[level-membership-for-neurology-category]
Chapter 35 Disorders of Glycosylation
Eukaryotic cells synthesize hundreds of types of sugar chains called glycans, which function within the cell, at the cell surface, and beyond. Within the cell, glycans influence protein folding, stability, turnover, and intracellular trafficking [Varki et al., 2009]. At the cell surface, they influence or determine cell-cell binding, receptor-ligand interactions, assembly of signaling complexes, binding to the extracellular matrix, tissue pattern formation, trafficking of lymphocytes, and much more [Varki et al., 2009]. The same glycan can function differently on different proteins or in different settings. This three-dimensional complexity makes understanding the roles of glycans challenging but provides the body with an extraordinarily sensitive fine-tuning mechanism for many physiologic functions. It is not surprising that disrupting normal glycosylation causes moderate to severe pathology in multiple human organ systems.
More than 40 rare inherited disorders of glycan biosynthesis have been identified [Jaeken et al., 2008; Grunewald, 2007; Freeze, 2007; Marquardt and Denecke, 2003]. Most of these are the newly defined congenital disorders of glycosylation (previously called carbohydrate-deficient glycoprotein syndrome and disialotransferrin developmental deficiency syndrome). Others, such as muscle-eye-brain disease and Walker–Warburg syndrome, were well known, but elucidation of their relation to glycosylation has provided new insights into their pathophysiology and opens possibilities for therapy.
N-Linked Glycosylation
Overview
Sugar chains are added to newly synthesized proteins in the lumen of the endoplasmic reticulum; they are quickly and extensively remodeled there, and later in the Golgi apparatus. All eukaryotic cells make a 14-sugar lipid-linked oligosaccharide in the endoplasmic reticulum membrane that is composed of Man, GlcNAc, and glucose (Glc). This entire chain is transferred to Asn within an Asn-X-Thr/Ser/Cys [Zielinska et al., 2010] consensus sequence (X is any amino acid except proline) as newly made proteins emerge from the ribosome into the endoplasmic reticulum lumen. Forty or more genes are required to synthesize and transfer this glycan to proteins [Freeze, 2001b].
Extensive remodeling begins soon after sugar chain transfer. Up to two-thirds of the original lipid-linked oligosaccharide glycan is discarded, and 6–15 other sugar units are added back to create a dazzling array of sugar chains. Why generate this complex process? The initial glycan helps proteins fold and also provides important checkpoints for monitoring of proper protein folding in the endoplasmic reticulum [Parodi, 2000]. The addition of more sugars in the Golgi usually imparts greater specificity to the sugar chain function. Understanding the biosynthetic pathways is important for appreciating the nature of the defects.
Biosynthesis
Individual monosaccharides are synthesized from glucose, derived from the diet, or salvaged from degraded glycans, and must be activated to their nucleotide sugar derivatives [Freeze, 1999]. Figure 35-1 depicts an abbreviated version of the known monosaccharide pathways. We employ widely used standard symbols to denote the different sugars [Varki et al., 2009]. Note that phosphorylation of the monosaccharide is the first step, and some pathways interconvert phosphorylated forms, such as Man-6-P→Man-1-P. Also, several alternative routes generate uridine diphosphate (UDP)-GlcNAc, UDP-galactose (Gal), and guanosine diphosphate (GDP)-fucose (Fuc). For some types of glycosylation, the nucleotide sugar donates the sugar to a lipid carrier derived from the polyprenol, dolichol phosphate (P-Dol) [Schenk et al., 2001a]. These products include Man-P-Dol and Glc-P-Dol. Dolichol itself is made from polyprenols using a specific reductase [Cantagrel et al., 2010].
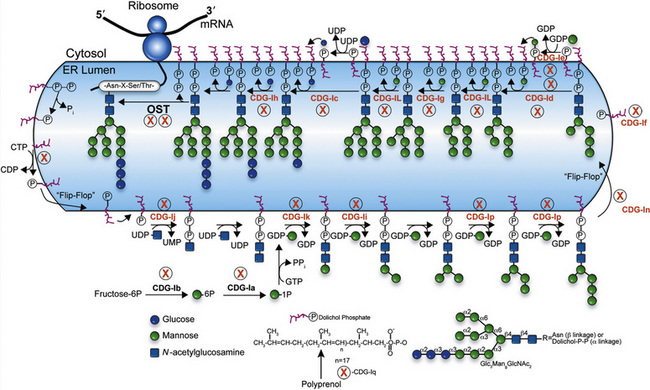
N-linked Glycan Biosynthesis
Biosynthesis of the lipid precursor oligosaccharide involves a series of steps and enzymes that add sugars in specific linkages and in a specific order [Freeze, 2001a]. It begins with P-Dol + UDP-GlcNAc forming GlcNAc-P-P-Dol on the cytosolic face of the endoplasmic reticulum. Another UDP-GlcNAc donates a second GlcNAc using a different GlcNAc transferase, and this is followed by the addition of five Man units derived from GDP-Man. At this point, a “flippase” reorients the entire molecule from the cytosolic face into the endoplasmic reticulum lumen. A series of Man transferases use Man-P-Dol to add four more Man units, making a three-branched structure. Three glucosyltransferases sequentially add Glc from Glc-P-Dol to one branch to complete the sugar chain. This 14-sugar unit molecule is the optimal substrate for the multisubunit oligosaccharyl transferase (OST) complex that recognizes the Asn-X-Thr/Ser consensus sequence on the protein and adds the sugar chain to Asn. Recent studies indicate that some Asn-X-Cys sites can also be glycosylated [Zielinska et al., 2010; Ostasiewicz et al., 2010]. Truncated glycans are poorly transferred and sometimes fail to occupy normal glycosylation sites. Following glycan transfer, P-P-Dol is converted back to P-Dol and then to Dol. Recycling of these carriers is extensive.
Within a few minutes of transfer to protein, the sugar chain is processed (Figure 35-2; see also Figure 35-1). A set of two glucosidases removes the Glc units. For some proteins, processing stops here, but for the great majority, a mannosidase in the endoplasmic reticulum removes a single Man unit. The proteins are carried to the Golgi, where another mannosidase called Golgi mannosidase I removes up to three more Man units. At this point, processing starts to add sugars. UDP-GlcNAc donates a GlcNAc to one branch, making the sugar chain an acceptable substrate for Golgi mannosidase II, which removes an additional two Man units. This process clears the way for the addition of a second GlcNAc to a Man on the remaining branch. Depending on the protein, up to three more GlcNAc transferases add GlcNAc units to the Man residues in a specific order to initiate up to five branches. These branches are then extended with Gal, derived from UDP-Gal, and sialic acid (Sia), derived from cytidine monophosphate (CMP)-Sia. In some cases, GDP-Fuc donates a Fuc to one or more of the branches or to the GlcNAc linked to the protein. In other cases, sulfates, phosphates, or glucuronic acids are added to selected sugars. Individual branches may accumulate several repeating units of Galβ1,4GlcNAc before being capped by Sia. These reactions occur in selected cisternae of the Golgi, and each nucleotide sugar donor must be translocated from its origin in the cytoplasm or nucleus to the Golgi by a substrate-selective transporter [Gerardy-Schahn et al., 2001].
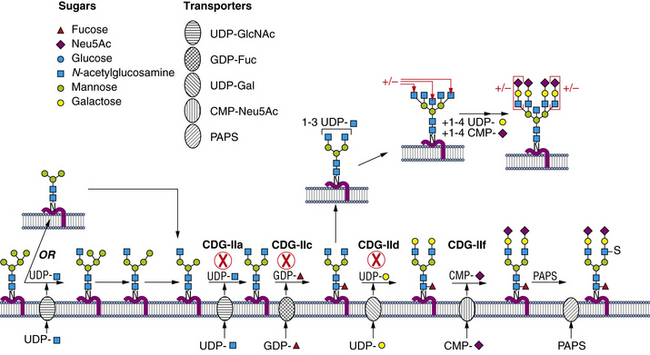
Enzymes and Cell Biology
The sugar-activating enzymes reside in either the cytoplasm or the nucleus. The N-linked biosynthetic enzymes all are in the endoplasmic reticulum or Golgi, with their active sites situated in the expected orientation. Most of the glycosyltransferases used for building the lipid-linked oligosaccharide and the Golgi transporters span the membrane many times [Ishida and Kawakita, 2004]. Most enzymes participating in glycan processing are attached to the membrane by a single membrane span. The transferases and nucleotide sugar transporters are constantly recycled in the dynamic Golgi to maintain their correct relationship to the maturing glycoproteins as they pass through the Golgi. Therefore, correct trafficking of the biosynthetic machinery is essential for optimal function.
Congenital Disorders of Glycosylation
Glycosylation is complex, and its disorders do not always lend themselves to phenotypic or symptomatic pigeonholing. Congenital disorders of glycosylation (CDG) nomenclature is still evolving as attempts are made to design a systematic, clinically useful system based on biochemical origins. Until 2008, CDGs resulting from defects in lipid-linked oligosaccharide biosynthesis and transfer to protein were defined as group I. The remaining CDGs that affect the biosynthesis and processing of the protein-bound sugar chains constituted group II. Individual disorders in group I or II were assigned lower-case letters (designating types, as noted earlier) when the genetic defect is proved. Several reviews provide comprehensive and up-to-date perspectives [Vodopiutz and Bodamer, 2008; Jaeken and Matthijs, 2007; Grunewald, 2007; Freeze, 2007; Marquardt and Denecke, 2003]. In 2008 a simplification of the nomenclature was proposed, as the growth in the number of disorders had produced a confusing series of letters and numbers that did not help the clinician or researcher. In the new system, diseases will be grouped in four categories: disorders of protein N-glycosylation, disorders of protein O-glycosylation, disorders of glycosphingolipid and glycosylphosphatidylinositol anchor glycosylation, and disorders of multiple glycosylation pathways. Individual disorders will be listed by the gene involved and, in parallel, its protein product. The authors suggest that the previous CDG designation may be listed in parentheses during the transitional period as the new nomenclature is adopted [Jaeken et al., 2008].
Diagnosis
In most patients with CDGs the diagnosis is straightforward and is based on the analysis of serum transferrin isoforms. Several methods are suitable and commercially available. These include isoelectric focusing [Freeze, 2001a; Jaeken and Matthijs, 2001], mass spectrometry [Wada, 2007; Kleinert et al., 2003; Lacey et al., 2001], zone electrophoresis, and high-pressure liquid chromatography [Quintana et al., 2009]. Electrospray ionization-mass spectrometry [Babovic and O’Brien, 2007] is the most informative because it easily distinguishes the absence of entire sugar chains, which characterizes group I CDGs, from the absence of one or more individual monosaccharide units typical of group II CDGs. The predominant isoform in normal transferrin has two sugar chains, each containing two negatively charged Sia molecules, designated tetrasialotransferrin. In group I disorders, isoelectric focusing analysis demonstrates loss of one or both entire glycan chains, producing “disialotransferrin” or “asialotransferrin,” respectively, but clearly this is an incomplete description. By contrast, electrospray ionization-mass spectrometry demonstrates that the loss of one or two chains reduces the mass by about 2200 or 4400 mass units, respectively. In group II disorders, isoelectric focusing may distinguish among “tri-,” “di-,” “mono-,” and “asialo” forms, reflecting variable loss of Sia, whereas electrospray ionization-mass spectrometry can indicate specific loss of Sia alone or combinations of Sia with additional sugars. This distinction is essential for determining assignment to group I or group II, and provides a signpost for specific identification of the defect. The single-step electrospray ionization-mass spectrometry technique can be accomplished using less than 5 mL of blood in less than 30 minutes [Lacey et al., 2001; Babovic and O’Brien, 2007]. This technique is therefore suitable for large-scale population screening for CDGs.
Transferrin isoform analysis produces few false-positive results. Uncontrolled fructose and galactose intolerance and recent heavy alcohol consumption produce a pattern typical of group I disorders [Kleinert et al., 2003]. In apparently rare instances, patients with genetically confirmed CDGs exhibited normal transferrin isoelectric focusing profiles, and in some patients, previously abnormal patterns normalized in preadolescence [Dupre et al., 2001; Fletcher et al., 2000]. Thus, a normal transferrin pattern should not exclude follow-up testing. Healthy neonates sometimes have a slightly abnormal transferrin pattern, which normalizes within a few weeks. Suspicious results in neonates should be repeated.
