Chapter 330 Disorders of Malabsorption
All disorders of malabsorption are associated with diminished intestinal absorption of one or more dietary nutrients. Malabsorption can result from a defect in the nutrient digestion in the intestinal lumen or from defective mucosal absorption. Malabsorption disorders can be categorized into generalized mucosal abnormalities usually resulting in malabsorption of multiple nutrients (Table 330-1) or malabsorption of specific nutrients (carbohydrate, fat, protein, vitamins, minerals, and trace elements) (Table 330-2). Almost all the malabsorption disorders are accompanied by chronic diarrhea (Chapter 333).
Table 330-1 MALABSORPTION DISORDERS AND CHRONIC DIARRHEA ASSOCIATED WITH GENERALIZED MUCOSAL DEFECT
IgA, immunoglobulin A.
Table 330-2 CLASSIFICATION OF MALABSORPTION DISORDERS AND CHRONIC DIARRHEA BASED ON THE PREDOMINANT NUTRIENT MALABSORBED
CARBOHYDRATE MALABSORPTION
FAT MALABSORPTION
AMINO ACID MALABSORPTION
MINERAL AND VITAMIN MALABSORPTION
DRUG INDUCED
Clinical Approach
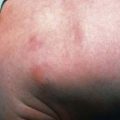
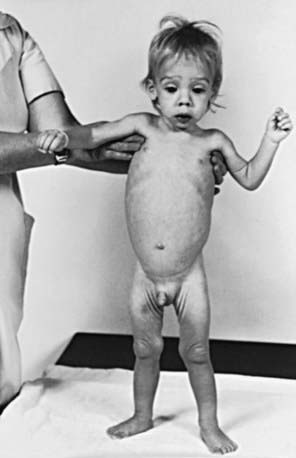
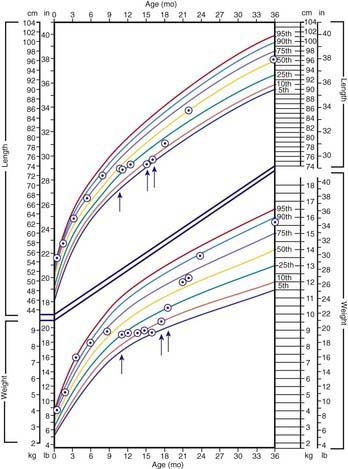
The clinical features depend on the extent and type of the malabsorbed nutrient. The common presenting features, especially in toddlers with malabsorption, are diarrhea, abdominal distention, and failure to gain weight, with a fall in growth chart percentiles. Physical findings include muscle wasting and the disappearance of the subcutaneous fat, with subsequent loose skinfolds (Fig. 330-1). The nutritional consequences of malabsorption are more dramatic in toddlers because of the limited energy reserves and higher proportion of calorie intake being used for weight gain and linear growth. In older children, malnutrition can result in growth retardation, as is commonly seen in children with late diagnosis of celiac disease. If malabsorption is untreated, linear growth slows, and with prolonged malnutrition, death can follow (Chapter 43). This extreme outcome is usually restricted to children living in the developing world, where resources to provide enteral and parenteral nutrition support may be limited. Specific findings on examination can guide toward a specific disorder; edema is usually associated with protein-losing enteropathy, digital clubbing with cystic fibrosis and celiac disease, perianal excoriation and gaseous abdominal distention with carbohydrate malabsorption, perianal and circumoral rash with acrodermatitis enteropathica, abnormal hair with Menkes syndrome, and the typical facial features diagnostic of the Johanson-Blizzard syndrome.
The nutritional assessment is an important part of clinical evaluation in children with malabsorptive disorders (Chapter 41). Long-term calcium and vitamin D malabsorption can lead to reduced bone mineral density and metabolic bone disease, with increased risk of bone fractures. Vitamin K malabsorption, irrespective of the underlying mechanism (fat malabsorption, mucosal atrophy), can result in coagulopathy. Severe protein-losing enteropathy is often associated with malabsorption syndromes (celiac disease, intestinal lymphangiectasia) and causes hypoalbuminemia and edema. Other nutrient deficiencies include iron malabsorption causing microcytic anemia and low reticulocyte count, low serum folate levels in conditions associated with mucosal atrophy, and low serum vitamin A and vitamin E concentrations in fat malabsorption.
Clinical history alone might not be sufficient to make a specific diagnosis, but it can direct the pediatrician toward a more structured and rational investigative approach. Diarrhea is the main clinical expression of malabsorption. Onset of diarrhea in early infancy suggests a congenital defect (Table 330-3). In secretory diarrhea due to disorders such as congenital chloride diarrhea and microvillus inclusion disease, the stool is watery and voluminous and can be mistaken for urine. Onset of symptoms after introduction of a particular food into a child’s diet can provide diagnostic clues, such as with sucrose in sucrase-isomaltase deficiency. The nature of the diarrhea may be helpful: explosive watery diarrhea suggests carbohydrate malabsorption; loose, bulky stools are associated with celiac disease; and pasty and yellowish offensive stools suggest an exocrine pancreatic insufficiency. Stool color is usually not helpful; green stool with undigested “peas and carrots” can suggest rapid intestinal transit in toddler’s diarrhea, which is a self-limiting condition unassociated with failure to thrive.
Table 330-3 DIARRHEAL DISEASES APPEARING IN THE NEONATAL PERIOD
| CONDITION | CLINICAL FEATURES |
|---|---|
| Microvillus inclusion disease | Secretory watery diarrhea |
| Tufting enteropathy | Secretory watery diarrhea |
| Congenital glucose-galactose malabsorption | Acidic diarrhea |
| Congenital lactase deficiency | Acidic diarrhea |
| Congenital chloride diarrhea | Hydramnion, secretory watery diarrhea Metabolic alkalosis |
| Congenital defective jejunal Na+/H+ exchange | Hydramnion, secretory watery diarrhea |
| Congenital bile acid malabsorption | Steatorrhea |
| Congenital enterokinase deficiency | Failure to thrive, edema |
| Congenital trypsinogen deficiency | Failure to thrive, edema |
| Congenital lipase and/or co-lipase deficiency | Failure to thrive, oily stool |
| Enteric anendocrinosis (NEUROG 3 mutation) | Hyperchloremic acidosis, failure to thrive |
Adapted from Schmitz J: Maldigestion and malabsorption. In Walker WA, Durie PR, Hamilton JR, et al, editors: Pediatric gastrointestinal disease, ed 3, Hamilton, Ontario, 2000, BC Decker, p 55.
