Chapter 22 Disorders of Neural Tube Development
Introduction
Neural tube defects (NTDs) are among the most frequently encountered congenital anomalies involving the central nervous system (CNS). Indeed, NTDs are second only to congenital heart defects as the most common serious birth defect, affecting between 0.5 and 10 per 1000 live births, depending on the population studied [Gelineau-van Waes and Finnell, 2001]. Rostral NTDs, such as anencephaly, that lead to an absent cranial vault and exposed brain are uniformly fatal. In contrast, most children born with posterior NTDs confined to the spinal column will survive with varying degrees of functional disability and shortened life span. NTDs may be classified in a number of ways but the most useful strategy is to assign the observed defect according to the suspected developmental abnormality. This allows a better appreciation of the severity of the CNS anomaly, which often correlates with the clinical picture and expected outcome. In this chapter, the term NTD will be used to encompass all defects that involve neural tube formation and differentiation. NTDs are also referred to as spinal or cranial dysraphism, depending on the level of the defect. Spina bifida formally refers to all types of spinal dysraphism, though it is often used synonymously with myelomeningocele (MMC), which is a severe and common form of spinal dysraphism in which meninges, along with spinal cord or axons, protrude outside of the vertebral column.
NTDs have been the subject of intensive research for decades and are considered to result from complex interactions of genes and environmental conditions. Indeed, over 200 gene defects in the mouse have now been associated with failed neural tube closure, and a number of these genes are likely to be involved in human NTDs as well [Harris and Juriloff, 2007; Gray and Ross, 2009; Harris, 2009]. This class of neurodevelopmental defect is particularly important to understand because a significant proportion of NTDs may be preventable with measures like prenatal maternal folic acid (FA) supplementation [MRC, 1991], by avoiding prenatal exposure to known teratogens like retinoids, and because medications often used in the treatment of epilepsy and neuropsychiatric disorders can increase the risk to pregnancies conceived while taking the drug [Ross, 2010]. This chapter will discuss the pathogenesis, risk factors, complications, and management of NTDs in children.
Anatomy and Embryology
Formation of the Neural Tube
Among the earliest morphological specializations in the embryo is the formation of the neural placode, followed by neural plate and then neural tube [Sadler, 2005]. At the end of the second gestational week, the human embryo is a bilaminar disc of epiblast cells overlying hypoblast cells [Sadler, 2004]. At the beginning of the third week, the disc develops a midline groove, the primitive streak, in the caudal third (Figure 22-1A), which marks the initiation of gastrulation and the formation of three germ layers – ectoderm (giving rise to skin and the nervous system), mesoderm (providing inductive signals to ectoderm and contributing to morphogenesis), and endoderm (giving rise to viscera). The primitive node at the cranial end of the streak contains cells that act to organize the embryonic axes (see Figure 22-1). At the same stage, thickening of the rostral ectoderm by the apical–basal elongation of cells into a pseudostratified columnar shape produces the neural placode that marks the initiation of neurulation (see Figure 22-1B) [Schoenwolf and Powers, 1987; Schoenwolf, 1988; Colas and Schoenwolf, 2001]. Cells migrating through the primitive streak and node displace the hypoblast cells to form endoderm and subsequently middle layer mesoderm. Cells that migrate through the node in the midline form the prechordal plate and notochord (Figure 22-2), which are important for induction of the ventral CNS structures, starting with the neural plate. The remainder of ectoderm surrounding the neural plate becomes epidermis.
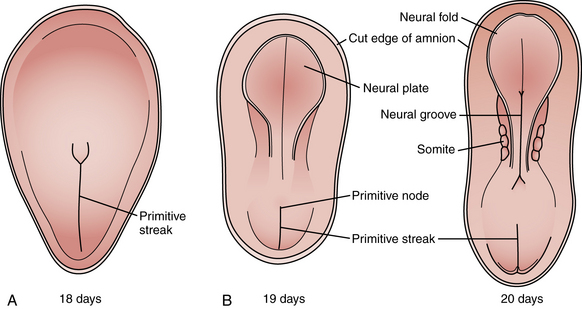
Fig. 22-1 Early stages of gastrulation and neurulation in human embryogenesis: views of the dorsal surface.
(Adapted from Sadler TW. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 2005;135C:2–8.)
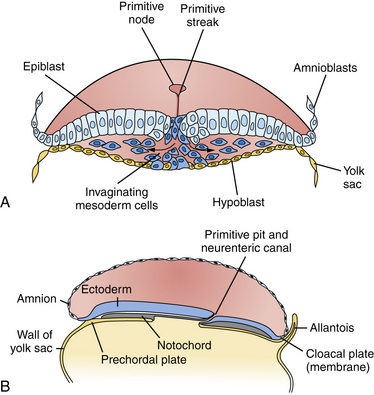
Fig. 22-2 Cross-sections through the primitive streak.
(Adapted from Sadler TW. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 2005;135C:2–8.)
Neural plate formation is actually a default state of the ectoderm, and formation of epidermis involves the inhibition of bone morphogenetic protein (BMP) and the wingless (Wnt) signaling pathway [Harland, 2000]. As the plate begins to emerge, morphogenesis, or shape changes, involving groups of cells in the neuroepithelium and surround, is essential to formation of brain and spinal cord (Figure 22-3). By 20 days of gestation, the neural plate appears indented in the midline, forming a groove or medial hinge region flanked by ridges – the neural folds. These folds elevate from the plane of the neural plate through the combined influences of proliferation of neural cells and underlying mesenchymal cells [Greene and Copp, 2009]. Bending inward of the neural folds at the dorsolateral hinge points occurs through morphological shape changes of the neural cells, which become radially elongated, while their apical (luminal) poles constrict, through a combination of cell-cycle regulation that moves nuclei to the basal end of cells in the hinge region, and actin-myosin contraction at the apical poles of cells in the hinge region, to bring the tips of the neural folds into apposition [Greene and Copp, 2009]. In the head region, proliferation and movement of epidermis and mesenchyme cells also help to push the neural folds into apposition, while at spinal levels, neighboring mesenchyme may be less critical to neural tube closure [Greene and Copp, 2009]. In addition to cell elongation and proliferation, the shape changes in the neural plate are affected by cell motility in the form of convergent extension, in which laterally placed cells move to the midline and migrate rostrocaudally in a process that is mediated by noncanonical Wnt signaling (Figure 22-4) [Wallingford and Harland, 2001; Wallingford et al., 2002]. Thus, once elevation and bending of the neural folds occur, the lateral margins or tips of the folds join and then fuse in the midline to become the neural tube.
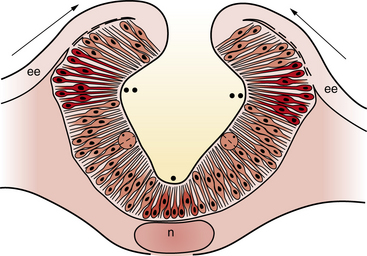
Fig. 22-3 A coronal cross-section through the cranial neural folds as they come together in the midline.
(Adapted from Sadler TW. Embryology of neural tube development. Am J Med Genet C Semin Med Genet 2005;135C:2–8.)
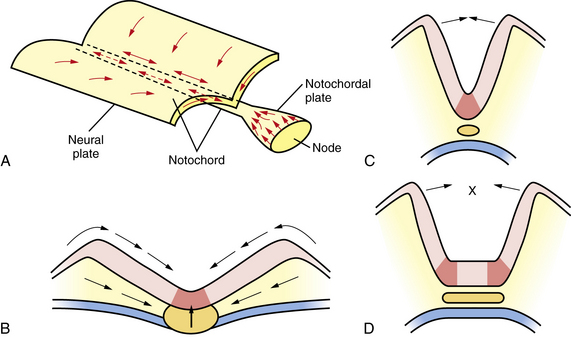
In order to achieve this rising and bending inward of the neural folds, the cells of the neural plate must proliferate in an ordered manner, called interkinetic nuclear migration of progenitors, forming a pseudostratified epithelium in which S-phase occurs at the basal (outer) surface of the neural folds, mitosis (M-phase) occurs at the apical (central or luminal) surface, and G1 and G2 phase nuclei are positioned at intermediate locations. In addition, in the process of convergent extension, cells move medially and through the medial hinge region to migrate rostrocaudally and elongate the neuraxis, narrowing the ventral floorplate (see Figure 22-3). If the floorplate is too wide or the neural folds fail to elevate and bend, the folds will not appose and NTDs will ensue (see Figure 22-4). The broadening of the ventral floorplate and shortening of the rostrocaudal axis reflect impaired convergent extension; they are demonstrated in the looptail mouse mutant, which bears a mutation in the Van Gogh-like 2 (Vangl2) gene that functions in the planar cell polarity (PCP) pathway [Ybot-Gonzalez et al., 2007]. When apposition is successful, midline fusion of the neural folds, a process known as neurulation, occurs first at primary closure points, and extends by adding multiple closure points like the teeth of a zipper rostrally and caudally from each node to complete neural tube closure [Pyrgaki et al., 2010]. In the mouse, there are typically three closure points:
Molecular Patterning of the Neural Tube
Work on frog, mouse, and zebrafish species has identified a host of factors that are necessary for proper neurulation and neural tube closure. These data have direct implications for human disease because vertebrates display a conserved body plan. A key observation was that a small fragment of non-neural tissue (mesoderm), when transplanted, was capable of duplicating the neural tube, indicating that secreted factors from mesoderm are sufficient to induce neurulation. Thus, the factors necessary for neurulation are intrinsic to and secreted from this mesodermal region. These factors include chordin, noggin, sonic hedgehog (SHH), and several others, which are both necessary and sufficient for proper neurulation and control of the amount and fate of the neuroectoderm (reviewed in De Robertis and Kuroda [2004]). In mammals, two distinct groups of non-neural cells appear to provide these early patterning signals: axial mesodermal cells of the notochord, which underlie the midline of the neural plate, and the cells of the epidermal ectoderm, which flank its lateral edges. The notochord is the source of ventralizing inductive factor, and the epidermal ectoderm is the source of dorsalizing factors. Opposing actions of these two signals appear to be critical in establishing the identity and pattern of cell types generated along the dorsal–ventral axis of the neural tube.
The nervous system is patterned along the anteroposterior, dorsal–ventral, and right–left axes in response to the expression of intrinsic patterning genes and critical embryonic signaling pathways involving secreted factors and cell–cell interactions [Altmann and Brivanlou, 2001]. The homeotic, Hox, transcription factor genes provide a positional “address” or identity of spinal cord and hindbrain neurons [Carpenter, 2002]. Fate determination along the dorsal–ventral axis also requires the action of three opposing signaling pathways:
In this model, SHH released by the notochord diffuses toward the ventral neural tube and induces the differentiation of the floor plate. The floor plate then becomes capable of producing additional SHH, which diffuses and establishes a gradient along the dorsal–ventral axis of the neural tube. Differentiation of cells in the dorsal half of the neural tube depends on signals such as BMPs provided by the lateral epidermal ectoderm. BMP4 and BMP7, released by the epidermal ectoderm, diffuse toward the dorsal neural tube and induce the differentiation of the roof plate. The roof plate then becomes capable of producing additional BMPs, which diffuse and establish a gradient along the ventral–dorsal axis of the neural tube. Regions of the spinal cord that are exposed to the highest concentrations of SHH and lowest concentrations of BMPs give rise to cells of ventral fate, including motor neurons, whereas cells exposed to lowest SHH and highest BMP concentrations give rise to cells of dorsal fate, such as commissural projection neurons [Cowan et al., 1997]. Fibroblast growth factor (FGF), NOTCH, and retinoic acid (RA) signaling is also known to act in dorsal–ventral patterning and neural fate determination, though their roles are less well understood. Rostrocaudally, the neural tube is regionalized into four major divisions: forebrain, midbrain, hindbrain, and spinal cord. Actions of FGF, Wnt and RA pathways confer a caudal identity [Diez del Corral and Storey, 2004; Aboitiz and Montiel, 2007] and also impact telencephalic patterning [Takahashi and Liu, 2006]. Homeotic Hox and Engrailed family transcription factors are particularly important for establishing segmental identities and morphogenetic features of hindbrain [Barrow et al., 2000; Cheng et al., 2010] (see also Chapter 24).
Epidemiology and Pathogenesis
Incidence
Among live births, females are more affected by NTDs than males by 2:1 [Elwood and Little, 1992; Little and Elwood, 1992; Shaw et al., 1994]. Additionally, the prevalence of NTDs varies across time, by region, and by ethnicity, and approximates 0.5 cases per 1000 in the United States but 10 per 1000 in parts of China and India [Northrup and Volcik, 2000; Gelineau-van Waes and Finnell, 2001]. These numbers do not reflect the decrease in incidence that has followed FA supplementation – with current estimates of 0.37 cases per 1000 reported in the US [Mills and Signore, 2004] – or the availability of prenatal diagnosis and the elective termination of some pregnancies with affected fetuses. The geographic and population variabilities in NTD rates are in keeping with the complexities of the etiology of neural tube closure failure, which involve both genetic and environmental factors [Janerich and Piper, 1978; Ross, 2010].
