Chapter 98 Disorders of the Autonomic Nervous System
Autonomic Dysfunction in Pediatric Practice
Introduction
The autonomic nervous system (ANS) is pervasive and affects every organ in the body, leading to its essential role in maintenance of homeostasis and stability (Figure 98-1). It is now appreciated that there is a wide spectrum of autonomic disorders affecting the pediatric population. The ANS is an integral part of the nervous system and acts to integrate multiple secondary functions, so that symptoms are widespread. Appreciation of the breadth of the ANS has increased since Langley originally proposed the generic term and designated its division into the sympathetic, parasympathetic, and enteric nervous systems [Langley, 1921]. Although the ANS is primarily an efferent system, it depends upon afferent information from various sources. Therefore, because development and maintenance of the autonomic and sensory systems are linked closely, many of the pediatric autonomic disorders are associated with sensory perturbations, especially those caused by genetic mutations. Furthermore, many functions of the peripheral ANS are regulated and integrated by a central autonomic network (CAN), whose extensive circuitry ranges from the forebrain to the brainstem, so that anatomical lesions, as well as emotions, can elicit autonomic symptoms.
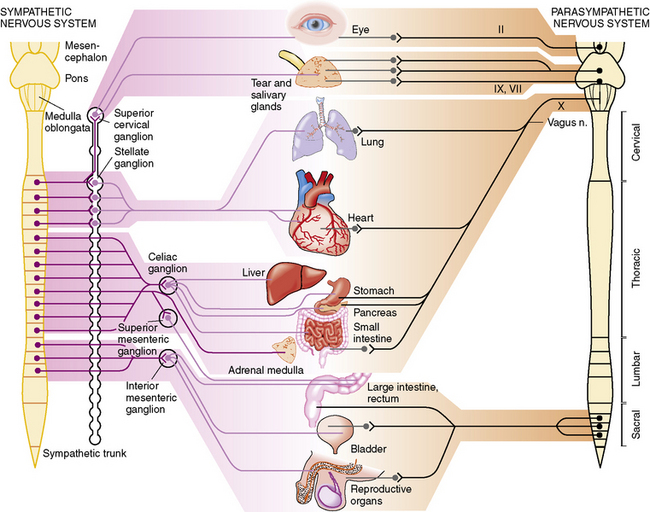
Fig. 98-1 Parasympathetic and sympathetic innervation of the major organs.
(From Janig W. Autonomic nervous system. In: Schmidt RF, Thews G, eds. Human Physiology, 2nd edn. Berlin: Springer, 1989;333.)
The scope of pediatric autonomic disorders is not well recognized [Axelrod et al., 2006]. Most texts that concern autonomic dysfunction focus on adult disorders; however, we now appreciate that there are a myriad of pediatric autonomic disorders, many of which present at birth or appear in early childhood. Although the basic anatomic, physiologic, and biochemical principles are the same for pediatric and adult autonomic disorders, there are some important differences in scope and presentation. Within the pediatric autonomic disorders there is a preponderance of genetic disorders with an intimate relationship to sensory dysfunction. Furthermore, the dynamic influence of neurologic maturation results in variations in clinical presentation and symptoms. This chapter will emphasize the breadth of pediatric disorders that feature autonomic dysfunction and highlight some of the more common disorders. The rationale and some techniques involved in evaluation and management also will be included.
Anatomic, Physiologic, and Biochemical Basis of Autonomic Nervous System Function
Embryologic Development
The ANS has its embryonic origins in the multipotential neural crest cells [Hamill and LaGamma, 2002]. The effector components of the ANS develop in the basal plate of the spinal cord and in the neural crest. By the 3rd week of human gestation, the neural plate begins to fuse and the neural crest cells start to migrate. These migrating cells eventually evolve into sensory and autonomic ganglia, as well as the adrenal chromaffin cells. The primitive sympathetic ganglia, in the thoracolumbar portion of the spinal cord, are formed from neural crest cells from the sixth somite to the posterior end of the neural crest and are recognizable by the 5th week of gestation. Preganglionic parasympathetic fibers arise from neural crest cells in the diencephalon and cranial part of the mesencephalon and rhombencephalon to form cranial nerves, and from neural crest cells in the second to fourth sacral segments to provide the parasympathetic supply to the pelvic viscera. The migrating neural crest cells also contribute to the formation of the enteric nervous system. After colonizing the gut, neural crest-derived cells within the gut wall then differentiate into glial cells plus many different types of neurons [Anderson et al., 2006]. Pupils, salivary glands, heart, gastrointestinal tract, and lungs become innervated from the 5th–7th week. In the 10th week, cardiac conducting fibers appear. Diseases due to defects in the neural crest induction, formation, or migration are referred to as neurocristopathies, and genes that cause some of these disorders, like Hirschsprung’s disease, have been cloned in mice models [Shahar and Shinawi, 2003].
Various factors promote normal progression from the embryonic to the mature autonomic and sensory nervous systems [Edlund and Jessell, 1999; Goridis and Brunet, 1999]. The process of differentiation is incumbent upon exposure of migrating neural crest cells to growth factors released by structures along the migratory route and then within the target tissue. Eventually, specificity will be determined by a neuronal cell’s ability to produce specific neurotransmitters. Several key transcription factors have been identified that play critical roles in the development of the ANS, such as the MASH1 and PHOX genes, which are necessary for differentiation of uncommitted neural crest cells to the developing ANS [Sommer et al., 1995; Tiveron et al., 1996]. Another important regulator of development and survival is nerve growth factor (NGF) [Levi-Montalcini, 1972; Thoenen and Barde, 1980]. In the embryonic neuron, NGF binding promotes migration from the neural crest and enhances maturation through neurite outgrowth. In the mature neuron, dependence on NGF decreases but it continues to enhance neurotransmitter synthesis [Thoenen and Barde, 1980].
The Peripheral Autonomic Nervous System
The peripheral ANS is a visceral and largely involuntary motor/effector (efferent) system. Its subdivisions can be defined by their anatomical locations or their function, as mediated by neurotransmitters. Traditionally, the ANS has been divided into sympathetic (thoracolumbar) and parasympathetic (craniosacral) divisions [Pick, 1970]. In addition, there is an important enteric division.
Sympathetic outflow emanates from major nuclei in the hypothalamus, midbrain, and brainstem, and descends through the cervical spinal cord, where axons synapse in the intermediolateral cell mass. From the thoracic and upper lumbar spinal segments, myelinated axons emerge in white rami and synapse in paravertebral ganglia, which are some distance from target organs. Postganglionic fibers, which are unmyelinated, rejoin the mixed nerve through the gray rami and innervate target organs, except for the adrenal medulla, which has only a preganglionic supply (see Figure 98-1). Parasympathetic outflow consists of cranial and sacral efferents. Cranial efferents accompany cranial nerves III, VII, IX, and X, and supply the eye, lacrimal and salivary glands, heart and lungs, and gastrointestinal tract, down to the level of the colon. The sacral outflow supplies the urinary tract and bladder, the large bowel, and the reproductive system. Most parasympathetic ganglia are close to target organs.
Although the traditional concept is that the sympathetic and parasympathetic systems are antagonistic, this is not always the case, as indicated in Table 98-1. Thus, when the sympathetic system is stimulated, a host of receptor systems are activated, including dilatation of the pupil, increase in glandular secretions, bronchodilatation, increase in heart rate and force of contraction, decrease in gastrointestinal tract motility, decrease in function of the reproductive organs, and mobilization of energy substrates. The parasympathetic system tends to have more focal responses, but some effects may be quite broad, particularly with the wide-ranging innervation of the vagus nerve. However, the parasympathetic system appears to have less influence on exocrine and endocrine function.
| Organ | Sympathetic | Parasympathetic |
|---|---|---|
| Eye | ||
| Pupil | Dilatation | Constriction |
| Ciliary muscle | Relax (far vision) | Constrict (near vision) |
| Lacrimal gland | Slight secretion | Secretion |
| Salivary glands | Slight secretion | Secretion |
| Heart | Increased rate | Decreased rate |
| Positive inotropism | Negative inotropism | |
| Lungs | Bronchodilatation | Bronchodilatation |
| Gastrointestinal | Decreased motility | Increased motility |
| Kidney | Decreased output | None |
| Bladder | Relax detrusor | Contract detrusor |
| Contract sphincter | Relax sphincter | |
| Penis | Ejaculation | Erection |
| Sweat glands | Secretion | Palmar sweating |
| Blood vessels | ||
| Arterioles | Constriction | None |
| Muscles | ||
| Arterioles | Constriction or dilatation | None |
| Metabolism | Glycogenolysis | None |
(Adapted from Axelrod FB et al. Pediatric autonomic disorders. State of the art. Pediatrics 2006;118:309.)
The Central Autonomic Nervous System
The peripheral ANS can operate independently but, to some extent, it is regulated and integrated by the CAN, whose circuitry ranges from the forebrain to the brainstem (Table 98-2) [Loewy, 1990]. The CAN maintains integral relationships with visceral sensory neurons via afferent input, primarily from cranial nerve X (vagus nerve), which is 80 percent afferent, transmitting visceral sensory information from the larynx, esophagus, trachea, and abdominal and thoracic viscera, as well as the stretch receptors of the aortic arch and chemoreceptors of the aortic bodies. This information is relayed through the nucleus tractus solitarius (NTS) to the major cerebral centers concerned with autonomic regulation, which include the amygdala, hypothalamus, midbrain, and forebrain [Loewy, 1990].
| Anatomic Area | General Function | Clinical Manifestations |
|---|---|---|
| Insular and medial prefrontal cortices | High-order autonomic control Input from gastric mechanoreceptors, arterial chemoreceptors, baroreceptors |
Cardiac arrhythmia |
| Extended amygdala | Expression of emotional states Integrates autonomic and motor responses |
Viscerosensory phenomena (e.g., unilateral hyperhidrosis) Vomiting (left temporal focus) Sexual arousal |
| Hypothalamus | Homeostasis Initiates and coordinates biologic rhythms, autonomic, neuroendocrine, and behavioral responses |
Hypo- or hyperthermia Poor stress response (autonomic storm) Insomnia |
| Midbrain | Coordinates autonomic, pain-controlling, and motor mechanisms for stress-related, aggressive, and reproductive behaviors | Hyper- or hypotension, arrhythmias Intractable vomiting and dysmotility Hypoventilation Urinary retention |
| Pons | Relays viscerosensory information to forebrain | |
| Nucleus of tractus solitarius | Relays viscerosensory information from vagus and glossopharyngeal nerves to other CAN regions | |
| Medulla | Cardiovascular and respiratory control via premotor autonomic and respiratory neurons controlling input to spinal, respiratory, and preganglionic motor neurons | Sleep disorders (e.g., apnea) |
CAN, central autonomic network.
(Adapted from Axelrod FB et al. Pediatric autonomic disorders. State of the art. Pediatrics 2006;118:309.)
Disorders in the forebrain circuits, such as ischemia secondary to blood flow disturbance or seizures, can cause cardiac arrhythmia [Hilz et al., 2002]. Within this circuitry, the NTS in the medulla oblongata, which receives input from the vagus and glossopharyngeal nerves, functions as a major relay station, allowing continuous feedback and integration. The hypothalamic area appears to have major influences on thermoregulation and sleep–wake cycling. Thus, the CAN serves many critical functions and affects visceromotor and neuroendocrine function, as well as motor and pain modulation. It aids in reflex adjustments of autonomic responses, and integrates autonomic, neuroendocrine, and behavioral responses that, in turn, maintain homeostasis, emotional expression, and response to stress [Benarroch and Chang, 1993; Critchley et al., 2001].
