Chapter 24 Disorders of Cerebellar and Brainstem Development
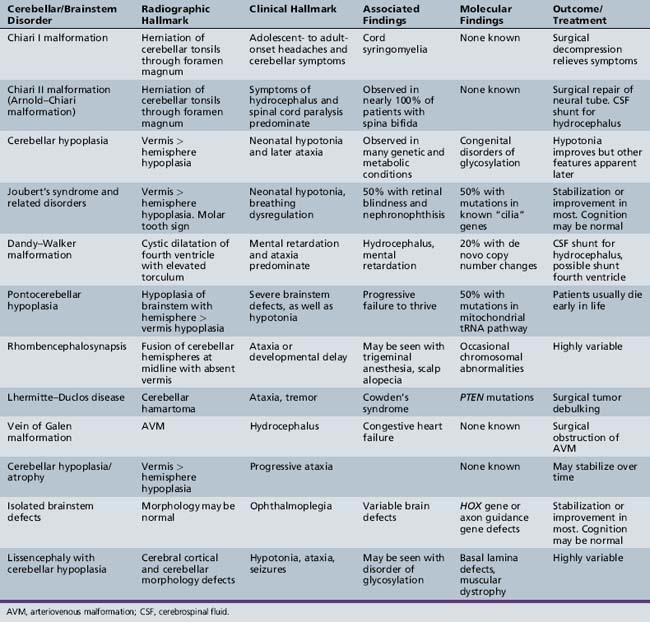
A number of newly described syndromes and diseases now allow for careful consideration of cerebellar and brainstem malformations in children. Because computed tomography (CT) scanning offers poor resolving power of the hindbrain due to tight encasement in the posterior fossa, the complete description of these diseases has lagged behind those involving the cerebral cortex. The introduction of magnetic resonance imaging (MRI) in the last 20 years has provided for the delineation of new syndromes, many of which were previously lumped together as “Dandy–Walker variants” (Table 24-1).
Chiari I Malformation
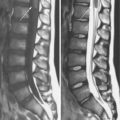
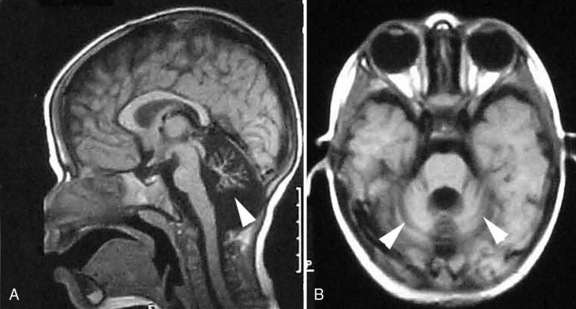
The Chiari I malformation is a disorder of uncertain origin that traditionally has been defined as a downward herniation of the cerebellar tonsils of at least 3–5 mm through the foramen magnum [Tubbs et al., 2007] (Figure 24-1). The anomaly is a leading cause of syringomyelia and occurs in association with osseous abnormalities of the craniovertebral junction. It must be distinguished from the Chiari II malformation (also known as Arnold–Chiari malformation) and the rare Chiari III malformation.
Pathophysiology
Although a number of theories as to the etiology of the Chiari I malformation have been suggested, no clear consensus has emerged. One popular theory that was dispelled was the idea that tethering of the spinal cord causes posterior fossa structures to be pulled into the spinal canal. Another theory gaining favor is that congenital hypoplasia of the posterior fossa occurs due to disrupted mesodermal development [Marin-Padilla and Marin-Padilla, 1981]. There is an increased risk of developing this malformation with growth hormone deficiency and bone defects like rickets. Female patients greatly outnumber male patients, and there is familial clustering, suggesting a genetic predisposition. Gene mutations of chromosomes 9 and 15 may contribute to development of this malformation [Boyles et al., 2006], and it also has recently been linked to Ehlers–Danlos syndrome [Castori et al., 2010]. In about 20 percent of individuals, there is identified trauma that precedes symptom onset.
Clinical Characteristics
Initial clinical features largely depend on whether syringomyelia is present, since this defect tends to dominate the clinical picture. Symptoms from syringomyelia, present in 30–70 percent of adult Chiari I patients but in only 12 percent of children, depend on the location and size of the syrinx and can occur as a result of stretching and distention of the cord [Milhorat et al., 1999; Aitken et al., 2009]. Long-tract findings include hyperreflexia, urinary incontinence, muscle wasting, loss of proprioception, and arm numbness/tingling.
Management
Surgical decompression of the posterior fossa with duraplasty is the treatment of choice in selected cases. The purpose of this surgery is to enlarge the bony compartment. It is important to evaluate for coexistent hydrocephalus before any correction is attempted. In patients with a coexistent syrinx, there is a clear indication for surgery, and in a series of 49 children with Chiari-associated syringomyelia, just over half demonstrated clinical and radiographic improvement after hindbrain decompression [Attenello et al., 2008]. In these patients, decompression of the posterior fossa alone is often sufficient to reduce the syringomyelia without directly shunting the fluid collection. In patients without a syrinx but with >5 mm of caudal displacement or with progressive features, surgery may be offered. In some individuals, clinical symptoms and herniation may spontaneously improve, so a conservative approach is generally adopted in minimally affected patients [Novegno et al., 2008] and in those with <3 mm displacement.
Cerebellar Hypoplasia
Cerebellar hypoplasia is generally an underappreciated entity until observed on brain MRI, and even then it may be missed, unless specifically looked for based upon symptomatology. Hypoplasia, as opposed to atrophy, excludes conditions in which cerebellar development was normal but then shows progressive degeneration, as in Friedreich’s ataxia, vitamin E ataxia, and ataxia with oculomotor apraxia. In patients with cerebellar hypoplasia, psychomotor delay is present in 70–85 percent, and epilepsy in 28 percent [Ventura et al., 2006], although the exact mechanism of the hypoplasia, when identified, can be a better predictor of outcome.