Until recently, specific diagnosis of the defect usually involved enzymatic, biochemical, and genetic analysis of the patient’s fibroblasts or leukocytes. Relatively few laboratories performed these analyses. With routine gene sequencing becoming more cost-effective, some genetic centers and commercial laboratories now offer various CDG gene diagnostic panels. In the coming years, decreasing costs, improved sequencing technology, and better informatics are likely to make this approach the first option. Prenatal testing is available for confirmed at-risk families [Matthijs et al., 1998, 2004].
General Clinical Features
About 1000 CDG patients have been identified, presenting with multiple organ dysfunction [Haeuptle and Hennet, 2009]. Patients with CDGs have protean presentations that, in some cases, may mimic mitochondrial (oxidative phosphorylation) disorders [Briones et al., 2001], but lack a history of maternal inheritance. An informal survey of CDG-affected families indicated that earlier nonspecific diagnoses frequently included a metabolic defect or cerebral palsy. Most patients first present to pediatric neurology or metabolic clinics. They frequently have combinations of liver, gastrointestinal, and coagulation disturbances [Jaeken and Matthijs, 2001; Marquardt and Denecke, 2003]. The possibility of a CDG should be investigated in any child presenting with developmental delay, seizures, hearing loss [Jaeken et al., 2009] or strabismus, particularly if any of these manifestations is accompanied by abnormal coagulation, liver dysfunction, or a gastrointestinal disorder. Most affected children are hypotonic and demonstrate failure to thrive.
Specific Disorders
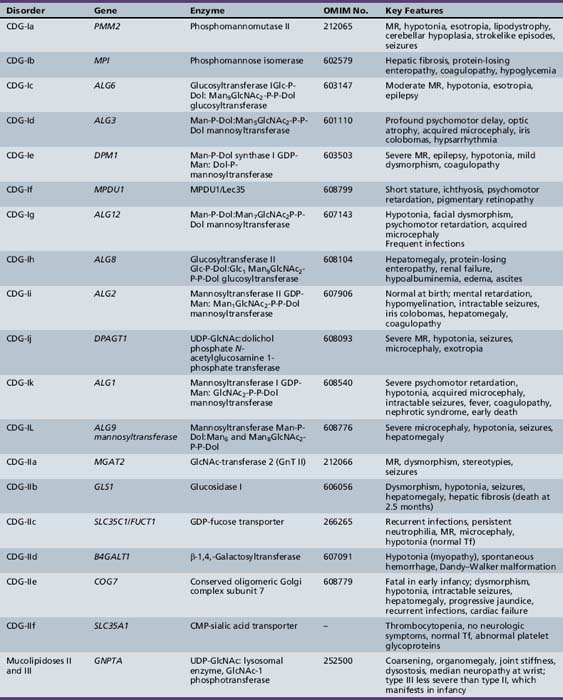
Table 35-1 summarizes the known CDG defects, utilizing the classification scheme proposed in 2008. This includes the mutated genes and major signs and symptoms. The known defects cover every aspect of the N-linked biosynthetic pathway. Activation or presentation of precursors (PMM2, PMI, DPM1, DPM3 MPDU1 [CDG-Ia, Ib, Ie, If]), glycosyltransferases for lipid-linked oligosaccharide biosynthesis (ALG6, NOT56L, ALG12, ALG8, ALG2, DPAGT1, HMT1, DOBD1 [CDG-Ic, Id, Ig, Ih, Ii, Ij, Ik, Il]), glycosidases that trim the protein-bound sugar chain (GLS1 [CDG-IIb]), Golgi-localized nucleotide sugar transporters (SLC35C1, SLC35A1 [CDG-IIc and IIf]), and glycosyltransferases that extend the trimmed chain (MGAT2, BG4ALT1 [CDG-IIa and IId]). DK1 (CDG-Im) impairs dolichol kinase function and impairs the final step of the de novo synthesis of dolichol phosphate [Denecke and Kranz, 2009]. SRD5A3 encodes the α-reductase that converts various polyprenols to dolichols [Cantagrel et al., 2010]. The conserved eight-subunit oligomeric Golgi (COG) complex that binds to the cytoplasmic face of the Golgi is needed for intra-Golgi or Golgi to endoplasmic reticulum retro-trafficking of multiple resident glycosyltransferases and nucleotide sugar transporters. Disorganized trafficking impairs multiple glycosylation pathways [Ungar et al., 2002]. Defects have now been identified in COG7, COG1, COG4 [CDG-IIe, IIg, IIj], COG8, COG5 and COG6 [Lübbehusen et al., 2010; Foulquier, 2009]. Appreciation of the importance of Golgi homeostasis in glycosylation led to the discovery of another disorder caused by mutations in a subunit of a vacuolar H+/ATPase that maintains appropriate pH of various organelles within the endocytic and exocytic pathways [Marshansky and Futai, 2008; Kornak et al., 2008; Guillard et al., 2009]. The intravesicular pH progressively decreases from the endoplasmic reticulum to Golgi, endosomes, and, finally, lysosomes.
The remainder of this chapter will focus on CDGs with significant neurologic manifestations.
Defects in Protein N-glycosylation
PMM2 (CDG-Ia)
PMM2 (CDG-Ia) is the best-known and most frequently recognized form of CDGs, first reported by Jaeken and co-workers in 1980 [Jaeken et al., 1980]. The defective gene was identified in 1995 as PMM2, which encodes the phosphomannomutase (PMM) that converts Man-6-P → Man-1-P. This defect results in insufficient production of lipid-linked oligosaccharide, leading to empty glycosylation sites. More than 800 patients are known worldwide, and more than 100 mutations have been cataloged [Barone et al., 2008; Jaeken, 2003; Matthijs et al., 2000; Haeuptle and Hennet, 2009].
Hagberg and associates described four stages of the typical (severe) phenotype [Hagberg et al., 1993]. The first is the infantile phase, marked by various combinations of dysmorphism, abnormal fat distribution (supragluteal and vulval fat pads, focal lipoatrophy), inverted nipples, cryptorchidism, esotropia, recurrent infections, cardiomyopathy or pericardial effusions, coagulopathies, nephrotic syndrome, hypothyroidism, life-threatening episodes of hepatic failure, and unexplained coma. As many as 20 percent of infants with CDG-Ia succumb in this phase [Jaeken and Matthijs, 2001]. In the second phase (comprising the remainder of the first decade), children experience seizures and strokelike episodes, often precipitated by intercurrent infections. The third phase (in the second decade of life) is marked by slowly progressive cerebellar ataxia and limb wasting, and by progressive visual loss secondary to pigmentary retinopathy. Adult survivors have moderate mental retardation with severe ataxia and hypogonadism, with or without skeletal deformities. Presentations are highly variable. In one girl with CDG-Ia, findings on computed tomography (CT) of the head were normal at 9 months, but subsequent imaging studies demonstrated progressive atrophy [Mader et al., 2002]. The investigators concluded that the cerebellar hypoplasia reported in infancy in most children with CDG-Ia likely reflects atrophy of antenatal onset, rather than hypoplasia.
More extensive testing for CDGs has led to the identification of milder CDG-Ia phenotypes [Briones et al., 2002; de Lonlay et al., 2001; Grünewald, 2009]. The patients often have high residual levels of PMM2 activity [Drouin-Garraud et al., 2001; Grünewald et al., 2001; Westphal et al., 2001b]. Some patients have only borderline cognitive impairment, but strabismus persists in these very mild cases. Few adult CDG-Ia patients are employed. A longitudinal study of eight Spanish patients confirmed the wide range of clinical manifestations, ranging from neonatal hemorrhage, non-immune hydrops, and death, through mental retardation and motor impairment without acute decompensation in patients in their 20s, to one individual with normal development and only gastrointestinal dysfunction in childhood [Perez-Duenas et al., 2009].
The carrier frequency of the most common mutant allele is about 1 in 70 in the northern European population [Schollen et al., 2000]. It is believed to be lethal in the homozygous state. Genotype-phenotype correlations have not been informative, but some evidence suggests that frequent polymorphisms in other glycosylation-related genes may influence the severity of the phenotype. No effective specific therapy for CDG-Ia exists. Experiments using CDG-Ia cells suggested that increasing dietary mannose might improve glycosylation in patients, but clinical trials demonstrated no benefit [Kjaergaard et al., 1998; Marquardt et al., 1997; Mayatepek et al., 1997].
Population studies find the risk of having a second child with CDG-Ia to be close to 1 in 3, rather than the expected mendelian ratio of 1 in 4, suggesting that reduced glycosylation may have some selective advantage [Schollen et al., 2004b]. At-risk couples should be counseled appropriately.
MPI-CDG (Ib)
MPI-CDG (Ib) is caused by mutations in MPI, the gene encoding phosphomannose isomerase, which interconverts Man-6-P and Fructose (Frc)-6-P. This reaction produces most of the mannose for glycoprotein synthesis [de Koning et al., 1998; Niehues et al., 1998]. About 25 patients have been identified since its discovery in 1998 [de Lonlay and Seta, 2009]. This phenotype is not associated with any primary neurologic symptoms. Gastrointestinal and hepatic pathology, with hypoglycemia, coagulopathy, and protein-losing enteropathy, is characteristic.
MPI-CDG (Ib) is unique in that simple dietary mannose therapy corrects the abnormalities, except for liver fibrosis [de Lonlay and Seta, 2009; Durand et al., 2003; Niehues et al., 1998]. The dietary mannose is taken up through transporters and converted to Man-6-P, thereby bypassing the metabolic block. Mannose has no known side effects at the concentrations used. Nonenzymatic protein glycation occurs at high concentrations of mannose, which is considerably more reactive than glucose, and can raise hemoglobin A1c (HbA1c) levels [Harms et al., 2002]. Some patients have received such treatment for longer than 10 years without complications [Westphal et al., 2001a; de Lonlay and Seta, 2009]. Twenty percent of the patients who were subsequently confirmed to have phosphomannose isomerase deficiency, however, died before discovery of the disorder [Freeze, 2001a]. The potentially fatal outcome and availability of simple, effective treatment mandate investigation for CDGs in suspected cases. One adult patient with MPI deficiency was not able to tolerate oral mannose, but her protein-losing enteropathy appeared to respond to heparin therapy [Liem et al., 2008].
ALG6-CDG (Ic)
ALG6-CDG (Ic) resembles, but is less severe than PMM2-CDG. It is characterized by moderate psychomotor retardation, hypotonia, esotropia, seizures, and ataxia. Nevertheless, at least five children have died of CDG-related complications [Marquardt and Denecke, 2003; Newell et al., 2003; Westphal et al., 2000]. The defect is in a glycosyltransferase, hALG6, that results in production of a truncated lipid-linked oligosaccharide sugar chain, which is inefficiently transferred to proteins. Patients sometimes experience life-threatening protein-losing enteropathy during bouts of gastroenteritis [Westphal et al., 2000]. Skeletal dysplasia, including a unique form associated with brachytelephalangy has been described in a compound heterozygote for ALG6 [Drijvers et al., 2010]. An adult woman has been identified in whom ALG6 deficiency was associated with mental retardation, skeletal anomalies, virilization, and deep vein thrombosis [Sun et al., 2005]. ALG6 deficiency was first identified in 1998 and subsequently in more than 35 patients [Haeuptle and Hennet, 2009; Burda et al., 1998; Grünewald et al., 2000; Imbach et al., 2000a], making it the second most common form of CDG.
NOT56L-CDG (Id)
Patients with NOT56L-CDG have mutations in NOT56L (ALG3), which encodes the mannosyltransferase used to synthesize Man6GlcNAc2 [Körner et al., 1999]. The index patient had microcephaly, optic nerve atrophy, iris colobomas, epilepsy, spastic quadriparesis, and profound psychomotor delay. Another patient had similar features plus Dandy–Walker malformation, with agenesis of the cerebellar vermis and corpus callosum. She also had recurrent hypoglycemia, thrombocytopenia, hypoalbuminemia, and coagulopathy. A total of eight children with NOT56L-CDG have now been described; all have a severe phenotype, with 7 of the 8 manifesting neurological impairments, including seizures, visual impairment, psychomotor retardation, and cerebral or cerebellar hypoplasia; there were varying combinations of hepatic, hematologic, endocrine, and dysmorphic manifestations, as well [Kranz et al., 2007b].