330.1 Evaluation of Children with Suspected Intestinal Malabsorption
Michael J. Lentze and David Branski
Investigations for Fat Malabsorption
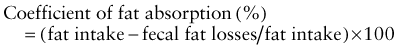
where fat intake and fat losses are in grams. Because fecal fat balance studies are cumbersome, expensive, and unpleasant to perform, simpler tests are often preferred. Among these stool tests, the acid steatocrit test is the most reliable. When bile acid deficiency is suspected of being the cause of fat malabsorption, the evaluation of bile acid levels in duodenal fluid aspirate may be useful.
Investigations for Exocrine Pancreatic Function (Fig. 330-2)
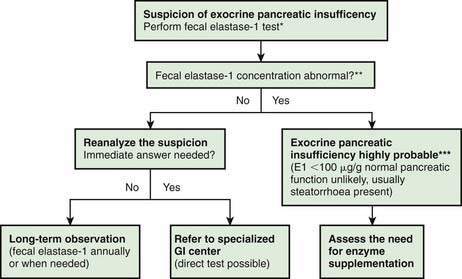
The gold standard test for exocrine pancreatic function is direct analysis of duodenal aspirate for volume, bicarbonate, trypsin and lipase upon secretin and pancreozymin/cholecysto-kinin stimulation. This involves duodenal intubation, and only a few centers perform this test (Chapter 340).
330.2 Gluten-Sensitive Enteropathy (Celiac Disease)
Etiology and Epidemiology
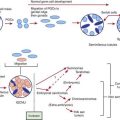
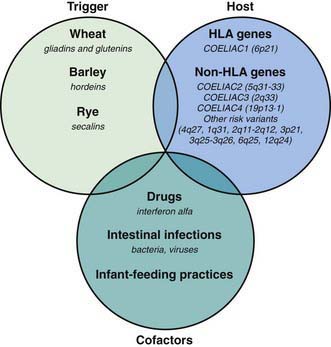
Celiac disease is a common disorder (1% prevalence of biopsy-proven disease). It is thought to be rare in Central Africa and East Asia. Environmental factors might affect the risk of developing celiac disease or the timing of its presentation. Prolonged breastfeeding has been associated with a reduced incidence of symptomatic disease. Less clear is the effect of the time of gluten introduction in the infant diet; the ingestion of increased amounts of gluten in the 1st year of life can increase the incidence. Infectious agents have been hypothesized to play a role because frequent rotavirus infections are associated with an increased risk. It is plausible that the contact with gliadin at a time when there is ongoing intestinal inflammation, altered intestinal permeability, and enhanced antigen presentation can increase the risk of developing the disease, at least in a subset of persons (Fig. 330-3).
Clinical Presentation and Associated Disorders
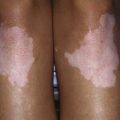
Clinical features of celiac disease vary considerably (Table 330-4). Intestinal symptoms are common in children whose disease is diagnosed within the 1st 2 years of life; failure to thrive, chronic diarrhea, vomiting, abdominal distention, muscle wasting, anorexia, and irritability are present in most cases (see Fig. 330-1). Occasionally there is constipation, rectal prolapse, or intussusception. As the age at presentation of the disease shifts to later in childhood, and with the more liberal use of serologic screening tests, extraintestinal manifestations and associated disorders, without any accompanying digestive symptoms, have increasingly become recognized, affecting almost all organs (Table 330-5).
Table 330-4 SOME CLINICAL MANIFESTATIONS OF CELIAC DISEASE IN CHILDREN AND ADOLESCENTS
| SYSTEM | MANIFESTATION | (POSSIBLE) CAUSE |
|---|---|---|
| Gastrointestinal | Diarrhea Distended abdomen Vomiting Anorexia Weight loss Failure to thrive Aphthous stomatitis |
Atrophy of the small bowel mucosa Malabsorption |
| Hematologic | Anemia | Iron malabsorption |
| Skeletal | Rickets Osteoporosis Enamel hypoplasia of the teeth |
Calcium/vitamin D malabsorption |
| Muscular | Atrophy | Malnutrition |
| Neurologic | Peripheral neuropathy Epilepsy Irritability |
Thiamine/vitamin B12 deficiency |
| Endocrinologic | Short stature Pubertas tarda Secondary hyperparathyroidism |
Malnutrition Calcium/vitamin D malabsorption |
| Dermatologic | Dermatitis herpetiformis Alopecia areata Erythema nodosum |
Autoimmunity |
| Respiratory | Idiopathic pulmonary hemosiderosis |
Adapted from Mearin ML: Celiac disease among children and adolescents, Curr Prob Pediatr Adolesc Health Care 37:81–112, 2007.
Table 330-5 RISK GROUPS FOR CELIAC DISEASE CASE-FINDING
IgA, immunoglobulin A.
Modified from Di Sabatino A, Corazza GR: Coeliac disease, Lancet 373:1480–1490, 2009.
Silent celiac disease is being increasingly recognized, mainly in asymptomatic 1st-degree relatives of celiac patients investigated during screening studies. However, small bowel biopsy in these people reveals severe mucosal damage consistent with celiac disease. Potential celiac disease is defined when patients are identified by positive screening studies but without documented celiac disease on small bowel biopsy. It is important to follow these patients because they can develop established celiac disease in the future (Table 330-6).
Table 330-6 CLINICAL SPECTRUM OF CELIAC DISEASE
SYMPTOMATIC
Frank malabsorption symptoms: chronic diarrhea, failure to thrive, weight loss
Extraintestinal manifestations: anemia, fatigue, hypertransaminasemia, neurologic disorders, short stature, dental enamel defects, arthralgia, aphthous stomatitis
SILENT
No apparent symptoms in spite of histologic evidence of villous atrophy
In most cases identified by serologic screening in at-risk groups (see Table 330-1)
LATENT
Subjects who have a normal histology, but at some other time, before or after, have shown a gluten-dependent enteropathy
POTENTIAL
Subjects with positive celiac disease serology but without evidence of altered jejunal histology
It might or might not be symptomatic
Diagnosis
The ultimate diagnosis of celiac disease relies on the demonstration of specific, though not pathognomonic, histopathologic abnormalities in the small bowel mucosa (Table 330-7). According to The European Society for Pediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) current criteria, the 2 requirements mandatory for the diagnosis of celiac disease are the finding of villous atrophy with hyperplasia of the crypts and abnormal surface epithelium, while the patient is eating adequate amounts of gluten, and a full clinical remission after withdrawal of gluten from the diet. The finding of circulating IgA celiac disease–associated antibodies at the time of diagnosis and their disappearance on a gluten-free diet adds weight to the diagnosis. A control biopsy to verify the consequences of the gluten-free diet on the mucosal architecture is considered mandatory only in patients with an equivocal clinical response to the diet. Gluten challenge is not considered mandatory except in situations where there is doubt about the initial diagnosis, for example, when an initial biopsy was not performed or when the biopsy specimen was inadequate or atypical of celiac disease.