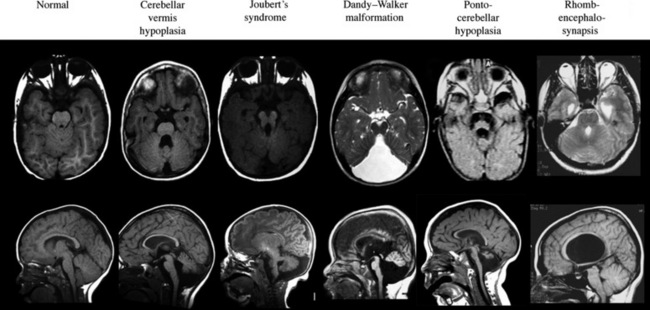
The term “global cerebellar hypoplasia” refers to a cerebellum of reduced volume (Figure 24-2). Typically, the vermis (cerebellar midline) is more severely affected than the hemispheres because the vermis is the last part of the cerebellum to form, and thus, it is probably more sensitive to alterations in genetic programs. The subarachnoid spaces may be passively enlarged. Typical causes include various chromosomal aberrations, such as trisomy 9, 13, and 18, several metabolic disorders including congenital disorders of glycosylation (CDGs) [Vermeer et al., 2007], prenatal teratogenic drugs (e.g., anticonvulsants), fetal infections, or isolated genetic cerebellar hypoplasia, such as that caused by mutations in the OPHN1 gene [Tentler et al., 1999]. Cerebellar hypoplasia can also be observed as part of other brain malformation syndromes, such as lissencephaly, pontocerebellar hypoplasia, or those associated with congenital muscular dystrophies.
Pathophysiology
The etiological basis of the cerebellar disorder predicts the pathophysiology, but generally follows some defect in cellular proliferation or differentiation of either the cerebellar Purkinje or granule cells. In the CDGs, as a result of insufficient protein glycosylation, there is probably an activation of the unfolded protein response, leading to an effect on the cerebellum (see Chapter 35). Likewise, teratogenic exposure to alcohol in animal models can affect neuronal migration by altering established pathways [Jiang et al., 2008].
Differential Diagnosis
It is critical to evaluate individuals with cerebellar hypoplasia for syndromic forms of this malformation, as well as to consider the various genetic etiologies (Table 24-2). Like most developmental brain anomalies, fetal infections, and toxic exposures, recessive and dominant inherited forms, as well as alterations in genetic copy number (using comparative genomic hybridization), should be considered. Normal transferrin isoelectric focusing can help exclude CDGs. Normal family history may help exclude genetic causes. Baseline laboratory tests, including creatine phosphokinase (CPK), vitamin E levels, and TORCH (toxoplasmosis, other infections, rubella, cytomegalovirus, and herpes simplex virus) titers, looking for prenatal viral infection, can help exclude muscle-eye-brain disease, vitamin E deficiency, and congenital infections. Mitochondrial disorders should be considered, especially in the setting of features such as growth retardation, hearing loss, acidosis, and respiratory or multi-organ involvement.
| Cause | Gene | Reference |
|---|---|---|
| Cerebellar ataxia, mental retardation and dysequilibrium syndrome | VLDLR, CA8 | Boycott et al. [2005]; Turkmen et al. [2009] |
| Cerebellar hypoplasia with pancytopenia | DKC1 | Pearson et al. [2008] |
| Cerebellar hypoplasia, Norman type (SCAR2) recessive | Unknown | Megarbane et al. [1999] |
| Cerebellar vermis aplasia (SCA29) dominant | Unknown | Rivier and Echenne [1992] |
| Cerebellar hypoplasia, mental retardation, distinctive facies | OPHN1 | des Portes et al. [2004] |
| Cerebellar aplasia with diabetes mellitus | PTF1A | Sellick et al. [2004] |
| Congenital disorders of glycosylation | CDG1 and 2 | Jaeken and Matthijs [2007] |
Joubert’s Syndrome and Related Disorders
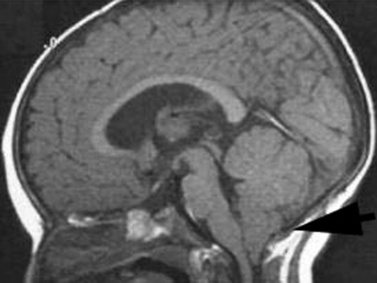
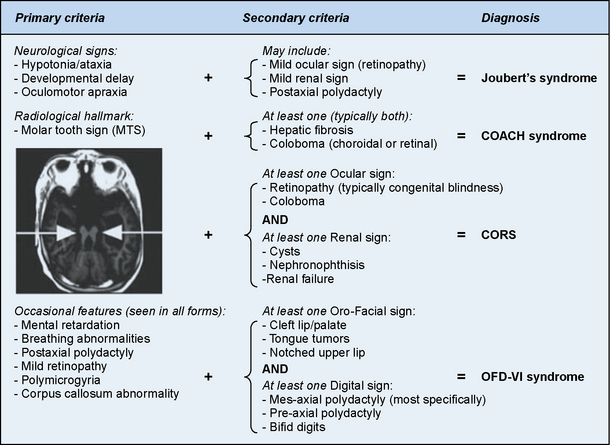
Joubert’s syndrome (JS) and related disorders (JSRDs) are a group of recessive congenital ataxia conditions, usually presenting with neonatal hypotonia, dysregulated breathing rhythms, oculomotor apraxia, and subsequently with mental retardation. JSRDs are the most common inherited congenital cerebellar malformation. The pathognomonic finding in JSRD is the unique molar tooth sign (MTS) on brain imaging (Figure 24-3). There is a tremendously broad spectrum of signs and symptoms, including kidney, retinal, and hepatic involvement, along with polydactyly and facial dysmorphisms.
Clinical Characteristics
The MTS is the result of cerebellar vermis hypoplasia, thick and maloriented superior cerebellar peduncles, and an abnormally deep interpeduncular fossa, which together give the appearance of a tooth. JS is the best-known and probably most common syndrome associated with the MTS. JS is notable for both intrafamilial and interfamilial phenotypic variability. Even in the original pedigree of four affected siblings, two had hypoplasia of the posterior inferior cerebellar vermis and two had complete agenesis of the cerebellar vermis [Joubert et al., 1968].
Clinical features are highly variable for patients with the MTS. Many patients with JS present in the newborn period with an unusual pattern of intermittent respiration, displaying alternating hyperpnea and apnea. In JS, there is no specific lung pathology to explain this respiratory abnormality. Instead, it likely relates to dysfunction of the respiratory centers located in the brainstem or cerebellum [Xu and Fraser, 2002]. Because some neonates have died from apnea, it is critical to institute careful respiratory care until this feature of JS passes, usually by 1 year of age. Some patients will have relatively mild disease, with congenital ataxia that lessens with age, and a delay in the ability to walk until age 4–5 years, but are otherwise healthy. Other patients will display congenital blindness and renal failure requiring dialysis or renal transplantation. It is important to differentiate the type of JSRD at the time of presentation, and then to monitor the patient periodically for development of any extra-central nervous system signs. If patients do not develop retinal or renal involvement by age 10–15 years, they are unlikely to do so.
The disorders related to JS include cerebellar vermis hypo/aplasia-oligophrenia-ataxia-ocular coloboma-hepatic fibrosis (COACH syndrome), cerebello-oculo-renal syndrome (CORS), and oro-facio-digital syndrome type VI (OFD-VI), all of which share the MTS, but additionally display features that distinguish them from JS [Zaki et al., 2008]. Diagnostic criteria can help distinguish each of the four major clinical entities within the JSRD spectrum.