Neurotransmitters
The peripheral ANS provides physiologic responses via multiple neurotransmitters and a chemical coding system of autonomic neurons. The neurotransmitters, which include amino acids, peptides, and monoamines, act by relaying, amplifying, and modulating signals between two neurons, or a neuron and another cell, such as the smooth muscle cell of blood vessel. Neurotransmitters are packaged into synaptic vesicles that cluster beneath the membrane on the presynaptic side of a synapse, and are released into the synaptic cleft, where they bind to receptors in the membrane on the postsynaptic side of the synapse (Figure 98-2). During the last two decades, it has become clear that, within a single neuron, multiple transmitter systems coexist and that, within a given ganglion, the variety and pattern of neurotransmitters is extensive. In turn, multiple organ systems then respond to the neurotransmitters released via various receptor systems.
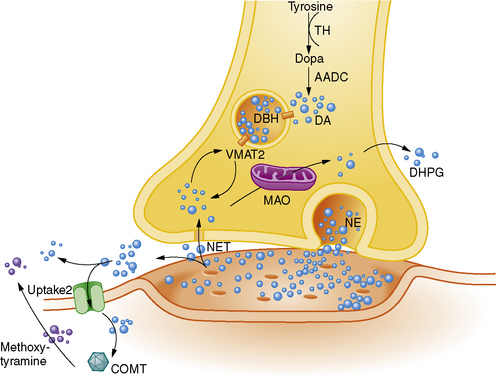
Under normal circumstances, the concentration of NE in a noradrenergic synapse is determined by the level of sympathetic activity. A steady state is maintained as a result of a careful balance among catecholamine synthesis, storage, release, and reuptake (see Fiugre 98-2) [Vincent and Robertson, 2002]. DA is the first catecholamine to be synthesized, with NE and epinephrine, in turn, being derived from further modifications of DA. Catecholamine synthesis also requires important co-factors; the enzyme dopamine β-hydroxylase (DβH) requires copper as a co-factor, and DOPA decarboxylase requires pyridoxal-phosphate. Approximately 70–90 percent of released NE is removed from the synaptic cleft through reuptake into the presynaptic neuron by the NE transporter. The remaining 10–30 percent is subject to extraneuronal uptake or spills over into the circulation [Eisenhofer et al., 2004].
The catecholamines also are vital neurotransmitters in the CAN, where they mediate such functions as attention, arousal, learning, memory, and mood. Dopamine is produced largely in neuronal cell bodies in two areas of the brainstem, the substantia nigra and the ventral tegmental area. Neurons containing NE and epinephrine also reside within several brainstem nuclei, including the locus ceruleus [Vincent and Robertson, 2002]. These efferent sympathetic neuronal pathways control autonomic reflexes at the level of the brainstem and spinal cord. They regulate postganglionic sympathetic synapses in the peripheral nervous system.
Genetic errors involved in synthesis, transport, storage, or metabolism of the neurotransmitters can result in a variety of autonomic disorders, such as DβH deficiency, in which lack of this essential enzyme results in inability to convert DA to NE, leading to symptoms consistent with sympathetic insufficiency [Robertson et al., 1986].
Clinical Features of Autonomic Dysfunction
As the ANS and its CAN component have pervasive effects affecting multiple other systems secondarily, there can be a myriad of clinical manifestations, depending upon which organ systems are perturbed. Rather than use an anatomic approach, one can use a functional or system approach, as listed in Table 98-3. Occasionally, there is autonomic dysfunction in a single system, with one or more isolated dysfunctions, such as children who have prominent enteric or gastrointestinal dysfunction or isolated gastroesophageal reflux disease. In such cases, the symptoms are limited to the particular system and investigations also should be restricted accordingly.
Table 98-3 Functional Organization of Symptoms Associated with Autonomic Disorders
| System | Dysfunction | Symptom |
|---|---|---|
| Vasomotor/cardiovascular | Hypertension Hypotension Arrhythmia Vascular changes |
Headache Dizziness, lightheadedness, blurred vision, loss of consciousness/syncope Palpitations, loss of consciousness/syncope Purple feet, mottled skin, acrocyanosis, blotching |
| Gastrointestinal | Oropharyngeal dysmotility Esophageal dysmotility Gastroesophageal reflux Bowel dysmotility |
Feeding problems (poor suck, drooling, aspiration pneumonia) Dysphagia (difficulty swallowing) Nausea, recurrent vomiting Bloating, profound constipation or diarrhea |
| Ophthalmologic | Alacrima Nonreactive/sluggish pupil Eyelid weakness |
Dry eye Dark/light intolerance Ptosis |
| Respiratory | Alveolar hypoventilation Apnea Insensitivity to hypoxia Insensitivity to hypercarbia |
Cyanosis with sleep Breath-holding spells Syncope at high altitudes/plane travel |
| Sudomotor | Altered sweating | Hypo- or hyperhidrosis, excessively dry skin, clammy hands and feet, unexplained fevers, heat intolerance |
| Urinary | Nocturia | Frequent awakenings to urinate or nocturnal enuresis >5 yrs of age |
| Central dysfunction | Thermoregulatory abnormalities Sleep–wake disturbance Altered affect and emotions Learning disability |
Decreased basal body temperature, unexplained high fevers Fractured sleep, insomnia, nocturnal enuresis >5 yrs of age Poor socialization skills, increased anxiety, emotional lability, tics/phobias Poor school performance, poor executive planning, attention problems, hyperactivity |
| Associated sensory dysfunction | Altered perception of pain | Decreased response to injury, injections, and dental procedures, self-mutilation, tactile defensiveness |
(Adapted from Axelrod FB et al. Pediatric autonomic disorders. State of the art. Pediatrics 2006;118:309.)
Vasovagal Syncope
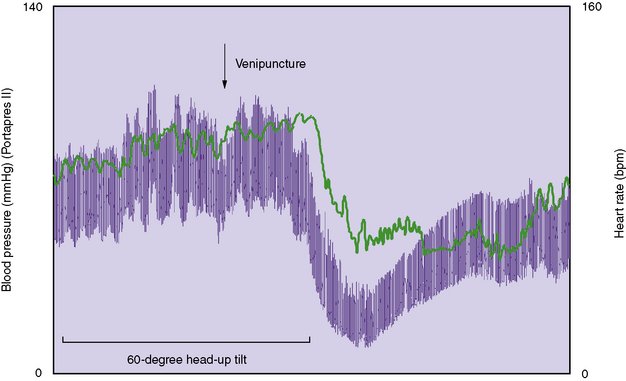
The most frequent noncardiac cause of unexplained transient loss of consciousness, or faint, is vasovagal syncope. It also is known as neurally mediated syncope and neurocardiogenic syncope, common or emotional fainting, or reflex syncope. During vasovagal syncope, vasodilatation and bradycardia occur simultaneously (Figure 98-3). The bradycardia is due to increased parasympathetic (vagal) outflow to the sinus node of the heart. The decrease in blood pressure is due to vasodilatation but the mechanism is not clear. Both reduction in sympathetic vasoconstrictor traffic (i.e., “passive” vasodilatation) and activation of a vasodilator system (i.e.,“active” vasodilatation) have been postulated [Kaufmann and Hainsworth, 2001]. Among the various postulated causes for active vasodilatation is activation of β2-adrenergic receptors secondary to epinephrine release from the adrenal medulla [Glover et al., 1962], and increased nitric oxide, a powerful vasodilator released by endothelial and other cells [Kaufmann et al., 1993].
Orthostatic Hypotension
Orthostatic hypotension, also called postural hypotension, is a temporary lowering of blood pressure (hypotension), usually due to standing up suddenly (orthostatic). The change in position causes a temporary reduction in blood flow and oxygen to the brain. When an individual stands up, gravity promotes the pooling of blood in the lower extremities, which decreases venous return of blood circulating back to the heart. Normally, cardiac and carotid sinus baroreceptors sense the decrease in blood volume and initiate increased heart rate and peripheral vasoconstriction. The latter increases resistance to blood flow and increases blood pressure. The underlying pathophysiology in individuals with orthostatic hypotension is an impaired efferent sympathetic signal to the arterioles, a failure to release norepinephrine appropriately upon standing, and consequent vasoconstrictive insufficiency resulting in blood pooling in the lower extremities, with subsequent decreased venous return to the heart and brain [Freeman, 2003]. Thus, orthostatic hypotension also is a common feature of a number of central autonomic neurodegenerative disorders, such as multiple system atrophy and Parkinson’s disease, and peripheral autonomic disorders, such as the autonomic peripheral neuropathies, in which there are decreased peripheral noradrenergic neurons or enzyme deficiencies resulting in low levels of NE – for example, in familial dysautonomia and dopamine β-hydroxylase deficiency, respectively. Furthermore, when blood volume is lowered by dehydration or shifted by food ingestion, orthostatic hypotension can be unmasked in physiologically intact individuals, especially older people.
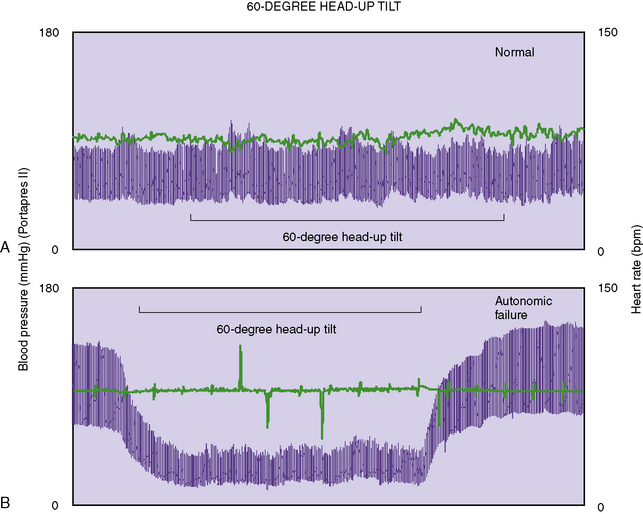
Orthostatic hypotension can be confirmed by measuring blood pressures and heart rate in supine and upright positions. Orthostatic hypotension is defined as a sustained drop in blood pressure of greater than 20 mmHg systolic or 10 mmHg diastolic within 3 minutes of being upright, associated with symptoms. The heart rate response to changes in blood pressure is minimal, although there may be a mild compensatory increase, i.e., below 30 beats per minute. (Figure 98-4). Presyncopal symptoms can appear, and include dizziness, lightheadedness, headache, and tunnel vision. Occasionally, individuals will faint and have syncope. The symptoms typically improve on lying down.
In a physiologically intact individual with no underlying condition, no medical treatment usually is needed for orthostatic hypotension. The most important treatments are assuring adequate fluids and avoiding potential triggers, such as prolonged sitting, quiet standing, warm environments, or vasodilating medications. Other measures that have been found helpful are use of postural maneuvers and physical therapy and exercise [van Lieshout et al., 1992]. When there is a coexisting underlying medical condition, then the orthostatic hypotension can be very debilitating and these supportive measures may not suffice [Freeman, 2003]. In such cases, various medications may be used, i.e., medications that increase blood volume (fludrocortisone, erythropoietin), medications that interfere with the release or action of epinephrine and NE (beta blockers, angiotensin-converting enzyme inhibitors), and medications that improve vasoconstriction (midodrine).
Orthostatic Intolerance/Postural Tachycardia Syndrome
Orthostatic intolerance (OI) is considered as a type of autonomic dysfunction that occurs as part of many autonomic disorders. As upright heart rate usually is increased greatly, the term postural tachycardia syndrome (POTS) also is used [Low et al., 2001].
Investigations demonstrate that, during postural challenge, the blood pressure does not fall, but the heart rate increases by more than 30 beats per minute or rises above 120 beats per minute (Figure 98-5). The supine heart rate usually is normal or slightly raised. Additional findings include a standing plasma NE level of at least 600 pg/mL, and blood volume usually is reduced. In the hyperadrenergic subgroup of OI patients, plasma renin activity and aldosterone are attenuated.