Over 30 years of clinical and basic science investigation are behind the insight that NTDs in humans are not caused by a single-gene defect but instead arise from the interplay of multiple genes, as well as gene–environment interactions [Holmes et al., 1976]. The twin concordance rate among same-sex twins (presumed monozygotic) is significantly increased (to 6.8 percent), supporting a genetic contribution [Janerich and Piper, 1978]. Furthermore, compared to an incidence in the general population of 1 per 1000, the risk of NTD recurrence in a family with one affected child increases to 1.8 per 100, but does not approach the 1 in 4 recurrence risk of an autosomal-recessive mutation with complete penetrance. Studies have documented the higher occurrence rate of NTDs in females compared to males, and the significantly higher incidence of consanguinity among parents of infants with NTDs [Elwood and Little, 1992; Shaw et al., 1994]. These and many other studies indicate prominent genetic, heritable contributions to failed neural tube closure that may well require the concerted action of multiple gene polymorphisms to manifest the disorder.
Complex Genetic Contributions
The complexity of the genetic underpinnings of neurulation is reflected in the NTD-prone mouse lines, for which over 200 mutations have been associated with failure of neural tube closure [Harris and Juriloff, 2007; Harris, 2009]. Despite this wealth of information, there is no single-gene polymorphism in the human homologs of these mouse genes that confers a robust, broadly reproducible enhanced risk of developing an NTD [Greene et al., 2009; Ross, 2010]. This has led to the supposition that either neurulation in the mouse is significantly different from that in humans, despite the similarities in morphogenesis and cell behaviors, or the genesis of NTDs requires the compounding of multiple gene polymorphisms. Supporting this latter premise, there are a number of examples in which modifier loci have been detected that increase the penetrance of NTD in mutant mouse models. A classic example is found the curly tail (ct) mouse, in which spina bifida is associated with a primary risk gene and at least three distinct modifier loci [Neumann et al., 1994]. The hypomorphic, partial loss-of-function mutation most responsible for NTDs in ct/ct mouse embryos occurs in a transcription factor called Grainy-head-like 3 (Grhl3). Another mutant with demonstrated multiple gene interactions is the SELHBc mouse, for which four loci have been mapped that conspire to result in exencephaly [Harris and Juriloff, 2007]. There are also examples of digenic mutations, or mutations in two genes, that must occur together to produce NTD in the mouse [Harris and Juriloff, 2007]. These digenic mutations in compound heterozygous mice affect processes from cell polarity (Cobl/Vangl2, Dvl1/Dvl2, Fzd3/Fzd6, Vangl2/Celsr1, Vangl2/Scrb, Vangl2/Ptk7), to actin cytoskeletal regulation (Enah/Vasp, Enah/Pfn1), to cell–cell adhesion (Gja1/Gja5, Itga1/Itga6), intracellular protein transport (Snx1/Snx2), intracellular signaling (Jnk1/Jnk2, Prkaca/Prkacb), or transcription regulation (Msx1/Msx2, Rara/Rarg). Genetic interactions that increase NTD risk certainly do occur in mice and can be expected to occur in humans as well [Ross, 2010].
The several hundred genes that have been associated with NTDs in mouse models are beginning to provide insights into molecular networks that are critically important for neurulation and may become clinically useful [Ross, in press]. For example, just as mutation in Vangl2 renders the looptail mouse prone to NTD, polymorphisms in human Vangl1 [Kibar et al., 2007] and Vangl2 [Lei et al., 2010] have recently been implicated in NTD patients. However, evidence that these single nucleotide polymorphisms (SNPs) can occur in unaffected individuals as well suggests that the picture is too incomplete at present to permit using these SNPs for risk assessment. The context of the SNP or genetic background and environmental factors must be taken into consideration. Toward establishing this capability, pathway relationships associated with NTDs are emerging that include PCP signaling pathways encompassing Wnt and SHH pathways, chromatin remodeling and DNA methylation pathways, cell-cycle regulation, apoptosis, and cytoskeletal regulation, as well as transcription factors [Harris and Juriloff, 2007; Harris, 2009; Ross, 2010]. The complexity of the genetic underpinnings of NTD indicates that meeting the challenge for determining individual risk – and the optimal preventative therapy – will require evaluation of multiple genes (in signaling, metabolic, and transcriptional pathways) in a single person to detect compounding effects of gene polymorphisms that alone might not be significant [Greene et al., 2009; Ross, 2010]. Advanced technologies for high-throughput genomic DNA sequencing and analysis, and detection of the epigenome and perhaps the microbiome, as well as untargeted metabolomic screening, will all play a role in the clinical evaluation of individual patient assessments of NTD risk and prevention.
Gene–Environment Interactions Influencing Neural Tube Defects
Environmental influences contributing to NTDs have long been recognized [Janerich and Piper, 1978; Zhu et al., 2009]. The variables that have been implicated as risk factors for nonsyndromic forms of spina bifida are listed in Table 22-1. Risk factors include maternal diabetes, in which both hyperglycemia and associated hyperinsulinemia increase NTD risk [Eriksson et al., 2003]. Maternal obesity and pre-pregnancy weight gain are also associated with increased NTD risk [Werler et al., 1996; Hendricks et al., 2001]. Moreover, maternal periconceptional elevations in simple sugars that raise the glycemic index have been associated with increased NTD risk, even among nondiabetic women [Shaw et al., 2003]. In animal models, exposing rat embryos to a hyperglycemic environment induces dysmorphisms, accompanied by increases in biomarkers of oxidative stress and inositol depletion [Wentzel et al., 2001].
| Risk Factor | Relative Risk (-Fold Increase) |
|---|---|
| Established Risk Factors | |
| History of previous affected pregnancy with same partner | 30 |
| Inadequate maternal intake of folic acid | 2–8 |
| Pregestational maternal diabetes | 2–10 |
| Valproic acid and carbamazepine | 10–20 |
| Suspected Risk Factors | |
| Maternal vitamin B12 status | 3 |
| Maternal obesity | 1.5–3.5 |
| Maternal hyperthermia | 2 |
| Maternal diarrhea | 3–4 |
| Gestational diabetes | NE |
| Fumonisins | NE |
| Paternal exposure to Agent Orange | NE |
| Chlorination disinfection byproducts in drinking water | NE |
| Electromagnetic fields | NE |
| Hazardous waste sites | NE |
| Pesticides | NE |
NE, not established.
(From Mitchell LE et al. Spina bifida. Lancet 2004;364:1885–1895.)
Gene–Diet Interactions in Neural Tube Defects: Role of Metabolism of Folic Acid and Other Nutrients
The important role of the folate pathway in the pathogenesis of NTDs has steadily gained acceptance since the landmark clinical investigations of Smithells and colleagues in the early 1980s prompted a large randomized trial of prenatal FA supplementation for women with previous NTD-affected pregnancies [Smithells et al., 1976; Smithells et al., 1983; MRC, 1991; Czeizel and Dudas, 1992]. Studies in the United Kingdom, later corroborated in Eastern Europe and elsewhere, indicated that the prevalence of NTDs could be reduced by 70 percent or more by prenatal supplementation with FA, even in the absence of maternal folate deficiency [MRC, 1991; Czeizel and Dudas, 1992]. However, some populations demonstrated only a small or no significant reduction in NTD rates with prenatal FA supplementation [Shaw et al., 1995], suggesting that differences in genetic background, diet, or other environmental exposures could impact the efficacy of folate supplementation. Inadequate intake of natural folate before and during early pregnancy is associated with a 2- to 8-fold increased risk of MMC and anencephaly, as indicated by several series of case-controlled, randomized clinical trials and community-based interventions [Wald, 1993]. Moreover, the risk of having a child affected by an NTD is indirectly related to both maternal folate intake and maternal folate intracellular level [Daly et al., 1995; Wald et al., 2001; Moore et al., 2003].
The mechanism underlying the association between NTDs and folate has not been established [Blom et al., 2006]. In view of the extraordinary variety of genes and molecular pathways contributing to NTD formation, perhaps the success of FA supplementation is due to the many biological functions to which folate metabolism contributes [Beaudin and Stover, 2009; Ross, 2010]. The exchange of single carbon units takes place in part in mitochondria, where serine cleavage generates glycine and formate, and glycine cleavage enzymes also generate formate that is transported to the cytoplasm. In the cytoplasm, formate enters the folate pathway for one-carbon metabolism (OCM) as formyl and then methenyl and methylene groups, added to or donated by dihydrofolates and tetrahydrofolates [Beaudin and Stover, 2007, 2009]. In the cytoplasm, the FA metabolic pathway is needed for physiological processes ranging from nucleotide biosynthesis, crucial for proliferation, to generation of pterin coactors impacting biochemical reactions, and generation of the body’s principal methyl donor, S-adenosyl methionine (SAM or Ado-met), which participates in methylation of DNA and histones modulating gene expression, and methylation of proteins and lipids [Blom et al., 2006; Beaudin and Stover, 2007; Miller, 2008].
Recent studies have demonstrated that NTDs can occur in the offspring of pregnant women who had maternal autoantibodies against folate receptors [Rothenberg et al., 2004]. These autoantibodies blocked the binding of 3HFA to folate receptors on placental membranes, presumably resulting in reduced levels of CNS folate that contributed to development of the NTD. This result was not replicated in a population study of a large Irish cohort [Molloy et al., 2009]. However, that research was weakened by the fact that a number of serum samples were taken from women up to 10 years after pregnancy. Another study, which was confined to measuring folate receptor antibodies in samples drawn during midgestation, found a significant association between antibody levels, the folate transport-blocking capability of the antibody, and NTD occurrence [Cabrera et al., 2008]. This may lead in future to clinical testing during first pregnancies that will be used to adjust the recommended level of FA supplementation in subsequent pregnancies.
Teratogens
RA is a well-known teratogen when administered to nonhuman embryos, in which one of its many effects is to induce NTDs, including spina bifida, exencephaly, and anencephaly in several different species [Shenefelt, 1972; Tibbles and Wiley, 1988; Yasuda et al., 1989]. Exposure to excess RA has also been implicated in human embryopathy [Lammer et al., 1985; Rosa et al., 1986]. In contrast, inactivation of RA-synthesizing enzyme genes or receptor genes in mice leads to significantly increased rates of NTDs. High doses of RA (vitamin A) or retinoids in medications such as isotretinoin (Accutane) are to be avoided during pregnancy, although vitamin A deficiency can also increase NTD risk [Azais-Braesco and Pascal, 2000].
Most, if not all, antiepileptic drugs (AEDs) are known teratogens [Ornoy, 2006]. Different AEDs, however, are associated with different constellations of malformations. An increased risk of MMC is associated with in utero exposure to valproic acid or carbamazepine alone, or in combination with other AEDs [Lammer et al., 1987; Dansky and Finnell, 1991; Matalon et al., 2002]. In infants exposed to valproic acid or carbamazepine, the risk of MMC can be as high as 1–2 percent [Koren and Kennedy, 1999; Zhu et al., 2009]. Women who use these drugs for indications other than epilepsy (e.g., bipolar disease, migraine, chronic pain) also are at increased risk of having a child with MMC if they become pregnant while taking these drugs. The mechanisms by which valproic acid and carbamazepine increase the risk of NTD have not been established, but there is general consensus that genetic predisposition to its teratogenic effect is required for valproate to promote NTDs. Among the pathogenic mechanisms associated with valproic acid are increased homologous recombination that induces mutations in the genome, generation of reactive oxygen species, and epigenetic reprogramming through direct inhibition of histone deacetylases by valproate (reviewed in Ross, 2010). Folate administration does not appear to protect against the effects of valproic acid or carbamazepine on neural tube closure.
Classification of Neural Tube Defects







Myelomeningocele
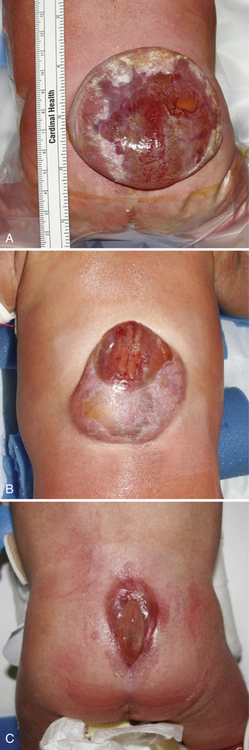
Myelomeningocele (MMC), the most complex of congenital spinal deformities, involves all tissue layers dorsal to and including the neural tube (i.e., spinal cord, nerve roots, meninges, vertebral bodies, skin). The dysplastic neural tube is a flat, disorganized segment of tissue located at the middle and most superficial portion of a cerebrospinal fluid-containing sac (Figure 22-5A, B). The surrounding epithelium may, in some cases, grow over the placode or, in some cases, the neural placode may remain in the spinal canal without a sac present. This condition is referred to as myeloschisis (Figure 22-5C), but probably is not substantially different from a typical MMC in embryologic terms. The absence of a sac probably occurs if cerebrospinal fluid flow in the spinal canal is obstructed, or if cerebrospinal fluid leaks from the spinal subarachnoid space into the amniotic cavity.
Antenatal Diagnosis
Maternal serum α-fetoprotein (AFP) determination and ultrasound examination are used to identify fetuses that have or are likely to have spina bifida or anencephaly [Drugan et al., 2001]. Positive findings from either of these two screening tests can be monitored with amniocentesis and detailed sonography [Kooper et al., 2007]. AFP is a component of fetal cerebrospinal fluid, and it may leak into the amniotic fluid from the open neural tube. Elevated amniotic AFP concentrations correlate with open NTDs, while closed lesions usually do not lead to increased AFP concentration. Detection of NTDs correlates with the magnitude of increase in the amniotic fluid AFP level; NTDs are associated in a minority of pregnancies with mildly elevated AFP levels, in a majority of those with moderately elevated levels, and overwhelmingly in those with very elevated AFP levels [Canick et al., 2003]. An AFP level of two times the normal median or higher is found in approximately 2 percent of unaffected pregnancies, 80 percent of open spina bifida pregnancies, and 95 percent of anencephalic pregnancies.