Pathophysiology
JSRDs fall within the “ciliopathy” spectrum of conditions, which are due to alterations in development or signaling within the cellular primary cilium. Motile cilia are well known for their roles in certain tissues, such as the sperm, where they propel cells, but there is another class of cilia, termed primary cilia, which are nonmotile, typically lacking the central pair of microtubules and outer dynein arms necessary for movement. Several genes associated with JS have now been established, and protein products of these genes are found specifically at the primary cilium in most cells throughout the body. Thus, JS is now included with other ciliopathies, including Bardet–Biedl syndrome, nephronophthisis, Leber congenital amaurosis, congenital hepatic fibrosis, Jeune asphyxiating thoracic dystrophy, and Ellis–van Creveld syndrome, as a disorder of the primary cilium [Tobin and Beales, 2009].
For some of these disorders, like Leber congenital amaurosis (congenital retinal blindness), the relation between primary cilia and pathogenesis is readily apparent, given the fact that the entire photoreceptor outer segment is a modified primary cilium. Within the developing cerebellum, primary cilia have been shown to be essential for reception of the cell signaling ligand sonic hedgehog, which in turn is essential for proliferation of cerebellar granule neurons [Chizhikov et al., 2007; Spassky et al., 2008]. Thus, it is postulated that reduced granule neuron production is the central aspect of the cerebellar hypoplasia.
Differential Diagnosis
JSRDs can be distinguished by the presence of the MTS, which can be detected prenatally and used for diagnosis [Saleem and Zaki, 2010]. In the absence of the MTS, other cerebellar malformations must be considered. In some families, there can be one child with JS and a second child with either a more severe disorder like Meckel–Gruber syndrome, or a less severe disease like Bardet–Biedl syndrome. This heterogeneity cannot be clearly explained by genetics alone, since even identical twins can show divergent severity [Raynes et al., 1999]. It is important to evaluate the whole family to determine if children with such divergent disease might share the same underlying genetic cause.
Management
The management of JSRD can be aided by anticipation of typical problems and the use of genetic testing to help determine prognosis. Patients identified in the neonatal intensive care unit should be monitored closely for apnea. Treatment with caffeine may help promote respiratory drive and an apnea monitor may be required for the first year of life. Because of the heterogeneity of these conditions, it is recommended that patients be evaluated by a geneticist and undergo screening for coexisting medical problems, including brain MRI, polysomnogram, echocardiogram, careful dysmorphology and retinal evaluation, and annual monitoring with liver function testing and renal ultrasound examinations [Parisi and Dobyns, 2003].
Prenatal diagnosis may be approached in one of several ways. JS is one of the disorders that may be encountered when cerebellar hypoplasia is detected on prenatal ultrasound. In this case, an ultrasonographer may observe nonspecific hypoplasia, and then will seek fetal brain MRI for more specific diagnosis. JS may diagnosed on brain MRI, based on the presence of the MTS [Saleem and Zaki, 2010]. Alternatively, prenatal diagnosis can be made genetically, especially in certain populations with founder mutations [Valente et al., 2010].
Genetic testing can predict disease severity, based upon established genotype–phenotype correlations. Mutations in certain genes for JS, such as AHI1, INPP5E, CC2D2A, and ARL13B, can be detected in patients with MTS who do not have involvement of other organs, or they can be detected in patients with MTS together with retinal blindness. Mutations in TMEM216 and RPGRIP1L genes are often encountered in those patients with MTS and renal involvement. Mutations in CEP290 are encountered in half of patients with MTS together with retinal blindness and renal involvement. Mutations in TMEM67 are the most common cause of MTS with liver involvement [Brancati et al., 2009]. Thus, the specific gene mutation identified in a patient can help predict the range of organ involvement.
Dandy–Walker Malformation
Dandy–Walker malformation (DWM) is defined by hypoplasia and upward rotation of the cerebellar vermis, accompanied by cystic dilatation of the fourth ventricle and frequently an enlarged posterior skull. It was first described by Dandy and Blackfan in 1914, and is the most common congenital malformation of the cerebellum with an incidence of about 1 in 5000 live births [Parisi and Dobyns, 2003]. Previous definitions of DWM have included features such as the presence of hydrocephalus or the presence of communication between the posterior fossa and the fourth ventricle. Furthermore, the now outdated term, Dandy–Walker variant, used to describe all cerebellar malformations before the routine use of brain MRI, should be replaced using stricter criteria.
This triad can be found in association with supratentorial hydrocephalus, which traditionally has been considered a complication rather than a part of the malformation complex [D’Agostino et al., 1963; Hart et al., 1972]. New data, however, suggest that the hydrocephalus may reflect a common developmental disruption affecting multiple brain regions [Aldinger et al., 2009].
Clinical Characteristics
The clinical spectrum associated with DWM is extremely broad, ranging from asymptomatic to severe mental retardation. Half of the patients have normal cognition, whereas others never achieve normal intellectual function, even when hydrocephalus is treated effectively. The vermis anatomy in DWM is statistically correlated with neurological and intellectual outcome [Klein et al., 2003]. Systemic malformations associated with DWM may include cardiac anomalies (ventriculoseptal defects, patent ductus arteriosus, transposition of the great arteries), urogenital anomalies (hydroceles, vesico-ureteral reflux, abnormal kidney shape), and other abnormalities (duodenal atresia, cleft palate, malformed limbs), and occur collectively in about half of patients [Sasaki-Adams et al., 2008]. Those with isolated DWM can have normal development.
Pathophysiology
The low empiric recurrence risk of approximately 1–2 percent for nonsyndromic DWM suggested that a simple mode of inheritance was unlikely [Murray et al., 1985], and the frequent association with chromosomal anomalies supported this hypothesis. Accumulating evidence suggests that many cases represent de novo alterations in genomic integrity, with focus on chromosome 3q2 and 6p25.3 associated with the Axenfeld–Rieger syndrome [Aldinger et al., 2009]. Additionally, a dominant form has been linked to chromosome 2q36.1 in association with occipital encephalocele.
The genes ZIC1, ZIC4, and FOXC1 when deleted can result in DWM. The ZIC1 [Grinberg et al., 2004; Aldinger et al., 2009] and ZIC4 genes are produced in cerebellar primordial cells, whereas the FOXC1 gene is produced by the developing mesenchyme, which gives rise to the meninges and posterior skull, overlying the developing cerebellum. This suggests at least two different mechanisms of disease, one in which some regulatory genes are required in the developing cerebellum itself, and others in the adjacent mesenchyme, with DWM a result of either defect. Because all genes identified to date are transcription factors, DWM is presumably a disorder associated with altered cellular programming during development; however, further research is required to delineate these mechanisms.