ALG12-CDG (Ig)
Eight cases of ALG12-CDG (Ig) have been reported [Chantret et al., 2002; Grubenmann et al., 2002; Thiel et al., 2002; Eklund et al., 2005; Kranz et al., 2007c]. The patients had typical CDG abnormalities, including hypotonia, generalized developmental delay, cerebellar hypoplasia, and decreased coagulation factors. Dysmorphic features included a long thin face, flat nasal bridge, and epicanthal folds. Frequent infections probably reflect reduced immunoglobulin G (IgG) concentrations. Affected males have genital hypoplasia, a feature not reported in other types of CDGs. More recent reports have emphasized features of skeletal dysplasia. These patients have mutations in hALG12, which encodes dolichol-P-mannose: Man7GlcNAc2-PP-dolichyl mannosyltransferase. Patients accumulate the truncated lipid-linked oligosaccharide, which is inefficiently transferred to proteins.
ALG8-CDG (Ih)
Eight patients have been described with ALG8-CDG (Ih), caused by a deficiency in hALG8, the gene encoding the second glucosyltransferase in lipid-linked oligosaccharide synthesis [Chantret et al., 2003; Schollen et al., 2004a; Stolting et al., 2009; Vesela et al., 2009]. Three patients had mild clinical presentations, including two siblings with pseudogynecomastia, epicanthus, hypotonia [Stolting et al., 2009], mental retardation, and ataxia, whereas the others all had severe multi-organ failure. Most died within a few months, but one patient with severe developmental delay survived a year before succumbing to renal failure. The others experienced hepatointestinal symptoms; one girl had cortical, cerebellar, and optic atrophy and intractable seizures before her death from systemic complications at 2 months [Vesela et al., 2009].
ALG2-CDG (Ii)
The only child with ALG2-CDG (Ii) recognized to date was normal at birth, except for an iris coloboma. Seizures, hepatomegaly, and coagulation abnormalities emerged in the first year. Imaging studies revealed cerebral hypomyelination. Pathologic mutations were found in hALG2, the enzyme that adds the second mannose to the growing lipid-linked oligosaccharide sugar chain [Thiel et al., 2003].
DPAGT1-CDG (Ij)
DPAGT1-CDG (Ij) is caused by a deficiency in UDP-GlcNAc:dolichol phosphate N-acetylglucosamine-1 phosphate transferase (GPT) activity encoded by DPAGT1. Two patients had severe hypotonia, intractable seizures, mental retardation, microcephaly, and exotropia [Wu et al., 2003].
ALG1/HMT1-CDG (Ik)
Ten children with HMT1-CDG (Ik) have been described [Kranz et al., 2004; Schwarz et al., 2004; Dupre et al., 2010]. Fifty percent had complications during pregnancy, and several had postnatal complications. Eighty percent were hypotonic, and all had at least one seizure, most being intractable; 8 of the 10 were dysmorphic, 7 of the 10 had visual impairment, and 5 of the 10 were microcephalic. Fifty percent had a fatal outcome. Patients with HMT1-CDG are deficient in GDP-Man:GlcNAc2-P-P-dolichol mannosyltransferase, encoded by the hALG1 gene, which adds the first Man to the lipid-linked oligosaccharide chain.
ALG9/DIBD1-CDG (Il)
Two patients with ALG9/DIBD1-CDG (Il) have been described. Abnormalities in the first case included severe microcephaly, central hypotonia, seizures, developmental delay, hepatomegaly, and bronchial asthma. This patient was found to carry a homozygous point mutation in human hALG9, whose product catalyzes the addition of both the seventh and ninth mannose units to the lipid-linked oligosaccharide chain [Frank et al., 2004]. The second patient was a girl with delayed development, epilepsy, hypotonia, failure to thrive, pericardial effusion, cystic renal disease, hepatosplenomegaly, esotropia, and inverted nipples. Neither lipodystrophy nor dysmorphic facial features were present. Magnetic resonance imaging (MRI) of the brain reflected cerebral and cerebellar atrophy and delayed myelination. Antithrombin III, factor XI, and cholesterol levels were low [Weinstein et al., 2005].
RFT1-CDG (In)
Six children have been described with RFT1-CDG (In) [Haeuptle et al., 2008; Clayton and Grunewald, 2009; Vleugels et al., 2009; Jaeken et al., 2009]. Most children have typical manifestations of the CDG spectrum: feeding problems, failure to thrive, severe developmental delay, poor to absent visual contact, epilepsy, and hypotonia, and variable respiratory, gastrointestinal, and coagulation abnormalities. All, however, have sensorineural deafness, which has only rarely been reported in other forms of CDG, and which is an important clue to this diagnosis. RFT1 is thought (but not absolutely proven) to be the flippase that transfers Man5GlcNAc2-PP-Dol from the cytoplasmic to the luminal face of the endoplasmic reticulum membrane.
MGAT2-CDG (IIa)
Three children with MGAT2-CDG (IIa) have been described [Cormier-Daire et al., 2000; Jaeken et al., 1994; Tan et al., 1996]. They were of Belgian, Iranian, and French descent, respectively, and all had severe psychomotor delay, acquired microcephaly and growth retardation, and variable combinations of hypotonia, ventricular septal defects, craniofacial dysmorphism (thin lips, hooked nose, large ears, hypertrophied gums, short neck), stereotypies, and coagulation defects. All were found to have impaired activity of the processing enzyme N-acetylglucosaminyltransferase II (MGAT2), which decreases the formation of multibranched N-linked glycans. A targeted disruption of this gene in the mouse faithfully recapitulates the human disorder [Wang et al., 2001].
GLS1-CDG (IIb)
Only one patient with GLS1-CDG (IIb) has been described [De Praeter et al., 2000]. Her parents were consanguineous. She had dysmorphic features, hypotonia, reduced nerve conduction velocity, seizures, and hepatomegaly with abnormal bile duct proliferation and fibrosis. The patient died at 2.5 months. This disorder was caused by mutations in α-glucosidase I, which trims the outermost glucose from oligosaccharides just after their transfer to nascent proteins. Although activity of the enzyme was severely decreased in this patient’s fibroblasts, transferrin glycosylation remained normal. This patient would be missed by the standard diagnostic assays. An endo-α-mannosidase apparently bypasses the glucosidase defect to permit normal oligosaccharide processing. Abnormal accumulation of fully glycosylated sugar chains may overwhelm the capacity of endoplasmic reticulum chaperone lectins.
TUSC3-CDG
Nine patients have been described with TUSC3-CDG. Seven were members of four sibships in a large consanguinous Iranian kindred, and all had nonsyndromic, moderate to severe mental retardation [Garshasbi et al., 2008]. Two were sibs from a small French family [Molinari et al., 2008]. TUSC3 codes for a subunit of the oligosaccharyltransferase complex (OST). It is not clear why these patients have no other systemic manifestations of hypoglycosylation, but it is theorized that differential tissue expression of another subunit associated with the OST (IAP) might compensate for TUSC3 deficiency in non-neurologic tissues [Garshasbi et al., 2008].
IAP-CDG
Screening of samples from 250 families with X-linked nonsyndromic mental retardation led to the identification of one patient who carried a c.932T/G, p.V311G mutation in the IAP gene [Molinari et al., 2008]. IAP (implantation-associated protein) is an ortholog to yeast Ost 6p. Like TUSC3, IAP is expressed in all tissues, including adult and fetal brain.
ALG11-CDG (Ip)
A brother and sister born to consanguineous Turkish parents were found to carry homozygous c.T257C mutations in the hALG11 gene; this disorder was designated ALG11-CDG, or CDG-Ip [Rind et al., 2010]. The proband was a girl who presented in the neonatal period with dysmorphism, vomiting, and hypotonia. She had visual and hearing impairment, intractable seizures, lipodystrophy, and elevated lactate without another metabolic explanation. She died at 2 years. Subsequently, a brother was born, who presented with hypotonia and vomiting at 6 weeks, and who was found to carry the same mutation. ALG11 encodes GDP-Man:Man3GlcNAc2-PP-dolichol mannosyltransferase. Deficiency of this enzyme impairs the elongation of lipid-linked oligosaccharides at the outer leaflet of the endoplasmic reticulum.
Defects in Protein O-Glycosylation
O-Xylosylglycan Synthesis
Glycosaminoglycans are very large glycan chains that usually are built on selected core proteins [Esko, 1999]. These molecules are located in the extracellular matrix, where they provide mechanical support and help to organize the matrix. At the cell surface, they bind growth factors and act as signaling molecules that enhance or inhibit cell proliferation. They are the most diverse and complex glycans. The protein-bound glycosaminoglycan molecules include heparan sulfate, chondroitin sulfate, dermatan sulfate, and keratan sulfate. All but the last are linked to protein through O-xylose, which is extended with two galactose units and a glucuronic acid. Each of these glycosaminoglycan chains is extended by one of several alternating disaccharides composed of GlcNAcα1,4GlcAβ1,4) (heparan sulfate) or GalNAcβ1,3GlcAβ1,3 (chondroitin sulfate and dermatan sulfate) or GlcNAcβ1,3Galβ1,4 (keratan sulfate). All of these glycosaminoglycans are partially sulfated. Hyaluronon, GlcNAcβ1,4GlcAβ1,3, is the only glycosaminoglycan that exists as a free glycan chain and is not sulfated.
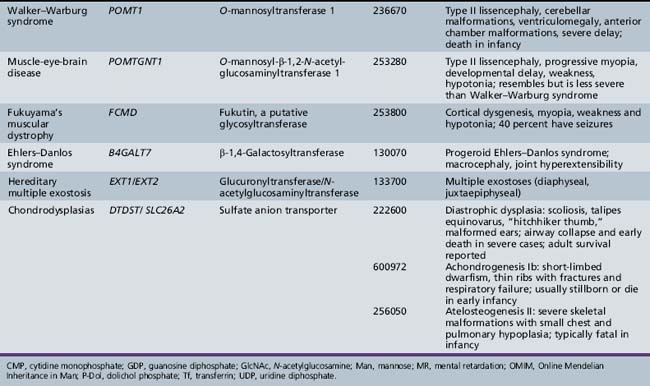
Three known defects in glycosaminoglycan synthesis cause clinical disorders, but none has neurologic manifestations. Mutations in the gene B4GALT7 cause abnormal synthesis of dermatan sulfate and are associated with the progeroid variant of Ehlers–Danlos syndrome [Quentin et al., 1990; Faiyaz-Ul-Haque et al., 2004]. This gene encodes an enzyme, xylosylprotein 4-β-galactosyltransferase, that adds the first galactose residue to xylose in the core of glycosaminoglycan chains. Patients exhibit multiple abnormalities in connective tissue, leading to short stature, diffuse osteopenia, loose but elastic skin, hypermobile joints, and hypotonia. Cognitive impairment has not been reported.
The condition multiple hereditary exostoses is inherited as an autosomal-dominant trait and characteristically affects the metaphyses of long bones [Zak et al., 2002]. Patients with multiple hereditary exostoses have mutations in the genes EXT1 and EXT2, both of which encode co-polymerase components that participate in the assembly of alternating residues of GlcNAc and GluA that form the backbone of the heparan sulfate glycosaminoglycan chains. Because heparan sulfate binds to many types of growth factors, a partial decrease in heparan sulfate is thought to upset the delicate regulation of chondrocyte proliferation. This situation in turn leads to enhanced chondrocyte growth and the formation of exostoses. Surprisingly, a significant proportion of patients fall within the autistic spectrum. Rigorous experiments in Ext-deficient mouse models indicate molecular alterations corresponding to dramatic behavioral changes that correlate with reduced heparan sulfate content of selected tissues.
Macular corneal dystrophy (types I and II) is caused by a deficiency in a specific sulfotransferase (CHST6) called corneal N-acetylglucosamine-6-sulfotransferase (GlcNAc6ST), which is responsible for the sulfation of corneal keratan sulfate [Akama et al., 2000]. The unsulfated keratan chains are poorly soluble, and their eventual precipitation disrupts the collagen network, leading to thinning and loss of transparency of the corneal stroma. This progressive disorder manifests between ages 5 and 9 with very small, punctate corneal opacities. Erosions, painful photophobia, and sensation of an ocular foreign body develop subsequently.
O–N-acetylgalactosamine Synthesis
These mucinous types of molecules are located at the surface of epithelial cells or in their secretions, and typically subserve barrier functions [Marth, 1999]. They usually are composed of large clusters of O-galactose N-acetylglucosamine (O-GalNAc)-linked sugar chains, each containing 2–10 sugars. Only a few are known to serve specific functions: for example, in lymphocyte recirculation and leukocyte extravasation [Lowe, 2003]. Few biosynthetic defects are known, and none has obvious neurologic manifestations.
