[level-membership-for-basic-science-category]
CHAPTER 13 DNA Packaging in Chromatin and Chromosomes
Chromosomal DNA molecules of eukaryotes are thousands of times longer than the diameter of the nucleus and must therefore be highly compacted throughout the cell cycle. This folding is accomplished by combining the DNA with structural proteins to make chromatin. A hierarchy of levels of chromatin folding compacts the DNA but permits transcriptional machinery access to those regions of the chromosome required for gene expression.
The First Level of Chromosomal DNA Packaging: The Nucleosome
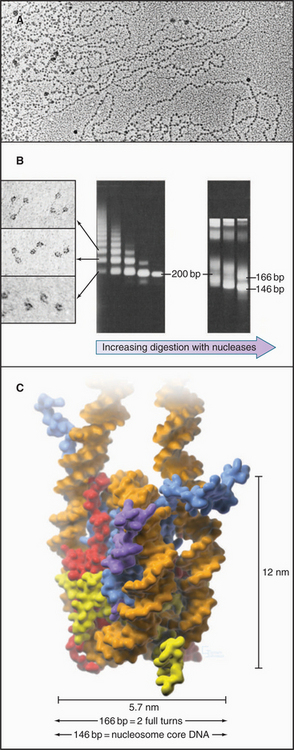
The continuous DNA fiber of each chromosome links hundreds of thousands of nucleosomes in series. Individual nucleosomes can be isolated following cleavage of DNA between neighboring particles. Random digestion of chromatin by DNA-cutting enzymes called nucleases initially yields a mixture of particles consisting of one or more nucleosomes containing multiples of about 200 base pairs of DNA (Fig. 13-1). Continued nuclease cleavage yields a stable particle with 146 base pairs of DNA (1.75 turns of the DNA around the protein core). This is called a nucleosome core particle.
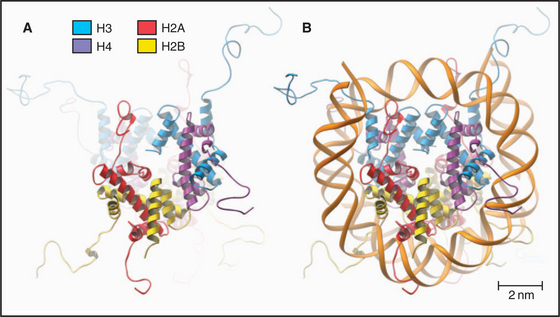
The nucleosome core particle is disk-shaped, with DNA coiled in a left-handed superhelix around an octamer of core histones. This octamer consists of a central tetramer composed of two closely linked H3:H4 heterodimers, flanked on either side by two H2A:H2B heterodimers. High-resolution crystal structures of nucleo-some core particles revealed that each core histone has a compact domain of 70 to 100 amino acid residues that adopts a characteristic Z-shaped “histone fold” consisting of a long α-helix flanked by two shorter α-helices (Fig. 13-2).
Epigenetics and the Histone Code
The revolution in biology that began with the structure of DNA and the realization that the sequence of bases in DNA provides a code that specifies the structure of proteins culminated 50 years later with the near complete sequencing of all the gene-rich portions of the human genome. To take advantage of this coding information, cells must control when to use it. Initial studies of the processes controlling gene expression focused on regulation of transcription by proteins that bind specific DNA sequences at the 5′ end of genes (see Chapter 15), as this is the way in which bacteria regulate their genes. This is now known to be only part of the story.
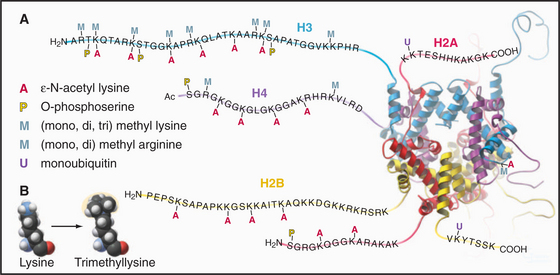
Eukaryotes impose another level of regulation on the utilization of their genes. This has been referred to as a histone code. The histone code hypothesis proposes that combinations of posttranslational modifications of histones are “read” by proteins that bind modified histones and then dictate whether particular regions of chromatin are transcribed by RNA polymerases or are held in an inactive state. Posttranslational modifications of histones include acetylation, phosphorylation, methylation, ubiquitination, and poly(ADP)ribosylation at many sites in the N-terminal tails and elsewhere (Fig. 13-3). Chromatin states created by histone modifications can be stably inherited through many rounds of cell division. Thus, this hypothesis can explain the phenomenon of epigenetic regulation (see Fig. 12-10): the stable, heritable regulation of chromosomal functions by information that is not simply encoded in the DNA sequence.
Regulation of Chromatin Structure by the Histone N-Terminal Tails
The N-terminal histone tails provide a molecular “handle” to manipulate DNA accessibility in chromatin (Fig. 13-3). This complex area can only be outlined here. The two key modifications contributing to the histone code are acetylation and methylation of lysine residues. Histones with acetylated lysines are generally associated with “open” chromatin that is permissive for RNA transcription, while histones with methylated lysines can be associated with either “open” or “closed” chromatin states. It should be emphasized that the histone code is complex and not fully understood. Since the histone modifications are read as combinations, individual modifications do not necessarily always have the same consequences. One example of this is the phosphorylation of histone H3 on serine 10 (H3-S10P). In mitotic cells, this correlates with a condensed and transcriptionally inactive chromatin structure, but when combined with acetylation of surrounding amino acid residues, it is also associated with the activation of gene transcription as nonproliferating cells reenter the cell cycle (see Chapter 41).
Acetylation reduces the net positive charge of the N-terminal domain, causing the chromatin to adopt an “open” conformation that is more favorable to transcription, as the histones bind less tightly to DNA. Acetylation also provides binding sites for a number of proteins with an approximately 100-amino-acid sequence motif called a bromodomain. Bromodomain binding to acetylated histone N-terminal tails is analogous to the binding of SH2 domains to phosphorylated tyrosine in cellular signaling pathways (see Fig. 25-10). Bromodomain-containing enzymes recruited to chromatin by acetylated histones often modify histones in other ways that promote or limit the accessibility of the DNA for transcription into RNA.
Proteins called transcription factors regulate gene expression by binding specific DNA sequences in promoter regions adjacent to the coding sequences of genes and recruiting transcriptional machinery (RNA polymerases and associated proteins) to the gene (see Fig. 15-19). Many transcription factors recruit a protein complex, called a coactivator, that facilitates loading of the transcriptional apparatus onto the gene. Often, coactivators are enzymes that modify N-terminal histone tails. One yeast coactivator contains over 10 proteins, including a histone acetyltransferase that transfers acetate groups from acetyl coenzyme A (CoA) to the ε-amino groups of lysine-14 and lysine-8 in the N-terminal tails of histone H3 (Fig. 13-4). Histone acetylation is crucial for life. Yeast cells die if these lysines are mutated to arginines, thus preserving their positive charge but preventing them from being acetylated.
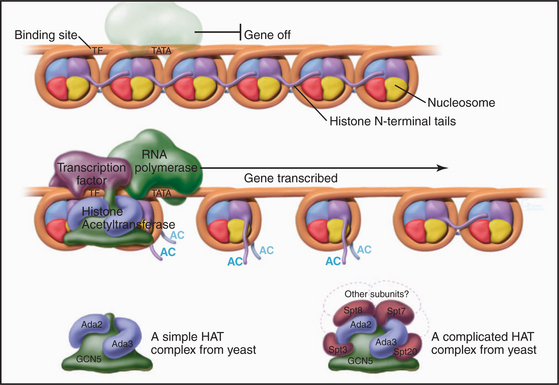
Figure 13-4 Acidic transcription factors (purple) bind specific DNA sequences and recruit coactivators to the 5′ ends of genes. Many of these coactivators have histone acetyltransferase activity and work by acetylating the N-terminal tails of the core histones, thereby loosening the chromatin structure and promoting the binding and activation of the RNA polymerase holoenzyme (see Chapter 15). The coactivators vary in composition and complexity from the relatively simple histone acetyltransferase complex (bottom left) to the huge and elaborate SAGA complex (bottom right). (AC, acetylation; TATA, DNA sequence in the gene promoter [see Chapter 15]). In this side view, only one of the two turns of DNA around the nucleosome is seen. GCN5, Ada2, Ada3, Spt3, Spt7, Spt8, and Spt20 are the names of budding yeast genes whose products are found in these complexes.
Histone acetylation is dynamic. Just as transcriptional coactivators contain histone acetyltransferases that add acetyl groups to nucleosomes and promote gene activation, so corepressors, which are recruited in a similar manner, can contain histone deacetylases that remove acetyl groups from selected lysine residues. This tends to inactivate gene expression. This mechanism regulates cell cycle progression during the G1 phase of the cell cycle (see Fig. 41-8).
