Chapter 25 Disorders of Brain Size
Introduction
The two obvious disorders of brain size – microcephaly (too small) and macrocephaly (too large) – are very common or relatively common disorders, depending largely on how they are defined. Microcephaly (MIC) and macrocephaly are usually defined as head circumference – or more formally, “occipitofrontal circumference” (OFC) – that is more than 2 standard deviations (SD) below or above the mean for age and gender [Opitz and Holt, 1990; Roche et al., 1987]. However, because this criterion includes many developmentally normal individuals and a host of underlying causes, researchers studying both usually define severe MIC or macrocephaly as OFC more than 3 or 4 SD below or above the mean [Barkovich et al., 1998; Dobyns, 1996; Woods et al., 2005; Jackson et al., 2002].
When defined as OFC smaller than 2 SD below the mean, approximately 2.3 percent of the population would be expected to have MIC if OFC is truly a normally distributed measurement [Ashwal et al., 2009]. The published estimates for OFC below −2 SD at birth are 55.8 per 10,000 [Vargas et al., 2001] and 54 per 10,000 [Dolk, 1991]. Based on 2004 census data of 3.7 million live births in the United States [Dye, 2005], this would predict that 25,000 neonates are born each year with MIC, far less than 2.3 percent of the population, which would be about 85,100 children. The difference may be accounted for by a non-normal distribution of neonatal head size, postnatal MIC, or incomplete ascertainment. If MIC is defined as OFC smaller than 3 SD below the mean, this would be expected to apply to only approximately 0.1 percent of the population, which agrees well with the published estimate of approximately 14 per 10,000 [Dolk, 1991].
Microcephaly
Microcephaly is a descriptive term that refers to a cranium that is significantly smaller than the standard for the individual’s age and sex. It should usually be considered as a neurologic sign rather than a disorder, as it may result from many different causes that affect several different stages of brain development [Ashwal et al., 2009]. MIC is a common neurological sign in isolation, and in association with other abnormalities. Across the literature and in practice, the definition of MIC and the approach to evaluation of affected individuals are not uniform [Leviton et al., 2002; Opitz and Holt, 1990]. About 1 percent of referrals to child neurologists are specifically for evaluation of MIC [Lalaguna-Mallada et al., 2004], and approximately 15 percent of children referred to child neurologists for evaluation of developmental disabilities have MIC [Watemberg et al., 2002].
Historically, a confusing plethora of terms have been used to describe and classify various types of MIC. When severe congenital MIC is seen without other major brain or somatic malformations it is known as primary microcephaly or microcephalia vera, a term first introduced by Giacomini in 1885 [Giacomini, 1885]. It is likely that primary MIC is not a distinct etiologic category, but a term that describes a group of disorders, many with etiologies not yet known. As MIC can conceivably result from any developmental defect or brain injury that disturbs prenatal or early postnatal brain growth, many different causes are known. Improvements in neuroimaging and genetic technologies have resulted in a better understanding of the types and causes of MIC, suggesting that a reappraisal of schemes for classification and diagnostic testing is warranted. We have chosen to separate MIC into two broad categories, congenital and postnatal onset.
Table 25-1 summarizes some of the common disorders associated with these two groups of microcephaly.
Table 25-1 Etiologies of Congenital and Postnatal Microcephaly
| Congenital | Postnatal Onset | |
|---|---|---|
| GENETIC | ||
| Isolated/Inborn errors of metabolism | Autosomal-recessive microcephaly Autosomal-dominant microcephaly X-linked microcephaly (uncertain) Chromosomal (rare: “apparently” balanced rearrangements and ring chromosomes) |
Congenital disorders of glycosylation Mitochondrial disorders Peroxisomal disorders Menkes’ disease Amino acidopathies and organic acidurias Glucose transporter defect |
| Syndromic | ||
| Chromosomal | Trisomy 21, 13, 18 Unbalanced rearrangements |
|
| Contiguous gene deletion | 4p deletion (Wolf–Hirschhorn syndrome) 5p deletion (cri du chat syndrome) 7q11.23 deletion (Williams’ syndrome) 22q11 deletion (velocardiofacial syndrome) |
17p13.3 deletion (Miller–Dieker syndrome) |
| Single-gene defects | Cornelia de Lange syndrome Holoprosencephaly (isolated or syndromic) Smith–Lemli–Opitz syndrome Seckel’s syndrome |
Rett’s syndrome Nijmegen breakage syndrome Ataxia-telangiectasia Cockayne’s syndrome Aicardi–Goutières syndrome XLAG syndrome |
| ACQUIRED | ||
| Disruptive injuries | Fetal death of a twin Ischemic stroke Hemorrhagic stroke |
Traumatic brain injury Hypoxic-ischemic encephalopathy Hemorrhagic and ischemic stroke |
| Infections | TORCHES syndrome and HIV | Meningitis and encephalitis Congenital HIV encephalopathy |
| Teratogens/Toxins | Alcohol, hydantoin, radiation Maternal phenylketonuria Poorly controlled maternal diabetes |
Lead poisoning Chronic renal failure |
| Deprivation | Maternal hypothyroidism Maternal folate deficiency Maternal malnutrition Placental insufficiency |
Hypothyroidism Anemia Malnutrition Congenital heart disease |
HIV, human immunodeficiency virus; TORCHES, toxoplasmosis, rubella, cytomegalovirus, herpes simplex, syphilis; XLAG, X-linked lissencephaly with abnormal genitalia.
(Adapted from Ashwal S, et al. Practice parameter: Evaluation of the child with microcephaly [an evidence-based review]: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Neurology 73:887–897, 2009.)
Pathology
The embryology relevant to neuronal proliferation and microcephaly is reviewed in the following chapter describing malformations of cortical development (Chapter 26). The pathological changes described in different types of MIC are diverse, which is not surprising, given the large number of associated conditions. Here we will confine our comments to severe congenital microcephaly. The macroscopic changes described in most pathological reports are subtle, consisting of very small cerebral volume, normal or minimally altered pattern of convolutions, and normal size of the third and lateral ventricles [Robain and Lyon, 1972]. However, our brain imaging experience shows that this is not quite true, as, in many forms, the frontal lobes are disproportionately small, and the number and complexity of the gyri and the depth of sulci are generally reduced.
The microscopic changes, especially those involving the cerebral cortex, are heterogeneous. In one group, the cortex has normal thickness and lamination, but the number of neurons in the brain is dramatically reduced. We suppose these to be the less severely affected individuals, although the available data are not clear on this point. In probably several other types of MIC, the cortex appears abnormally thin, presumably resulting from premature exhaustion of the germinal zone [Barkovich et al., 1992; Evrard et al., 1989].
In the latter, abnormalities of cellular architecture predominate in the first two layers of the cortex, referred to as “type I familial MIC” by Robain [Robain and Lyon, 1972]. Layer two is almost devoid of granule neurons, and may be fragmented into small nests (sometimes called “glomeruli”) or small columns that protrude into the molecular layer. In a few individuals, the vertical bands of neurons arising in layer two cross the molecular layer to protrude into the meninges. Neurons may be seen in the molecular layer, either as scattered large pyramidal or stellate neurons, or as persistence of a fetal monolayer of granule neurons found just beneath the pia. The lower cortical layers were less affected, but with abnormal distribution of cells in some areas. In some brains, persistence of fetal wavy or “combed” monocellular bands in the middle of the cortex has been seen [Robain and Lyon, 1972]. In these types of MIC, the cerebellum is typically small but proportionate to the reduced size of the cerebrum or relatively larger.
Severe congenital MIC has been observed in combination with several other types of brain malformations, including holoprosencephaly, disproportionate brainstem and cerebellar hypoplasia, true lissencephaly with widespread malformation of neuronal migration, diffuse periventricular nodular heterotopia, and diffuse polymicrogyria (Table 25-2).
Table 25-2 Severe Congenital Microcephaly Types by Imaging or Pathology
| Microcephaly Type | References |
|---|---|
| MIC WITH SIMPLIFIED GYRAL PATTERN ONLY (PRIMARY MIC) | |
| MIC with normal six-layer cortex (probably high-functioning) | Barkovich et al. [1992] |
| MIC with layer two cortical dysplasia | Robain and Lyon [1972] |
| MIC with simplified gyri and enlarged extra-axial space (may also be associated with postnatal MIC) | Basel-Vanagaite and Dobyns [2010] |
| MIC WITH DISPROPORTIONATE PONTOCEREBELLAR HYPOPLASIA (MIC-PCH) | |
| MIC with simplified gyri and pontocerebellar hypoplasia, NOS | Basel-Vanagaite and Dobyns [2010] |
| MIC with simplified gyri and pontocerebellar hypoplasia and enlarged extra-axial space | Basel-Vanagaite and Dobyns [2010] |
| Von Monakow type MIC-PCH | Thurel and Gruner [1960] |
| MICROLISSENCEPHALY (MLIS) WITH TRUE AGYRIA-PACHYGYRIA | |
| Barth MLIS syndrome | Barth et al. [1982] |
| Microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1) | Juric-Sekhar et al. [2010] |
| Norman–Roberts MLIS syndrome | Dobyns et al. [1984] |
| MIC WITH DIFFUSE PERIVENTRICULAR NODULAR HETEROTOPIA (MIC-PNH) | |
| MIC-PNH | Robain and Lyon [1972] |
| MIC WITH DIFFUSE POLYMICROGYRIA (MDP) | |
| MDP isolated | Barkovich et al. [1992] |
| MDP with other congenital anomalies (somatic) | Pavone et al. [2000] |
| MIC WITH OTHER CORTICAL MALFORMATIONS | |
| MIC with cortical malformations, NOS (not well defined) | |
MIC, microcephaly; NOS, not otherwise specified.
Brain Imaging
In most patients with primary MIC, brain imaging reveals characteristic abnormalities that we designated “microcephaly with simplified gyral pattern” [Barkovich et al., 1998; Dobyns and Barkovich, 1999]. This pattern consists of a reduced number of gyri separated by abnormally shallow sulci. Common associated abnormalities include foreshortened frontal lobes, mildly enlarged lateral ventricles, and sometimes a thin corpus callosum or even partial agenesis of the corpus callosum.
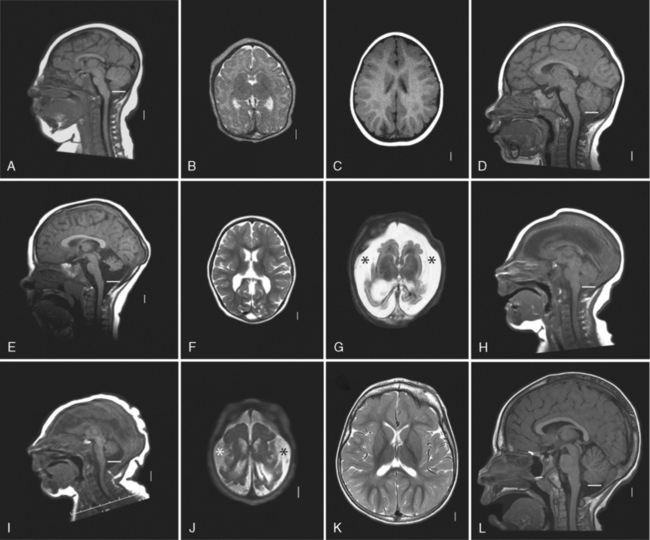
While the first several genes associated with severe congenital MIC were associated with nonspecific brain imaging patterns (as described above), several recently identified MIC genes are associated with recognizable patterns of abnormalities. Focusing on children with severe congenital MIC, the authors recently reviewed brain imaging in approximately 250 children with MIC, most of whom (230 of 247) had MIC without associated somatic anomalies [Basel-Vanagaite et al., 2010]. Among this group of patients, four relatively common brain imaging patterns were found, which involved abnormalities in the gyral pattern, size of extra-axial space, and relative size of the brainstem and cerebellum in comparison to the cerebrum. The four groups were:
Examples are shown in Figure 25-1. Rare forms of severe MIC are associated with additional brain malformations, as listed in Table 25-2.
Clinical Features
In general, all forms of MIC are associated with below-average intelligence [Dolk, 1991; Nelson and Deutschberger, 1970]. However, mild MIC with OFC between −2 and −3 SD is not inevitably linked with mental retardation; 7.5 percent of a large group of microcephalic children had normal intelligence [Martin, 1970; Sells, 1977]. However, some patients with mild MIC have severe or profound mental retardation. Their intellectual disability may be partly explained by associated brain abnormalities, whether developmental or destructive, as brain imaging frequently reveals additional abnormalities [Sugimoto et al., 1993].
Cognitive Impairment
A correlation between MIC and mental retardation has been recognized since studies in the late 1800s, and subsequent research has explored the strength of this correlation in a number of ways, although rarely in a prospective manner among a broad sample of subjects. In reported studies, the incidence of MIC has varied, depending on the population studied. Prevalence estimates of MIC in institutionalized patients have reported a rate of MIC ranging from 6.5 percent [Krishnan et al., 1989] to 53 percent [Roboz, 1973]. In contrast, for children seen in neurodevelopmental clinics, the prevalence of microcephaly averages 24.7 percent (range 6–40.4 percent) [Smith, 1981; Martin, 1970; Desch et al., 1990].
Other studies have looked at the incidence and significance of MIC in children who were functioning normally or had normal intelligence. In one report of 1006 students in mainstream classrooms it was found that 1.9 percent had mild microcephaly (−2–3 SD) and none had severe microcephaly (below −3 SD) [Sells, 1977]. The microcephalic subjects had a similar mean IQ (99.5) to the normocephalic group (105), but lower mean academic achievement scores (49 vs. 70). Another report, looking at the records of 1775 normally intelligent patients aged 11–21 years, followed in adolescent medicine clinics, found 11 (0.6 percent) with severe MIC (below −3 SD) [Barmeyer, 1971]. Among a separate sample of 106 retarded adolescents, the incidence of severe MIC was 11 percent.
A related issue concerns the incidence of developmental disability in individuals with MIC. Several investigations based on the United States National Collaborative Perinatal Project (1959–1974) have data regarding the degree of developmental disability in children with MIC. In an early report, OFC measurements of less than 43 cm (−2.3 SD) for males and 42 cm (−2.4 SD) for females at 1 year of age were associated with IQ <80 at 4 years in half the individuals [Nelson and Deutschberger, 1970]. A second study using these data found congenital MIC (<2 SD) in 1.3 percent that was associated with a greater risk of mental retardation at 7 years (15.3 vs. 7 percent) in selected populations [Camp et al., 1998]. A third study found that, of normocephalic children, 2.6 percent were mentally retarded (IQ ≤70) and 7.4 percent had borderline IQ scores (71–80). Of the 114 (0.4 percent) children with mild microcephaly (2–3 SD), 10.5 percent were mentally retarded and 28 percent had borderline IQ scores [Dolk, 1991]. Severe MIC (below −3 SD) was found in 41 (0.14 percent) children, and 51.2 percent were mentally retarded and 17 percent had borderline IQ scores. These reports have been supported by findings in several other studies [O’Connell et al., 1965; Watemberg et al., 2002].