O-mannosylglycan Synthesis: Congenital Muscular Dystrophy and Limb-Girdle Spectrum
Overview
The dystrophin glycoprotein complex assembles on the sarcolemma of skeletal muscle cells. Mutations that affect the integrity of this complex cause congenital muscular dystrophies by compromising the integrity of the basement membrane [Martin, 2003]. Alpha-dystroglycan is a major component of this complex and contains O-mannose-linked glycans (i.e., those with an O linkage between Ser/Thr and mannose residues). Mutations in genes encoding enzymes involved in this O-linked glycosylation cause a group of rare congenital muscular dystrophies with an autosomal-recessive inheritance pattern and a variable degree of brain involvement [Martin and Freeze, 2003]. Additional types of congenital muscular dystrophy likely will be found to result from as yet unrecognized defects in this glycosylation pathway (Figure 35-3).
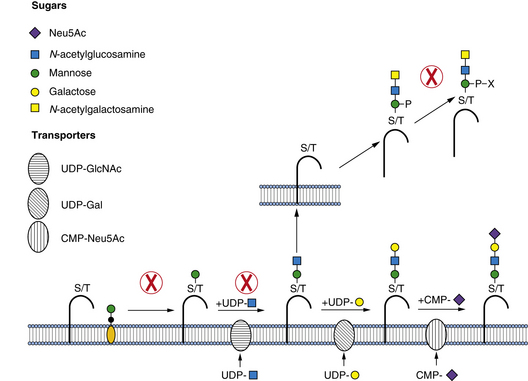
Biosynthesis of O-Mannose Glycans
POMT1 (protein-O-mannosyltransferase 1) and POMT2 (protein-O-mannosyltransferase 2) form a complex and use Man-P-Dol to form the Man-O-Ser/Thr linkage, in the endoplasmic reticulum lumen. This linkage is extended by addition of β1,2-GlcNAc through a pathway-specific GlcNAc transferase (POMGnT1). Both POMT1/2 and POMGnT1 have proven enzymatic activities [Endo, 2004]. In α-dystroglycan, this disaccharide can be extended with β1,4-Gal and capped by α-2,3-Sia. Alternatively, the GlcNAc is extended by GalNAc, Man is converted to Man-6-P and further to an undefined Man-6-P-diester. The presence of Man-6-P diester is considered essential for normal binding of α-dystroglycan to the matrix [Yoshida et al., 2010]. The biosynthetic pathway and specific structures are unknown, but it is clear that this pathway is not the same one as that used for lysosomal enzyme targeting. More complex branched glycans with glucuronic acid (GlcA) or sulfate can be found in the brain, suggesting that this pathway is much more complex [Yuen et al., 1997]. About one-third of all O-linked chains in the brain are built on O-Man, and α-dystroglycan is clearly not the only protein with these glycans, since brain-specific deletion of α-dystroglycan does not significantly change the amount of O-Man glycans [Yoshida et al., 2010].
Functional Defects
Defects in this pathway are depicted in Table 35-1. Deficiencies in POMT1 and POMT2 impair the addition of the linkage Man unit to the protein, preventing further elongations at that site. These mutations account for about 30 percent of the diagnosed cases of Walker–Warburg syndrome [Beltran-Valero de Bernabe et al., 2002; Muntoni et al., 2004]. Defects in POMGNT1 impair addition of GlcNAc to Man and cause muscle-eye-brain disease [Zhang et al., 2003].
Other congenital muscular dystrophies also have been linked to glycosylation abnormalities, but the specific molecular mechanisms are not known. These proteins include fukutin, fukutin-related protein, and LARGE. Mutations in FCMD cause Fukuyama congenital muscular dystrophy, common in Japan. Mutations in the fukutin-related protein gene, FKRP, cause CMD1C and limb-girdle muscular dystrophy [Muntoni et al., 2004]. The LARGE gene causes a form of murine muscular dystrophy, and mutations in its human homolog were found in a patient with a congenital muscular dystrophy and mental retardation [Longman et al., 2003]. The clinical presentations of congenital muscular dystrophies reflect the manifestations associated with specific mutations and may overlap. Mutations in POMT1, FKRP, and FCMD all have been associated with a Walker–Warburg syndrome phenotype [Muntoni et al., 2004]. FCMD, FKRP, and LARGE all have glycosyltransferase-like domains and characteristic catalytic residues, but transferase activity has not been demonstrated. Sequence homologies usually are insufficient to predict the specific transferase reactions because sugar-specific signatures are few, and the acceptor substrate may require both peptide and glycan recognition. Overexpression of LARGE can functionally bypass multiple glycosylation defects in α-dystroglycan, offering a potentially broad therapeutic approach [Barresi et al., 2004].
Clinical Features
The European Neuromuscular Center proposed a set of diagnostic criteria for congenital muscular dystrophies. These include hypotonia beginning in the first 6 months, early multiple contractures, diffuse weakness and muscular atrophy with sparing of extraocular muscles, normal mental development (in many), variable course, early elevation of serum creatine kinase, myopathic changes on electromyography (EMG), and necrotic-regenerative changes on muscle biopsy [Dubowitz, 1997].
The three forms of congenital muscular dystrophy associated with impaired O-glycosylation have considerable phenotypic overlap, as mentioned earlier. Fukuyama congenital muscular dystrophy is characterized by cortical dysgenesis and simple myopia. Affected children have global developmental delay, with regression in motor skills in the latter half of the first decade and death in adolescence [Messina et al., 2010]. Almost all of the patients with Fukuyama congenital muscular dystrophy have seizures. Fukuyama congenital muscular dystrophy is the most common form of congenital muscular dystrophy in Japan, in contrast with Western populations, in which merosin deficiency predominates.
Walker–Warburg syndrome also features type II lissencephaly, in combination with ventriculomegaly, cerebellar malformations, retinal and anterior chamber malformations, and congenital cataracts. Walker–Warburg syndrome represents the most severe congenital muscular dystrophy phenotype, with congenital blindness, hypotonia, profound developmental delay, and failure to thrive. Most affected children die in the first 6 months of life [Messina et al., 2010].
Biochemical and Genetic Tests
Several monoclonal antibodies recognize some feature of the O-Man chain on α-dystroglycan, and their binding is greatly reduced or eliminated in all of these disorders [Michele et al., 2002]. Antibody against the peptide portion binds normally, but the loss of the glycan chains reduces the apparent size of the protein. Specific enzymatic assays can be used to diagnose muscle-eye-brain disease [Zhang et al., 2003], and for POMT1 [Lommel et al., 2010]. Sequencing of the genes is needed to identify specific mutations. It is likely that additional biosynthetic genes will be identified soon.
Defects in Glycosphingolipid and Glycosylphosphatidylinositol Glycosylation
Developmental delay, seizures, and blindness are found in autosomal-recessive Amish infantile epilepsy. A large Amish family was identified with a nonsense mutation in SIAT9 that truncated protein [Simpson et al., 2004]. SIAT9 is a sialyltransferase needed for synthesis of gangliosides GM3 (Siaα2-3Galβ1-4Glc-ceramide) from lactosylceramide (Galβ1-4Glc-ceramide). Patients accumulate nonsialylated plasma glycosphingolipids, such as GM3, and also lack downstream GM3-dependent molecules.
Only one defect in glycophosphatidylinositol anchor synthesis with neurological features is known, and has been reported as Mabry’s syndrome [Thompson et al., 2010]. Genetic mapping demonstrated that patients with mental retardation, hyperphosphatasia, unusual facial features, hypotonia, and seizures have mutations in PIGV. The gene encodes the second mannosyltransferase used for GPI-anchor synthesis. Total surface anchors are reduced, including glycophosphatidylinositol-anchored alkaline phosphatase, which is instead found at very high levels in the plasma [Krawitz et al., 2010].
Defects in Multiple Glycosylation and Other Pathways
DPM1-CDG (Ie)
DPM1-CDG is another severe phenotype, characterized by psychomotor delay, profound hypotonia, microcephaly, cortical blindness, and intractable seizures [Imbach et al., 2000b; Kim et al., 2000]. Laboratory testing revealed elevated creatine kinase and depressed antithrombin III. Some children have dysmorphic features, including downslanting palpebral fissures, flat occiput and nasal bridge, hemangiomas of the occiput and sacrum, a high narrow palate, and mild limb shortening. Dolichol phosphate mannose synthase (DPM1) activity is markedly diminished in all reported cases.
DPM3-CDG (Io)
A 27-year-old woman has been reported with DPM3 deficiency, whose phenotype includes short stature, dilated cardiomyopathy, a strokelike episode, and a myopathy characterized by mild proximal weakness and absent glycosylated α-dystroglycan on muscle biopsy [Lefeber et al., 2009]. DPM activity is required to provide Man-P-Dol precursors for N-, C-, and O-linked glycosylation and for glycophosphatidylinositol anchor biosynthesis. In this case, a primary defect in the N-glycosylation pathway has led to an O-linked disorder – an α-dystroglycanopathy.
MPDU1-CDG (If)
Four children with MPDU1-CDG have been described. Abnormalities included severe psychomotor retardation and variable features, including growth retardation, optic nerve atrophy, icthyosis, dysmorphism (parietal bossing, thin lips), hypo- or hypertonia, enlarged extra-axial spaces, thrombocytopenia, transient deficiency of growth hormone and insulin-like growth factor 1, and mild elevations of creatine kinase [Kranz et al., 2001; Schenk et al., 2001b]. All have mutations in MPDU1/Lec35, leading to deficient function of the Lec35 protein. Lec35 may act as a chaperone for P-Dol in the endoplasmic reticulum membrane, ensuring appropriate lateral spacing of Man-P-Dol and Glc-P-Dol. In the absence of such spacing, these compounds may form rafts that alter local concentration gradients, impairing synthesis of the lipid-linked oligosaccharide and its accessibility to the oligosaccharyltransferase complex.
B4GALT1-CDG (IId)
A single patient with B4GALT1-CDG (IId) has been described. Abnormalities included psychomotor retardation, a Dandy–Walker malformation, progressive hydrocephalus, hypotonia, and myopathy [Hansske et al., 2002]. CDG-IId was caused by mutations in B4GALT1, encoding one of the isozymes that add β-1,4-galactose units to N-linked glycans during processing. The other isozymes do not compensate for this transferase deficiency.
GNE-CDG
Mutations in GNE, which encodes UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase, cause both hereditary inclusion body myopathy (IBM2) and distal myopathy with rimmed vacuoles [Grandis et al., 2010]. IBM2 presents with a distal myopathy that later spreads proximally, but characteristically spares the quadriceps. Creatine kinase is mildly elevated. The disorder is most frequent among Iranian Jews, who carry an M712 T mutation in GNE. Nonaka myopathy, or distal myopathy with rimmed vacuoles, has a similar presentation, but is usually associated with different mutations.
SLC35A1-CDG (IIf)
The sole reported patient with SLC35A1-CDG (IIf) had severe thrombocytopenia and complete loss of the sialyl-Lewis-X antigen on leukocytes [Willig et al., 2001; Martinez-Duncker et al., 2005]. This disorder was associated with mutations in the gene encoding the Golgi cytidine monophosphate (CMP)-sialic acid transporter [Martinez-Duncker et al., 2005]. Although platelet membrane proteins exhibited altered glycosylation, transferrin and other serum glycoproteins were normal. This finding underscores the limitations of transferrin analysis.
SLC35A1-CDG (IIc)
SLC35A1-CDG (IIc) was originally described as leukocyte adhesion deficiency II and is characterized by moderate to severe mental retardation, rhizomelic short stature, a broad flat nasal bridge, microcephaly, elevated leukocytes, frequent infections, persistent marked neutrophilia, and periodontitis. The disorder is caused by mutations in the GDP-fucose transporter, which limits the synthesis of fucosylated glycans [Lübke et al., 2001; Lühn et al., 2001]. One of these is sialyl Lewis-X (sLeX), a glycan essential for leukocyte rolling before extravasation. Oral fucose supplements effectively reduced leukocytosis in two patients by allowing synthesis of sufficient sLeX [Hidalgo et al., 2003; Marquardt et al., 1999]. These patients also lack fucosylated H-antigen, the precursor for the ABO blood group. Fucose supplements have not provoked antigen synthesis or immunologic reactions. Transferrin glycosylation is normal in this type.
