Disorders that primarily affect white matter
The leukodystrophies can be distinguished on the basis of their clinical, etiologic, and neuropathologic characteristics (Fig. 5.1, Tables 5.1–5.3). This chapter covers the leukodystrophies that are not known to be due to lysosomal or peroxisomal disorders. Those that are caused by lysosomal or peroxisomal disorders are described in more detail in Chapter 23.
Table 5.1
Clinical features of leukodystrophies

CACH, childhood ataxia with central hypomyelination syndrome.
Table 5.2
Etiologic classification of leukodystrophies
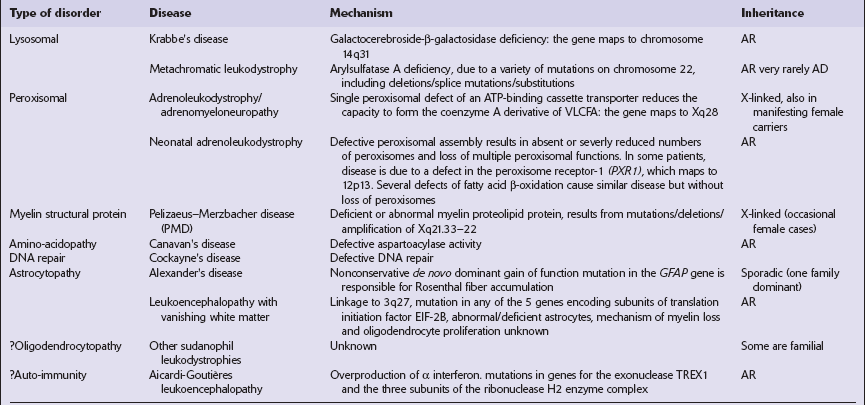
AR, autosomal recessive; AD, autosomal dominant; VLCFA, very long chain fatty acids.
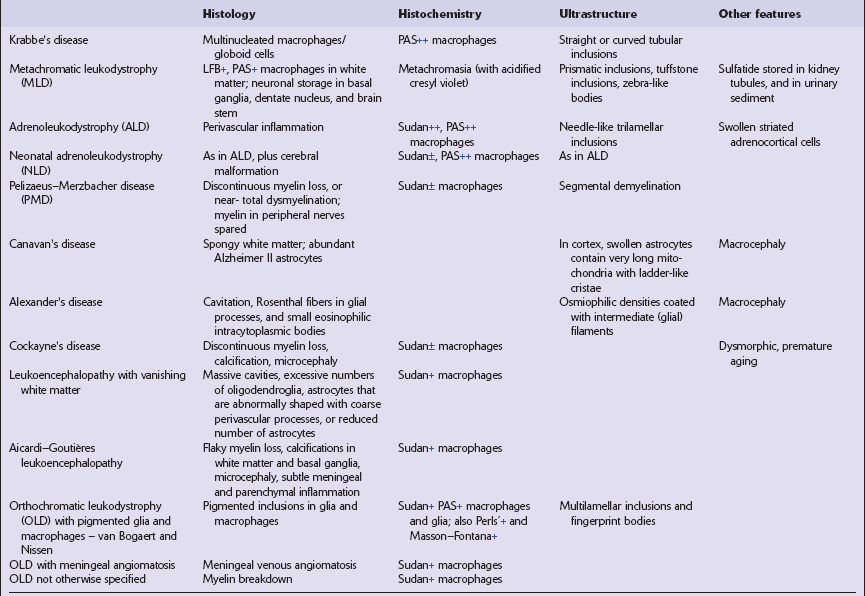
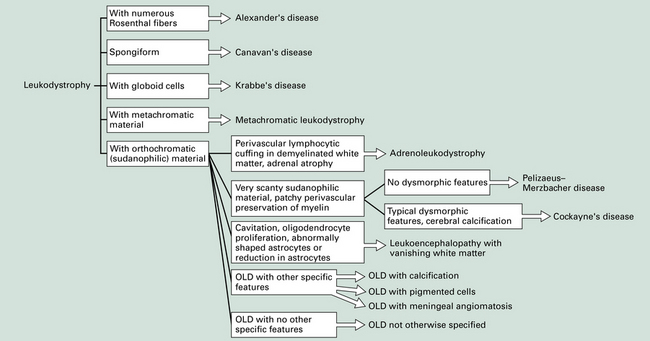
5.1 Descriptive classification of leukodystrophies.
This provides a morphologic approach to the differential diagnosis of the leukodystrophies. OLD, orthochromatic leukodystrophy.