Table 330-7 OTHER CAUSES OF FLAT MUCOSA
Modified from Di Sabatino A, Corazza GR: Coeliac disease, Lancet 373:1480–1490, 2009.
Treatment
The only treatment for celiac disease is lifelong strict adherence to a gluten-free diet (Fig. 330-4). This requires a wheat-, barley-, and rye-free diet. Despite evidence that oats are safe for most patients with celiac disease, there is concern regarding the possibility of contamination of oats with gluten during harvesting, milling, and shipping. Nevertheless, it seems wise to add oats to the gluten-free diet only when the latter is well established, so that possible adverse reactions can be readily identified. There is a consensus that all celiac disease patients should be treated with a gluten-free diet regardless of the presence of symptoms. However, whereas it is relatively easy to assess the health improvement after treatment of celiac disease in patients with clinical symptoms of the disease, it proves difficult in persons with asymptomatic celiac disease. The nutritional risks, particularly osteopenia, are those mainly feared for subjects who have silent celiac disease and continue on a gluten-containing diet. Little is known about the health risks in untreated patients with minor enteropathy, which may be clinically silent. There are no guidelines concerning the need for a gluten-free diet in subjects with “potential” celiac disease (patients with positive celiac disease–associated serology but without enteropathy).
Branski D, Fasano A, Troncone R. Latest development in the pathogenesis and treatment of celiac disease. J Pediatr. 2006;149:295-300.
Branski D, Troncone R. Celiac disease: a reappraisal. J Pediatr. 1998;133:181-187.
Collin P, Huhtala H, Virta L, et al. Diagnosis of celiac disease in clinical practice: physician’s alertness to the condition essential. J Clin Gastroenterol. 2007;41:152-156.
Di Sabatino A, Corazza RG. Coeliac disease. Lancet. 2009;373:1480-1493.
Dubois PC, Van Heel DA. Translational mini-review series on the immunogenetics of gut disease: immunogenetics of celiac disease. Clin Exp Immunol. 2008;153:162-173.
Ford AC, Chey WD, Talley NJ, et al. Yield of diagnostic tests for celiac disease in individuals with symptoms suggestive of irritable bowel syndrome. Arch Intern Med. 2009;169:651-658.
Green PHR, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731-1743.
Jones R, Sleet S. Coeliac disease. BMJ. 2009;338:539-540.
Kline RM, Neudorf SML, Baron HI. Correction of celiac disease after allogeneic hematopoietic stem cell transplantation for acute myelogenous leukemia. Pediatrics. 2007;120:e1120-e1122.
Kurppa K, Ashorn M, Iltanen S, et al. Celiac disease without villous atrophy in children: a prospective study. J Pediatr. 2010;157:373-380.
Ludvigsson JF, Montgomery SM, Ekbom A, et al. Small-intestinal histopathology and mortality risk in celiac disease. JAMA. 2009;302:1171-1178.
McGowan KE, Castiglione DA, Butzner JD. The changing face of childhood celiac disease in North America: Impact of serological testing. Pediatrics. 2009;124:1572-1578.
Mearin ML. Celiac disease among children and adolescents. Curr Prob Pediatr Adolesc Health Care. 2007;37:81-112.
Meresse B, Ripoche J, Heyman M, et al. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009;2:8-23.
Olsson C, Hernell O, Hornell A, et al. Difference in celiac disease risk between Swedish birth cohorts suggests an opportunity for primary prevention. Pediatrics. 2008;122:528-534.
Rashtak S, Murray JA. Tailored testing for celiac disease. Ann Intern Med. 2007;147:339-340.
Richey R, Howdle P, Shaw E, et al. Recognition and assessment of coeliac disease in children and adults: summary of NICE guidance. BMJ. 2009;338:1386-1388.
Rodrigues AF, Jenkins HR. Investigation and management of coeliac disease. Arch Dis Child. 2008;93:251-254.
Sattar N, Lazare F, Kacer M, et al. Celiac disease in children, adolescents, and young adults with autoimmune disease. J Pediatr. 2011;158:272-275.
Simmons JH, Klingensmith GJ, McFann K, et al. Celiac autoimmunity in children with type 1 diabetes: a two-year follow-up. J Pediatr. 2011;158:276-281.
Smyth DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767-2776.
Sollid LM, Lundin KEA. Diagnosis and treatment of celiac disease. Mucosal Immunol. 2009;2:3-7.
Telega G, Bennet TR, Welin S. Emerging new clinical patterns in the presentation of celiac disease. Arch Pediatr Adolesc Med. 2008;162:164-168.
Troncone R, Ivarsson A, Szajewska H, et al. Review article: future research on celiac disease—a position report from the European multistakeholder platform on celiac disease (CDEUSSA). Aliment Pharmacol Ther. 2008;27:1030-1043.
van Koppen EJ, Schweizer JJ, Csizmadia CGDS, et al. Long-tern health and quality-of-life consequences of mass screening for childhood celiac disease: a 10-year follow-up study. Pediatrics. 2009;123:e582-e588.
Zanchi C, Di Leo G, Ronfani L, et al. Bone metabolism in celiac disease. J Pediatr. 2008;153:262-265.