A number of additional studies of microcephalic children have examined other clinical factors. Available data are conflicting as to whether having proportionate MIC is less predictive of developmental and learning disabilities [Sells, 1977] or not [Nelson and Deutschberger, 1970]. Other studies have shown that early severe medical illness or acquired brain injury can be associated with MIC and a future risk of retardation [Avery et al., 1972]. The pattern of head growth can also be a significant predictor of outcome. Infants whose birth OFCs were normal but who acquired MIC by age 1 year were likely to be severely delayed. On the other hand, when MIC and developmental delay were acquired as a consequence of the combined deprivations of early childhood malnutrition, poverty, and lack of stimulation, as frequently occurs in emerging countries [Grantham-McGregor et al., 2007], significant potential for physical and cognitive recovery exists [Rutter, 1998].
There is also some evidence to support the generally held belief that there is a correlation between the severity of MIC and degree of developmental disability. One study of 212 children with MIC, seen in either a birth defects or a child development clinic, found a significant correlation between the degree of MIC and severity of mental retardation. Among the 113 subjects with mild MIC (2–3 SD below the mean), mental retardation was found in just 11 percent. The mean IQ of the children with the most normal OFC, between 2.0 and 2.1 SD below the mean, was 63. Mental retardation was diagnosed in 50 percent of the 99 subjects with more severe MIC (≥3 SD), and in all of those with an OFC more than 7 SD below the mean. The mean IQ of the children with an OFC between 5 and 7 SD below the mean was 20 [Pryor and Thelander, 1968].
Epilepsy
The relation between MIC and epilepsy is of great clinical importance for several reasons:
One study involving 66 children with MIC (<−2 SD) found an overall prevalence of epilepsy of 40.9 percent [Abdel-Salam et al., 2000]. It has also been suggested that epilepsy is more common in postnatal-onset than in congenital MIC. In one study, epilepsy occurred in 50 percent of children with postnatal-onset microcephaly compared to only 35.7 percent of those with congenital MIC [Abdel-Salam et al., 2000]. A second study found that epilepsy was four times more common in postnatal-onset MIC [Qazi and Reed, 1973].
MIC also is a significant risk factor for medically refractory epilepsy [Berg et al., 1996; Chawla et al., 2002; Aneja et al., 2001]. In one study of 30 children, MIC was found in 58 percent of those with medically refractory epilepsy compared to 2 percent in whom seizures were controlled [Chawla et al., 2002].
Although children with MIC are at greater risk for epilepsy, many do not have epilepsy. There are, however, certain MIC syndromes in which epilepsy is a prominent feature. Knowledge of these disorders and their genetic basis can help establish a diagnosis and determine prognosis. Some of the more commonly recognized entities are summarized in Table 25-3.
Table 25-3 Severe Epilepsy and Microcephaly Associated Genetic Syndromes*
| Disorder | Gene(s) or Locus |
|---|---|
| STRUCTURAL MALFORMATIONS | |
| Classic lissencephaly (isolated LIS sequence) | LIS1, DCX, TUBA1A |
| Lissencephaly: X-linked with abnormal genitalia | ARX |
| Lissencephaly: autosomal-recessive with cerebellar hypoplasia | RELN, VLDLR |
| Bilateral frontoparietal polymicrogyria (COB) | GPR56 |
| Periventricular heterotopia with microcephaly | ARFGEF2 |
| Holoprosencephaly-associated genes | SHH, SIX3, GLI2, TDGF1, PTCH1, FOXH1, ZIC2, TFIF1, SMAD2 |
| Holoprosencephaly phenotypes-associated loci | HPE1 21q22.3 HPE2 2p21 HPE3 7q36 HPE4 18p11.3 HPE5 13q32 HPE 6 2q37.1 HPE7 9q22.3 HPE 8 14q13 HPE9 2q14 |
| SYNDROMES | |
| Wolf–Hirschhorn syndrome | 4p16 |
| Angelman’s syndrome | UBE3A, 15q11–q13 |
| Rett’s syndrome | Xp22, Xq28 |
| MEHMO (mental retardation, epilepsy, hypogonadism, microcephaly, obesity) | Xp22.13–p21.1 |
| Mowat–Wilson syndrome (microcephaly, mental retardation, distinct facial features with/without Hirschsprung’s disease) | ZFHX1B, 2q22 |
* Adapted from Ashwal S, et al. Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Neurology 73:887–897, 2009. Data extracted from OMIM (http://www.ncbi.nlm.nih.gov/omim); the reader is referred to that source for updated information as new entries are added and data are revised. The reader can also go directly to GeneTests (http://www.genetests.org), to which OMIM links, for updated information regarding the availability of genetic testing on a clinical or research basis.
Studies have not examined the role of obtaining a routine electroencephalography (EEG) in children with MIC to determine their risk for developing epilepsy. In one study of children with MIC, EEG abnormalities were found in 51 percent of 39 children who either had no seizures or occasional febrile seizures [Abdel-Salam et al., 2000]. EEG abnormalities (focal, generalized, or mixed epileptiform discharges) were present in 78 percent of 18 children with medically refractory epilepsy.
Cerebral Palsy
Not unexpectedly, many children with MIC are diagnosed later in infancy with cerebral palsy, and likewise, children with cerebral palsy are frequently found to be microcephalic. Data from one study of 216 children with MIC and developmental disabilities found a rate of cerebral palsy of 21.4 percent compared to 8.8 percent in a population of normocephalic developmentally disabled children (p <0.001) [Watemberg et al., 2002]. In contrast, several studies have examined the incidence of MIC in children with cerebral palsy. Three studies of children with cerebral palsy found congenital MIC in 1.8 percent of cases [Croen et al., 2001; Pharoah, 2007; Laisram et al., 1992]. In three other studies, the combined incidence of congenital and postnatal-onset MIC ranged between 32.5 percent and 81 percent, and averaged 47.9 percent [Edebol-Tysk, 1989; Lubis et al., 1990; Suzuki et al., 1999]. In one of these studies, 68 percent were diagnosed with secondary (i.e., acquired microcephaly) and 13 percent had congenital MIC [Edebol-Tysk, 1989]. Others have shown that the yield of determining the etiology of cerebral palsy is improved if MIC is present [Shevell et al., 2003]. These data suggest that it is important for physicians and others caring for children with MIC to monitor for the development of cerebral palsy, so that appropriate physical and occupational therapeutic interventions can be initiated.
Ophthalmological Disorders
No studies have surveyed the incidence of vision loss or specific ophthalmological disorders in children with MIC. One study found an incidence of 145 cases of congenital eye malformations (microphthalmia, anophthalmia, cataracts, coloboma, etc.) in 212,479 consecutive births [Stoll et al., 1997]. MIC was among the malformations in 56 percent of these children. Another study (n = 360) with severe MIC (below −3 SD) found eye abnormalities in 6.4 percent, but in only 0.2 percent of 3600 age-matched normocephalic controls [Kraus et al., 2003]. Other reported eye abnormalities in children with MIC that have been reported when searching the OMIM database for MIC have found associations with anophthalmia, blindness or visual loss, cataracts, colobomas, microphthalmia, nystagmus, optic atrophy, ptosis, and retinal disorders. Table 25-4 lists some of the more common MIC syndromes associated with ophthalmological disorders.
Table 25-4 Microcephaly Disorders with Prominent Ophthalmologic Involvement*
| Syndrome (OMIM Number) | Ophthalmologic Abnormality |
|---|---|
| Aicardi–Goutières syndrome (225750) | Visual inattention, abnormal eye movements |
| Allan–Herndon–Dudley syndrome (300523) | Rotary nystagmus, disconjugate eye movements |
| Alpers’ syndrome (203700) | Blindness, visual disturbances; microcephaly occasional |
| Borjeson–Forssman–Lehmann syndrome (301900) | Deep-set eyes, nystagmus, ptosis, poor vision, narrow palpebral fissures |
| Branchial clefts with characteristic facies, growth retardation, imperforate nasolacrimal duct, and premature aging (113620) | Upslanting palpebral fissures, telecanthus, hypertelorism, ptosis, lacrimal duct obstruction, coloboma Coloboma, microphthalmia, cataract |
| Cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome (609528) | Downslanting palpebral fissures, hypertelorism, hypoplastic optic discs; described in two families |
| Cerebro-oculofacioskeletal syndrome (214150) | Cataracts, blepharophimosis Microphthalmia, deep-set eyes, nystagmus |
| **CHARGE syndrome (214800) | Colobomas, anophthalmia, ptosis, hypertelorism, downslanting palpebral fissures |
| Cockayne’s syndrome (216400) | Pigmentary retinopathy, optic atrophy, corneal opacity, decreased lacrimation, nystagmus, cataracts |
| Cohen’s syndrome (216550) | Downslanting palpebral fissures, chorioretinal dystrophy, myopia, decreased visual acuity, optic atrophy |
| Down syndrome (190685) | Upslanting palpebral fissures, epicanthal folds, iris Brushfield spots |
| Fraser’s syndrome (219000) | Cryptophthalmos, malformed lacrimal ducts, hypertelorism, blindness |
| Glucose transport defect (606777) | Abnormal paroxysmal eye movements; eye findings rare |
| Holoprosencephaly (236100) | Cyclopia, ethmocephaly, cebocephaly, hypotelorism |
| Incontinentia pigmenti (308300) | Microphthalmia, cataract, optic atrophy, retinal vascular proliferation, retinal fibrosis, retinal detachment, uveitis, keratitis |
| Jacobsen’s syndrome (147791) | Epicanthal folds, hypertelorism Ptosis, strabismus, coloboma, optic atrophy |
| Kabuki syndrome (147920) | Long palpebral fissures, eversion of lateral third of lower eyelids, ptosis, blue sclerae, broad/arched/sparse eyebrows |
| Mental retardation with optic atrophy, deafness, and seizures (309555) | Optic atrophy, severe visual impairment |
| Mental retardation, microcephaly, growth retardation, and joint contractures (606240) | Ptosis; single case report of two sisters |
| Microcephaly, hiatus hernia, and nephrotic syndrome (251300) | Absent cleavage of eye anterior chamber; described in one case report |
| Microphthalmia, syndromic (309800) | Microphthalmia, optic nerve hypoplasia, coloboma, pigmentary retinopathy |
| Mitochondrial DNA depletion syndrome (251880) | Nystagmus, disconjugate eye movements, optic dysplasia; microcephaly occasional |
| Mosaic variegated aneuploidy syndrome (257300) | Hypertelorism, upslanting palpebral fissures, epicanthal folds, cataracts, nystagmus |
| Mucolipidosis IV (252650) | Corneal clouding, corneal opacities, fibrous dysplasia of the cornea, progressive retinal degeneration, optic atrophy, strabismus, decreased electroretinogram |
| Neuronal ceroid-lipofuscinosis (256730) | Progressive visual loss, optic atrophy, retinal degeneration, macular degeneration, abnormal electroretinogram |
| Norrie’s disease (310600) | Blindness, retinal dysgenesis/dysplasia/detachment, cataracts, optic atrophy, other ocular abnormalities |
| Oculodentodigital dysplasia (164200) | Microcornea, short palpebral fissures, epicanthal folds, glaucoma, cataract, iris anomalies |
| Oculopalatocerebral syndrome (257910) | Persistent hypertrophic primary vitreous Microphthalmos, leukocoria, retrolental fibrovascular membrane; rarely reported |
| Oculopalatoskeletal syndrome (257920) | Blepharophimosis, blepharoptosis, epicanthus inversus, hypertelorism, conjunctival telangiectasia, glaucoma, anterior chamber anomalies, abnormal eye motility; rare |
| Osteoporosis-pseudoglioma syndrome (259770) | Pseudoglioma, blindness, microphthalmia, vitreoretinal abnormalities, cataract, iris atrophy |
| Pelizaeus–Merzbacher disease (312080) | Rotary nystagmus, optic atrophy |
| Peters plus syndrome (261540) | Hypertelorism, Peters anomaly, anterior chamber cleavage disorder, nystagmus, ptosis, glaucoma, cataract, myopia, coloboma |
| Pyridoxamine 5′-phosphate oxidase deficiency (610090) | Rotary eye movements; rare disorder |
| Pyruvate decarboxylase deficiency (312170) | Episodic ptosis, abnormal eye movements |
| Pyruvate dehydrogenase deficiency (312170) | Nystagmus, ptosis, saccade initiation failure, oculomotor apraxia |
| Rhizomelic chondrodysplasia punctata (215100) | Cataract |
| Roberts’ syndrome (268300) | Hypertelorism, shallow orbits, prominent eyes, bluish sclerae, corneal clouding, microphthalmia, cataract, lid coloboma |
| Smith–Lemli–Opitz syndrome (270400) | Ptosis, epicanthal folds, cataracts, hypertelorism, strabismus |
| Spastic paraplegia, optic atrophy, microcephaly, and XY sex reversal (603117) | Optic atrophy and poor vision; single case report |
| Syndactyly with microcephaly and mental retardation (272440) | One family of several described had optic atrophy and poor vision |
| Townes–Brocks syndrome (107480) | Chorioretinal coloboma, Duane anomaly; both of these are rare |
| Velocardiofacial syndrome (192430) | Narrow palpebral fissures, small optic discs, tortuous retinal vessels, posterior embryotoxon |
| Walker-Warburg syndrome (236670) | Multiple ocular findings including retinal detachment, cataracts, microphthalmia, hyperplastic primary vitreous, optic nerve hypoplasia, colobomata, glaucoma |
| Warburg micro syndrome (600118) | Multiple ocular findings, including microphthalmia, microcornea, congenital cataracts, optic atrophy, ptosis |
| Wolf–Hirschhorn syndrome (194190) | Hypertelorism, exophthalmos, ptosis, Rieger anomaly, nystagmus, iris coloboma |
* Adapted from Ashwal S, et al. Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Neurology 73:887–897, 2009. From OMIM (http://www.ncbi.nlm.nih.gov/sites/entrez). Gene map loci are listed in each OMIM entry. Disorders are listed alphabetically; prevalence data are not known.
** CHARGE (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities and deafness.
Audiological Disorders
No studies have surveyed the incidence of hearing loss or audiological disorders in children with MIC. One study of 100 children with complex ear anomalies recorded that 85 had neurological involvement and 13 children had MIC [Wiznitzer et al., 1987]. Hearing loss is likely the most common audiological disorder associated with MIC, and Table 25-5 summarizes some of the common MIC syndromes listed in OMIM in which prominent audiological involvement is reported.