PELIZAEUS–MERZBACHER DISEASE
 Differential Diagnosis of Leukodystrophy
Differential Diagnosis of Leukodystrophy
 Immunogenic demyelination (see Chapters 19 and 20); such as multiple sclerosis and other inflammatory demyelinating diseases (acute disseminated encephalomyelitis).
Immunogenic demyelination (see Chapters 19 and 20); such as multiple sclerosis and other inflammatory demyelinating diseases (acute disseminated encephalomyelitis).
 Progressive multifocal leukoencephalopathy (see Chapter 13).
Progressive multifocal leukoencephalopathy (see Chapter 13).
 Toxic leukoencephalopathy (see Chapter 25).
Toxic leukoencephalopathy (see Chapter 25).
 Demyelination associated with systemic disorders; such as central pontine myelinolysis (see Chapter 22) and Marchiafava–Bignami syndrome (see Chapter 21).
Demyelination associated with systemic disorders; such as central pontine myelinolysis (see Chapter 22) and Marchiafava–Bignami syndrome (see Chapter 21).
unremarkable in the connate cases, but generally appears gray and firm with a blurred gray–white matter interface (see Fig. 5.2a,b). Cerebellar atrophy can be marked. Involvement of brain stem and cord tracts renders them a dull yellow, contrasting with the whiteness of the preserved peripheral myelin in cranial and spinal roots.
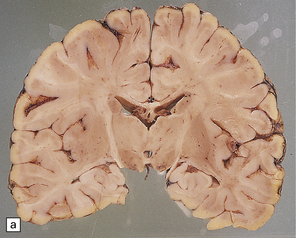
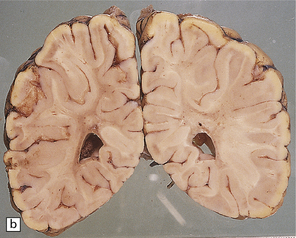
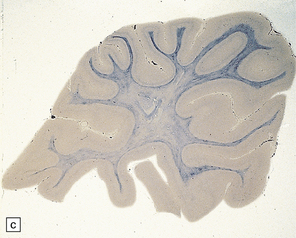
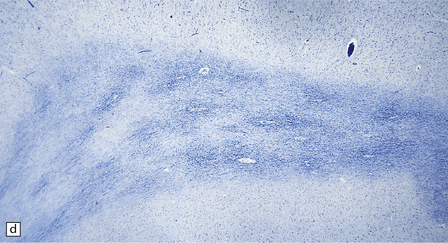
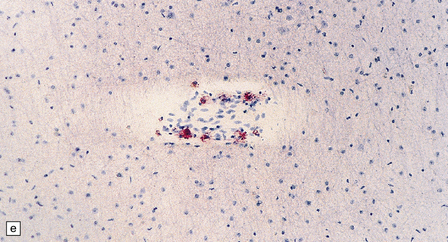
5.2 Pelizaeus–Merzbacher disease.
(a,b) Sections of the brain from a 15-year-old male with a typical slowly progressive clinical disorder. Much of the cerebral white matter appears gray and granular, but the U-fibers are relatively spared. The internal capsules and corpus callosum are streaked with gray. (a) Coronal section through the basal ganglia. (b) Coronal section through the parieto-occipital region. (c) In the occipital lobe from this patient there is only scant myelin staining. (d) At higher magnification the white matter takes on a tigroid appearance as islands of residual myelin surround blood vessels. (e) In this frozen section there is only sparse orthochromatic lipid.
MICROSCOPIC APPEARANCES
Myelin staining may be absent (connate variant) or severely reduced but leaving a flaky or tigroid pattern where residual myelin islets have a tendency to hug blood vessels (Figs 5.2–5.4). Axons are relatively preserved, though oligodendroglia are reduced and astrocytosis is marked. All central myelin is affected, while spinal and cranial nerve roots and peripheral nerves are normal (Fig. 5.5). Sudanophilic lipid is sparse and contained in perivascular macrophages. Cerebellar cortical degeneration is quite common (Fig. 5.6).
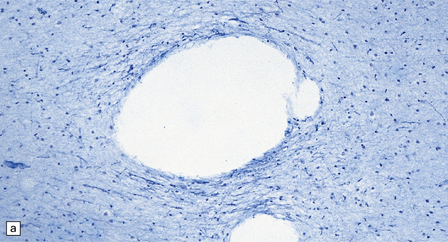
5.3 Pelizaeus–Merzbacher disease.
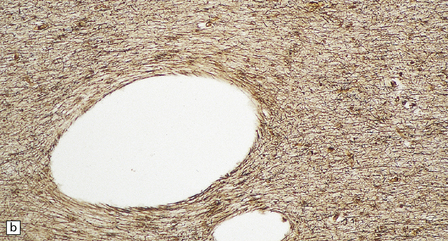
As with other leukodystrophies there is a relatively spared population of axons in demyelinated regions. (a) Occipital white matter stained for myelin. (b) An adjacent serial section stained for axons.