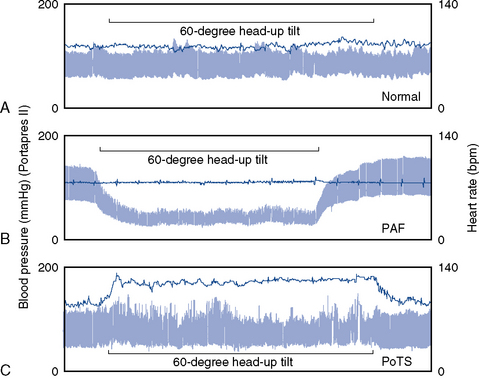
Fig. 98-5 Blood pressure and heart rate measured continuously before, during and after 60-degree head-up tilt.
(From Mathias CJ. To stand on one’s own legs. Clin Med 2002;2:237.)
The etiology of OI is unknown. It has been observed predominantly in young postpubertal females (female to male ratio at least 4:1) and the onset often is predated by a recent viral infection. Further supporting the hypothesis that OI is an immunologically mediated phenomenon is the fact that it often is a prominent feature of other secondary autonomic disorders that are believed to have an autoimmune etiology, such as idiopathic autonomic neuropathy and chronic fatigue syndrome. Increased propensity to venous pooling [Streeten et al., 1998] and a strong association with the joint hypermobility syndrome have been reported [Gazit et al., 2003].
Autonomic Disorders in Children and Adolescents
Pediatric autonomic disorders can be thought of as either primary (inherited) or secondary (acquired or associated with other systemic disorders). Within the primary or inherited disorders are many genetic disorders that result in incomplete development of the ANS or biochemical errors that affect neurotransmitter production or efficiency; for some of these disorders, the specific genetic mutations have been identified (Table 98-4). Clinical manifestations of the developmental disorders are noted early, usually within the first year of life. Those disorders that occur as a result of a biochemical error leading to inefficient mitochondrial metabolism or to storage of deleterious material within the neuron can be more insidious and later in their presentation.
| Disorders | Category | Disorders |
|---|---|---|
| PRIMARY | ||
| Developmental disorders | Hereditary sensory and autonomic disorders (HSAN) | Familial dysautonomia (HSAN III) Congenital sensory neuropathy with anhidrosis (HSAN IV) Congenital sensory neuropathy (HSAN II) |
| Allgrove’s syndrome | ||
| Chromosomal disorders | Rett’s syndrome Prader–Willi syndrome Fragile X/fragile X-associated tremor/ataxia syndrome |
|
| Prematurity | ||
| Dysregulation disorders | Cardiorespiratory and cardiac | Congenital central hypoventilation syndrome (CCHS) Reflex anoxic seizures |
| Biochemical errors | Myopathies | Mitochondrial myopathies Leber’s hereditary optic neuropathy Leigh’s syndrome Kearns–Sayre syndrome Myoneurogastrointestinal disorder with encephalopathy |
| Neurotransmitter deficiencies | Dopamine β-hydroxylase deficiency Norepinephrine transporter deficiency Menkes’ disease |
|
| Storage disorders | Fabry’s disease Familial amyloid polyneuropathy (FAP) |
|
| SECONDARY | ||
| Metabolic | Endocrine disorders | Diabetes Addison’s/Cushing’s disease Thyroid disorders |
| Autoimmune | Postinfectious? | Idiopathic autonomic neuropathy Paraneoplastic disorders Chronic fatigue syndrome |
| Neoplasia | Secreting tumors | Pheochromocytoma Carcinoid tumors |
| Intoxications | Drugs that modify sympathetic or parasympathetic activity | |
| Psychosocial/unknown | Functional gastrointestinal disorders | Cyclic vomiting syndrome Functional abdominal pain |
| Autism | ||
The secondary autonomic disorders include a number of broad and heterogeneous categories. For some of the secondary autonomic disorders, autoimmune mechanisms have been postulated [Etienne and Weimer, 2006; Low et al., 2003]. Other autonomic disorders are associated secondarily with metabolic diseases, such as diabetes; others are secondary to deleterious effects of drugs or toxins; and still others are idiopathic or unknown. Table 98-4 lists some of these disorders, but the number of primary and secondary disorders continues to expand. A few representative disorders that are present in childhood will be described.
Primary (Inherited) Autonomic Disorders
Autonomic Disorders Associated with Developmental Arrest
Hereditary sensory and autonomic neuropathies
The complexities of the ANS and its intimate relationship with sensory function are illustrated especially well in the group of genetic disorders known as hereditary sensory and autonomic neuropathies (HSAN) [Axelrod, 2002a, b; Dyck, 1993; Minde et al., 2004] (see also Chapter 89 for further description of these neuropathies). Each HSAN disorder is caused by a different genetic error affecting a specific aspect of small-fiber neurodevelopment and resulting in variable phenotypic expression. With the exception of hereditary sensory radicular neuropathy (HSAN type I), which is a dominant disorder presenting in the second decade, the other HSANs are autosomal-recessive disorders presenting at birth. For each HSAN type, penetrance is complete but there can be marked variability in expression. Characteristic to all HSANs is the fact that intradermal injection of histamine phosphate fails to elicit a normal axon flare response. Of the five HSAN subtypes, autonomic dysfunction is associated with II, III, and IV only.
Familial Dysautonomia
Of all the HSAN disorders, familial dysautonomia is the most extensively studied and described, and often is used as the prototype with which to compare other HSAN disorders [Axelrod 2002b]. In familial dysautonomia, the gene is IKBKAP, and over 99 percent of individuals with familial dysautonomia are homozygous for a mutation in intron 20, suggesting that there was a founder effect and explaining why the disorder occurs almost exclusively in individuals of Ashkenazi Jewish extraction [Anderson et al., 2001; Slaugenhaupt et al., 2001]. The IKBKAP gene codes for the IKAP/hELP1 protein, which is associated with the human elongator complex that aids in transcriptional elongation [Hawkes et al., 2002]. Studies suggest that familial dysautonomia mutations lead to misspliced messenger RNA and tissue-specific reductions in normal IKAP/hELP1 protein, with subsequent downregulation of genes involved in neuronal migration and function. Downregulation of these critical target genes results in aberrant neurogenic differentiation and migration behavior of neural crest cells, which eventually impacts the sympathetic and sensory systems [Close et al., 2006; Chariot et al., 2007; Lee et al., 2009; Slaugenhaupt et al., 2001]. In familial dysautonomia, there is inadequate development, as well as limited survival, of sensory and autonomic neurons, with the sympathetic population affected more widely than the parasympathetic. Pathologic studies have demonstrated decreased unmyelinated and small myelinated neuronal populations in the peripheral sensory nervous system and the ANS [Grover-Johnson and Pearson, 1976; Pearson et al., 1974]. Although central autonomic symptoms are present, no consistent central neuropathology has been described yet.
Although patients with familial dysautonomia have decreased pain and temperature perception, the sensory perturbations are not as profound as in the other HSAN disorders [Hilz and Axelrod, 2000]. Bone and skin pain is diminished but not absent; sensitivity to visceral pain is intact. Corneal and tendon reflexes are hypoactive, and taste appreciation is diminished, consistent with absence of lingual fungiform papillae. With age, vibratory sensory loss and impaired coordination appear [Axelrod et al., 1981].
The autonomic disturbances, however, are pervasive and impose the greatest impediments to function and survival [Axelrod, 2002a; Axelrod et al., 2002]. In addition to absence of tears (alacrima) with emotional crying, a cardinal feature of the disorder, feeding difficulties are frequent due to poor oral coordination and hypotonia. Recurrent misdirection, especially of liquids, and frequent gastroesophageal reflux put the patient at risk for aspiration and chronic lung disease. Protracted episodes of nausea and vomiting can be triggered by emotional or physical stress, and even arousal from sleep. These episodes, also termed the dysautonomic crisis, usually are associated with a constellation of signs, including agitation, tachycardia, and hypertension. Vasomotor and cardiovascular perturbations manifest as erythematous skin blotching and hyperhidrosis with excitation or even eating. Patients can exhibit both extreme hypertension and profound and rapid postural hypotension without compensatory tachycardia. Consistent with decreased autonomic neurons, patients exhibit denervation hypersensitivity to cholinergic and adrenergic agents [Bickel et al., 2002; Smith et al., 1965]. Patients have baroreflex failure and chemoreceptor dysfunction, so they are relatively insensitive to hypoxemia [Bernardi et al., 2002; Edelman et al., 1970; Filler et al., 1965; Maayan et al., 1992], which limits ability to cope with pneumonia or travel to high altitudes. Ensuing hypoxemia may lead to hypotension, bradyarrhythmia, and even syncope. Developmental milestones commonly are delayed, but intelligence usually is within normal ranges [Welton et al., 1979].
Although the gene has been identified and there are early reports of nutritional supplements that may be able to modify genetic expression in cell culture [Anderson et al., 2003a, b; Slaugenhaupt et al., 2004; Lee et al., 2009], the mainstay of treatment remains preventative and supportive. These treatments have included measures to maintain eye moisture, fundoplication with gastrostomy to provide nutrition and avoid risk of aspiration, use of central agents such as benzodiazepines and clonidine to control vomiting and the dysautonomic crisis, and fludrocortisone and midodrine to combat cardiovascular lability [Axelrod, 2005]. As a result of improved supportive measures, about half of the patients now reach adulthood [Axelrod et al., 2002a, b].
Hereditary Sensory and Autonomic Neuropathy Type IV (Congenital Insensitivity to Pain with Anhidrosis)
Congenital insensitivity to pain with anhidrosis is caused by mutations in the neurotrophic tyrosine kinase receptor type 1 (NTRK1) gene, located on chromosome 1 (1q21–q22) [Indo et al., 1996]. As a result of loss-of-function mutations, signal transduction at the NGF receptor is impeded, and NGF-dependent neurons – the nociceptive sensory and sympathetic neurons – fail to survive. There is no particular ethnic distribution for this disorder but one-half of reported cases have occurred in consanguineous marriages [Axelrod, 2002a, b].
HSAN type IV is characterized by anhidrosis – absent or markedly decreased sweating, which probably is secondary to impaired thoracolumbar sympathetic outflow. It is the anhidrosis that causes episodic fevers and extreme hyperpyrexia, which are usually the earliest signs of the disorder. Anhidrosis also contributes to the thick and calloused appearance of the skin, with lichenification of palms, dystrophic nails, and areas of hypotrichosis on the scalp [Pinsky and DiGeorge, 1966]. As evidence of parasympathetic dysfunction, patients exhibit miosis with dilute intraocular mecholyl. Cardiovascular responses are normal in the early years but some of the adolescents and adults manifest mild postural hypotension. Severe learning problems, often associated with hyperactivity, are frequent and may be due to central autonomic involvement [Indo et al., 1996; Vassella et al., 1968]. In contrast to patients with familial dysautonomia, emotional tearing is normal, there is no acrocyanosis, and gastrointestinal dysmotility is infrequent. Cyclical crises do not occur. Insensitivity to hypoxia and hypercapnia has not been noted.
Neuropathological studies have demonstrated decreased neuronal populations. Sural nerve biopsies show that myelinated nerve fibers are present but unmyelinated fibers are absent [Dyck, 1993]. Skin biopsy morphology of HSAN IV patients reveals deficient C and Aδ fibers in the epidermis, and absent or hypoplastic dermal sweat glands without innervation [Goebel et al., 1980; Langer et al., 1981; Nolano et al., 2000]. The lack of sweat gland innervation accounts for the clinical manifestation of severe anhidrosis and the objective absence of sympathetic skin responses [Hilz et al., 1999].
Congenital Sensory Neuropathy (Hereditary Sensory and Autonomic Neuropathy Type II)
HSAN II, also known as “Morvan’s disease,” is a recessive neuropathy typically diagnosed in the first decade [Barry et al., 1974; Hilz, 2002]. A cohort of HSAN II patients, mainly of French Canadian kinship, have loss-of-function mutations in the single-exon HSN2 gene [Lafreniere et al., 2004; Riviere et al., 2004; Roddier et al., 2005]. However, there is also a large number of patients who appear to have clinical features consistent with HSAN II, in whom these genetic mutations have not been identified, suggesting that there is at least one other cohort with similar phenotype but a different genotype [Axelrod and Gold von Simson, 2007].