Sonography also can be used to differentiate between ventral wall defects and NTDs [Lennon and Gray, 1999], and to identify additional structural malformations that are characteristic of fetuses with chromosomal abnormalities [Sepulveda et al., 2004]. When a diagnosis of spina bifida is confirmed, ultrasound examination is used to assess spontaneous leg and foot motion and to screen for leg and spine deformities, a Chiari II malformation, and other physical defects. Ultrasonography can detect or confirm the extent of the NTDs [Watson et al., 1991] and has had an enormous impact on the number of liveborn infants among populations in which termination of pregnancy is accepted [Zlotogora et al., 2002]. It is 60 percent accurate in low-risk pregnancies, which is equivalent to the accuracy of serum AFP screening (64 percent), 89 percent accurate in high-risk pregnancies, and 100 percent accurate for women referred for confirmation of a suspected spina bifida by another ultrasonographer [Chan et al., 1993, 1995]. The data indicate that neither sonography nor AFP screening alone provides sufficient sensitivity or specificity, but that, when these studies are used together, the predictive value is much higher [Chan et al., 1995].
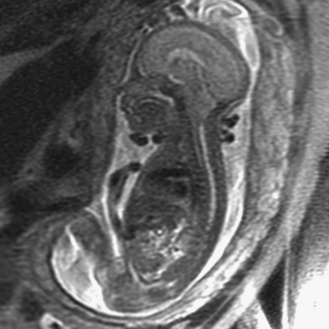
Prenatal magnetic resonance imaging (MRI), with ultra-fast T2-weighted sequences, also can be used to characterize the Chiari II and other malformations (Figure 22-6). Such prenatal imaging studies might help to predict neurologic deficit [Cochrane et al., 1996] and ambulatory potential [Biggio et al., 2001]. Most fetuses with spina bifida that are not electively terminated receive no treatment until after birth. Several studies have investigated whether method of delivery influences the outcome for infants with the disorder. A study based on a review of this work concluded that, in general, conclusive evidence is lacking that, relative to vaginal delivery, cesarean section improves the outcome in children with spina bifida [Anteby and Yagel, 2003]. Cesarean section, however, might be justified for large lesions, to reduce the risk of trauma, and is done after in utero treatment of spina bifida because the forces of labor are likely to produce a wound dehiscence.
Prevention
NTDs occur during early pregnancy, often before a woman knows she is pregnant; 50–70 percent of these defects can be prevented if a woman consumes sufficient FA daily before conception and throughout the first trimester of her pregnancy. Two landmark studies in the early 1990s found that administration of FA had a profound effect on the risk of NTDs [MRC, 1991; Czeizel and Dudas, 1992]. In 1992, to reduce the number of cases of MMC, the U.S. Public Health Service recommended that all women capable of becoming pregnant consume 400 μg of FA daily. Because approximately 50 percent of pregnancies in the United States are unplanned and the neural tube develops before most women know they are pregnant, it was also recommended that women consume this amount of FA routinely. Three approaches to increase FA consumption were cited:
Mandatory fortification of cereal grain products was legislated in the U.S. in 1996 and fully implemented as of January 1998. The estimated number of NTD-affected pregnancies in the U.S. declined from 4000 in the period 1995–1996 to 3000 in 1999–2000 [CDC, 2004; Mills and Signore, 2004]. This analysis controlled for declines in live births with NTDs as a result of recently implemented screening programs. Since the overall reduction of 25–30 percent in NTD rates in the U.S. since fortification was put into effect is below the expected 70 percent reduction, the debate continues regarding whether the fortification standard should be raised [Pitkin, 2007; Smith et al., 2008; Oakley, 2010].
Regardless of whether the optimal regimen for prenatal FA supplementation has been determined, there is still a significant proportion of pregnancies at risk for NTD for which FA will not effectively prevent NTD. Evidence in mouse models indicates that certain genetic backgrounds may include partial block in folate metabolism that can be circumvented by using alternative supplements. For example, the NTDs in the mouse mutant, Axial defects (Axd), are resistant to FA supplementation, but methionine – which will contribute to the methylation arm of the folate metabolic pathway – can reduce the occurrence of neurulation failure [Essien and Wannberg, 1993]. Another mouse mutant, curly tail (ct), which is prone to spina bifida that is not rescued by FA, can be protected against NTD by supplementation with myo-inositol [Greene and Copp, 1997]. Currently, clinical trials of prenatal inositol supplementation for the prevention of NTD are under way in the U.K. It appears likely that additional options for prevention of NTD will be available in the foreseeable future.
Clinical Features
The mortality rate for MMC is approximately 50 percent in the absence of therapy. Early surgery for closure of the lumbosacral defect is required to prevent meningitis. Later causes of death include hydrocephalus and renal failure. The renal complications are induced by chronic urinary tract infections, abnormal urodynamic function, and genitourinary tract abnormalities, such as progressive hydronephrosis. Patients at particular risk are the subgroup with high pressure within the bladder [Snodgrass and Adams, 2004].
MMCs may be situated at any longitudinal level of the neuroaxis. The location and extent of the defect determine the nature and degree of neurologic impairment; rating scales have been developed in an attempt to standardize the evaluation of affected children [Oi and Matsumoto, 1992]. Lumbosacral involvement is most common. Thoracic defects are the most complex and frequently are associated with serious complications. Cervical cord involvement is different from MMC of the lower spine and can be differentiated into two types:
Cervical lesions are clearly protuberant, covered at the base by full-thickness skin, and covered on the dome by a thick epithelium. Neural tissue is not superficially exposed but usually is tethered to adjacent dural or intrasaccular tissues. These differences likely are responsible for the more favorable outcome in cervical lesions [Meyer-Heim et al., 2003]. Varying degrees of paresis of the legs, usually profound, and sphincter dysfunction are the major clinical manifestations. Congenital dislocation of the hips or deformities of the feet, such as clubbing, may also occur. Severe sensory loss and accompanying trophic ulcers may complicate the condition. Occasionally, only sphincter disturbances are present. Radiographs reveal the primary defect of the vertebral arch.
Hydrocephalus is present in about 70–85 percent of patients with MMC, and occurs most frequently when the lesion is situated in the thoracolumbar area [Rintoul et al., 2002], which is the case in 90 percent of patients. Most persons with spina bifida have normal intelligence, but specific cognitive disabilities and language difficulties are common and can adversely affect educational and occupational achievements and the ability to live independently [Northrup and Volcik, 2000; Vachha and Adams, 2003].
Secondary Abnormalities
Central Nervous System Complications
Seizures have been reported in up to 17 percent of patients with MMC and almost always occur in those with shunted hydrocephalus [Talwar et al., 1995]. Electroencephalographic (EEG) abnormalities are nonspecific. Additional CNS abnormalities seen in these patients are believed to explain the cause of seizures, and include encephalomalacia, previous stroke, malformations, and intracranial calcifications. Seizures may be difficult to control, and frequently, exacerbation of seizures is associated with shunt malfunction or ventriculitis.
Bladder and Bowel Dysfunction
Bladder dysfunction and urinary incontinence pose major management problems and may be present at birth in the form of hydronephrosis [Stone, 1995; Silveri et al., 1997]. Interruption of sacral nerve roots and fiber connections between the brainstem and sacral cord causes the dysfunction. Loss of sphincter tone, overflow incontinence, sacral and rectal loss of sensation, and loss of detrusor activity on cystometry are seen. In other patients with higher lesions, dyssynergia of reflex pathways results in irregular contractions of the bladder in conjunction with outlet obstruction. Normal bladder control occurs in 10 percent of children with MMC. Prevention of bladder infection requires intermittent catheterization to maintain low residual urine volumes and prophylactic antibacterial drugs [Buyse et al., 1995]. Vesicoureteral reflux often develops during the second and third years of life, and assessment for this problem must be on-going. Re-implantation of the ureters into the bladder, or external drainage of the ureters either directly or through an ileal conduit, may be helpful. Transurethral resection of the external sphincter has been recommended when ureteral dilatation occurs. The use of prosthetic devices emulating sphincters has been useful in highly selected patients [Simeoni et al., 1996], as has selective sacral rhizotomy [Schneidau et al., 1995].
Constipation and fecal incontinence are common problems in children with spinal dysraphism and usually can be managed medically [Knab et al., 2001]. Dietary manipulation, oral and rectal laxatives, and manual evacuation are common treatments. Retrograde colonic enemas also may be successful.
Orthopedic Problems
It is important to consider that the paralysis below the cord lesion associated with spina bifida will be hypotonic, which predicts the types of orthopedic problems. Orthopedic defects associated with this paralysis, muscle imbalance, and accompanying regional spasticity may be severe and necessitate early intervention. Hip subluxation usually is treated with prosthetic splints or plaster casting [Heeg et al., 1998]. Sensory deficits of the casted skin areas frequently enhance the likelihood of skin ulcers. Severe foot deformities are seen in up to 80 percent of children and are treated with splinting or casting; neither age nor segmental level influences outcome of the foot deformity [Flynn et al., 2004]. Physical therapy may help to preserve and extend the range of motion of the joints [McDonald, 1995].
In infants and children, progressive leg or foot deformity, weakness, pain, or deterioration of gait or bladder function implies restricted growth or tethering of the spinal cord. Cord tethering may be found in many older children with spina bifida who are neurologically stable, however. Evidence of brainstem dysfunction may be recorded by means of respiratory pneumograms with a carbon dioxide challenge [Petersen et al., 1995] or by using brainstem auditory-evoked response testing [Docherty et al., 1987]. These signs and symptoms indicate the possibility of a Chiari II malformation or a tethered spinal cord.
Considerations in the differential diagnosis of delayed deterioration in a child with repaired MMC include a malfunctioning or infected shunt, seizures, scoliosis, hydrocephalus, hydromyelia, and an undetected second lesion of occult spinal dysraphism, such as a dermoid, epidermoid, or arachnoid cyst. Of course, intercurrent illness or drug toxicity should be considered. Surgical repair of a worsening tethered spinal cord and shunting or fenestration of syringomyelia can prevent decline of function [Iskandar et al., 1994].
Chiari II Malformation
Classification
Chiari was among the first to recognize multiple hindbrain malformations associated with congenital hydrocephalus [Arnett, 2003]. Four types of Chiari malformation have been characterized. Chiari I malformation is a downward displacement of the cerebellum and cerebellar tonsils. Chiari II malformation, also known as the Arnold–Chiari malformation, is a complex malformation that includes downward displacement of the cerebellar vermis and tonsils, and is encountered almost exclusively in patients with MMC. Chiari III is an encephalocervical meningocele, and Chiari IV refers to hypoplasia of the cerebellum. The differentiating features are indicated in Table 22-2.
| Type | Anatomic Abnormalities | Neurologic Findings |
|---|---|---|
| I | Downward displacement of the cerebellum and cerebellar tonsils, elongated brainstem fourth ventricle | Mild and delayed onset of headache and brainstem symptoms, usually beginning in adolescence; symptoms secondary to hydrocephalus or syringomyelia |
| II (Arnold–Chiari malformation) | Downward displacement of the cerebellar vermis and cerebellar tonsils; thinned and elongated medulla may actually be positioned side by side with upper segments of the atrophic cervical spinal cord | Associated with myelomeningocele in more than 95 percent of patients; symptoms resulting from progressive hydrocephalus and secondary brainstem dysfunction; feeding and respiratory complications are common, including apnea |
| III | Encephalocervical meningocele with spina bifida over the cervical area and protrusion of cerebellum through posterior encephalocele | Features as for Chiari II, without the same degree of association with myelomeningocele |
| IV | Hypoplasia of the cerebellum | Variable, ranging from asymptomatic to classic cerebellar dysfunction |
(Data from Friede [1989]; Norman et al. [1995]; Sarnat [1992].)
The major features of the Chiari II malformation include:
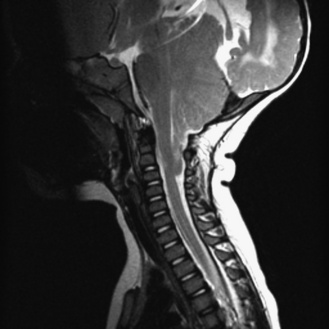
Fig. 22-7 A postnatal sagittal T2-weighted magnetic resonance image of a child with a Chiari II malformation.
Hydromyelia and syringomyelia of the cervical spinal cord occur in 20–50 percent of patients.
Pathophysiology
The precise mechanism causing Chiari II malformation is unknown. The most parsimonious theory is that the open neural tube fusion to the overlying skin produces downward traction on the neural axis, resulting in herniation of the brainstem and cerebellum through the foramen magnum. This herniation results in obstruction of hindbrain cerebrospinal fluid outflow, which leads to hydrocephalus. This theory is in keeping with the observation that the Chiari II malformation precedes the development of hydrocephalus. Hydrocephalus associated with the Chiari II malformation probably results primarily from one or both of two basic causes. The first is the hindbrain malformation that blocks either the fourth ventricular outflow or the cerebrospinal fluid flow through the posterior fossa. The second is aqueductal stenosis or atresia that may be associated with the malformation in approximately 50–85 percent of cases [Peach, 1965; Stein and Schut, 1979; Bell et al., 1987]. Further insights into the pathogenesis of the Chiari II malformation will come from study of animal models of NTDs. It has been possible to examine the developmental progression of this constellation of malformations in the Splotch animal model of spina bifida due to mutations in the PAX3 gene [McLone and Dias, 2003].