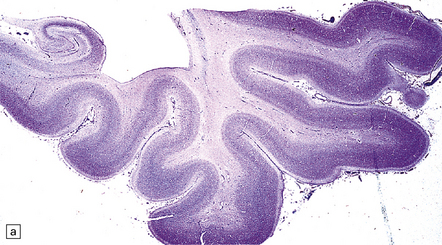
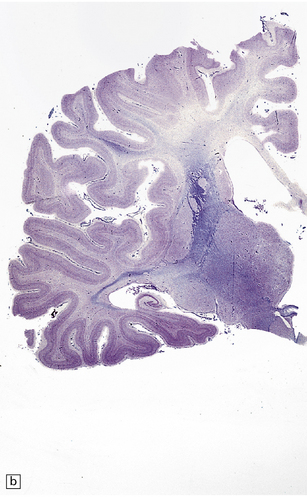
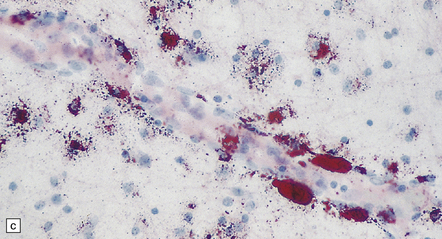
5.4 Myelin and sudanophilic lipid in Pelizaeus–Merzbacher disease.
(a,b) Coronal sections from a 6-month-old child with the connate form of Pelizaeus–Merzbacher disease. Myelin is demonstrable in the internal capsule and optic radiation only. (a) Temporal lobe. (b) Coronal section at mid-thalamic level. (c) Moderate amounts of orthochromatic lipid can be demonstrated.

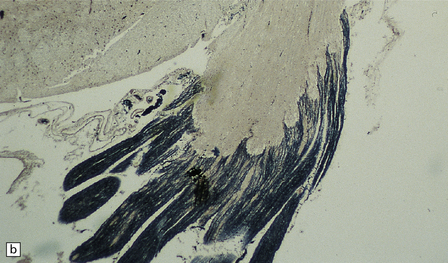
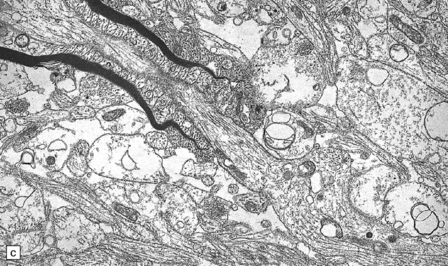
5.5 Sparing of myelin in peripheral nerves in Pelizaeus–Merzbacher disease.
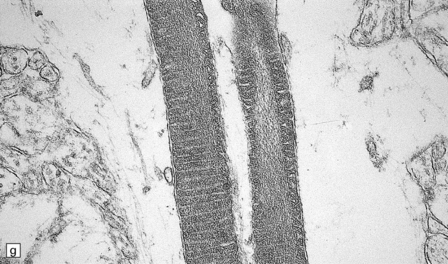
This is a characteristic finding. (a) Anterior aspect of the spinal cord showing the marked contrast between the normal white roots and the gray (demyelinated) cord. (b) Lateral pons showing the fifth cranial nerve root. Compare the normal peripheral myelin with the unmyelinated central structures. (c) Ultrastructural examination of the white matter from a cerebral biopsy showing segmental demyelination. The myelin stops abruptly at a heminode, leaving a naked axon continuing through the neuropil.