Table 25-5 Microcephaly Syndromes with Prominent Ear or Auditory Impairments*
| Syndrome (OMIM Number) | Ear or Audiologic Abnormality |
|---|---|
| Allan–Herndon–Dudley syndrome (300523) | Large ears, simple ears, pinna modeling anomalies, prominent antihelix, flattened antihelix |
| Alpha-thalassemia/mental retardation syndrome (309580) | Small ears, low-set ears, posteriorly rotated ears, sensorineural hearing loss |
| Brachyphalangy, polydactyly, tibial aplasia/hypoplasia (609945) | Overfolded helices, hearing loss, cleft lobules, preauricular tags, cup-shaped ears |
| Branchial arch syndrome (301950) | Hearing loss and external ear anomalies |
| Branchial clefts with characteristic facies, growth retardation, imperforate nasolacrimal duct, and premature aging (113610) | Low-set ears, posteriorly rotated ears, hypoplastic superior helix, microtia, ear pits, overfolded ears, supra-auricular sinuses, conductive hearing loss |
| Camptodactyly, tall stature, and hearing loss syndrome (610474) | Microcephaly occurs occasionally |
| Cerebrocostomandibular syndrome (117650) | Low-set ears, conductive hearing loss, posteriorly rotated ears |
| Cerebro-oculofacioskeletal syndrome 1 (214150) | Large ear pinnae |
| CHARGE syndrome (214800) | Small ears, lop ears, deafness (sensorineural ± conductive), Mondini defect |
| Chondrodysplasia punctata (215100) | Hearing loss |
| Chromosome 18 deletion syndrome (601808) | External ear abnormalities |
| Chromosome 9q subtelomeric deletion syndrome (610253) | Malformed ears, hearing loss |
| Cockayne’s syndrome (216400) | Malformed ears, sensorineural hearing loss |
| Coffin–Lowry syndrome (303600) | Prominent ears, sensorineural hearing loss |
| Cornelia de Lange syndrome (122470) | Low-set ears, hearing loss |
| Cutis verticis gyrate, retinitis pigmentosa, and sensorineural deafness (605685) | Sensorineural hearing loss; only one case report |
| Deafness, conductive, with malformed external ear (221300) | Conductive hearing loss, malformed external ears, low-set external ears, malformed ossicles |
| Deafness, congenital, and onychodystrophy (220500) | Sensorineural hearing loss |
| Dislocated elbows, bowed tibias, scoliosis, deafness, cataracts, microcephaly, and mental retardation (603133) | Single case report of 4 siblings in consanguineous family |
| Ear, patella, and short stature syndrome (24690) | Bilateral microtia, hearing loss, Mondini malformation, low-set ears, atretic auditory canal |
| Feingold’s syndrome (164280) | “Ear abnormalities” common in one description |
| Focal dermal hypoplasia (305600) | Protruding, simple ears, low-set ears, narrow auditory canals, mixed hearing loss |
| Genitopatellar syndrome (606170) | One case report with hearing loss as an associated finding |
| ***GOMBO syndrome (233270) | One case report with conductive hearing loss |
| Iris coloboma with ptosis, hypertelorism, and mental retardation (243310) | Low-set ears, overfolded helices, sensorineural hearing loss |
| Johanson–Blizzard syndrome (243800) | Sensorineural hearing loss, cystic dilatation of cochlea and vestibular structures |
| Kabuki syndrome (147920) | Large prominent ears, recurrent otitis media in infancy, posteriorly rotated ears, hearing loss, preauricular pit |
| Kearns–Sayre syndrome (530000) | Sensorineural hearing loss |
| Klippel–Feil syndrome (118100) | One case reported with microcephaly; hearing loss of any type common; external ear abnormalities occasional |
| Lathosterolosis (607330) | Conductive hearing loss |
| Mental retardation, with optic atrophy, deafness and seizures (309555) | Hearing loss; described in one family |
| Mental retardation–hypotonic facies syndrome, X-linked (309580) | Deafness |
| Microphthalmia, syndromic (601186) | Simple anteverted ears, hearing loss |
| Monosomy 1p36 syndrome (607872) | Sensorineural hearing loss, external ear abnormalities |
| Oculodentodigital dysplasia (164200) | Conductive hearing loss |
| Oculopalatoskeletal syndrome (257920) | Conductive hearing loss |
| Otopalatodigital syndrome (311300) | Low-set ears, conductive hearing loss, posteriorly rotated ears |
| POR** deficiency (201750) | Conductive hearing loss, simple ears |
| Progeroid facial appearance with hand anomalies (602249) | Prominent ears, conductive hearing loss; one case report |
| Renpenning’s syndrome 1 (309500) | Cupped ears |
| Rubinstein–Taybi syndrome (180849) | Low-set ears, hearing loss |
| Shprinzten–Goldberg craniosynostosis (182212) | Low-set ears, posteriorly rotated ears, conductive hearing loss (rare) |
| Townes–Brocks syndrome (107480) | Multiple external ear abnormalities; sensorineural hearing loss |
| Trichorhinophalangeal syndrome type II (15030) | Hearing loss, large protruding ears |
| Velocardiofacial syndrome (192430) | Occasional microcephaly and minor auricular abnormalities seen |
| Waardenburg’s syndrome (148820) | Hearing loss |
| Williams–Beuren syndrome (194050) | Early-onset progressive sensorineural hearing loss |
| Wolf–Hirschhorn syndrome (194190) (602952) | Preauricular tags, preauricular pits, hearing loss, narrow external auditory canals |
* Adapted from Ashwal S, et al. Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Neurology 73:887–897, 2009. From OMIM (http://www.ncbi.nlm.nih.gov/sites/entrez). Gene map loci are listed in each OMIM entry. Disorders are listed alphabetically; prevalence data are not known.
** POR – cytochrome P450 oxido reductase deficiency.
*** GOMBO – Growth retardation, ocular abnormalities, Microcephaly, Brachydactyly and Oligophrenia.
Etiology
Extrinsic Causes
Extrinsic injuries before birth or early in life can certainly lead to MIC. The developing nervous system is highly vulnerable to infections, including cytomegalovirus, toxoplasmosis, rubella, herpes simplex, and group B coxsackievirus. Intrauterine infections with these can result in MIC [Evrard, 1992; Norman et al., 1995; Volpe, 2000]. MIC also has been reported in infants of women exposed to ionizing radiation, as shown in studies following exposure to atomic bomb radiation or to radium implantation in the cervix during the first trimester [Dekaban, 1968; Wood et al., 1967]. Maternal metabolic disorders during pregnancy, such as diabetes mellitus, uremia, and undiagnosed or inadequately treated phenylketonuria, may result in neonatal MIC [Levy et al., 1996; Rouse et al., 1997]. Malnutrition, hypertension, and placental insufficiency may all result in intrauterine growth retardation and MIC.
Maternal alcoholism during pregnancy has also been linked with MIC as part of the fetal alcohol syndrome [Clarren et al., 1978; Loebstein and Koren, 1997; Ouellette et al., 1977; Spohr et al., 1993]. The clinical features include growth and mental retardation, midfacial hypoplasia, short palpebral fissures, epicanthal folds, and behavioral disturbances. Neuropathologic findings include MIC, heterotopia, widespread cortical and white-matter dysplasias, and defects of neuronal and glial migration [Wisniewski et al., 1983]. MIC has also been reported with maternal exposure to cocaine [Loebstein and Koren, 1997]. Some other reports are largely anecdotal, so the associations are often not proven.
Familial Mild Microcephaly
Mild MIC may have either complex (polygenic) or autosomal-dominant inheritance. The autosomal-recessive forms typically present with severe primary MIC and many reviews have not clearly separated patients with mild and severe MIC, often making clinical data difficult to interpret. The polygenic or autosomal-dominant forms are generally associated with mild to moderate cognitive problems, with epilepsy being uncommon. The risk of recurrence in siblings may be as high as 50 percent with the assumption of autosomal-dominant inheritance, but is probably lower, considering that polygenic inheritance may be involved. The genetic basis for familial mild MIC is not known, but several genes have been identified that cause mild MIC, as indicated in Table 25-6.
Patients with primary MIC tend to fall into two further, albeit somewhat heterogeneous, subgroups [Dobyns, 2002]. The first subgroup includes children with severe MIC but only moderate neurologic problems, usually with moderate mental retardation and with no spasticity or epilepsy. The second subgroup consists of severe MIC with a much more severe neurologic phenotype that consists of abnormal neonatal reflexes, generalized spasticity, and epilepsy [Barkovich et al., 1998; Dobyns, 2002; Sztriha et al., 1999; ten Donkelaar et al., 1999]. These children have poor feeding and recurrent vomiting, leading to poor weight gain, profound mental retardation, and severe spastic quadriparesis. Most of these children also have early-onset intractable epilepsy. The wide clinical spectrum suggests pathogenetically heterogeneous conditions, and several syndromes and genes have been identified (see Table 25-6).
Primary Microcephaly
When congenital MIC is the only abnormality on evaluation, the disorder has been designated primary MIC. As discussed previously, this designation becomes much more useful when restricted to children with birth occipitofrontal circumference below −3 SD. Most patients with primary MIC also have mild growth deficiency, with stature typically −2 to −3 SD, which may be part of the syndrome or partly nutritional. This deficiency is much less striking than their head size, which is typically −4 to −8 SD after early childhood. Most affected persons fall into one of two groups described below [Dobyns, 2002].
The first group is composed of children with extreme MIC but only moderate neurologic problems, usually with only moderate mental retardation without spasticity or epilepsy [Barkovich et al., 1998; Peiffer et al., 1999; Tolmie et al., 1987]. Their neonatal examinations are usually normal, except for MIC, but many children initially have poor feeding and weight gain. They may have normal tone or mild distal spasticity, but do not have moderate or severe spasticity. Seizures are uncommon and are easily controlled. Febrile seizures occur and should be managed as in any other child. Early development is only mildly delayed and many infants progress to walking between 1 and 2 years of age and develop limited language skills. Several genes have been identified from studies of patients with this disorder (see Table 25-6).
The second group consists of primary MIC with a severe neurologic phenotype that includes severe spasticity and epilepsy [Barkovich et al., 1998; Dobyns, 2002; Sztriha et al., 1999; ten Donkelaar et al., 1999; Tolmie et al., 1987]. Neonatal examination demonstrates abnormal neonatal reflexes and generalized spasticity, and these children subsequently develop impaired feeding and recurrent vomiting, leading to poor weight gain, severe developmental delay, profound mental retardation, and severe spastic quadriparesis. Most of these infants have early-onset intractable epilepsy. In addition to a simplified gyral pattern, brain magnetic resonance imaging (MRI) may demonstrate other abnormalities, as summarized above (see Figure 25-1). Children with Amish lethal microcephaly have this phenotype, except that hypotonia predominates rather than spasticity, and seizures are not prominent [Kelley et al., 2002; Rosenberg et al., 2002].
The term radial microbrain was introduced by Evrard to describe the brain in some patients with severe mental retardation, profound MIC, and early death, describing an abnormally small brain that has a normal gyral pattern, normal cortical thickness, and normal cortical lamination, although the number of cortical neurons was only 30 percent of normal [Evrard et al., 1989; Evrard, 1992]. He hypothesized that a decreased number of radial neuronal-glial units was responsible for this form of MIC. This subgroup fits into the lower-functioning group of patients with primary MIC, rather than comprising an independent syndrome. However, multiple causes with different pathologic changes and clinical courses are likely to emerge from this group.
Severe Microcephaly with Cortical Malformation
Although still incompletely delineated, several syndromes with severe congenital microcephaly and additional severe brain malformations are known. The combination of severe microcephaly and true lissencephaly (with an abnormally thick cortex) has been reported, with at least three different patterns [Barth et al., 1982; Dobyns and Barkovich, 1999; Sztriha et al., 1998]. The most common of these very rare syndromes is probably the Barth microlissencephaly syndrome, which consists of severe microcephaly, diffuse complete agyria, and severe brainstem and cerebellar hypoplasia [Barth et al., 1982; Kroon et al., 1996]. Severe microcephaly with diffuse periventricular nodular heterotopia has been described, and clearly differs from other forms of heterotopia [Robain and Lyon, 1972; Sheen et al., 2004]. Some patients with severe microcephaly also have had diffuse polymicrogyria [Dobyns and Barkovich, 1999].
Severe Microcephaly with Proportionate Growth Deficiency
Seckel’s syndrome consists of severe intrauterine and postnatal growth deficiency and microcephaly, and abnormal facial features. including large eyes, beaklike protrusion of the nose, narrow face, and receding lower jaw [Majewski and Goecke, 1982; Seckel, 1960]. All affected individuals have severe mental retardation, although the severity varies considerably and some patients live to adulthood. Abnormalities of the brain seen on postmortem examination or brain imaging demonstrate pure microcephaly with deficient production of neurons and other cell types in some patients [Hori et al., 1987], whereas other patients have severe brain malformations, including lissencephaly [Capovilla et al., 2001; Shanske et al., 1997; Sugio et al., 1993]. Some patients have had various hematological disorders, such as pancytopenia or acute myeloid leukemia [Butler et al., 1987; Hayani et al., 1994].
MOPD1, or Taybi–Linder syndrome, consists of similar severe intrauterine and postnatal growth deficiency and microcephaly, combined with abnormal body proportions and short limbs. Typical skeletal changes consist of a low and broad pelvis with poor formation of the acetabulum, short and bowed humerus and femur, dislocated hips and elbows, retarded epiphyseal maturation, cleft vertebral arches, platyspondyly, horizontal acetabular roofs, and short long bones with enlarged metaphyses. Patients with MOPD1 also may have skin abnormalities, including hyperkeratosis and sparseness of hair and eyebrows [Meinecke et al., 1991; Sigaudy et al., 1998; Taybi, 1992]. Brain malformations, in addition to the severe microcephaly, are common and include lissencephaly, heterotopia, callosal agenesis, and cerebellar vermis hypoplasia [Klinge et al., 2002; Sigaudy et al., 1998].
MOPD2 consists of similar severe intrauterine and postnatal growth deficiency, proportionate microcephaly at birth that progresses to disproportionate microcephaly, shortening of the middle and distal segments of the limbs, a progressive bony dysplasia, abnormal facial appearance, including prominent nose and malformed ears, and a high squeaky voice [Hall et al., 2004; Majewski and Goecke, 1998; Majewski et al., 1982]. These patients may have dilated arteries in the brain that resemble aneurysms or moyamoya disease [Kannu et al., 2004; Young et al., 2004]. Although all affected individuals have severe microcephaly, no other brain malformations have been described [Fukuzawa et al., 2002].