Clinical features
The symptoms associated with Chiari II malformations include apnea, swallowing difficulties, and stridor in the newborn, and headache, quadriparesis, scoliosis, and balance and coordination difficulties in the older child; they are present in up to one-third of persons with the disorder. It is often difficult to differentiate between symptoms related to the hydrocephalus and those related to the Chiari II malformation, but several features are uniformly seen in this condition (Box 22-1). These symptoms are directly referable to cerebellar, brainstem, and cranial nerve dysfunction. More than one-third of affected infants display feeding disturbances associated with reflux and aspiration. Other features include vocal cord paralysis with stridor and abnormalities of ventilation, including both obstructive and central apnea.
Box 22-1 Clinical Manifestations of Chiari II Malformation

















(From McLone DG, Dias MS. The Chiari II malformation: Cause and impact. Childs Nerv Syst 2003;19:540–550.)
The causes of the clinical abnormalities of brainstem function are threefold. First, they relate in part to the brainstem malformation, which involves cranial nerves and other nuclei, and which are present in the large majority of cases [Gilbert et al., 1986]. Second, compression and traction of the anomalous caudal brainstem by hydrocephalus and increased intracranial pressure also may play a role, especially in the vagal nerve disturbance that results in vocal cord paralysis and stridor. Third, ischemic and hemorrhagic necrosis of the brainstem often is present also, and may be secondary to the disturbed arterial architecture of the caudally displaced vertebrobasilar circulation [Charney et al., 1987].
Management
Fetal Repair of Myelomeningocele
Animal studies, in which a model for spina bifida is created by laminectomy and exposure of the spinal cord to amniotic fluid, demonstrate that function can be retained if the lesion is closed before birth [Michejda and Bacher, 1985; Meuli et al., 1995]. Other evidence indicates that the Chiari II malformation, which occurs in almost all persons with spina bifida, is acquired and could potentially be prevented by in utero closure [Osaka et al., 1978; Paek et al., 2000].
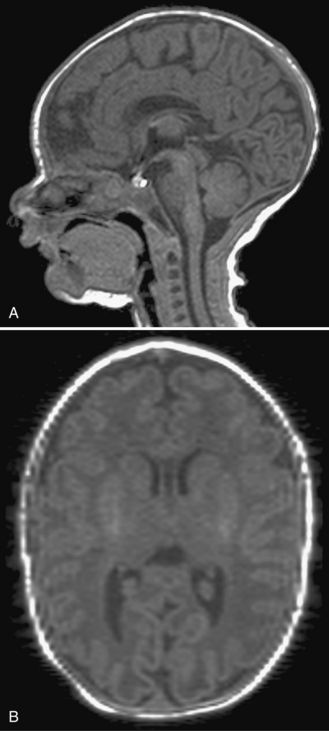
Some evidence suggests that surgical treatment in utero results in improved outcomes. In 1998, in utero repair of MMC by hysterotomy was reported [Adzick et al., 1998; Tulipan and Bruner, 1998]. Early experience suggested that infants with MMC treated by hysterotomy had at least partial correction of hindbrain herniation [Tulipan et al., 1998], and possibly a diminished need for shunting relative to infants who received postnatal treatment [Bruner et al., 1999]. Compared with historical controls, infants given treatment in utero have a lower incidence of moderate to severe hindbrain herniation and hydrocephalus requiring shunting (Figure 22-8) [Tulipan and Bruner, 1999]. In a series of 50 MMC cases treated in utero, reversal of hindbrain herniation was reported in all patients, and the proportion requiring shunting was less than in historical control subjects (43 versus 85 percent) [Johnson et al., 2003]. A similar proportion requiring shunting (54 percent) also was noted in a second series of 116 spina bifida cases treated in utero at Vanderbilt University Medical Center [Bruner et al., 2004]. Comparisons between infants with spina bifida treated in utero and historical control subjects are, however, subject to substantial bias. The approach is being tested in a prospective randomized clinical trial.
Management in the Newborn Period
The value of immediate correction of the defect within 48 hours of birth is widely accepted. Even when cerebrospinal fluid leakage occurs, however, a delay in closure for up to 48 hours does not increase the risk of infection or worsen the neurologic deficit [McComb, 1997]. In such cases, the patient is given antibiotics, and the exposed placode is kept clean and moist. When primary closure is impossible because of the large cutaneous area involved, epithelialization must be encouraged, and surgery performed later. The decision to give vigorous therapy for the most severely affected infants with MMC is beset by moral and medical considerations; restricted therapy often is associated with survival but poor outcome. Appropriate treatment of newborns with MMC increases the number of survivors and perhaps the quality of survival. Early surgical repair is appropriate for most newborns with myelodysplasia [McComb, 1997]. Each patient must be assessed individually in the context of advances in technology and medical care.
Treatment of Chiari II Malformation
Careful monitoring of infants with known Chiari II malformations is advocated [Cai and Oakes, 1997; Stevenson, 2004]. Evaluation with MRI is the procedure of choice. Before surgical decompression of the posterior fossa in symptomatic patients is considered, it is important to treat hydrocephalus if present. A properly functioning ventricular shunt often can obviate the need for decompression of hindbrain herniation. Many patients will have resolution of brainstem symptomatology once a shunt is placed [Caldarelli et al., 1995]. Significant improvement in the size of the accompanying spinal syrinx may occur after ventriculoperitoneal shunting or shunt revision [Milhorat et al., 1992].
Suboccipital craniectomy to decompress the suboccipital and cervical areas may be warranted to reduce the progressive compression of neural and vascular structures. Studies have suggested that early surgical intervention to alleviate brainstem compression is associated with improved long-term neurologic outcome and a decreased need for tracheostomy or gastrostomy [Pollack et al., 1992, 1996]. Alleviation of fourth ventricle outflow obstruction by foraminal fenestration or shunting may be required in rare cases. In one series of 50 symptomatic Chiari II patients, surgical treatment resulted in complete relief of symptoms in 20 percent, improvement in 66 percent, stabilization in 8 percent, and worsening in 6 percent [Dyste et al., 1989]. Despite these data, no standards for treatment have yet been established because the current neurosurgical literature is characterized by significant debate and variable results [Tubbs et al., 2004].
Outcome
Short-term and long-term survival of patients with spina bifida has increased with improvements in medical and surgical management. The most recent population-based data indicate that the 1-year survival rate is about 87 percent, and that roughly 78 percent of all persons with MMC survive to the age of 17 years [Wong and Paulozzi, 2001]. Unfortunately, these patients continue to be subject to excess morbidity and mortality throughout adulthood [Singhal and Mathew, 1999; McDonnell and McCann, 2000; Bowman et al., 2001]. Patients with MMC are at substantial risk for leg weakness and paralysis, sensory loss, bowel and bladder dysfunction, and orthopedic abnormalities (e.g., clubfoot, contractures, hip dislocation, scoliosis, kyphosis). In general, the functional level of the defect corresponds to the anatomic level of the bony spinal defect, as determined by radiologic studies. Patients with MMC also develop symptoms from associated malformations of the CNS, including hydrocephalus, syringomyelia, and Chiari II malformations. Almost all patients with thoracic level lesions require a ventricular shunt, whereas shunting is required in less than 70 percent of those with sacral level lesions [Rintoul et al., 2002]. Radiographic evidence of Chiari II malformations is present in more than 75 percent of affected persons [Just et al., 1990].
Virtually all infants and children born with NTDs are provided with surgical and medical treatment. Previous studies reported that the 5-year survival rate for children with sacral lesions was in the range of 80–90 percent, with increased mortality among children with lesions located more superiorly along the neuraxis [Honein et al., 2001]. Studies of the long-term outcome in children born with MMC have demonstrated an improved outcome that coincides with medical advances [Wong and Paulozzi, 2001]. Among 101 patients assessed at birth, the mortality rate was 18 percent; 53 percent were community ambulators, 58 percent attended normal school and were grade-appropriate, 75 percent were continent of urine, and 86 percent were continent of stool. Hydrocephalus was present in 93 children, and 85 required shunting. The shunt revision rate in the first year of life was 50 percent and, after 2 years, decreased to 10 percent per year [Steinbok et al., 1992]. Of note, an increased risk of shunt infections follows new abdominal surgery for renal complications [Aldana et al., 2002].
Counseling
Counseling is essential to ensure that parents understand the nature and severity of the deformities and the necessary surgical and long-term rehabilitative efforts [Liptak et al., 1988]. Parents also should be aware of the patient’s potential for intellectual and physical development. In addition, parents should be educated about the higher incidence of late medical complications that can arise, such as latex allergy, and their warning signs in children with spina bifida [Konz et al., 1995]. It is important to communicate these potential problems to family members and others involved in their care.
Anencephaly
Anencephaly is a congenital malformation in which both cerebral hemispheres are absent [MTFA, 1990]. Most anencephalic infants are stillborn, and those infants born alive die shortly after birth [Walters et al., 1997]. Epidemiologic studies demonstrate a striking variation in prevalence rates. The highest incidence is in Great Britain and Ireland, and the lowest incidence is in Asia, Africa, and South America. Other countries have intermediate incidence rates. Anencephaly occurs six times more frequently in whites than in blacks. Female fetuses are more often affected than males.
Antenatal diagnosis is possible using assays of AFP or acetylcholinesterase, which are increased in maternal serum and amniotic fluid [Brock et al., 1985; Wald et al., 1989; Loft et al., 1993; Crandall and Chua, 1995]. Antenatal diagnosis also is feasible through the use of fetal ultrasonography [Birnbacher et al., 2002]. In the past two decades, prenatal screening with ultrasound examination during the first trimester, which is nearly 100 percent accurate, and maternal AFP determinations have resulted in earlier detection of anencephaly [Aubry et al., 2003]. Earlier detection has resulted in a dramatic decrease in the average gestational age at birth, from 35.6 weeks in the 1970s to 19.6 weeks in 1988 to 1990, with virtually no term liveborn anencephalic infants born after 1990 in those pregnancies in which a prenatal diagnosis of anencephaly had been made [Drugan et al., 2001].
Differential Diagnosis
In anencephaly, the absence of the brain and calvaria can be total or partial. Acrania is defined as congenital partial or total absence of the skull. Craniorachischisis is characterized by anencephaly, accompanied by a contiguous bony defect of the spine and exposure of neural tissue. Craniorachischisis is typically, an extremely thin and flattened spinal cord, accompanied by foreshortening of the neuraxis and is thought to result from impaired convergent extension during embryogenesis. In iniencephaly, dysraphism in the occipital region is accompanied by severe retroflexion of the neck and trunk, with three cardinal features: deficiency of the occipital bone, cervicothoracic spinal retroflexion, and rachischisis. Iniencephaly differs from anencephaly in that the cranial cavity is present and skin covers the head and retroflexed region. Severe retroflexion of the neck present on fetal ultrasound examination may suggest the diagnosis. A majority of the patients also have visceral and other severe CNS malformations. Another related condition is meroanencephaly. Meroanencephaly is a rare form of anencephaly characterized by malformed cranial bones and a median cranial defect, through which protrudes the area cerebrovasculosa. In encephalocele, the brain and meninges herniate through a defect in the calvaria [Botto et al., 1999].
Encephalocele
An encephalocele is a herniation of intracranial contents through a midline skull defect. Also known as cephaloceles, these lesions are classified by their contents and location [David, 1993]. Cranial meningoceles contain only leptomeninges and cerebrospinal fluid, whereas encephaloceles also contain brain parenchyma. The incidence of cephaloceles is approximately 0.8–5 per 10,000 live births, with encephaloceles being the most common form [Siffel et al., 2003].
Encephaloceles usually are located in the occipital (75 percent) or frontal areas (25 percent). Basal and trans-sphenoidal encephaloceles are rare; they may appear between the ethmoid and sphenoid bones and extend into the upper pharynx [Harley, 1991]. Encephaloceles that extend from the area of the orbit, nose, or forehead are termed sincipital encephaloceles; those in the occipital region are termed notencephaloceles. Exencephaly consists of a large outpouching of brain tissue with surrounding thick walls. This defect may involve the spinal cord, forming an encephalomyelocele. Cranial encephalocele may contain a combination of meninges, ventricles, and brain parenchyma [Wininger and Donnenfeld, 1994].
Etiology
The etiology of encephaloceles is not known but likely is multifactorial [Van Allen et al., 1993; Golden and Chernoff, 1995; Martinez-Lage et al., 1996; McComb, 1997]. Studies have indicated that consumption of FA during the periconceptional period can reduce the risk of anencephaly, as well as spina bifida [Botto et al., 1999; Honein et al., 2001]. A similar protective effect, however, has not been noted for encephalocele [Stevenson et al., 2000], suggesting that efforts to increase FA consumption among women of reproductive age are not likely to have any influence on the incidence of encephaloceles. Although some affected fetuses are stillborn, or some affected pregnancies are terminated as a consequence of increased prenatal diagnosis of NTDs, a substantial proportion of affected infants are live-born [Allen et al., 1996]. Although the etiology in most cases likely is multifactorial, some cases display likely monogenetic causes [Van Esch et al., 2004) that are associated with well-defined syndromes (Table 22-3).