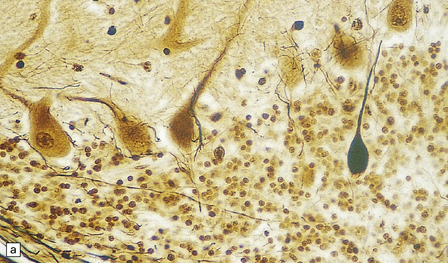
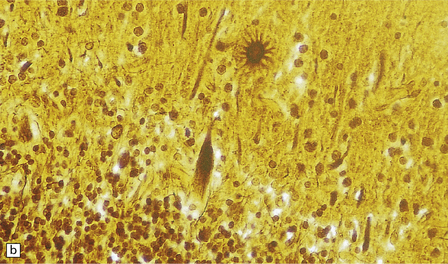
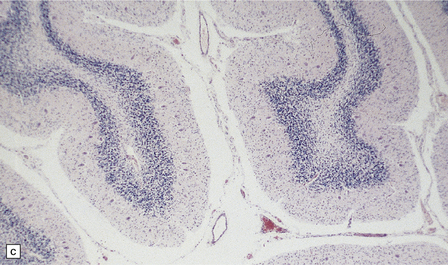
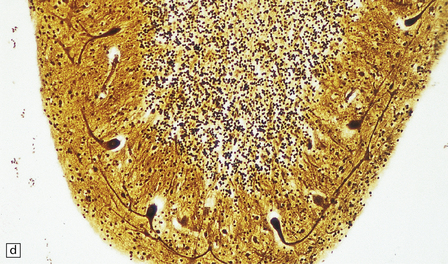
5.6 Cerebellar degeneration in Pelizaeus–Merzbacher disease.
Cerebellar degeneration is common in Pelizaeus–Merzbacher disease. (a) An example of cortical degeneration with loss of Purkinje cells and formation of axonal ‘torpedoes’. (b) Surviving Purkinje cells have abnormal dendritic swellings – ‘asteroid’ bodies. (c) An example of granule cell deficiency and malpositioned Purkinje cells in the molecular layer. (d) The ectopic Purkinje cells have ‘weeping willow’ dendrites.
CANAVAN’S DISEASE
Megalencephaly and increased brain weight may occur in the first 2 years of life, but become less apparent in older patients. At brain cut there is a very poor distinction between cerebral gray and white matter (see Fig. 5.7a,b). The soft gelatinous white matter does not cavitate, but the involved subcortical U-fibers may sink below the surface of the brain slices.
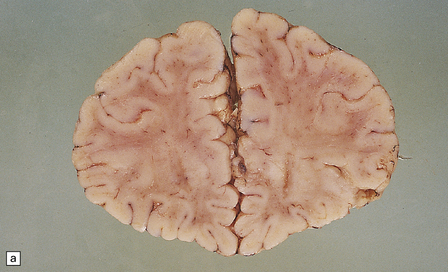
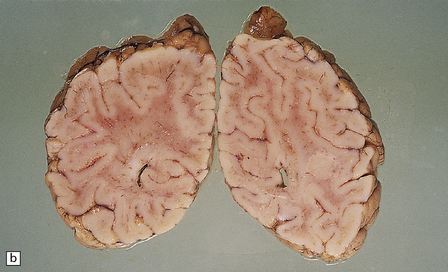
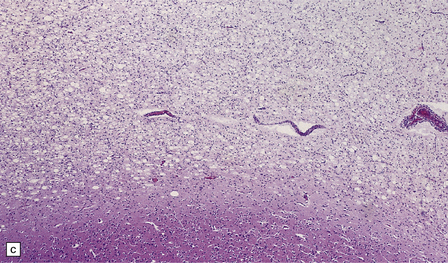
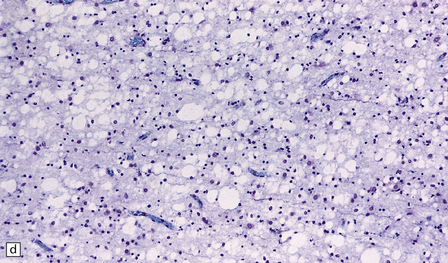
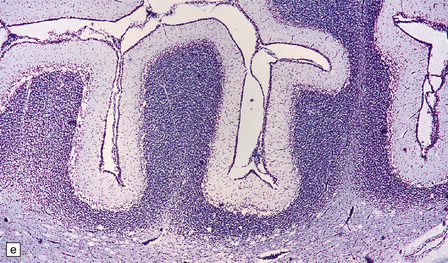
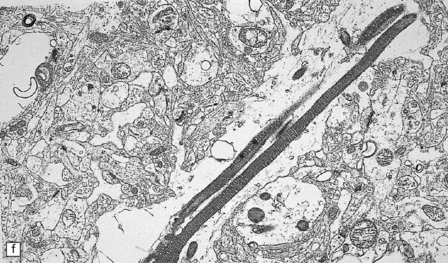
5.7 Spongiform leukodystrophy: Canavan’s disease.
In a 5-month-old infant the large brain weighed 900 g, and coronal sections showed discolored gelatinous white matter. (a) Frontal lobes. (b) Occipital lobes. (c) Section from the frontal lobe showing replacement of all the white matter, including the U-fibers, by spongy tissue. (d) Higher magnification of (c). (e) Spongy degeneration of the cerebellar white matter encroaches on the inner aspect of the cortex. (f) Electron microscopy of the cerebral cortex showing a hydropically swollen astrocytic process containing abnormally long mitochondria. (g) The mitochondria have a granular filamentous core and dilated ladder-like cristae.
