Chapter 23 Disorders of Forebrain Development
Introduction
The prosencephalon forms at the end of primary neurulation as one of three principal vesicles: the hindbrain, the midbrain, and the forebrain (prosencephalon). Detailed embryology of the normal prosencephalon is beyond the scope of this chapter, but is covered comprehensively in Rao and Jacobson [2005]. The primary disorders of prosencephalic formation, prosencephaly and atelencephaly, are extremely rare and not further discussed in this chapter. The major disorders of prosencephalic formation, holoprosencephaly, agenesis of the corpus callosum, and septo-optic dysplasia, are discussed below, after a brief introduction to prosencephalic development.
Prosencephalon Patterning
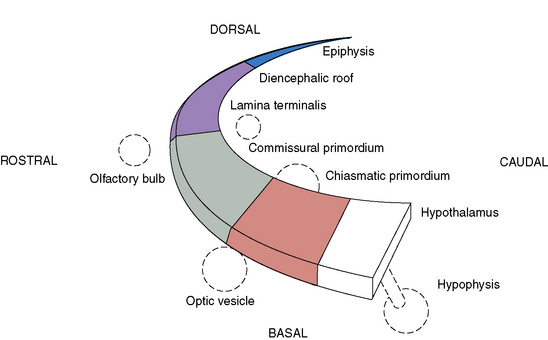
Prosencephalic development occurs by inductive interactions under the primary influence of the prechordal mesoderm. The peak time period of development is the second and third months of gestation, with the earliest prominent phases in the fifth and sixth weeks of gestation. The major inductive relationship of concern is between the notochord/prechordal mesoderm and the forebrain. This interaction occurs ventrally at the rostral end of the embryo, and thus the term ventral induction is sometimes used. The inductive interaction influences formation of much of the face and of the forebrain; accordingly, severe disorders of brain development at this time usually result in striking facial anomalies. Development of the prosencephalon is considered best in terms of three sequential events: prosencephalic formation, prosencephalic cleavage, and midline prosencephalic development (Figure 23-1). Prosencephalic formation begins at the rostral end of the neural tube at the end of the first month and the beginning of the second month of gestation. It consists of segmentation of the prosencephalon into three major domains, termed prosomeres (P1–P3). P1 gives rise to the pretectum, the region of brain immediately rostral to the midbrain-derived tectum. P2 is associated with development of the thalamus, and P3 gives rise to the prethalamus. More rostral brain regions, including the telencephalon, are also divided into prosomeric boundaries, the clearest of which are the anatomic boundaries of the developing basal ganglia, the medial and lateral basal ganglia, which arise as bulges along the ventrolateral telencephalon and express the markers Dlx and Arx. The neocortex itself exhibits regionally restricted gene expression. Manipulation of these expression patterns results in shifting of cellular identities of the neocortex (FGF8, EMX2). The evidence to date, however, argues against anatomically and regionally restricted boundaries, because cell lineage experiments demonstrate that sibling cells can occupy multiple nuclei throughout the anteroposterior axis.
Prosencephalic Cleavage
Three crucial thickenings or plates of tissue become apparent around the end of the second month; these are the commissural, the chiasmatic, and the hypothalamic plates. These structures are important in the formation, respectively, of the corpus callosum and septum pellucidum, the optic nerve chiasm, and the hypothalamic structures. The most prominent of these embryologic changes is the formation of the corpus callosum, the earliest component of which appears at 9 weeks. Disorders associated with abnormal development of the prosencephalon are outlined in Table 23-1.
| Region Affected | Disorder |
|---|---|
| Commissural plate | Agenesis of the corpus callosum and/or septum pellucidum |
| Commissural and chiasmatic plates | Septo-optic dysplasia |
| Commissural, chiasmatic, and hypothalamic plates | Septo-optic-hypothalamic dysplasia |
Holoprosencephaly
Holoprosencephaly (HPE) is a complex brain malformation characterized by a failure of the forebrain (prosencephalon) to separate completely into two distinct cerebral hemispheres (i.e., distinct telencephalon and diencephalon). This process is normally completed by the fifth week of gestation [Golden, 1999].
HPE is typically associated with midline facial anomalies.
Historical Background
Individuals with cyclopia have been described for centuries in mythology and in the scientific literature since the late 18th century [Siebert et al., 1990]. In 1882, Kundrat first described the cerebral changes of HPE, including absent olfactory nerves. Believing the absence of olfactory lobes and bulbs was the cardinal feature, he termed the condition “arhinencephaly” [Kundrat, 1882]. Yakovlev recognized involvement of the entire telencephalon and called the single telencephalic ventricle a “holosphere” and the malformation “holotelencephaly” [Yakovlev, 1959]. DeMyer found that the thalamus and other diencephalic structures were also involved and coined the still-favored term “holoprosencephaly” to indicate that the defect involved the entire prosencephalon [DeMyer and Zeman, 1963].
Epidemiology
HPE is the most common developmental defect of the forebrain and midface in humans, and occurs in 1 in 250 pregnancies [Matsunaga and Shiota, 1977], but because only 3 percent of the fetuses with HPE survive to delivery, the incidence in live births is only approximately 1:10,000 [Bullen et al., 2001; Croen et al., 2000; Forrester and Merz, 2000; Rasmussen et al., 1996]. Two-thirds of affected patients have the most severe subtype of HPE [Ming and Muenke, 1998]. However, high-resolution magnetic resonance imaging (MRI) has increased identification of children with less severe forms who are not diagnosed until later in infancy. Therefore, the true live birth prevalence of HPE may be higher than previously estimated, and the proportion of cases with milder subtypes appears to be increasing in more recently reported series [Stashinko et al., 2004]. There also appears to be a slight female preponderance in some case series. A few studies of limited size suggest a higher than average prevalence of HPE in Far East Asians and Filipinos [Forrester and Merz, 2000].
Definition and Subtypes of Holoprosencephaly
The sine qua non of HPE is an incomplete separation of the cerebral hemispheres that results in lack of cleavage (nonseparation) of midline structures involving the telencephalon and diencephalon. HPE typically is divided into three main subtypes, delineated by DeMyer and co-workers [DeMyer, 1987; DeMyer and Zeman, 1963] and distinguished by the degree of separation of the cerebral hemispheres (Figure 23-2).
Fig. 23-2 Subtypes of holoprosencephaly.
(From Hahn JS, Plawner LL. Evaluation and management of children with holoprosencephaly. Pediatr Neurol 2004;31:79.)
In addition to DeMyer’s classic HPE types, another subtype is identifiable, the middle interhemispheric variant [Simon et al., 2002]. In this variant, the midportion of the cerebral hemispheres is continuous across the midline, with absence of the corpus callosum seen only in this region. There is separation of the anterior frontal lobes, basal forebrain, and occipital lobes. Evidence for this malformation being a subtype of HPE is bolstered by the finding that a mutation in the ZIC2 gene, which has been implicated in causing the classic forms of HPE, also has been found in patients with the middle interhemispheric variant [Brown et al., 2001; Solomon et al., 2010].
In addition to nonseparation of the cerebral hemispheres, failure of separation also is common in the hypothalamic, caudate, lentiform, and thalamic nuclei. About one-quarter of patients have some degree of midbrain nonseparation [Simon et al., 2000]. Occasionally, isolated neuronal heterotopias are seen, particularly in the middle interhemispheric variant. The gyri often are normally developed, although in alobar and semilobar HPE the gyri may be excessively smooth or broad [Barkovich et al., 2002].
Although HPE is typically divided into these subtypes, the degree of malformation and brain regions involved occur along a spectrum, and individual patients frequently do not fall neatly into any one category [Hahn and Barnes, 2010]. Milder cases are being increasingly identified with use of high-resolution MRI. For example, a mild form of lobar HPE (septo-preoptic type) can be recognized, with minimal cortical fusion restricted to the preoptic regions (involving the suprachiasmic region and anterior hypothalamus) and/or the septal region (subcallosal region) [Hahn et al., 2010].
Neuropathological Findings
Cytoarchitecturally, the brain usually shows normal cortical layering; in some rare cases, the histopathology may show disorganization or abnormal lamination [Golden, 1999]. The defects in cortical organization may represent secondary injury to the cerebral cortex or an abnormality in connections into and out of the cerebral cortex. Periventricular and white-matter glioneuronal heterotopia are also encountered in rare cases. The hippocampus is virtually always present, although it may show incomplete or abnormal development [Golden, 1999]. The cerebellum may show various degrees of cell migration abnormalities, such as misplaced nodules that are aberrantly located in the white matter or within the dentate nuclei [Larroche, 1977].
Etiology
Multiple environmental and genetic factors have been implicated in causing HPE. Prenatal exposures to a variety of toxins, medications, and infections also have been reported. Anecdotal reports suggest viruses, low-calorie diets, hypocholesterolemia, maternal diabetes, and use of salicylates, alcohol, and contraceptives as possible causes [Johnson and Rasmussen, 2010]. The strongest teratogenic evidence exists for maternal diabetes [Barr et al., 1983] and exposure to alcohol [Sulik and Johnston, 1982] and retinoic acid [Croen et al., 2000]. A diabetic mother’s risk of having a child with HPE is approximately 1 percent, a greater than 100-fold increase over the general population. A recent population study confirmed the risks of pre-existing maternal diabetes and salicylates (aspirin), but also noted an increased risk with artificial reproductive therapy [Miller et al., 2010]. Some teratogens are thought to produce HPE via interference with the sonic hedgehog gene signaling pathways, mediated by perturbations of either cholesterol biosynthesis or the ability of target tissue to sense or transduce the sonic hedgehog signal [Cohen and Shiota, 2002].
Approximately 30–50 percent of live births with HPE have chromosomal abnormalities [Bullen et al., 2001; Croen et al., 1996], but this is likely an overestimation based on under-reporting of milder cases. HPE can be seen in association with trisomy 13, trisomy 18, or triploidy. HPE can also be seen in several malformation syndromes having normal karyotypes, such as Pallister–Hall syndrome, Rubinstein–Taybi syndrome, and Smith–Lemli–Opitz syndrome [Siebert et al., 1990]. An updated list of genetic disorders associated with HPE can be found on the Online Mendelian Inheritance in Man (OMIM) website (http://www.ncbi.nlm.nih.gov/omim).