HSAN II is characterized by profound sensory loss involving large- and small-fiber modalities. All peripheral sensations are affected but distribution of somatic involvement may vary. Pain, temperature, and position senses are involved. Unrecognized injuries and fractures of hands, feet, and limbs, as well as Charcot joints, are frequent. Self-mutilation may begin as early as 4 months of age and usually is associated with eruption of primary dentition [Zaenglein et al., 2002]. Tendon reflexes are diminished. Sensory nerve action potentials are absent [Dyck et al., 1983; Ohta et al., 1973]. Taste sensation is diminished and lingual fungiform papillae are hypotrophic. Corneal and gag reflexes may be diminished. Deep tendon reflexes are decreased and hypotonia is common, which delays attainment of developmental milestones and may contribute to scoliosis. Despite the marked sensory abnormalities, other aspects of the neurologic examination may be normal, including mental function and cerebellar and motor functions.
In the French Canadian HSAN II patients, autonomic dysfunction is described as minimal [Lafreniere et al., 2004; Riviere et al., 2004; Roddier et al., 2005]. However, for many other patients who are presumed to have HSAN II, the autonomic dysfunction is complex and evident from birth [Axelrod and Pearson, 1984; Dyck, 1993; Thomas, 1992]. For these latter patients, the neonatal course is characterized by severe feeding problems and frequent apnea [Axelrod, 2002b; Dyck, 1993; Thomas, 1992]. Gastroesophageal reflux occurs commonly. Episodic hyperhidrosis, as well as patchy areas of anhidrosis, can occur in the same patient. Overflow tearing frequently is delayed but eventually is normal. Pupils have an exaggerated response to parasympathomimetic agents. Blood pressure lability, including postural hypotension, has not been observed. Sural nerve biopsy reveals marked reduction in nerve size and depletion of large and small myelinated fibers, but only slightly decreased number of unmyelinated fibers [Dyck, 1993; Hilz, 2002]. No cutaneous sensory receptors or nerve fibers are seen, but catecholaminergic sympathetic fibers can be demonstrated by aldehyde-induced fluorescence [Hilz, 2002].
Allgrove’s syndrome
Allgrove’s syndrome, or the triple A syndrome, is a rare autosomal-recessive disorder that was first described in1978 [Allgrove et al., 1978]. The original clinical triad included adrenocorticotropic hormone (ACTH)-resistant adrenal insufficiency, achalasia, and alacrima. Over time, it was appreciated that autonomic dysfunction often was present and prominent [Chu et al., 1996; Kimber et al., 2003]. All four clinical features may not be present in every patient and time of onset for each is variable. The syndrome can present in the first decade of life with severe hypoglycemic episodes, which can cause seizures or death, or dysphagia secondary to achalasia and decreased oral secretions. It also is possible that achalasia and ACTH insensitivity are not recognized until adolescence or even adulthood [Moore et al., 1991; Mullaney et al., 1998]. The major impact of autonomic dysfunction is upon postganglionic cholinergic functions, so that sweating and oral secretions are diminished and males suffer sexual impotence [Houlden et al., 2002]. The autonomic ocular findings include alacrima, keratoconjunctivitis sicca, lacrimal gland atrophy, and pupillary abnormalities, with hypersensitivity to dilute pilocarpine and inappropriate accommodation [Brooks et al., 2004; Mullaney et al., 1998; Tsilou et al., 2001]. Patients can exhibit orthostatic hypotension with preservation of compensatory tachycardia. Many patients have progressive neurologic findings consisting of sensorimotor degeneration, optic neuropathy, and cerebellar features, as well as predominant abnormalities in the parasympathetic ANS [Chu et al., 1996; Kimber et al., 2003]. The Allgrove locus is on chromosome 12q13 [Brooks et al., 2004; Houlden et al., 2002; Weber et al., 1996]. Mutations have been found in the AAAS gene that codes for the WD-repeat containing ALADIN (alacrima-achalasia-adrenal insufficiency-neurologic disorder) protein [Sandrini et al., 2001]. Interestingly, there is significant clinical variability between patients with the same AAAS mutation, suggesting genetic heterogeneity.
Chromosomal disorders
Rett’s Syndrome
Rett’s syndrome is a neurodevelopmental disorder predominantly affecting females. Mutations in the gene encoding methyl-CpG-binding protein 2 (MECP2) on the X chromosome have been identified in 95 percent of girls with the Rett’s syndrome phenotype [Amir et al., 1999; Fang et al., 2004]. Typically, development is normal until 6–8 months of age, but then there is regression, with slowing of head circumference growth, loss of language, development of stereotypical hand movements, and gait and truncal apraxia. Some girls also develop electroencephalogram abnormalities, seizures, spasticity, and scoliosis. The autonomic features include cardiorespiratory dysregulation and abnormal blood pressure responses. Respiratory dysregulation includes hyperventilation, apnea, breath holding, and rapid shallow breathing [Cirignotta et al., 1986; Elian and Rudolf, 1991; Julu et al., 1997, 2001; Kerr, 1992; Nomura et al., 1997; Southall et al., 1988; Weese-Mayer et al., 2005]. During wakefulness, breathing dysrhythmias are associated with agitation or excitement, as well as other motor functions. During sleep, polysomnography has documented increased frequency of desaturation events and periodic breathing, and treatment is required to protect the child from the sequelae of acute and chronic intermittent hypoxemia and hypercarbia. The abnormal blood pressure responses include episodic extreme hypertension and tachycardia due to decreased cardiac vagal tone and subsequent unopposed sympathetic activity [Julu et al., 2001; Nomura et al., 1997]. Further support for autonomic dysregulation comes from observations of decreased heart rate variability, prolongation of corrected QT intervals, and sinus bradycardia [Ellaway et al., 1999; Guideri et al., 1999, 2001; Sekul et al., 1994]. Despite survival into adulthood, 26 percent of all deaths from Rett’s syndrome are sudden and unexpected, and cardiac causes for sudden death have been suggested due to autonomic dysregulation [Guideri et al., 1999; Sekul et al., 1994].
Prader–Willi Syndrome
Prader–Willi syndrome is a multisystem genetic disorder caused by deletion or suppressed expression of genes on chromosome 15q. Due to imprinting, the maternally inherited copies of these genes are virtually silent; only the paternal copies of the genes are expressed. Prader–Willi syndrome results from the loss of paternal copies of this region. Necdin, one of the several proteins genetically inactivated in individuals with Prader–Willi syndrome, has a role in promoting cellular migration and axon outgrowth, and is important for the differentiation of central and sensory neurons [Tennese et al., 2008]. In addition to hypotonia, short stature, polyphagia, obesity, small hands and feet, hypogonadism, and mild mental retardation, there are clinical features suggesting central and peripheral ANS involvement. Hypothalamic dysfunction is suggested by abnormalities in appetite regulation and pubertal development, and growth hormone serum levels are depressed. Patients with Prader–Willi syndrome appear to have diminished parasympathetic nervous system activity, based on studies demonstrating lower resting diastolic blood pressures and significantly less change in diastolic blood pressure with head-up tilt [DiMario et al., 1994].
Fragile X Syndrome
Fragile X syndrome is caused by a change in the FMR1 gene, located on the X chromosome. The expansion of a single trinucleotide gene sequence (CGG) results in methylation of that portion of the DNA and silencing of the FMR1 protein that is required for normal neural development. Normally, the FMR1 gene contains between 6 and 55 repeats of the CGG codon. In people with fragile X syndrome, the FMR1 allele has more than 200 CGG repeats of this codon [Nolin et al., 2003; Sherman, 2002]. The degree of severity correlates with the number of repeats.
Fragile X-Associated Tremor/Ataxia Syndrome
Fragile X-associated tremor/ataxia syndrome has been reported in carriers of permutation alleles (55–200 CGG repeats). Clinical features include cerebellar ataxia, neuropathy, autonomic dysfunction, severe intention tremor, and other signs of neurodegeneration, such as brain atrophy, memory loss and dementia, anxiety, and irritability. Premature ovarian failure is reported in 25 percent of women with permutations. Women with permutations have been noted to have increased prevalence of autoimmune diseases, such as hypothyroidism and fibromyalgia [Coffey et al., 2008]. Here, the clinical problems are thought due to toxic gain of function of the elevated levels of the expanded repeat FMR1 messenger RNA [Hessl et al., 2007]. Affected individuals have been shown to have diminished amygdala and autonomic responsiveness to social-emotional stimuli, as measured by functional MRI; lack of eye blink potentiation; and reduced sympathetic outflow, as measured by skin conductance [Hessl et al., 2007].
Prematurity
There is a close link between maturation degree of the ANS and gestational or postnatal age, with substantial autonomic dysfunction in preterm infants. At equivalent postconceptional ages, prematurely born infants have decreased ventilatory responses to hypoxia and have decreased heart rate variability, which may explain their increased prevalence of apnea and bradycardia, and increased risk for unexplained sudden death [Hunt, 2006; Witcombe et al., 2008]. Both the presence and severity of apnea of prematurity progressively decrease, the higher the postmenstrual age. However, premature birth may exert long-lasting effects on central and peripheral mechanisms that control cardiovascular activity. Even in the late preterm infant born at 34–37 weeks, autonomic abnormalities may be evident and, importantly, these abnormalities may persist. It has been noted that sleep state and age continue to affect heart rate and blood pressure patterns in prematurely born infants over the first 6 months of term-corrected age. In addition, when compared with age-matched term infants, preterm infants had persistently lower blood pressure, signifying long-term alterations in cardiovascular control in infants born prematurely. What is still needed are standardized and widely available means to assess autonomic maturation and determine risk stratification in the preterm infant.
Dysregulation disorders
Congenital Central Hypoventilation Syndrome
Disorders with cardiorespiratory dysregulation as their prominent feature affect breathing control. Thus, their consequences can be fatal. One of these disorders, congenital central hypoventilation syndrome, is now recognized as a specific genetic entity. First described in 1970 and often referred to as “Ondine’s curse” [Mellins et al., 1970; Weese-Mayer et al., 1999], it typically presents in the newborn period with cyanosis during sleep. More severely affected cases hypoventilate when awake and asleep, with resultant hypercarbia and hypoxemia [Weese-Mayer et al., 1999]. With compromised ventilatory and arousal responses to hypercarbia and hypoxemia, patients do not increase their minute ventilation nor perceive the physiologic compromise from breath holding or exercise. In addition to its prominent effect on cardiorespiratory regulation, children with congenital central hypoventilation syndrome often have diffuse ANS dysfunction, affecting heart rate and blood pressure responses, gastrointestinal motility, and other homeostatic functions, including sweating and body temperature regulation [Faure et al., 2002; Trang et al., 2005; Weese-Mayer et al., 2001]. Altered perceptions of pain and anxiety, and ophthalmologic abnormalities, including strabismus, altered pupillary responses, and accommodation, also have been described [Goldberg and Ludwig, 1996; Pine et al., 1994; Weese-Mayer et al., 1992, 2001]. Hirschsprung’s disease occurs in about 20 percent of cases and various tumors of neural crest origin in about 5 percent of cases [Bower and Adkins, 1980; Haddad et al., 1978; Weese-Mayer et al., 1992]. For children with congenital central hypoventilation syndrome, neurodevelopmental outcome depends on the ability to compensate for ANS dysregulation and the impact of chronic/intermittent hypoxemia [Weese-Mayer et al., 2001]. The mainstay of treatment is mechanical ventilation, with or without tracheostomy. Diaphragm pacing, mask ventilation, and negative pressure ventilation are other options.
