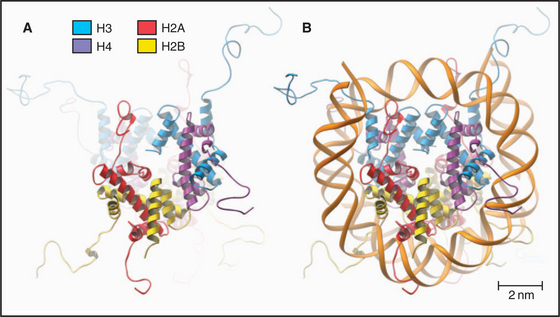
The nucleosome core particle is disk-shaped, with DNA coiled in a left-handed superhelix around an octamer of core histones. This octamer consists of a central tetramer composed of two closely linked H3:H4 heterodimers, flanked on either side by two H2A:H2B heterodimers. High-resolution crystal structures of nucleo-some core particles revealed that each core histone has a compact domain of 70 to 100 amino acid residues that adopts a characteristic Z-shaped “histone fold” consisting of a long α-helix flanked by two shorter α-helices (Fig. 13-2).
Epigenetics and the Histone Code
The revolution in biology that began with the structure of DNA and the realization that the sequence of bases in DNA provides a code that specifies the structure of proteins culminated 50 years later with the near complete sequencing of all the gene-rich portions of the human genome. To take advantage of this coding information, cells must control when to use it. Initial studies of the processes controlling gene expression focused on regulation of transcription by proteins that bind specific DNA sequences at the 5′ end of genes (see Chapter 15), as this is the way in which bacteria regulate their genes. This is now known to be only part of the story.
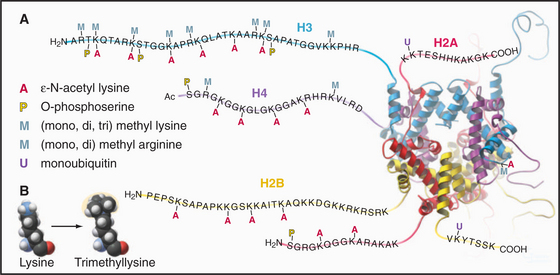
Eukaryotes impose another level of regulation on the utilization of their genes. This has been referred to as a histone code. The histone code hypothesis proposes that combinations of posttranslational modifications of histones are “read” by proteins that bind modified histones and then dictate whether particular regions of chromatin are transcribed by RNA polymerases or are held in an inactive state. Posttranslational modifications of histones include acetylation, phosphorylation, methylation, ubiquitination, and poly(ADP)ribosylation at many sites in the N-terminal tails and elsewhere (Fig. 13-3). Chromatin states created by histone modifications can be stably inherited through many rounds of cell division. Thus, this hypothesis can explain the phenomenon of epigenetic regulation (see Fig. 12-10): the stable, heritable regulation of chromosomal functions by information that is not simply encoded in the DNA sequence.
Regulation of Chromatin Structure by the Histone N-Terminal Tails
The N-terminal histone tails provide a molecular “handle” to manipulate DNA accessibility in chromatin (Fig. 13-3). This complex area can only be outlined here. The two key modifications contributing to the histone code are acetylation and methylation of lysine residues. Histones with acetylated lysines are generally associated with “open” chromatin that is permissive for RNA transcription, while histones with methylated lysines can be associated with either “open” or “closed” chromatin states. It should be emphasized that the histone code is complex and not fully understood. Since the histone modifications are read as combinations, individual modifications do not necessarily always have the same consequences. One example of this is the phosphorylation of histone H3 on serine 10 (H3-S10P). In mitotic cells, this correlates with a condensed and transcriptionally inactive chromatin structure, but when combined with acetylation of surrounding amino acid residues, it is also associated with the activation of gene transcription as nonproliferating cells reenter the cell cycle (see Chapter 41).
Acetylation reduces the net positive charge of the N-terminal domain, causing the chromatin to adopt an “open” conformation that is more favorable to transcription, as the histones bind less tightly to DNA. Acetylation also provides binding sites for a number of proteins with an approximately 100-amino-acid sequence motif called a bromodomain. Bromodomain binding to acetylated histone N-terminal tails is analogous to the binding of SH2 domains to phosphorylated tyrosine in cellular signaling pathways (see Fig. 25-10). Bromodomain-containing enzymes recruited to chromatin by acetylated histones often modify histones in other ways that promote or limit the accessibility of the DNA for transcription into RNA.