In nonsyndromic and nonchromosomal HPE, multiple pedigrees have been described, manifesting various inheritance patterns, most commonly autosomal-dominant and autosomal-recessive inheritance. At least nine genes have been associated with HPE, including SHH (7q36), ZIC2 (13q32), SIX3 (2p21), TGIF (18p11.3), PATCHED-1 (9q22), GLI2 (2q14), DISP1 (1q24), NODAL (10q), and FOXH1 (8q24.3) [Roessler and Muenke, 2010]. Of these genes, the four most commonly affected (SHH, ZIC2, SIX3, and TGIF) account for only 25 percent of the cases of HPE with normal chromosomes [Dubourg et al., 2004; Roessler and Muenke, 2010] and approximately 5–10 percent of all HPE patients.
SHH was the first identified HPE-associated mutated gene and has been the most extensively studied [Belloni et al., 1996]. The Shh protein is a secreted intercellular signaling molecule involved in establishing cell fates at several points during development. Shh is expressed early in development in the ventral forebrain and is critical for ventral patterning of the developing neural tube. It also is expressed in the ectoderm of the frontonasal and maxillary processes. Disruption of Shh signaling in several animal models produces the brain and facial malformations seen in HPE.
The SHH signaling network is the common pathway through which multiple environmental and genetic influences interact to cause HPE. Several of the other HPE genes are important components of the SHH signaling network; PTCH is a receptor for SHH and GLI2 is a mediator of SHH target gene transcription. A well-studied example of environmental influences acting through the SHH signaling network is the association of HPE with various causes of hypocholesterolemia [Ming and Muenke, 2002]. SHH is modified by cholesterol, which is felt to be essential for its proper functioning. Maternal hypocholesterolemia has been implicated in causing HPE. The incidence of HPE is also increased in Smith–Lemli–Opitz syndrome, which is due to a defect in 7-dehydrocholesterol reductase, the final enzyme in cholesterol synthesis [Kelley et al., 1996]. In addition, an outbreak of cyclopia (severe midline facial malformation) in sheep was believed to be caused by ingestion of plants containing cyclopamine and jervine, compounds that interfere with sonic hedgehog signaling by binding to and thereby sequestering the protein “smoothened” [Keeler and Binns, 1968].
In addition to the SHH gene, the Nodal signaling pathway also appears to be implicated in HPE. This nodal pathway includes TGIF, NODAL, GDF1, TDGF1, and FOXH1. A complex genetic interaction involving these gene products and which causes a reduction in nodal signaling may produce HPE [Roessler and Muenke, 2010; Roessler et al., 2008].
Studies of genotype–phenotype correlation in mutations of SHH have found that the same mutation can result in extremely variable phenotypes. Even within the same family, an identical mutation in SHH may result in a severe brain malformation, a “mild sign” known as a microform (such as a single median maxillary central incisor), or no phenotypic abnormality. It is estimated that, among carriers of an abnormal gene, 37 percent will have HPE, 27 percent will have a microform, and 36 percent will have no clinical abnormality [Cohen, 1989]. This great phenotypic variability with the same genetic mutation has suggested that, although HPE was thought of as a single-gene disorder, it should be considered a multifactorial disorder, with the specific phenotype being the result of multiple genetic and environmental influences [Ming and Muenke, 2002]. A large majority of HPE cases, therefore, appear likely to be due to a disturbance of a complex network of interacting genes and signaling centers [Roessler and Muenke, 2010]. In addition, the timing of teratogenic exposure appears to be critical to the specific resulting phenotype. An experimental model using cyclopamine, an inhibitor of cholesterol synthesis, has revealed that varying the time of exposure resulted in a continuum of midline facial malformation [Cordero et al., 2004].
Clinical Manifestations and Outcomes
Along with the midline brain malformation seen in HPE, a corresponding midline facial malformation may be present. In its most severe and usually lethal form, cyclopia, with the presence of a single midline eye and a proboscis (rudimentary single-nostril nose) above the eye, can be present. Survivors may have hypotelorism, a flattened nasal bridge, median cleft lip and palate, or a single median maxillary central incisor. The oft-quoted statement “the face predicts the brain” [DeMyer et al., 1964] refers to the observation that the degree of facial malformation frequently reflects the degree of brain malformation. This was amended later to “the face predicts the brain approximately 80 percent of the time,” in recognition of individuals with alobar HPE with a normal facial appearance, as well as cases of milder brain malformation associated with abnormal facies [Cohen, 1989].
Previous studies indicated that children with HPE do not survive beyond early infancy. This may have been due to the identification of only the most severe cases. Early death is typical for most cytogenetically abnormal children and those individuals with the most severe facial features (cyclopia or ethmocephaly) [Croen et al., 1996]. In children without these risk factors, more recent studies have indicated that long-term survival is not uncommon. Among cytogenetically normal patients with all types of HPE, more than 50 percent are alive at 12 months [Barr and Cohen, 1999; Olsen et al., 1997]. In one series of 104 HPE patients, the mean age was 4 years, and 15 percent were between 10 and 19 years of age [Hahn and Plawner, 2004]. Even with the most severe type, alobar HPE, children may survive. In an observational study of alobar HPE, 30 percent were alive at 12 months, with long-term survival noted as well [Barr and Cohen, 1999]. When death did occur in the alobar group, causes were associated with brainstem dysfunction, pneumonia, dehydration from diabetes insipidus, and rarely, intractable seizures.
Children with HPE may experience a variety of medical and neurologic problems. A significant proportion develop hydrocephalus. The likelihood of requiring ventriculoperitoneal shunting is much higher in alobar than in semilobar or lobar HPE (60 percent versus 8 percent, respectively) [Plawner et al., 2002]. The risk of developing hydrocephalus also is higher when a dorsal cyst is present [Simon et al., 2001]. The dorsal cyst is thought to form as a result of the thalami being fused across the midline, thereby blocking cerebrospinal fluid egress from the third ventricle and disrupting cerebrospinal fluid. Most children that have HPE without hydrocephalus are microcephalic.
As in children with other midline brain defects, endocrinologic problems are very common. Diabetes insipidus is particularly frequent, and in one series of 68 children, three-quarters of individuals were affected [Plawner et al., 2002]. Diabetes insipidus can be insidious in presentation; serum sodium concentrations of 160 mEq/L or greater have been occasionally detected on routine electrolyte evaluation and were not accompanied by any acute symptoms. Diabetes insipidus also can have a fluctuating course. Growth hormone deficiency, hypocortisolism, and hypothyroidism also may occur. The endocrinopathies may be due to midline defects involving the hypothalamus and are rarely due to a dysgenetic (e.g., hypoplastic or ectopic) pituitary gland.
Approximately half of the children with HPE have epilepsy, and the likelihood of developing seizures does not correlate with the severity of the brain malformation. The most common seizure type is complex partial seizures, with or without secondary generalization. Seizure type, however, can be variable, including generalized tonic-clonic, tonic, atonic, myoclonic seizures or infantile spasms [Hahn and Plawner, 2004]. In 50 percent of affected children, the seizures are relatively easy to control with antiepileptic medication. Having a concurrent area of cortical dysplasia is a risk factor for having difficult-to-control seizures.
Feeding problems and swallowing dysfunction are common in children with HPE, and are correlated with the severity of the brain malformation. Two-thirds of patients with alobar and semilobar HPE require gastrostomy tubes [Plawner et al., 2002]. Abnormalities of muscle tone are also very common in HPE, and the severity correlates with the severity of the brain malformation. Many children have early hypotonia, followed by the development of upper limb and oromotor dystonia, and lower limb spasticity [Hahn and Plawner, 2004].
Developmental disability affects nearly all patients with HPE. The severity of the brain malformation determines the degree of delay and neurological impairments. Severe developmental delay is present in alobar HPE [Barr and Cohen, 1999]. There is no reported case of a child with alobar HPE who is able to sit independently. In lobar HPE, approximately 50 percent of children ambulate (with or without assistance), use their hands functionally, and have some verbal communication [Plawner et al., 2002]. Formal neuropsychologic evaluation in all types of HPE demonstrates relative strengths in receptive language and socialization, and weaknesses in visual reasoning and nonverbal problem skills [Kovar et al., 2001].
Management
Children with alobar HPE, a dorsal cyst, or normo- or macrocephaly should be closely observed for the development of hydrocephalus. Most experts recommend ventriculoperitoneal shunting for hydrocephalus, even in severe HPE, because failure to institute this measure will lead to progressive head enlargement and greater difficulty in caring for the child [Barr and Cohen, 1999; Hahn and Plawner, 2004]. Electrolyte screening should be performed to assess for the subacute development of diabetes insipidus. Screening for other endocrine abnormalities should be considered less frequently, with assays of cortisol, thyroid-stimulating hormone, free thyroxine, and insulin-like growth factor-1 (IGF-1). Because seizures may not develop in half of the children with HPE, prophylactic antiepileptic drugs are not recommended. If seizures do develop, the possibility of acute reactive seizures should be evaluated for and a serum sodium level checked. MRI also should be considered to evaluate for focal heterotopia. Gastrostomy tubes often are necessary to address the complex management issues of ensuring adequate calories related to feeding dysfunction and delivery of adequate free water necessary for management of diabetes insipidus. Motor difficulties and dystonia may be partially responsive to trihexyphenidyl. This agent may improve upper extremity and oromotor function [Hahn and Plawner, 2004]. Motor dysfunction, including hypertonia and dystonia, is common and may require physical, pharmacological, or surgical therapies.
Prenatal Diagnosis and Imaging
The sensitivity of first-trimester ultrasound examination in the detection of HPE remains unclear. It can detect alobar HPE [Filly et al., 1984; Sepulveda et al., 2004], but may be much less sensitive in detecting milder cases. A recent study from Germany reported a series of 51 fetuses (79 percent having a chromosomal abnormality) diagnosed with HPE by ultrasound at a mean age of 22 weeks [Wenghoefer et al., 2010]. The presence of large dorsal cysts, hydrocephalus, or midline craniofacial defects may provide clues that eventually lead to the recognition of the associated HPE.