In addition to marking nucleosomes by modification of their N-terminal tails, cells also use the energy provided by ATP hydrolysis to actively remodel nucleosomes. This involves complex protein “machines” that can alter nucleosome structure, move nucleosomes around, or both. Two large “machines” in yeast—RSC (remodels the structure of chromatin) with 15 subunits and SWI/SNF (switch/sniff) with 11 subunits—each has a key subunit that utilizes ATP hydrolysis to translocate along the DNA helix. One proposal is that these “machines” use ATP hydrolysis to force an extra 40 to 60 base pairs of DNA onto the nucleosome. Since this excess DNA cannot fit smoothly against the surface of the histone octamer, it presumably bulges out in a loop from the nucleosomal surface. If the position of this loop migrates around the surface of the nucleosome, the nucleosome will “jump” 40 to 60 base pairs along the DNA. This process can uncover sequences that are important for gene regulation that had been hidden by association with a nucleosome. Alternatively, this mechanism may be used to loosen the nucleosome and allow the exchange of histone dimers in and out.
Histone Acetylation and Nucleosome Assembly
During DNA replication, existing nucleosomes are partitioned randomly between daughter DNA strands. Newly assembled nucleosomes then fill the gaps. When not associated with DNA, histones are always bound to protein chaperones. Newly translated H3 and H4, which are acetylated on lysine-9 of H3 and lysine-5 and lysine-12 of H4, associate with a chromatin assembly factor, called CAF1. One of the three subunits of CAF1 is a chaperone called retinoblastoma-associated protein of 48 kD (RbAp48). CAF1 is targeted to sites of DNA replication by interaction with proliferating cell nuclear antigen (PCNA), a doughnut-shaped protein that helps DNA polymerase to slide along the DNA during replication (see Fig. 42-11). Thus, CAF1 delivers newly synthesized histones to sites on the chromosome where new nucleosomes are required as DNA is synthesized during the S phase of the cell cycle (see Chapter 42). H3 and H4 are deposited first on the new DNA, followed by two H2A:H2B heterodimers to complete the assembly of the nascent nucleosome.
Linker DNA and the Linker Histone H1
When examined by electron microscopy at low ionic strength, nucleosomal chromatin resembles a string of beads with diameters of about 10 nm and linker DNA extended between adjacent nucleosomes (Fig. 13-1). Each nucleosome in chromosomes is typically associated with about 200 base pairs of DNA. With subtraction of 166 base pairs for two turns around the histone octamer, this leaves 34 base pairs of linker DNA between adjacent nucleosomes. Linker DNA can vary widely in length in different tissues and cell types.
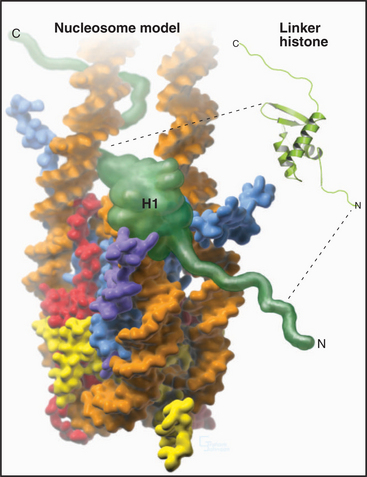
A fifth histone, H1 or linker histone, is thought to bind to linker DNA at the side of each nucleosome core where the DNA molecule enters and exits the structure (Fig. 13-5). H1 histones have a “winged helix” central domain flanked by unstructured basic domains at both the N- and C-termini (Fig. 13-3). Mammals have at least eight variant forms (called subtypes) of H1 histones (H1a–e, H10, H1t, and H1oo). The amino acid sequences of these variants differ by 40% or more. Of these, H10 is found in cells entering the nondividing Go state (see Chapter 41), while H1t and H1oo are found exclusively in developing sperm and oocytes, respectively.
The Second Level of Chromosomal DNA Packaging: The 30-nm Fiber
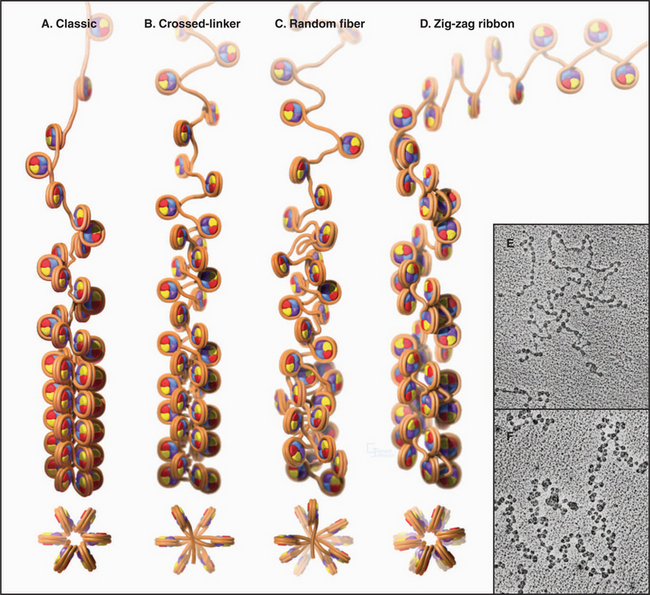
Levels of chromatin structure beyond the nucleosome are poorly understood. One job of linker histone H1 is to promote the packaging of chromatin into the 30-nm fiber, a condensed filament of nucleosomes that can be observed by electron microscopy. Investigators now agree that the 30-nm fiber is unlikely to be a simple helix (solenoid) of nucleosomes. More complex models, similar to those shown in Figure 13-6B and D, are now favored.
Higher Levels of Chromosomal DNA Packaging in Interphase Nuclei
Dense packing of macromolecules in the nucleus makes it very difficult to observe the details of higher-level folding of chromatin fibers directly. Visualization of specific DNA loci within fixed interphase nuclei by in situ hybridization (see Fig. 13-15) can be used to estimate the degree of chromatin compaction by comparing the physical distance between two DNA sequences with a known number of base pairs between them. For regions of DNA up to about 250,000 base pairs apart, the chromatin fiber is shortened about twofold to threefold relative to the 30-nm fiber. When sequences are separated by tens of millions of base pairs, the shortening increases by another 20-fold to 30-fold. This suggests that there are at least two levels of chromatin folding beyond the 30-nm fiber.
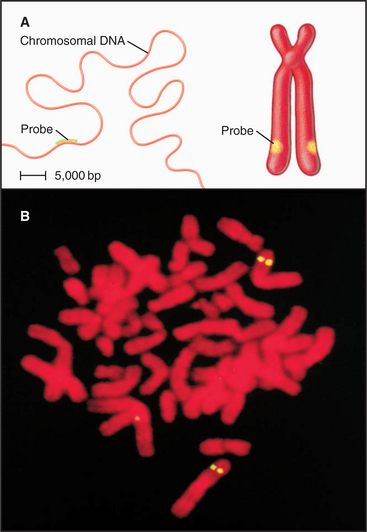
Figure 13-15 fluorescence in situ hybridization performed on mitotic chromosomes. A., Chromosomes are spread on a slide as in Figure 13-14. Following chemical fixation steps to preserve the chromosomal structure, the chromosomal proteins are removed by digestion with proteases and the genomic DNA strands are melted (separated) by heating. Next, a “probe DNA” (yellow) is added. This probe DNA is single-stranded so that it can base-pair (hybridize) to its complementary sequences in the chromosome. The probe DNA is chemically labeled with biotin. Next, the sites of hybridization on the chromosomes are detected with fluorescently labeled avidin, a protein from egg white that binds to biotin with extremely high affinity. The sites of avidin-binding appear yellow, whereas the remainder of the chromosomal DNA is counterstained with a red dye. B, The micrograph shows FISH analysis using a probe from near the von Hippel Lindau locus on chromosome 3.
(B, Courtesy of Jeanne Lawrence, University of Massachusetts, Amherst.)
The organization of chromatin fibers can be observed by fluorescence microscopy of living cells after labeling with a fluorescent marker, such as the jellyfish green fluorescent protein (GFP [see Fig. 6-3]) (Fig. 13-7). These labeled chromosome arms are dynamic, changing both their structure and location as cells traverse the cell cycle. At times in the cycle when a chromosome arm becomes relatively more decondensed, it is possible to observe the presence of a fiber, 100 to 300 nm in diameter, called a chromonema fiber. Similar fibers are seen in electron micrographs of interphase cells. It is not yet known whether the chromonema fiber is the next level of chromatin packing above the 30-nm fiber.