Proteins called transcription factors regulate gene expression by binding specific DNA sequences in promoter regions adjacent to the coding sequences of genes and recruiting transcriptional machinery (RNA polymerases and associated proteins) to the gene (see Fig. 15-19). Many transcription factors recruit a protein complex, called a coactivator, that facilitates loading of the transcriptional apparatus onto the gene. Often, coactivators are enzymes that modify N-terminal histone tails. One yeast coactivator contains over 10 proteins, including a histone acetyltransferase that transfers acetate groups from acetyl coenzyme A (CoA) to the ε-amino groups of lysine-14 and lysine-8 in the N-terminal tails of histone H3 (Fig. 13-4). Histone acetylation is crucial for life. Yeast cells die if these lysines are mutated to arginines, thus preserving their positive charge but preventing them from being acetylated.
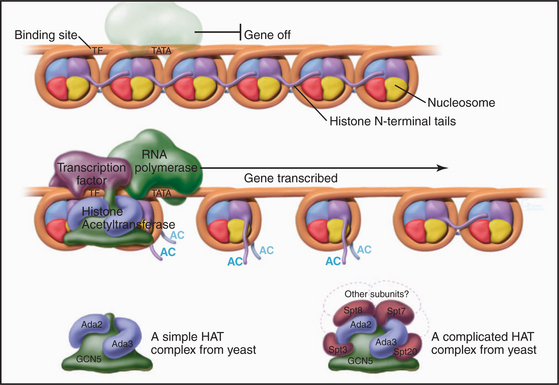
Figure 13-4 Acidic transcription factors (purple) bind specific DNA sequences and recruit coactivators to the 5′ ends of genes. Many of these coactivators have histone acetyltransferase activity and work by acetylating the N-terminal tails of the core histones, thereby loosening the chromatin structure and promoting the binding and activation of the RNA polymerase holoenzyme (see Chapter 15). The coactivators vary in composition and complexity from the relatively simple histone acetyltransferase complex (bottom left) to the huge and elaborate SAGA complex (bottom right). (AC, acetylation; TATA, DNA sequence in the gene promoter [see Chapter 15]). In this side view, only one of the two turns of DNA around the nucleosome is seen. GCN5, Ada2, Ada3, Spt3, Spt7, Spt8, and Spt20 are the names of budding yeast genes whose products are found in these complexes.
Histone acetylation is dynamic. Just as transcriptional coactivators contain histone acetyltransferases that add acetyl groups to nucleosomes and promote gene activation, so corepressors, which are recruited in a similar manner, can contain histone deacetylases that remove acetyl groups from selected lysine residues. This tends to inactivate gene expression. This mechanism regulates cell cycle progression during the G1 phase of the cell cycle (see Fig. 41-8).
In addition to marking nucleosomes by modification of their N-terminal tails, cells also use the energy provided by ATP hydrolysis to actively remodel nucleosomes. This involves complex protein “machines” that can alter nucleosome structure, move nucleosomes around, or both. Two large “machines” in yeast—RSC (remodels the structure of chromatin) with 15 subunits and SWI/SNF (switch/sniff) with 11 subunits—each has a key subunit that utilizes ATP hydrolysis to translocate along the DNA helix. One proposal is that these “machines” use ATP hydrolysis to force an extra 40 to 60 base pairs of DNA onto the nucleosome. Since this excess DNA cannot fit smoothly against the surface of the histone octamer, it presumably bulges out in a loop from the nucleosomal surface. If the position of this loop migrates around the surface of the nucleosome, the nucleosome will “jump” 40 to 60 base pairs along the DNA. This process can uncover sequences that are important for gene regulation that had been hidden by association with a nucleosome. Alternatively, this mechanism may be used to loosen the nucleosome and allow the exchange of histone dimers in and out.
Histone Acetylation and Nucleosome Assembly
During DNA replication, existing nucleosomes are partitioned randomly between daughter DNA strands. Newly assembled nucleosomes then fill the gaps. When not associated with DNA, histones are always bound to protein chaperones. Newly translated H3 and H4, which are acetylated on lysine-9 of H3 and lysine-5 and lysine-12 of H4, associate with a chromatin assembly factor, called CAF1. One of the three subunits of CAF1 is a chaperone called retinoblastoma-associated protein of 48 kD (RbAp48). CAF1 is targeted to sites of DNA replication by interaction with proliferating cell nuclear antigen (PCNA), a doughnut-shaped protein that helps DNA polymerase to slide along the DNA during replication (see Fig. 42-11). Thus, CAF1 delivers newly synthesized histones to sites on the chromosome where new nucleosomes are required as DNA is synthesized during the S phase of the cell cycle (see Chapter 42). H3 and H4 are deposited first on the new DNA, followed by two H2A:H2B heterodimers to complete the assembly of the nascent nucleosome.