Differential Diagnosis
Previous literature also used the term Dandy–Walker variant, which included any of the various cerebellar hypoplasias; this should be dropped in favor of more specific terminology. It is important to consider mega cisterna magna, retrocerebellar cysts, and Blake’s pouch cyst in the differential diagnoses [Nelson et al., 2004]. In each of these disorders, there is a fluid collection in the posterior fossa adjacent to the cerebellum, but not within the fourth ventricle, without elevation of the torcula; they thus do not meet the criteria for DWM. Additionally, in these three conditions, which are often found incidentally, the prognosis is usually excellent. In some cases, imaging findings are intermediate between one of these conditions and DWM, and expert review might be necessary to arrive at a precise diagnosis. As mentioned, the association with hydrocephalus, congenital heart disease, urogenital anomalies, and cleft lip/palate can support the diagnosis of DWM.
Pontocerebellar Hypoplasia
Pontocerebellar hypoplasia (PCH) is distinguished from other cerebellar and brainstem defects by the presence of hypoplasia of the brainstem structures, as well as of the cerebellum [Barth, 1993]. In the PCH spectrum, reductions in the size of these structures can vary widely, the degree of atrophy being associated with a worse prognosis.
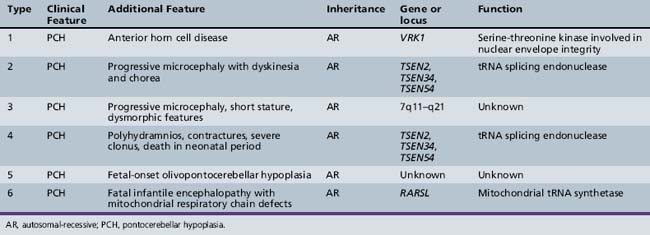
Six forms of PCH have been identified, each with unique clinical and genetic features. In PCH1, there is central and peripheral motor dysfunction from birth, leading to early death, usually before 1 year of age. PCH and gliosis are seen, in association with anterior horn cell degeneration resembling infantile spinal muscular atrophy (SMA) [Rudnik-Schoneborn et al., 2003]. PCH2 is associated with progressive microcephaly from birth, combined with extrapyramidal dyskinesias. Motor and mental development does not progress, and severe chorea and epilepsy are frequent. In contrast to PCH1, signs of anterior horn cell disease are absent. PCH3 is characterized by progressive microcephaly and linkage to chromosome 7q11–q21. PCH4, 5, and 6 are all clinically similar, some with features of mitochondrial encephalopathy.
Clinical Characteristics
Patients typically present at birth with difficulty feeding, with maintaining their airway, or with temperature regulation. Labor may be associated with failure to progress through the birth canal. Patients display global hypotonia with a reduced level of alertness. Brainstem features predominate and include difficulty with coordination of sucking and swallowing and with handling oral secretions. These may be a reflection of defects in brainstem circuitry from development or may represent progressive degeneration of these circuits. There are some clinical features that can help distinguish the various PCH subtypes, but in actuality it is difficult to distinguish precisely between these subtypes solely on clinical grounds, so molecular testing can be helpful (Table 24-3).
Pathophysiology
The genetic etiologies of several of the PCH subtypes recently have been identified. One of the genes for PCH1 (VRK1) encodes for vaccinia-related kinase 1, a serine-threonine kinase required for nuclear envelope formation [Renbaum et al., 2009]. Absence of VRK1 suggests the presence of a defect in cellular integrity of posterior fossa structures, as well as anterior horn cells, which causes this condition.
Using homozygosity mapping, several of the genes for other forms of PCH have been identified in three of the four subunits of the tRNA splicing endonuclease. These genes, TSEN2, TSEN34, and TSEN54, cause PCH2 and PCH4 when mutated [Budde et al., 2008]. An additional gene mutation in the RARSL gene, encoding a mitochondrial arginine tRNA synthase, is mutated in a severe form of PCH, termed PCH6 [Edvardson et al., 2007]. The function of the splicing endonuclease is to remove introns from pre-RNAs, including tRNAs. Since in humans there are several tRNAs that require splicing, these genes presumably help promote maturation of tRNAs that are essential for mitochondrial protein synthesis. These findings suggest that a defect in mitochondrial function underlies some forms of PCH. This has led to the suggestion that PCH represents a form of mitochondrial disorder, although there is yet no evidence of generalized mitochondrial disturbances or accumulation of metabolites typically associated with mitochondrial dysfunction. Furthermore, there is no clear evidence for alterations in muscle integrity or visual or hearing deficits that are often associated with mitochondrial disorders. This raises the possibility of unique functions of these genes in modifying mitochondrial function during brainstem and cerebellar development.
Rhombencephalosynapsis
Clinical Characteristics
The clinical characteristics of Rho are mild and nonspecific, and include signs of developmental delay, hypotonia, and ataxia [Kruer et al., 2009]. Most children can learn to walk and speak, but usually these skills develop between 3 and 6 years of age. There is no evidence of developmental regression and epilepsy is uncommon.
Pathophysiology
In one series of 40 cases, Rho was always associated with other brain abnormalities, including Purkinje cell heterotopias, collicular and thalamic fusion, agenesis of the corpus callosum, lobar holoprosencephaly, and neural tube defects [Pasquier et al., 2009].
Differential Diagnosis
Cerebellotrigeminal-dermal dysplasia, also known as Gomez–Lopez–Hernandez (GLH) syndrome, displays Rho, and should be distinguished from isolated forms of this disorder. GLH represents a rare neurocutaneous syndrome of craniosynostosis, ataxia, trigeminal anesthesia, scalp alopecia, cerebellar anomalies, midface hypoplasia, corneal opacities, mental retardation, and short stature. Rho is the most consistent finding in GLH [Poretti et al., 2008a], but most frequently it is seen in isolation from these other findings.
Rho has also been reported in Vacterl-H syndrome [Pasquier et al., 2009], comprising a combination of vertebral, anal, cardiac, tracheoesophageal, renal, limb, and other anomalies, with associated hydrocephalus.
Lhermitte–Duclos Disease
Clinical Characteristics
Patients with LDD present with ataxia, signs and symptoms of increased intracranial pressure, and seizures [Abel et al., 2005]. LDD is associated with the familial hamartoma-neoplasia syndrome, Cowden’s disease (CD), an inherited cancer/hamartoma syndrome involving breast, thyroid, and other organs. Both conditions are linked to germline heterozygous mutations in the PTEN gene on chromosome 10 [Backman et al., 2001; Kwon et al., 2001]. LDD is frequently associated with features of CD. In one series of 31 patients with PTEN mutations, approximately 10 percent met full criteria for CD; another 30 percent had thyroid disease [Abel et al., 2005].