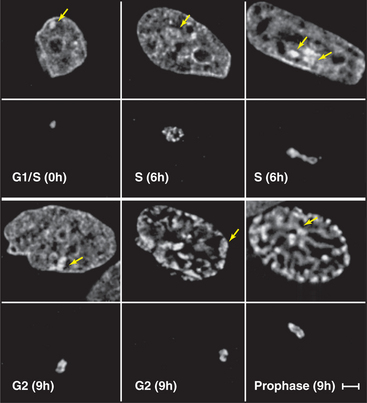
Figure 13-7 direct visualization of changes in the compaction and location of a chromosome arm in a living cell. DNA molecules carrying the binding sites for a specific DNA-binding protein were integrated into the chromosomes of a cell at random and caused to amplify into large arrays, which, in some cases, corresponded to whole chromosome arms. These cells were then induced to express the DNA-binding protein as a fusion to jellyfish green fluorescent protein (GFP). In the lower panel at each time point, the fluorescently labeled chromosome arm can be seen to change both its degree of condensation and its position within the nucleus as a function of the cell cycle. The upper panels show total DNA stained with DAPI. DNA replication occurs in the S phase, which is separated from mitosis (cell division) by the G1 and G2 gap phases (see Chapter 40). These studies are potentially revealing, but they should be interpreted cautiously, as the labeled regions represent artificial arrays of DNA sequence rich in particular binding sites and may not exactly mimic the behavior of natural segments of chromosomes.
(From Li G, Sudlow G, Belmont AS: Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: Precise choreography of condensation/decondensation and nuclear positioning. J Cell Biol 140:975–989, 1998.)
Functional Compartmentation of the Nucleus: Heterochromatin and Euchromatin
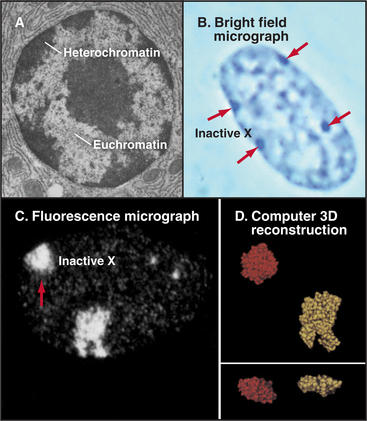
Chromatin has traditionally been divided into two main classes based on structural and functional criteria. Euchromatin contains almost all of the genes, both actively transcribed and quiescent. Heterochromatin is transcriptionally inert and is generally more condensed than the euchromatin; it was initially recognized because it stains more darkly with DNA-binding dyes than the remainder of the nucleus. A typical nucleus has both euchromatin and heterochromatin, the latter usually being concentrated near the nuclear envelope and around nucleoli. Much of the interior of nuclei is occupied by pale-staining euchromatin rich in actively transcribing genes. Nuclei that are less active in transcription have relatively more heterochromatin (Fig. 13-8). Two types of heterochromatin are recognized.
Constitutive heterochromatin is associated with special types of DNA sequences, such as satellite DNAs (see Fig. 12-9), that are packaged into a particular type of “closed” conformation in every cell. Establishment of constitutive heterochromatin involves transcription of these repeated DNA elements to produce double-stranded RNAs that are cleaved into short fragments by the RNAi machinery (see Fig. 16-12). The resulting short RNAs are thought to target components that promote heterochromatin formation to their sites of transcription in the chromosome (see Fig. 16-13).
Facultative heterochromatin is epigenetic: Rather than showing an invariant link with particular DNA sequences, it consists of sequences that are in heterochromatin in some cells and in euchromatin in others. X chromosome inactivation is the classic example of facultative heterochromatin in mammals. In females, one X chromosome in each cell (selected at random) is inactivated early in development prior to implantation of the embryo. The inactivated X chromosome forms a discrete patch of heterochromatin at the nuclear periphery known as the Barr body (Fig. 13-8). Because most genes carried on the inactivated X chromosome become transcriptionally silent, females with two X chromosomes have the same levels of gene expression as do males with a single X chromosome.
The epigenetic mark that best defines heterochromatin is methylation of histone H3 on lysine 9 (H3-K9me). This modification acts as a specific binding site for heterochromatin protein 1 (HP1). The amino-terminal region of HP1 contains a motif of 50 amino acids called a chromodomain (chromatin modification organizer) that binds to histone H3 methylated on lysine 9. HP1 recruits other proteins, including histone methyltransferases and deacetylases, that further adjust the array of posttranslational modifications on the amino-terminal tails of the histones in order to establish a “closed” chromatin conformation that represses transcription.
HP1 also recruits DNA methyltransferases that modify the underlying DNA by adding a methyl group to the 5′ position on cytosine in the dinucleotide CpG. If methylation occurs near the 5′ promoters of genes (see Chapter 15), in regions with an above average concentration of CpG and referred to as CpG islands, it can inactivate gene transcription and promote formation of heterochromatin. Several specific binding proteins recognize DNA containing 5-methyl-cytosine. One of these, methyl cytosine–binding protein (MeCP2), represses expression of nearby genes by recruiting a histone deacetylase complex that removes acetyl groups from the core histone N-terminal tails (Fig. 13-9). A second methyl-CpG-binding protein also binds to HP1, which in turn recruits histone deacetylases and enzymes that methylate histone H3 on lysine 9. This creates more HP1-binding sites and causes the heterochromatin to spread laterally along the chromosome.
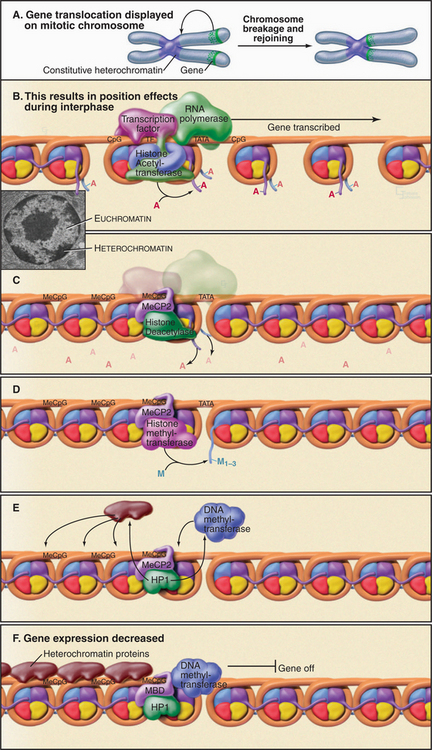
Figure 13-9 use of position effect to illustrate the steps in formation of heterochromatin. If a transcriptionally active gene is moved next to a region of heterochromatin, it is repressed as the heterochromatin spreads. A, The relative position of the gene is shown on mitotic chromosomes as it would be determined by in situ hybridization (Fig. 13-15). B–F, Diagrammatic representation of the effects of this gene translocation on transcription of the gene during interphase. Stages in the process of heterochromatin formation involve the following: removal of acetyl groups from the histones by a histone deacetylase (B–C); addition of two or three methyl groups to lysine 9 of H3 (D); binding of HP1, which recruits a DNA methyltransferase plus other heterochromatin proteins (E); in heterochromatin the methylation of DNA and the subsequent binding of HP1 plus other heterochromatin proteins (E–F). The gene is silent. In this side view, only one of the two turns of DNA around the nucleosome is seen. AC, acetylation; Me, methylation.
(Inset, From Fawcett DW: The Cell. Philadelphia, WB Saunders, 1981.)
Not all silent chromatin is classical heterochromatin. For example, the polycomb group proteins, which help to confirm the identity of particular body segments during development by regulating the expression of a number of homeodomain transcription factors (see Fig. 15-17), do so by creating an alternative type of “closed” euchromatin environment that is unfavorable for gene transcription.
Polycomb group proteins are found in two complexes: PRC1 and PRC2 (polycomb repressive complex). Polycomb silencing starts with methylation of histone H3 lysine 27 (H3-K27me) by the PRC2 complex. The polycomb protein itself, in the PRC1 complex, has a chromodomain that binds specifically to H3-K27me. Two functions have been proposed for the PRC1 complex. First, its binding causes nucleosomes to form dense clumps that are resistant to remodeling and “opening” by ATP-dependent remodeling “machines.” Second, PRC1 contains an E3 ubiquitin ligase activity (see Chapter 23) that transfers a single ubiquitin molecule to lysine119 of histone H2A. Together, these activities do not turn genes on and off; instead, they apparently lock genes that are already off in a silent epigenetic state that is stable even through many generations of cell division.
Controlling the Influence and Spread of Heterochromatin
Most HP1 is highly mobile in nuclei, moving on a time frame of seconds. Moreover, it recruits chromatin and DNA-modifying enzymes that can act on multiple substrates. As a result, heterochromatin is not a static “closed” chromatin compartment but can “invade” nearby genes along the chromosome. If an actively transcribed gene is moved into close proximity to constitutive heterochromatin by a chromosomal rearrangement, transcription from the gene is repressed as heterochromatin spreads across it (Fig. 13-9). This is called position effect.