Table 22-3 Selected Syndromes Associated with Encephaloceles
| Syndrome | Clinical Features |
|---|---|
| Walker–Warburg syndrome | Hydrocephalus, severe neurologic impairment, vermian agenesis, type II lissencephaly, autosomal-recessive inheritance |
| Meckel–Gruber syndrome | Polycystic kidney, sloping forehead, polydactyly, hepatobiliary fibrosis, cleft lip and palate, autosomal-recessive inheritance |
| Dandy–Walker syndrome | Hydrocephalus, partial or complete absence of cerebellar vermis, posterior fossa cyst contiguous with fourth ventricle, cranial nerve palsies, nystagmus, truncal ataxia, sporadic occurrence |
| Joubert’s syndrome | Cerebellar vermian aplasia, episodic hyperpnea, abnormal eye movements, rhythmic protrusion of tongue, ataxia, retardation, coloboma, retinal dystrophy, renal cysts, autosomal-recessive inheritance |
| Goldenhar–Gorlin syndrome | Orofacial abnormalities, pre-auricular tags, epibulbar dermoids, sporadic occurrence |
| Knobloch’s syndrome | Vitreoretinal degeneration with retinal detachment, high myopia, and occipital encephalocele, normal intelligence, gene localized to 21q22.3 |
| Robert’s syndrome | Anterior encephalocele, autosomal-recessive inheritance |
| Median cleft face syndrome | Anterior encephalocele, autosomal-dominant inheritance |
(Data from Online Mendelian Inheritance in Man [OMIM], http://www.ncbi.nim.nih.gov/omim,accessed2005.)
Clinical Characteristics
A fluctuant, round, balloon-like mass that protrudes from the cranium, usually posteriorly, is the most typical manifestation of encephaloceles (Figure 22-9). The mass may pulsate and be covered by an erythematous, translucent, or opaque membrane, or by normal skin. The covering may not be uniform throughout its surface. The amount of compromised and deformed neural tissue and the degree of resultant microcephaly determine the extent of cerebral dysfunction [Lorber and Schofield, 1979], but otherwise little has been published about prognosis. Brain tissue not extending into the encephalocele (i.e., retained within the intracranial cavity) may be deformed and functionally impaired.
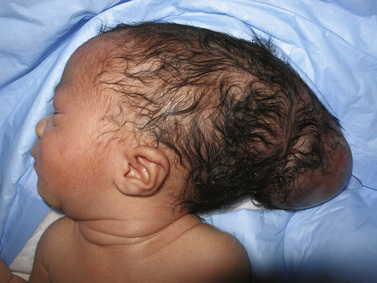
Severe intellectual and motor delays typically occur in association with microcephaly; motor delay is accompanied by weakness and spasticity. Intellectual impairment is more prevalent in patients with posterior encephaloceles than in those with anterior encephaloceles [Hockley et al., 1990]. Some patients, however, may have fairly normal development. Occipital lobe destruction is associated with various degrees of visual impairment. When the deformity extends into the ventricle, hydrocephalus is almost inevitable. Because of increased intracranial pressure, the encephalocele may become stretched until the rim of neuronal tissue is infarcted, with resultant infection and rupture if the problem is not surgically corrected.
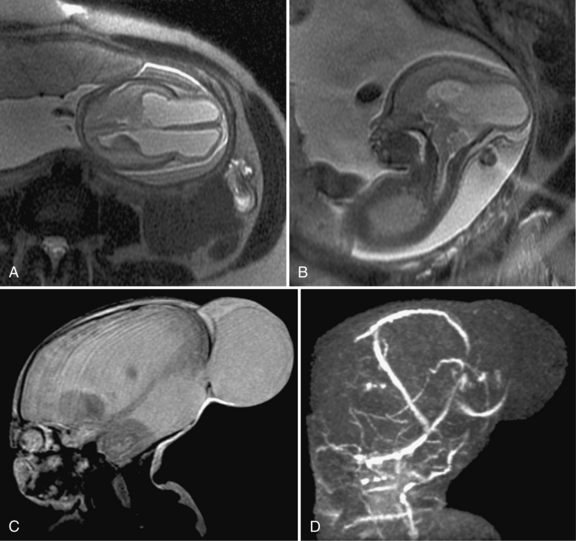
Other malformations may accompany encephaloceles, including Dandy–Walker syndrome, Klippel–Feil syndrome, Arnold–Chiari malformation, porencephaly, agenesis of the corpus callosum, myelodysplasia, optic nerve dysplasia, cleft palate, and others [Mecke and Passarge, 1971; Lieblich et al., 1978; Pagon et al., 1978; Fisher and Smith, 1981; Cohen and Lemire, 1982; Koenig et al., 1982; Aleksic et al., 1983; Waterson et al., 1985; Sertie et al., 1996; Wang et al., 1999; Joy et al., 2001; Van Esch et al., 2004] (see Table 22-3). Although ultrasonography may be helpful, the clinical diagnosis is confirmed by computed tomography (CT) or MRI (Figure 22-10).
Neuroendocrine disturbances occur, particularly with basal encephaloceles that involve the sella turcica or sphenoid sinus [Lieblich et al., 1978]. These conditions may be undetectable by gross inspection. Intranasal mass or endocrine dysfunction is the cardinal feature. Encephaloceles are seen with a variety of chromosomal disorders (e.g., trisomy 13 or 18; 13q or 16q deletion) and syndromes (see Table 22-3). Occipital encephalocele, microcephaly, cleft palate or lip, polydactyly, holoprosencephaly, and polycystic kidneys constitute Meckel–Gruber syndrome [Chao et al., 2005]. Retinal degeneration with detachment and occipital encephalocele (Knobloch’s syndrome) is another autosomal-recessive condition [Wilson et al., 1998].
Nasal glioma, a congenital tumor, appears as a frontonasal mass that mimics an encephalocele [Rahbar et al., 2003]. The tumor is derived from herniated brain tissue that has lost its connection to the brain; this relation can be demonstrated by neuroimaging techniques.
Management
Prenatal diagnosis of encephaloceles may be established with determination of increased amniotic AFP content and ultrasound studies [Graham et al., 1982]. Surgical correction of all but the smallest encephaloceles is necessary. Accompanying hydrocephalus may require ventriculoperitoneal shunting. Associated systemic abnormalities are present in approximately half of the patients, depending on the syndromic nature of the condition. A full battery of endocrinologic screens should be performed to evaluate basal encephaloceles.
Occult Forms of Spinal Dysraphism
The spectrum of occult spinal dysraphism includes anomalies where the overlying skin is mostly normal and covers the underlying NTD. These anomalies include distortion of the spinal cord or roots by fibrous bands and adhesions, intraspinal lipomas, dermoid or epidermoid cysts, fibrolipomas, spinal cord lipomas, lipomyelomeningocele, and split cord malformations [Anderson, 1975; Byrd et al., 1991; Kriss et al., 1995].
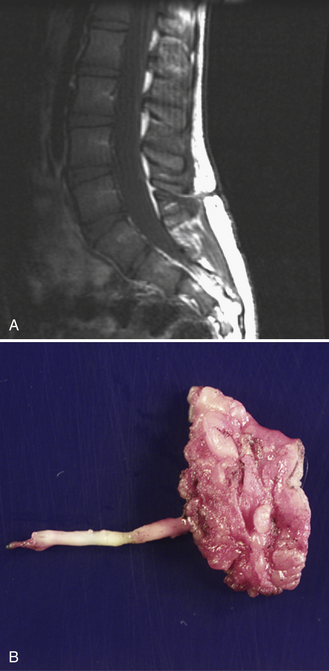
Spinal Cord Lipoma
Spinal cord lipomas are a continuum of developmental anomalies that range from a small fatty mass attached to the distal spinal cord to very complex anomalies that involve all spinal structures. In some cases, the lipoma is entirely intraspinal and extends through a limited defect in the posterior elements of the spine into the subcutaneous tissues (Figure 22-11). In other cases, the spinal cord lipoma is continuous with the caudal spinal cord, and the spinal cord itself extends into the subcutaneous tissues, along with the meninges. If the spinal cord extends into the subcutaneous tissues along with a cerebrospinal fluid-containing space, this is usually referred to as a lipomyelomeningocele.
Dermal Sinus Tract
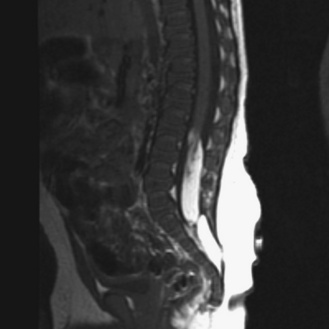
Recurrent meningitis from external contamination of cerebrospinal fluid may result from occult congenital malformations along the spinal canal and neuroaxis. These external connections include midline dermal sinus; temporal bone fistula to the middle ear, eustachian tube, or nasopharynx; neurenteric fistula; and basal encephalocele or meningocele involving the cribriform plate, sphenoid bone, or clivus. From an embryological perspective, sinus tracts consist of epithelial-lined canals that probably represent persistence of an ectodermal-derived pathway from the skin to the CNS (Figure 22-12). In most cases, the spinal cord appears largely normal, implying that neural tube formation is complete but a persistent communication is still present. The spinal cord may be low or deformed by the presence of the sinus tract.
Spina Bifida Occulta
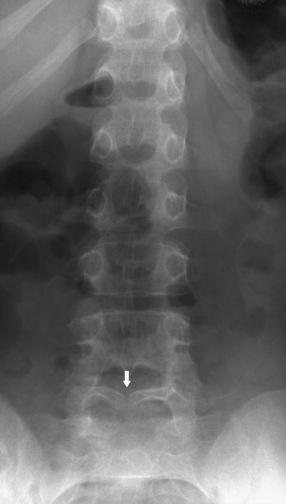
Spina bifida occulta, a confusing term, is defined as a defect in the posterior bony components of the vertebral column without involvement of the cord or meninges (Figure 22-13). It occurs in at least 5 percent of the population but most often is asymptomatic. This defect often is found incidentally on radiographic studies, or is diagnosed because of a subtle clinical finding such as a tuft of hair or a cutaneous angioma or lipoma in the midline of the back, marking the location of the defect. The presence of such a cutaneous lesion is associated with spina bifida occulta in only approximately 10 percent of cases, although the percentage increases to approximately 50 percent when two or more skin lesions are present [Guggisberg et al., 2004]. This defect is located most often in the lower lumbar area, involving the lamina of L5 and S1. The presence of a bifid spinous process is not associated with neurologic deficits or spinal instability. No treatment or intervention is required.
Meningocele
Meningocele, a protrusion of meninges without accompanying nervous tissue, is not associated with neurologic deficit. The mass usually is evident as a fluid-filled protrusion covered by skin or membrane in the midline. Membranous lesions are found rostrally, and skin-covered lesions are more evenly distributed along the neuraxis. MRI analysis is helpful in determining the contents of a mass along the spine and in differentiating meningocele from MMC (Figure 22-14). Very small subcutaneous lesions may remain undetected for prolonged periods and typically require no specific treatment, but must be differentiated from small encephaloceles.
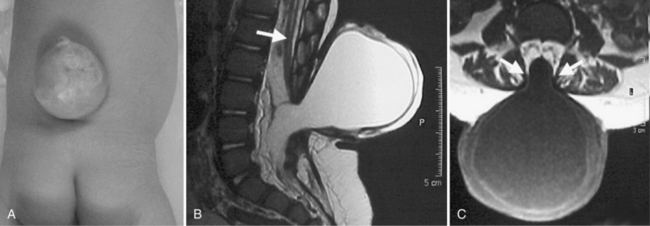
Fig. 22-14 Lumbar meningocele in a newborn.
(From http://www.neurocirugia.com/intervenciones/ meningocele/Meningocele.htm, with permission.)
A meningocele in the cranial or high cervical area may coexist with aqueductal stenosis, hydromyelia, or a Chiari II malformation. Membrane-covered meningoceles are more likely to be accompanied by severe abnormalities; lesions covered with normal skin often are free of associated abnormalities. Elective surgical treatment is recommended, except for very small lesions [Feltes et al., 2004].
Split Cord Malformations
Embryology
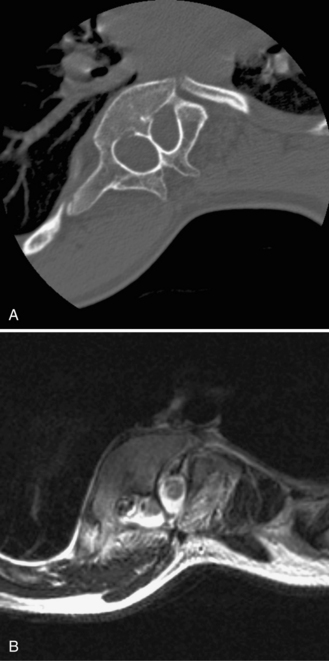
Split cord malformations can be divided into two different types. In type I, present in 50 percent of cases, a split spinal cord is surrounded by a normal undivided arachnoid-dural sleeve without a septum. In type II, present in the other 50 percent, each hemicord is invested by a separate dural sleeve, divided by a fibrous, cartilaginous, or bony septum [Pang et al., 1992]. The bony septum can be visualized on CT, which may be helpful if surgery is required (Figure 22-15). These conditions are differentiated from diplomyelia (see below) on the basis of the integrity of the spinal cord; in diastematomyelia, only a single central canal that is split at or near the midline is present.
Diplomyelia is a side-by-side or anteroposterior duplication of the spinal cord [Dias and Pang, 1995]. It is differentiated from diastematomyelia by the presence of two central canals, each surrounded by gray and white matter arranged in the normal pattern. The two cords often are completely reunited caudally but may remain separated to the tip of the conus medullaris. A bony septum may partly intervene between the duplicated cords. The two cords are often unequal and may be side by side (the most common position), or one may be dorsal to the other. These malformations are compatible with normal function; deterioration suggests the presence of diastematomyelia or tethering. The etiology of this condition also remains unknown.