Although these syndromes dominate the literature concerning intrauterine and postnatal growth deficiency and microcephaly, review of many reports suggests an overall substantial causal heterogeneity, with probable confusion among these and other syndromes in this group. In support of this likelihood, several novel syndromes have been reported [Kantaputra, 2002; Okajima et al., 2002].
MLIS MOPD1-type
MLIS occurs in some patients with microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1), a syndrome that is difficult to distinguish from severe forms of Seckel’s syndrome [Juric-Sekhar et al., 2010; Klinge et al., 2002; Meinecke et al., 1991; Ozawa et al., 2005]. The phenotype consists of severe prenatal growth deficiency and microcephaly, sparse hair and dry scaling skin, skeletal anomalies such as platyspondyly, slender ribs, short and bowed proximal humeri and femurs, small iliac wings, dysplastic acetabulum and small hands and feet, and profound developmental handicaps. A few have developed aplastic anemia, another overlap with Seckel’s syndrome. The neuropathology consists of a variant form of LIS-3L with frontal predominance.
MLIS Barth-type
The Barth-type of MLIS is possibly the most severe of all the known LIS syndromes. The phenotype consists of polyhydramnios, probably due to poor fetal swallowing, severe congenital microcephaly (birth OFC approximately 28 cm), weak respiratory effort, and survival for only a few hours or days [Barth et al., 1982; Dobyns and Barkovich, 1999; Kroon et al., 1996]. The neuropathology consists of a variant form of LIS-4L with extreme hypoplasia of many structures, as described above.
Genetics
At least five loci for primary microcephaly with mild or moderate phenotype have been mapped to 1q31, 8p22-pter, 9q34, 15q, and 19q13.1–q13.2, and two of these loci (MCPH1 and ASPM) have now been cloned. Two genes causing severe microcephaly with severe phenotype have also been identified, those being the causal genes for Amish lethal microcephaly and microcephaly with periventricular nodular heterotopia. In Seckel’s syndrome, defects in DNA repair were suggested by chromosome instability with exposure to mitomycin C [Abou-Zahr et al., 1999; Bobabilla-Morales et al., 2003]. Subsequently, at least three loci for Seckel’s syndrome have been confirmed, mapped to chromosomes 3q22.1–q24 and 18p11.31–q11.2, and at least one additional unknown locus [Faivre et al., 2002]. Mutations of the ATR gene in 3q2 were recently identified [Alderton et al., 2004; O’Driscoll and Jeggo, 2003]. All of these genes are listed in Table 25-6, with references. Of interest, the two genes associated with microcephaly and mild–moderate phenotype both reveal an evolutionary signature of rapid evolution [Evans et al., 2004a, b].
Antenatal Diagnosis
Microcephaly can often, but not always, be diagnosed by second-trimester fetal ultrasonography [Bromley and Benacerraf, 1995]. This likely is due to variable onset of the deceleration in head growth. When this occurs early, as it often does for severe microcephaly, ultrasound examination should be able to detect the abnormality, but not when it begins in the late second or third trimester.
Genetic Counseling
Some older references cite a 6 percent risk of a family’s having a second microcephalic child, but these sources do not consistently address severity of the microcephaly. This percentage may be useful for mild and borderline microcephaly with birth occipitofrontal circumference between −2 and −3 SD below the mean. On the basis of findings in many families with two or more affected siblings with primary microcephaly and other forms of severe microcephaly with birth occipitofrontal circumference below −3 SD, counseling for autosomal-recessive inheritance is appropriate in this group. Thus, most forms of severe congenital MIC (with or without intrauterine growth retardation) are genetic, most if not all having autosomal-recessive inheritance. Disorders associated with postnatal MIC are much more heterogeneous, with examples of autosomal-dominant (familial or sporadic), autosomal-recessive, and X-linked inheritance (see Table 25-6).
Chromosome Disorders
An OMIM search lists more than 400 syndromes with microcephaly, making this an unhelpful search term. Some of the better-known disorders include Angelman’s, Cornelia de Lange (Brachmann–de Lange), and Dubowitz’s syndromes [Opitz and Holt, 1990].
With potentially hundreds of causes of microcephaly, including prenatal and postnatal onset, as well as genetic and acquired etiologies, diagnostic evaluations may be complex. Investigation of patients with microcephaly includes evaluation for prenatal exposure to teratogens, especially alcohol, drugs, and isotretinoin (a vitamin A analog), and assessment of the family history, birth history, and associated malformations. Laboratory studies should include titers for toxoplasmosis, syphilis, rubella virus, cytomegalovirus, and herpes simplex viruses; neuroimaging [Sugimoto et al., 1993]; evaluation for maternal and childhood metabolic disorders; and genetic testing, including chromosome analysis and testing for small deletions or duplications, which currently is performed by fluorescence in situ hybridization with subtelomeric probes [Knight et al., 2000]. Algorithms for the evaluation of the infant and child with congenital (Figure 25-2) and postnatal (Figure 25-3) microcephaly have recently been published and serve as a generalized approach to the diagnostic evaluation [Ashwal et al., 2009].
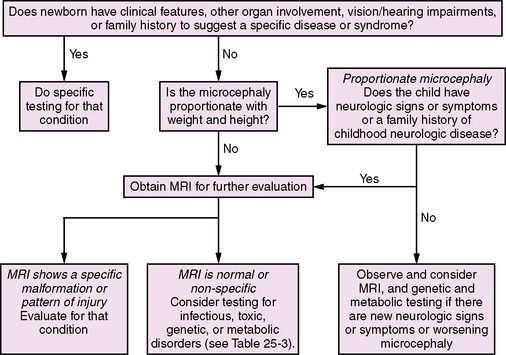
Fig. 25-2 Algorithm for the diagnostic evaluation of the infant or child with congenital microcephaly.
(Adapted from Ashwal S, et al. Practice parameter: Evaluation of the child with microcephaly [an evidence-based review]: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society, Neurology 73:887–897, 2009.)
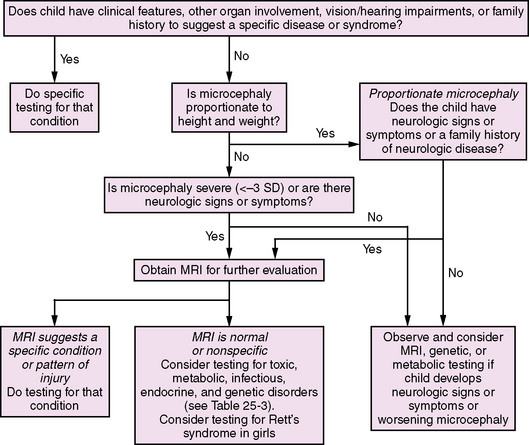
Fig. 25-3 Algorithm for the diagnostic evaluation of the infant or child with postnatal-onset microcephaly.
(Adapted from Ashwal S, et al. Practice parameter: Evaluation of the child with microcephaly [an evidence-based review]: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 73:887–897, 2009.)
Megalencephaly (and Macrocephaly)
Macrocephaly is defined as an OFC of 2 SDs or more above the mean for age, gender, and gestation, measured over the greatest frontal circumference. It is caused by a myriad of conditions, such as hydrocephalus, cerebral edema, space-occupying lesions, subdural fluid collection, thickening or enlargement of the skull (or hyperostosis), and a truly enlarged brain or megalencephaly (Box 25-1). The classic definition of megalencephaly (MEG) stands as an oversized and overweight brain (or an increased brain mass) that exceeds the mean by 2 SD for age and gender [DeMyer, 1986].
Box 25-1 Causes of Macrocephaly






















































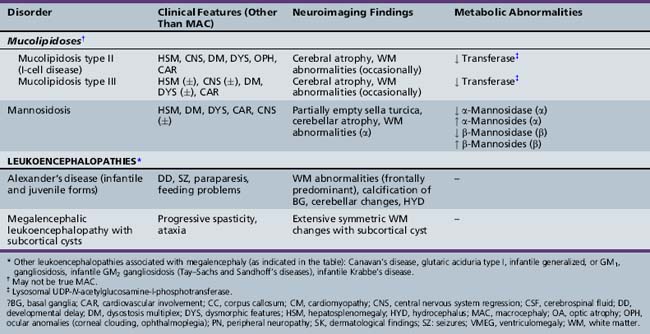
The classification of megalencephaly has been challenging due to its association with a large number of diverse syndromes and etiologies. DeMyer first divided it in 1972 into anatomic and metabolic types [DeMyer, 1972]. Metabolic megalencephalies result from cellular edema or abnormal accumulation of metabolic substrates within the neurons and glia secondary to an underlying biochemical defect (most commonly an enzyme deficiency), without an increase in cell number. The various causes of metabolic megalencephalies are listed in Tables 25-7 and 25-8, and include cerebral organic acid disorders (such as Canavan’s disease, glutaric aciduria type I) and lysosomal storage disorders (such generalized, or GM1, gangliosidosis, Tay–Sachs disease, Krabbe’s disease, some mucopolysaccharidoses), among others. A number of these disorders (most notably, Canavan’s, Krabbe’s, and Alexander’s diseases, and megalencephalic leukoencephalopathy with subcortical cysts) are leukoencephalopathies, i.e., demyelinating disorders whereby the underlying biochemical or genetic defect alters myelin formation and function. The metabolic megalencephalies are not true cortical malformations and will not be discussed further in this chapter, but are discussed in other sections of this book.
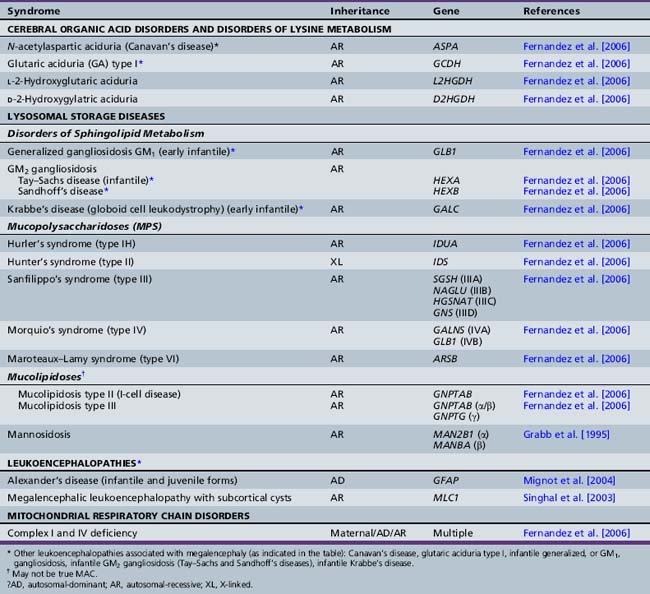
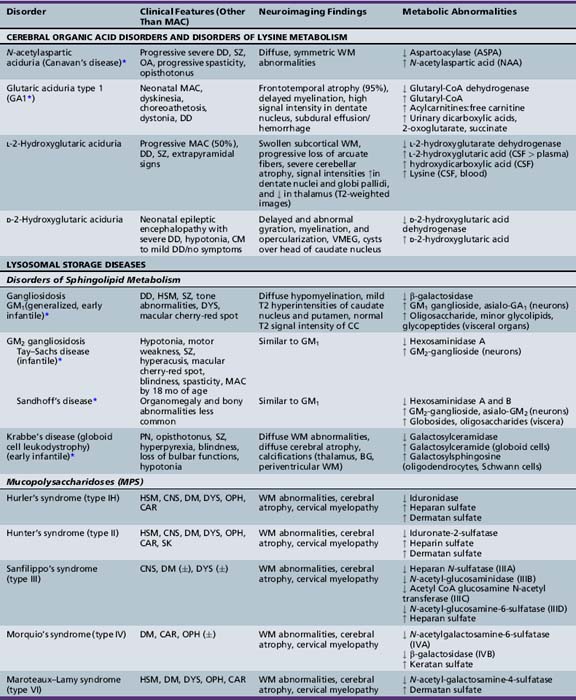
With the above-mentioned considerations in mind, Tables 25-9 and 25-10 list the most common syndromes and disorders in which macrocephaly is a defining feature or is of diagnostic significance. The presence of true megalencephaly (vs. absolute or relative macrocephaly) is indicated in the second column of Table 25-9.