Linker DNA and the Linker Histone H1
When examined by electron microscopy at low ionic strength, nucleosomal chromatin resembles a string of beads with diameters of about 10 nm and linker DNA extended between adjacent nucleosomes (Fig. 13-1). Each nucleosome in chromosomes is typically associated with about 200 base pairs of DNA. With subtraction of 166 base pairs for two turns around the histone octamer, this leaves 34 base pairs of linker DNA between adjacent nucleosomes. Linker DNA can vary widely in length in different tissues and cell types.
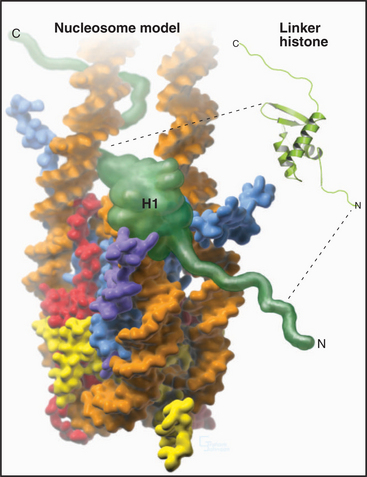
A fifth histone, H1 or linker histone, is thought to bind to linker DNA at the side of each nucleosome core where the DNA molecule enters and exits the structure (Fig. 13-5). H1 histones have a “winged helix” central domain flanked by unstructured basic domains at both the N- and C-termini (Fig. 13-3). Mammals have at least eight variant forms (called subtypes) of H1 histones (H1a–e, H10, H1t, and H1oo). The amino acid sequences of these variants differ by 40% or more. Of these, H10 is found in cells entering the nondividing Go state (see Chapter 41), while H1t and H1oo are found exclusively in developing sperm and oocytes, respectively.
The Second Level of Chromosomal DNA Packaging: The 30-nm Fiber
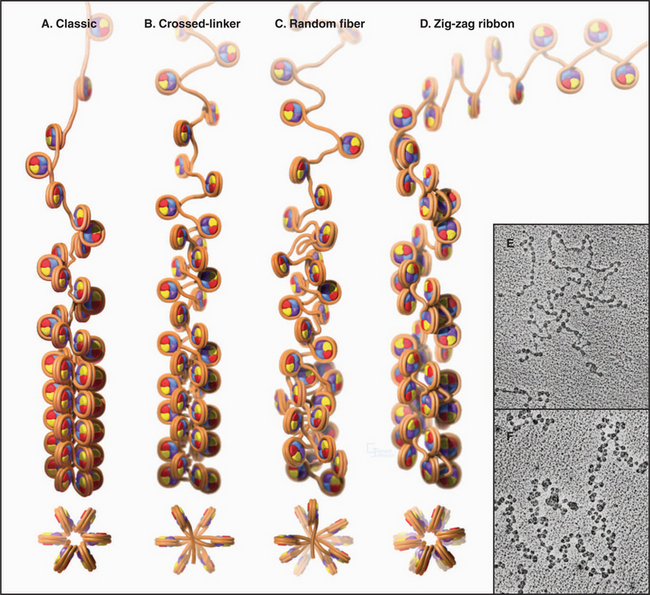
Levels of chromatin structure beyond the nucleosome are poorly understood. One job of linker histone H1 is to promote the packaging of chromatin into the 30-nm fiber, a condensed filament of nucleosomes that can be observed by electron microscopy. Investigators now agree that the 30-nm fiber is unlikely to be a simple helix (solenoid) of nucleosomes. More complex models, similar to those shown in Figure 13-6B and D, are now favored.
Higher Levels of Chromosomal DNA Packaging in Interphase Nuclei
Dense packing of macromolecules in the nucleus makes it very difficult to observe the details of higher-level folding of chromatin fibers directly. Visualization of specific DNA loci within fixed interphase nuclei by in situ hybridization (see Fig. 13-15) can be used to estimate the degree of chromatin compaction by comparing the physical distance between two DNA sequences with a known number of base pairs between them. For regions of DNA up to about 250,000 base pairs apart, the chromatin fiber is shortened about twofold to threefold relative to the 30-nm fiber. When sequences are separated by tens of millions of base pairs, the shortening increases by another 20-fold to 30-fold. This suggests that there are at least two levels of chromatin folding beyond the 30-nm fiber.