Clinical Characteristics
Patients with split cord malformations present with a congenital scoliosis, hydrocephalus, or cutaneous lesion such as hairy patch, dimple, hemangioma, subcutaneous mass, or teratoma [Kothari and Bauer, 1997]. A progressive myelopathy, with deformities of the feet, scoliosis, kyphosis, or discrepancy in leg length, may develop. Resection of the spur frequently does not result in clinical improvement, but may prevent further deterioration. The intervening mesenchymal elements appear to contribute to progressive neurologic, urologic, and orthopedic deterioration from spinal cord tethering. Resection of the spur should be performed in patients who have progressive neurologic manifestations; those without worsening symptoms should be observed until progression occurs and then resection performed [Miller et al., 1993]. Split cord malformations can be detected prenatally by ultrasonography [Anderson et al., 1994; Sonigo-Cohen et al., 2003]. MRI is the preferred neuroimaging study for the evaluation of patients suspected of having this condition. Although most lesions occur in the lumbosacral region, presentations involving the cervical cord are reported. Split cord malformations are more common in females (female to male ratio of greater than 3:1) [Mathieu et al., 1982]. Urodynamic and electrophysiologic studies are abnormal in approximately 80 percent of patients [Kothari and Bauer, 1997].
Disorders of Secondary Neurulation
Fibrofatty Filum Terminale
The filum terminale is the nonfunctional continuation of the end of the spinal cord. It usually consists of fibrous tissue without functional nervous tissue. Although its embryologic origin is unclear, it probably represents the termination of the neural tube and its most caudal link to the rest of the embryonic tissues. The filum can be enlarged either with fibrous tissue only or with fat (Figure 22-16). If the filum is thicker than 2 mm, it is usually defined by radiologists as being “associated with tethered spinal cord.” A thickened or fatty filum terminale may be associated with a low conus and a spectrum of clinical findings, including bladder dysfunction, leg numbness and weakness, and scoliosis. In the presence of neurologic findings and a fatty filum, an untethering procedure may be considered.
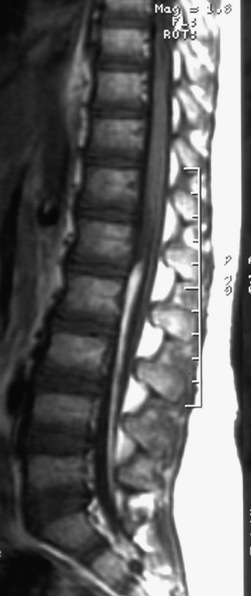
Sacral Agenesis
Sacral agenesis is defined as congenital absence of the whole or part of the sacrum (Davidoff et al., 1991; Towfighi and Housman, 1991). It has a heterogeneous etiology. In its classic form, often described as the caudal regression syndrome, malformations of most or all structures derived from the caudal region of the embryo, including the urogenital system, the hindgut, caudal spine, spinal cord, and the lower limbs, may be seen. Approximately 15 percent to 25 percent of mothers of these children have insulin-dependent diabetes mellitus.
An autosomal-dominant form of sacral agenesis, sometimes described as a partial sacral dysgenesis, ASP (anal atresia, sacral anomalies, presacral mass) syndrome, or Currarino’s syndrome, also exists. Typical features include hemisacrum with preservation of the first sacral vertebra [Lynch et al., 2000]. The defect usually takes the form of a sickle-shaped or crescent-shaped deformity of the sacrum; the hemisacrum or so-called scimitar sign is present in approximately 75 percent of patients (Figure 22-17). In its most severe presentation, the hemisacrum is associated with a presacral mass (anterior meningocele, enteric cyst, and/or presacral teratoma) and anorectal stenosis. Autosomal-dominant inheritance, localization to 7q36, and mutations in a homeobox gene, HLXB9, have been identified in several affected patients [Ross et al., 1998; Belloni et al., 2000]. Mutations in the coding sequence of HLXB9 have been identified in nearly all cases of familial Currarino’s syndrome and in approximately 30 percent of patients with sporadic Currarino’s syndrome.
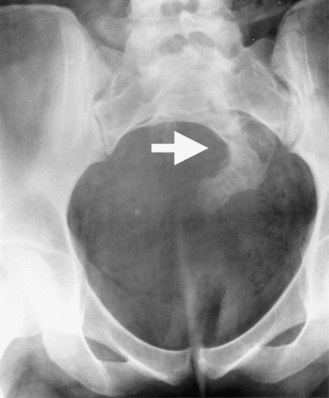
Fig. 22-17 Partial sacral dysgenesis, or Currarino’s syndrome.
Note the scimitar sacrum (arrow) with a right-sided defect.
(From Lynch SA et al. Autosomal dominant sacral agenesis: Currarino syndrome. J Med Genet 2000;37:561–566. Reproduced by permission from BMJ Publishing Group.)
The neurologic findings are similar to those in MMC, ranging from a minimal deficit to equinovarus deformity of the feet, to more extensive sensory and motor deficits of the lower extremities. The level of bone anomaly corresponds well to the level of weakness but not to sensory loss, as sensation usually is preserved. The caudal spinal cord often is truncated, dysraphic, and tethered [Estin and Cohen, 1995]. Most patients have neurogenic urinary tract and bowel impairment, visceral abnormalities, flattened buttocks, and prominent iliac crests. Constipation and perianal sepsis are common complaints. Ascending infection resulting in bacterial meningitis also has been reported.
Some patients experience progressive neurologic deficits, demonstrating that sacral agenesis is not always a static disability [O’Neill et al., 1995]. Slow deterioration of neurologic function may masquerade as an orthopedic or urologic problem, unless the potential for progressive lesions is appreciated. Dural sac stenosis, tethered spinal cord, diastematomyelia, and cauda equina lipomas and dermoids also have been associated with sacral agenesis. Although plain radiographs demonstrate the degree of sacral agenesis, CT or MRI is necessary to delineate the underlying spinal cord anomalies (Figure 22-18). Surgical intervention is indicated in patients with progressive neurologic deficits associated with occult spinal cord lesions [Pang, 1993]. Treatment is similar to that for patients with myelodysplasia, particularly for the problems of urinary incontinence, constipation, progressive urinary tract and renal dysfunction, and orthopedic abnormalities.
Sirenomelia is a condition typically characterized by fusion of the lower limbs, single umbilical artery, and severe malformations of the urogenital and lower gastrointestinal tracts. Controversy exists in the literature regarding whether sirenomelia occurs as a separate entity or represents the extreme form of caudal regression syndrome. The presence of two umbilical arteries, nonlethal renal anomalies, nonfused lower limbs, abdominal wall defects, and abnormalities of tracheoesophageal tree, neural tube, and heart serves to differentiate caudal regression syndrome from sirenomelia, however. In addition, caudal regression syndrome is strongly associated with maternal diabetes, whereas sirenomelia is typically due to bilateral renal agenesis and associated severe pulmonary hypoplasia [Das et al., 2002].
References
The complete list of references for this chapter is available online at www.expertconsult.com.
Aboitiz F., Montiel J. Co-option of signaling mechanisms from neural induction to telencephalic patterning. Rev Neurosci. 2007;18:311-342.
Adzick N.S., Sutton L.N., Crombleholme T.M., et al. Successful fetal surgery for spina bifida. Lancet. 1998;352:1675-1676.
Aldana P.R., Ragheb J., Sevald J., et al. Cerebrospinal fluid shunt complications after urological procedures in children with myelodysplasia. Neurosurgery. 2002;50:313-318. discussion 318–320
Aleksic S., Budzilovich G., Greco M.A., et al. Encephalocele (cerebellocele) in the Goldenhar-Gorlin syndrome. Eur J Pediatr. 1983;140:137-138.
Allen W.P., Stevenson R.E., Thompson S.J., et al. The impact of prenatal diagnosis on NTD surveillance. Prenat Diagn. 1996;16:531-535.
Altmann C.R., Brivanlou A.H. Neural patterning in the vertebrate embryo. Int Rev Cytol. 2001;203:447-482.
Anderson F.M. Occult spinal dysraphism: a series of 73 cases. Pediatrics. 1975;55:826-835.
Anderson N.G., Jordan S., MacFarlane M.R., et al. Diastematomyelia: diagnosis by prenatal sonography. Am J Roentgenol. 1994;163:911-914.
Anteby E.Y., Yagel S. Route of delivery of fetuses with structural anomalies. Eur J Obstet Gynecol Reprod Biol. 2003;106:5-9.
Arnett B. Arnold-Chiari malformation. Arch Neurol. 2003;60:898-900.
Aubry M.C., Aubry J.P., Dommergues M. Sonographic prenatal diagnosis of central nervous system abnormalities. Childs Nerv Syst. 2003;19:391-402.
Azais-Braesco V., Pascal G. Vitamin A in pregnancy: requirements and safety limits. Am J Clin Nutr. 2000;71:1325S-1333S.
Barrow J.R., Stadler H.S., Capecchi M.R. Roles of Hoxa1 and Hoxa2 in patterning the early hindbrain of the mouse. Development. 2000;127:933-944.
Beaudin A.E., Stover P.J. Folate-mediated one-carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Res C Embryo Today. 2007;81:183-203.
Beaudin A.E., Stover P.J. Insights into metabolic mechanisms underlying folate-responsive neural tube defects: a minireview. Birth Defects Res A Clin Mol Teratol. 2009;85:274-284.
Bell W.O., Sumner T.E., Volberg F.M. The significance of ventriculomegaly in the newborn with myelodysplasia. Childs Nerv Syst. 1987;3:239-241.
Belloni E., Martucciello G., Verderio D., et al. Involvement of the HLXB9 homeobox gene in Currarino syndrome. Am J Hum Genet. 2000;66:312-319.
Biggio J.R.Jr, Owen J., Wenstrom K.D., et al. Can prenatal ultrasound findings predict ambulatory status in fetuses with open spina bifida? Am J Obstet Gynecol. 2001;185:1016-1020.
Birnbacher R., Messerschmidt A.M., Pollak A.P. Diagnosis and prevention of neural tube defects. Curr Opin Urol. 2002;12:461-464.
Blom H.J., Shaw G.M., den Heijer M., et al. Neural tube defects and folate: case far from closed. Nat Rev Neurosci. 2006;7:724-731.
Botto L.D., Moore C.A., Khoury M.J., et al. Neural-tube defects. N Engl J Med. 1999;341:1509-1519.
Bowman R.M., McLone D.G., Grant J.A., et al. Spina bifida outcome: a 25-year prospective. Pediatr Neurosurg. 2001;34:114-120.
Brock D.J., Barron L., van Heyningen V. Prenatal diagnosis of neural-tube defects with a monoclonal antibody specific for acetylcholinesterase. Lancet. 1985;1:5-8.
Bruner J.P., Tulipan N., Reed G., et al. Intrauterine repair of spina bifida: preoperative predictors of shunt-dependent hydrocephalus. Am J Obstet Gynecol. 2004;190:1305-1312.
Bruner J.P., Tulipan N., Paschall R.L., et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
Buyse G., Verpoorten C., Vereecken R., et al. Treatment of neurogenic bladder dysfunction in infants and children with neurospinal dysraphism with clean intermittent (self)catheterisation and optimized intravesical oxybutynin hydrochloride therapy. Eur J Pediatr Surg. 1995;5(Suppl 1):31-34.
Byrd S.E., Darling C.F., McLone D.G. Developmental disorders of the pediatric spine. Radiol Clin North Am. 1991;29:711-752.
Cabrera R.M., Shaw G.M., Ballard J.L., et al. Autoantibodies to folate receptor during pregnancy and neural tube defect risk. J Reprod Immunol. 2008;79:85-92.
Cai C., Oakes W.J. Hindbrain herniation syndromes: the Chiari malformations (I and II). Semin Pediatr Neurol. 1997;4:179-191.
Caldarelli M., Di Rocco C., Colosimo C.Jr, et al. Surgical treatment of late neurological deterioration in children with myelodysplasia. Acta Neurochir (Wien). 1995;137:199-206.
Canick J.A., Kellner L.H., Bombard A.T. Prenatal screening for open neural tube defects. Clin Lab Med. 2003;23:385-394. ix
Carpenter E.M. Hox genes and spinal cord development. Dev Neurosci. 2002;24:24-34.
CDC. Spina bifida and anencephaly before and after folic acid mandate–United States, 1995-1996 and 1999-2000. MMWR Morb Mortal Wkly Rep. 2004;53:362-365.
Chan A., Robertson E.F., Haan E.A., et al. Prevalence of neural tube defects in South Australia, 1966-91: effectiveness and impact of prenatal diagnosis. BMJ. 1993;307:703-706.
Chan A., Robertson E.F., Haan E.A., et al. The sensitivity of ultrasound and serum alpha-fetoprotein in population-based antenatal screening for neural tube defects. South Australia 1986-1991. Br J Obstet Gynaecol. 1995;102:370-376.
Chao A., Wong A.M., Hsueh C., et al. Integration of imaging and pathological studies in Meckel-Gruber syndrome. Prenat Diagn. 2005;25:267-268.
Charney E.B., Rorke L.B., Sutton L.N., et al. Management of Chiari II complications in infants with myelomeningocele. J Pediatr. 1987;111:364-371.
Cheng Y., Sudarov A., Szulc K.U., et al. The Engrailed homeobox genes determine the different foliation patterns in the vermis and hemispheres of the mammalian cerebellum. Development. 2010;137:519-529.
Cochrane D.D., Wilson R.D., Steinbok P., et al. Prenatal spinal evaluation and functional outcome of patients born with myelomeningocele: information for improved prenatal counselling and outcome prediction. Fetal Diagn Ther. 1996;11:159-168.