330.3 Other Malabsorptive Syndromes
Congenital Intestinal Mucosal Defects
Microvillus Inclusion Disease (Congenital Microvillus Atrophy)
Microvillus inclusion disease is an autosomal recessive disorder, which manifests at birth with profuse watery secretory diarrhea. It is the most commonly recognized cause of congenital diarrhea. Light microscopy of the small bowel mucosa demonstrates diffuse thinning of the mucosa, with hypoplastic villus atrophy and no inflammatory infiltrate. Diagnosis may be easily performed with light microscopy using PAS and CD10 staining, which shows a very thin or absent brush border, together with positive PAS and CD10 intracellular inclusions. Electron microscopy shows enterocytes with absent or sparse microvilli. The apical cytoplasm of the enterocytes contains electron-dense secretory granules; the hallmark is presence of microvilli within involutions of the apical membrane. Polyhydramnios is not a classic presentation of MID. Neonates usually present with dehydration and failure to thrive. Despite parenteral nutrition, diarrhea continues and initial fluid management is difficult. The disease is fatal without long-term parenteral nutrition support. Some infants present with rapid onset of liver disease, which is associated with pruritus. Most children die in infancy or early childhood. The long-acting somatostatin analog octreotide has been used as treatment and can reduce the volume of stool in some infants (Chapter 331). Intestinal transplantation is the only definitive treatment for this rare disease. Rarely, in milder forms of the disease, the patient can reach young adulthood and enjoy partially oral feeding. The underlying gene defect is a mutation in MYO5B, which encodes a protein involved in subcellular protein trafficking. Several types of mutations are involved.
Tufting Enteropathy
No treatment has been effective, so management requires permanent parenteral nutrition with possible intestinal transplantation (Chapter 331). Several types of mutations are involved, opening the door to genotype-phenotype analysis.
Carbohydrate-Deficient Glycoprotein Syndrome and Enterocyte Heparan Sulfate Deficiency
Congenital disorders of glycosylation (also carbohydrate-deficient glycoprotein, CDG) are genetic disorders of assembly of N-glycans in the cytosol and endoplasmic reticulum, resulting in a variety of manifestations (Chapter 81.6). The subtypes of CDG I are all associated with protein-losing enteropathy. Diagnosis can be established by isoelectric focusing of serum transferrin, enzyme analysis, and/or DNA analysis. Oral mannose can provide effective therapy in CDG Ib, so early identification of children presenting with hypoglycemia, hypothyroidism, and/or thyroid binding globulin deficiency is beneficial.
Congenital enterocyte heparan deficiency (CEHD) is a rare cause of intractable diarrhea with protein-losing enteropathy, which may be an unusual presentation of the carbohydrate-deficient glycoprotein syndrome (CDGS) type 1 (also known as Jaeken syndrome) (Chapter 81.6). Heparan sulfate is a glycosaminoglycan with multiple roles in the intestine, including restriction of charged macromolecules, such as albumin, in the vascular lumen.
Intestinal Lymphangiectasia
Obstruction of the lymphatic drainage of the intestine can be due to either congenital defects in lymphatic duct formation or to secondary causes (Table 330-8). The congenital form is often associated with lymphatic abnormalities elsewhere in the body, as occur with Turner, Noonan, and Klippel-Trenaunay-Weber syndromes. Causes of secondary lymphangiectasia include constrictive pericarditis, heart failure, retroperitoneal fibrosis, abdominal tuberculosis, and retroperitoneal malignancies. Lymph rich in proteins, lipids, and lymphocytes leaks into the bowel lumen, resulting in protein-losing enteropathy, steatorrhea, and lymphocyte depletion. Hypoalbuminemia, hypogammaglobulinemia, edema, lymphopenia, malabsorption of fat and fat-soluble vitamins, and chylous ascites often occur. Intestinal lymphangiectasia can also manifest with ascites, peripheral edema, and a low serum albumin.
Autoimmune Enteropathy
Extraintestinal autoimmune disorders are usual and include arthritis, membranous glomerulonephritis, insulin-dependent diabetes, thrombocytopenia, autoimmune hepatitis, hypothyroidism, and hemolytic anemia. It is essential to exclude underlying primary immune deficiency, particularly in boys with other autoimmune features (diabetes mellitus), because a proportion have underlying Immune dysregulation, Polyendocrinopathy, Enteropathy, X linked (IPEX) syndrome (Chapter 120.5). This systemic autoimmune disorder is due to mutations in FOXP3, a transcriptional regulator essential for the normal development of regulatory T cells (Tregs). Autoimmune enteropathy is reported in cases of Schimke immunoosseous dysplasia.
Abetalipoproteinemia
Abetalipoproteinemia is a rare autosomal recessive disorder of lipoprotein metabolism (Bassen Kornsweig syndrome) (Chapter 80). It is associated with severe fat malabsorption from birth. Children fail to thrive during the 1st year of life, with stools that are pale, foul smelling, and bulky. The abdomen is distended and deep tendon reflexes are absent as a result of peripheral neuropathy, which is secondary to vitamin E (fat-soluble vitamin) deficiency. Intellectual development tends to be slow. After 10 yr of age, intestinal symptoms are less severe, ataxia develops, and there is a loss of position and vibration sensation with the onset of intention tremors unless vitamin E levels are maintained in the normal range. These latter symptoms reflect involvement of the posterior columns, cerebellum, and basal ganglia. In adolescence, atypical retinitis pigmentosa develops without adequate supplemental of vitamin E; for instance, using a TPGS formulation of the vitamin.
Homozygous Hypobetalipoproteinemia
Homozygous hypobetalipoproteinemia (Chapter 80) is transmitted as an autosomal dominant trait. The homozygous form is indistinguishable from abetalipoproteinemia. The parents of these patients, as heterozygotes, have reduced plasma LDL and apoprotein-β concentrations, whereas the parents of patients with abetalipoproteinemia have normal levels. On transmission electron microscopy of small bowel biopsies, the size of lipid vacuoles in enterocytes differentiates between abetalipoproteinemia and hypobetalipoproteinemia: Many small vacuoles are present in hypobetalipoproteinemia, and larger vacuoles are seen in abetalipoproteinemia.
Wolman Disease
Wolman disease is a rare, lethal lipid storage disease that leads to lipid accumulation in multiple organs, including the small intestine. In addition to vomiting, severe diarrhea, and hepatosplenomegaly, patients have steatorrhea as a result of lymphatic obstruction. Deficiency of lysosomal acid lipase is the underlying cause of disease (Chapter 80). Successful long-term bone marrow engraftment has resulted in normalization of peripheral blood leukocyte lysosomal enzyme acid lipase activity, with subsequent resolution of diarrhea and the restoration of developmental milestones.