About 20 human LCRs have been identified to date. They generally regulate the proper expression of genes that are expressed only in particular tissues and at particular times. How they do this is not entirely clear. LCRs typically consist of short regions (often a 150 to 300 base pair stretch of DNA) that are rich in binding sites for transcriptional regulators (see Chapter 14). Somehow, the concerted action of these factors promotes formation of an “open” chromatin region containing acetylated histones that may stretch for thousands of base pairs away from the LCR. Experiments in which a single LCR drives the expression of a cluster of several genes reveal that the LCR stimulates the expression of only one gene at a time. Thus, LCRs appear to work by physically associating with a gene, forming a loop in the chromatin and actively turning on its expression rather than by setting up a broad domain in which gene expression is insulated from the surrounding chromosome.
Imprinting: A Specialized Type of Gene Silencing
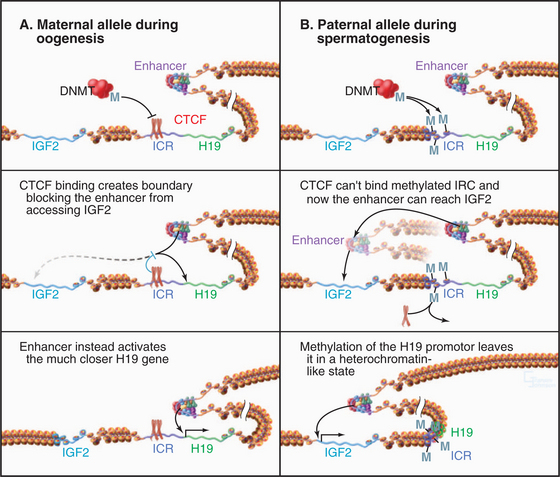
One well-studied system involves the insulin-like growth factor-2 (IGF2) and H19 genes of the mouse (H19 is an RNA that does not encode for a protein; Fig. 13-10). The imprinting control region (ICR) of the DNA that controls these two genes has binding sites for the CTCF protein discussed previously. When CTCF binds to the chromosome derived from the egg, it acts as an insulator element, preventing access of the IGF2 gene to a transcriptional enhancer, keeping it turned off. Under these conditions, the H19 gene is expressed. In chromosomes derived from the sperm, the control region is methylated on CpG sequences. This stops CTCF from binding, and as a result, the paternal copy of IGF2 has access to its enhancer and is expressed. Under these conditions, the H19 gene is not expressed. This simple switch ensures that only the paternal copy of the IGF2 gene and the maternal copy of H19 are expressed in the offspring.
Large-Scale Structural Compartmentation of the Nucleus
Although interphase nuclei lack a high degree of order, a number of general organizational principles are recognized. First, individual chromosomes tend to concentrate within discrete territories and intermingle with one another only to a limited extent. This is seen most clearly in human somatic cell nuclei when individual chromosomes are visualized by a special type of in situ hybridization called chromosome painting (Fig. 13-11). Actively transcribing genes are frequently located on the surface of territories occupied by individual chromosomes. However, in some cases, active genes can be located well outside of the territories, as though their activation involved looping out a much larger domain from the remainder of the chromosome. In some cases, this movement during gene activation involves relocation from regions (“compartments”) of the nucleus where transcription is relatively infrequent into areas where transcription is favored (Fig. 13-12C–D).
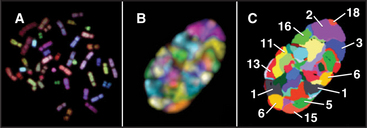
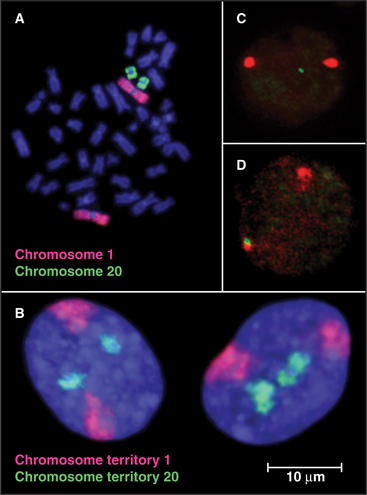
Second, heterochromatin tends to be concentrated near the nuclear periphery in a wide range of cell types. This was the first indication that particular chromosomal regions might have preferred locations within the nucleus. Subsequent studies confirmed that the distribution of chromosomes within interphase nuclei is not random. Rather, chromosomes that are rich in actively transcribed genes tend to be localized toward the interior of the nucleus, while chromosomes with a lower gene content tend to be found near the nuclear periphery (Fig. 13-12A).
A much less well resolved issue is whether chromosomes change their positions within nuclei as cells traverse the cell cycle or undergo more long-term de-velopmental programs. In human lymphocytes, centro-meres tend to cluster together near the periphery of the nucleus during the G1 phase of the cell cycle. During the S phase, they tend to be more dispersed and in the nuclear interior. These movements of chromosome domains depend on actin and myosin, but the detailed mechanism is not known.
Higher-Order Structure of Chromosomes
Special Interphase Chromosomes with Clearly Resolved Structures
Studies of specialized chromosomes from organisms ranging from flies to mammals suggest that chromosomes have large-scale structural domains composed of loops containing thousands to millions of base pairs. Such loop domains are clearly seen in lampbrush chromosomes, found during meiotic prophase in oocytes of many species (Fig. 13-13A). Lampbrush chromosome loops are sites of intense transcriptional activity as oocytes stockpile huge stores of the components needed for rapid cell division during early development of the fertilized egg. The loops are easily seen because the DNA is coated with many RNA transcripts, together with proteins that package them.
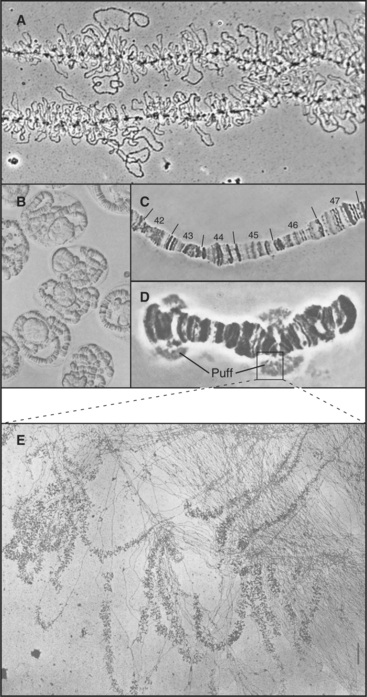
Similar loops can be seen in the giant polytene chromosomes found in some tissues of Drosophila larvae. Each polytene chromosome consists of more than 1000 identical DNA molecules packed side by side in precise linear register. Light microscopy reveals that polytene chromosomes have a complex pattern of thousands of bands (Fig. 13-13B–D) representing domains of differentially compacted chromatin. Each band contains one or several genes and potentially constitutes a domain for gene expression. Stress or stimulation of gene expression by hormones causes certain bands to lose their compact shape and puff out laterally. Such puffs are composed of hundreds of identical chromatin loop domains, all being actively transcribed (Fig. 13-13E).
Higher-Order Structure of Mitotic Chromosomes
Although polytene chromosomes provide the clearest demonstration of structural domains along an interphase chromosome, the arms of typical diploid mitotic chromosomes also appear to have a domain substructure. This can be seen when mammalian chromosomes are subjected to G-banding (Fig. 13-14). G-banded human chromosomes from the early (prometaphase) stage of mitosis have up to 2000 discrete bands. Although the structural basis for the bands is not known, dark G-bands tend to be relatively enriched for A:T residues, poor in genes, and rich in LINE elements (see Fig. 12-5) and tend to replicate later in S phase than light G-bands (also called R, or reverse, bands). Cytogeneticists have used these highly reproducible banding patterns for years to identify individual human chromosomes and even portions thereof.
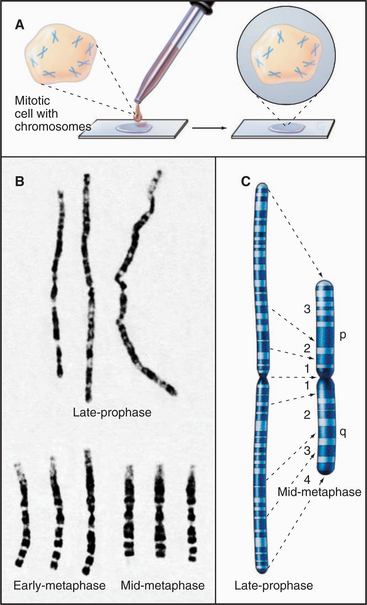
This reproducibility of higher-order structure in mitotic chromosomes is also easily seen when specific DNA sequences are marked by in situ hybridization. When individual loci are highlighted by this method, they appear as pairs of spots on the sister chromatids (Fig. 13-15). The two spots are distributed symmetrically, indicating that the chromatin fiber is folded similarly in both chromatids.