Individuals with the congenital central hypoventilation syndrome phenotype are heterozygous for a PHOX2B gene mutation, located on chromosome 4p12 [Amiel et al., 2003; Matera et al., 2004; Sasaki et al., 2003; Weese-Mayer et al., 2003]. In 90–95 percent of congenital central hypoventilation syndrome cases, there is a polyalanine expansion mutation in exon 3, and in 5–10 percent of cases there is a unique mutation. A relationship has been noted between polyalanine repeat length and the severity of autonomic dysfunction, as indicated by number of associated autonomic symptoms [Berry-Kravis et al., 2005; Matera et al., 2004; Trochet et al., 2005; Weese-Mayer et al., 2003]. Subjects with unique mutations in PHOX2B have a higher rate of Hirschsprung’s disease, higher frequency of 24-hour/day ventilation, and more frequent neural crest tumors than the polyalanine expansion mutation group.
Reflex Anoxic Seizures
When vasovagal episodes begin in the very young (ages 6 months to 2 years) and are recurrent, they may induce reflex anoxic seizures [Appleton, 1993; Stephenson, 1978]. Like seizures, the attacks can occur in clusters. However, the episodes are not true seizures, as they are induced by vagal stimulation with the neurologic manifestation secondary to cerebral anoxia. Children who exhibit reflex anoxic seizures are believed to have vagal hypersensitivity or increased vagotonia that results in cardioinhibition. The vagal stimulus causes a period of asystole, which is brief, usually lasting less than 15 seconds. During an asystolic phase, the child is limp and pale. Frequently, following the limp period, there is stiffening of the body or opisthotonus, and there even may be urinary incontinence. Use of anticholinergic agents and pacemakers has been beneficial for some patients with extremely frequent episodes.
Autonomic Disorders Associated with Biochemical Errors
Mitochondrial encephalomyopathies
Mitochondrial encephalomyopathies are heterogeneous multisystem disorders characterized by structural or biochemical defects in the mitochondria, which impair normal oxidative phosphorylation [DiMauro, 1993]. In general, involvement of the ANS has not been stressed; however, on occasion, the autonomic abnormalities can be so severe that they can overshadow the myopathic features and may delay definitive diagnosis [Zelnik et al., 1996]. In a review of 37 confirmed cases, Nissenkorn et al. noted that the most frequent neurologic manifestations were abnormal tone, seizures, extrapyramidal movements, and autonomic dysfunction; the latter was present in 43 percent of patients [Nissenkorn et al., 2000]. Nerve deafness was found in 7 patients and myopathy was found in only 6 patients. The most frequent autonomic manifestations were gastrointestinal motility, presenting either as difficulty swallowing, pseudo-obstruction, or chronic diarrhea. Among the other autonomic features were apnea, intracardiac conduction defects, and neurogenic bladder. Autonomic features also have been reported with Leigh’s syndrome, Kearns–Sayre syndrome, and myoneurogastrointestinal disorder with encephalopathy (MNGIE) [Bardosi et al., 1987; Berenberg et al., 1977; Pincus, 1972]. In addition, autonomic or visceral features, such as cardiac conduction defects or hypothermia, and feeding problems occasionally may occur in other mitochondrial diseases, including Leber’s hereditary optic neuropathy [Newman and Wallace, 1990] and Menkes’ disease [Kodama, 1993]. Additionally, decreased lacrimation, vasomotor disturbances characterized by blotchy erythema and skin mottling, altered sweating, and postural hypotension also have been noted in individuals in whom muscle biopsy has verified abnormal respiratory chain enzymes [Zelnik et al., 1996].
Neurotransmitter deficiencies
Dopamine-β-Hydroxylase Deficiency
DβH deficiency is a mendelian recessive disorder that demonstrates the critical role of catecholamines in controlling central and peripheral autonomic functions [Robertson et al., 1986]. The enzyme DβH is required for conversion of DA to NE, so that loss of DβH leads to an inability of the neuron to synthesize NE, resulting in accumulation of DOPA and DA and its metabolites. As DβH is located almost exclusively in the chromaffin granules of the adrenal medulla and the large dense-core synaptic vesicles of noradrenergic neurons, clinical features of DβH deficiency are consistent with normal parasympathetic and sympathetic cholinergic function, but there is a lack of sympathetic noradrenergic function and adrenomedullary failure. Affected individuals experience orthostatic hypotension, exercise intolerance, fatigue, episodes of fainting, and syncope. Ptosis, hyperextensible joints, sluggish deep-tendon reflexes, mild facial weakness, and hypotonic skeletal muscles occur in some patients [Man in’t Veld et al., 1987; Robertson et al., 1986, 1991; Timmers et al., 2004]. Although DβH deficiency is present from birth, the diagnosis may not be recognized until adulthood, despite ptosis of eyelids being a frequent occurrence. In some patients, the perinatal period can be complicated as a result of episodic vomiting, dehydration, hypotension, hypothermia, and profound hypoglycemia. Children have markedly reduced exercise capacity and, by adulthood, orthostatic hypotension can be debilitating.
The biochemical hallmark of this syndrome consists of minimal or undetectable plasma NE and epinephrine levels, in conjunction with a 5- to 10-fold elevation of plasma DA level. Diagnosis can be made based on this unique catecholamine profile, as DβH molecular genetic testing is available on a research basis only. The only effective treatment is l-threo-3,4-dihydroxyphenylserine (DOPS), which is converted peripherally directly into NE. Treatment with DOPS results in a sustained relief of orthostatic symptoms [Timmers et al., 2004].
Norepinephrine Transporter Deficiency
The norepinephrine transporter (NET) is a sodium-dependent 12-transmembrane-spanning protein that is located presynaptically on noradrenergic axons and varicosities, and serves to inactivate NE following release in the central and peripheral nervous system. The gene encoding NET is located on chromosome 16q12.2 [Robertson et al., 2001; Shannon et al., 2000]. Shannon et al. found that a missense mutation in exon 9 in one family resulted in nonfunctional NET and clinical symptoms of orthostatic intolerance, with postural tachycardia. Consistent with the inability to inactivate NE, the probands had elevated plasma NE and diminished conversion to the intraneuronal metabolite, dihydroxyphenylglycol (DHPG) [Shannon et al., 2000]. Treatment of NET deficiency is directed at attenuating the postural tachycardia.
Menkes’ Disease
Menkes’ disease, also known as kinky hair disease, is an infantile-onset, X-linked recessive neurodegenerative disorder caused by deficiency or dysfunction of a copper-transporting ATPase, ATP7A [Kaler et al., 2008; Menkes et al., 1962; Vulpe et al., 1993]. It was Danks et al. who noted the similarity of kinky hair to the brittle wool of Australian sheep raised in areas with copper-deficient soil, and demonstrated abnormal levels of copper and ceruloplasmin in these patients [Danks et al., 1973]. As the result of a mutation in the ATP7A gene, copper is distributed poorly to cells in the body. Copper accumulates in some tissues, such as the small intestine and kidneys, while the brain and other tissues have unusually low levels. The decreased supply of copper can reduce the activity of numerous copper-containing enzymes that are necessary for the structure and function of bone, skin, hair, blood vessels, and the nervous system. Children with the classic form of Menkes’ disease are fair-complexioned and usually present at 2–3 months of age with loss of developmental milestones, profound truncal hypotonia, and seizures; death usually occurs by 3 years of age. People with milder variants may have minimal neurologic symptoms, with normal intelligence or only mild mental retardation and autonomic dysfunction.
The clinical and pathologic features of this condition reflect decreased activities of enzymes that require copper as a co-factor, including DβH, cytochrome c oxidase, and lysyl oxidase. Deficient cytochrome c oxidase probably accounts for most of the neurologic symptoms, similar to patients with Leigh’s disease (subacute necrotizing encephalomyelopathy) who have reduced or absent cytochrome c oxidase activity and similar neuropathologic changes. Decreased lysyl oxidase activity accounts for the connective-tissue fragility and vascular abnormalities in Menkes’ disease, since lysyl oxidase deaminates lysine and hydroxylysine in the first step of collagen cross-linkage. Lysyl oxidase localizes to the trans-Golgi network, so subcutaneous injections of copper-histidine do not improve the activity of lysyl oxidase, as the copper is not delivered into the Golgi apparatus. Tyrosinase deficiency (involved in melanin biosynthesis) most likely accounts for the hypopigmentation of the hair and skin seen in Menkes’ disease. Due to the partial deficiency of DβH, patients with Menkes’ disease have a unique catecholamine profile; the ratio of dihydroxyphenylacetic acid to dihydroxyphenylglycol is distinctively elevated and there is a high ratio of dopamine to norepinephrine [Kaler et al., 1993]. These findings can be detected in the neonatal period, allowing treatment with daily copper injections to commence within days after birth. Although survival appears to be improved, clinical outcomes are variable; it has been suggested that the response to early copper treatment depends on the ATP7A genotype and that optimal outcomes occur only in patients with mutations that permit some residual copper transport [Kaler et al., 2008].
Storage disorders
Fabry’s Disease
Fabry’s disease, or angiokeratoma corporis diffusum, is an X-linked, recessively inherited disorder that is associated with deficiency of the enzyme α-galactosidase A (ceramide trihexosidase). The enzyme deficiency results in the accumulation of ceramide trihexoside and other neutral glycosphingolipids. The condition affects hemizygous males, as well as both heterozygous and homozygous females; males tend to experience the most severe clinical symptoms, while females vary from virtually no symptoms to those as serious as in males. This variability is thought to be due to X-inactivation patterns during embryonic development of the female. There is extensive lipid deposition in various tissues that include the skin, nervous system, vascular endothelium, kidney, cardiovascular system, and eye [Brady, 1993]. There is preferential involvement of small nerve fibres and the accumulation of storage product in the central ANS and autonomic ganglia. Sural nerve biopsies have demonstrated degeneration and loss of unmyelinated fibres [Toyooka and Said, 1997], and skin biopsies show decreased intraepidermal small nerve fibers [Cable et al., 1982]. Fabry’s disease can be diagnosed by assaying the enzyme α-galactosidase A in leukocytes or skin fibroblasts.
The autonomic manifestations include hypohidrosis or anhidrosis, reduced saliva and tear formation, impaired cutaneous flare response to scratch and intradermal histamine, and disordered intestinal motility. Patients’ gastrointestinal symptoms may be as severe as their sensory complaints. Pupillary constriction to dilute pilocarpine has been documented, suggesting denervation supersensitivity, although the cardiovascular autonomic reflexes in one series were normal [Cable et al., 1982].
Until recently, treatment of Fabry’s disease targeted the symptomatic effects. Currently, however, it is being treated at the cellular level through costly enzyme replacement therapy using agalsidase alpha and agalsidase beta [Hilz, 2007]. Unfortunately, enzyme replacement therapy is not a cure, and must be infused recurrently for maximum benefit.
Secondary Autonomic Disorders
Autonomic Disorders Associated with Metabolic Diseases
Diabetes Mellitus
Diabetes mellitus is associated with autonomic neuropathy in adults. However, this association is less well recognized in the pediatric population. In one study that utilized cardiovascular reflex tests to assess autonomic involvement in children newly diagnosed with insulin-dependent diabetes mellitus, almost one-fifth of the diabetic children had cardiovascular autonomic dysfunction [Verrotti et al., 1995]. Thus, the probability of autonomic involvement in pediatric patients may be higher than previously considered. In the adult with diabetic autonomic neuropathy, morbidity and mortality are considerably higher than in those without neuropathy, and it has been implicated in the increased risk of ischemic heart disease and sudden death from cardiac arrhythmias. Other clinical features of diabetic neuropathy seen in adult patients include hypertension, as well as postural hypotension, gastrointestinal dysmotility (gastroparesis and diarrhea), and sudomotor abnormalities such as gustatory sweating. These areas need better evaluation in the pediatric population.