Fetal MRI has been used to diagnosis various forms of HPE, including alobar, semilobar, lobar [Dill et al., 2009; Wong et al., 2005], and middle interhemispheric variants [Pulitzer et al., 2004] (Figure 23-3). Other midline anomalies, such as agenesis of the corpus callosum (isolated or with interhemispheric cysts), absence of the septum pellucidum, and hydrocephalus with communication of the lateral ventricles, are sometimes misdiagnosed prenatally as HPE [Malinger et al., 2005].
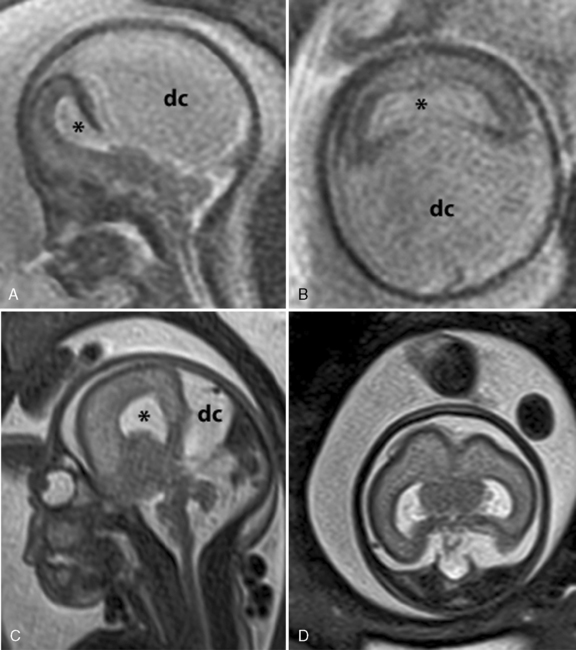
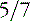 -week gestational age fetus with alobar HPE. The thalami, basal ganglia, and midbrain structures are incompletely delineated on the midline sagittal image (A). A large dorsal cyst (dc) communicates with the monoventricle (asterisk). The hemispheres are not separated, resulting in a holosphere (B). C and D, Fetal MRI of a 26-week gestational age fetus with trisomy 13 and semilobar HPE. The HASTE fetal sequence in the midsagittal plane (C) shows a monoventricle (asterisk), a moderate size dorsal cyst (dc), and inferior cerebellar vermis hypoplasia (Dandy–Walker complex). On the axial image (D), the thalami and basal ganglia appear fused, while the posterior hemispheres are separated.
-week gestational age fetus with alobar HPE. The thalami, basal ganglia, and midbrain structures are incompletely delineated on the midline sagittal image (A). A large dorsal cyst (dc) communicates with the monoventricle (asterisk). The hemispheres are not separated, resulting in a holosphere (B). C and D, Fetal MRI of a 26-week gestational age fetus with trisomy 13 and semilobar HPE. The HASTE fetal sequence in the midsagittal plane (C) shows a monoventricle (asterisk), a moderate size dorsal cyst (dc), and inferior cerebellar vermis hypoplasia (Dandy–Walker complex). On the axial image (D), the thalami and basal ganglia appear fused, while the posterior hemispheres are separated.Genetic Counseling and Testing
The recurrence risk of isolated HPE is estimated to be 6 percent [Roach et al., 1975]. Special attention should be given to the family history, to identify “microforms,” such as anosmia or a single central incisor. Such a finding would indicate a substantially higher recurrence risk. Genetic testing for SIX3, SHH, TGIF, and ZIC2 is commercially available and should be considered if familial HPE is suspected. Prenatal testing for these genes is possible by means of amniocentesis or chorionic villus sampling. The gene tests, in conjunction with fetal MRI, have been found to be helpful in prenatal diagnosis and counseling in a series of pregnancies [Mercier et al., 2010]. High-resolution cytogenetic analysis is important in every fetus or newborn with HPE, since abnormalities can be identified in 24–45 percent of all individuals. Newer molecular methods, including subtelomeric multiplex ligation-dependent probe amplification and comparative genomic hybridization microarray testing, have identified chromosomal deletions, duplications, and unbalanced rearrangements in some patients with HPE. These tests may be useful as an addition to the chromosomal and HPE gene mutational analyses [Pineda-Alvarez et al., 2010].
Agenesis of the Corpus Callosum
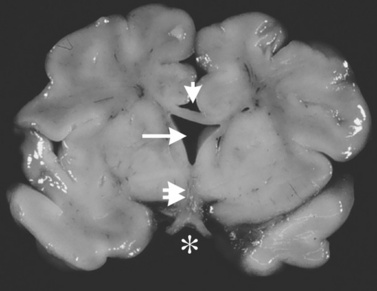
The corpus callosum forms between the 8th and 14th weeks of fetal development. Nearly 200 million axons course through this structure and innervate each opposing hemisphere in a homotopic manner; that is, each axon innervates structures in the mirror location in the opposing hemisphere. Absence or diminution of this structure is found in over 1 in 3000 live births, and is the most common birth defect of the central nervous system after spina bifida [Glass et al., 2008]. Callosal abnormalities are frequently seen with other malformations of brain development, including polymicrogyria, heterotopia, and midbrain and hindbrain abnormalities, but can present in an isolated manner (Figure 23-4). Many cases are also associated with birth defects in other organ systems, including ophthalmologic, cardiac, and renal defects. Agenesis of the corpus callosum (ACC) can be caused by chromosomal disorders, and there is increasing recognition that subtle copy number variants (CNVs) of particular genes may play a critical role in the etiology of ACC [Sherr et al., 2005; O’Driscoll et al., 2010]. Single-gene mutations and metabolic disorders can also disrupt callosal development. The best-known and most studied environmental cause of ACC, fetal alcohol syndrome, can also result in a significant reduction in white-matter volume. There is a great diversity of clinical outcomes for patients with ACC. Many individuals with ACC have deficits in social cognition, even with a normal IQ. Many of these individuals carry clinical diagnoses that place their phenotype on the autism spectrum. Conversely, individuals with normal IQ and social deficits are often found on MRI to have isolated ACC; no other brain malformations are found. In contrast, in children with ACC and associated brain anomalies, many have seizures and significant developmental impairment, including intellectual disabilities and cerebral palsy. In some individuals with ACC due to chromosomal, metabolic, or single-gene disorders, the outcome can be quite severe, including a significantly shortened life expectancy.
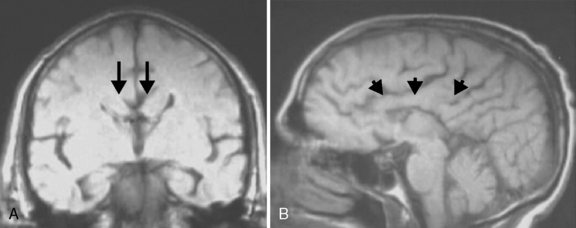
Fig. 23-4 Agenesis of the corpus callosum without other associated central nervous system malformations.
Historical Background
The corpus callosum was mentioned in the literature as early as the 2nd century by Galen, but the first clear description came in 1543 from Vesalius [Barkovich, 1996], who hypothesized that it served mainly as mechanical support for the ventricles and the fornices [Greenblatt and Dagi, 1997]. In addition to this “architectural” function, in the 17th century, Thomas Willis, Giovanni Lancisi, François de la Peyronie, and others thought that the corpus callosum was the “seat of the soul,” perhaps analogous to a more recent search for the “seat of consciousness” [Turner, 1955]. In 1908, Liepmann and colleagues reported a patient with unilateral apraxia and agraphia due to a callosal lesion. This was perhaps the first suggestion that callosal communication was necessary to integrate function of the two cerebral hemispheres. This concept was elegantly investigated by Sperry, Gazzaniga, and others. They demonstrated the concept of the “split brain” in patients who had undergone a callosotomy for treatment of intractable epilepsy [Gazzaniga, 2000]. These seminal studies presented clear evidence for lateralization and specialization of cerebral function and dependence on integration of the two hemispheres for many cognitive and behavioral tasks [Bloom and Hynd, 2005]. Surprising, however, was the additional observation that severing the corpus callosum, even in a young child, did not necessarily impact development or cognition significantly. None the less, the presence of altered cognitive abilities, even in individuals with ACC having normal IQ, suggests that the corpus callosum plays an important role in brain development, a process that is being better defined by on-going investigation [Paul et al., 2007]. This also underscores the likely significant differences between severing the corpus callosum after birth (and presumably after much of synaptogenesis) and preventing its formation from the onset.
Epidemiology
Agenesis of the corpus callosum is found in 1 in 4000 live births, as assessed by a population-based birth defect study in California [Glass et al., 2008]. This screening method only tracks birth defects (including ACC) that are identified in the first year of life. Because many cases of ACC are detected after the first year, realistic estimates of ACC incidence hover around 1 in 3000. Smaller studies support an incidence ranging from 1 in 1000 to 1 in 7000 [Cockerell et al., 1995; Wang et al., 2004]. In the California cohort, there was a slightly higher prevalence of ACC in African American babies and a lower prevalence in babies of Asian mothers [Glass et al., 2008]. Like many birth defects, the incidence rate of ACC is higher in births from mothers over age 40, with a nearly 3-fold increased risk compared to women in their 20s. There is also a slightly higher prevalence in children born to older fathers, as has also been reported recently for autism [Durkin et al., 2008]. Babies with ACC are nearly four times more likely to be born prematurely than the general population. It should be noted that detection of ACC occurs more commonly with better ultrasound surveillance, and its detection in utero may alert physicians to the increased probability of premature birth [Plasencia et al., 2007]. In addition, knowing about ACC should also alert the clinician to screen for other organ malformations. In infants with nonchromosomal ACC, approximately 20 percent have cardiac malformations, and this has been shown to increase to 61 percent if karyotype-visible cytogenetic abnormalities are present. Musculoskeletal, renal, and gastrointestinal abnormalities were also commonly observed in this series [Glass et al., 2008].