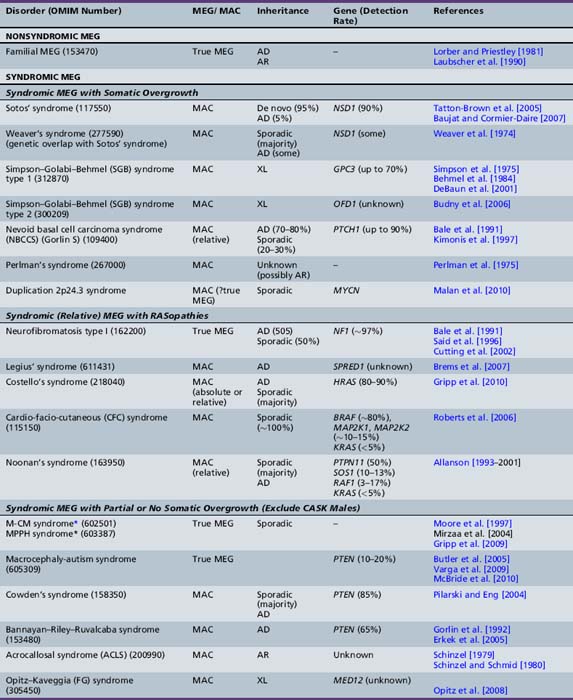
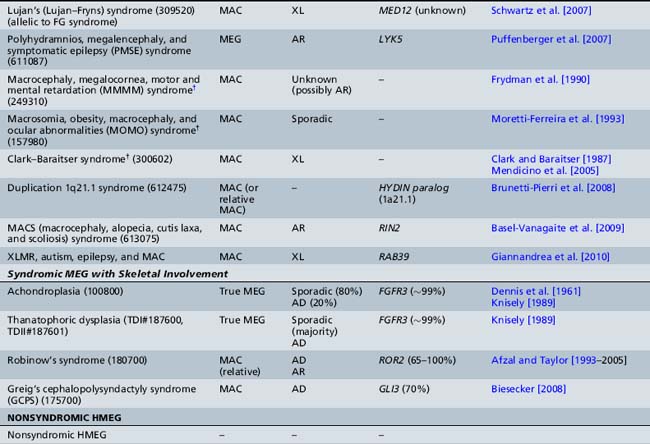
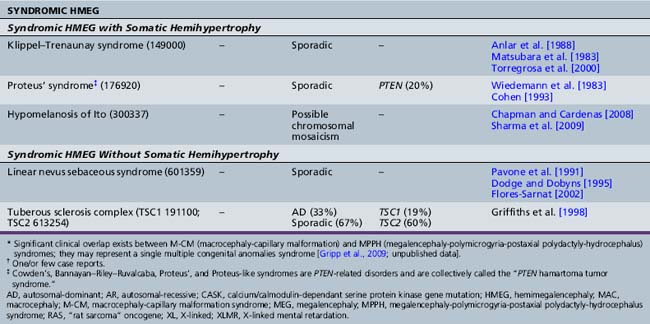
Table 25-10 Syndromic and Nonsyndromic Megalencephaly and Hemimegalencephaly: Clinical and Neuroimaging Features
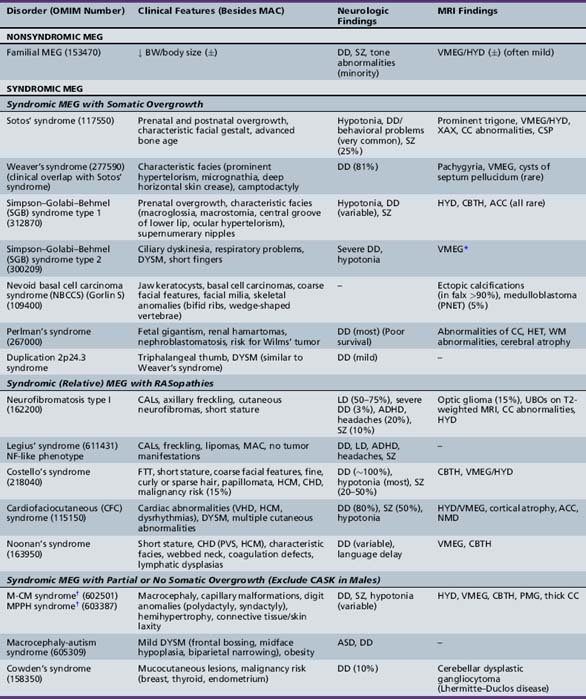
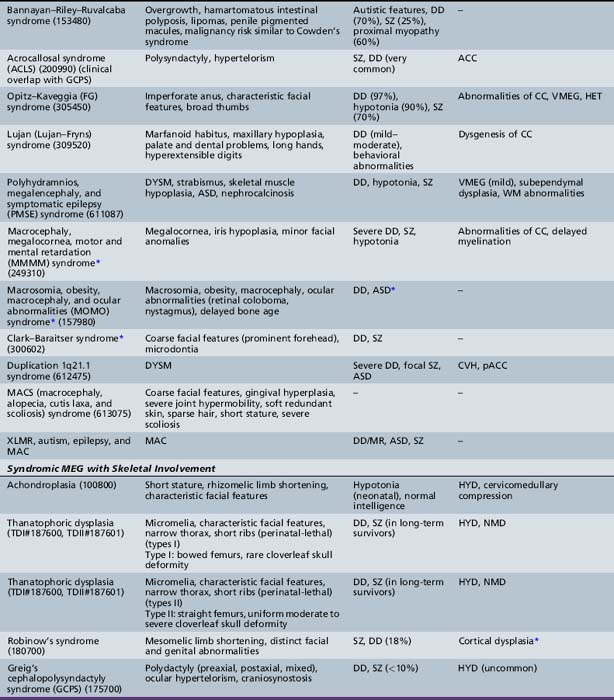
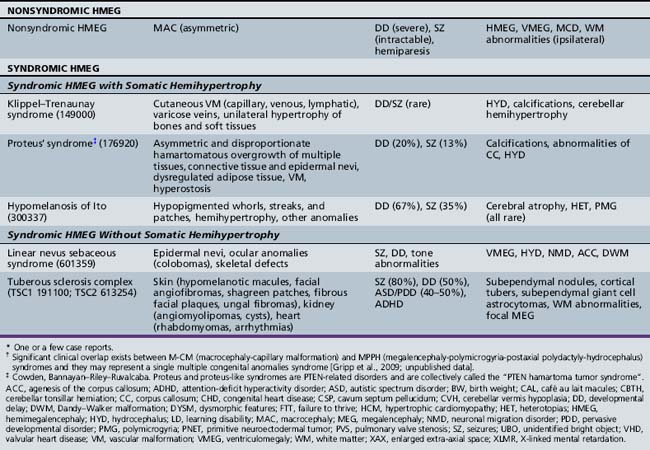
Unilateral megalencephaly (or hemimegalencephaly) is a rare diffuse enlargement of one cerebral hemisphere, with unique clinical and neuroimaging characteristics and syndromic associations. The most common causes of hemimegalencephaly are outlined in Tables 25-9 and 25-10 as well.
Pathology and Pathogenesis
Numerous animal models of syndromic and nonsyndromic megalencephaly display neuronal and glial hypertrophy. Pten (phosphatase and tensin homolog on chromosome ten) mutant mice were found to develop macrocephaly and behavioral abnormalities reminiscent of human autistic spectrum disorder, such as reduced social activity, increased anxiety, and sporadic seizures [Kwon et al., 2001; Kwon et al., 2006; Ogawa et al., 2007], closely resembling the human phenotype of PTEN-related disorders that are described later in this chapter. At the cellular level, in vivo effects of loss of Pten include loss of neuronal polarity, neuronal hypertrophy, and, in one study, increased astrocyte proliferation and hypertrophy [Kwon et al., 2001; Fraser et al., 2004]. Increasing attention has been paid to the role of the mammalian target of rapamycin (mTOR), a serine/threonine kinase that has well-known functions in regulation of cellular proliferation and growth, a crucial role in neuronal development and synaptic plasticity [Jaworski and Sheng, 2006], and a contribution to Pten-mediated growth regulation in the mammalian nervous system. mTOR inhibition reversed neuronal hypertrophy in Pten-deficient mice and also resulted in amelioration of a subset of Pten-associated abnormal behaviors, thereby substantiating evidence that the mTOR pathway downstream of PTEN is critical for its complex phenotype [Kwon et al., 2003; Zhou et al., 2009].
Loss of Tsc1 and Tsc2, two downstream negative regulators of the mTOR pathway [Inoki et al., 2002; Manning et al., 2002; Potter et al., 2002], has been shown to cause neuronal hypertrophy in vitro and in vivo [Jaworski et al., 2005; Tavazoie et al., 2005; Meikle et al., 2007], supporting a role of TSC1 and TSC2 in neuronal growth regulation and synaptic function. Interruptions of TSC1 and TSC2 cause tuberous sclerosis complex, known to be associated with megalencephaly, hemimegalencephaly, and focal megalencephaly [Choi et al., 2008]. Zhou et al. suggested that there is a common signal transduction pathway potentially responsible for the autism-like symptoms in individuals bearing TSC1/2 and/or PTEN mutations, and proposed that mTOR inhibitors are potential therapeutic agents for this subset of patients [Meikle et al., 2008; Zhou et al., 2009]. The Nf1 knockout mouse was found to have increased neuroglial progenitor/stem cell (NSC) proliferation and gliogenesis in the brainstem, also driven by mTOR-mediated activation [Lee et al., 2010].
Other animal models of megalencephaly include mouse mutants with loss-of-function mutations in genes regulating programmed cell death, or apoptosis, such as Caspase-3, Caspase-9, and Apaf-1, which were found to have gross brain malformations and neuronal hyperplasia. However, these mutations, when germline, are embryonically lethal [Kuida et al., 1996, 1998; Cecconi et al., 1998; Yoshida et al., 1998; Hakem et al., 1998; Marks and Berg, 1999]. Transgenic mice overexpressing insulin-like growth factor (IGF)-I exhibit brain overgrowth characterized by increased numbers of neurons and oligodendrocytes, as well as excessive myelin formation [Carson et al., 1993; Donahue et al., 1996; Petersson et al., 1999; D’Ercole et al., 2002]. IGF-1 stimulates:
As a result of these events, brain growth is increased with IGF-I overexpression and reduced with decreased IGF-I signaling. Although much less information is available in humans, individuals with IGF-I gene deletions or mutations that result in severe deficits in IGF-1 expression are microcephalic and mentally retarded [Walenkamp and Wit, 2007]. Little evidence supporting comparable actions for IGF-II is available.
The CD81 null mouse has a markedly increased brain size (up to 30 percent larger) due to an increased number of astrocytes and microglia throughout the brain, possibly through regulation of cell proliferation by a contact inhibition-dependent mechanism. CD81 is a member of the tetraspanin family of small membrane proteins associated with the regulation of cell migration and mitotic activity [Geisert et al., 2002]. In yet another animal model, transgenic mice expressing a stabilized β-catenin in neural precursors develop enlarged brains with increased cerebral cortical surface area and folds resembling sulci and gyri of higher mammals [Chenn and Walsh, 2002, 2003]. Brains from these animals have enlarged lateral ventricles lined with neuroepithelial precursor cells that are derived from an expanded precursor population. Compared with the wild type of precursors, a greater proportion of transgenic precursors re-enter the cell cycle after mitosis, which suggests that β-catenin regulates cerebral cortical size by controlling the generation of neural precursor cells.
Among the few models with postnatal progressive megalencephaly are the epileptic megalencephaly BALB/cByJ-Kv1.1mceph/mceph (called mceph/mceph) mice [Donahue et al., 1996] and the epileptic (epi/epi) chicken [George et al., 1990a]. The mceph/mceph mice carry a spontaneous germline mutation in a gene encoding a potassium ion channel subunit. This mutation makes the channel protein, Kv1.1, nonfunctional and causes complex partial epilepsy with the limbic system as the major focus (temporal lobe epilepsy [TLE]); interestingly, in parallel to progressive epileptic behavior, the mceph/mceph brains show progressive overgrowth, in the absence of other structural brain abnormalities. This excessive brain enlargement is restricted to the hippocampus and ventral cortical structures, including the piriform/entorhinal cortex and amygdala, whereas the thalamus, olfactory bulb, and cerebellum have wild-type sizes. The volume increase in the mceph/mceph hippocampus is due to a doubling of the number of neurons and astrocytes. In humans, Kv1.1 mutations, where only one amino acid is changed, have been found in patients with epilepsy or episodic ataxia type 1 (EA1). From extensive studies of the mceph/mceph mouse, it has been hypothesized that some human idiopathic megalencephalies with severe early-onset seizures are caused by such severe ion channelopathies [Almgren et al., 2008].
Clinical Features
Nonsyndromic (Idiopathic or Familial) Megalencephaly
The most common and largest group of anatomic megalencephaly is idiopathic megalencephaly that runs in families, the so-called “familial megalencephaly.” In one large retrospective series of 557 children referred for macrocephaly, idiopathic megalencephaly was diagnosed in 109, with a familial incidence of at least 50 percent of cases [Lorber and Priestley, 1981]. In a similarly large study, Laubscher et al. observed a familial incidence of 50 out of 71 cases (70 percent) with primary megalencephaly. There are multiple additional reports of familial megalencephaly in the older literature [DeMyer, 1972; Platt and Nash, 1972; Schreier et al., 1974; Asch and Myers, 1976; Day and Schutt, 1979]. This is generally a diagnosis of exclusion following the identification of macrocephaly in a family member, most often a parent, and the absence of an identifiable disorder known to be associated with macrocephaly. Box 25-2 lists the original diagnostic criteria for familial megalencephaly, developed by DeMyer in 1986 [DeMyer, 1986]. The onset of megalencephaly in idiopathic familial and nonfamilial MEG may be congenital or postnatal. OFCs and the progression and velocity of brain growth tend to vary, but the OFC curve generally levels off to parallel the normal one. While most children are neurodevelopmentally normal (and hence the previous designation of “benign” megalencephaly), a wide range of developmental disorders, tone abnormalities, and seizures are present in familial and nonfamilial cases. Clearly, individuals with idiopathic megalencephaly range from those who have fully normal cognitive and motor function to those with substantial neurologic disability [DeMyer, 1972; Schreier et al., 1974; Alvarez et al., 1986; Lewis et al., 1989]. Mild dysmorphic features related to excessive head growth (such as dolichocephaly and frontal bossing) are frequently observed. Neuroradiologically, megalencephaly may be associated with mild or borderline ventriculomegaly, or an enlarged extra-axial space [Alvarez et al., 1986; Laubscher et al., 1990]. A few familial cases have been complicated by hydrocephalus requiring neurosurgical intervention [Schreier et al., 1974; Day and Schutt, 1979]. Most reported cases of familial megalencephaly appear to be autosomal-dominant, with a strong sex predilection for males; however, very few reports of autosomal-recessive types exist [Gragg, 1971; Härtel et al., 2005].
Box 25-2 Diagnostic Criteria for Familial Megalencephaly
* Some familial cases may have tone abnormalities, variable degrees of developmental delay and/or seizures.
(Criteria developed by DeMyer: Megalencephaly: types, clinical syndromes, and management, [1986].)
The clinical features of the most common megalencephaly and hemimegalencephaly syndromes are outlined in Table 25-10, and are discussed briefly below.
Etiology
The most significant macrocephaly (and/or megalencephaly) syndromes are listed in Tables 25-9 and 25-10, with a brief overview of their clinical features, MRI findings, and genetic bases. These include classic overgrowth syndromes, such as Sotos’, Weaver’s, and Simpson–Golabi–Behmel syndromes; PTEN-related disorders, such as Cowden’s and Bannayan–Riley–Ruvalcaba syndromes; the macrocephaly-capillary malformation (M-CM) syndrome (previously termed macrocephaly cutis marmorata telangiectatica congenita, or CMTC); and skeletal dysplasias, such as achondroplasia and thanatophoric dysplasia, as well as a number of chromosomal disorders. By far, the majority of these disorders are inherited as an autosomal-dominant trait. Their clinical features and neurodevelopmental outcome are quite variable and dependent on the ensuing neuronal dysfunction caused by the specific underlying disorder. The most notable megalencephaly/macrocephaly disorders are discussed briefly below.
Overgrowth Syndromes
Sotos’ syndrome is an autosomal-dominant disorder due to mutations or deletions of NSD1 (nuclear receptor-binding SET domain protein-1). Macrocephaly is usually present at all ages in more than 90 percent of children and is considered to be a cardinal feature [Agwu et al., 1999; Rio et al., 2003; Tatton-Brown et al., 2005]. In some series, macrocephaly was present at birth in 50 percent of children, with birth OFCs as high as +4 above the mean, and later OFCs ranging between +2 and +7 SD. Most patients have a nonprogressive neurologic dysfunction characterized by clumsiness and poor coordination [Cole and Hughes, 1994]. Delays in expressive language and motor development during infancy are particularly common and, in some instances, may be followed by attainment of normal or near-normal intelligence. Several patients with Sotos’ syndrome and autistic features have been reported [Morrow et al., 1990; Battaglia and Carey, 2006]. Seizures and tone abnormalities are occasionally present [Cohen, 1989, 1999; Cole and Hughes, 1990, 1994]. Brain MRI abnormalities present in patients with Sotos’ syndrome and an NSD1 mutation include enlarged extra-axial fluid and lateral ventricles in 70 percent and 60 percent of patients, respectively, and it has been suggested that these increased CSF spaces are primarily responsible for macrocephaly in Sotos’ syndrome, rather than true megalencephaly [Schaefer et al., 1997]. Between 80 and 90 percent of patients have a demonstrable NSD1 abnormality. NSD1 is involved in an intricate regulatory network of genes that appear to have a concerted role in various processes, including cell growth and tumorigenesis [Lucio-Eterovic et al., 2010].