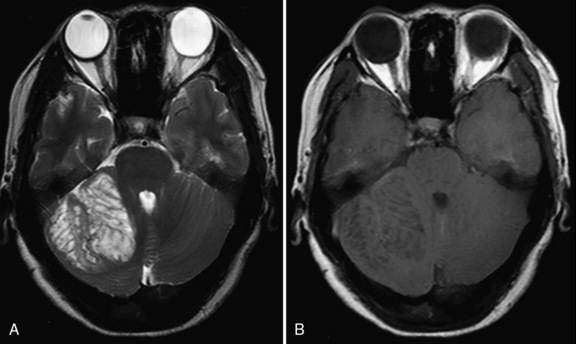
Brain MRI shows a nonenhancing unilateral lesion in the cerebellum, with mass effect on surrounding structures. The lesion characteristically is hypointense on T1 and hyperintense on T2 images, with alternating parallel hyperintense and isointense stripes that correspond to the inner molecular and granular layers of the cerebellum (Figure 24-4).
Management
The typical course consists of an insidious expansion of a posterior fossa mass. Decompressive surgery for symptomatic patients has been successful in improving long-term survival but the lack of clear tumor margins makes resection challenging. Chemotherapy is ineffective in treating these hamartomatous-like lesions. Although surgical management and anticipation of other PTEN-associated symptoms are the mainstays of treatment, it is possible that use of mammalian target of rapamycin (mTOR) inhibitors (e.g., rapamycin) might be effective, as preliminary studies show hyperactivation of this pathway in LDD [Abel et al., 2005].
Vein of Galen Malformation
Clinical Characteristics
There are three typical clinical presentations reported within the spectrum of VGAM [Gold et al., 1964]. The neonatal presentation consists of severe cardiorespiratory failure, including hydrops fetalis and renal failure secondary to aortic flow reversal. Of the vast majority of patients presenting in the newborn period, most show high-output cardiac failure with severe pulmonary hypertension, which may be mistaken for congenital heart disease. The infantile form presents with smaller and less hemodynamically apparent cardiac involvement. These patients can present with increasing head circumference due to aqueductal compression by the dilated vein of Galen with resultant hydrocephalus, or can present with nonspecific developmental delay. Other features may include hypothalamic and hypophyseal dysfunction, intracranial or carotid bruit, dilated scalp veins, proptosis, or epistaxis. In the childhood form, the typical presentation is headaches and seizures caused by subarachnoid and intraparenchymal hemorrhages. Such patients usually have insignificant cardiac symptoms [Heuer et al., 2010].
Pathophysiology
The vascular malformation itself is thought to originate around 2–3 months of gestation from the choroidal arteries. The high blood flow, coupled with dural sinus stenosis, causes the anterior segment of the median prosencephalic vein of Markowski (which normally regresses) to progressively enlarge instead, forming the aneurysmal component [Gailloud et al., 2005]. The subsequent left-to-right shunt causes a noncyanotic high-flow state with resultant heart failure and accompanying macrocephaly.
Differential Diagnosis
Two types of arteriovenous connections have been recognized. The simplest (or “choroidal”) type consists of direct, high-flow shunts located within the wall of the venous aneurysm. The second (or “mural”) type involves the interposition of an arterial network between arterial feeders and the venous aneurysm itself. This network is usually located in the quadrigeminal cistern and results in less significant blood flow [Gailloud et al., 2005].
Management
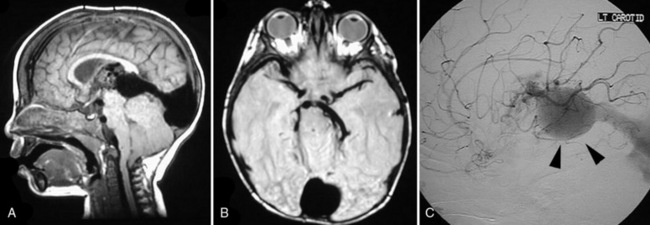
Patients with neonatal-onset VGAM historically suffered from a poor prognosis, which has significantly improved with the recent development of standard endovascular embolization. Prenatal ultrasound can detect VGAMs, but MRI and MR angiography can better delineate the anatomic boundaries and blood flow. Conventional angiography is the gold standard for precise evaluation of VGAM architecture, including evaluation of arterial feeders and venous drainage (Figure 24-5). Following radiographic studies, the goal is to stabilize patients with medications until they are old enough to tolerate endovascular surgery. The complete removal of the lesion is rarely achieved because of hemodynamic instability of the infant and location of the lesion. As a result, surgical treatment is now reserved for the evacuation of intracranial hematomas and treatment of hydrocephalus. Embolization is achieved using embolic glue or microcoils. One of the goals is to prevent the development of cerebral venous hypertension, so stepwise embolization to allow for collateral formation is advised [Hoang et al., 2009]. Recently, a 21-point scale was developed to help guide therapy [Lasjaunias et al., 2006], and most children survive, frequently with some neuromotor sequelae.
Cerebellar Atrophy
Clinical and Radiographic Characteristics
It can be difficult to separate CA from cerebellar hypoplasia, the former being characterized primarily by the full complement of folia, but with increases in the interfolial cerebrospinal fluid spaces, implying shrinkage of the parenchyma (Figure 24-6). Separation of CA from cerebellar hypoplasia is not always possible and the two can coexist in several different diseases, such as the CDGs.
Pathophysiology
While precise mechanisms are specific to each cause, CA is usually accompanied by some combination of degeneration of the Purkinje and granular cell layer, as well as some involvement of the cerebellar white matter. For instance, ataxia telangiectasia and spinocerebellar ataxias (see Chapter 67) are correlated with Purkinje cell degeneration, whereas in other conditions there is a predilection for the granular layer [Pascual-Castroviejo et al., 1994]. Purkinje cells are extremely vulnerable to various cytotoxic and metabolic derangements, and when these cells are damaged, the whole cerebellum can become atrophic.
Differential Diagnosis
The clinical presentation and appearance of the cerebellum on MRI may suggest the specific etiology of CA (Box 24-1). In some individuals there may be clinical findings to suggest a specific entity, such as the presence of oculocutaneous telangiectasias (e.g., ataxia telangiectasia), cataracts (Marinesco–Sjögren syndrome), progressive myoclonic seizures (neuronal ceroid-lipofuscinosis), or peripheral nerve involvement (infantile neural axonal dystrophy). However, in the majority of patients, such diagnostic findings are usually not present. When they are absent, it is wise to consider a broad differential to include serum albumin and α-fetoprotein (which can be abnormal in ataxia telangiectasia and ataxia-oculomotor apraxia), a lysosomal enzyme panel, liver enzymes, serum copper and ceruloplasmin levels, and a metabolic profile (including a chemistry profile, plasma amino acids, urine organic acids, and serum lactate, pyruvate, and ammonia levels).