Three broad classes of models try to explain how the chromatin fiber is organized in mitotic chromosomes. Hierarchical coiling models suggest that the 30-nm chromatin fiber coils on itself, reaching larger and larger diameters and higher degrees of compaction. Large chromatin fibers (˜100 nm in diameter) can be seen in early prophase, as chromosome condensation begins (see Chapter 44). These models postulate that the final condensed mitotic chromosome forms by coiling up this fiber.
Loop domain models suggest that chromatin loops, containing an average of 15,000 to 100,000 base pairs, provide the structural basis for large-scale chromatin compaction in mitotic chromosomes. If metaphase chromosomes are swelled in hypotonic solutions, it is readily apparent that loops of chromatin radiate outward from the central chromatid axis (Fig. 13-16C). Similarly, if stripped of histones, isolated metaphase chromosomes consist of an enormous pool of DNA surrounding a residual structure that retains the general shape of chromosome arms. In certain cases, it is possible to trace individual loops of DNA radiating outward from the central structure (Fig. 13-16B). Similar looped structures can be seen if interphase nuclei are depleted of histones.
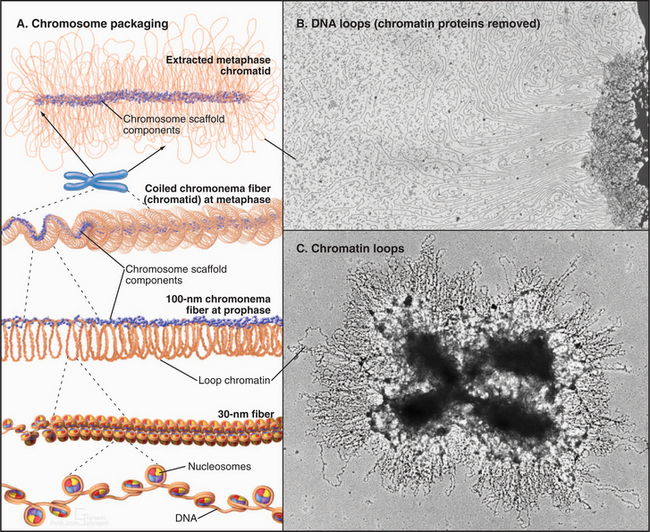
A third class of model combines aspects of the first two models. This proposes that most of the condensation of the chromatin occurs through hierarchical coiling or folding of the ˜100-nm fiber. During the final stages of folding, key proteins that later make up the mitotic chromosome scaffold (see later) become concentrated along the axial regions of the condensing chromosome arms (Fig. 13-16A). These proteins cross-link chromatin into a network that stabilizes the overall structure. If cross-links form at intervals, the chromatin between the cross-links forms loops that are seen if the chromosome is expanded or extracted. This model explains most available data, but the question of chromatin folding in chromosomes remains an area of active investigation and controversy.
Role of Nonhistone Proteins in Mitotic Chromosome Structure and Function
Early evidence suggesting that nonhistone proteins might contribute to mitotic chromosome structure came from experiments in which chromosomes were treated with nucleases to remove the DNA and extracted to remove most chromosomal proteins, including essentially all of the histones. The surviving remnant of the chromosome contained about 5% of the proteins and less than 0.1% of the DNA but still looked like a chromosome (Fig. 13-17). If the DNA was not digested, loops of DNA protruded from the protein mass (Fig. 13-16B). This protein remnant was called the chromosome scaffold because it looked like the structural backbone for the metaphase chromosome. This mechanical function is still disputed, but chromosome scaffold preparations contain a number of proteins with essen-tial roles in the structure and maintenance of chromo-somes.
The Nuclear Matrix
DNA Sequences Associated with the Chromosome Scaffold and Nuclear Matrix
Loop domain models of mitotic chromosomes predict that the DNA might attach to the scaffold at specific sites. Scaffold/matrix attachment regions (S/MARs) are regions of DNA that associate preferentially with the nuclear matrix and chromosome scaffold in biochemical fractionation experiments (Fig. 13-18). In one example, when over 900,000 contiguous base pairs of the genome of the fruit fly Drosophila melanogaster were examined for the presence of S/MARs, 16 DNA regions, spaced 15,000 to 115,000 base pairs apart, showed a strong interaction with the nuclear matrix in vitro. The spacing to these sites roughly corresponds to the predicted size of loop domains in nuclei and chromosomes.
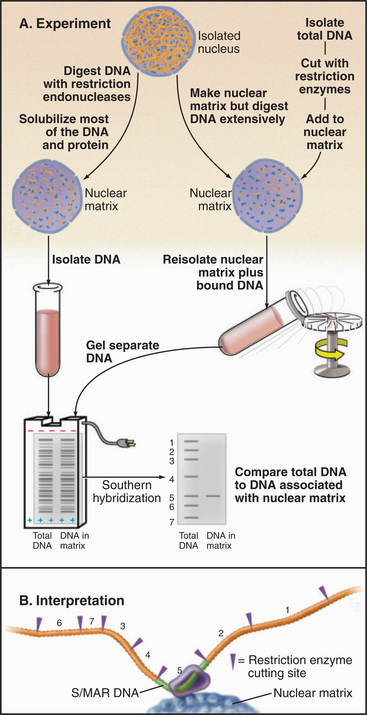
Figure 13-18 Experimental procedures used to identify S/MARs. These are regions associated with the nuclear matrix or chromosome scaffold in vitro. A, left, DNA of isolated nuclei is digested with restriction endonucleases. Most DNA and proteins are solubilized; the DNA that remains is separated by agarose gel electrophoresis and transferred to a membrane for analysis by Southern blotting. In this method, restriction fragments from a cloned region of genomic DNA (numbers in A and B) are labeled with radioactivity and hybridized to DNA on the membrane, similar to the method in Fig. 13-15. Right, DNA of isolated nuclei is digested extensively with restriction endonucleases. Most of the DNA and proteins are solubilized. Next, the same restriction fragments from the cloned region of genomic DNA used in the experiment on the left are mixed with the nuclear matrix under conditions where they can rebind to matrix proteins. Unbound DNA is washed away, and the bound sequences are detected by Southern blotting. B, From the restriction map of the chromosomal region used to prepare the probes, it is possible to identify the fragment(s) that bind preferentially to the nuclear matrix. Such fragments contain S/MARs. Both ap-proaches give the same answer.
Proteins of the Mitotic Chromosome and Chromosome Scaffold
The name condensin is self-explanatory but is actually misleading. This complex of five polypeptides was originally thought to be essential for mitotic chromosome condensation. The role of the complex is now known to be more subtle, since chromosomes can condense in its absence. Condensin is composed of two SMC proteins (SMC2 and SMC4), plus three auxiliary subunits. Vertebrates have two condensin complexes containing alternative sets of auxiliary subunits. Each SMC molecule folds back on itself at a hinge region to form an antiparallel coiled-coil. This brings together two globular domains, each with half of an ATP-binding site (Fig. 13-19C). ATP binding is thought to cause the two globular domains to come together. Condensin is formed when the two SMC proteins associate with each other via their hinges and the auxiliary subunits bind to the globular regions of the molecule.
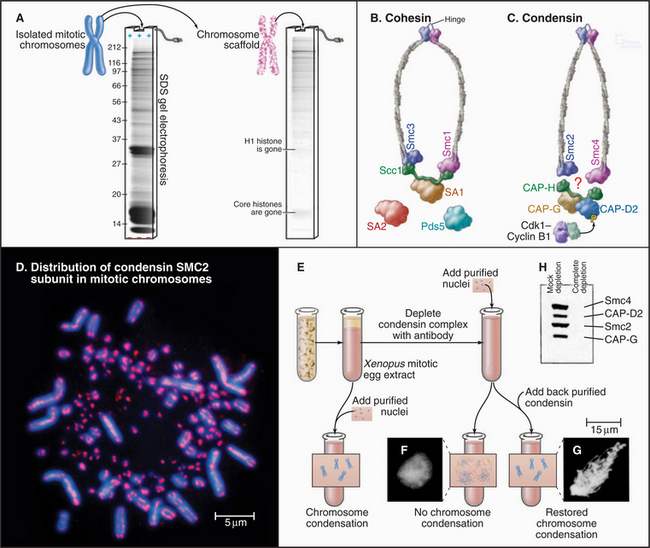
Figure 13-19 condensin and cohesin complexes have essential roles in chromosome structure and function. A., Isolation of the mitotic chromosome scaffold. Left, SDS polyacrylamide gel of total chromosomal proteins (see Box 6-3). Right, Most proteins are solubilized by the protocol used to make chromosome scaffolds. Remaining proteins include components of the condensin complex and DNA topoisomerase IIa. B–C, Subunit composition and structural organization of the cohesin and condensin complexes. Condensin is regulated by phosphorylation of the CAP-D2 subunit by Cdk1:cyclin B kinase. D, Immunofluorescence micrograph showing the distribution of condensin subunit SMC2 on mitotic chromosomes of the chicken. The tiny chromosomes, called microchromosomes, are commonly seen in normal bird chromosomes. E, The experimental protocol showing that condensin is required for mitotic chromosome condensation in vitro. F, Chromatin lacking condensin does not form mitotic chromosomes in vitro, and this is restored by adding back condensin. G–H, SDS polyacrylamide gel reveals the members of the condensin complex and demonstrates that they can be depleted from egg extract using a specific antibody.