Prenatal Diagnosis and Prediction of Outcomes
The majority of information on the epidemiology of callosal agenesis comes from studies of postnatally diagnosed cases. Because callosal agenesis can be observed only with a brain imaging study, individuals who have a clinical phenotype warranting imaging (ranging from head trauma to epilepsy and global developmental delay) will be diagnosed, while those who are asymptomatic will not be ascertained at the same rate. From these postnatal cases it is unclear what the full spectrum of outcomes is for callosal agenesis. Moreover, there are anecdotal case studies reporting normal outcomes in individuals with callosal agenesis, further underscoring the need to understand outcomes from a perspective free of ascertainment bias [Ramelli et al., 2006]. Routine fetal imaging, particularly as it becomes more common, provides that opportunity. In the studies that investigated all types of ACC in large cohorts, approximately 70 percent of individuals had other associated anomalies in the central nervous system, including cortical malformations, cysts, and posterior fossa abnormalities [Tang et al., 2009; Glenn et al., 2005; Brisse et al., 1998]. Isolated ACC only accounted for approximately 30 percent of the total number of abnormalities. This distribution also roughly correlates with ratios seen in postnatal cases [Hetts et al., 2006]. The clinical outcomes of these patients have, however, only been studied in case series with a small number of patients, as many of the fetally detected cases result in pregnancy termination [Guillem et al., 2003]. In cases of prenatally detected, isolated ACC, approximately 50 percent show neurological impairment in the first few years of life, but many more show cognitive and behavioral deficits during school years [Moutard et al., 2003]. There is a small subset of individuals who are reportedly normal. These studies also suggest that children with isolated ACC are more likely to have a favorable outcome [Chadie et al., 2008], yet no data are currently available to allow clinicians to stratify prognosis within the isolated ACC group. More studies of large cohorts are needed to address this critical point, and a better understanding of the genetics and other causes of ACC will help in assessing outcomes. Current standard ultrasonography can reliably detect callosal agenesis at 22 weeks’ gestation [Bennett et al., 1996]. however, many pregnant women receive a single ultrasound at 18 weeks to evaluate fetal anatomy. There are recommendations within the obstetrical community to perform two ultrasounds, one transvaginally at 14 weeks to assess nuchal translucency and other early visible anatomic changes, and a second study at 20–22 weeks to better visualize later-developing structures, such as the corpus callosum [Timor-Tritsch, 2006].
Development of the Corpus Callosum
The corpus callosum is the largest white-matter tract in the brain, with 190 million axons crossing the midline to innervate homotopic structures. The disproportionate increase in white-matter volume in mammalian, and specifically primate, evolution points to the importance of long-range connectivity (particularly of the frontal lobes) in brain evolution and function [Smaers et al., 2010]. The first callosal fibers can be seen crossing the midline around the 14th week of gestation. The midline is composed of a number of structures that likely assist in guiding these callosal fibers, including the glial wedge, midline zipper glia, glial sling, and indusium griseum glia (Figure 23-5). Some of these structures are known to secrete guidance molecules, such as netrin and Slit2, and the tips of the callosal axons have guidance receptors for these molecules, such as DCC and ROBO [Donahoo and Richards, 2009]. The full complexity of molecular and cellular events necessary for callosal development is not yet known, but these events can be seen within a general framework of developmental steps that include: birth and specification of commissural neurons, guidance of these neurons to the midline, midline fusion and development of key midline structures (as outlined in Figure 23-5), and axonal crossing of the midline with guidance of crossed neurons to final site of connectivity. In patients with ACC, it is difficult to determine which one of these steps is altered, with the exception of patients with microcephaly and absent Probst bundles. These individuals likely have defects in the initial birth and specification of commissural neurons, as found in patients with 1q44 deletions [Boland et al., 2007]. In contrast, it is more likely that patients who have Probst bundles (and normal or near-normal cerebral white-matter volume) have deficits in axonal guidance or midline fusion.
Imaging and the Corpus Callosum
As outlined above, the initial axons that cross the midline to constitute the corpus callosum do so around 14 weeks of gestation. This cannot reliably be seen by ultrasound until after 20 weeks [Bennett et al., 1996]. Fetal MRI has been used recently as a supplement to ultrasound, and the data suggest that this technology provides greater imaging precision, which may have prognostic implications [Glenn et al., 2005; Malinger et al., 2004]. Imaging of children and adults with ACC has shown that the missing corpus callosum is frequently only one component of the spectrum of brain malformations found in an individual patient. In a retrospective study of 142 cases of callosal agenesis (82 had agenesis and 60 had hypoplasia), over half of the patients had malformations of cortical development, one-third had cerebellar malformations, and one-quarter had brainstem anomalies. Almost all had reductions in white-matter volume outside of the commissural tracts. Among these 142 patients, only 5 had “isolated” ACC, underscoring the connection between callosal anomalies and other malformations of brain development [Hetts et al., 2006]. It has been observed anecdotally that many patients with ACC have small additional malformations, such as periventricular nodular heterotopia, which are not seen in low-resolution scans and subsequently are only discerned using high-resolution volumetric scans at 3.0 Tesla. Given the high correlation between associated anomalies and seizures, this is clearly important prognostic information to ascertain.
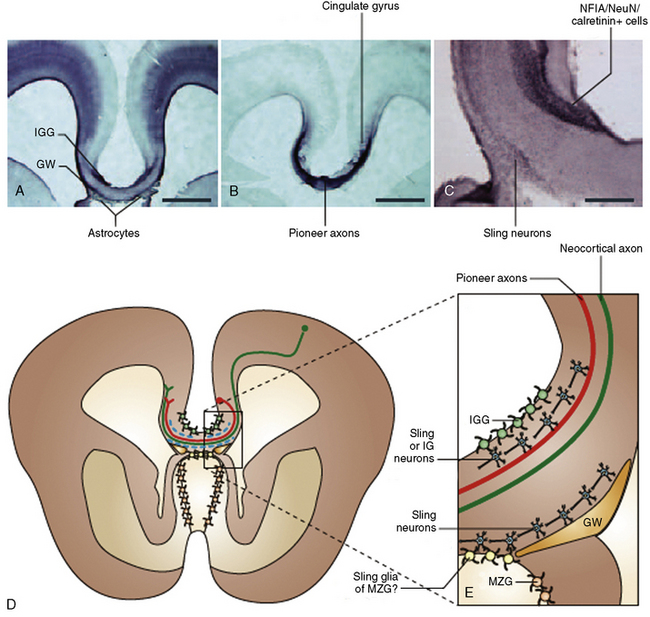
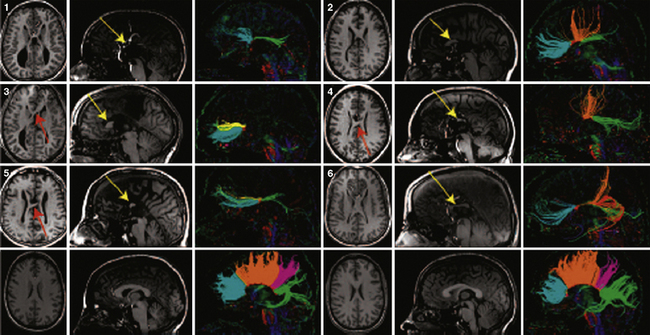
In addition to the knowledge gained from high-resolution conventional structural imaging, new techniques such as diffusion tensor imaging (DTI) have provided novel insights into the full spectrum of changes in callosal agenesis. Evidence suggests that the ventral cingulum bundle (CB) is smaller and has lower fractional anisotropy in ACC patients [Nakata et al., 2009]. Since the cingulum bundle connects the cingulate cortex with the limbic system, reduction in the CB may explain some of the behavioral deficits observed in patients with ACC. DTI can not only measure the integrity of white-matter tracts but, coupled with tractography, can delineate the anatomical features of major white-matter tracts. This approach has been used in animal models and patients. In experimental studies, conventional dye labeling approaches have shown that the tracts identified by this noninvasive MR technique correlates well with the white-matter histological measurements [Ren et al., 2007]. Because DTI and tractography are noninvasive, these approaches have been applied to many patient populations in the last few years, including patients with partial callosal agenesis (pACC). Two studies have shown that the callosal remnant contains both the normally observed homotopic fibers and aberrant heterotopic fibers. These heterotopic fibers (also called a sigmoid bundle) are fibers that course from the frontal lobe in one hemisphere to the contralateral parietal or occipital lobes [Wahl et al., 2009; Tovar-Moll et al., 2006] (Figure 23-6). Indeed, it appears that many combinations of “rewiring” are observed in these patients. In a few individuals, these heterotopic fibers can be visualized by conventional T1 imaging, supporting the benefits of using tractography. The precise significance of these “heterotopic” fibers remains unclear, but they do suggest that aberrant connectivity may be more widespread than just involvement of the corpus callosum. Indeed, an early postmortem pathology study of callosal agenesis cases found evidence for lack of pyramidal tract decussation [Parrish et al., 1979]. Recent investigations in patients with Joubert’s syndrome and horizontal gaze palsy with progressive scoliosis lend further support to the notion that DTI coupled with tractography can lead to noninvasive measurements of aberrant axonal tracts [Poretti et al., 2007; Sicotte et al., 2006]. Tractography of patients with a partial corpus callosum also have provided new insight into which regions of the cerebral hemispheres are connected through the residual callosum. The prevailing assumption had been that, if the residual callosum was more anterior, the frontal lobes were likely to be the connected structures. However, data from multiple individuals with pACC appear to contradict this assumption. Rather, tractography data suggest that there is not an obvious correlation between callosal remnant location and the cortical regions innervated (Figure 23-7). These findings suggest that the corpus callosum can be disrupted in more specific ways than previously anticipated and that other white-matter tracts may also be affected, even if none of these findings is detectable on conventional imaging.