COG complex
COG7-CDG (IIe)
The first report of COG7-CDG (IIe) described two siblings. Manifestations included perinatal asphyxia and dysmorphic features, including low-set dysplastic ears, micrognathia, short neck, and loose wrinkled skin [Wu et al., 2004]. They had generalized hypotonia, hepatosplenomegaly, and progressive jaundice that appeared shortly after birth. A CT scan revealed an enlarged cisterna cerebelli superior in one patient; severe epilepsy developed in both. They both died by 10 weeks from recurrent infections and cardiac insufficiency. A total of 8 patients are now known, most with similar presentations, although one [Zeevaert et al., 2009] was less severely affected. This type is caused by a mutation in COG7, which encodes one of the eight subunits of the COG complex [Oka et al., 2004; Ungar et al., 2002]. This cytoplasmic complex associates with the cytoplasmic face of the Golgi, and is involved in the shuttling of glycosyltransferases and nucleotide sugar transporters between the Golgi and other intracellular compartments. The mutation destabilizes the complex and slows the trafficking of these molecules, presumably leading to their degradation or mislocalization.
COG1-CDG (IIg)
The sole COG1-CDG (IIg) patient described to date is a girl with hypotonia, rhizomelic short stature and acquired microcephaly [Foulquier et al., 2006]. She had mild dysmorphic features, subtle hepatosplenomegaly, mild developmental delay at 21 months, and slight cerebral and cerebellar atrophy. Transferrin isoelectric focusing demonstrated a type II pattern; ApoC-III isoelectric focusing also demonstrated a hypoglycosylated pattern, confirming abnormal O-linked glycosylation. The patient was found to have a homozygous insertion (c.2659–2660insC) in the hCOG1 gene, which would predict a truncated COG1 protein.
COG8-CDG (IIh)
Two patients have been described with COG8-CDG (IIh). The first had a severe phenotype, characterized by delayed development and hypotonia, first appreciated in the latter half of the first year, with the subsequent evolution of myoclonic seizures and growth failure, with a height and weight below the first percentile for age [Kranz et al., 2007a]. At last follow-up at 8.5 years, the child suffered from severe retardation, double incontinence, intolerance of wheat and dairy products, markedly reduced muscle mass, mild spasticity and contractures of the lower extremities, and esotropia. Laboratory studies had demonstrated evidence of chronic axonal neuropathy. This child was heterozygous for two COG8 mutations: IVS3+1G>A and 1687–1688 del TT. Both mutations produce a truncated protein. The second case was a girl who presented at 6 months with an acute encephalopathy after initial normal development [Foulquier et al., 2007]. The child regressed, and manifested hypotonia and alternating esotropia and pseudoptosis. She subsequently developed a progressive cerebellar syndrome, accompanied by action myoclonus. Over time, the deep tendon reflexes disappeared, and oculomotor apraxia has emerged. Laboratory abnormalities have included abnormal levels of coagulation factors, transaminases, and creatine kinase. In both cases, transferrin isoelectric focusing documented a type II pattern, and Apo C-III was hypoglycosylated. This child was homozygous for a nonsense mutation (C to G) at position c.1611 in the COG8 cDNA.
COG4-CDG (IIj)
There is a single report of a child with COG4-CDG (IIj) [Reynders et al., 2009]. This child became symptomatic around 4 months of age, following immunization, when he experienced fever, irritability, and the onset of complex partial seizures. He had dysmorphic features, including thick hair and unusual facies, axial hypotonia, and mild limb spasticity and hyperreflexia. From 12 months, he experienced frequent infections, and by 3 years was microcephalic, with ataxia, delayed milestones (including absent speech), and frontotemporal atrophy on imaging studies. Laboratory investigations indicated elevated transaminases, alkaline phosphatase, LDL cholesterol, and low levels of coagulation factors and platelets. Studies of the COG4 gene identified a point mutation of the paternal allele (C2185T) and a microdeletion of the maternal allele.
COG5-CDG
The sole patient described with COG5 deficiency was a girl who presented at 8 years with mild mental retardation (IQ 50–55), mild dysarthria, truncal and appendicular ataxia, and hypotonia [Paesold-Burda et al., 2009]. She had no systemic abnormalities and extensive metabolic investigation was negative. MRI documented diffuse cerebellar and brainstem atrophy. Subsequently, she made slow developmental progress. Investigation at 12 years revealed evidence of abnormal N-linked and O-linked glycosylation. Sequencing of the COG5 gene identified a homozygous mutation (c.1669–15T>C), which explained the observed altered splicing of the transcript.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Akama T.O., Nishida K., Nakayama J., et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet. 2000;26:237.
Babovic-Vuksanovic D., O’Brien J.F. Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferrin glycoforms. Mol Diagn Ther. 2007;11:303-311.
Barone R., Sturiale L., Sofia V., et al. Clinical phenotype correlates to glycoprotein phenotype in a sib pair with PMM2-CDG. Am J Med Genet A. 2008;146A:2103-2108.
Barresi R., Michele D.E., Kanagawa M., et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med. 2004;10:696.
Beltran-Valero de Bernabe D., Currier S., Steinbrecher A., et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033.
Briones P., Vilaseca M.A., Garcia-Silva M.T., et al. Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. Eur J Paediatr Neurol. 2001;5:127.
Briones P., Vilaseca M.A., Schollen E., et al. Biochemical and molecular studies in 26 Spanish patients with congenital disorder of glycosylation type Ia. J Inherit Metab Dis. 2002;25:635.
Burda P., Borsig L., de Rijk-van Andel J., et al. A novel carbohydrate-deficient glycoprotein syndrome characterized by a deficiency in glucosylation of the dolichol-linked oligosaccharide. J Clin Invest. 1998;102:647.
Cantagrel V., Lefeber D.J., Ng B.G., et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142(2):203-217.
Chantret I., Dancourt J., Dupre T., et al. A deficiency in dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new subtype of congenital disorders of glycosylation. J Biol Chem. 2003;278:9962.
Chantret I., Dupre T., Delenda C., et al. Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose:Man7GlcNAc2-PP-dolichyl mannosyltransferase. J Biol Chem. 2002;277:25815.
Clayton P.T., Grünewald S. Comprehensive description of the phenotype of the first case of congenital disorder of glycosylation due to RFT1 deficiency (CDG-In). J Inherit Metab Dis. 2009. doi: 10.1007/s10545-009-1108-x
Cormier-Daire V., Amiel J., Vuillaumier-Barrot S., et al. Congenital disorders of glycosylation IIa cause growth retardation, mental retardation, and facial dysmorphism. J Med Genet. 2000;37:875.
de Koning T.J., Dorland L., van Diggelen O.P., et al. A novel disorder of N-glycosylation due to phosphomannose isomerase deficiency. Biochem Biophys Res Commun. 1998;245:38.
de Lonlay P., Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for MPI-CDG. Biochim Biophys Acta. 2009;1792(9):841-843.
de Lonlay P., Seta N., Barrot S., et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: A series of 26 cases. J Med Genet. 2001;38:14.
Denecke J., Kranz C. Hypoglycosylation due to dolichol metabolism defects. Biochim Biophys Acta. 2009;1792:888-895.
De Praeter C.M., Gerwig G.J., Bause E., et al. A novel disorder caused by defective biosynthesis of N-linked oligosaccharides due to glucosidase I deficiency. Am J Hum Genet. 2000;66:1744.
Drijvers J.M., Lefeber D.J., de Munnik S.A., et al. Skeletal dysplasia with brachytelephalangy in a patient with a congenital disorder of glycosylation due to ALG6 gene mutations. Clin Genet. 2010;77(5):507-509.
Drouin-Garraud V., Belgrand M., Grünewald S., et al. Neurological presentation of a congenital disorder of glycosylation PMM2-CDG: Implications for diagnosis and genetic counseling. Am J Med Genet. 2001;101:46.
Dubowitz V. Congenital muscular dystrophy. In: Emery A.E.H., editor. Diagnostic criteria for neuromuscular disorders. London: Royal Society of Medicine Press, 1997.
Dupre T., Cuer M., Barrot S., et al. Congenital disorder of glycosylation Ia with deficient phosphomannomutase activity but normal plasma glycoprotein pattern. Clin Chem. 2001;47:132.
Dupre T., Vuillaumier-Barrot S., Chantret I., et al. Guanosine diphosphate-mannose:GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. J Med Genet. 2010. Aug 2. [Epub ahead of print]
Durand G., Dupre T., Vuillaumier-Barrot S., et al. Congenital disorders of glycosylation. Ann Pharm Fr. 2003;61:330.
Eklund E.A., Newell J.L., Sun L., et al. Molecular and clinical description of the first US patients with congenital disorder of glycosylation Ig. Mol Genet Metab. 2005;84(1):25-31.
Endo T. Structure, function and pathology of O-mannosyl glycans. Glycoconj J. 2004;21:3.
Esko J. Proteoglycans and glycosaminoglycans. In: Varki A., Cummings R., Esko J.D., et al, editors. Essentials of Glycobiology. New York: Spring Harbor Laboratory Press, 1999.
Faiyaz-Ul-Haque M., Zaidi S.H.E., Al-Ali M., et al. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am J Med Genet. 2004;128A:39-45.
Fletcher J.M., Matthijs G., Jaeken J., et al. Carbohydrate-deficient glycoprotein syndrome: Beyond the screen. J Inherit Metab Dis. 2000;23:396.
Foulquier F. COG defects, birth and rise!. Biochim Biophys Acta. 2009;1792(9):896-902.
Foulquier F., Ungar D., Reynders E., et al. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum Mol Genet. 2007;16(7):717-730.
Foulquier F., Vasile E., Schollen E., et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci USA. 2006;103(10):3764-3769.
Frank C.G., Grubenmann C.E., Eyaid W., et al. Identification and functional analysis of a defect in the human ALG9 gene: Definition of congenital disorder of glycosylation type IL. Am J Hum Genet. 2004;75:146.
Freeze H.H. Glycosylation precursors. In: Varki A., Cummings R., Esko J.D., et al, editors. Essentials of glycobiology. New York: Spring Harbor Laboratory Press, 1999.
Freeze H.H. Congenital disorders of glycosylation and the pediatric liver. Semin Liver Dis. 2001;21:501.
Freeze H.H. Update and perspectives on congenital disorders of glycosylation. Glycobiology. 2001;11:129R.
Freeze H.H. Congenital Disorders of Glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007;7:389-396.
Garshasbi M., Hadavi V., Habibi H., et al. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am J Hum Genet. 2008;82(5):1158-1164.
Gerardy-Schahn R., Oelmann S., Bakker H. Nucleotide sugar transporters: Biological and functional aspects. Biochimie. 2001;83:775.
Grandis M., Gulli R., Cassindrini D., et al. The spectrum of GNE mutations: allelic heterogeneity for a common phenotype. Neurol Sci. 2010;31:377-380.
Grubenmann C.E., Frank C.G., Kjaergaard S., et al. ALG12 mannosyltransferase defect in congenital disorder of glycosylation type lg. Hum Mol Genet. 2002;11:2331.
Grunewald S. Congenital disorders of glycosylation: rapidly enlarging group of (neuro)metabolic disorders. Early Hum Dev. 2007;83:825-830.
Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta. 2009;1792:827-834.
Grünewald S., Imbach T., Huijben K., et al. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol. 2000;47:776.
Grünewald S., Schollen E., Van Schaftingen E., et al. High residual activity of PMM2 in patients’ fibroblasts: Possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am J Hum Genet. 2001;68:347.
Guillard M., Dimopoulou A., Fischer B., et al. Vacuolar H+-ATPase meets glycosylation in patients with cutis laxa. Biochim Biophys Acta. 2009;1792(9):903-914.
Haeuptle M.A., Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30(12):1628-1641.
Haeuptle M.A., Pujol F.M., Neupert C., et al. Human RFT1 deficiency leads to a disorder of N-linked glycosylation. Am J Hum Genet. 2008;82:600-606.
Hagberg B.A., Blennow G., Kristiansson B., et al. Carbohydrate-deficient glycoprotein syndromes: Peculiar group of new disorders. Pediatr Neurol. 1993;9:255.
Hansske B., Thiel C., Lubke T., et al. Deficiency of UDP-galactose: N-acetylglucosamine beta-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J Clin Invest. 2002;109:725.
Harms H.K., Zimmer K.P., Kurnik K., et al. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. 2002;91:1065.