(A [gel] and D [micrograph], Courtesy of William C. Earnshaw. F–H, From Hirano T, Kobayashi R, Hirano M: Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 89:511–521, 1997.)
Condensin binds to chromosomes only during mitosis when it is concentrated along the central axis of chromosome arms. The cell cycle kinase Cdk1:cyclin B (see Chapter 40) regulates condensin binding to chromosomes by phosphorylation of an auxiliary subunit. When condensin binds to naked DNA in a test tube, it is able to use the energy of ATP hydrolysis to supercoil the DNA, possibly by coiling DNA around itself. The role of this activity is unknown, because vertebrate chromosomes can condense to a normal extent in the absence of condensin. However, those chromosomes are fragile, lack a chromosome scaffold if extracted as described previously, and lose their orderly structure while trying to segregate in anaphase. It now appears that condensin regulates the timing of chromosome condensation and cooperates with other nonhistone proteins to stabilize chromosome structure throughout mitosis. Chromosomes lacking condensin can segregate normally at anaphase under specialized conditions.
Cohesin is the second major SMC-containing protein complex of mitotic chromosomes. Cohesin is a tetramer containing SMC1 and SMC3 plus two auxiliary subunits. One of these, Scc1, is cleaved by a protease called separase to initiate the separation of sister chromatids in mitotic anaphase (see Fig. 44-16). Cohesin, like condensin, is a ring-like molecule (Fig. 13-19). How cohesin holds the two sister chromatids together is not known, though given its ring-like structure, it might physically encircle two sister DNA molecules. Cohesin assembles on chromosomes during DNA replication and is recruited to regions of heterochromatin by HP1. Sister chromatid cohesion and mitotic segregation are defective in fission yeast with mutations in their HP1 ortholog.
Specialized Chromosomal Substructures: The Kinetochore
Embedded in the surface of the centromeric heterochromatin of most eukaryotes is a button-like structure called the kinetochore, which directs chromosomal movements in mitosis (Fig. 13-20). When a thin section of the centromere is examined by electron microscopy, the kinetochore appears to have a number of layers. The inner kinetochore is continuous with the surface of the centromeric heterochromatin and is composed of a specialized form of chromatin. The outer kinetochore consists of an outer plate with a fibrous corona on its outer surface. It is constructed from protein complexes that link the chromatin to microtubules of the mitotic spindle. Kinetochores also include protein complexes that form signaling pathways to regulate the progression of the cell through mitosis without errors (see Fig. 44-11).
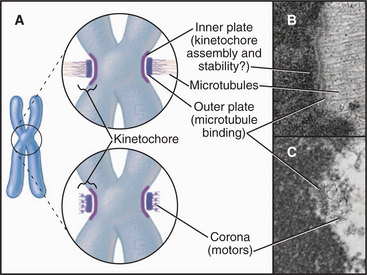
The multilayered kinetochore structure is visible only during mitosis. During interphase, the centromere persists as a condensed ball of heterochromatin that resembles other areas of condensed chromatin within the nucleus. The distinct kinetochore structure forms on the surface of the centromere during an early stage of mitosis called prophase (see Chapter 44), reaching its mature state following nuclear envelope breakdown when the chromosome comes into contact with microtubules at the onset of mitotic prometaphase.
Chapter 12 describes the three types of centromeres known in eukaryotes. Point centromeres found in budding yeasts assemble on defined DNA sequences and do not require epigenetic activation to function. They bind one microtubule. Regional centromeres, found in organisms ranging from fission yeast to humans, are based on preferred DNA sequences but require epigenetic activation in order to function. They bind 2 to 20 or more microtubules. In holocentromeres, as found in Caenorhabditis elegans and many insects, the underlying DNA sequences are unknown, and the microtubules (roughly 20 in C. elegans) bind all along the poleward-facing surface of the mitotic chromosome. Given this diversity of centromeres, it is remarkable that the proteins responsible for centromere assembly and function are well conserved across evolution.
Centromere Proteins of the Budding Yeast
Two sorts of factors bind to centromere DNA. First are CBFs, DNA-binding complexes that recognize the specific CDE I and CDE III DNA sequences that specify the point centromere (see Fig. 12-7). This binding occurs on the surface of a specialized nucleosome, which is marked by a centromere-specific histone H3 variant related to CENP-A (Fig. 13-21). CDE I and CDE III are juxtaposed on this nucleosome and linked by a stretch of A:T-rich DNA called CDE II, which completes one turn around the nucleosome.
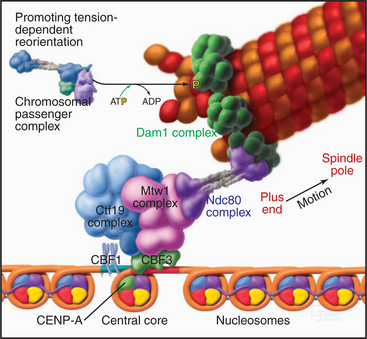
Figure 13-21 hypothetical model for the organization of the budding yeast kinetochore. The organization of the DNA in the budding yeast point kinetochore is discussed in Chapter 12.
Several large complexes bind to this nucleosome/CBF platform (Fig. 13-21). Since the kinetochore is a large integrated structure, the exact complexes that are obtained when it is fractionated vary from study to study as different methods are employed. The 11-subunit Ctf19 complex links the inner and outer kinetochore. The four components of the NDC80 complex are highly conserved in organisms from budding yeast to humans and are required for chromosomes to make robust attachments to microtubules. The Dam1 complex (10 subunits) appears to make the final attachment to the microtubule. This complex has been poorly conserved during evolution, and its vertebrate counterpart has yet to be identified.
Another conserved group of proteins that bind to the kinetochore includes components of the mitotic checkpoint pathway—a signaling network that causes mitotic progression to pause until all chromosomes make suitable connections to both spindle poles of the mitotic spindle (see Fig. 44-11).
Mammalian Centromere Proteins
The first three specific centromere proteins identified in any species were discovered in humans using autoantibodies present in the sera of certain individuals with rheumatic disease (Figs. 13-22 and 13-23). These proteins, designated CENPs A–C, are conserved from humans to yeasts. CENPs A–C are components of the inner kinetochore, to which they remain bound throughout the cell cycle. As was mentioned previously, CENP-A is a histone H3 variant, so the regional centromere, like the point centromere, must be based on modified nucleosomes. How CENP-A selects the DNA that it assembles into kinetochore-specific nucleosomes is unknown.
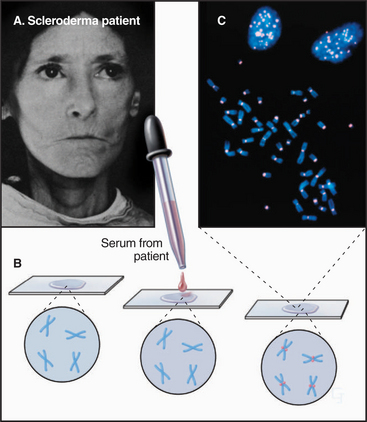
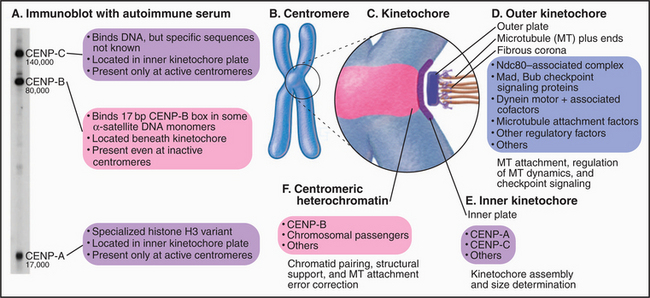
CENP-B probably originated as the enzyme responsible for movement of an ancient transposon. CENP-B binds specifically to a 17–base pair sequence (the CENP-B box) in α-satellite DNA (Fig. 13-23A). CENP-B is required for the efficient establishment of the epigenetic state that favors kinetochore assembly on α-satellite DNA arrays, but how it acts is unknown.