Cohen M.M.Jr, Lemire R.J. Syndromes with cephaloceles. Teratology. 1982;25:161-172.
Colas J.F., Schoenwolf G.C. Towards a cellular and molecular understanding of neurulation. Dev Dyn. 2001;221:117-145.
Cowan W.M., Jessell T.M., Zipursky S.L. Molecular and cellular approaches to molecular medicine. New York: Oxford Univesity Press; 1997.
Crandall B.F., Chua C. Detecting neural tube defects by amniocentesis between 11 and 15 weeks’ gestation. Prenat Diagn. 1995;15:339-343.
Czeizel A.E., Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832-1835.
Daly L.E., Kirke P.N., Molloy A., et al. Folate levels and neural tube defects. Implications for prevention. JAMA. 1995;274:1698-1702.
Dansky L.V., Finnell R.H. Parental epilepsy, anticonvulsant drugs, and reproductive outcome: epidemiologic and experimental findings spanning three decades; 2: Human studies. Reprod Toxicol. 1991;5:301-335.
Das B.B., Rajegowda B.K., Bainbridge R., et al. Caudal regression syndrome versus sirenomelia: a case report. J Perinatol. 2002;22:168-170.
David D.J. Cephaloceles: classification, pathology, and management – a review. J Craniofac Surg. 1993;4:192-202.
Davidoff A.M., Thompson C.V., Grimm J.M., et al. Occult spinal dysraphism in patients with anal agenesis. J Pediatr Surg. 1991;26:1001-1005.
De Robertis E.M., Kuroda H. Dorsal-ventral patterning and neural induction in Xenopus embryos. Annu Rev Cell Dev Biol. 2004;20:285-308.
Dias M.S., Pang D. Split cord malformations. Neurosurg Clin N Am. 1995;6:339-358.
Diez del Corral R., Storey K.G. Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays. 2004;26:857-869.
Docherty T.B., Herbaut A.G., Sedgwick E.M. Brainstem auditory evoked potential abnormalities in myelomeningocoele in the older child. J Neurol Neurosurg Psychiatry. 1987;50:1318-1322.
Drugan A., Weissman A., Evans M.I. Screening for neural tube defects. Clin Perinatol. 2001;28:279-287. vii
Dyste G.N., Menezes A.H., VanGilder J.C. Symptomatic Chiari malformations. An analysis of presentation, management, and long-term outcome. J Neurosurg. 1989;71:159-168.
Elwood M., Little J. Distribution by sex. In: Elwood J.M., Little J., Elwood J.H., editors. Epidemiology and Control of Neural Tube Defects. New York: Oxford University Press; 1992:306-324.
Eriksson U.J., Cederberg J., Wentzel P. Congenital malformations in offspring of diabetic mothers – animal and human studies. Rev Endocr Metab Disord. 2003;4:79-93.
Essien F.B., Wannberg S.L. Methionine but not folinic acid or vitamin B-12 alters the frequency of neural tube defects in Axd mutant mice. J Nutr. 1993;123:27-34.
Estin D., Cohen A.R. Caudal agenesis and associated caudal spinal cord malformations. Neurosurg Clin N Am. 1995;6:377-391.
Feltes C.H., Fountas K.N., Dimopoulos V.G., et al. Cervical meningocele in association with spinal abnormalities. Childs Nerv Syst. 2004;20:357-361.
Fisher N.L., Smith D.W. Occipital encephalocele and early gestational hyperthermia. Pediatrics. 1981;68:480-483.
Flynn J.M., Herrera-Soto J.A., Ramirez N.F., et al. Clubfoot release in myelodysplasia. J Pediatr Orthop B. 2004;13:259-262.
Friede R.L. Developmental Neuropathology, ed 2. NY: Springer-Verlag; 1989.
Gelineau-van Waes J., Finnell R.H. Genetics of neural tube defects. Semin Pediatr Neurol. 2001;8:160-164.
Gilbert J.N., Jones K.L., Rorke L.B., et al. Central nervous system anomalies associated with meningomyelocele, hydrocephalus, and the Arnold-Chiari malformation: reappraisal of theories regarding the pathogenesis of posterior neural tube closure defects. Neurosurgery. 1986;18:559-564.
Golden J.A., Chernoff G.F. Multiple sites of anterior neural tube closure in humans: evidence from anterior neural tube defects (anencephaly). Pediatrics. 1995;95:506-510.
Graham D., Johnson T.R.Jr, Winn K., et al. The role of sonography in the prenatal diagnosis and management of encephalocele. J Ultrasound Med. 1982;1:111-115.
Gray J.D., Ross M.E. Mechanistic insights into folate supplementation from Crooked tail and other NTD-prone mutant mice. Birth Defects Res A Clin Mol Teratol. 2009;85:314-321.
Greene N.D., Copp A.J. Inositol prevents folate-resistant neural tube defects in the mouse. Nat Med. 1997;3:60-66.
Greene N.D., Copp A.J. Development of the vertebrate central nervous system: formation of the neural tube. Prenat Diagn. 2009;29:303-311.
Greene N.D., Stanier P., Copp A.J. Genetics of human neural tube defects. Hum Mol Genet. 2009;18:R113-R129.
Guggisberg D., Hadj-Rabia S., Viney C., et al. Skin markers of occult spinal dysraphism in children: a review of 54 cases. Arch Dermatol. 2004;140:1109-1115.
Harland R. Neural induction. Curr Opin Genet Dev. 2000;10:357-362.
Harley E.H. Pediatric congenital nasal masses. Ear Nose Throat J. 1991;70:28-32.
Harris M.J. Insights into prevention of human neural tube defects by folic acid arising from consideration of mouse mutants. Birth Defects Res A Clin Mol Teratol. 2009;85:331-339.
Harris M.J., Juriloff D.M. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2007;79:187-210.
Heeg M., Broughton N.S., Menelaus M.B. Bilateral dislocation of the hip in spina bifida: a long-term follow-up study. J Pediatr Orthop. 1998;18:434-436.
Hendricks K.A., Nuno O.M., Suarez L., et al. Effects of hyperinsulinemia and obesity on risk of neural tube defects among Mexican Americans. Epidemiology. 2001;12:630-635.
Hockley A.D., Goldin J.H., Wake M.J. Management of anterior encephalocele. Childs Nerv Syst. 1990;6:444-446.
Holmes L.B., Driscoll S.G., Atkins L. Etiologic heterogeneity of neural-tube defects. N Engl J Med. 1976;294:365-369.
Honein M.A., Paulozzi L.J., Mathews T.J., et al. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA. 2001;285:2981-2986.
Iskandar B.J., Oakes W.J., McLaughlin C., et al. Terminal syringohydromyelia and occult spinal dysraphism. J Neurosurg. 1994;81:513-519.
Janerich D.T., Piper J. Shifting genetic patterns in anencephaly and spina bifida. J Med Genet. 1978;15:101-105.
Johnson M.P., Sutton L.N., Rintoul N., et al. Fetal myelomeningocele repair: short-term clinical outcomes. Am J Obstet Gynecol. 2003;189:482-487.
Joy H.M., Barker C.S., Small J.H., et al. Trans-sphenoidal encephalocele in association with Dandy-Walker complex and cardiovascular anomalies. Neuroradiology. 2001;43:45-48.
Just M., Schwarz M., Ludwig B., et al. Cerebral and spinal MR-findings in patients with postrepair myelomeningocele. Pediatr Radiol. 1990;20:262-266.
Kibar Z., Torban E., McDearmid J.R., et al. Mutations in VANGL1 associated with neural-tube defects. N Engl J Med. 2007;356:1432-1437.
Knab K., Langhans B., Behrens R., et al. The neuropathic bowel in spina bifida – a cross-sectional study in 226 patients. Eur J Pediatr Surg. 2001;11(Suppl 1):S41-S42.
Koenig S.B., Naidich T.P., Lissner G. The morning glory syndrome associated with sphenoidal encephalocele. Ophthalmology. 1982;89:1368-1373.
Konz K.R., Chia J.K., Kurup V.P., et al. Comparison of latex hypersensitivity among patients with neurologic defects. J Allergy Clin Immunol. 1995;95:950-954.
Kooper A.J., de Bruijn D., van Ravenwaaij-Arts C.M., et al. Fetal anomaly scan potentially will replace routine AFAFP assays for the detection of neural tube defects. Prenat Diagn. 2007;27:29-33.
Koren G., Kennedy D. Safe use of valproic acid during pregnancy. Can Fam Physician. 1999;45:1451-1453.
Kothari M.J., Bauer S.B. Urodynamic and neurophysiologic evaluation of patients with diastematomyelia. J Child Neurol. 1997;12:97-100.
Kriss V.M., Kriss T.C., Desai N.S., et al. Occult spinal dysraphism in the infant. Clin Pediatr (Phila). 1995;34:650-654.
Lammer E.J., Chen D.T., Hoar R.M., et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837-841.
Lammer E.J., Sever L.E., Oakley G.P.Jr. Teratogen update: valproic acid. Teratology. 1987;35:465-473.
Lei Y.P., Zhang T., Li H., et al. VANGL2 mutations in human cranial neural-tube defects. N Engl J Med. 2010;362:2232-2235.
Lennon C.A., Gray D.L. Sensitivity and specificity of ultrasound for the detection of neural tube and ventral wall defects in a high-risk population. Obstet Gynecol. 1999;94:562-566.
Lieblich J.M., Rosen S.E., Guyda H., et al. The syndrome of basal encephalocele and hypothalamic-pituitary dysfunction. Ann Intern Med. 1978;89:910-916.
Liptak G.S., Bloss J.W., Briskin H., et al. The management of children with spinal dysraphism. J Child Neurol. 1988;3:3-20.
Little J., Elwood J.M. Geographical variation. In: Elwood J.M., Little J., EJ H., editors. Epidemiology and Control of Neural Tube Defects. New York: Oxford University Press, Inc; 1992:96-146.
Loft A.G., Hogdall E., Larsen S.O., et al. A comparison of amniotic fluid alpha-fetoprotein and acetylcholinesterase in the prenatal diagnosis of open neural tube defects and anterior abdominal wall defects. Prenat Diagn. 1993;13:93-109.
Lorber J., Schofield J.K. The prognosis of occipital encephalocele. Z Kinderchir Grenzgeb. 1979;28:347-351.
Lynch S.A., Wang Y., Strachan T., et al. Autosomal dominant sacral agenesis: Currarino syndrome. J Med Genet. 2000;37:561-566.
Martinez-Lage J.F., Poza M., Sola J., et al. The child with a cephalocele: etiology, neuroimaging, and outcome. Childs Nerv Syst. 1996;12:540-550.
Matalon S., Schechtman S., Goldzweig G., et al. The teratogenic effect of carbamazepine: a meta-analysis of 1255 exposures. Reprod Toxicol. 2002;16:9-17.
Mathieu J.P., Decarie M., Dube J., et al. [Diastematomyelia. Study of 69 cases (author’s transl)]. Chir Pediatr. 1982;23:29-35.
McComb J.G. Spinal and cranial neural tube defects. Semin Pediatr Neurol. 1997;4:156-166.
McDonald C.M. Rehabilitation of children with spinal dysraphism. Neurosurg Clin N Am. 1995;6:393-412.
McDonnell G.V., McCann J.P. Why do adults with spina bifida and hydrocephalus die? A clinic-based study. Eur J Pediatr Surg. 2000;10(Suppl 1):31-32.
McLone D.G., Dias M.S. The Chiari II malformation: cause and impact. Childs Nerv Syst. 2003;19:540-550.
Mecke S., Passarge E. Encephalocele, polycystic kidneys, and polydactyly as an autosomal recessive trait simulating certain other disorders: the Meckel syndrome. Ann Genet. 1971;14:97-103.
Meuli M., Meuli-Simmen C., Hutchins G.M., et al. In utero surgery rescues neurological function at birth in sheep with spina bifida. Nat Med. 1995;1:342-347.
Meyer-Heim A.D., Klein A., Boltshauser E. Cervical myelomeningocele – follow-up of five patients. Eur J Paediatr Neurol. 2003;7:407-412.
Michejda M., Bacher J. Functional and anatomic recovery in the monkey brain following excision of fetal encephalocele. Pediatr Neurosci. 1985;12:90-95.
Milhorat T.H., Johnson W.D., Miller J.I., et al. Surgical treatment of syringomyelia based on magnetic resonance imaging criteria. Neurosurgery. 1992;31:231-244. discussion 244–235
Miller A., Guille J.T., Bowen J.R. Evaluation and treatment of diastematomyelia. J Bone Joint Surg Am. 1993;75:1308-1317.
Miller A.L. The methylation, neurotransmitter, and antioxidant connections between folate and depression. Altern Med Rev. 2008;13:216-226.
Mills J.L., Signore C. Neural tube defect rates before and after food fortification with folic acid. Birth Defects Res A Clin Mol Teratol. 2004;70:844-845.
Molloy A.M., Quadros E.V., Sequeira J.M., et al. Lack of association between folate-receptor autoantibodies and neural-tube defects. N Engl J Med. 2009;361:152-160.
Moore L.L., Bradlee M.L., Singer M.R., et al. Folate intake and the risk of neural tube defects: an estimation of dose-response. Epidemiology. 2003;14:200-205.
MRC V. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet. 1991;338:131-137.
MTFA. The infant with anencephaly. The Medical Task Force on Anencephaly. N Engl J Med. 1990;322:669-674.