Hidalgo A., Ma S., Peired A.J., et al. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood. 2003;101:1705.
Imbach T., Grünewald S., Schenk B., et al. Multi-allelic origin of congenital disorder of glycosylation (CDG)-Ic. Hum Genet. 2000;106:538.
Imbach T., Schenk B., Schollen E., et al. Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J Clin Invest. 2000;105:233.
Ishida N., Kawakita M. Molecular physiology and pathology of the nucleotide sugar transporter family (SLC35). Pflugers Arch. 2004;447:768.
Jaeken J., Hennet T., Freeze H.H., et al. On the nomenclature of congenital disorders of glycosylation (CDG). J Inherit Metab Dis. 2008;31:669-672.
Jaeken J. Komrower Lecture. Congenital disorders of glycosylatic (CDG): Its all in it!. J Inherit Metab Dis. 2003;26:99-118.
Jaeken J., Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261-278.
Jaeken J., Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet. 2001;2:129-151.
Jaeken J., Schachter H., Carchon H., et al. Carbohydrate deficient glycoprotein syndrome type II: A deficiency in Golgi localised N-acetyl-glucosaminyltransferase II. Arch Dis Child. 1994;71:123.
Jaeken J., Vanderschueren-Lodewyckx M., Caeaer P., et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG deficiency, increased serum arysulphatase A and increased CSF protein: A new syndrome? Pediatr Res. 1980;14:179.
Jaeken J., Vleugels W., Régal L., et al. RFT1-CDG: Deafness as a novel feature of congenital disorders of glycosylation. J Inherit Metab Dis. 2009. Oct 24. [Epub ahead of print]
Kim S., Westphal V., Srikrishna G., et al. Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (DPM1-CDG). J Clin Invest. 2000;105:191.
Kjaergaard S., Kristiansson B., Stibler H., et al. Failure of short-term mannose therapy of patients with carbohydrate-deficient glycoprotein syndrome type 1A. Acta Paediatr. 1998;87:884.
Kleinert P., Kuster T., Durka S., et al. Mass spectrometric analysis of human transferrin in different body fluids. Clin Chem Lab Med. 2003;41:1580.
Kornak U., Reynders E., Dimopoulou A., et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40(1):32-34.
Körner C., Knauer R., Stephani U., et al. Carbohydrate deficient glycoprotein syndrome type IV: Deficiency of dolichyl-P-Man:Man5GlcNAc2-PP-dolichyl mannosyltransferase. EMBO J. 1999;18:6816.
Kranz C., Ng B.G., Sun L., et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum Mol Genet. 2007;16(7):731-741.
Kranz C., Sun L., Eklund E.A., et al. CDG-Id in two siblings with partially different phenotypes. Am J Med Genet A. 2007;143A(13):1414-1420.
Kranz C., Basinger A.A., Gucsavas-Calkoglu M., et al. Expanding spectrum of congenital disorder of glycosylation Ig CDG-Ig): Sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am J Med Genet A. 2007;143A:1371-1378.
Kranz C., Denecke J., Lehle L., et al. Congenital disorder of glycosylation type Ik (CDG-Ik): A defect of mannosyltransferase I. Am J Hum Genet. 2004;74:545.
Kranz C., Denecke J., Lehrman M.A., et al. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If). J Clin Invest. 2001;108:1613.
Krawitz P.M., Schweiger M.R., Rodelsperger C., et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010. Aug 29. [Epub ahead of print]
Lacey J.M., Bergen H.R., Magera M.J., et al. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem. 2001;47:513.
Lefeber D.J., Schönberger J., Morava E., et al. Deficiency of Man-P-Dol synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. 2009;85(1):76-86.
Liem Y.S., Bode L., Freeze H.H., et al. Using heparin therapy to reverse protein-losing enteropathy in a patient with MPI-CDG. Nat Clin Pract Gastroenterol Hepatol. 2008;5(4):220-224.
Lommel M., Cirak S., Willer T., et al. Correlation of enzyme activity and clinical phenotype in POMT1-associated dystroglycanopathies. Neurology. 2010;74:157-164.
Longman C., Brockington M., Torelli S., et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12:2853.
Lowe J.B. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr Opin Cell Biol. 2003;15:531.
Lübbehusen J., Thiel C., Rind N., et al. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum Mol Genet. 2010;19(18):3623-3633.
Lübke T., Marquardt T., Etzioni A., et al. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet. 2001;28:73.
Lühn K., Wild M.K., Eckhardt M., et al. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet. 2001;28:69.
Mader I., Dobler-Neumann M., Kuker W., et al. Congenital disorder of glycosylation type Ia: Benign clinical course in a new genetic variant. Childs Nerv Syst. 2002;18:77.
Marquardt T., Denecke J. Congenital disorders of glycosylation: Review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003;162:359.
Marquardt T., Hasilik M., Niehues R., et al. Mannose therapy in carbohydrate-deficient glycoprotein syndrome type I – first results of the German Multicenter Study. Amino Acids. 1997;12:389.
Marquardt T., Luhn K., Srikrishna G., et al. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976.
Marshansky V., Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20(4):415-426.
Marth J.D. O-glycans. In: Varki A., Cummings R., Esko J.D., et al, editors. Essentials of glycobiology. New York: Spring Harbor Laboratory Press, 1999.
Martinez-Duncker I., Dupre T., Piller V., et al. Genetic complementation reveals a novel human congenital disorder of glycosylation of type II, due to inactivation of the Golgi CMP-sialic acid transporter. Blood. 2005;105(7):2671-2676.
Martin P.T. Dystroglycan glycosylation and its role in matrix binding in skeletal muscle. Glycobiology. 2003;13:55R.
Martin P.T., Freeze H.H. Glycobiology of neuromuscular disorders. Glycobiology. 2003;13:67R.
Matthijs G., Schollen E., Bjursell C., et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (PMM2-CDG). Hum Mutat. 2000;16:386.
Matthijs G., Schollen E., Cassiman J.J., et al. Prenatal diagnosis in CDG1 families: Beware of heterogeneity. Eur J Hum Genet. 1998;6:99.
Matthijs G., Schollen E., Van Schaftingen E. The prenatal diagnosis of congenital disorders of glycosylation (CDG). Prenat Diagn. 2004;24:114.
Mayatepek E., Schröder M., Kohlmuller D., et al. Continuous mannose infusion in carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr. 1997;86:1138.
Messina S., Bruno C., Moroni I., et al. Congenital muscular dystrophies with cognitive impairment: A population study. Neurology. 2010;75(10):898-903.
Michele D.E., Barresi R., Kanagawa M., et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417.
Molinari F., Foulquier F., Tarpey P.S., et al. Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am J Hum Genet. 2008;82:1150-1157.
Muntoni F., Brockington M., Torelli S., et al. Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol. 2004;17:205.
Newell J.W., Seo N.S., Enns G.M., et al. Congenital disorder of glycosylation Ic in patients of Indian origin. Mol Genet Metab. 2003;79:221.
Niehues R., Hasilik M., Alton G., et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414.
Oka T., Ungar D., Hughson F.M., et al. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004;15:2423.
Ostasiewicz P., Zielinska D.F., Mann M., et al. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J Proteome Res. 2010;9(7):3688-3700.
Paesold-Burda P., Maag C., Troxler H., et al. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum Mol Genet. 2009;18:4350-4356.
Parodi A.J. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69.
Perez-Duenas B., Garcia-Cazorla A., Pineda M., et al. Long-term evolution of eight Spanish patients with CDG type Ia: typical and atypical manifestations. Eur J Paediatr Neurol. 2009;13(5):444-451.
Quentin E., Gladen A., Roden L., et al. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc Natl Acad Sci USA. 1990;87:1342.
Quintana E., Montero R., Casado M., et al. Comparison between high performance liquid chromatography and capillary zone electrophoresis for the diagnosis of congenital disorders of glycosylation. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2513-2518.
Reynders E., Foulquier F., Leao Teles E., et al. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum Mol Genet. 2009;18:3244-3256.
Rind N., Schmeiser V., Thiel C., et al. A severe human metabolic disease caused by deficiency of the endoplasmatic mannosyltransferase hALG11 leads to congenital disorder of glycosylation-Ip. Hum Mol Genet. 2010;19(8):1413-1424. Epub 2010 Jan 15
Schenk B., Fernandez F., Waechter C.J. The ins(ide) and outs(ide) of dolichyl phosphate biosynthesis and recycling in the endoplasmic reticulum. Glycobiology. 2001;11:61R.
Schenk B., Imbach T., Frank C.G., et al. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J Clin Invest. 2001;108(11):1687-1695.
Schollen E., Frank C.G., Keldermans L., et al. Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency). J Med Genet. 2004;41:550.
Schollen E., Kjaergaard S., Legius E., et al. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in PMM2-CDG (congenital disorders of glycosylation type Ia). Eur J Hum Genet. 2000;8:367.
Schollen E., Kjaergaard S., Martinsson T., et al. Increased recurrence risk in congenital disorders of glycosylation type Ia (PMM2-CDG) due to a transmission ratio distortion. J Med Genet. 2004;41:877.
Schwarz M., Thiel C., Lubbehusen J., et al. Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylation type Ik. Am J Hum Genet. 2004;74:472.
Simpson M.A., Cross H., Proukakis C., et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225-1229.
Stolting T., Omran H., Erlekotte A., et al. Novel ALG8 mutations expand the clinical spectrum of congenital disorder of glycosylation type Ih. Mol Genet Metab. 2009;98(3):305-309.
Sun L., Eklund E.A., Van Hove J.L., et al. Clinical and molecular characterization of the first adult congenital disorder of glycosylation (CDG) type Ic patient. Am J Med Genet A. 2005;137(1):22-26.
Tan J., Dunn J., Jaeken J., et al. Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. Am J Hum Genet. 1996;59:810.
Thiel C., Schwarz M., Hasilik M., et al. Deficiency of dolichyl-P-Man: Man7GlcNAc2-PP-dolichyl mannosyltransferase causes congenital disorder of glycosylation type Ig. Biochem J. 2002;367:195.
Thiel C., Schwarz M., Peng J., et al. A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. J Biol Chem. 2003;278:22498.
Thompson M.D., Nezarati M.M., Gillessen-Kaesbach G., et al. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am J Med Genet A. 2010;152A:1661-1669.
Ungar D., Oka T., Brittle E.E., et al. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J Cell Biol. 2002;157:405.
Varki A., Cummings R., Esko J.D., et al, editors. Essentials of glycobiology, ed 2, New York: Spring Harbor Laboratory Press, 2009.
Vesela K., Honzik T., Hansikova H., et al. A new case of ALG8 deficiency (CDG Ih). J Inherit Metab Dis. 2009. Aug 18. [Epub ahead of print]
Vleugels W., Haeuptle M.A., Ng B.G., et al. RTF1 deficiency in three novel CDG patients. Hum Mutat. 2009;30(10):1428-1434.
Vodopiutz J., Bodamer O.A. Congenital disorders of glycosylation-a challenging group of IEMs. J Inherit Metab Dis. 2008.
Wada Y. Mass spectrometry in the detection and diagnosis of congenital disorders of glycosylation. Eur J Mass Spectrom (Chichester, Eng). 2007;13:101-103.
Wang Y., Tan J., Sutton-Smith M., et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: Conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology. 2001;11:1051.
Weinstein M., Schollen E., Matthijs G., et al. CDG-IL: an infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am J Med Genet A. 2005;136(2):194-197.
Westphal V., Kjaergaard S., Davis J.A., et al. Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: Long-term outcome and effects of mannose supplementation. Mol Genet Metab. 2001;73:77.
Westphal V., Peterson S., Patterson M., et al. Functional significance of PMM2 mutations in mildly affected patients with congenital disorders of glycosylation Ia. Genet Med. 2001;3:393.
Westphal V., Murch S., Kim S., et al. Reduced heparan sulfate accumulation in enterocytes contributes to protein-losing enteropathy in a congenital disorder of glycosylation. Am J Pathol. 2000;157:1917.
Willig T.B., Breton-Gorius J., Elbim C., et al. Macrothrombocytopenia with abnormal demarcation membranes in megakaryocytes and neutropenia with a complete lack of sialyl-Lewis-X antigen in leukocytes – a new syndrome? Blood. 2001;97:826.
Wu X., Rush J.S., Karaoglu D., et al. Deficiency of UDP-GlcNAc:dolichol phosphate N-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum Mutat. 2003;22:144.