At present, at least 40 mammalian kinetochore proteins are known (Fig. 13-23B). As in the budding yeast, these are organized into protein complexes. One such complex of 10 proteins contains all four members of the NDC10 complex plus six other components. This NDC10-associated complex is found at both human and C. elegans kinetochores. Under some conditions, it is possible to find an association of CENP-C with this complex, raising the possibility that CENP-C might anchor NDC10 to the specialized chromatin of the inner kinetochore.
Human NDC10-associated complex members associate with kinetochores during prophase, as cells are about to enter mitosis, and disappear from kinetochores at the end of mitosis. Functional analysis reveals that the NDC10-associated complex links the inner centromere to the microtubules of the mitotic spindle. Components of the complex are also required for the signaling components of the mitotic checkpoint to associate with kinetochores (see Fig. 44-11). The proteins that actually link kinetochores to the spindle microtubules remain unknown.
Amor DJ, Kalitsis P, Sumer H, Choo KH. Building the centromere: From foundation proteins to 3D organization. Trends Cell Biol. 2004;14:359-368.
Belmont AS. Visualizing chromosome dynamics with GFP. Trends Cell Biol. 2001;11:250-257.
Belmont AS. Mitotic chromosome scaffold structure: New approaches to an old controversy. Proc Natl Acad Sci U S A. 2002;99:15855-15857.
Bird AP, Wolffe AP. Methylation-induced repression: Belts, braces, and chromatin. Cell. 1999;99:451-454.
Flaus A, Owen-Hughes T. Mechanisms for ATP-dependent chromatin remodelling: Farewell to the tuna-can octamer? Curr Opin Genet Dev. 2004;14:165-173.
Francis NJ, Kingston RE. Mechanisms of transcriptional memory. Nat Rev Mol Cell Biol. 2001;2:409-421.
Gassmann R, Vagnarelli P, Hudson D, Earnshaw WC. Mitotic chromosome formation and the condensin paradox. Exp Cell Res. 2004;296:35-42.
Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798-802.
Henikoff A, Ahmad K. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol. 2005;21:133-153.
Hirano T. The ABCs of SMC proteins: Two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev. 2002;16:399-414.
Khorasanizadeh S. The nucleosome: From genomic organization to genomic regulation. Cell. 2004;116:259-272.
Labrador M, Corces VG. Setting the boundaries of chromatin domains and nuclear organization. Cell. 2002;111:151-154.
Maiato H, Deluca J, Salmon ED, Earnshaw WC. The dynamic kinetochore-microtubule interface. J Cell Sci. 2004;117:5461-5477.
Naar AM, Lemon BD, Tjian R. Transcriptional coactivator complexes. Annu Rev Biochem. 2001;70:475-501.
Otte AP, Kwaks TH. Gene repression by polycomb group protein complexes: A distinct complex for every occasion? Curr Opin Genet Dev. 2003;13:448-454.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41-45.
Swedlow JR, Hirano T. The making of the mitotic chromosome: Modern insights into classical questions. Mol Cell. 2003;11:557-569.
West AG, Gaszner M, Felsenfeld G. Insulators: Many functions, many mechanisms. Genes Dev. 2002;16:271-288.
Workman JL, Kingston RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem. 1998;67:545-579.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 13 DNA Packaging in Chromatin and Chromosomes
Chromosomal DNA molecules of eukaryotes are thousands of times longer than the diameter of the nucleus and must therefore be highly compacted throughout the cell cycle. This folding is accomplished by combining the DNA with structural proteins to make chromatin. A hierarchy of levels of chromatin folding compacts the DNA but permits transcriptional machinery access to those regions of the chromosome required for gene expression.
The First Level of Chromosomal DNA Packaging: The Nucleosome
The continuous DNA fiber of each chromosome links hundreds of thousands of nucleosomes in series. Individual nucleosomes can be isolated following cleavage of DNA between neighboring particles. Random digestion of chromatin by DNA-cutting enzymes called nucleases initially yields a mixture of particles consisting of one or more nucleosomes containing multiples of about 200 base pairs of DNA (Fig. 13-1). Continued nuclease cleavage yields a stable particle with 146 base pairs of DNA (1.75 turns of the DNA around the protein core). This is called a nucleosome core particle.
The nucleosome core particle is disk-shaped, with DNA coiled in a left-handed superhelix around an octamer of core histones. This octamer consists of a central tetramer composed of two closely linked H3:H4 heterodimers, flanked on either side by two H2A:H2B heterodimers. High-resolution crystal structures of nucleo-some core particles revealed that each core histone has a compact domain of 70 to 100 amino acid residues that adopts a characteristic Z-shaped “histone fold” consisting of a long α-helix flanked by two shorter α-helices (Fig. 13-2).
Epigenetics and the Histone Code
The revolution in biology that began with the structure of DNA and the realization that the sequence of bases in DNA provides a code that specifies the structure of proteins culminated 50 years later with the near complete sequencing of all the gene-rich portions of the human genome. To take advantage of this coding information, cells must control when to use it. Initial studies of the processes controlling gene expression focused on regulation of transcription by proteins that bind specific DNA sequences at the 5′ end of genes (see Chapter 15), as this is the way in which bacteria regulate their genes. This is now known to be only part of the story.
Eukaryotes impose another level of regulation on the utilization of their genes. This has been referred to as a histone code. The histone code hypothesis proposes that combinations of posttranslational modifications of histones are “read” by proteins that bind modified histones and then dictate whether particular regions of chromatin are transcribed by RNA polymerases or are held in an inactive state. Posttranslational modifications of histones include acetylation, phosphorylation, methylation, ubiquitination, and poly(ADP)ribosylation at many sites in the N-terminal tails and elsewhere (Fig. 13-3). Chromatin states created by histone modifications can be stably inherited through many rounds of cell division. Thus, this hypothesis can explain the phenomenon of epigenetic regulation (see Fig. 12-10): the stable, heritable regulation of chromosomal functions by information that is not simply encoded in the DNA sequence.
Regulation of Chromatin Structure by the Histone N-Terminal Tails
The N-terminal histone tails provide a molecular “handle” to manipulate DNA accessibility in chromatin (Fig. 13-3). This complex area can only be outlined here. The two key modifications contributing to the histone code are acetylation and methylation of lysine residues. Histones with acetylated lysines are generally associated with “open” chromatin that is permissive for RNA transcription, while histones with methylated lysines can be associated with either “open” or “closed” chromatin states. It should be emphasized that the histone code is complex and not fully understood. Since the histone modifications are read as combinations, individual modifications do not necessarily always have the same consequences. One example of this is the phosphorylation of histone H3 on serine 10 (H3-S10P). In mitotic cells, this correlates with a condensed and transcriptionally inactive chromatin structure, but when combined with acetylation of surrounding amino acid residues, it is also associated with the activation of gene transcription as nonproliferating cells reenter the cell cycle (see Chapter 41).
Acetylation reduces the net positive charge of the N-terminal domain, causing the chromatin to adopt an “open” conformation that is more favorable to transcription, as the histones bind less tightly to DNA. Acetylation also provides binding sites for a number of proteins with an approximately 100-amino-acid sequence motif called a bromodomain. Bromodomain binding to acetylated histone N-terminal tails is analogous to the binding of SH2 domains to phosphorylated tyrosine in cellular signaling pathways (see Fig. 25-10). Bromodomain-containing enzymes recruited to chromatin by acetylated histones often modify histones in other ways that promote or limit the accessibility of the DNA for transcription into RNA.
Proteins called transcription factors regulate gene expression by binding specific DNA sequences in promoter regions adjacent to the coding sequences of genes and recruiting transcriptional machinery (RNA polymerases and associated proteins) to the gene (see Fig. 15-19). Many transcription factors recruit a protein complex, called a coactivator, that facilitates loading of the transcriptional apparatus onto the gene. Often, coactivators are enzymes that modify N-terminal histone tails. One yeast coactivator contains over 10 proteins, including a histone acetyltransferase that transfers acetate groups from acetyl coenzyme A (CoA) to the ε-amino groups of lysine-14 and lysine-8 in the N-terminal tails of histone H3 (Fig. 13-4). Histone acetylation is crucial for life. Yeast cells die if these lysines are mutated to arginines, thus preserving their positive charge but preventing them from being acetylated.