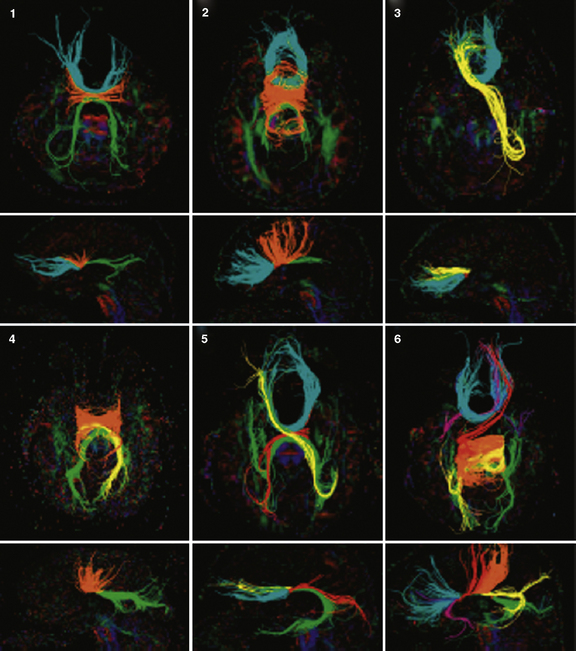
Fig. 23-6 Q-ball tractography of subjects with partial agenesis of the corpus callosum.
(From Wahl M et al. Variability of homotopic and heterotopic callosal connectivity in partial agenesis of the corpus callosum: A 3T diffusion tensor imaging and Q-ball tractography study. AJNR 2009;30:282–289.)
Etiology
Genetic
As outlined above, many molecular and cellular processes are necessary for normal callosal formation. Therefore, disruption of any of these processes can lead to callosal agenesis. There are many single-gene recessive disorders that are associated with callosal agenesis (Table 23-2), but it is currently unclear what percentage of cases can be accounted for by single-gene recessive disorders. The rate of familial recurrence has not been studied, but even in large research cohorts the number of multiplex families is small. This suggests that de novo genetic events may play an important role. In the California cohort, 17 percent of ACC patients had a causative chromosomal disorder, ranging from aneuploidies to translocations, deletions, and duplications, which were all cytogenetically visible [Glass et al., 2008]. A recent publication catalogs de novo deletions and duplications at recurrent genetic loci from nearly 400 ACC patients [O’Driscoll et al., 2010]. These include regions in 1p36, 1q4, 6p25, 6q2, 8p, 13q, and 14q. There are also unpublished data demonstrating smaller chromosomal CNVs in ACC patients. Preliminary data from that study suggest that over 15 percent of ACC patients have a large de novo CNV that may be causative. Other modes of inheritance are also likely, such as autosomal-recessive with multiple interacting loci or autosomal-dominant with partial penetrance. Although the initial presumption has been that most cases of ACC have a genetic etiology, it is possible that nongenetic causes, although less well studied and understood, may play an important etiologic role.
Table 23-2 Disorders Associated with Agenesis of the Corpus Callosum*
| Disorder | Salient Features |
|---|---|
| With Identified Genes† | |
| Andermann’s syndrome (KCC3) | ACC, progressive neuropathy and dementia |
| Donnai–Barrow syndrome (LRP2) | Diaphragmatic hernia, exomphalos, ACC, deafness |
| Frontonasal dysplasia (ALX1) | ACC, bilateral extreme microphthalmia, bilateral oblique facial cleft |
| XLAG (ARX) | Lissencephaly, ACC, intractable epilepsy |
| Microcephaly (TBR2) | ACC, polymicrogyria |
| Microcephaly with simplified gyral pattern and ACC (WDR62) | |
| Mowat–Wilson syndrome (ZFHX1B) | Hirschsprung’s disease, ACC |
| Pyridoxine-dependent epilepsy (ALDH7A1) | ACC, seizures, other brain malformations |
| Pyruvate dehydrogenase deficiency (PDHA1, PDHB, PDHX) | ACC with other brain changes |
| ACC with fatal lactic acidosis (MRPS16) | Complex I and IV deficiency, ACC, brain malformations |
| HSAS/MASA syndromes (L1CAM) | Hydrocephalus, adducted thumbs, ACC, MR |
| ACC Seen Consistently (No Gene Yet Identified) | |
| Acrocallosal syndrome | ACC, polydactyly, craniofacial changes, MR |
| Aicardi’s syndrome | ACC, chorioretinal lacunae, infantile spasms, MR |
| Chudley–McCullough syndrome | Hearing loss, hydrocephalus, ACC, colpocephaly |
| FG syndrome | MR, ACC, craniofacial changes, macrocephaly |
| Genitopatellar syndrome | Absent patellae, urogenital malformations, ACC |
| Temtamy’s syndrome | ACC, optic coloboma, craniofacial changes, MR |
| Toriello–Carey syndrome | ACC, craniofacial changes, cardiac defects, MR |
| Vici’s syndrome | ACC, albinism, recurrent infections, MR |
| ACC Seen Occasionally (Partial List)‡ | |
| ACC with spastic paraparesis (SPG11; SPG15) | Progressive spasticity and neuropathy, thin corpus callosum |
| Craniofrontonasal syndrome | Coronal craniosynostosis, facial asymmetry, bifid nose |
| Fryns’ syndrome | CDH, pulmonary hypoplasia, craniofacial changes |
| Marden–Walker syndrome | Blepharophimosis, micrognathia, contractures, ACC |
| Meckel–Gruber syndrome | Encephalocele, polydactyly, polycystic kidneys |
| Nonketotic hyperglycinemia (GLDC, GCST, GCSH) | ACC, cerebral and cerebellar atrophy, myoclonus, progressive encephalopathy |
| Microphthalmia with linear skin defects | Microphthalmia, linear skin markings, seizures |
| Opitz G syndrome | Pharyngeal cleft, craniofacial changes, ACC, MR |
| Orofaciodigital syndrome | Tongue hamartoma, microretrognathia, clinodactyly |
| Pyruvate decarboxylase deficiency | Lactic acidosis, seizures, severe MR and spasticity |
| Rubinstein–Taybi syndrome | Broad thumbs and great toes, MR, microcephaly |
| Septo-optic dysplasia (DeMorsier’s syndrome) | Hypoplasia of septum pellucidum and optic chiasm |
| Sotos’ syndrome | Physical overgrowth, MR, craniofacial changes |
| Warburg micro syndrome | Microcephaly, micropthalmia, microgenitalia, MR |
| Wolf–Hirschhorn syndrome | Microcephaly, seizures, cardiac defects, 4p− |
4p−, deletion of the terminal region of the short arm of chromosome 4, defines the genotype for Wolf–Hirschhorn patients; ACC, agenesis of the corpus callosum; ARX, Aristaless-related homeobox gene; CDH, congenital diaphragmatic hernia; HSAS/MASA, X-linked hydrocephalus/mental retardation, aphasia, shuffling gait, and adducted thumbs; KCC3, KCl co-transporter 3; L1CAM, L1 cell adhesion molecule; MR, mental retardation; MRPS16, mitochondrial ribosomal protein S16; SPG11, spastic paraplegia 11; XLAG, X-linked lissencephaly with absent corpus callosum and ambiguous genitalia; ZFHX1B, zinc finger homeobox 1b.
* Reliable incidence data are unavailable for these very rare syndromes.
‡ Many of these may also consistently have a thin or dysplastic corpus callosum, such as Sotos’ syndrome or agenesis of the corpus callosum (ACC) with spastic paraparesis (SPG11). The overlap between ACC and these conditions is still under investigation. Other gene symbols are omitted from this section.
Nongenetic
The best example of environmental causes of ACC is fetal alcohol syndrome. A number of studies report both loss of white-matter volume alone, and callosal agenesis without appreciable white-matter volume loss in fetal alcohol syndrome [Riley et al., 1995; Swayze et al., 1997; Bookstein et al., 2002; Spadoni et al., 2006]. There is clearly variability in the expression of callosal deficits in fetal alcohol syndrome, but it is unclear whether this relates to timing and amount of alcohol consumption or whether genetic factors in the mother or baby may play a role. In mouse models, genetic background does influence severity of brain injury [Wainwright and Gagnon, 1985]. Additionally, the cell surface molecule L1, which is involved in both axon guidance and fasciculation, has been proposed to be a target for alcohol toxicity on the brain by directly inhibiting cell–cell adhesion [Ramanathan et al., 1996]. There are also case reports of herpes simplex virus and cytomegalovirus infections resulting in ACC [Jayaram and Wake, 2010; Chiappini et al., 2007], but these are not well documented. Similarly, there are a few reports of heavy metal toxicity and ACC [Barone et al., 1998], but without many reports, it is difficult to attribute a significant percentage of ACC cases to these causes.
Clinical Manifestations
Association of Agenesis of the Corpus Callosum with Autism and Related Neurodevelopmental Disorders
As in many children with other brain malformations, epilepsy, mental retardation, and cerebral palsy are common [Shevell, 2002]. However, it is difficult to know the precise prevalence of these associated features because most individuals with ACC are evaluated specifically because of their clinical difficulties, introducing ascertainment bias. With that limitation in mind, many studies have reported that patients with ACC have significant cognitive and neuromotor impairment [Lacey, 1985]. Epilepsy is common and is more prevalent in patients who have other associated brain malformations, such as periventricular nodular heterotopia or polymicrogyria. In addition to cognitive impairment, many patients with ACC have behavioral difficulties, such as autism and attention-deficit hyperactivity disorder [Badaruddin et al., 2007]. Autistic features are present in approximately 40 percent of patients with ACC who have normal IQs, and many of these individuals have autism by strict research criteria (i.e., using standardized neuropsychological tests such as the Autism Diagnostic Interview – Revised and the Autism Diagnostic Observation Schedule; see Chapter 48) [Lau et al., 2010]. A more detailed analysis shows that these “high-functioning” individuals with ACC have problems with social cognition, paralinguistic communication, and executive function skills [Paul et al., 2007; Symington et al., 2010; Brown et al., 2005]. These deficits help to explain how individuals with ACC have difficulties holding down jobs, finding partners, and living independently, even though they may have normal intelligence. Indeed, Kim Peek, whose life was fictionalized in the movie Rain Man, had callosal agenesis [Ross, 2006]. There is also literature evidence to support the association of schizophrenia with ACC [David, 1994].