Cormier-Daire V, Bonnefont JP, Rustin P, et al. Mitochondrial DNA rearrangements with onset as chronic diarrhea with villous atrophy. J Pediatr. 1994;124:63-70.
Cuenod B, Brousse N, Goulet O, et al. Classification of intractable diarrhea in infancy using clinical and immunohistological criteria. Gastroenterology. 1990;99:1037-1043.
Fabre A, Roquelaure B, Lacoste C, et al. Exclusion of EGFR, HRAS, DSP, JUP, CTNNB1, PLEC1, and EPPK1 as functional candidate genes in 7 families with syndromic diarrhoea. J Pediatr Gastroenterol Nutr. 2009;48:501-503.
Girault D, Goulet O, Ledeist F, et al. Intractable diarrhea syndrome associated with phenotypic abnormalities and immune deficiency. J Pediatr. 1994;125:36-42.
Glocker EO, Frede N, Perro M, et al. Infant colitis—it’s in the genes. Lancet. 2010;376:1272.
Goulet O, Kedinger M, Brousse N, et al. Intractable diarrhea of infancy: a new entity with epithelial and basement membrane abnormalities. J Pediatr. 1995;127:212-219.
Goulet O, Ruemmele F. Causes and management of intestinal failure in children. Gastroenterology. 2006;130:S16-S28.
Goulet O, Salomon J, Ruemmele F, et al. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;20:20.
Goulet O, Vinson C, Roquelaure B, et al. Syndromic (phenotypic) diarrhea in early infancy. Orphanet J Rare Dis. 2008;3:6.
Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology. 2010;138:2388-2398.
Jaeken J, Matthij G, Saudubray JM, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoproteins syndrome with hepatic-intestinal presentation. Am J Hum Genet. 1998;62:1535-1539.
Müller T, Hess MW, Schiefermeier N, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40:1163-1165.
Murch S, Winyard PJD, Koletzki S, et al. Congenital enterocyte heparan sulphate deficiency with massive albumin loss, secretory diarrhoea and malnutrition. Lancet. 1996;347:1299-1301.
Oren A, Houwen RH. Phosphomannoseisomerase deficiency as the cause of protein-losing enteropathy and congenital liver fibrosis. J Pediatr Gastroenterol Nutr. 1999;29:231-232.
Patey N, Scoazec JY, Cuenod-Jabri B, et al. Distribution of cell adhesion molecules in infants with intestinal epithelial dysplasia (tufting enteropathy). Gastroenterology. 1997;113:833-843.
Patey-Mariaud de Serre N, Canioni D, Ganousse S, et al. Digestive histopathological presentation of IPEX syndrome. Mod Pathol. 2009;22:95-102.
Phillips AD, Schmitz J. Familial microvillous atrophy: a clinicopathological survey of 23 cases. J Pediatr Gastroenterol Nutr. 1992;14:380-396.
Reifen RM, Cutz E, Griffiths AM, et al. Tufting enteropathy: a newly recognized clinicopathological entity associated with refractory diarrhea in infants. J Pediatr Gastroenterol Nutr. 1994;18:379-385.
Ruemmele FM, Brousse N, Goulet O. Autoimmune enteropathy—molecular concepts. Curr Opinion in Gastroenterol. 2004;20:587-591.
Ruemmele FM, Jan D, Lacaille F, et al. New perspectives for children with microvillous inclusion disease: early small bowel transplantation. Transplantation. 2004;15:1024-1028.
Ruemmele FM, Moes N, de Serre NP, et al. Clinical and molecular aspects of autoimmune enteropathy and immune dysregulation, polyendocrinopathy autoimmune enteropathy X-linked syndrome. Curr Opin Gastroenterol. 2008;24:742-748.
Ruemmele FM, Schmitz J, Goulet O. Microvillous inclusion disease (microvillous atrophy). Orphanet J Rare Dis. 2006 Jun;26(1):22.
Sivagnanam M, Mueller JL, Lee H, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135:429-437.
Torgerson TR, Linane A, Moes N, et al. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. 2007;132:1705-1717.
Wang J, Cortina G, Wu SV, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med. 2006;355:270-280.
330.4 Intestinal Infections and Infestations Associated with Malabsorption
330.5 Immunodeficiency Disorders
Abonia JP, Castells MC. Common variable immunodeficiency. Allergy Asthma Proc. 2002;23:53-57.
Aslam A, Misbah SA, Talbot K, et al. Vitamin E deficiency induced neurological disease in common variable immunodeficiency: two cases and a review of the literature of vitamin E deficiency. Clin Immunol. 2004;112:24-29.
Aydogan M, Eifan AO, Gocmen I, et al. Clinical and immunologic features of pediatric patients with common variable immunodeficiency and respiratory complications. J Invest Allerg Clin Immunol. 2008;18:260-265.
Detkova D, de Gracia J, Lopes da Silva S, et al. Common variable immunodeficiency. Chest. 2007;131:1883-1889.
Onigbanjo MT, Orange JS, Perez EE, et al. Hypogammaglobulinemia in a pediatric tertiary setting. Clin Immunol. 2007;125:52-59.
330.6 Immunoproliferative Small Intestinal Disease
Al-Saleem T, Al-Mondhiry H. Immunoproliferative small intestinal disease (IPSID): a model for mature B-cell neoplasms. Blood. 2005;105:2274-2280.
Economidou I, Manousos ON, Triantafillidis JK, et al. Immunoproliferative small intestinal disease in Greece: presentation of 13 cases including two from Albania. Eur J Gastroenterol Hepatol. 2006;18:1029-1038.
Lecuit M, Abachin E, Martin A, et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med. 2004;350:239-248.
Salem PA, Estephan HH. Immunoproliferative small intestinal disease, Curr Concepts. Cancer J. 2005;11:374-382.
Witzig TE, Wahner-Roedler DL. Heavy chain disease. Curr Treat Options Oncol. 2002;3:247-254.
Zamir A, Parasher G, Moukarzel A, et al. Immunoproliferative small intestinal disease in a 16-year-old boy presenting as severe malabsorption with excellent response to tetracycline treatment. J Clin Gastroenterol. 1998;27:85-89.