NSD1 mutations have also been found in a significant proportion of patients with Weaver’s syndrome, a rarer overgrowth disorder characterized by macrocephaly, dysmorphic facial features (especially prominent hypertelorism), metaphyseal flaring of the femurs, camptodactyly, deep-set nails, and hoarse, low-pitched cry. Therefore, significant clinical and genetic overlap exists between these two disorders of macrocephaly and overgrowth [Proud et al., 1998; Rio et al., 2003; Cecconi et al., 2005].
Simpson–Golabi–Behmel syndrome (SGBS) is an X-linked complex congenital overgrowth syndrome characterized by macroglossia, macrosomia, renal and skeletal abnormalities, and an increased risk of embryonal tumors. Macrocephaly is often congenital. Patients may have hypotonia and mild developmental delay, although most have normal intelligence [Neri et al., 1998]. Most cases of SGBS are due to mutations or deletions of the glypican-3 (GPC3) gene at Xq26, a member of a multigene family encoding at least six distinct glycosylphosphatidylinositol-linked cell-surface heparan sulfate proteoglycans (HSPGs); these act as co-receptors for multiple families of growth factors that have been shown to regulate cell proliferation, differentiation, and patterning, including that of the brain. In support of the glypicans’ role in development, mice with null mutations in glypican-1 (Gpc1) have a severely reduced brain size and an abnormally small-sized cerebellum. Therefore, Gpc1 may have a role in early neurogenesis, possibly through regulation of fibroblast growth factor (fgf) signaling [Jen et al., 2009].
SGBS type 2 is an X-linked mental retardation syndrome with macrocephaly (OFCs +2 to +6 SD above the mean) and ciliary dysfunction, manifesting as recurrent respiratory tract infections, with abnormal functional studies of the respiratory cilia. Recently, a family with this syndrome co-segregating with a frameshift mutation in the oral-facial-digital type 1 (OFD1) gene was reported [Budny et al., 2006].
RASopathies
NF1 shares features of other overgrowth syndromes, such as the presence of macrocephaly, various types of tumors, and, occasionally, hemihyperplasia of a limb or digit, despite an increased incidence of short stature. Macrocephaly in the absence of hydrocephalus occurs in 50 percent of individuals with NF1 [Tonsgard, 2006]. Quantitative MRI studies have demonstrated the presence of true megalencephaly, largely secondary to increased white-matter volume [Bale et al., 1991; Said et al., 1996; Steen et al., 2001; Cutting et al., 2002]. Learning disabilities have been reported in up to 70 percent of individuals, and 3 percent have severe developmental delay. Their neurocognitive profile may also include easy distractibility, impulsiveness, and deficient visual-motor coordination. Seizures occur in approximately 6–7 percent of patients. Frank hydrocephalus with aqueductal stenosis, as well as asymptomatic ventricular dilatation, has been observed in approximately 4 percent of patients. NF1 is a tumor suppressor gene, expressed in neurons and glial cells, which encodes neurofibromin, one of the earliest identified regulators of the RAS-MAPK pathway; it thus has important roles in cellular proliferation and differentiation [Daston et al., 1992; Nordlund et al., 1993; Cichowski and Jacks, 2001].
Recently, a dominant condition that overlaps with NF1 clinically (with macrocephaly, café au lait lesions, and axillary freckling) has been described in association with heterozygous mutations in SPRED1, a member of the SPROUTY/SPREAD family of proteins that are also regulators of RAS–RAF interaction and MAPK signaling [Brems et al., 2007] (see Legius’ syndrome in Tables 25-9 and 25-10).
Costello’s syndrome is a unique combination of failure to thrive, cardiac abnormalities, and a predisposition to papillomata and malignant tumors. In a systematic review of 28 patients, absolute or relative macrocephaly was found in 100 percent of patients, and, more specifically an evolving megalencephaly and cerebellar enlargement, overlapping with M-CM syndrome [Gripp et al., 2010]. Neurologic abnormalities include developmental delay/mental retardation, nystagmus, and hypotonia [Quezada and Gripp, 2007; Gripp and Lin, 2009].
Macrocephaly-Capillary Malformation Syndrome
The macrocephaly-capillary malformation (MCAP) syndrome is a distinct syndrome characterized by megalencephaly, vascular malformations (most often cutis marmorata), hemihypertrophy, digit anomalies, and skin and connective tissue laxity. More than 100 patients with M-CM syndrome have been reported [Clayton-Smith et al., 1997; Moore et al., 1997; Vogels et al., 1998; Thong et al., 1999; Franceschini et al., 2000; Robertson et al., 2000; Giuliano et al., 2004; Lapunzina et al., 2004; Canham and Holder, 2008], and its neuroimaging findings were reviewed by Garavelli et al. [2005] and Conway et al. [2007]. The megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus (MPPH) syndrome is a more recently described syndrome in an initial cohort of five patients [Mirzaa et al., 2004], and four subsequent single cases [Colombani et al., 2006; Garavelli et al., 2007; Tohyama et al., 2007; Pisano et al., 2008]. Since then, a marked increase in ascertainment of patients with overlapping features of both syndromes has been witnessed, and it is proposed that they represent a single megalencephaly syndrome [Gripp et al., 2009; unpublished data]. MEG is most often congenital, with OFCs ranging from +2 to +4 SD above the mean at birth, and reaching up to +8 SD later in life. Variable degrees of developmental delay, hypotonia, and seizures occur. Vascular anomalies are a characteristic and defining feature and most commonly consist of cutis marmorata, the cutaneous marbled appearance frequently seen in Caucasian newborns that tends to fade with time but often persists. Other vascular anomalies include a midline nevus flammeus, various types of hemangiomas in any location, vascular rings, and telangiectasias. Digit anomalies include the common 2–3 toe syndactyly (>25 percent syndactyly), 2–3–4 finger syndactyly, and postaxial polydactyly. Common MRI abnormalities (Figure 25-4A) include diffuse megalencephaly that is symmetric or mildly asymmetric, a very high rate of hydrocephalus that is often shunted, or ventriculomegaly, progressive posterior fossa crowding with cerebellar tonsillar herniation that may require decompression, polymicrogyria that is by far bilateral perisylvian in distribution, and white-matter abnormalities. A distinct subset of patients has a very thick (or mega-) corpus callosum [Conway et al., 2007; unpublished data]. Serial neuroimaging has demonstrated that, despite shunting procedures, OFCs continue to follow an accelerated growth rate, thereby demonstrating the presence of true megalencephaly. All reported cases to date appear sporadic.
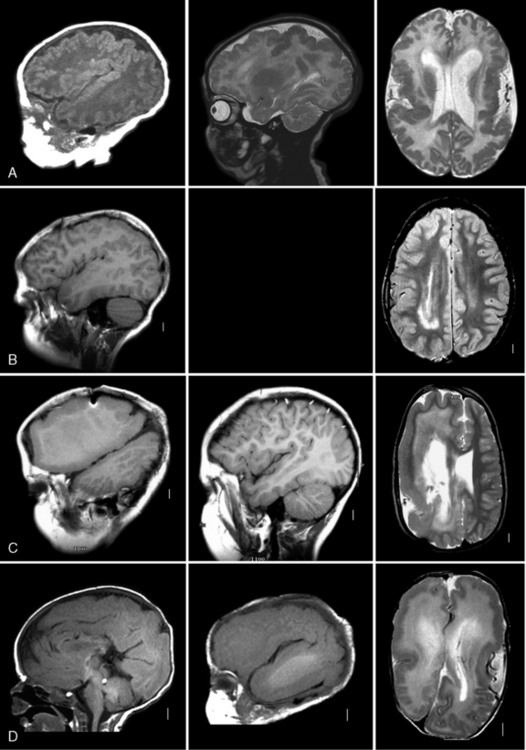
Fig. 25-4 Subtypes of megalencephaly and hemimegalencephaly.
Right parasagittal (left column, except midsagittal in D), left parasagittal (middle column), and axial (right column) magnetic resonance images from four patients with megalencephaly (MEG) or hemimegalencephaly (HMEG) variants. A, The top row images depict symmetric MEG and perisylvian polymicrogyria with normal white matter. The patient was a female with the originally described “megalencephaly polymicrogyria polydactyly hydrocephalus” (MPPH) syndrome [Mirzaa et al., 2004]. The symmetry and normal white matter distinguish this malformation from HMEG. B, The second row images show partial HMEG, with enlargement of the posterior frontal, temporal, and parietal lobes on the right. The abnormal white matter typical of HMEG is seen circling the back of the right lateral ventricle. C, The third row images demonstrate severe HMEG involving the entire right hemisphere, but sparing the left. The central and deep white matter has diffusely bright signal, sparing only the superficial U fibers. D, The bottom row images show a very rare malformation consisting of bilateral HMEG that is more severe on the left side. The patient survived only a few months.
(Courtesy of Dr. William B Dobyns, University of Washington and Principal Investigator, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA.)
Perhaps as a severe variant of this syndrome, Gohlich-Ratmann et al. reported three sporadic cases with congenital megalencephaly, a greatly hypertrophied corpus callosum, and complete lack of motor development [Gohlich-Ratmann et al., 1998]. Cranial MRI demonstrated bilateral and symmetric megalencephaly and polymicrogyria. Two cases were subsequently reported with similar features, one with minimal motor development (with the ability roll sideways only) [Dagli et al., 2008; Hengst et al., 2010]. Two children from a consanguineous family were similarly reported with mega-corpus callosum, polymicrogyria, and moderate psychomotor retardation, suggestive of autosomal-recessive inheritance. These patients additionally exhibited pontine and cerebellar vermis hypoplasia [Bindu et al., 2010].
PTEN-Related Disorders
PTEN is a tumor suppressor gene, somatic mutations of which have been reported to varying degrees in multiple sporadic malignancies (such as glioblastoma multiforme, among others) [Eng, 2000, 2003]. Germline mutations of PTEN have been found in a set of disorders of macrocephaly and hamartomatous overgrowth, namely Cowden’s (CS), Bannayan–Riley–Ruvalcaba (BRRS), and Proteus’ syndromes, and in a subset of patients with a “Proteus-like” phenotype. CS and BRSS have a high degree of clinical overlap and are believed to constitute a single clinical spectrum (CS-BRRS). Macrocephaly is a prominent and progressive feature, with OFCs typically +4.5 SD or more above the mean, and reaching up to +8 SD. Hypotonia and delayed gross motor skills are common findings. Around 60 percent of patients have a mild proximal myopathy, and 25 percent have seizures. Additional features include hamartomas, lipomas, intestinal polyps, and various types of cutaneous vascular malformations. PTEN mutation carriers are at increased risk for various tumors (most notably of the breast, thyroid, and endometrium).
Proteus’ syndrome (PS) is a rare and highly variable disorder with relentless asymmetric and disproportionate overgrowth of body parts, vascular malformations, cerebriform connective tissue nevi, epidermal nevi, and dysregulated adipose tissue [Cohen and Hayden, 1979; Wiedemann et al., 1983; Cohen et al., 2002], which has been reported in association with HMEG or unilateral MEG. Given the genetic overlap between these disorders of dysregulated cellular proliferation, the term “PTEN hamartoma tumor syndrome” (PHTS) has been coined for this group of distinct conditions [Marsh et al., 1999; Eng, 2000]. PTEN mediates cell cycle arrest and/or apoptosis by negatively regulating the phosphinositide-3-kinase-Akt serine/threonine protein kinase (PI3K/Akt) pathway [Furnari et al., 1998; Li et al., 1998; Weng et al., 1999]. Accumulating evidence suggests that PTEN also regulates cell survival pathways, such as the MAPK pathway [Gu et al., 1998; Simpson and Parsons, 2001; Weng et al., 2001, 2002]. PTEN mutations have recently been identified in patients with isolated macrocephaly and autistic spectrum disorders (ASDs), and/or developmental delay, as discussed below.
Macrocephaly-Autism Syndrome
An increased rate of macrocephaly is a consistent and replicated biological finding in ASD, and appears to be the single most consistent physical characteristic of children with autism. Multiple studies of OFC in persons with ASD have shown that macrocephaly occurs more frequently than expected [Bailey et al., 1993; Bolton et al., 2001; Davidovitch et al., 1996; Woodhouse et al., 1996; Lainhart et al., 1997; Stevenson et al., 1997; Fombonne et al., 1999; Fidler et al., 2000; Miles et al., 2000; Aylward et al., 2002; Gillberg and De Souza, 2002; Deutsch and Joseph, 2003; Dementieva et al., 2005; Lainhart et al., 2006; Courchesne et al., 2010]. These studies show, on average, a rate of macrocephaly of 20 percent in patients with ASD [Fombonne et al., 1999]. Neuroimaging studies of autism have found increased mean total brain volume in children by 2–4 years of age [Courchesne et al., 2001; Sparks et al., 2002; Hazlett et al., 2005]. Furthermore, postmortem studies show increased brain weight in children with autism, with frank megalencephaly in some [Bailey et al., 1998; Kemper and Bauman, 1998]. Causes of macrocephaly other than increased brain volume are rarely found in patients with idiopathic autism [Lainhart et al., 1997; Stevenson et al., 1997; Bailey et al., 1998; Bigler et al., 2003]. Given these data, this common association has been termed the “macrocephaly-autism syndrome.”
Butler and colleagues [2005] published the first report directly linking PTEN and isolated ASDs, identifying mutations in 3 of 18 individuals. These 3 subjects were boys, 2 of whom had the largest OFCs in the cohort, of +7 SD and +8 SD above the mean [Butler et al., 2005]. A similar study of 71 patients with isolated ASD (57 of whom met the Diagnostic and Statistical Manual of Mental Disorders [DSM]-IV criteria for autism) identified PTEN mutations in 2 out of 16 tested individuals who also had macrocephaly [Herman et al., 2007]. Using individuals with ASD and macrocephaly drawn from the Paris Autism Research International Sibpair (PARIS) study, the Autism Genetic Research Exchange [AGRE], and separately recruited patients, Buxbaum et al. found a PTEN mutation in 1 of 88 subjects tested [Buxbaum et al., 2007].