Neumann P.E., Frankel W.N., Letts V.A., et al. Multifactorial inheritance of neural tube defects: localization of the major gene and recognition of modifiers in ct mutant mice. Nat Genet. 1994;6:357-362.
Norman M.G., McGillvary B.C., Kalousek D.K., et al. Congenital Malformations of the Brain: Pathological, Embryological, Clinical, Radiological and Genetic Aspects. New York: Oxford University Press; 1995.
Northrup H., Volcik K.A. Spina bifida and other neural tube defects. Curr Probl Pediatr. 2000;30:313-332.
O’Neill O.R., Piatt J.H.Jr, Mitchell P., et al. Agenesis and dysgenesis of the sacrum: neurosurgical implications. Pediatr Neurosurg. 1995;22:20-28.
Oakley G.P.Jr. Folic acid-preventable spina bifida: a good start but much to be done. Am J Prev Med. 2010;38:569-570.
Oi S., Matsumoto S. A proposed grading and scoring system for spina bifida: Spina Bifida Neurological Scale (SBNS). Childs Nerv Syst. 1992;8:337-342.
Ornoy A. Neuroteratogens in man: an overview with special emphasis on the teratogenicity of antiepileptic drugs in pregnancy. Reprod Toxicol. 2006;22:214-226.
Osaka K., Tanimura T., Hirayama A., et al. Myelomeningocele before birth. J Neurosurg. 1978;49:711-724.
Paek B.W., Farmer D.L., Wilkinson C.C., et al. Hindbrain herniation develops in surgically created myelomeningocele but is absent after repair in fetal lambs. Am J Obstet Gynecol. 2000;183:1119-1123.
Pagon R.A., Chandler J.W., Collie W.R., et al. Hydrocephalus, agyria, retinal dysplasia, encephalocele (HARD +/− E) syndrome: an autosomal recessive condition. Birth Defects Orig Artic Ser. 1978;14:233-241.
Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-778. discussion 778–759
Pang D., Dias M.S., Ahab-Barmada M. Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
Peach B. Arnold-Chiari malformation: Anatomic features of 20 cases. Arch Neurol. 1965;12:613-621.
Petersen M.C., Wolraich M., Sherbondy A., et al. Abnormalities in control of ventilation in newborn infants with myelomeningocele. J Pediatr. 1995;126:1011-1015.
Pitkin R.M. Folate and neural tube defects. Am J Clin Nutr. 2007;85:285S-288S.
Pollack I.F., Losken H.W., Biglan A.W. Incidence of increased intracranial pressure after early surgical treatment of syndromic craniosynostosis. Pediatr Neurosurg. 1996;24:202-209.
Pollack I.F., Pang D., Albright A.L., et al. Outcome following hindbrain decompression of symptomatic Chiari malformations in children previously treated with myelomeningocele closure and shunts. J Neurosurg. 1992;77:881-888.
Pyrgaki C., Trainor P., Hadjantonakis A.K., et al. Dynamic imaging of mammalian neural tube closure. Dev Biol. 2010.
Rahbar R., Resto V.A., Robson C.D., et al. Nasal glioma and encephalocele: diagnosis and management. Laryngoscope. 2003;113:2069-2077.
Rintoul N.E., Sutton L.N., Hubbard A.M., et al. A new look at myelomeningoceles: functional level, vertebral level, shunting, and the implications for fetal intervention. Pediatrics. 2002;109:409-413.
Rosa F.W., Wilk A.L., Kelsey F.O. Teratogen update: vitamin A congeners. Teratology. 1986;33:355-364.
Ross A.J., Ruiz-Perez V., Wang Y., et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358-361.
Ross M.E. Gene-environment interactions: folate metabolism and the embryonicnervous system. Wiley Interdiscip Rev Syst Biol Med. 2010.
Rothenberg S.P., da Costa M.P., Sequeira J.M., et al. Autoantibodies against folate receptors in women with a pregnancy complicated by a neural-tube defect. N Engl J Med. 2004;350:134-142.
Sadler T.W. Langman’s Medical Embryology. Philadelphia: Lippincott, Williams & Wilkins, 2004;65-115.
Sadler T.W. Embryology of neural tube development. Am J Med Genet C Semin Med Genet. 2005;135C:2-8.
Sarnat H.B. Cerebral dysgenesis: embryology and clinical expression. New York: Oxford University Press; 1992.
Schneidau T., Franco I., Zebold K., et al. Selective sacral rhizotomy for the management of neurogenic bladders in spina bifida patients: long-term followup. J Urol. 1995;154:766-768.
Schoenwolf G.C. Microsurgical analyses of avian neurulation: separation of medial and lateral tissues. J Comp Neurol. 1988;276:498-507.
Schoenwolf G.C., Powers M.L. Shaping of the chick neuroepithelium during primary and secondary neurulation: role of cell elongation. Anat Rec. 1987;218:182-195.
Sepulveda W., Dezerega V., Be C. First-trimester sonographic diagnosis of holoprosencephaly: value of the “butterfly” sign. J Ultrasound Med. 2004;23:761-765. quiz 766–767
Sertie A.L., Quimby M., Moreira E.S., et al. A gene which causes severe ocular alterations and occipital encephalocele (Knobloch syndrome) is mapped to 21q22.3. Hum Mol Genet. 1996;5:843-847.
Shaw G.M., Jensvold N.G., Wasserman C.R., et al. Epidemiologic characteristics of phenotypically distinct neural tube defects among 0.7 million California births, 1983–1987. Teratology. 1994;49:143-149.
Shaw G.M., Schaffer D., Velie E.M., et al. Periconceptional vitamin use, dietary folate, and the occurrence of neural tube defects. Epidemiology. 1995;6:219-226.
Shaw G.M., Quach T., Nelson V., et al. Neural tube defects associated with maternal periconceptional dietary intake of simple sugars and glycemic index. Am J Clin Nutr. 2003;78:972-978.
Shenefelt R.E. Morphogenesis of malformations in hamsters caused by retinoic acid: relation to dose and stage at treatment. Teratology. 1972;5:103-118.
Siffel C., Wong L.Y., Olney R.S., et al. Survival of infants diagnosed with encephalocele in Atlanta, 1979-98. Paediatr Perinat Epidemiol. 2003;17:40-48.
Silveri M., Capitanucci M.L., Capozza N., et al. Occult spinal dysraphism: neurogenic voiding dysfunction and long-term urologic follow-up. Pediatr Surg Int. 1997;12:148-150.
Simeoni J., Guys J.M., Mollard P., et al. Artificial urinary sphincter implantation for neurogenic bladder: a multi-institutional study in 107 children. Br J Urol. 1996;78:287-293.
Singhal B., Mathew K.M. Factors affecting mortality and morbidity in adult spina bifida. Eur J Pediatr Surg. 1999;9(Suppl 1):31-32.
Smith A.D., Kim Y.I., Refsum H. Is folic acid good for everyone? Am J Clin Nutr. 2008;87:517-533.
Smithells R.W., Nevin N.C., Seller M.J., et al. Further experience of vitamin supplementation for prevention of neural tube defect recurrences. Lancet. 1983;1:1027-1031.
Smithells R.W., Sheppard S., Schorah C.J. Vitamin dificiencies and neural tube defects. Arch Dis Child. 1976;51:944-950.
Snodgrass W.T., Adams R. Initial urologic management of myelomeningocele. Urol Clin North Am. 2004;31:427-434. viii
Sonigo-Cohen P., Schmit P., Zerah M., et al. Prenatal diagnosis of diastematomyelia. Childs Nerv Syst. 2003;19:555-560.
Stein S.C., Schut L. Hydrocephalus in myelomeningocele. Childs Brain. 1979;5:413-419.
Steinbok P. Dysraphic lesions of the cervical spinal cord. Neurosurg Clin N Am. 1995;6:367-376.
Steinbok P., Cochrane D.D. Cervical spinal dysraphism. J Neurosurg. 1995;83:569-570.
Steinbok P., Irvine B., Cochrane D.D., et al. Long-term outcome and complications of children born with meningomyelocele. Childs Nerv Syst. 1992;8:92-96.
Stevenson K.L. Chiari Type II malformation: past, present, and future. Neurosurg Focus. 2004;16:E5.
Stevenson R.E., Allen W.P., Pai G.S., et al. Decline in prevalence of neural tube defects in a high-risk region of the United States. Pediatrics. 2000;106:677-683.
Stone A.R. Neurourologic evaluation and urologic management of spinal dysraphism. Neurosurg Clin N Am. 1995;6:269-277.
Takahashi H., Liu F.C. Genetic patterning of the mammalian telencephalon by morphogenetic molecules and transcription factors. Birth Defects Res C Embryo Today. 2006;78:256-266.
Talwar D., Baldwin M.A., Horbatt C.I. Epilepsy in children with meningomyelocele. Pediatr Neurol. 1995;13:29-32.
Tibbles L., Wiley M.J. A comparative study of the effects of retinoic acid given during the critical period for inducing spina bifida in mice and hamsters. Teratology. 1988;37:113-125.
Towfighi J., Housman C. Spinal cord abnormalities in caudal regression syndrome. Acta Neuropathol. 1991;81:458-466.
Tubbs R.S., Webb D.B., Oakes W.J. Persistent syringomyelia following pediatric Chiari I decompression: radiological and surgical findings. J Neurosurg. 2004;100:460-464.
Tulipan N., Bruner J.P. Myelomeningocele repair in utero: a report of three cases. Pediatr Neurosurg. 1998;28:177-180.
Tulipan N., Bruner J.P. Fetal surgery for spina bifida. Lancet. 1999;353:406-407.
Tulipan N., Hernanz-Schulman M., Bruner J.P. Reduced hindbrain herniation after intrauterine myelomeningocele repair: A report of four cases. Pediatr Neurosurg. 1998;29:274-278.
Ulloa F., Marti E. Wnt won the war: antagonistic role of Wnt over Shh controls dorso-ventral patterning of the vertebrate neural tube. Dev Dyn. 2010;239:69-76.
Vachha B., Adams R. Language differences in young children with myelomeningocele and shunted hydrocephalus. Pediatr Neurosurg. 2003;39:184-189.
Van Allen M.I., Kalousek D.K., Chernoff G.F., et al. Evidence for multi-site closure of the neural tube in humans. Am J Med Genet. 1993;47:723-743.
Van Esch H., Poirier K., de Zegher F., et al. ARX mutation in a boy with transsphenoidal encephalocele and hypopituitarism. Clin Genet. 2004;65:503-505.
Wald N. Folic acid and the prevention of neural tube defects. Ann N Y Acad Sci. 1993;678:112-129.
Wald N., Cuckle H., Nanchahal K. Amniotic fluid acetylcholinesterase measurement in the prenatal diagnosis of open neural tube defects. Second report of the Collaborative Acetylcholinesterase Study. Prenat Diagn. 1989;9:813-829.
Wald N.J., Law M.R., Morris J.K., et al. Quantifying the effect of folic acid. Lancet. 2001;358:2069-2073.
Wallingford J.B., Fraser S.E., Harland R.M. Convergent extension: the molecular control of polarized cell movement during embryonic development. Dev Cell. 2002;2:695-706.
Wallingford J.B., Harland R.M. Xenopus Dishevelled signaling regulates both neural and mesodermal convergent extension: parallel forces elongating the body axis. Development. 2001;128:2581-2592.
Walters J., Ashwal S., Masek T. Anencephaly: where do we now stand? Semin Neurol. 1997;17:249-255.
Wang P., Chang F.M., Chang C.H., et al. Prenatal diagnosis of Joubert syndrome complicated with encephalocele using two-dimensional and three-dimensional ultrasound. Ultrasound Obstet Gynecol. 1999;14:360-362.
Waterson J.R., DiPietro M.A., Barr M. Apert syndrome with frontonasal encephalocele. Am J Med Genet. 1985;21:777-783.
Watson W.J., Chescheir N.C., Katz V.L., et al. The role of ultrasound in evaluation of patients with elevated maternal serum alpha-fetoprotein: a review. Obstet Gynecol. 1991;78:123-128.
Wentzel P., Wentzel C.R., Gareskog M.B., et al. Induction of embryonic dysmorphogenesis by high glucose concentration, disturbed inositol metabolism, and inhibited protein kinase C activity. Teratology. 2001;63:193-201.
Werler M.M., Louik C., Shapiro S., et al. Prepregnant weight in relation to risk of neural tube defects. JAMA. 1996;275:1089-1092.
Wilson C., Aftimos S., Pereira A., et al. Report of two sibs with Knobloch syndrome (encephalocoele and vitreoretinal degeneration) and other anomalies. Am J Med Genet. 1998;78:286-290.
Wininger S.J., Donnenfeld A.E. Syndromes identified in fetuses with prenatally diagnosed cephaloceles. Prenat Diagn. 1994;14:839-843.
Wong L.Y., Paulozzi L.J. Survival of infants with spina bifida: a population study, 1979–94. Paediatr Perinat Epidemiol. 2001;15:374-378.
Yasuda Y., Konishi H., Matsuo T., et al. Aberrant differentiation of neuroepithelial cells in developing mouse brains subsequent to retinoic acid exposure in utero. Am J Anat. 1989;186:271-284.
Ybot-Gonzalez P., Savery D., Gerrelli D., et al. Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development. 2007;134:789-799.
Zhu H., Kartiko S., Finnell R.H. Importance of gene-environment interactions in the etiology of selected birth defects. Clin Genet. 2009;75:409-423.
Zlotogora J., Amitai Y., Kaluski D.N., et al. Surveillance of neural tube defects in Israel. Isr Med Assoc J. 2002;4:1111-1114.