Figure 13-4 Acidic transcription factors (purple) bind specific DNA sequences and recruit coactivators to the 5′ ends of genes. Many of these coactivators have histone acetyltransferase activity and work by acetylating the N-terminal tails of the core histones, thereby loosening the chromatin structure and promoting the binding and activation of the RNA polymerase holoenzyme (see Chapter 15). The coactivators vary in composition and complexity from the relatively simple histone acetyltransferase complex (bottom left) to the huge and elaborate SAGA complex (bottom right). (AC, acetylation; TATA, DNA sequence in the gene promoter [see Chapter 15]). In this side view, only one of the two turns of DNA around the nucleosome is seen. GCN5, Ada2, Ada3, Spt3, Spt7, Spt8, and Spt20 are the names of budding yeast genes whose products are found in these complexes.
Histone acetylation is dynamic. Just as transcriptional coactivators contain histone acetyltransferases that add acetyl groups to nucleosomes and promote gene activation, so corepressors, which are recruited in a similar manner, can contain histone deacetylases that remove acetyl groups from selected lysine residues. This tends to inactivate gene expression. This mechanism regulates cell cycle progression during the G1 phase of the cell cycle (see Fig. 41-8).
In addition to marking nucleosomes by modification of their N-terminal tails, cells also use the energy provided by ATP hydrolysis to actively remodel nucleosomes. This involves complex protein “machines” that can alter nucleosome structure, move nucleosomes around, or both. Two large “machines” in yeast—RSC (remodels the structure of chromatin) with 15 subunits and SWI/SNF (switch/sniff) with 11 subunits—each has a key subunit that utilizes ATP hydrolysis to translocate along the DNA helix. One proposal is that these “machines” use ATP hydrolysis to force an extra 40 to 60 base pairs of DNA onto the nucleosome. Since this excess DNA cannot fit smoothly against the surface of the histone octamer, it presumably bulges out in a loop from the nucleosomal surface. If the position of this loop migrates around the surface of the nucleosome, the nucleosome will “jump” 40 to 60 base pairs along the DNA. This process can uncover sequences that are important for gene regulation that had been hidden by association with a nucleosome. Alternatively, this mechanism may be used to loosen the nucleosome and allow the exchange of histone dimers in and out.
Histone Acetylation and Nucleosome Assembly
During DNA replication, existing nucleosomes are partitioned randomly between daughter DNA strands. Newly assembled nucleosomes then fill the gaps. When not associated with DNA, histones are always bound to protein chaperones. Newly translated H3 and H4, which are acetylated on lysine-9 of H3 and lysine-5 and lysine-12 of H4, associate with a chromatin assembly factor, called CAF1. One of the three subunits of CAF1 is a chaperone called retinoblastoma-associated protein of 48 kD (RbAp48). CAF1 is targeted to sites of DNA replication by interaction with proliferating cell nuclear antigen (PCNA), a doughnut-shaped protein that helps DNA polymerase to slide along the DNA during replication (see Fig. 42-11). Thus, CAF1 delivers newly synthesized histones to sites on the chromosome where new nucleosomes are required as DNA is synthesized during the S phase of the cell cycle (see Chapter 42). H3 and H4 are deposited first on the new DNA, followed by two H2A:H2B heterodimers to complete the assembly of the nascent nucleosome.
Linker DNA and the Linker Histone H1
When examined by electron microscopy at low ionic strength, nucleosomal chromatin resembles a string of beads with diameters of about 10 nm and linker DNA extended between adjacent nucleosomes (Fig. 13-1). Each nucleosome in chromosomes is typically associated with about 200 base pairs of DNA. With subtraction of 166 base pairs for two turns around the histone octamer, this leaves 34 base pairs of linker DNA between adjacent nucleosomes. Linker DNA can vary widely in length in different tissues and cell types.
A fifth histone, H1 or linker histone, is thought to bind to linker DNA at the side of each nucleosome core where the DNA molecule enters and exits the structure (Fig. 13-5). H1 histones have a “winged helix” central domain flanked by unstructured basic domains at both the N- and C-termini (Fig. 13-3). Mammals have at least eight variant forms (called subtypes) of H1 histones (H1a–e, H10, H1t, and H1oo). The amino acid sequences of these variants differ by 40% or more. Of these, H10 is found in cells entering the nondividing Go state (see Chapter 41), while H1t and H1oo are found exclusively in developing sperm and oocytes, respectively.
The Second Level of Chromosomal DNA Packaging: The 30-nm Fiber
Levels of chromatin structure beyond the nucleosome are poorly understood. One job of linker histone H1 is to promote the packaging of chromatin into the 30-nm fiber, a condensed filament of nucleosomes that can be observed by electron microscopy. Investigators now agree that the 30-nm fiber is unlikely to be a simple helix (solenoid) of nucleosomes. More complex models, similar to those shown in Figure 13-6B and D, are now favored.
Higher Levels of Chromosomal DNA Packaging in Interphase Nuclei
Dense packing of macromolecules in the nucleus makes it very difficult to observe the details of higher-level folding of chromatin fibers directly. Visualization of specific DNA loci within fixed interphase nuclei by in situ hybridization (see Fig. 13-15) can be used to estimate the degree of chromatin compaction by comparing the physical distance between two DNA sequences with a known number of base pairs between them. For regions of DNA up to about 250,000 base pairs apart, the chromatin fiber is shortened about twofold to threefold relative to the 30-nm fiber. When sequences are separated by tens of millions of base pairs, the shortening increases by another 20-fold to 30-fold. This suggests that there are at least two levels of chromatin folding beyond the 30-nm fiber.
Figure 13-15 fluorescence in situ hybridization performed on mitotic chromosomes. A., Chromosomes are spread on a slide as in Figure 13-14. Following chemical fixation steps to preserve the chromosomal structure, the chromosomal proteins are removed by digestion with proteases and the genomic DNA strands are melted (separated) by heating. Next, a “probe DNA” (yellow) is added. This probe DNA is single-stranded so that it can base-pair (hybridize) to its complementary sequences in the chromosome. The probe DNA is chemically labeled with biotin. Next, the sites of hybridization on the chromosomes are detected with fluorescently labeled avidin, a protein from egg white that binds to biotin with extremely high affinity. The sites of avidin-binding appear yellow, whereas the remainder of the chromosomal DNA is counterstained with a red dye. B, The micrograph shows FISH analysis using a probe from near the von Hippel Lindau locus on chromosome 3.
(B, Courtesy of Jeanne Lawrence, University of Massachusetts, Amherst.)
The organization of chromatin fibers can be observed by fluorescence microscopy of living cells after labeling with a fluorescent marker, such as the jellyfish green fluorescent protein (GFP [see Fig. 6-3]) (Fig. 13-7). These labeled chromosome arms are dynamic, changing both their structure and location as cells traverse the cell cycle. At times in the cycle when a chromosome arm becomes relatively more decondensed, it is possible to observe the presence of a fiber, 100 to 300 nm in diameter, called a chromonema fiber. Similar fibers are seen in electron micrographs of interphase cells. It is not yet known whether the chromonema fiber is the next level of chromatin packing above the 30-nm fiber.
Figure 13-7 direct visualization of changes in the compaction and location of a chromosome arm in a living cell. DNA molecules carrying the binding sites for a specific DNA-binding protein were integrated into the chromosomes of a cell at random and caused to amplify into large arrays, which, in some cases, corresponded to whole chromosome arms. These cells were then induced to express the DNA-binding protein as a fusion to jellyfish green fluorescent protein (GFP). In the lower panel at each time point, the fluorescently labeled chromosome arm can be seen to change both its degree of condensation and its position within the nucleus as a function of the cell cycle. The upper panels show total DNA stained with DAPI. DNA replication occurs in the S phase, which is separated from mitosis (cell division) by the G1 and G2 gap phases (see Chapter 40). These studies are potentially revealing, but they should be interpreted cautiously, as the labeled regions represent artificial arrays of DNA sequence rich in particular binding sites and may not exactly mimic the behavior of natural segments of chromosomes.
(From Li G, Sudlow G, Belmont AS: Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: Precise choreography of condensation/decondensation and nuclear positioning. J Cell Biol 140:975–989, 1998.)
Functional Compartmentation of the Nucleus: Heterochromatin and Euchromatin
Chromatin has traditionally been divided into two main classes based on structural and functional criteria. Euchromatin contains almost all of the genes, both actively transcribed and quiescent. Heterochromatin is transcriptionally inert and is generally more condensed than the euchromatin; it was initially recognized because it stains more darkly with DNA-binding dyes than the remainder of the nucleus. A typical nucleus has both euchromatin and heterochromatin, the latter usually being concentrated near the nuclear envelope and around nucleoli. Much of the interior of nuclei is occupied by pale-staining euchromatin rich in actively transcribing genes. Nuclei that are less active in transcription have relatively more heterochromatin (Fig. 13-8). Two types of heterochromatin are recognized.