Autoimmune Diseases
Postinfectious disorders
Idiopathic Autonomic Neuropathy and Paraneoplastic Disorders
These two disorders are more likely to occur in the adult population but are worth noting as a model of how an autoimmune process can be associated with autonomic failure. Idiopathic autonomic neuropathy and the paraneoplastic disorders are characterized by seropositivity for antibodies that bind to or block ganglionic acetylcholine receptors [Vernino et al., 2000]. In patients with idiopathic autonomic neuropathy, severe panautonomic failure develops in a subacute manner (over a period of days or weeks). Recovery is slow and usually incomplete. Sympathetic failure is manifested as severe orthostatic hypotension and anhidrosis, and parasympathetic failure as dry mouth, sexual dysfunction, an impaired pupillary response to light and accommodation, and a fixed heart rate [Suarez et al., 1994]. Many patients have gastrointestinal dysmotility and present with anorexia, early satiety, abdominal pain and vomiting after eating, constipation, or diarrhea. Motor-nerve and sensory-nerve abnormalities are minimal or absent. Similarly, with some types of cancer, especially small-cell carcinoma of the lung, a syndrome of subacute autonomic failure can occur, which is an indirect immunologic effect of the cancer [Khurana, 1997]. Vernino et al. were able to demonstrate a significant correlation between the serum level of ganglionic-receptor-binding antibody and the severity of autonomic dysfunction in both disorders [Vernino et al., 2000]. In addition, they noted that decreases in antibody levels were accompanied by improvement in clinical and laboratory measures of autonomic function.
Chronic Fatigue Syndrome
Chronic fatigue syndrome is characterized by chronic or relapsing fatigue, lasting for at least 6 months, causing impaired overall physical and mental functioning [Fukuda et al., 1994]. As physical findings are limited, chronic fatigue syndrome often is a diagnosis of exclusion. Self-reported symptoms can include cognitive difficulties, muscle pain, joint pain, headache, sleep disturbance, poor sleep, and post-exercise malaise, as well as a variety of gastrointestinal symptoms [Carter et al., 1995; Fukuda et al., 1994; Rowe et al., 1995]. As the onset of symptoms often follows an infectious disease, an autoimmune mechanism has been postulated [Buchwald et al., 1997; Clements et al., 1995]. According to a report generated from a Centers for Disease Control and Prevention workshop [Fukuda et al., 1994], pediatric chronic fatigue syndrome patients mostly are teenage females who report a preceding inflammatory condition. As in the adult experience, adolescents with chronic fatigue syndrome frequently exhibit orthostatic intolerance with POTS [Rowe et al., 1995; Stewart et al., 1999]. Stewart et al. have demonstrated loss of heart rate variability consistent with vagal withdrawal, increased blood pressure variability consistent with enhanced modulation of sympathetic tone, and impaired baroreflex [Stewart et al., 1999].
Psychosocial/Unknown
Functional gastrointestinal disorders
Functional gastrointestinal disorders lack anatomical, inflammatory, or biochemical abnormalities to explain the symptoms [Drossman, 1999]. Although their pathophysiology generally is unknown, it is hypothesized that the interaction between specific psychosocial factors and gut innervation through the brain–gut axis, including both neuroendocrine system and ANS, may produce an abnormality of gut function. The gut dysfunction may be expressed by either abnormal motility or visceral hyperalgesia, or both. According to the Rome II criteria, there are four major groups:
Cyclic Vomiting Syndrome
Cyclic vomiting syndrome (CVS) is characterized by severe, discrete episodes of nausea, vomiting, and lethargy of unclear etiology, with baseline return to health between episodes [Stein et al., 2001; Stickler, 2005]. Although CVS occurs in all age groups, it predominantly affects children with symptoms appearing between 3 and 7 years, and frequently evolves into migraine headaches in adulthood [Stickler, 2005]. Common triggers are emotional or physical stress, including exercise, menstruation, motion, and even particular foods, such as chocolate. CVS has been described as having four phases. The symptom-free interval phase is the period between episodes. The prodrome phase precedes the onset of vomiting and is characterized by nausea, usually with abdominal pain and occasionally with headache, photophobia, or vertigo. The length of the prodrome phase varies from moments to several hours. The vomiting phase consists of nausea and vomiting, and patients may require hospitalization for rehydration. In addition, autonomic symptoms usually are exhibited, including pallor, increased salivation, diaphoresis, increased blood pressure, diarrhea, dizziness, and lethargy. The final phase is the recovery phase, which begins when the nausea and vomiting stop and there is a return of appetite and energy.
Autonomic testing in patients with CVS has demonstrated increased sympathetic modulation, as reflected in heart-rate variability and postural intolerance [Rashed et al., 1999; To et al., 1999], but the diagnosis of CVS is made by history rather than by specific tests. Criteria include five vomiting episodes, or a minimum of three episodes over a 6-month period, and stereotypical pattern and symptoms in the individual patient, with a return to baseline health between episodes and no other existing condition. The cause of CVS is unknown, but genetic factors have been suggested, as a subset of children with CVS has maternal inheritance and an associated mitochondrial DNA variation [Wang et al., 2004]. Furthermore, children with mitochondrial myopathy frequently have both severe migraines and episodic vomiting.
In the absence of known pathophysiology, treatment of CVS remains empiric. In addition to avoidance of triggers, pharmacologic therapy may be used in patients with more than a single episode of CVS per month. The preventative treatments that have been tried include cyproheptadine, amitriptyline, propranolol, phenobarbital, and erythromycin [Li and Balint, 2000]. If abortive therapy fails, supportive combinations, such as ondansetron plus lorazepam, or chlorpromazine plus diphenhydramine, may attenuate a cyclic vomiting attack. If psychological stressors trigger episodes, stress management techniques or benzodiazepine anxiolytics may help to abort attacks in the early stages. A family history positive for migraines predicts a high response rate (80 percent) to antimigraine medications [Li and Balint, 2000].
Functional Abdominal Pain
The association of functional abdominal pain and autonomic dysfunction in children still is poorly understood. It is not uncommon for children with functional gastrointestinal disorders to report various autonomic symptoms, including dizziness, headaches, flushing, sweating, Raynaud’s phenomenon, and severe fatigue [Chelimsky et al., 2001]. In addition, a subset of children with functional abdominal pain has been noted to have POTS and mild peripheral neuropathy. Patients with functional abdominal pain often respond favorably to treatments directed towards the ANS dysfunction, such as relaxation and guided imagery, and medical treatment, such as increasing dietary salt, fludrocortisone, and beta blockers [Sandroni et al., 1999].
Autism
Autism is a complex neurodevelopmental disorder that produces social, behavioral, and language impairment. In addition, it is now appreciated that autism also produces symptoms attributable to other organ systems. Some of these manifestations, including unexplained constipation or diarrhea, urinary retention, cold and clammy extremities, and sleep disturbances, suggest underlying autonomic dysfunction [Ming et al., 2005]. Autonomic tests have demonstrated blunted autonomic arousal responses to visual and auditory social stimuli [Hirstein et al., 2001; Palkovitz and Wiesenfeld, 1980]. In addition, there is low baseline cardiac vagal tone and low cardiac baroreceptor sensitivity, resulting in hyperactive heart rate and blood pressure responses [Ming et al., 2005]. Furthermore, children with autistic behavior are less flexible in their autonomic adaptation to attention-demanding tasks, and demonstrate less decrease in heart rate variability than normal controls during periods of task performance [Ming et al., 2005].
In support of central autonomic dysfunction, pathological examinations have revealed abnormalities in central structures often associated with autonomic control, such as the brainstem, the amygdala, the limbic system, the cerebellum, and the prefrontal lobes [Courchesne 1997]. In addition, abnormal levels of monaminergic and cholinergic neurotransmitters, including NE, DA, ACh, serotonin, and various neuropeptides, have been reported [Ming et al., 2005]. Additionally, secretin and oxytocin, both polypeptide neurotransmitters that cross the blood–brain barrier, each have been reported to improve some of the symptoms in different autistic subgroups [Hollander et al., 2003; Lamson and Plaza, 2001], which supports central autonomic dysfunction in autism.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Allgrove J., Clayden G.S., Grant D.B., et al. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet. 1978;1:1284.
Amiel J., Laudier B., Attie-Bitach T., et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459.
Amir R.E., Van den Veyver I.B., Wan M., et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185.
Anderson R.B., Newgreen D.F., Young H.M. Neural crest and the development of the enteric nervous system. Adv Exp Med Biol. 2006;589:181.
Anderson S.L., Coli R., Daly I.W., et al. Familial dysautonomia is caused by mutations of the IKAP gene. Am J Hum Genet. 2001;68:753.
Anderson S.L., Qiu J., Rubin B.Y. EGCG corrects aberrant splicing of IKBKAP mRNA in cells from patients with familial dysautonomia. Biochem Biophys Res Commun. 2003;310(2):627.
Anderson S.L., Qui J., Rubin B. Tocotrienols induce IKBKAP expression: a possible therapy for familial dysautonomia. Biochem Biophys Res Commun. 2003;306:303.
Appleton R.E. Reflex anoxic seizures. Br Med J. 1993;307:214.
Axelrod F.B. Familial dysautonomia: A review of the current pharmacological treatments. Expert Opin Pharmacother. 2005;6:561.
Axelrod F.B., Goldberg J.D., Ye X.Y., et al. Survival in familial dysautonomia: Impact of early intervention. J Pediatr. 2002;141:518.
Axelrod F.B. Genetic disorders as models to understand autonomic dysfunction. Clin Auton Res. 2002;12(Suppl 1):1.
Axelrod F.B. Hereditary Sensory and Autonomic Neuropathies: Familial Dysautonomia and other HSANs. Clin Auton Res. 2002;12(Suppl 1):2.
Axelrod F.B., Chelimsky G., Weese-Mayer D. Pediatric Autonomic Disorders. State of the Art. Pediatrics. 2006;118:309.
Axelrod F.B., Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet Journal of Rare Diseases. 2007;2(39):39.
Axelrod F.B., Iyer K., Fish I., et al. Progressive sensory loss in familial dysautonomia. Pediatrics. 1981;65:517.
Axelrod F.B., Pearson J. Congenital sensory neuropathies. Diagnostic distinction from familial dysautonomia. Am J Dis Child. 1984;138:947.
Bardosi A., Creutzfeld W., DiMauro S., et al. Myo-neurogastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome c-oxidase. A new mitochondrial multisystem disorder. Acta Neuropathol (Berl). 1987;74:248.
Barry J.E., Hopkins I.J., Neal B.W. Congenital sensory neuropathy. Arch Dis Child. 1974;49:128.
Benarroch E.E., Chang F.L. Central autonomic disorders. J Clin Neurophysiol. 1993;10(1):39.
Berenberg R.A., Pellock J.M., DiMauro S., et al. Lumping or splitting? “Ophthalmoplegia plus” or Kearns-Sayre syndrome? Ann Neurol. 1977;1:37.
Bernardi L., Hilz M., Stemper B., et al. Respiratory and cerebrovascular responses to hypoxia and hypercapnia in familial dysautonomia. Am J Respir Crit Care Med. 2002;167:141.
Berry-Kravis E.M., Zhou L., Rand C.M., et al. Unique PHOX2B mutations in children with congenital central hypoventilation syndrome (CCHS). Pediatr Res. 2005:2289. PAS 57
Bickel A., Axelrod F.B., Schmetz M., et al. Dermal microdialysis provides evidence for hypersensitivity to noradrenaline in patients with familial dysautonomia. J Neurol Neurosurg Psychiatry. 2002;73:299.
Bower R.J., Adkins J.C. Ondine’s curse and neurocristopathy. Clin Pediatr(Bologna). 1980;19:665.
Brady R.O. Fabry disease. In: Dyck P.J., Thomas P.K., Griffin J.W., Low P.A., Podulso J.F., editors. Peripheral neuropathy. Philadelphia: WB Saunders; 1993:1169.
Brooks B.P., Kleta R., Caruso R.C., et al. Triple-A syndrome with prominent ophthalmic features and a novel mutation in the AAAS gene: a case report. BMC Ophthalmol. 2004;4:7.