330.7 Short Bowel Syndrome
Jon A. Vanderhoof and David Branski
Short bowel syndrome results from congenital malformations or resection of the small bowel. Causes of short bowel syndrome are listed in Table 330-9. Loss of >50% of the small bowel, with or without a portion of the large intestine, can result in symptoms of generalized malabsorption disorder or in specific nutrient deficiencies, depending on the region of the bowel resected. At birth, the length of small bowel is 200-250 cm; by adulthood, it grows to 300-800 cm. Bowel resection in an infant has a better prognosis than in an adult because of the potential for intestinal growth. An infant with as little as 15 cm of bowel with an ileocecal valve, or 20 cm without, has the potential to survive and be eventually weaned from total parenteral nutrition (TPN).
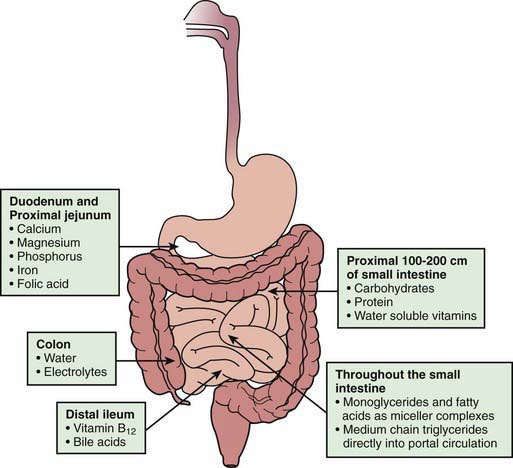
In addition to the length of the bowel, the anatomic location of the resection is also important. The jejunum has more circular folds and longer villi. The proximal 100-200 cm of jejunum is the main site for carbohydrate, protein, iron, and water-soluble vitamin absorption, whereas fat absorption occurs over a longer length of the small bowel. Depending on the region of the bowel resected, specific nutrient malabsorption can result. Vitamin B12 and bile salts are only absorbed in the distal ileum (Fig. 330-5). Jejunal resections are generally tolerated better than ileal resections because the ileum can adapt to absorb nutrients and fluids. Net sodium and water absorption is relatively much higher in the ileum. Ileal resection has a profound effect on fluid and electrolyte absorption due to malabsorption of sodium and water by the remaining ileum; ileal malabsorption of bile salts stimulates increased colonic secretion of fluid and electrolytes.
Complications
In patients who are unable to achieve full enteral feeding after several years of nutritional rehabilitation, surgical bowel lengthening procedures may be considered. In some children with complications of parenteral nutrition, especially impending liver failure, small intestinal and liver transplantation may be considered (Chapter 331).
330.8 Chronic Malnutrition
Primary malnutrition (i.e., undernutrition) is very common in developing countries and is directly related to increased disease burden and mortality (Chapter 43). In developed countries, chronic malnutrition occurs mainly as a result of decreased food intake, malabsorption syndromes, and increased nutritional needs in children with chronic diseases, and it affects 11-50% of hospitalized children. Child neglect and improper preparation of formula can result in severe malnutrition. Malnutrition can be identified by evaluating dietary intake, by medical history (anorexia, vomiting, dysphagia, mood and behavioral changes, abdominal pain, diarrhea), by anthropometric measurements (e.g., reduced weight per age and weight per height, BMI <5th percentile) by clinical signs of nutrient deficiencies (atrophic tongue in iron deficiency anemia or alopecia in zinc deficiency).
Nutritional rehabilitation in malnourished children is discussed in Chapter 43.
Black RE, Allen LH, Bhutta ZA, et al. Maternal and child undernutrition: global and regional exposures and health consequences. Lancet. 2008;371:243-260.
Brown KH. Diarrhea and malnutrition. J Nutr. 2003;133:328S-332S.
Caulfield LE, de Onis M, Blossner M, et al. Undernutrition as an underlying cause of child deaths associated with diarrhea, pneumonia, malaria, and measles. Am J Clin Nutr. 2004;80:193-198.
Fonseca BK, Holdgate A, Craig JC. Enteral vs. intravenous rehydration therapy for children with gastroenteritis: a meta-analysis of randomized controlled trials. Arch Pediatr Adolesc Med. 2004;158:483-490.
Guarino A, Albano F, Ashkenazi S, et al. ESPGHAN/ESPID guidelines for the management of acute gastroenteritis in children in Europe. J Pediatr Gastroenterol Nutr. 2008;46:S81-S122.
World Health Organization. Management of severe malnutrition: a manual for physicians and other senior health workers. Geneva: World Health Organization; 1999.
330.9 Enzyme Deficiencies
Michael J. Lentze and David Branski
Exocrine Pancreatic Insufficiency
Disorders of exocrine pancreatic insufficiency are discussed in Chapter 341. Cystic fibrosis is the most common congenital disorder associated with exocrine pancreatic insufficiency. Although rare, the next most common cause of pancreatic insufficiency in children is Shwachman-Diamond syndrome. Other rare disorders causing exocrine pancreatic insufficiency are Blizzard-Johanson syndrome (severe steatorrhea, aplasia of alae nasi, deafness, hypothyroidism, scalp defects), Pearson bone marrow syndrome (sideroblastic anemia, variable degree of neutropenia, thrombocytopenia), and isolated pancreatic enzyme deficiency (lipase, colipase and lipase-colipase, trypsinogen, amylase). Deficiency of enterokinase—a key enzyme that is produced in the proximal small bowel and is responsible for the activation of trypsinogen to trypsin—manifests clinically as exocrine pancreatic insufficiency.
330.11 Rare Inborn Defects Causing Malabsorption
Disorders of Fat Transport
Abetalipoproteinemia, hypobetalipoproteinemia, and chylomicron retention disease are described in Chapter 80. The long-chain fatty acid (FATP4) and cholesterol transporters, the latter being called Niemann-Pick C1-like protein (NPC1L1), have been characterized at the intestinal brush border in knock-out mice models showing a hyperproliferative hyperkeratosis and an impaired fatty acid and cholesterol uptake. NPC1L1 is inhibited by ezetimibe, which is used to restrict the absorption of dietary cholesterol.