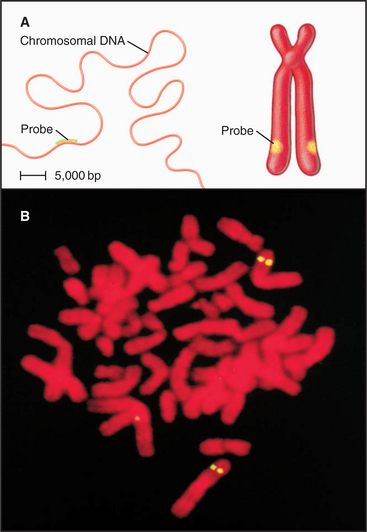
Figure 13-15 fluorescence in situ hybridization performed on mitotic chromosomes. A., Chromosomes are spread on a slide as in Figure 13-14. Following chemical fixation steps to preserve the chromosomal structure, the chromosomal proteins are removed by digestion with proteases and the genomic DNA strands are melted (separated) by heating. Next, a “probe DNA” (yellow) is added. This probe DNA is single-stranded so that it can base-pair (hybridize) to its complementary sequences in the chromosome. The probe DNA is chemically labeled with biotin. Next, the sites of hybridization on the chromosomes are detected with fluorescently labeled avidin, a protein from egg white that binds to biotin with extremely high affinity. The sites of avidin-binding appear yellow, whereas the remainder of the chromosomal DNA is counterstained with a red dye. B, The micrograph shows FISH analysis using a probe from near the von Hippel Lindau locus on chromosome 3.
(B, Courtesy of Jeanne Lawrence, University of Massachusetts, Amherst.)
The organization of chromatin fibers can be observed by fluorescence microscopy of living cells after labeling with a fluorescent marker, such as the jellyfish green fluorescent protein (GFP [see Fig. 6-3]) (Fig. 13-7). These labeled chromosome arms are dynamic, changing both their structure and location as cells traverse the cell cycle. At times in the cycle when a chromosome arm becomes relatively more decondensed, it is possible to observe the presence of a fiber, 100 to 300 nm in diameter, called a chromonema fiber. Similar fibers are seen in electron micrographs of interphase cells. It is not yet known whether the chromonema fiber is the next level of chromatin packing above the 30-nm fiber.
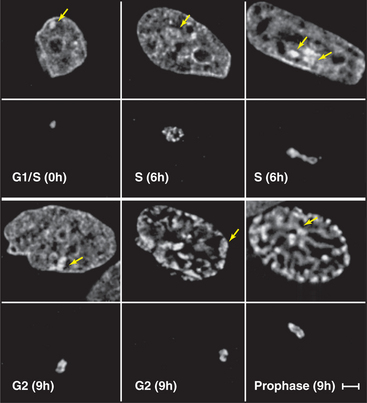
Figure 13-7 direct visualization of changes in the compaction and location of a chromosome arm in a living cell. DNA molecules carrying the binding sites for a specific DNA-binding protein were integrated into the chromosomes of a cell at random and caused to amplify into large arrays, which, in some cases, corresponded to whole chromosome arms. These cells were then induced to express the DNA-binding protein as a fusion to jellyfish green fluorescent protein (GFP). In the lower panel at each time point, the fluorescently labeled chromosome arm can be seen to change both its degree of condensation and its position within the nucleus as a function of the cell cycle. The upper panels show total DNA stained with DAPI. DNA replication occurs in the S phase, which is separated from mitosis (cell division) by the G1 and G2 gap phases (see Chapter 40). These studies are potentially revealing, but they should be interpreted cautiously, as the labeled regions represent artificial arrays of DNA sequence rich in particular binding sites and may not exactly mimic the behavior of natural segments of chromosomes.
(From Li G, Sudlow G, Belmont AS: Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: Precise choreography of condensation/decondensation and nuclear positioning. J Cell Biol 140:975–989, 1998.)
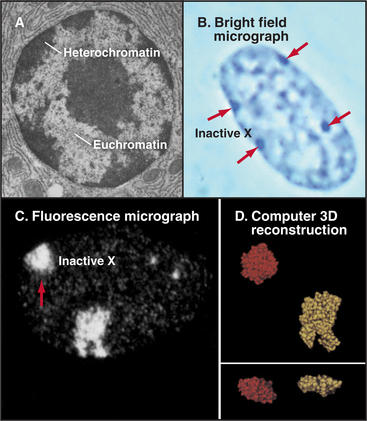
Functional Compartmentation of the Nucleus: Heterochromatin and Euchromatin
Chromatin has traditionally been divided into two main classes based on structural and functional criteria. Euchromatin contains almost all of the genes, both actively transcribed and quiescent. Heterochromatin is transcriptionally inert and is generally more condensed than the euchromatin; it was initially recognized because it stains more darkly with DNA-binding dyes than the remainder of the nucleus. A typical nucleus has both euchromatin and heterochromatin, the latter usually being concentrated near the nuclear envelope and around nucleoli. Much of the interior of nuclei is occupied by pale-staining euchromatin rich in actively transcribing genes. Nuclei that are less active in transcription have relatively more heterochromatin (Fig. 13-8). Two types of heterochromatin are recognized.