Buchwald D., Wener M.H., Pearlman T., et al. Markers of inflammation and immune activation in chronic fatigue and chronic fatigue syndrome. J Rheumatol. 1997;24:372.
Cable W.J., Kolodny E.H., Adams R.D. Fabry disease: impaired autonomic function. Neurology. 1982;32:498.
Carter B.D., Edwards J.F., Kronenberger W.G., et al. Case control study of chronic fatigue in pediatric patients. Pediatrics. 1995;95:179.
Chariot A., Creppe C., Cornez I., et al. Molecular and cellular characterization of IKAP protein and the Elongator complex. Implications for familial dysautonomia. Bull Mem Acad R Med Belg. 2007;162(5–6):315.
Chelimsky G., Boyle J.T., Tusing L., et al. Autonomic abnormalities in children with functional abdominal pain: coincidence or etiology? J Pediatr Gastroenterol Nutr. 2001;33:47.
Chu M.L., Berlin D., Axelrod F.B. Allgrove syndrome: documenting cholinergic dysfunction by autonomic tests. J Pediatr. 1996;129:156.
Cirignotta F., Lugaresi E., Montagna P. Breathing impairment in Rett syndrome. Am J Med Genet. 1986;24(Suppl 1):167.
Clements G.B., McGarry F., Nairn C., et al. Detection of enterovirus-specific RNA in serum: the relationship to chronic fatigue. J Med Virol. 1995;45:156.
Close P., Hawkes N., Cornez I., et al. Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell. 2006;22:521.
Coffey S.M., Cook K., Tartaglia N., et al. Expanded Clinical Phenotype of Women with the FMR1 Premutation. Am J Med Genet. 2008;146A:1009.
Courchesne E. Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr Opin Neurobiol. 1997;7:269.
Critchley H.D., Mathias C.J., Dolan R.J. Neural activity relating to reward anticipation in human brain. Neuron. 2001;29:537.
Danks D.M., Cartwright E., Stevens B.J., et al. Menkes’ Kinky Hair Disease: Further Definition of the Defect in Copper Transport. Science. 1973;179:1140.
DiMario F.J., Dunham B., Burleson J.A., et al. An evaluation of autonomic nervous system function in patients with Prader-Willi Syndrome. Pediatrics. 1994;93:76.
DiMauro S. Mitochondrial myopathies. In: Rosenberg R.N., Pruisner S.B., DiMauro S., Barachi R.L., Kunkel L.M., editors. The molecular and genetic basis of neurological diseases. Boston: Butterworth-Heinemann; 1993:665.
Drossman D.A. The functional gastrointestinal disorders and the Rome II process. Gut. 1999;45(Suppl 2):II1.
Dyck P.J.. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. Dyck P.J., Thomas P.K., Griffin J.W., Low P.A., Podulso J.F., editors. Peripheral Neuropathy. vol 2. Philadelphia: WB Saunders; 1993:1065.
Dyck P.J., Mellinger J.F., Reagan T.J. Not “indifference to pain” but varieties of hereditary sensory and dysautonomic neuropathy. Brain. 1983;106:373.
Edelman N.H., Cherniack N.S., Lahiri S., et al. The effects of abnormal sympathetic nervous function upon the ventilatory response to hypoxia. J Clin Invest. 1970;41:1153.
Edlund T., Jessell T.M. Progression from extrinsic to intrinsic signaling in cell fate specification: A view from the nervous system. Cell. 1999;96:211.
Eisenhofer G., Kopn I.J., Goldstein D.S. Catecholamine Metabolism: A Contemporary View with Implications for Physiology and Medicine. Pharmacol Rev. 2004;56:331.
Elian M., Rudolf N.M. EEG and respiration in Rett syndrome. Acta Neurol Scand. 1991;83(2):123.
Ellaway C.J., Sholler G., Leonard H., et al. Prolonged QT interval in Rett syndrome. Arch Dis Child. 1999;80:470.
Etienne M., Weimer L.H. Immune-mediated autonomic neuropathies. Curr Neurol Neurosci Rep. 2006;6:57.
Fang P., Jin W., Glaze D.G., et al. MECP2 gene deletions account for ~10 percent of Rett syndrome cases. Am Soc Hum Gen. 2004;476:2652.
Faure C., Viarme F., Cargill G., et al. Abnormal esophageal motility in children with congenital central hypoventilation syndrome. Gastroenterology. 2002;122:1258.
Filler J., Smith A.A., Stone S., et al. Respiratory control in familial dysautonomia. J Pediatr. 1965;81:509.
Freeman V. Treatment of Orthostatic Hypotension. Semin Neurol, The Autonomic Nervous System. 2003;23(4):435.
Fukuda K., Straus S.E., Hickie I., et al. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med. 1994;121:953.
Gazit Y., Nahir A.M., Grahame R., et al. Dysautonomia in the joint hypermobility syndrome. Am J Med. 2003;115:33.
Glover W., Greenfield A., Shanks R. The contribution made by adrenaline to the vasodilation in the human forearm during emotional stress. J Physiol. 1962;161:42P.
Goebel H.H., Veit S., Dyck P.J. Confirmation of virtual unmyelinated fiber absence in hereditary sensory neuropathy type IV. J Neuropathol Exp Neurol. 1980;39(6):670.
Goldberg D.S., Ludwig I.H. Congenital central hypoventilation syndrome: ocular findings in 37 children. J Pediatr Ophthalmol Strabismus. 1996;33:175.
Goridis C., Brunet J.F. Transcriptional control of neurotransmitter phenotype. Curr Opin Neurobiol. 1999;9:47.
Grover-Johnson N., Pearson J. Deficient vascular innervation in familial dysautonomia, an explanation for vasomotor instability. Neuropathol Appl Neurobiol. 1976;2:217.
Guideri F., Acampa M., DiPerri T., et al. Progressive cardiac dysautonomia observed in patients affected by classic Rett Syndrome and not in the preserved speech variant. J Child Neurol. 2001;16(5):370.
Guideri F., Acampa M., Hayek G., et al. Reduced heart rate variability in patients affected with Rett syndrome: A possible explanation for sudden death. Neuropediatrics. 1999;30(3):146.
Haddad G.G., Mazza N.M., Defendini R., et al. Congenital failure of automatic control of ventilation, gastrointestinal motility and heart rate. Medicine. 1978;57:517.
Hamill R., LaGamma E.F. Autonomic nervous system development. In: Mathias C.J., Bannister R., editors. Autonomic Failure. A textbook of clinical disorders of the autonomic nervous system. ed 4. Oxford: Oxford University Press; 2002:16.
Hawkes N.A., Otero E., Winkler E.S., et al. Purification and characterization of the human elongator complex. J Biol Chem. 2002;277(4):3047.
Hessl D., Rivera S., Koldewyn K., et al. Amygdala dysfunction in men with the fragile X permutation. Brain. 2007;130:404.
Hilz M.J.. Assessment and evaluation of hereditary sensory and autonomic neuropathies with autonomic and neurophysiological examinations. Clin Auton Res. 2002(Suppl 1):33.
Hilz M.J. Evaluation of peripheral and autonomic nerve function in Fabry disease. Acta Pediatrica. 2007;91:38.
Hilz M.J., Axelrod F.B. Quantitative sensory testing of thermal and vibratory perception in familial dysautonomia. Clin Auton Res. 2000;10:177.
Hilz M.J., Devinsky O., Doyle W., et al. Decrease of sympathetic cardiovascular modulation after temporal lobe epilepsy surgery. Brain. 2002;25:985.
Hilz M.J., Stemper B., Axelrod F.B. Sympathetic skin response differentiates hereditary sensory autonomic neuropathies III and IV. Neurology. 1999;52(8):1652.
Hirstein W., Iversen P., Ramachandran V.S. Autonomic responses of autistic children to people and objects. Proc Biol Sci. 2001;268:1883.
Hollander E., Novotny S., Hanratty M., et al. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology. 2003;28:193.
Houlden H., Smith S., De Carvalho M., et al. Clinical and genetic characterization of families with triple A (Allgrove) syndrome. Brain. 2002;125:2681.
Hunt C.E. Ontogeny of autonomic regulation in late preterm infants born at 34–37 weeks postmenstrual age. Semin Perinatol. 2006;30:73.
Indo Y., Tsuruta M., Hayashida Y., et al. Mutations in the NTRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13:485.
Janig W. Autonomic nervous system. In: Schmidt R.F., Thews G., editors. Human Physiology. ed 2. Berlin: Springer; 1989:333.
Julu P.O.O., Kerr A.M., Apartopoulos F., et al. Characterization of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child. 2001;85(1):29.
Julu P.O.O., Kerr A.M., Hansen S., et al. Functional evidence of brain stem immaturity in Rett syndrome. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):47.
Kaler S.G., Goldstein D.S., Holmes C., et al. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann Neurol. 1993;33:171.
Kaler S.G., Holmes C.S., Goldstein D.S., et al. Neonatal Diagnosis and Treatment of Menkes Disease. N Engl J Med. 2008;358:605.
Kaufmann H., Berman J., Oribe E., et al. Possible increase in the synthesis of endothelial derived relaxing factor (EDRF) during vasovagal syncope. Clin Auton Res. 1993;3:69.
Kaufmann H., Hainsworth R. Why do we faint? Muscle Nerve. 2001;24:981.
Kerr A.M. A review of the respiratory disorder in the Rett syndrome. Brain Dev. 1992;14:S43.
Khurana R.K. Paraneoplastic autonomic dysfunction. In: Low P.A., editor. Clinical autonomic disorders: evaluation and management. ed 2. Philadelphia: Lippincott-Raven; 1997:545.
Kimber J., McLean B.N., Prevett M., et al. Allgrove or 4 “A” syndrome: an autosomal recessive syndrome causing multisystem neurological disease. J Neurol Neurosurg Psychiatry. 2003;74:654.
Kodama H. Recent developments in Menkes disease. J Invest Metab Dis. 1993;16:791.
Lafreniere R.G., MacDonald M.L., Dube M.P., et al. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am J Hum Genet. 2004;74(5):1064.
Lamson D.W., Plaza S.M. Transdermal secretin for autism – a case report. Altern Med Rev. 2001;6:311.
Langer J., Goebel H.H., Veit S. Eccrine sweat glands are not innervated in hereditary sensory neuropathy type IV. An electron-microscopic study. Acta Neuropathol (Berl). 1981;54(3):199.
Langley J.N. The autonomic nervous system, part I. Cambridge: Heffer; 1921.
Lee G., Papapetrou E.P., Kim H., et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402.
Levi-Montalcini R. The morphological effects of immunosympathectomy. In: Steiner G., Schonbam E., editors. Immunosympathectomy. Amsterdam: Elsievier; 1972:55.
Li B.U., Balint J.P. Cyclic vomiting syndrome: evolution in our understanding of a brain-gut disorder. Adv Pediatr. 2000;47:117.
Li B.U., Issenman R.M., Sarna S.K. Consensus statement – 2nd International Scientific Symposium on CVS. The Faculty of the 2nd International Scientific Symposium on Cyclic Vomiting Syndrome. Dig Dis Sci. 1999;44:9S-11S.
Loewy A.S. Central autonomic pathways. In: Loewy A.S., Spyer K.M., editors. Central Regulation of Autonomic Functions. New York, NY: Oxford University Press; 1990:88.
Low P.A., Schondorf R., Rummans T.A. Why do patients have orthostatic symptoms in POTS? Clin Auton Res. 2001;11:223.
Low P.A., Vernino S., Suarez G. Autonomic dysfunction in peripheral nerve disease. Muscle Nerve. 2003;27:646.
Maayan Ch, Carley D.W., Axelrod F.B., et al. Respiratory system stability and abnormal carbon dioxide homeostasis. J Appl Physiol. 1992;72(3):1186.