Disorders of Vitamin Absorption
Vitamin B12 (cobalamin) is used exclusively by microorganisms and is acquired mostly from meat and milk. Its absorption starts with the removal of cobalamin from dietary protein by gastric acidity and its binding to haptocorrin. In the duodenum, pancreatic proteases hydrolyze the cobalamin-haptocorrin complex, allowing the binding of cobalamin to intrinsic factor (IF), which originates from parietal cells. The receptor of the cobalamin-IF complex is located at the apical membrane of the ileal enterocytes and represents a heterodimer consisting of cubulin and amnionless, with endocytic uptake of this ligand into endosomes, where it binds to megalin and forms a cobalamin–transcobalamin-2 complex (after cleavage of IF) for further transcytosis. As a cofactor for methionine synthase, cobalamin converts homocysteine to methionine. Cobalamin deficiency can be caused by inadequate intake of the vitamin (e.g., breast-feeding by mothers on a vegetarian diet), primary or secondary achlorhydria including autoimmune gastritis, exocrine pancreatic insufficiency, bacterial overgrowth (Chapter 330.4), ileal disease (Crohn disease, Chapter 328), ileal (or gastric) resection, infections (fish tapeworm), and Whipple disease (Chapter 333).
Folate is an essential vitamin required to synthesize methionine from homocysteine. It is found mainly in green leafy vegetables, legumes, and oranges. It is converted to 5-methyltetrahydrofolate (5MTHF) after its uptake by enterocytes. Secondary folate deficiency is caused by insufficient folate intake, villous atrophy (e.g., celiac disease, IBD), treatment with phenytoin, and trimethoprim among others (Chapter 448.1). Several inherited disorders of folate metabolism and transport have been described.
Disorders of Electrolyte and Mineral Absorption
Prenatal clinical signs of this disorder are a dilated small bowel that can mislead to a diagnosis of intestinal obstruction. Newborns with congenital chloride diarrhea present with severe life-threatening secretory diarrhea during the 1st weeks of life. Laboratory findings are metabolic alkalosis, hypochloremia, hypokalemia, and hyponatremia (with high plasma renin and aldosterone activities). Fecal chloride concentrations are >90 mmol/L and exceed the sum of fecal sodium and potassium. Early diagnosis and aggressive lifelong enteral substitution of KCl in combination with NaCl (chloride doses of 6-8 mmol/kg/day for infants and 3-4 mmol/kg/day for older patients) prevent mortality and long-term complications (such as urinary infections, hyperuricemia with renal calcifications, renal insufficiency, and hypertension) and allow normal growth and development. Orally administered proton pump inhibitors, cholestyramine, and butyrate can reduce the severity of diarrhea. The diarrheal symptoms usually tend to regress with age. However, febrile diseases are likely to exacerbate symptoms as a consequence of severe dehydration and electrolyte imbalances. (See Chapter 52 for fluid and electrolyte management.)
The congenital form of acrodermatitis enteropathica manifests with severe deficiency of body zinc soon after birth in bottle-fed children or after weaning from breastfeeding. Clinical signs of this disorder are anorexia, diarrhea, failure to thrive, humoral and cell-mediated immunodeficiency (poor wound healing, recurrent infections), male hypogonadism, skin lesions (vesicobullous dermatitis on the extremities and perirectal, perigenital, and perioral regions, and alopecia), and neurologic abnormalities (tremor, apathy, depression, irritability, nystagmus, photophobia, night blindness, and hypogeusia). The genetic defect of acrodermatitis enteropathica is caused by a mutation in the Zrt-Irt-lik protein 4 (ZIP4, SLC39A4), normally expressed on the apical membrane, which enables the uptake of zinc into the cytosol of enterocytes. The zinc-dependent alkaline phosphatase and plasma zinc levels are low. Paneth cells in the crypt of the small intestinal mucosa show inclusion bodies. Acrodermatitis enteropathica requires long-term treatment with elemental zinc 1 mg/kg/day. Maternal zinc deficiency impairs embryonic, fetal, and postnatal development. Acquired forms of zinc deficiency are described in Chapter 51.
Andrews GK. Regulation and function of Zip4, the acrodermatitis enteropathica gene. Biochem Soc Trans. 2008;36:1242-1246.
Baerlocher K, Solioz M. Disorders of copper, zinc and iron metabolism. In: Babu E, Duran M, Blaskovics ME, Gibson KM, editors. Physician’s guide to the laboratory diagnosis of metabolic diseases. ed 2. Heidelberg: Springer-Verlag; 2003:631-658.
Booth IW, Stange G, Murer H, et al. Defective jejunal brush-border Na+/H+ exchange: a cause of congenital secretory diarrhoea. Lancet. 1985;1:1066-1069.
Bor MV, Cetin M, Aytac S, et al. Long term biweekly 1 mg oral vitamin B12 ensures normal hematological parameters, but does not correct all other markers of vitamin B12 deficiency. A study in patients with inherited vitamin B12 deficiency. Haematologica. 2008;93:1755-1758.
Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008;88:249-286.
Broer S, Bailey CG, Kowalczuk S, et al. Iminoglycinuria and hyperglycinuria are discrete human phenotypes resulting from complex mutations in proline and glycine transporters. J Clin Invest. 2008;118:3881-3892.
Brunham LR, Singaraja RR, Hayden MR. Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu Rev Nutr. 2006;26:105-129.
Camargo SM, Singer D, Makrides V, et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with Hartnup mutations. Gastroenterology. 2009;136:872-882.
Drummond KN, Michael AF, Ulstrom RA, Good RA. The blue diaper syndrome: familial hypercalcemia with nephrocalcinosis and indicanuria; a new familial disease, with definition of the metabolic abnormality. Am J Med. 1964;37:928-948.
Grasbeck R. Imerslund-Grasbeck syndrome (selective vitamin B12 malabsorption with proteinuria). Orphanet J Rare Dis. 2006;1:17-23.
Heinz-Erian P, Muller T, Krabichler B, et al. Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am J Hum Genet. 2009;84:188-196.
Hihnala S, Hoglund P, Lammi L, et al. Long-term clinical outcome in patients with congenital chloride diarrhea. J Pediatr Gastroenterol Nutr. 2006;42:369-375.
Hoglund P, Auranen M, Socha J, et al. Genetic background of congenital chloride diarrhea in high-incidence populations: Finland, Poland, and Saudi Arabia and Kuwait. Am J Hum Genet. 1998;63:760-768.
Kamoun P, Parvy P, Rabier D, et al. Dicarboxylic aminoaciduria. J Inherit Metab Dis. 1994;17:758.
Kellett GL, Brot-Laroche E, Mace OJ, Leturque A. Sugar absorption in the intestine: the role of GLUT2. Annu Rev Nutr. 2008;28:35-54.
Kleta R, Romeo E, Ristic Z, et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet. 2004;36:999-1002.