MICROSCOPIC APPEARANCES
Extensive vacuolation of the white matter, which is particularly prominent at the deep gray–white matter junction, is associated with diffuse myelin loss (Fig. 5.7). Although cortical neurons are normal there are numerous Alzheimer type II astrocytes within the cortex. The vacuoles measure up to 100 μm, and appear empty. There is no sudanophilia and peripheral nerves apear unaffected. Ultrastructural examination reveals swelling of astrocyte processes and splitting of thin myelin lamellae in the white matter. Cortical changes include enlarged pale astrocytes in deeper cortical layers that contain abnormally elongated mitochondria with abnormal ladder-like cristae, unique to Canavan’s disease.
ALEXANDER’S DISEASE
The brain is usually, though not always, enlarged. The cerebral white matter is diffusely discolored, very soft and gelatinous, and often cavitated, especially in the frontal lobes (see Fig. 5.8a,b).
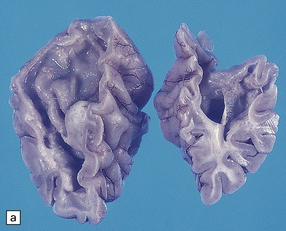
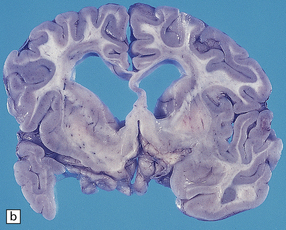
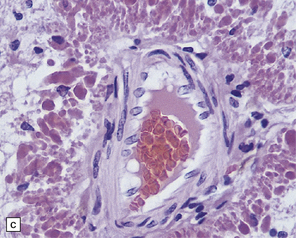
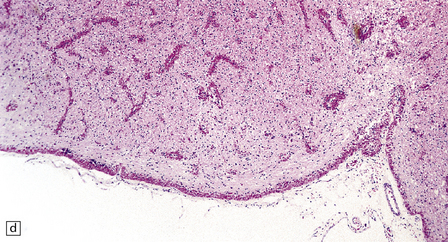
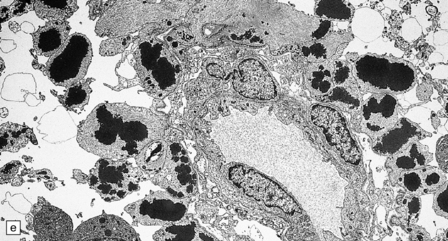
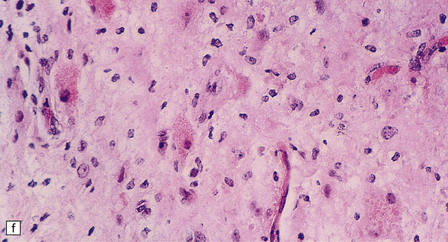
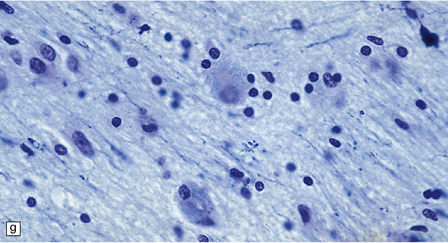
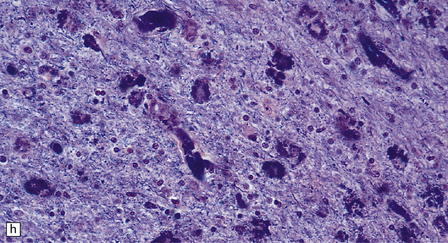
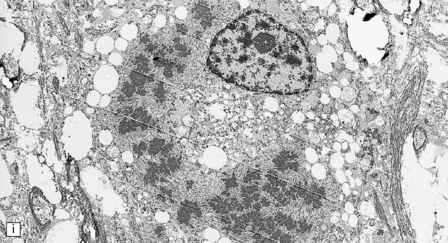
5.8 Alexander’s disease.
(a,b) Coronal sections of the cerebrum. Gray gelatinous and collapsed periventricular white matter more posteriorly (a). There is cavitation of the white matter anteriorly (b). (c) Rosenthal fibers are eosinophilic club-shaped bodies. Typically, they are clustered around blood vessels. (d) Rosenthal fibers also accumulate close to the pia. Large numbers are present in this demyelinated pyramid in the ventral medulla. (e) Ultrastructurally, the perivascular bodies are composed of astrocytic processes distended by irregular osmiophilic densities covered by glial filaments. (f) Astrocyte cell bodies frequently contain small inclusions, which are smaller than typical Rosenthal fibers, but ultrastructurally and tinctorially similar. (g) These smaller inclusions stain in a similar fashion to Rosenthal fibers. (h) Within the astrocyte cell body the small inclusions often have a signet ring appearance. (i) This shape is paralleled by the ultrastructural arrangement of osmiophilic densities.