Management
Currently, the mainstay of management for patients with ACC includes symptomatic measures similar to those described above for patients with HPE. These may include use of antiepileptic drugs for epilepsy, physical and occupational therapy for hypotonia and cerebral palsy, and speech therapy. Patients with ACC, including apparently high-functioning individuals, often have difficulty with more complex cognitive and behavioral tasks [Paul et al., 2007; Symington et al., 2010; Brown et al., 2005; Brown and Paul, 2000; Paul et al., 2003, Paul et al., 2004]. Anecdotal reports suggest that these individuals do better with therapy targeted at simplifying these tasks, providing repetition, and supporting a slower learning pace. Many individuals with ACC have social deficits, but appear to desire social interactions [Symington et al., 2010]. With recent reports of the potential benefits of intranasal oxytocin treatment for autistic individuals with social deficits, some have hypothesized that similar approaches may have long-lasting benefits in ACC patients. This approach remains to be tested.
Septo-Optic Dysplasia
The constellation of symptoms that comprise septo-optic dysplasia (SOD) is presumed to result from failure of formation of the optic nerves, septum pellucidum, pituitary gland, and all midline structures within the prosencephalon (Figure 23-8). Patients typically present with pituitary hormone abnormalities that can result in hypoglycemia or microphallus at birth, or growth failure and other endocrine manifestations throughout childhood. It is a rare condition, with an estimated incidence of 1–10 in 100,000; known genetic etiologies contribute to only a small percentage of cases. Also, data suggest that young maternal age contributes to the risk of developing SOD. SOD is better viewed as a complex (SOD complex), with variable etiology and clinical presentation.
Definition and Subtypes
There are three cardinal features of SOD: optic nerve hypoplasia, pituitary abnormalities, and midline brain defects (involving primarily the septum pellucidum and, at times, the corpus callosum). The diagnosis of SOD is typically made when two of the three features are present; however, given the heterogeneity intrinsic to this constellation of symptoms, most investigators have initiated their analysis by ascertaining patients with optic nerve hypoplasia (ONH) and then grouping those that had both pituitary dysfunction and absence of the septum pellucidum. From a clinical perspective, those that had ONH and pituitary anomalies but a normal septum pellucidum will have similar management issues. In contrast, those that have ONH and an absent septum pellucidum without pituitary abnormalities often have other brain malformations and likely represent a separate group [Riedl et al., 2008].
Epidemiology
The incidence of SOD has been estimated at between 1 and 10 per 100,000 [Tornqvist et al., 2002 #5096; Patel et al., 2006 #5092; Murray et al., 2005 #5094]. This large range arises from difficulty in fully ascertaining these patients and in utilizing a common definition, as some patients just have bilateral ONH without absence of the septum pellucidum.
Etiology
SOD and ONH have been associated with primiparous birth and young maternal age [Tornqvist et al., 2002; Patel et al., 2006; Murray et al., 2005; Elster and McAnarney, 1979]. Additional risk factors that have been consistently reported include maternal smoking and low socioeconomic status. Other associations, such as in utero exposure to alcohol, cocaine, other drugs of abuse, and antidepressants, have been suggested in case reports but have not been adequately sampled in larger cohort studies. Two genes have been found in association with SOD. The homeobox gene, HESX1, was shown to cause anterior prosencephalic disruption in mice and humans, including disruption of anterior pituitary development [Dattani et al., 1998]. Heterozygous mutations in HESX1 have also been found in approximately 1 percent of SOD patients, although these mutations are inherited and show incomplete penetrance [Thomas et al., 2001; McNay et al., 2007]. Another transcription factor gene, SOX2, also has been found to be mutated (mostly de novo single-nucleotide changes) in patients with variable presentations in the SOD spectrum [Kelberman et al., 2006]. Chromosomal changes also have rarely been reported in SOD cases [Singh et al., 2004]. Thus, most patients with SOD are hypothesized to result from a combination of genetic and environmental factors, recognizing that other mechanisms are yet to be discovered.
Clinical Manifestations
SOD can present at birth with manifestations of pituitary insufficiency, including hypoglycemia (with seizures leading to death if not rapidly identified), microphallus and undescended testes (from hypogonadotropic hypogonadism), and midline birth defects, including cleft lip and palate, as well as other brain malformations. Because of disruption in anterior pituitary development, SOD patients are at risk for adrenocorticotropic hormone, thyroid stimulating hormone, and growth hormone deficiency, in addition to lack of gonadotropic hormones. Many patients with SOD have additional central nervous system malformations. In the largest study of MRI findings in SOD, approximately half the patients had brain malformations. These included a thin corpus callosum, hippocampal abnormalities, and other cortical malformations, such as polymicrogyria and schizencephaly. As with HPE and ACC, many patients with SOD have developmental delay and cerebral palsy. This appears to correlate with the involvement of additional brain structures [Riedl et al., 2008].
Management
Symptom management is essential in individuals with SOD and careful attention should be paid to the heterogeneity of this complex disorder. Any child presenting with nystagmus should be evaluated for optic nerve involvement, and if ONH is detected, the patient should be assessed for anterior pituitary hormone deficiency. Additionally, many children with SOD can have developmental delay and seizures, and should be evaluated for these possible concerns [Webb and Dattani, 2010]. Because children are at risk for adrenocorticotropic hormone, thyroid stimulating hormone, and growth hormone deficiency (as well as hypothalamic dysfunction and poor production of the posterior pituitary hormones, antidiuretic hormone and oxytocin), they can present with hypoglycemia, diabetes insipidus, and poor thermoregulation. Multiple case reports of sudden death in SOD patients have described these complications [Brodsky et al., 1997].
Isolated Septum Pellucidum Dysplasias
The septum pellucidum can be divided into two parts: the “true” septum, or septum verum, which is a nerve cell-containing area, and the septum pellucidum [Andy and Stephan, 1968]. The septum pellucidum is a thin translucent plate that forms the medial wall of the lateral ventricle. Its development is linked to the development of the corpus callosum, which accounts for the common association of developmental anomalies of both structures. The two major defects include absence of the septum pellucidum and absence of the cavum septum pellucidum, which are easily differentiated on brain MRI (Figure 23-9).
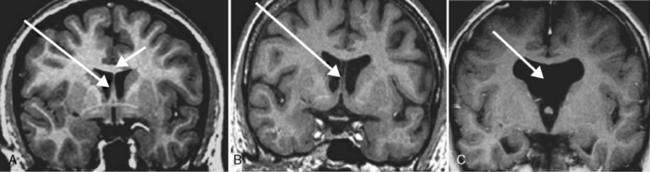
Fig. 23-9 Septum pellucidum defect.
(From Born CM et al. The septum pellucidum and its variants. An MRI study. Eur Arch Psychiatry Clin Neurosci 2004;254:295.)
Absence of the Septum Pellucidum
Absence of the septum pellucidum may be due to primary agenesis or to secondary mechanisms, such as hydrocephalus, with subsequent damage. The prevalence of absence or hypoplasia of the septum pellucidum has been estimated at 2–3 per 100,000 persons, although this is heavily biased by the reason for imaging and by imaging technique detection rates. Absence nearly always is associated with other central nervous system anomalies, such as SOD, schizencephaly, ACC, hydrocephalus, or encephaloceles [Barkovich and Norman, 1989]. In the rare instance in which no other major central nervous system malformations are identified, then epilepsy and mental retardation are infrequently seen. Absence of the septum pellucidum occasionally is identified on prenatal ultrasound examination, where it may indicate a brain malformation. Most fetuses in which this feature is seen are later found to have a more severe brain malformation, including HPE, hydrocephalus, or SOD [Lepinard et al., 2005; Malinger et al., 2004].
Cavum Septum Pellucidum
Cavum septum pellucidum is identified in approximately 20 percent of normal control subjects [Born et al., 2004]. Controversy exists in the literature over whether this represents a true malformation or just a variant of normal. An elevated prevalence of cavum septum pellucidum has been reported in several psychiatric conditions, including schizophrenia and bipolar disorder. Widely separated pellucidum leaflets greater than 1 cm across occur much less commonly and may be a marker of disturbed brain development, when this finding takes on the same significance as absence of the septum pellucidum. This finding is clearly abnormal only after the neonatal period, because all premature infants exhibit an ultrasonographically demonstrable cavum at up to 34 weeks of gestation. Nevertheless, a large cavum (more than 1 cm in diameter) in a term newborn should be viewed as suggestive of other brain malformations. In one series, all children with such findings had abnormal neurologic outcome [Bodensteiner and Schaefer, 1990].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Andy O.J., Stephan H. The septum in the human brain. J Comp Neurol. 1968;133:383-410.
Badaruddin D.H., Andrews G.L., Bolte S., et al. Social and Behavioral Problems of Children with Agenesis of the Corpus Callosum. Child Psychiatry Hum Dev. 2007.
Barkovich A.J. Analyzing the corpus callosum. Am J Neuroradiol. 1996;17:1643-1645.
Barkovich A.J., Norman D. Absence of the septum in the diagnosis of congenital brain malformations. Am J Roentgenol. 1989;152:843-852.
Barkovich A.J., Simon E.M., Clegg N.J., et al. Analysis of the cerebral cortex in holoprosencephaly with attention to the sylvian fissures. Am J Neuroradiol. 2002;23(1):143-150.
Barone S.Jr, Haykal-Coates N., Parran D.K., et al. Gestational exposure to methylmercury alters the developmental pattern of trk-like immunoreactivity in the rat brain and results in cortical dysmorphology. Brain Res Dev Brain Res. 1998;109:13-31.
Barr M.Jr, Cohen M.M.Jr. Holoprosencephaly survival and performance. Am J Med Genet. 1999;89(2):116-120.
Barr M.Jr, Hanson J.W., Currey K., et al. Holoprosencephaly in infants of diabetic mothers. J Pediatr. 1983;102(4):565-568.
Belloni E., Muenke M., Roessler E., et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14(3):353-356.