Box 24-1 Types of Cerebellar Atrophy




















Specific considerations for CA include diseases such as toxic exposures to phenytoin or lithium (which are unusual in childhood), postinfectious cerebellar ataxia (usually presents with acute rather than chronic ataxia), and many autosomal-dominant and recessive genetic disorders, as reviewed in Chapter 67. Relatively recently recognized syndromes include cerebellar ataxia with mental retardation, optic atrophy, and skin abnormalities (CAMOS) syndrome, due to mutations in the ZNF592 gene [Nicolas et al., 2010], and ataxia oculomotor apraxia (AOA) syndrome, which presents very similarly to ataxia telangiectasia (e.g. with progressive ataxia) but backs telangectasias, increased risk for infections, leukemias and lymphomas seen in ataxia telangectasia. Mutations in the APTX and SETX genes have emerged as the major causes.
CA also may be seen in later stages of Friedreich’s ataxia (FA), one of the most common forms of autosomal-recessive ataxia (reviewed in detail in Chapter 67). It usually presents before adolescence with generalized ataxia and absent tendon reflexes, Babinski signs, impaired position and vibration sense, and scoliosis. The triad of hypoactive knee and ankle reflexes and ataxia in childhood is generally sufficient for diagnosis. In FA, it is important to recognize the clinically significant hypertrophic cardiomyopathy and diabetes, as well as the causative trinucleotide expansion in the FXN gene, which is involved in mitochondrial function and iron homeostasis. Treatments are emerging for many of the anticipated problems of FA, specifically targeted at underlying pathogenic mechanisms [Schmucker and Puccio, 2010].
Also reviewed in Chapter 67, the various autosomal-dominant forms of spinocerebellar ataxia (SCA) are a cause of CA in children. Although most SCAs present post adolescence, there are a number that can manifest at earlier ages, including SCA13, SCA17, SCA21, SCA25, and the related disorder known as juvenile Huntington’s disease. Many of the SCAs are due to CAG trinucleotide repeat expansions within the coding sequence of their respective genes; because the CAG tract encodes for glutamine, such disorders have also been called polyglutamine disorders. Due to the mutability of these expansions, the dominant forms display anticipation, referring to earlier onset and increasing severity of disease in subsequent generations of a family. SCA13 displays childhood onset associated with mild mental retardation and short stature, and is one of the rare dominant SCAs not associated with repeat expansion. SCA17 displays childhood or adult onset associated with mental deterioration, occasional chorea, dystonia, myoclonus, and epilepsy. The other forms of SCA listed above have been observed only in single publications.
Management
Except for vitamin E therapy for ataxia with vitamin E deficiency (AVED) (see Chapter 103) and some on-going but as yet inconclusive trials in FA, there are no specific treatments available for the conditions causing CA. Aggravating medications should be avoided. Physical medicine management is helpful (e.g., physical, occupational, and speech therapy; use of canes, walkers, writing instruments, or weights to minimize tremor) and can improve the activities of daily living (see Chapter 105).
Isolated Brainstem Defects
The advent of higher-resolution MRI, as well as diffusion tensor imaging, has allowed for the delineation of a number of developmental brainstem defects [Barkovich et al., 2009]. As the brainstem contains nuclei for oculomotor and facial movements, as well transit and decussation of ascending and descending tracts, the major symptoms of the disorders of brainstem development described to date involve coordination defects, abnormal oculomotor function, and abnormal lateralization of function. The two most important diseases to consider are horizontal gaze palsy with progressive scoliosis (HGPPS) and pontine tegmental cap dysplasia (PTCD) (Figure 24-7). HGPPS patients have congenital horizontal gaze palsy; MRI shows quite characteristic brainstem hypoplasia with absence of the facial colliculi, presence of a deep midline dorsal pontine cleft (split pons sign), and a “butterfly” configuration of the medulla [Rossi et al., 2004]. Neurophysiological studies have demonstrated that there is failure of decussation of the corticospinal and spinocortical tracts [Jen et al., 2004].
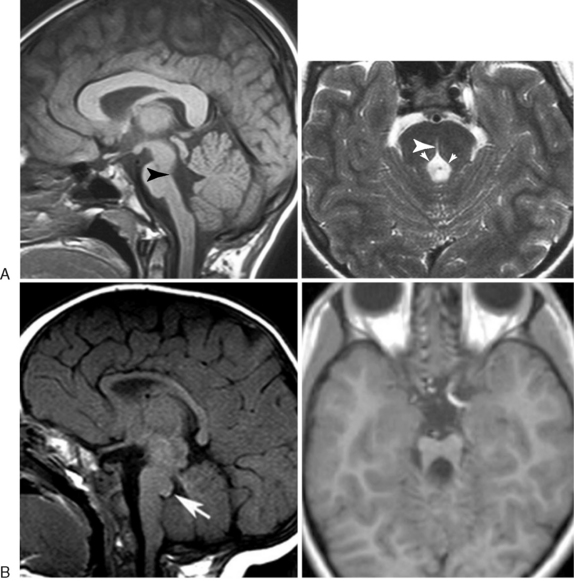
Fig. 24-7 Magnetic resonance imaging analysis of isolated brainstem defects.
(Used by permission from Barth PG et al. Pontine tegmental cap dysplasia: a novel brain malformation with a defect in axonal guidance. Brain 2007;130:2258–2266; Rossi A et al. MR imaging of brain-stem hypoplasia in horizontal gaze palsy with progressive scoliosis. AJNR Am J Neuroradiol 2004;25:1046–1048.)
PTCD patients display peripheral auditory dysfunction, impaired swallowing, facial palsy, and ataxia. MRI analysis shows a unique protrusion from the dorsal pons into the fourth ventricle, flattened ventral pons, vermis hypoplasia, and absence of the middle cerebellar peduncles [Barth et al., 2007]. Diffusion tensor imaging shows ectopic transverse fiber bundles at the site of the pontine tegmentum and complete absence of transverse fibers in the ventral pons. Because brainstem defects described to date involve unique sets of axonal tracts or brainstem nuclei, clinical characteristics are highly variable. The pathophysiology of some of these diseases has uncovered a role for axon guidance molecules that have been previously identified from mouse genetic studies, including the ROBO3 and DCC genes [Jen et al., 2004; Srour et al., 2010]. The differential diagnosis must include the other hindbrain defects discussed in this chapter, and careful review of the structural magnetic resonance, diffusion tensor imaging studies and clinical features can differentiate these from previously described conditions. No known treatment exists for these disorders but rehabilitation medicine management is helpful.