Wu X., Steet R.A., Bohorov O., et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518.
Yoshida-Moriguchi T., Yu L., Stalnaker S.H., et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88-92.
Yuen C.T., Chai W., Loveless R.W., et al. Brain contains HNK-1 immunoreactive O-glycans of the sulfoglucuronyl lactosamine series that terminate in 2-linked or 2,6-linked hexose (mannose). J Biol Chem. 1997;272:8924.
Zak B.M., Crawford B.E., Esko J.D. Hereditary multiple exostoses and heparan sulfate polymerization. Biochim Biophys Acta. 2002;1573:346.
Zeevaert R., Foulquier F., Cheillan D., et al. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur J Med Genet. 2009;52(5):303-305.
Zhang W., Vajsar J., Cao P., et al. Enzymatic diagnostic test for muscle-eye-brain type congenital muscular dystrophy using commercially available reagents. Clin Biochem. 2003;36:339.
Zielinska D.F., Gnad F., Wiśniewski J.R., et al. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141(5):897-907.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 35 Disorders of Glycosylation
Eukaryotic cells synthesize hundreds of types of sugar chains called glycans, which function within the cell, at the cell surface, and beyond. Within the cell, glycans influence protein folding, stability, turnover, and intracellular trafficking [Varki et al., 2009]. At the cell surface, they influence or determine cell-cell binding, receptor-ligand interactions, assembly of signaling complexes, binding to the extracellular matrix, tissue pattern formation, trafficking of lymphocytes, and much more [Varki et al., 2009]. The same glycan can function differently on different proteins or in different settings. This three-dimensional complexity makes understanding the roles of glycans challenging but provides the body with an extraordinarily sensitive fine-tuning mechanism for many physiologic functions. It is not surprising that disrupting normal glycosylation causes moderate to severe pathology in multiple human organ systems.
More than 40 rare inherited disorders of glycan biosynthesis have been identified [Jaeken et al., 2008; Grunewald, 2007; Freeze, 2007; Marquardt and Denecke, 2003]. Most of these are the newly defined congenital disorders of glycosylation (previously called carbohydrate-deficient glycoprotein syndrome and disialotransferrin developmental deficiency syndrome). Others, such as muscle-eye-brain disease and Walker–Warburg syndrome, were well known, but elucidation of their relation to glycosylation has provided new insights into their pathophysiology and opens possibilities for therapy.
N-Linked Glycosylation
Overview
Sugar chains are added to newly synthesized proteins in the lumen of the endoplasmic reticulum; they are quickly and extensively remodeled there, and later in the Golgi apparatus. All eukaryotic cells make a 14-sugar lipid-linked oligosaccharide in the endoplasmic reticulum membrane that is composed of Man, GlcNAc, and glucose (Glc). This entire chain is transferred to Asn within an Asn-X-Thr/Ser/Cys [Zielinska et al., 2010] consensus sequence (X is any amino acid except proline) as newly made proteins emerge from the ribosome into the endoplasmic reticulum lumen. Forty or more genes are required to synthesize and transfer this glycan to proteins [Freeze, 2001b].
Extensive remodeling begins soon after sugar chain transfer. Up to two-thirds of the original lipid-linked oligosaccharide glycan is discarded, and 6–15 other sugar units are added back to create a dazzling array of sugar chains. Why generate this complex process? The initial glycan helps proteins fold and also provides important checkpoints for monitoring of proper protein folding in the endoplasmic reticulum [Parodi, 2000]. The addition of more sugars in the Golgi usually imparts greater specificity to the sugar chain function. Understanding the biosynthetic pathways is important for appreciating the nature of the defects.
Biosynthesis
Individual monosaccharides are synthesized from glucose, derived from the diet, or salvaged from degraded glycans, and must be activated to their nucleotide sugar derivatives [Freeze, 1999]. Figure 35-1 depicts an abbreviated version of the known monosaccharide pathways. We employ widely used standard symbols to denote the different sugars [Varki et al., 2009]. Note that phosphorylation of the monosaccharide is the first step, and some pathways interconvert phosphorylated forms, such as Man-6-P→Man-1-P. Also, several alternative routes generate uridine diphosphate (UDP)-GlcNAc, UDP-galactose (Gal), and guanosine diphosphate (GDP)-fucose (Fuc). For some types of glycosylation, the nucleotide sugar donates the sugar to a lipid carrier derived from the polyprenol, dolichol phosphate (P-Dol) [Schenk et al., 2001a]. These products include Man-P-Dol and Glc-P-Dol. Dolichol itself is made from polyprenols using a specific reductase [Cantagrel et al., 2010].
N-linked Glycan Biosynthesis
Biosynthesis of the lipid precursor oligosaccharide involves a series of steps and enzymes that add sugars in specific linkages and in a specific order [Freeze, 2001a]. It begins with P-Dol + UDP-GlcNAc forming GlcNAc-P-P-Dol on the cytosolic face of the endoplasmic reticulum. Another UDP-GlcNAc donates a second GlcNAc using a different GlcNAc transferase, and this is followed by the addition of five Man units derived from GDP-Man. At this point, a “flippase” reorients the entire molecule from the cytosolic face into the endoplasmic reticulum lumen. A series of Man transferases use Man-P-Dol to add four more Man units, making a three-branched structure. Three glucosyltransferases sequentially add Glc from Glc-P-Dol to one branch to complete the sugar chain. This 14-sugar unit molecule is the optimal substrate for the multisubunit oligosaccharyl transferase (OST) complex that recognizes the Asn-X-Thr/Ser consensus sequence on the protein and adds the sugar chain to Asn. Recent studies indicate that some Asn-X-Cys sites can also be glycosylated [Zielinska et al., 2010; Ostasiewicz et al., 2010]. Truncated glycans are poorly transferred and sometimes fail to occupy normal glycosylation sites. Following glycan transfer, P-P-Dol is converted back to P-Dol and then to Dol. Recycling of these carriers is extensive.
Within a few minutes of transfer to protein, the sugar chain is processed (Figure 35-2; see also Figure 35-1). A set of two glucosidases removes the Glc units. For some proteins, processing stops here, but for the great majority, a mannosidase in the endoplasmic reticulum removes a single Man unit. The proteins are carried to the Golgi, where another mannosidase called Golgi mannosidase I removes up to three more Man units. At this point, processing starts to add sugars. UDP-GlcNAc donates a GlcNAc to one branch, making the sugar chain an acceptable substrate for Golgi mannosidase II, which removes an additional two Man units. This process clears the way for the addition of a second GlcNAc to a Man on the remaining branch. Depending on the protein, up to three more GlcNAc transferases add GlcNAc units to the Man residues in a specific order to initiate up to five branches. These branches are then extended with Gal, derived from UDP-Gal, and sialic acid (Sia), derived from cytidine monophosphate (CMP)-Sia. In some cases, GDP-Fuc donates a Fuc to one or more of the branches or to the GlcNAc linked to the protein. In other cases, sulfates, phosphates, or glucuronic acids are added to selected sugars. Individual branches may accumulate several repeating units of Galβ1,4GlcNAc before being capped by Sia. These reactions occur in selected cisternae of the Golgi, and each nucleotide sugar donor must be translocated from its origin in the cytoplasm or nucleus to the Golgi by a substrate-selective transporter [Gerardy-Schahn et al., 2001].
Enzymes and Cell Biology
The sugar-activating enzymes reside in either the cytoplasm or the nucleus. The N-linked biosynthetic enzymes all are in the endoplasmic reticulum or Golgi, with their active sites situated in the expected orientation. Most of the glycosyltransferases used for building the lipid-linked oligosaccharide and the Golgi transporters span the membrane many times [Ishida and Kawakita, 2004]. Most enzymes participating in glycan processing are attached to the membrane by a single membrane span. The transferases and nucleotide sugar transporters are constantly recycled in the dynamic Golgi to maintain their correct relationship to the maturing glycoproteins as they pass through the Golgi. Therefore, correct trafficking of the biosynthetic machinery is essential for optimal function.
Congenital Disorders of Glycosylation
Glycosylation is complex, and its disorders do not always lend themselves to phenotypic or symptomatic pigeonholing. Congenital disorders of glycosylation (CDG) nomenclature is still evolving as attempts are made to design a systematic, clinically useful system based on biochemical origins. Until 2008, CDGs resulting from defects in lipid-linked oligosaccharide biosynthesis and transfer to protein were defined as group I. The remaining CDGs that affect the biosynthesis and processing of the protein-bound sugar chains constituted group II. Individual disorders in group I or II were assigned lower-case letters (designating types, as noted earlier) when the genetic defect is proved. Several reviews provide comprehensive and up-to-date perspectives [Vodopiutz and Bodamer, 2008; Jaeken and Matthijs, 2007; Grunewald, 2007; Freeze, 2007; Marquardt and Denecke, 2003]. In 2008 a simplification of the nomenclature was proposed, as the growth in the number of disorders had produced a confusing series of letters and numbers that did not help the clinician or researcher. In the new system, diseases will be grouped in four categories: disorders of protein N-glycosylation, disorders of protein O-glycosylation, disorders of glycosphingolipid and glycosylphosphatidylinositol anchor glycosylation, and disorders of multiple glycosylation pathways. Individual disorders will be listed by the gene involved and, in parallel, its protein product. The authors suggest that the previous CDG designation may be listed in parentheses during the transitional period as the new nomenclature is adopted [Jaeken et al., 2008].
Diagnosis
In most patients with CDGs the diagnosis is straightforward and is based on the analysis of serum transferrin isoforms. Several methods are suitable and commercially available. These include isoelectric focusing [Freeze, 2001a; Jaeken and Matthijs, 2001], mass spectrometry [Wada, 2007; Kleinert et al., 2003; Lacey et al., 2001], zone electrophoresis, and high-pressure liquid chromatography [Quintana et al., 2009]. Electrospray ionization-mass spectrometry [Babovic and O’Brien, 2007] is the most informative because it easily distinguishes the absence of entire sugar chains, which characterizes group I CDGs, from the absence of one or more individual monosaccharide units typical of group II CDGs. The predominant isoform in normal transferrin has two sugar chains, each containing two negatively charged Sia molecules, designated tetrasialotransferrin. In group I disorders, isoelectric focusing analysis demonstrates loss of one or both entire glycan chains, producing “disialotransferrin” or “asialotransferrin,” respectively, but clearly this is an incomplete description. By contrast, electrospray ionization-mass spectrometry demonstrates that the loss of one or two chains reduces the mass by about 2200 or 4400 mass units, respectively. In group II disorders, isoelectric focusing may distinguish among “tri-,” “di-,” “mono-,” and “asialo” forms, reflecting variable loss of Sia, whereas electrospray ionization-mass spectrometry can indicate specific loss of Sia alone or combinations of Sia with additional sugars. This distinction is essential for determining assignment to group I or group II, and provides a signpost for specific identification of the defect. The single-step electrospray ionization-mass spectrometry technique can be accomplished using less than 5 mL of blood in less than 30 minutes [Lacey et al., 2001; Babovic and O’Brien, 2007]. This technique is therefore suitable for large-scale population screening for CDGs.
Transferrin isoform analysis produces few false-positive results. Uncontrolled fructose and galactose intolerance and recent heavy alcohol consumption produce a pattern typical of group I disorders [Kleinert et al., 2003]. In apparently rare instances, patients with genetically confirmed CDGs exhibited normal transferrin isoelectric focusing profiles, and in some patients, previously abnormal patterns normalized in preadolescence [Dupre et al., 2001; Fletcher et al., 2000]. Thus, a normal transferrin pattern should not exclude follow-up testing. Healthy neonates sometimes have a slightly abnormal transferrin pattern, which normalizes within a few weeks. Suspicious results in neonates should be repeated.
Until recently, specific diagnosis of the defect usually involved enzymatic, biochemical, and genetic analysis of the patient’s fibroblasts or leukocytes. Relatively few laboratories performed these analyses. With routine gene sequencing becoming more cost-effective, some genetic centers and commercial laboratories now offer various CDG gene diagnostic panels. In the coming years, decreasing costs, improved sequencing technology, and better informatics are likely to make this approach the first option. Prenatal testing is available for confirmed at-risk families [Matthijs et al., 1998, 2004].