Bennett G.L., Bromley B., Benacerraf B.R. Agenesis of the corpus callosum: prenatal detection usually is not possible before 22 weeks of gestation. Radiology. 1996;199:447-450.
Bloom J.S., Hynd G.W. The role of the corpus callosum in interhemispheric transfer of information: excitation or inhibition? Neuropsychol Rev. 2005;15:59-71.
Bodensteiner J.B., Schaefer G.B. Wide cavum septum pellucidum: A marker of disturbed brain development. Pediatr Neurol. 1990;6:391.
Boland E., Clayton-Smith J., Woo V.G., et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet. 2007;81:292-303.
Bookstein F.L., Sampson P.D., Connor P.D., et al. Midline corpus callosum is a neuroanatomical focus of fetal alcohol damage. Anat Rec. 2002;269:162-174.
Born C.M., Meisenzahl E.M., Frodl T., et al. The septum pellucidum and its variants. An MRI study. Eur Arch Psychiatry Clin Neurosci. 2004;254:295.
Brisse H., Sebag G., Fallet C., et al. [Prenatal MRI of corpus callosum agenesis. Study of 20 cases with neuropathological correlations]. J Radiol. 1998;79:659-666.
Brodsky M.C., Conte F.A., Taylor D., et al. Sudden death in septo-optic dysplasia. Report of 5 cases. Arch Ophthalmol. 1997;115:66-70.
Brown L.Y., Odent S., David V., et al. Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination. Hum Mol Genet. 2001;10(8):791-796.
Brown W.S., Paul L.K. Cognitive and psychosocial deficits in agenesis of the corpus callosum with normal intelligence. Cognit Neuropsychiatry. 2000;5:135-157.
Brown W.S., Paul L.K., Symington M., et al. Comprehension of humor in primary agenesis of the corpus callosum. Neuropsychologia. 2005;43:906-916.
Bullen P.J., Rankin J.M., Robson S.C. Investigation of the epidemiology and prenatal diagnosis of holoprosencephaly in the North of England. Am J Obstet Gynecol. 2001;184(6):1256-1262.
Chadie A., Radi S., Trestard L., et al. Neurodevelopmental outcome in prenatally diagnosed isolated agenesis of the corpus callosum. Acta Paediatr. 2008.
Chiappini E., Galli L., Paganelli S., et al. Congenital cytomegalovirus infection associated with corpus callosum agenesis. Pediatr Neurol. 2007;36:277.
Cockerell O.C., Gupta S., Catchpole M., et al. The British Neurological Surveillance Unit: a nation-wide scheme for the ascertainment of rare neurological disorders. Neuroepidemiology. 1995;14:182-187.
Cohen M.M.Jr. Perspectives on holoprosencephaly: Part III. Spectra, distinctions, continuities, and discontinuities. Am J Med Genet. 1989;34(2):271-288.
Cohen M.M.Jr, Shiota K. Teratogenesis of holoprosencephaly. Am J Med Genet. 2002;109(1):1-15.
Cordero D., Marcucio R., Hu D., et al. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J Clin Invest. 2004;114(4):485-494.
Croen L.A., Shaw G.M., Lammer E.J. Holoprosencephaly: epidemiologic and clinical characteristics of a California population. Am J Med Genet. 1996;64(3):465-472.
Croen L.A., Shaw G.M., Lammer E.J. Risk factors for cytogenetically normal holoprosencephaly in California: a population-based case-control study. Am J Med Genet. 2000;90(4):320-325.
Dattani M.T., Martinez-Barbera J.P., Thomas P.Q., et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125-133.
David A.S. Schizophrenia and the corpus callosum: developmental, structural and functional relationships. Behav Brain Res. 1994;64:203-211.
DeMyer W. Holoprosencephaly (cyclopia-arhinencephaly). In: Vinken P.J., Bruyn G.W., Klawans H.L., editors. Handbook of Clinical Neurology. ed 6. Amsterdam: Elsevier Science Publishers; 1987:225-244. Revised series
DeMyer W., Zeman W. Alobar holoprosencephaly (arhinencephaly) with median cleft lip and palate: clinical, electroencephalographic and nosologic considerations. Confin Neurol. 1963;23:1-36.
DeMyer W., Zeman W., Palmer C.G. The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics. 1964;34:256-263.
Dill P., Poretti A., Boltshauser E., et al. Fetal magnetic resonance imaging in midline malformations of the central nervous system and review of the literature. J Neuroradiol. 2009;36(3):138-146.
Donahoo A.L., Richards L.J. Understanding the mechanisms of callosal development through the use of transgenic mouse models. Semin Pediatr Neurol. 2009;16:127-142.
Dubourg C., Lazaro L., Pasquier L., et al. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum Mutat. 2004;24(1):43-51.
Durkin M.S., Maenner M.J., Newschaffer C.J., et al. Advanced parental age and the risk of autism spectrum disorder. Am J Epidemiol. 2008;168:1268-1276.
Elster A.B., McAnarney E.R. Maternal age re septo-optic dysplasia. J Pediatr. 1979;94:162-163.
Filly R.A., Chinn D.H., Callen P.W. Alobar holoprosencephaly: ultrasonographic prenatal diagnosis. Radiology. 1984;151(2):455-459.
Forrester M.B., Merz R.D. Epidemiology of holoprosencephaly in Hawaii, 1986-97. Paediatr Perinat Epidemiol. 2000;14(1):61-63.
Gazzaniga M.S. Cerebral specialization and interhemispheric communication: does the corpus callosum enable the human condition? Brain. 2000;123(Pt 7):1293-1326.
Glass H.C., Shaw G.M., Ma C., et al. Agenesis of the corpus callosum in California 1983–2003: a population-based study. Am J Med Genet A. 2008;146A:2495-2500.
Glenn O.A., Goldstein R.B., Li K.C., et al. Fetal magnetic resonance imaging in the evaluation of fetuses referred for sonographically suspected abnormalities of the corpus callosum. J Ultrasound Med. 2005;24:791-804.
Golden J.A. Towards a greater understanding of the pathogenesis of holoprosencephaly. Brain Dev. 1999;21(8):513-521.
Greenblatt S., Dagi T.F.. A history of neurosurgery: in its scientific and professional contexts. 1997.
Guillem P., Fabre B., Cans C., et al. Trends in elective terminations of pregnancy between 1989 and 2000 in a French county (the Isere). Prenat Diagn. 2003;23:877-883.
Hahn J.S., Barnes P.D. Neuroimaging advances in holoprosencephaly: Refining the spectrum of the midline malformation. Am J Med Genet C Semin Med Genet. 2010;154C(1):120-132.
Hahn J.S., Barnes P.D., Clegg N.J., et al. Septopreoptic holoprosencephaly: a mild subtype associated with midline craniofacial anomalies. Am J Neuroradiol. 2010;31(9):1596-1601.
Hahn J.S., Plawner L.L. Evaluation and management of children with holoprosencephaly. Pediatr Neurol. 2004;31(2):79-88.
Hetts S.W., Sherr E.H., Chao S., et al. Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations. Am J Roentgenol. 2006;187:1343-1348.
Jayaram P.M., Wake C.R. A rare case of absent corpus callosum with severe ventriculomegaly due to congenital herpes simplex infection. J Obstet Gynaecol. 2010;30:316-1348.
Johnson C.Y., Rasmussen S.A. Non-genetic risk factors for holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):73-85.
Keeler R.F., Binns W. Teratogenic compounds of Veratrum californicum (Durand). V. Comparison of cyclopian effects of steroidal alkaloids from the plant and structurally related compounds from other sources. Teratology. 1968;1(1):5-10.
Kelberman D., Rizzoti K., Avilion A., et al. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116:2442-2455.
Kelley R.L., Roessler E., Hennekam R.C., et al. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am J Med Genet. 1996;66(4):478-484.
Kovar C., Plawner L., Sweet V., et al. Cognitive profiles of children with holoprosencephaly. Arch Clin Neuropsychol. 2001;16(8):781.
Kundrat H. Arhinencephalie als typische Art von Missbildung. Graz: von Leuschner and Lubensky; 1882.
Lacey D.J. Agenesis of the corpus callosum. Clinical features in 40 children. Am J Dis Child. 1985;139:953-955.
Larroche J.C. Malformations of the CNS. In: Larroche J.C., editor. Developmental pathology of the neonate. Amsterdam: Excerpta Medica; 1977:473-516.
Lau Y., Marco E., Hinkley L.B., et al. Agenesis of the Corpus Callosum and the Autism Spectrum. 9th International Meeting for Autism Research (IMFAR). 2010. May 20–22, 2010, Philadelphia, 2010
Lepinard C., Coutant R., Boussion F., et al. Prenatal diagnosis of absence of the septum pellucidum associated with septo-optic dysplasia. Ultrasound Obstet Gynecol. 2005;25:73-75.
Malinger G., Ben-Sira L., Lev D., et al. Fetal brain imaging: a comparison between magnetic resonance imaging and dedicated neurosonography. Ultrasound Obstet Gynecol. 2004;23:333-340.
Malinger G., Lev D., Kidron D., et al. Differential diagnosis in fetuses with absent septum pellucidum. Ultrasound Obstet Gynecol. 2004;25:42.
Malinger G., Lev D., Kidron D., et al. Differential diagnosis in fetuses with absent septum pellucidum. Ultrasound Obstet Gynecol. 2005;25(1):42-49.
Matsunaga E., Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16(3):261-272.
McNay D.E., Turton J.P., Kelberman D., et al. HESX1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol Metab. 2007;92:691-697.
Mercier S., Dubourg C., Belleguic M., et al. Genetic counseling and “molecular” prenatal diagnosis of holoprosencephaly (HPE). Am J Med Genet C Semin Med Genet. 2010;154C(1):191-196.
Miller E.A., Rasmussen S.A., Siega-Riz A.M., et al. Risk factors for non-syndromic holoprosencephaly in the National Birth Defects Prevention Study. Am J Med Genet C Semin Med Genet. 2010;154C(1):62-72.