MICROSCOPIC APPEARANCES
Diffuse demyelination leading to rarefaction of CNS white matter, with an abundance of Rosenthal fibers clustered most densely around blood vessels and in subpial and subependymal regions, is the principal feature (Fig. 5.8). All white matter is at risk, but the cerebellum is less often affected. As in other pathologic processes the Rosenthal fibers are irregularly elongated or rounded, hyaline eosinophilic bodies which ultrastructurally take the form of dense round osmiophilic structures coated with thickened glial fibrils within astrocytic processes. Similar but smaller inclusions are also present in the cell bodies of astrocytes, surrounding the nucleus, an appearance apparently restricted to Alexander’s disease and therefore of diagnostic utility. Although megalencephaly is a feature up to 2 years of age with some increase in brain weight, there is a decline to normal weight with longer survival.
OTHER LEUKODYSTROPHIES
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Apart from Cockayne’s syndrome, the etiologies of these rare conditions are largely unknown, and so morphologic classification remains paramount (Figs 5.9–5.12). Although the tigroid demyelination in Cockayne’s syndrome mimics Pelizaeus–Merzbacher there are many differences in this etiologically distinct disorder, notably profound microcephaly and vasocentric calcification in the cerebral cortex and basal ganglia.
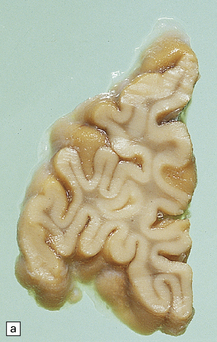
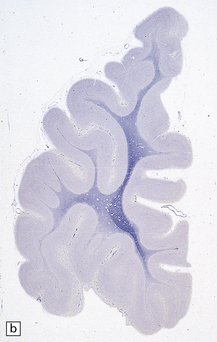
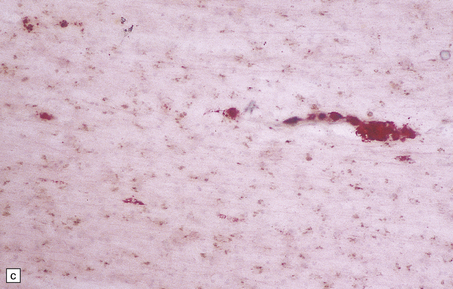
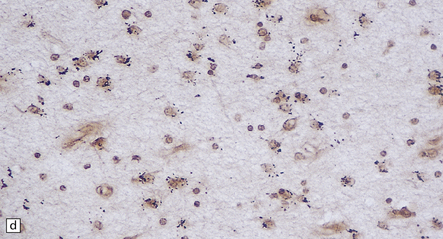
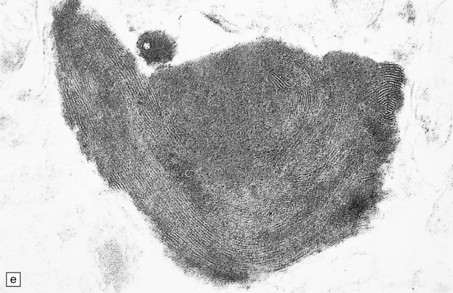
5.9 Leukodystrophy with pigmented glia and macrophages: van Bogaert and Nissen disease.
(a) Coronal section from the frontal pole showing diffuse white matter disease. (b) Myelin-stained counterpart of (a). (c) Histochemically, there is scanty lipid deposition. (d) There is a positive reaction for melanin. (e) Inclusions within the macrophages are multilamellar and include fingerprint profiles.
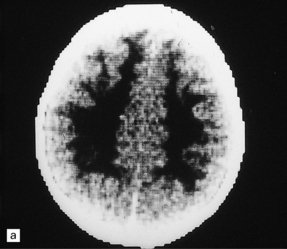
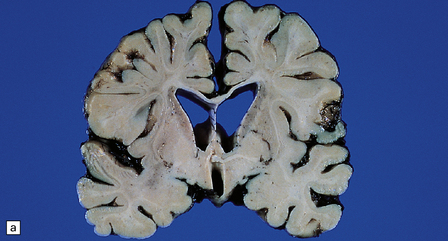
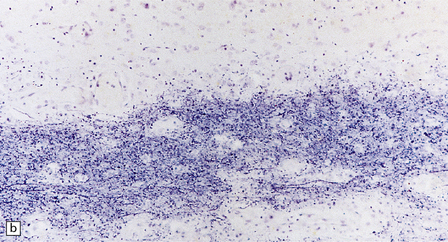
5.10 Leukoencephalopathy with vanishing white matter (VWM).
(a) Computerized tomographic scan indicates massive white matter cavitation in a child with familial VWM. (b) Coronal slice through the brain of a 60-year-old man with a long history of familial VWM of adult onset. (c) Histology of the residual white matter around the cavities shows hypercellular tissue containing numerous oligodendrocytes.