Lissencephaly with Cerebellar Hypoplasia
A number of conditions are connected with cerebellar and brainstem developmental defects associated with cerebral cortical anomalies. These fall into three predominant categories, with unique clinical presentations and imaging anomalies [Jissendi-Tchofo et al., 2009] (Figure 24-8). Many of these conditions are reviewed in other chapters of Part V.
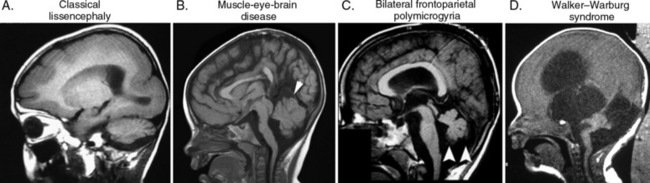
The tubulin pathway defects, associated with mutations in the LIS1 and DCX genes, show predominantly classical lissencephaly with mild cerebellar hypoplasia. The clinical picture is dominated by defects in cerebral cortical lamination, that produce profound mental impairment and intractable epilepsy. The overall cerebellar architecture and brainstem are intact, but they are mildly hypoplastic (see Figure 24-8). Mutation in the tubulin TUBA1A shows a wide spectrum of cerebellar and brainstem involvement [Kumar et al., 2010; Lecourtois et al., 2010].
The reelin signaling pathway defects, associated with mutations in RELN and VLDLR, show cortical pachygyria and severe cerebellar and brainstem hypoplasia. Both the RELN and VLDLR mutations are associated with unique hindbrain imaging findings, and are discernible with careful review of imaging. The clinical picture of RELN is similar to that of classical lissencephaly, with profound mental impairment and intractable epilepsy. Mutations in VLDLR have been reported in familial dysequilibrium syndrome and Uner Tan’s syndrome (quadripedilism), with variable effects on cognition [Boycott et al., 2005; Ozcelik et al., 2008].
The congenital muscular dystrophies typically show brainstem involvement, although the degree of impairment is highly variable. Walker–Warburg syndrome, due to mutations in POMT1 and POMT2 genes, shows the most severe clinical and imaging findings, with hydrocephalus, retinal dysplasia, cataracts, severe cobblestone lissencephaly, diffuse central nervous system white-matter signal abnormalities, and severe brainstem and cerebellar hypoplasia [Beltran-Valero de Bernabe et al., 2002; van Reeuwijk et al., 2005]. Muscle-eye-brain disease, due to mutations in POMGNT1, shows intermediate phenotypes with ventriculomegaly, mild cobblestone lissencephaly, and less severe brainstem and cerebellar hypoplasia and cerebellar cysts [Hehr et al., 2007]. Bilateral frontoparietal polymicrogyria, due to mutations in the GPR56 gene, shows the cobblestone-like appearance of polymicrogyria, as well as cerebellar and brainstem involvement that is similar to that observed in muscle-eye-brain disease [Piao et al., 2002].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abel T.W., Baker S.J., Fraser M.M., et al. Lhermitte-Duclos disease: a report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J Neuropathol Exp Neurol. 2005;64:341-349.
Aitken L.A., Lindan C.E., Sidney S., et al. Chiari type I malformation in a pediatric population. Pediatr Neurol. 2009;40:449-454.
Aldinger K.A., Lehmann O.J., Hudgins L., et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41:1037-1042.
Attenello F.J., McGirt M.J., Gathinji M., et al. Outcome of Chiari-associated syringomyelia after hindbrain decompression in children: analysis of 49 consecutive cases. Neurosurgery. 2008;62:1307-1313. discussion 1313
Backman S.A., Stambolic V., Suzuki A., et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396-403.
Barkovich A.J., Millen K.J., Dobyns W.B. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132:3199-3230.
Barth P.G. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411-422.
Barth P.G., Majoie C.B., Caan M.W., et al. Pontine tegmental cap dysplasia: a novel brain malformation with a defect in axonal guidance. Brain. 2007;130:2258-2266.
Beltran-Valero de Bernabe D., Currier S., Steinbrecher A., et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033-1043.
Boycott K.M., Flavelle S., Bureau A., et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77:477-483.
Boyles A.L., Enterline D.S., Hammock P.H., et al. Phenotypic definition of Chiari type I malformation coupled with high-density SNP genome screen shows significant evidence for linkage to regions on chromosomes 9 and 15. Am J Med Genet A. 2006;140:2776-2785.
Brancati F., Iannicelli M., Travaglini L., et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat. 2009;30:E432-E442.
Budde B.S., Namavar Y., Barth P.G., et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40:1113-1118.
Castori M., Camerota F., Celletti C., et al. Natural history and manifestations of the hypermobility type Ehlers-Danlos syndrome: a pilot study on 21 patients. Am J Med Genet A. 2010;152A:556-564.
Chizhikov V.V., Davenport J., Zhang Q., et al. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci. 2007;27:9780-9789.
D’Agostino A.N., Kernohan J.W., Brown J.R. The Dandy-Walker Syndrome. J Neuropathol Exp Neurol. 1963;22:450-470.
des Portes V., Boddaert N., Sacco S., et al. Specific clinical and brain MRI features in mentally retarded patients with mutations in the Oligophrenin-1 gene. Am J Med Genet. 2004;124A:364-371.
Edvardson S., Shaag A., Kolesnikova O., et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857-862.
Gailloud P., O’Riordan D.P., Burger I., et al. Diagnosis and management of vein of galen aneurysmal malformations. J Perinatol. 2005;25:542-551.
Gold A., Ransohoff J., Carter S. Vein of Galen Malformation. Acta Neurol Scand Suppl. 1964;40(Suppl 11):11-31.
Grinberg I., Northrup H., Ardinger H., et al. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. 2004;36:1053-1055.
Hart M.N., Malamud N., Ellis W.G. The Dandy-Walker syndrome. A clinicopathological study based on 28 cases. Neurology. 1972;22:771-780.
Hehr U., Uyanik G., Gross C., et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics. 2007;8:279-288.
Heuer G.G., Gabel B., Beslow L.A., et al. Diagnosis and treatment of vein of Galen aneurysmal malformations. Childs Nerv Syst. 2010;26:879-887.
Hoang S., Choudhri O., Edwards M., et al. Vein of Galen malformation. Neurosurg Focus. 2009;27:E8.
Jaeken J., Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261-278.
Jen J.C., Chan W.M., Bosley T.M., et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509-1513.
Jiang Y., Kumada T., Cameron D.B., et al. Cerebellar granule cell migration and the effects of alcohol. Dev Neurosci. 2008;30:7-23.