Man in’t Veld A.J., Bloosma F., Moleman P., et al. Congenital dopamine-β-hydroxylase deficiency: a novel orthostatic syndrome. Lancet. 1987;1:183.
Matera I., Bachetti T., Puppo F., et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset central hypoventilation syndrome. J Med Genet. 2004;41:373.
Mathias C.J. To stand on one’s own legs. Clin Med. 2002;2:237.
Mellins R.B., Balfour H.H.Jr, Turino G.M., et al. Failure of automatic control of ventilation (Ondine’s curse). Report of an infant born with this syndrome and review of the literature. Medicine. 1970;49:487.
Menkes J.H., Alter M., Steigleder G.K., et al. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics. 1962;29:764.
Minde J., Toolanen G., Andersson T., et al. Familial insensitivity to pain (HSAN V) and a mutation in the NGFB gene. A neurophysiological and pathological study. Muscle Nerve. 2004;30:752.
Ming X., Julu P., Brimacombe M., et al. Reduced cardiac parasympathetic activity in children with autism. Brain and Development. 2005;27:509.
Moore P.S., Couch R.M., Perry Y.S., et al. Allgrove syndrome: an autosomal recessive syndrome of ACTH insensitivity, achalasia and alacrima. Clin Endocrinol (Oxf). 1991;34:107.
Mullaney P.B., Weatherhead R., Millar L., et al. Keratoconjunctivitis sicca associated with achalasia of the cardia, adrenocortical insufficiency, and lacrimal gland degeneration: Keratoconjunctivitis sicca secondary to lacrimal gland degeneration may parallel degenerative changes in esophageal and adrenocortical function. Ophthalmology. 1998;105:643.
Newman N.J., Wallace D.C. Mitochondria and Leber’s hereditary optic neuropathy. Am J Ophthalmol. 1990;109:726.
Nissenkorn A., Zeharia A., Lev D., et al. Neurologic Presentations of Mitochondrial Disorders. J Child Neurol. 2000;15:44.
Nolano M., Crisci C., Santoro L., et al. Absent innervation of skin and sweat glands in congenital insensitivity to pain with anhidrosis. Clin Neurophysiol. 2000;111:1596.
Nolin S.L., Brown W.T., Glicksman A., et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72(2):454.
Nomura Y., Kimura K., Arai H., et al. Involvement of the autonomic nervous system in the pathophysiology of Rett syndrome. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):42.
Ohta M., Ellefson R.D., Lambert E.H., et al. Hereditary sensory neuropathy, type II: clinical, electrophysiologic, histologic and biochemical studies of a Quebec kinship. Arch Neurol. 1973;29:23.
Palkovitz R.J., Wiesenfeld A.R. Differential autonomic responses of autistic and normal children. J Autism Dev Disord. 1980;10:347.
Pearson J., Axelrod F., Dancis J. Current concepts of dysautonomia: neuropathological defects. Ann N Y Acad Sci. 1974;228:288.
Pick J. The Autonomic Nervous System. Philadelphia, PA: Lippincott; 1970.
Pincus J.H. Subacute necrotizing encephalomyopathy (Leigh’s disease): a consideration of clinical features and etiology. Dev Med Neurol. 1972;14:87.
Pine D.S., Weese-Mayer D.E., Silvestri J.M., et al. Anxiety and congenital central hypoventilation syndrome. Am J Psychiatry. 1994;151:864.
Pinsky L., Di George A.M. Congenital famillial sensory neuropathy with anhidrosis. J Pediatr. 1966;68(1):1.
Rashed H., Abell T.L., Familoni B.O., et al. Autonomic function in cyclic vomiting syndrome and classic migraine. Dig Dis Sci. 1999;44:74S.
Riviere J.B., Verlaan D.J., Shekarabi M., et al. A mutation in the HSN2 gene causes sensory neuropathy type II in a Lebanese family. Ann Neurol. 2004;56:572.
Robertson D., Flattem N., Tellioglu T., et al. Familial orthostatic tachycardia due to norepinephrine transporter deficiency. Ann N Y Acad Sci. 2001;940:527.
Robertson D., Goldberg M.R., Hollister A.S., et al. Isolated failure of autonomic noradrenergic neurotransmission: evidence of impaired [beta]-hydroxylation of dopamine. N Engl J Med. 1986;314:1494.
Robertson D., Perry S.E., Hollister A.S., et al. Dopamine-[beta]-hydroxylase deficiency: a genetic disorder of cardiovascular regulation. Hypertension. 1991;18:1.
Roddier K., Thomas T., Marleau G., et al. Two mutations in the HSN2 gene explain the high prevalence of HSAN2 in French Canadians. Neurology. 2005;64:1762.
Rowe P.C., Bou-Holaigah I., Kan J.S., et al. Is neurally mediated hypotension an unrecognised cause of chronic fatigue? Lancet. 1995;345:623.
Sandrini F., Farmakidis C., Kirschner L.S., et al. Spectrum of mutations of the AAAS gene in Allgrove syndrome: Lack of mutations in six kindreds with isolated resistance to corticotropin. J Clin Endocrin Metab. 2001;86:5433.
Sandroni P., Opfer-Gehrking T.L., McPhee B.R., et al. Postural tachycardia syndrome: clinical features and follow-up study. Mayo Clin Proc. 1999;74:1106.
Sasaki A., Kanai M., Kijima K., et al. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet. 2003;114:22.
Sekul E.A., Moak J.P., Schultz R.J., et al. Electrocardiographic findings in Rett syndrome: An explanation for sudden death? J Pediatr. 1994;125:80.
Shahar E., Shinawi M. Neurocristopathies presenting with neurologic abnormalities associated with Hirschsprung’s disease. Pediatr Neurol. 2003;28:385.
Shannon J.R., Flattem N.L., Jordan J., et al. Orthostatic intolerance and tachycardia associated with norepinephrine-transporter deficiency. N Engl J Med. 2000;342:541.
Sherman S. Epidemiology. In Hagerman R.J., Hagerman P.J., editors: Fragile X Syndrome, Diagnosis Treatment and Research, ed 3, Baltimore: Johns Hopkins University Press, 2002.
Slaugenhaupt S.A., Blumenfeld A., Gill S.P., et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet. 2001;68:598.
Slaugenhaupt S.A., Mull J., Leyne M., et al. Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum Mol Genet. 2004;13(4):429.
Smith A.A., Hirsch J.I., Dancis J. Responses to infused methacholine in familial dysautonomia. Pediatrics. 1965;36:225.
Sommer L., Shah N., Rao M., et al. The cellular function of MASH1 in autonomic neurogenesis. Neuron. 1995;15:1245.
Southall D.P., Kerr A.M., Tirosh E., et al. Hyperventilation in the awake state: potentially treatable component of Rett syndrome. Arch Dis Child. 1988;63:1039.
Stein M.T., Katz R.M., Jellinek M.S., et al. Cyclic vomiting. J Dev Behav Pediatr. 2001;22:S139.
Stephenson J.B.P. Reflex anoxic seizures (“white breath-holding”): non-epileptic vagal attacks. Arch Dis Child. 1978;53:193.
Stewart J.M., Gewitz M.H., Weldon A. Orthostatic intolerance in adolescent chronic fatigue syndrome. Pediatrics. 1999;103:116.
Stickler G.B. Relationship between cyclic vomiting syndrome and migraine. Clin Pediatr (Bologna). 2005;44:505.
Streeten D.H., Anderson G.H.Jr, Richardson R., et al. Abnormal orthostatic changes in blood pressure and heart rate in subjects with intact sympathetic nervous functions: evidence for excessive venous pooling. J Lab Clin Med. 1998;111:326.
Suarez G.A., Fealey R.D., Camilleri M., et al. Idiopathic autonomic neuropathy: clinical, neurophysiologic, and follow-up studies on 27 patients. Neurology. 1994;44:1675.
Tennese A.A., Gee C.B., Wevrick R. Loss of the Prader-Wili syndrome protein necdin causes defective migration, axonal outgrowth and survival of embryonic sympathetic neurons. Dev Dyn. 2008;237:1935.
Thoenen H., Barde Y.A. Physiology of nerve growth factor. Physiol Rev. 1980;60:1284.
Thomas P.K. Autonomic involvement in inherited neuropathies. Clin Auton Res. 1992;2:51.
Timmers H.J., Deinum J., Wevers R.A., et al. Congenital dopamine-beta-hydroxylase deficiency in humans. Ann N Y Acad Sci. 2004;1018:520.
Tiveron M., Hirsch M., Brunet J. The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. J Neurosci. 1996;16:7649.
To J., Issenman R.M., Kamath M.V. Evaluation of neurocardiac signals in pediatric patients with cyclic vomiting syndrome through power spectral analysis of heart rate variability. J Pediatr. 1999;135:363.
Toyooka K., Said G. Nerve biopsy findings in hemizygous and heterozygous patients with Fabry’s disease. J Neurol. 1997;244:464.
Trang H., Girard A., Laude D., et al. Short-term blood pressure and heart rate variability in congenital central hypoventilation syndrome. Clin Sci. 2005;108:225.
Trochet D., O’Brien L.M., Gozal D., et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet. 2005;76:421.
Tsilou E., Stratakis C.A., Rubin B.I., et al. Ophthalmic manifestations of Allgrove syndrome: report of a case. Clin Dysmorphol. 2001;10:231.
van Lieshout J.J., ten Harkel A.D., Wieling W. Physical manoeuvres for combating orthostatic dizziness in autonomic failure. Lancet. 1992;339:897.
Vassella F., Emrich H.M., Kraus-Ruppert R., et al. Congenital sensory neuropathy with anhidrosis. Arch Dis Child. 1968;43:124.
Vernino S., Low P.A., Fealey R.D., et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847.
Verrotti A., Chiarelli F., Blasetti A., et al. Autonomic neuropathy in diabetic children. J Pediatrics and Child Health. 1995;31:545.
Vincent S., Robertson D. The broader view: catecholamine abnormalities. Clin Auton Res. 2002;1:44.
Vulpe C., Levinson B., Whitney S., et al. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7.
Wang Q., Ito M., Adams K., et al. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A. 2004;131:50.
Weber A., Wienker T.F., Jung M., et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster. Hum Mol Genet. 1996;5:2061.
Weese-Mayer D.E., Berry-Kravis E.M., Zhou L., et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2B. Am J Med Genet. 2003;123A:267.
Weese-Mayer D.E., Boothby C.M., Lieske S.P., et al. Autonomic nervous system dysregulation in breathing and heart rate in girls with Rett Syndrome. Pediatr Res PAS. 2005;57:1210.
Weese-Mayer D.E., Shannon D.C., Keens T.G., et al. American Thoracic Society Statement on the diagnosis and management of idiopathic congenital central hypoventilation syndrome. Am J Respir Crit Care Med. 1999;160:368.
Weese-Mayer D.E., Silvestri J.M., Huffman A.D., et al. Case/control family study of autonomic nervous system dysfunction in idiopathic congenital central hypoventilation syndrome. Am J Med Genet. 2001;100:237.
Weese-Mayer D.E., Silvestri J.M., Menzies L.J., et al. Congenital central hypoventilation syndrome: diagnosis, management, and long-term outcome in thirty-two children. J Pediatr. 1992;120:381.
Welton W., Clayson D., Axelrod F.B., et al. Intellectual development and familial dysautonomia. Pediatrics. 1979;63:708.
Witcombe N.B., Yiallourou S.R., Walker A.M., et al. Blood pressure and heart rate patterns during sleep are altered in preterm-born infants: implications for sudden infant death syndrome. Pediatrics. 2008;122:e1242.
Zaenglein A.L., Chang M.W., Meehan S.A., et al. Extensive Riga-Fede disease of the lip and tongue. J Am Acad Dermatol. 2002;47:445.
Zelnik N., Axelrod F.B., Leshinsky E., et al. Mitochondrial encephalopathies presenting with features of autonomic and visceral dysfunction. Pediatr Neurol. 1996;14:251.