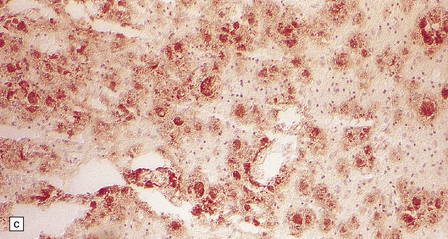
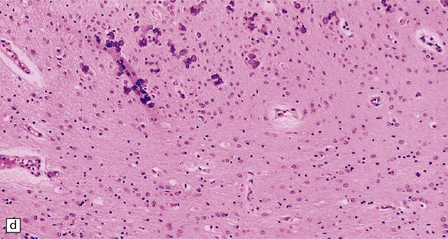
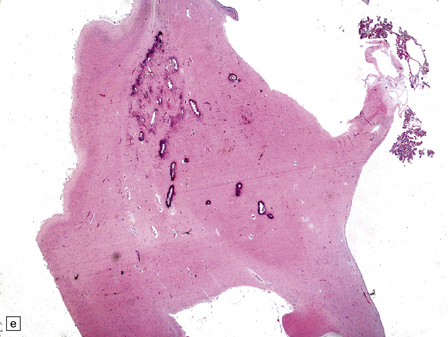
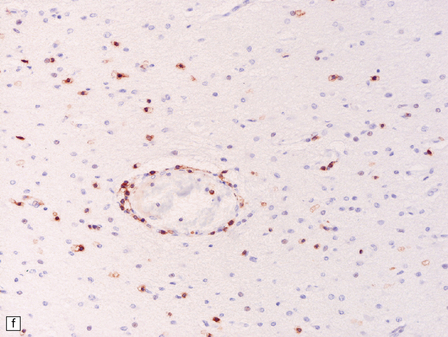
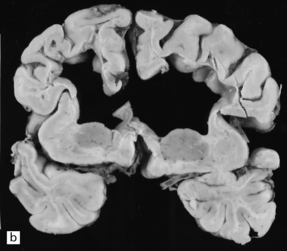
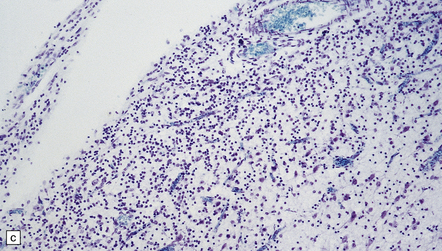
5.11 Leukodystrophy with microcephaly and calcifications: Aicardi–Goutières leukoencephalopathy.
(a) Coronal section of the microencephalic brain (550 g at 20 months) in which there is marked ventricular dilatation and reduction in the bulk of the white matter, which is gray and rubbery. The corpus callosum is very thin. (b) The white matter shows poorly stained myelin. (c) In a frozen section the white matter contains plenty of sudanophilic lipid. (d) The white matter also contains scattered calcifications and is markedly gliotic. (e) Extensive calcification in the basal ganglia. (f) Subtle perivascular lymphocytic inflammation in the white matter.
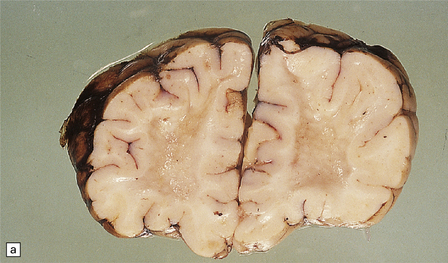
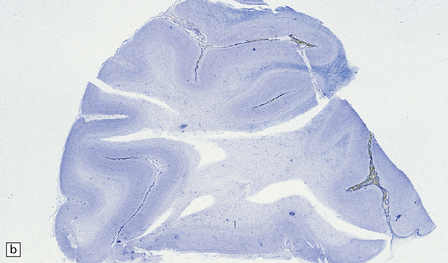
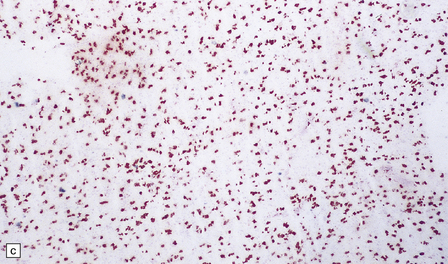
5.12 Sudanophilic leukodystrophy with no other specific features (familial example).
Despite extensive clinical, biochemical and morphologic examinations, some cases of both familial and sporadic sudanophilic leukodystrophy cannot be classified and remain a major challenge for neuropathologists. (a) Coronal section through the frontal lobes of a nine-month-old child showing brown gelatinous white matter. (b) Histology reveals a lack of myelin staining. (c) There are prominent orthochromatic lipid deposits.