Jissendi-Tchofo P., Kara S., Barkovich A.J. Midbrain-hindbrain involvement in lissencephalies. Neurology. 2009;72:410-418.
Joubert M., Eisenring J.J., Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology. 1968;18:302-303.
Klein O., Pierre-Kahn A., Boddaert N., et al. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst. 2003;19:484-489.
Kruer M.C., Blasco P.A., Anderson J.C., et al. Truncal ataxia, hypotonia, and motor delay with isolated rhombencephalosynapsis. Pediatr Neurol. 2009;41:229-231.
Kumar R.A., Pilz D.T., Babatz T.D., et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010.
Kwon C.H., Zhu X., Zhang J., et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404-411.
Lasjaunias P.L., Chng S.M., Sachet M., et al. The management of vein of Galen aneurysmal malformations. Neurosurgery. 2006;59:S184-S194. discussion S183–S113
Lecourtois M., Poirier K., Friocourt G., et al. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010;119:779-789.
Marin-Padilla M., Marin-Padilla T.M. Morphogenesis of experimentally induced Arnold–Chiari malformation. J Neurol Sci. 1981;50:29-55.
Megarbane A., Delague V., Salem N., et al. Autosomal recessive congenital cerebellar hypoplasia and short stature in a large inbred family. Am J Med Genet. 1999;87:88-90.
Milhorat T.H., Chou M.W., Trinidad E.M., et al. Chiari I malformation redefined: clinical and radiographic findings for 364 symptomatic patients. Neurosurgery. 1999;44:1005-1017.
Murray J.C., Johnson J.A., Bird T.D. Dandy-Walker malformation: etiologic heterogeneity and empiric recurrence risks. Clin Genet. 1985;28:272-283.
Nelson M.D.Jr, Maher K., Gilles F.H. A different approach to cysts of the posterior fossa. Pediatr Radiol. 2004;34:720-732.
Nicolas E., Poitelon Y., Chouery E., et al. CAMOS, a nonprogressive, autosomal recessive, congenital cerebellar ataxia, is caused by a mutant zinc-finger protein, ZNF592. Eur J Hum Genet. 2010.
Novegno F., Caldarelli M., Massa A., et al. The natural history of the Chiari Type I anomaly. J Neurosurg Pediatr. 2008;2:179-187.
Ozcelik T., Akarsu N., Uz E., et al. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc Natl Acad Sci USA. 2008;105:4232-4236.
Parisi M.A., Dobyns W.B. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab. 2003;80:36-53.
Pascual-Castroviejo I., Gutierrez M., Morales C., et al. Primary degeneration of the granular layer of the cerebellum. A study of 14 patients and review of the literature. Neuropediatrics. 1994;25:183-190.
Pasquier L., Marcorelles P., Loget P., et al. Rhombencephalosynapsis and related anomalies: a neuropathological study of 40 fetal cases. Acta Neuropathol. 2009;117:185-200.
Pearson T., Curtis F., Al-Eyadhy A., et al. An intronic mutation in DKC1 in an infant with Hoyeraal-Hreidarsson syndrome. Am J Med Genet A. 2008;146A:2159-2161.
Piao X., Basel-Vanagaite L., Straussberg R., et al. An autosomal recessive form of bilateral frontoparietal polymicrogyria maps to chromosome 16q12.2–21. Am J Hum Genet. 2002;70:1028-1033.
Poretti A., Bartholdi D., Gobara S., et al. Gomez-Lopez-Hernandez syndrome: an easily missed diagnosis. Eur J Med Genet. 2008;51:197-208.
Poretti A., Wolf N.I., Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood. Eur J Paediatr Neurol. 2008;12:155-167.
Raynes H.R., Shanske A., Goldberg S., et al. Joubert syndrome: monozygotic twins with discordant phenotypes. J Child Neurol. 1999;14:649-654.
Renbaum P., Kellerman E., Jaron R., et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85:281-289.
Rivier F., Echenne B. Dominantly inherited hypoplasia of the vermis. Neuropediatrics. 1992;23:206-208.
Rossi A., Catala M., Biancheri R., et al. MR imaging of brain-stem hypoplasia in horizontal gaze palsy with progressive scoliosis. Am J Neuroradiol. 2004;25:1046-1048.
Rudnik-Schoneborn S., Sztriha L., Aithala G.R., et al. Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. Am J Med Genet A. 2003;117A:10-17.
Saleem S.N., Zaki M.S. Role of MR imaging in prenatal diagnosis of pregnancies at risk for Joubert syndrome and related cerebellar disorders. Am J Neuroradiol. 2010;31:424-429.
Sasaki-Adams D., Elbabaa S.K., Jewells V., et al. The Dandy-Walker variant: a case series of 24 pediatric patients and evaluation of associated anomalies, incidence of hydrocephalus, and developmental outcomes. J Neurosurg Pediatr. 2008;2:194-199.
Schmucker S., Puccio H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum Mol Genet. 2010;19:R103-R110.
Sellick G.S., Barker K.T., Stolte-Dijkstra I., et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36:1301-1305.
Spassky N., Han Y.G., Aguilar A., et al. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol. 2008;317:246-259.
Srour M., Riviere J.B., Pham J.M., et al. Mutations in DCC cause congenital mirror movements. Science. 2010;328:592.
Tentler D., Gustavsson P., Leisti J., et al. Deletion including the oligophrenin-1 gene associated with enlarged cerebral ventricles, cerebellar hypoplasia, seizures and ataxia. Eur J Hum Genet. 1999;7:541-548.
Tobin J.L., Beales P.L. The nonmotile ciliopathies. Genet Med. 2009;11:386-402.
Tubbs R.S., Lyerly M.J., Loukas M., et al. The pediatric Chiari I malformation: a review. Childs Nerv Syst. 2007;23:1239-1250.
Turkmen S., Guo G., Garshasbi M., et al. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet. 5, 2009. e1000487
Valente E.M., Logan C.V., Mougou-Zerelli S., et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010.
van Reeuwijk J., Janssen M., van den Elzen C., et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005;42:907-912.
Ventura P., Presicci A., Perniola T., et al. Mental retardation and epilepsy in patients with isolated cerebellar hypoplasia. J Child Neurol. 2006;21:776-781.
Vermeer S., Kremer H.P., Leijten Q.H., et al. Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J Neurol. 2007;254:1356-1358.
Xu F., Fraser D.T. Role of the cerebellar deep nuclei in respiratory modulation. Cerebellum. 2002;1(1):35-40.
Zaki M.S., Abdel-Aleem A., Abdel-Salam G., et al. The molar tooth sign: a new Joubert syndrome and related cerebellar disorders classification system tested in Egyptian families. Neurology. 2008;70:556-565.