Subsequently, PTEN mutations were found in 5 of 60 (8.3 percent) individuals with macrocephaly and ASD, and 6 of 49 (12.2 percent) individuals with macrocephaly and developmental delay (DD)/mental retardation (MR) [Varga et al., 2009]; these results were extended by a cohort study whereby PTEN mutations were found in 7 of 99 (7.1 percent) individuals with MAC/ASD and 8 of 100 (8 percent) of individuals with MAC/DD [McBride et al., 2010]. Therefore, the estimated PTEN mutation frequency in macrocephaly-autism syndrome is approximately 20 percent overall. It is interesting to note that, from a molecular standpoint, mutations in TSC1/TSC2, NF1, or PTEN activate the mTOR/PI3K pathway and lead to syndromic ASD with tuberous sclerosis, neurofibromatosis, or macrocephaly, respectively, as mentioned above.
Disorders of Skeletal Involvement
Achondroplasia and thanatophoric dysplasia are well known to be associated with true megalencephaly from multiple reports in the early literature [Dennis et al., 1961; Cohen et al., 1967; Priestley and Lorber, 1981; Knisely, 1989]. MEG frequently coexists with mild ventriculomegaly, with or without hydrocephalus related to stenosis of the sigmoid sinus at the level of narrowed jugular foramina that rarely requires surgical intervention [Pierre-Kahn et al., 1980]. Robinow’s syndrome is a genetically heterogeneous condition characterized by mesomelic limb shortening and facial and genital anomalies. The estimated frequency of MEG (which may be congenital) is up to 64 percent in the dominant form and 25 percent in the recessive form [Mazzeu et al., 2007]. Greig’s cephalopolysyndactyly syndrome (GCPS) is a rare pleiotropic, multiple congenital anomaly syndrome characterized by the triad of polysyndactyly, hypertelorism, and macrocephaly, which is not typically associated with other central nervous system anomalies [Biesecker, 2008]. The acrocallosal syndrome resembles GCPS, with the presence of preaxial polysyndactyly and macrocephaly, but is distinguished by agenesis of the corpus callosum, and a much higher incidence of seizures and mental retardation [Johnston et al., 2003].
Chromosomal Abnormalities
Macrocephaly has been reported in several chromosomal disorders, such as trisomy 5p [Leschot and Lim, 1979; Reichenbach et al., 1999], partial duplications of 12p [Rauch et al., 1996], proximal and distal 15q duplications [Hood et al., 1986; Roggenbuck et al., 2004], deletions of 22q13 [Phelan et al., 1992; Tabolacci et al., 2005], and Pallister–Killian syndrome due to mosaic isochromosome 12p [Smigiel et al., 2008]. A number of reciprocal microdeletion and microduplication syndromes are associated with MIC/MAC, such as those involving 1q21.1 [Brunetti-Pierri et al., 2008], 2p24.3 (the locus for the MYCN gene) [Malan et al., 2010], and 5q35.3 (the NSD1 locus) [Lucio-Eterovic et al., 2010]. MEG has also been reported in two patients with Klinefelter’s syndrome. One had frank, bilateral, and symmetric MEG, in association with polymicrogyria and neuronal heterotopias [Budka, 1978]; the other had mild, focal, and unilateral MEG, in association with macrogyria [Choi et al., 1980].
Other Megalencephaly/Macrocephaly Syndromes
Other rarer macrocephaly syndromes with few reports in the literature include the macrocephaly, megalocornea, motor and mental retardation (MMMM) syndrome, macrosomia, obesity, macrocephaly, and ocular abnormalities (MOMO) syndrome, and Clark–Baraitser syndrome. A single old report described a patient with ataxia telangiectasia, megalencephaly, and intestinal lymphangiectasia [Scott, 1969].
Unilateral Megalencephaly (or Hemimegalencephaly)
Hemimegalencephaly (HMEG) is a brain malformation characterized by hamartomatous overgrowth of all or part of the cerebral hemisphere, and often associated with ipsilateral cortical malformations. The involved hemisphere is frequently enlarged, with cortical dysgenesis, white-matter hypertrophy, and a dilated and dysmorphic lateral ventricle. There is no clear predilection for the right or left side [Barkovich and Chuang, 1990].
HMEG traditionally has been regarded as the result of an early disturbance in neuronal proliferation and migration. The known associations of HMEG with other disorders of cellular proliferation (such as tuberous sclerosis complex) support this hypothesis. A relation between epidermal growth factor and excessive proliferation in HMEG has been suggested [Takashima et al., 1991; Kato et al., 1996].
Clinically, macrocephaly is usually apparent at birth, and cranial asymmetry dependent on the degree of HMEG may be evident. OFC may increase rapidly during the first few months, raising the possibility of obstructive hydrocephalus or a space-occupying lesion. However, subsequently, and possibly as a result of intractable seizures, the head size diminishes relative to the normal curve and eventually the patients may become normocephalic or microcephalic. Intractable epilepsy usually begins within the first few months of life, and is the most frequent and severe neurologic manifestation, occurring in up to 93 percent of cases [Vigevano et al., 1989; George et al., 1990b]. Partial, motor, or partial complex seizures are the most frequent types of epilepsy in HMEG, and are associated with infantile spasms in 50 percent of patients. Developmental delay is often early and severe, although in a few cases it can be mild to near normal. Different grades of hemiparesis, ranging from none or mild to overt hemiplegia, are seen contralateral to the HMEG [Flores-Sarnat, 2002].
Neuroimaging shows moderate to marked enlargement of the effected cerebral hemisphere [Fitz et al., 1978; Kalifa et al., 1987], with enlargement of the lateral ventricle. Occasionally, enlargement maybe localized to the frontal or temporoparietal regions. Anomalies of cortical development, including polymicrogyria, pachygyria, and gray-matter heterotopias, are always seen in the affected portions of the hemisphere, but can be seen in the “unaffected” hemisphere as well [Barkovich and Kuzniecky, 1996]. The underlying hemispheric white matter is usually abnormal, with abnormal signal characteristics and/or alteration in volume (increased or decreased) in some individuals. Figure 25-4 (B–D) shows variable degrees of HMEG. EEG abnormalities are often extensive throughout the abnormal hemisphere and a suppression-burst pattern can be observed early on in the most severe cases. Predictors of poor outcome are severity of hemiparesis, the degree of cortical dysplasia on brain MRI, and abnormal EEG activity. Both the epilepsy and the degree of developmental handicap may be improved in selected patients by anatomical or functional hemispherectomy [Delvin et al., 2003; Di Rocco et al., 2006; Kwan et al., 2008].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abdel-Salam G.M., Halasz A.A., Czeizel A.E. Association of epilepsy with different groups of microcephaly. Dev Med Child Neurol. 2000;42:760-767.
Abidi F., Holloway L., Moore C.A., et al. Novel human pathological mutations. Gene symbol: JARID1C. Disease: mental retardation, X-linked. Hum Genet. 2009;125(3):344.
Abou-Zahr F., Bejjani B., Kruyt F.A., et al. Normal expression of the Fanconi anemia proteins FAA and FAC and sensitivity to mitomycin C in two patients with Seckel syndrome. Am J Med Genet. 1999;83:388-391.
Afzal A.R., Taylor R. ROR2-related Robinow syndrome. In: Pagon R.A., Bird T.C., Dolan C.R., et al, editors. GeneReviews [Internet]. Seattle [WA]: University of Washington, Seattle, 1993–2005. Jul 28
Agwu J.C., Shaw N.J., Kirk J., et al. Growth in Sotos syndrome. Arch Dis Child. 1999;80(4):339-342.
Alderton G.K., Joenje H., Varon R., et al. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet. 2004;13:3127-3138.
Al-Dosari M.S., Shaheen R., Colak D., et al. Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47(6):411-414.
Allanson J.E.. Noonan syndrome. Nov 15. Pagon R.A., Bird T.C., Dolan C.R., et al, editors. GeneReviews [Internet]. Seattle, [WA]: University of Washington, Seattle, 1993–2001. [updated 2008 Oct 07]
Almgren M., Schalling M., Lavebratt C. Idiopathic megalencephaly – possible cause and treatment opportunities: from patient to lab. Eur J Paediatr Neurol. 2008;12(6):438-445.
Alvarez L.A., Maytal J., Shinnar S. Idiopathic external hydrocephalus: natural history and relationship to benign familial macrocephaly. Pediatrics. 1986;77(6):901-907.
Amiel J., Rio M., de Pontual L., et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988-993.
Amir R.E., Van den Veyver I.B., Wan M., et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl – cpg-binding protein 2. Nat Genet. 1999;23:185-188.
Aneja S., Ahuja B., Taluja V., et al. Epilepsy in children with cerebral palsy. Indian J Pediatr. 2001;68:111-115.
Anlar B., Yalaz K., Erzen C. Klippel-Trenaunay-Weber syndrome: a case with cerebral and cerebellar hemihypertrophy. Neuroradiology. 1988;30(4):360.
Antoccia A., Kobayashi J., Tauchi H., et al. Nijmegen breakage syndrome and functions of the responsible protein, NBS1. Genome Dynamics. 2006;1:191-205.
Asch A.J., Myers G.J. Benign familial macrocephaly: report of a family and review of the literature. Pediatrics. 1976;57(4):535-539.
Ashwal S., Michelson D., Plawner L., et al. Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2009;73:887-897.
Avery G.B., Meneses L., Lodge A. The clinical significance of “measurement microcephaly.”. Amer J Dis Child. 1972;123:214-217.
Aylward E.H., Minshew N.J., Field K., et al. Effects of age on brain volume and head circumference in autism. Neurology. 2002;59(2):175-183.
Baala L., Briault S., Etchevers H.C., et al. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet. 2007;39(4):454-456.
Bailey A., Luthert P., Bolton P., et al. Autism and megalencephaly. Lancet. 1993;341(8854):1225-1226.
Bailey A., Luthert P., Dean A., et al. A clinicopathological study of autism. Brain. 1998;121(Pt 5):889-905.
Bale S.J., Anmos C.I., Parry D.M., et al. Relationship between head circumference and height in normal adults and in the nevoid basal cell carcinoma syndrome and neurofibromatosis type I. Am J Med Genet. 1991;40(2):206-210.
Barkovich A., Ferriero D., Barr R., et al. Microlissencephaly: a heterogeneous malformation of cortical development. Neuropediatr. 1998;29:113-119.
Barkovich A., Gressens P., Evrard P. Formation, maturation, and disorders of brain neocortex. J Neuroradiol. 1992;13:423-446.
Barkovich A.J., Chuang S.H. Unilateral megalencephaly: correlation of MR imaging and pathologic characteristics. Am J Neuroradiol. 1990;11(3):523-531.
Barkovich A.J., Kuzniecky R.I. Neuroimaging of focal malformations of cortical development. J Clin Neurophysiol. 1996;13(6):481-494.
Barmeyer G.H. Magic, science, and head circumference. Rocky Mt Med J. 1971;68(5):42-44.
Barth P.G., Mullaart R., Stam F.C., et al. Familial lissencephaly with extreme neopallial hypoplasia. Brain Dev. 1982;4:145-151.
Basel-Vanagaite L., Dobyns W.B. Clinical and brain imaging heterogeneity of severe microcephaly. Pediatr Neurol. 2010;43:7-16.
Basel-Vanagaite L., Raas-Rotchild A., Kornreich L., et al. Familial hydrocephalus with normal cognition and distinctive radiological features. Am J Med Genet A. 2010;152A:2743-2748.
Basel-Vanagaite L., Sarig O., Hershkovitz D., et al. RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am J Hum Genet. 2009;85(2):254-263.
Battaglia A., Carey J.C. Etiologic yield of autistic spectrum disorders: a prospective study. Am J Med Genet C Semin Med Genet. 2006;142C(1):3-7.
Baujat G., Cormier-Daire V. Sotos syndrome. Orphanet J Rare Dis. 2007;7(2):36.
Behmel A., Plöchl E., Rosenkranz W. A new X-linked dysplasia gigantism syndrome: identical with the Simpson dysplasia syndrome? Hum Genet. 1984;67(4):409-413.
Berg A.T., Levy S.R., Novotny E.J., et al. Predictors of intractable epilepsy in childhood: a case-control study. Epilepsia. 1996;37:24-30.
Biesecker L.G. The Greig cephalopolysyndactyly syndrome. Orphanet J Rare Dis. 2008;24(3):10.
Bigler E.D., Tate D.F., Neeley E.S., et al. Temporal lobe, autism, and macrocephaly. Am J Neuroradiol. 2003;24(10):2066-2076.
Bindu P.S., Taly A.B., Sinha S., et al. Mega-corpus callosum, polymicrogyria, and psychomotor retardation syndrome. Pediatr Neurol. 2010;42(2):129-132.
Bobabilla-Morales L., Corona-Rivera A., Corona-Rivera J.R., et al. Chromosome instability induced in vitro with mitomycin C in five Seckel syndrome patients. Am J Med Genet. 2003;123A:148-152.
Boland E., Clayton-Smith J., Woo V.G., et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet. 2007;81:292-303.
Bolton P.F., Roobol M., Allsopp L., et al. Association between idiopathic infantile macrocephaly and autism spectrum disorders. Lancet. 2001;358(9283):726-727.
Bond J., Roberts E., Mochida G.H., et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316-320.
Bond J., Roberts E., Springell K., et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37:353-355.
Borck G., Wunram H., Steiert A., et al. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum Genet. 2011;129(1):45-50.
Brems H., Chmara M., Sahbatou M., et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39(9):1120-1126.
Bromley B., Benacerraf B.R. Difficulties in the prenatal diagnosis of microcephaly. J Ultrasound Med. 1995;14:303-306.
Brunetti-Pierri N., Berg J.S., Scaglia F., et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40(12):1466-1471.
Budka H. Megalencephaly and chromosomal anomaly. Acta Neuropathol. 1978;43(3):263-266. Sep 15
Budny B., Chen W., Omran H., et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120(2):171-178. Sep
Butler M.G., Dasouki M.J., Zhou X.P., et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42(4):318-321.
Butler M.G., Hall B.D., Maclean R.N., et al. Do some patients with Seckel syndrome have hematological problems and/or chromosome breakage? Am J Med Genet. 1987;27:645-649.
Buxbaum J.D., Cai G., Chaste P., et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(4):484-491.
Camp B.W., Broman S.H., Nichols P.L., et al. Maternal and neonatal risk factors for mental retardation: defining the ‘at-risk’ child. Early Hum Dev. 1998;50:159-173.
Canham N.L., Holder S.E. Macrocephaly-cutis marmorata telangiectatica congenita: a report on the natural history of a mild case. Clin Dysmorphol. 2008;17(4):279-281.