REFERENCES
Aicardi, J., Goutières, F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol.. 1984;15:49–54.
Biffi, A., Aubourg, P., Cartier, N. Gene therapy for leukodystrophies. Hum Mol Genet.. 2011;20:R42–R53.
Borrett, D., Becker, L.E. Alexander’s disease; a disease of astrocytes. Brain.. 1985;108:367–385.
Brenner, M., Johnson, A.B., Boespflug-Tanguy, O., et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander’s disease. Nat Genet.. 2001;27:117–120.
Cleaver, J.E., Thompson, L.H., Richardson, A.S., et al. A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat.. 1999;14:9–22.
Friede, R.L. Developmental neuropathology. Berlin: Springer-Verlag; 1989.
Goutières, F., Aicardi, J., Barth, P.G., et al. Aicardi–Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol.. 1998;44:900–907.
Graveleau, P., Gray, F., Plas, J., et al. Cavitary orthochromatic leukodystrophy with oligodendroglial changes. A sporadic adult case. Rev Neurol Paris.. 1985;141:713–718.
Gray, F., Destee, A., Bourre, J.M., et al. Pigmentary type of orthochromatic leukodystrophy (OLD): a new case with ultrastructural and biochemical study. J Neuropathol Exp Neurol.. 1987;46:585–596.
Harding, B.N., Malcolm, S., Ellis, D., et al. A case of Pelizaeus–Merzbacher disease showing increased dosage of the proteolipid protein gene. Neuropathol Appl Neurobiol.. 1995;21:111–115.
Leegwater, P.A., Konst, A.A., Kuyt, B., et al. The gene for leukoencephalopathy with vanishing white matter is located on chromosome 3q27. Am J Hum Genet.. 1999;65:728–734.
Matalon, R., Michals, K., Kaul, R. Canavan disease: from spongy degeneration to molecular analysis. J Pediatr.. 1995;127:511–517.
Nance, M.A., Berry, S.A. Cockayne syndrome: review of 140 cases. Am J Med Genet.. 1992;42:68–84.
Olivier, M., Lenard, H.G., Aksu, F., et al. A new leukoencephalopathy with bilateral anterior temporal lobe cysts. Neuropediatrics.. 1998;29:225–228.
Pridmore, C.L., Baraitser, M., Harding, B., et al. Alexander’s disease: clues to diagnosis. J Child Neurol.. 1993;35:727–741.
Razavi, E.F., Larroche, J.C., Gaillard, D. Infantile familial encephalopathy with cerebral calcifications and leukodystrophy. Neuropediatrics.. 1988;19:72–79.
Rodriguez, D., Gelot, A., Della, G.B., et al. Increased density of oligodendrocytes in childhood ataxia with diffuse central hypomyelination (CACH) syndrome: neuropathological and biochemical study of two cases. Acta Neuropathol.. 1999;97:469–480.
Schiffmann, R., van der Knaap, M.S. The latest on leukodystrophies. Curr Opin Neurol.. 2004;17:187–192.
Seiser, A., Jellinger, K., Brainin, M. Pigmentary type of orthochromatic leukodystrophy with early onset and protracted course. Neuropediatrics.. 1990;21:48–52.
Takada, K., Becker, L.E. Cockayne’s syndrome: report of two autopsy cases associated with neurofibrillary tangles. Clin Neuropathol.. 1986;5:64–68.
Tunon, T., Ferrer, I., Gallego, J., et al. Leucodystrophy with pigmented glial and scavenger cells (pigmentary type of orthochromatic leucodystrophy). Neuropathol Appl Neurobiol.. 1988;14:337–344.
van der Knaap, M.S., Barth, P.G., Gabreels, F.J., et al. A new leukoencephalopathy with vanishing white matter. Neurology.. 1997;48:845–855.
van der Knaap, M.S., Barth, P.G., Vrensen, G.F., et al. Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol.. 1996;92:206–212.
Yool, D.A., Edgar, J.M., Montague, P., et al. The proteolipid protein gene and myelin disorders in man and animal models. Hum Mol Genet.. 2000;9:987–992.