General Clinical Features
About 1000 CDG patients have been identified, presenting with multiple organ dysfunction [Haeuptle and Hennet, 2009]. Patients with CDGs have protean presentations that, in some cases, may mimic mitochondrial (oxidative phosphorylation) disorders [Briones et al., 2001], but lack a history of maternal inheritance. An informal survey of CDG-affected families indicated that earlier nonspecific diagnoses frequently included a metabolic defect or cerebral palsy. Most patients first present to pediatric neurology or metabolic clinics. They frequently have combinations of liver, gastrointestinal, and coagulation disturbances [Jaeken and Matthijs, 2001; Marquardt and Denecke, 2003]. The possibility of a CDG should be investigated in any child presenting with developmental delay, seizures, hearing loss [Jaeken et al., 2009] or strabismus, particularly if any of these manifestations is accompanied by abnormal coagulation, liver dysfunction, or a gastrointestinal disorder. Most affected children are hypotonic and demonstrate failure to thrive.
Specific Disorders
Table 35-1 summarizes the known CDG defects, utilizing the classification scheme proposed in 2008. This includes the mutated genes and major signs and symptoms. The known defects cover every aspect of the N-linked biosynthetic pathway. Activation or presentation of precursors (PMM2, PMI, DPM1, DPM3 MPDU1 [CDG-Ia, Ib, Ie, If]), glycosyltransferases for lipid-linked oligosaccharide biosynthesis (ALG6, NOT56L, ALG12, ALG8, ALG2, DPAGT1, HMT1, DOBD1 [CDG-Ic, Id, Ig, Ih, Ii, Ij, Ik, Il]), glycosidases that trim the protein-bound sugar chain (GLS1 [CDG-IIb]), Golgi-localized nucleotide sugar transporters (SLC35C1, SLC35A1 [CDG-IIc and IIf]), and glycosyltransferases that extend the trimmed chain (MGAT2, BG4ALT1 [CDG-IIa and IId]). DK1 (CDG-Im) impairs dolichol kinase function and impairs the final step of the de novo synthesis of dolichol phosphate [Denecke and Kranz, 2009]. SRD5A3 encodes the α-reductase that converts various polyprenols to dolichols [Cantagrel et al., 2010]. The conserved eight-subunit oligomeric Golgi (COG) complex that binds to the cytoplasmic face of the Golgi is needed for intra-Golgi or Golgi to endoplasmic reticulum retro-trafficking of multiple resident glycosyltransferases and nucleotide sugar transporters. Disorganized trafficking impairs multiple glycosylation pathways [Ungar et al., 2002]. Defects have now been identified in COG7, COG1, COG4 [CDG-IIe, IIg, IIj], COG8, COG5 and COG6 [Lübbehusen et al., 2010; Foulquier, 2009]. Appreciation of the importance of Golgi homeostasis in glycosylation led to the discovery of another disorder caused by mutations in a subunit of a vacuolar H+/ATPase that maintains appropriate pH of various organelles within the endocytic and exocytic pathways [Marshansky and Futai, 2008; Kornak et al., 2008; Guillard et al., 2009]. The intravesicular pH progressively decreases from the endoplasmic reticulum to Golgi, endosomes, and, finally, lysosomes.
The remainder of this chapter will focus on CDGs with significant neurologic manifestations.
Defects in Protein N-glycosylation
PMM2 (CDG-Ia)
PMM2 (CDG-Ia) is the best-known and most frequently recognized form of CDGs, first reported by Jaeken and co-workers in 1980 [Jaeken et al., 1980]. The defective gene was identified in 1995 as PMM2, which encodes the phosphomannomutase (PMM) that converts Man-6-P → Man-1-P. This defect results in insufficient production of lipid-linked oligosaccharide, leading to empty glycosylation sites. More than 800 patients are known worldwide, and more than 100 mutations have been cataloged [Barone et al., 2008; Jaeken, 2003; Matthijs et al., 2000; Haeuptle and Hennet, 2009].
Hagberg and associates described four stages of the typical (severe) phenotype [Hagberg et al., 1993]. The first is the infantile phase, marked by various combinations of dysmorphism, abnormal fat distribution (supragluteal and vulval fat pads, focal lipoatrophy), inverted nipples, cryptorchidism, esotropia, recurrent infections, cardiomyopathy or pericardial effusions, coagulopathies, nephrotic syndrome, hypothyroidism, life-threatening episodes of hepatic failure, and unexplained coma. As many as 20 percent of infants with CDG-Ia succumb in this phase [Jaeken and Matthijs, 2001]. In the second phase (comprising the remainder of the first decade), children experience seizures and strokelike episodes, often precipitated by intercurrent infections. The third phase (in the second decade of life) is marked by slowly progressive cerebellar ataxia and limb wasting, and by progressive visual loss secondary to pigmentary retinopathy. Adult survivors have moderate mental retardation with severe ataxia and hypogonadism, with or without skeletal deformities. Presentations are highly variable. In one girl with CDG-Ia, findings on computed tomography (CT) of the head were normal at 9 months, but subsequent imaging studies demonstrated progressive atrophy [Mader et al., 2002]. The investigators concluded that the cerebellar hypoplasia reported in infancy in most children with CDG-Ia likely reflects atrophy of antenatal onset, rather than hypoplasia.
More extensive testing for CDGs has led to the identification of milder CDG-Ia phenotypes [Briones et al., 2002; de Lonlay et al., 2001; Grünewald, 2009]. The patients often have high residual levels of PMM2 activity [Drouin-Garraud et al., 2001; Grünewald et al., 2001; Westphal et al., 2001b]. Some patients have only borderline cognitive impairment, but strabismus persists in these very mild cases. Few adult CDG-Ia patients are employed. A longitudinal study of eight Spanish patients confirmed the wide range of clinical manifestations, ranging from neonatal hemorrhage, non-immune hydrops, and death, through mental retardation and motor impairment without acute decompensation in patients in their 20s, to one individual with normal development and only gastrointestinal dysfunction in childhood [Perez-Duenas et al., 2009].
The carrier frequency of the most common mutant allele is about 1 in 70 in the northern European population [Schollen et al., 2000]. It is believed to be lethal in the homozygous state. Genotype-phenotype correlations have not been informative, but some evidence suggests that frequent polymorphisms in other glycosylation-related genes may influence the severity of the phenotype. No effective specific therapy for CDG-Ia exists. Experiments using CDG-Ia cells suggested that increasing dietary mannose might improve glycosylation in patients, but clinical trials demonstrated no benefit [Kjaergaard et al., 1998; Marquardt et al., 1997; Mayatepek et al., 1997].
Population studies find the risk of having a second child with CDG-Ia to be close to 1 in 3, rather than the expected mendelian ratio of 1 in 4, suggesting that reduced glycosylation may have some selective advantage [Schollen et al., 2004b]. At-risk couples should be counseled appropriately.
MPI-CDG (Ib)
MPI-CDG (Ib) is caused by mutations in MPI, the gene encoding phosphomannose isomerase, which interconverts Man-6-P and Fructose (Frc)-6-P. This reaction produces most of the mannose for glycoprotein synthesis [de Koning et al., 1998; Niehues et al., 1998]. About 25 patients have been identified since its discovery in 1998 [de Lonlay and Seta, 2009]. This phenotype is not associated with any primary neurologic symptoms. Gastrointestinal and hepatic pathology, with hypoglycemia, coagulopathy, and protein-losing enteropathy, is characteristic.
MPI-CDG (Ib) is unique in that simple dietary mannose therapy corrects the abnormalities, except for liver fibrosis [de Lonlay and Seta, 2009; Durand et al., 2003; Niehues et al., 1998]. The dietary mannose is taken up through transporters and converted to Man-6-P, thereby bypassing the metabolic block. Mannose has no known side effects at the concentrations used. Nonenzymatic protein glycation occurs at high concentrations of mannose, which is considerably more reactive than glucose, and can raise hemoglobin A1c (HbA1c) levels [Harms et al., 2002]. Some patients have received such treatment for longer than 10 years without complications [Westphal et al., 2001a; de Lonlay and Seta, 2009]. Twenty percent of the patients who were subsequently confirmed to have phosphomannose isomerase deficiency, however, died before discovery of the disorder [Freeze, 2001a]. The potentially fatal outcome and availability of simple, effective treatment mandate investigation for CDGs in suspected cases. One adult patient with MPI deficiency was not able to tolerate oral mannose, but her protein-losing enteropathy appeared to respond to heparin therapy [Liem et al., 2008].
ALG6-CDG (Ic)
ALG6-CDG (Ic) resembles, but is less severe than PMM2-CDG. It is characterized by moderate psychomotor retardation, hypotonia, esotropia, seizures, and ataxia. Nevertheless, at least five children have died of CDG-related complications [Marquardt and Denecke, 2003; Newell et al., 2003; Westphal et al., 2000]. The defect is in a glycosyltransferase, hALG6, that results in production of a truncated lipid-linked oligosaccharide sugar chain, which is inefficiently transferred to proteins. Patients sometimes experience life-threatening protein-losing enteropathy during bouts of gastroenteritis [Westphal et al., 2000]. Skeletal dysplasia, including a unique form associated with brachytelephalangy has been described in a compound heterozygote for ALG6 [Drijvers et al., 2010]. An adult woman has been identified in whom ALG6 deficiency was associated with mental retardation, skeletal anomalies, virilization, and deep vein thrombosis [Sun et al., 2005]. ALG6 deficiency was first identified in 1998 and subsequently in more than 35 patients [Haeuptle and Hennet, 2009; Burda et al., 1998; Grünewald et al., 2000; Imbach et al., 2000a], making it the second most common form of CDG.
NOT56L-CDG (Id)
Patients with NOT56L-CDG have mutations in NOT56L (ALG3), which encodes the mannosyltransferase used to synthesize Man6GlcNAc2 [Körner et al., 1999]. The index patient had microcephaly, optic nerve atrophy, iris colobomas, epilepsy, spastic quadriparesis, and profound psychomotor delay. Another patient had similar features plus Dandy–Walker malformation, with agenesis of the cerebellar vermis and corpus callosum. She also had recurrent hypoglycemia, thrombocytopenia, hypoalbuminemia, and coagulopathy. A total of eight children with NOT56L-CDG have now been described; all have a severe phenotype, with 7 of the 8 manifesting neurological impairments, including seizures, visual impairment, psychomotor retardation, and cerebral or cerebellar hypoplasia; there were varying combinations of hepatic, hematologic, endocrine, and dysmorphic manifestations, as well [Kranz et al., 2007b].
ALG12-CDG (Ig)
Eight cases of ALG12-CDG (Ig) have been reported [Chantret et al., 2002; Grubenmann et al., 2002; Thiel et al., 2002; Eklund et al., 2005; Kranz et al., 2007c]. The patients had typical CDG abnormalities, including hypotonia, generalized developmental delay, cerebellar hypoplasia, and decreased coagulation factors. Dysmorphic features included a long thin face, flat nasal bridge, and epicanthal folds. Frequent infections probably reflect reduced immunoglobulin G (IgG) concentrations. Affected males have genital hypoplasia, a feature not reported in other types of CDGs. More recent reports have emphasized features of skeletal dysplasia. These patients have mutations in hALG12, which encodes dolichol-P-mannose: Man7GlcNAc2-PP-dolichyl mannosyltransferase. Patients accumulate the truncated lipid-linked oligosaccharide, which is inefficiently transferred to proteins.
ALG8-CDG (Ih)
Eight patients have been described with ALG8-CDG (Ih), caused by a deficiency in hALG8, the gene encoding the second glucosyltransferase in lipid-linked oligosaccharide synthesis [Chantret et al., 2003; Schollen et al., 2004a; Stolting et al., 2009; Vesela et al., 2009]. Three patients had mild clinical presentations, including two siblings with pseudogynecomastia, epicanthus, hypotonia [Stolting et al., 2009], mental retardation, and ataxia, whereas the others all had severe multi-organ failure. Most died within a few months, but one patient with severe developmental delay survived a year before succumbing to renal failure. The others experienced hepatointestinal symptoms; one girl had cortical, cerebellar, and optic atrophy and intractable seizures before her death from systemic complications at 2 months [Vesela et al., 2009].
ALG2-CDG (Ii)
The only child with ALG2-CDG (Ii) recognized to date was normal at birth, except for an iris coloboma. Seizures, hepatomegaly, and coagulation abnormalities emerged in the first year. Imaging studies revealed cerebral hypomyelination. Pathologic mutations were found in hALG2, the enzyme that adds the second mannose to the growing lipid-linked oligosaccharide sugar chain [Thiel et al., 2003].