Ming J.E., Muenke M. Holoprosencephaly: from Homer to Hedgehog. Clin Genet. 1998;53(3):155-163.
Ming J.E., Muenke M. Multiple Hits during Early Embryonic Development: Digenic Diseases and Holoprosencephaly. Am J Hum Genet. 2002;71(5):1017-1032.
Moutard M.L., Kieffer V., Feingold J., et al. Agenesis of corpus callosum: prenatal diagnosis and prognosis. Childs Nerv Syst. 2003;19:471-476.
Murray P.G., Paterson W.F., Donaldson M.D. Maternal age in patients with septo-optic dysplasia. J Pediatr Endocrinol Metab. 2005;18:471-476.
Nakata Y., Barkovich A.J., Wahl M., et al. Diffusion abnormalities and reduced volume of the ventral cingulum bundle in agenesis of the corpus callosum: a 3T imaging study. Am J Neuroradiol. 2009;30:1142-1148.
O’Driscoll M.C., Black G.C., Clayton-Smith J., et al. Identification of genomic loci contributing to agenesis of the corpus callosum. Am J Med Genet A. 2010;152A:2145-2159.
Olsen C.L., Hughes J.P., Youngblood L.G., et al. Epidemiology of holoprosencephaly and phenotypic characteristics of affected children: New York State, 1984-1989. Am J Med Genet. 1997;73(2):217-226.
Parrish M.L., Roessmann U., Levinsohn M.W. Agenesis of the corpus callosum: a study of the frequency of associated malformations. Ann Neurol. 1979;6:349-354.
Patel L., McNally R.J., Harrison E., et al. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J Pediatr. 2006;148:85-88.
Paul L.K., Brown W.S., Adolphs R., et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8:287-299.
Paul L.K., Schieffer B., Brown W.S. Social processing deficits in agenesis of the corpus callosum: narratives from the Thematic Appreciation Test. Arch Clin Neuropsychol. 2004;19:215-225.
Paul L.K., Van Lancker-Sidtis D., Schieffer B., et al. Communicative deficits in agenesis of the corpus callosum: nonliteral language and affective prosody. Brain Lang. 2003;85:313-324.
Pineda-Alvarez D.E., Dubourg C., David V., et al. Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients. Am J Med Genet C Semin Med Genet. 2010;154C(1):93-101.
Plasencia W., Dagklis T., Borenstein M., et al. Assessment of the corpus callosum at 20-24 weeks’ gestation by three-dimensional ultrasound examination. Ultrasound Obstet Gynecol. 2007;30(2):169-172.
Plawner L.L., Delgado M.R., Miller V.S., et al. Neuroanatomy of holoprosencephaly as predictor of function: beyond the face predicting the brain. Neurology. 2002;59:1058-1066.
Poretti A., Boltshauser E., Loenneker T., et al. Diffusion tensor imaging in Joubert syndrome. Am J Neuroradiol. 2007;28:1929-1933.
Pulitzer S.B., Simon E.M., Crombleholme T.M., et al. Prenatal MR findings of the middle interhemispheric variant of holoprosencephaly. Am J Neuroradiol. 2004;25(6):1034-1036.
Ramanathan R., Wilkemeyer M.F., Mittal B., et al. Alcohol inhibits cell-cell adhesion mediated by human L1. J Cell Biol. 1996;133:381-390.
Ramelli G., Zanda N., Wyttenbach M., et al. The prognosis of agenesis of the corpus callosum might mostly be favourable. Swiss Med Wkly. 2006;136:404-405.
Rao M.S., Jacobson M., editors. Developmental Neurobiology, ed 4, NY: Kluwer Academic, 2005.
Rasmussen S.A., Moore C.A., Khoury M.J., et al. Descriptive epidemiology of holoprosencephaly and arhinencephaly in metropolitan Atlanta, 1968-1992. Am J Med Genet. 1996;66(3):320-333.
Ren T., Zhang J., Plachez C., et al. Diffusion tensor magnetic resonance imaging and tract-tracing analysis of Probst bundle structure in Netrin1- and DCC-deficient mice. J Neurosci. 2007;27:10345-10349.
Riedl S., Vosahlo J., Battelino T., et al. Refining clinical phenotypes in septo-optic dysplasia based on MRI findings. Eur J Pediatr. 2008;167:1269-1276.
Riley E.P., Mattson S.N., Sowell E.R., et al. Abnormalities of the corpus callosum in children prenatally exposed to alcohol. Alcohol Clin Exp Res. 1995;19:1198-1202.
Roach E., DeMyer W., Conneally P.M., et al. Holoprosencephaly: birth data, genetic and demographic analyses of 30 families. Birth Defects Orig Artic Ser. 1975;11(2):294-313.
Roessler E., Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):52-61.
Roessler E., Ouspenskaia M.V., Karkera J.D., et al. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet. 2008;83(1):18-29.
Ross P.E. Half-brained schemes. Sci Am. 2006;294:24-26.
Sepulveda W., Dezerega V., Be C. First-trimester sonographic diagnosis of holoprosencephaly: value of the “butterfly” sign. J Ultrasound Med. 2004;23(6):761-765. quiz 766–767
Sherr E.H., Owen R., Albertson D.G., et al. Genomic microarray analysis identifies candidate loci in patients with corpus callosum anomalies. Neurology. 2005;65:1496-1498.
Shevell M.I. Clinical and diagnostic profile of agenesis of the corpus callosum. J Child Neurol. 2002;17:896-900.
Sicotte N.L., Salamon G., Shattuck D.W., et al. Diffusion tensor MRI shows abnormal brainstem crossing fibers associated with ROBO3 mutations. Neurology. 2006;67:519-521.
Siebert J.R., Cohen M.M.Jr, Sulik K.K., et al. Syndromes. Holoprosencephaly An overview and atlas of cases. New York: Wiley-Liss; 1990:337-385.
Simon E.M., Hevner R., Pinter J.D., et al. Assessment of the deep gray nuclei in holoprosencephaly. Am J Neuroradiol. 2000;21(10):1955-1961.
Simon E.M., Hevner R.F., Pinter J.D., et al. The dorsal cyst in holoprosencephaly and the role of the thalamus in its formation. Neuroradiology. 2001;43(9):787-791.
Simon E.M., Hevner R.F., Pinter J.D., et al. The middle interhemispheric variant of holoprosencephaly. Am J Neuroradiol. 2002;23(1):151-155.
Singh V., Boesel C.P., Baker P. Septo-optic dysplasia and dentato-olivary dysplasia in a case of 18q deletion/3p trisomy. Clin Neuropathol. 2004;23:28-33.
Smaers J.B., Schleicher A., Zilles K., et al. Frontal white matter volume is associated with brain enlargement and higher structural connectivity in anthropoid primates. PLoS ONE. 2010;5:e9123.
Solomon B.D., Mercier S., Velez J.I., et al. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C(1):133-141.
Spadoni A.D., McGee C.L., Fryer S.L., et al. Neuroimaging and fetal alcohol spectrum disorders. Neurosci Biobehav Rev. 2006.
Stashinko E.E., Clegg N.J., Kammann H.A., et al. A retrospective survey of perinatal risk factors of 104 living children with holoprosencephaly. Am J Med Genet. 2004;128A:114-119.
Sulik K.K., Johnston M.C.. Embryonic origin of holoprosencephaly: interrelationship of the developing brain and face. Scan Electron Microsc. 1982(Pt 1):309-322.
Swayze V.W.2nd, Johnson V.P., Hanson J.W., et al. Magnetic resonance imaging of brain anomalies in fetal alcohol syndrome. Pediatrics. 1997;99:232-240.
Symington S.H., Paul L.K., Symington M.F., et al. Social cognition in individuals with agenesis of the corpus callosum. Soc Neurosci. 2010:1-13.
Tang P.H., Bartha A.I., Norton M.E., et al. Agenesis of the corpus callosum: an MR imaging analysis of associated abnormalities in the fetus. Am J Neuroradiol. 2009;30:257-263.
Thomas P.Q., Dattani M.T., Brickman J.M., et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10:39-45.
Timor-Tritsch I.E. As technology evolves, so should its application: shortcomings of the “18-week anatomy scan”. J Ultrasound Med. 2006;25:423-428.
Tornqvist K., Ericsson A., Kallen B. Optic nerve hypoplasia: Risk factors and epidemiology. Acta Ophthalmol Scand. 2002;80:300-304.
Tovar-Moll F., Moll J., de Oliveira-Souza R., et al. Neuroplasticity in Human Callosal Dysgenesis: A Diffusion Tensor Imaging Study. Cereb Cortex. 2006.
Turner E. The seat of consciousness. Lancet. 1955;269:1305-1307.
Wahl M., Strominger Z., Jeremy R.J., et al. Variability of homotopic and heterotopic callosal connectivity in partial agenesis of the corpus callosum: a 3T diffusion tensor imaging and Q-ball tractography study. Am J Neuroradiol. 2009;30:282-289.
Wainwright P., Gagnon M. Moderate prenatal ethanol exposure interacts with strain in affecting brain development in BALB/c and C57BL/6 mice. Exp Neurol. 1985;88:84-94.
Wang L.W., Huang C.C., Yeh T.F. Major brain lesions detected on sonographic screening of apparently normal term neonates. Neuroradiology. 2004;46:368-373.
Webb E.A., Dattani M.T. Septo-optic dysplasia. Eur J Hum Genet. 2010;18:393-397.
Wenghoefer M., Ettema A.M., Sina F., et al. Prenatal ultrasound diagnosis in 51 cases of holoprosencephaly: craniofacial anatomy, associated malformations, and genetics. Cleft Palate Craniofac J. 2010;47(1):15-21.
Wong A.M., Bilaniuk L.T., Ng K.K., et al. Lobar holoprosencephaly: prenatal MR diagnosis with postnatal MR correlation. Prenat Diagn. 2005;25(4):296-299.
Yakovlev P.I. Pathoarchitectonic studies of cerebral malformations. III. Arrhinencephalies (holotelencephalies). J Neuropathol Exp Neurol. 1959;18:22-25.