Lee PJ, Van’t Hoff WG, Leonard JV. Catch-up growth in Fanconi-Bickel syndrome with uncooked cornstarch. J Inherit Metab Dis. 1995;18:153-156.
Martens K, Jaeken J, Matthijs G, Creemers JW. Multi-system disorder syndromes associated with cystinuria type I. Curr Mol Med. 2008;8:544-550.
Nyhan WL, Ozand PT. Atlas of metabolic diseases. London: Chapman & Hall Medical; 1998.
Palacin M, Nunes V, Font-Llitjos M, et al. The genetics of heteromeric amino acid transporters. Physiology (Bethesda). 2005;20:112-124.
Patel SB, Zablocki CJ. Plant sterols and stanols: their role in health and disease. J Clin Lipidol. 2008;2:S11-S19.
Pietrangelo A. Hereditary hemochromatosis—a new look at an old disease. N Engl J Med. 2005;350:2383-2397.
Qiu A, Jansen M, Sakaris A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917-928.
Santer R, Groth S, Kinner M, et al. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi-Bickel syndrome. Hum Genet. 2002;110:21-29.
Santer R, Hillebrand G, Steinmann B, et al. Intestinal glucose transport: evidence for a membrane traffic-based pathway in humans. Gastroenterology. 2003;124:34-39.
Sperandeo MP, Andria G, Sebastio G. Lysinuric protein intolerance: update and extended mutation analysis of the SLC7A7 gene. Hum Mutat. 2008;29:14-21.
Veldhuis NA, Gaeth AP, Pearson RB, et al. The multi-layered regulation of copper translocating P-type ATPases. Biometals. 2009;22:177-190.
Whitehead VM. Acquired and inherited disorders of cobalamin and folate in children. Br J Haematol. 2006;134:125-136.
330.12 Malabsorption in Eosinophilic Gastroenteritis
Furuta GT, Forbes D, Boey C, et al. Eosinophilic gastrointestinal diseases. J Pediatr Gastroenterol Nutr. 2008;47:234-238.
Mueller S. Classification of eosinophilic gastrointestinal diseases, Best Prac Res. Clin Gastroenterol. 2008;22:425-440.
Oh HE, Chetty R. Eosinophilic gastroenteritis: a review. J Gastroenterol. 2008;43:741-750.
Pratt CA, Demain JG, Rathkopf MM. Food allergy and eosinophilic gastrointestinal disorders: Guiding our diagnosis and treatment. Curr Probl Pediatr Adolesc Health Care. 2008;38:170-188.
Stone KD, Prussin C. Immunomodulatory therapy of eosinophil-associated gastrointestinal diseases. Clin Exper Allergy. 2008;38:1858-1865.
Tsibouris P, Galeas T, Moussia M, et al. Two cases of eosinophilic gastroenteritis and malabsorption due to Enterobious vermicularis. Dig Dis Sci. 2005;50:2389-2392.
Yan BN, Shaffer EA. Primary eosinophilic disorders of the gastrointestinal tract. Gut. 2009;58:721-732.
330.13 Malabsorption in Inflammatory Bowel Disease
Crohn disease and ulcerative colitis represent the 2 forms of chronic, immune-mediated IBD that commonly affect pediatric patients (Chapter 328). Because the small bowel is involved in the majority of pediatric Crohn disease patients, malabsorption of nutrients is far more of a problem than in ulcerative colitis. At the time of diagnosis, significant weight loss is observed in up to 85% of pediatric patients with Crohn disease and in about 65% with ulcerative colitis, due to inadequate intake of energy and micronutrients as well as diarrhea and malabsorption. Consequently, growth failure due to chronic undernutrition is far more common in Crohn disease than in ulcerative colitis, affecting up to 40% of cases.
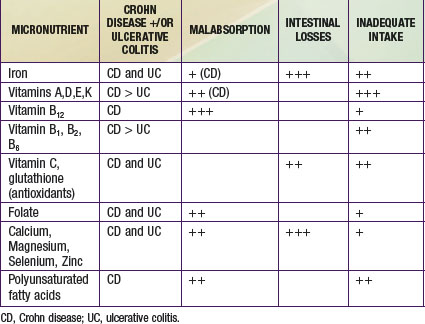
Optimizing nutritional status and growth are key priorities in the management of IBD in children and adolescents. Energy intake should meet the added costs of catch-up growth and are usually in the range of 40-70 kcal/kg ideal body weight per day. Protein requirements are higher in Crohn disease (1-1.5 g/kg/day). Bone mineral density deficit is common, even in pediatric patients who have not been exposed to systemic corticosteroid therapy. Osteoporosis or osteopenia is best assessed by bone densitometry, and levels of vitamin 25-hydroxyvitamin D should be monitored. Other micronutrient deficiencies that result from inadequate intake, malabsorption, and gut losses are shown in Table 330-10.
Day AS, Whitten KE, Sidler M, et al. Systematic review: nutritional therapy in paediatric Crohn’s disease. Aliment Pharmacol Ther. 2008;27:293-307.
Hernando A, Bretón I, Marín-Jimenez I, et al. Refeeding syndrome in a patient with Crohn’s disease. J Clin Gastroenterol. 2008;42:430-431.
Hueschkel R, Salvestrini C, Beattie RM, et al. Guidelines for the management of growth failure in childhood inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:839-849.
Kappelman MD, Bousvaros A. Nutritional concerns in pediatric inflammatory bowel disease patients. Molec Nutr Food Res. 2008;52:867-874.
Kleinman RE, Baldassano RN, Caplan A, et al. Nutrition support for pediatric patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2004;39:15-27.
Moorthy D, Cappellano KL, Rosenberg IH. Nutrition and Crohn’s disease: an update of print and Web-based guidance. Nutr Rev. 2008;66:387-397.
Pons R, Whitten KE, Woodhead H, et al. Dietary intakes of children with Crohn’s disease. Br J Nutr. 2009;30:1-6.
Ruemmele F, Roy CC, Levy E, et al. Nutrition as primary therapy in pediatric Crohn’s disease: fact or fantasy? J Pediatr. 2000;136:285-291.
Shamir R. Nutritional issues in inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2009;48:S86-S88.
Wiskin AE, Wootten SA, Beattie RM. Nutrition issues in pediatric Crohn’s disease. Nutr Clin Prac. 2007;22:214-222.