Capovilla G., Lorenzetti M.E., Montagnini A., et al. Seckel’s syndrome and malformations of cortical development: report of three new cases and review of the literature. J Child Neurol. 2001;16:382-386.
Carson M.J., Behringer R.R., Brinster R.L., et al. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10(4):729-740.
Cecconi F., Alvarez-Bolado G., Meyer B.I., et al. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94(6):727-737.
Cecconi M., Forzano F., Garavelli L., et al. Recurrence of Mowat-Wilson syndrome in siblings with a novel mutation in the ZEB2 gene. Am J Med Genet A. 2008;146A(23):3095-3099.
Cecconi M., Forzano F., Milani D., et al. Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am J Med Genet A. 2005;134(3):247-253.
Chapman K., Cardenas J.F. Hemimegalencephaly in a patient with a neurocutaneous syndrome. Semin Pediatr Neurol. 2008;15(4):190-193.
Chawla S., Aneja S., Kashyap R., et al. Etiology and clinical predictors of intractable epilepsy. Pediatr Neurol. 2002;27:186-191.
Chenn A., Walsh C.A. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13(6):599-606.
Chenn A., Walsh C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297(5580):365-369.
Choi H., Ho K.C., Luprecht G.L. Chromatin-negative Klinefelter’s syndrome with focal megalencephaly. Acta Neurol Scand. 1980;62(6):357-367. Dec
Choi Y.J., Di Nardo A., Kramvis I., et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22(18):2485-2495.
Cichowski K., Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104(4):593-604.
Clark R.D., Baraitser M. A new X-linked mental retardation syndrome. Am J Med Genet. 1987;26(1):13-15. Jan
Clarren S.K., Alvord S.E., Sumi S.M., et al. Brain malformations related to prenatal exposure to ethanol. J Pediatr. 1978;92:64-67.
Clayton-Smith J., Kerr B., Brunner H., et al. Macrocephaly with cutis marmorata, haemangioma and syndactyly – a distinctive overgrowth syndrome. Clin Dysmorphol. 1997;6(4):291-302.
Cohen M.E., Rosenthal A.D., Matson D.D. Neurological abnormalities in achondroplastic children. J Pediatr. 1967;71(3):367-376.
Cohen M.M.Jr. Overgrowth syndromes: an update. Adv Pediatr. 1999;46:441-491.
Cohen M.M.Jr. Proteus syndrome: clinical evidence for somatic mosaicism and selective review. Am J Med Genet. 1993;47(5):645-652.
Cohen M.M.Jr, Hayden P.W. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):291-296.
Cohen M.M.Jr, Neri G., Weksberg R.. Beckwith-Wiedemann syndrome. Overgrowth syndromes. Oxford: Oxford University Press; 2002:11-31. Chapter 2, Oxford Monographs on Medical Genetics no. 43
Cole T.R., Hughes H.E. Sotos syndrome. J Med Genet. 1990;27:571-576.
Cole T.R., Hughes H.E. Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet. 1994;31(1):20-32.
Colombani M., Chouchane M., Pitelet G., et al. A new case of megalencephaly and perisylvian polymicrogyria with post-axial polydactyly and hydrocephalus: MPPH syndrome. Eur J Med Genet. 2006;49(6):466-471.
Conway R.L., Pressman B.D., Dobyns W.B., et al. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet A. 2007;143A(24):2981-3008.
Courchesne E., Campbell K., Solso S. Brain growth across the life span in autism: Age-specific changes in anatomical pathology. Brain Res. 2010. Oct 1
Courchesne E., Karns C.M., Davis H.R., et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245-254.
Croen L.A., Grether J.K., Curry C.J., et al. Congenital abnormalities among children with cerebral palsy: more evidence for prenatal antecedents. J Pediatr. 2001;138:804-810.
Cutting L.E., Cooper K.L., Koth C.W., et al. Megalencephaly in NF1: predominantly white matter contribution and mitigation by ADHD. Neurology. 2002;59(9):1388-1394.
D’Ercole A.J., Ye P., O’Kusky J.R. Mutant mouse models of insulin-like growth factor actions in the central nervous system. Neuropeptides. 2002;36(2–3):209-220.
Dagli A.I., Stalker H.J., Williams C.A. A patient with the syndrome of megalencephaly, mega corpus callosum and complete lack of motor development. Am J Med Genet A. 2008;146A(2):204-207.
Daston M.M., Scrable H., Nordlund M., et al. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8(3):415-428.
Davidovitch M., Patterson B., Gartside P. Head circumference measurements in children with autism. J Child Neurol. 1996;11(5):389-393.
Day R.E., Schutt W.H. Normal children with large heads – benign familial megalencephaly. Arch Dis Child. 1979;54(7):512-517. Jul
DeBaun M.R., Ess J., Saunders S. Simpson Golabi Behmel syndrome: progress toward understanding the molecular basis for overgrowth, malformation, and cancer predisposition. Mol Genet Metab. 2001;72(4):279-286.
Dekaban A.S. Abnormalities in children exposed to x-radiation during various stages of gestation: tentative timetable of radiation injury to the human fetus. I. J Nucl Med. 1968;9:471-477.
Dementieva Y.A., Vance D.D., Donnelly S.L., et al. Accelerated head growth in early development of individuals with autism. Pediatr Neurol. 2005;32(2):102-108.
DeMyer W. Megalencephaly in children: clinical syndromes, genetic patterns, and differential diagnosis from other causes of megalencephaly. Neurology. 1972;22:634-643.
DeMyer W. Megalencephaly: types, clinical syndromes and management. Pediatr Neurol. 1986;2:321-328.
Dennis J.P., Rosenberg H.S., Alvord E.C. Megalencephaly, internal hydrocephalus and other neurological aspects of achondroplasia. Brain. 1961;84:427-445.
Desch L.W., Anderson S.K., Snow J.H. Relationship of head circumference to measures of school performance. Clin Pediatr. 1990;29:389-392.
Deutsch C.K., Joseph R.M. Brief report: cognitive correlates of enlarged head circumference in children with autism. J Autism Dev Disord. 2003;33(2):209-215. Apr
Devlin A.M., Cross J.H., Harkness W., et al. Clinical outcomes of hemispherectomy for epilepsy in childhood and adolescence. Brain. 2003;126(Pt 3):556-566. Mar
de Wit M.C., de Coo I.F., Halley D.J., et al. Movement disorder and neuronal migration disorder due to ARFGEF2 mutation. Neurogenetics. 2009;10(4):333-336.
Di Rocco C., Battaglia D., Pietrini D., et al. Hemimegalencephaly: clinical implications and surgical treatment. Childs Nerv Syst. 2006;22(8):852-866.
Dobyns W.B. Lessons from the Lissencephaly Research Project: five new malformation syndromes (abstract). Am J Hum Genet. 1996;59:A21.
Dobyns W.B. Primary microcephaly: new approaches for an old disorder. Am J Med Genet. 2002;112:315-317.
Dobyns W.B., Barkovich A.J. Microcephaly with simplified gyral pattern (oligogyric microcephaly) and microlissencephaly: reply. Neuropediatr. 1999;30:104-106.
Dobyns W.B., Curry C., Hoyme H., et al. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991;48:584-594.
Dobyns W.B., Stratton R.F., Greenberg F. Syndromes with lissencephaly. I: Miller-Dieker and Norman-Roberts syndromes and isolated lissencephaly. Am J Med Genet. 1984;18:509-526.
Dodge N.N., Dobyns W.B. Agenesis of the corpus callosum and Dandy-Walker malformation associated with hemimegalencephaly in the sebaceous nevus syndrome. Am J Med Genet. 1995;56(2):147-150. Mar 27
Dolk H. The predictive value of microcephaly during the first year of life for mental retardation at seven years. Dev Med Child Neurol. 1991;33:974-983.
Donahue L.R., Cook S.A., Johnson K.R., et al. Megencephaly: a new mouse mutation on chromosome 6 that causes hypertrophy of the brain. Mamm Genome. 1996;7(12):871-876.
Dubourg C., Lazaro L., Pasquier L., et al. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum Mutat. 2004;24:43-51.
Dye J.. Fertility of American Women: June 2004. 2005.
Edebol-Tysk K. Epidemiology of spastic tetraplegic cerebral palsy in Sweden. I. Impairments and disabilities. Neuropediatrics. 1989;20:41-45.
Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183-198.
Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37(11):828-830.
Erkek E., Hizel S., Sanlý C., et al. Clinical and histopathological findings in Bannayan-Riley-Ruvalcaba syndrome. J Am Acad Dermatol. 2005;53(4):639-643.
Evans P.D., Anderson J.R., Vallender E.J., et al. Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet. 2004;13:489-494.
Evans P.D., Anderson J.R., Vallender E.J., et al. Reconstructing the evolutionary history of microcephalin, a gene controlling human brain size. Hum Mol Genet. 2004;13:1139-1145.
Evrard P.. Normal and abnormal development of the brain. Handbook of Neuropsychology. Amsterdam: Elsevier; 1992;6:11-44.
Evrard P., de Saint-Georges P., Kadhim H., et al. Pathology of prenatal encephalopathies. In: Child Neurology and Developmental Disabilities. Baltimore MD: Paul H. Brookes; 1989:153-176.
Faivre L., Le Merrer M., Lyonnet S., et al. Clinical and genetic heterogeneity of Seckel syndrome. Am J Med Genet. 2002;112:379-383.
Fernandez J., Saudubray J.-M., Van Den Berghe G., et al. Inborn Metabolic Diseases: Diagnosis and Treatment. NY: Springer; 2006.
Fidler D.J., Bailey J.N., Smalley S.L. Macrocephaly in autism and other pervasive developmental disorders. Dev Med Child Neurol. 2000;42(11):737-740.
Fitz C.R., Harwood-Nash D.C., Boldt D.W. The radiographic features of unilateral megalencephaly. Neuroradiology. 1978;15(3):145-148.
Flores-Sarnat L. Hemimegalencephaly: part 1. Genetic, clinical, and imaging aspects. J Child Neurol. 2002;17(5):373-384. discussion 384
Fombonne E., Rogé B., Claverie J., et al. Microcephaly and macrocephaly in autism. J Autism Dev Disord. 1999;29(2):113-119.
Franceschini P., Licata D., Di Cara G., et al. Macrocephaly-Cutis marmorata telangiectatica congenita without cutis marmorata? Am J Med Genet. 2000;90(4):265-269.
Fraser M.M., Zhu X., Kwon C.H., et al. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64(21):7773-7779.
Frydman M., Berkenstadt M., Raas-Rothschild A., et al. Megalocornea, macrocephaly, mental and motor retardation (MMMM). Clin Genet. 1990;38(2):149-154.
Fukuzawa R., Sato S., Sullivan M.J., et al. Autopsy case of microcephalic osteodysplastic primordial “dwarfism” type II. Am J Med Genet. 2002;113:93-96.
Furnari F.B., Huang H.J., Cavenee W.K. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58(22):5002-5008.
Garavelli L., Guareschi E., Errico S., et al. Megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus (MPPH): report of a new case. Neuropediatrics. 2007;38(4):200-203.
Garavelli L., Leask K., Zanacca C., et al. MRI and neurological findings in macrocephaly-cutis marmorata telangiectatica congenita syndrome: report of ten cases and review of the literature. Genet Couns. 2005;16(2):117-128.
Geisert E.E.Jr, Williams R.W., Geisert G.R., et al. Increased brain size and glial cell number in CD81-null mice. J Comp Neurol. 2002;453(1):22-32.
George D.H., Munoz D.G., McConnell T., et al. Megalencephaly in the epileptic chicken: a morphometric study of the adult brain. Neuroscience. 1990;39(2):471-477.
George R.E., Hoffman H.J., Hwang P.A., et al. Management of intractable seizures in unilateral megalencephaly. J Epilepsy. 1990;3(Suppl):305-313.
Giacomini C. Contributo allo studio della microcefalia. Arch Psychiatr. 1885;6:63-81.
Giannandrea M., Bianchi V., Mignogna M.L., et al. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet. 2010;86(2):185-195.
Gilfillan G.D., Selmer K.K., Roxrud I., et al. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet. 2008;82:1003-1010.
Gillberg C., de Souza L. Head circumference in autism, Asperger syndrome, and ADHD: a comparative study. Dev Med Child Neurol. 2002;44(5):296-300.
Giuliano F., David A., Edery P., et al. Macrocephaly-cutis marmorata telangiectatica congenita: seven cases including two with unusual cerebral manifestations. Am J Med Genet A. 2004;126A(1):99-103.
Göhlich-Ratmann G., Baethmann M., Lorenz P., et al. Megalencephaly, mega corpus callosum, and complete lack of motor development: a previously undescribed syndrome. Am J Med Genet. 1998;79(3):161-167.
Gorlin R.J., Cohen M.M.Jr, Condon L.M., et al. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44(3):307-314.
Grabb P.A., Albright A.L., Zitelli B.J. Multiple suture synostosis, macrocephaly, and intracranial hypertension in a child with alpha-D-mannosidase deficiency. Case report. J Neurosurg. 1995;82:647-649.
Gragg G.W. Familial megalencephaly. Birth Defects Orig Artic Ser. 1971;7(1):228-230.
Grantham-McGregor S., Cheung Y.B., Cueto S., et alInternational Child Development Steering Group. Developmental potential in the first 5 years for children in developing countries. Lancet. 2007;369(9555):60-70.
Griffith E., Walker S., Martin C., et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet. 2008;40(2):232-236.
Griffiths P.D., Gardner S., Smith M., et al. Hemimegalencephaly and Focal Megalencephaly in Tuberous Sclerosis Complex. Am J Neuroradiol. 1998;19(10):1935-1938.
Gripp K.W., Hopkins E., Doyle D., et al. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A. 2010;152A(5):1161-1168.
Gripp K.W., Hopkins E., Vinkler C., et al. Significant overlap and possible identity of macrocephaly capillary malformation and megalencephaly polymicrogyria-polydactyly hydrocephalus syndromes. Am J Med Genet A. 2009;149A(5):868-876.
Guernsey D.L., Jiang H., Hussin J., et al. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet. 2010;87(1):40-51.
Gu J., Tamura M., Yamada K.M. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol. 1998;143(5):1375-1383.
Hakem R., Hakem A., Duncan G.S., et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94(3):339-352.
Hall J.G., Flora C., Scott C.I., et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am J Med Genet. 2004;130A:55-72.
Härtel C., Bachmann S., Bönnemann C., et al. Familial megalencephaly with dilated Virchow-Robin spaces in magnetic resonance imaging: an autosomal recessive trait? Clin Dysmorphol. 2005;14(1):31-34.
Hassan M.J., Khurshi M., Azeem Z., et al. Previously described sequence variant in CDK5RAP2 gene in a Pakistani family with autosomal recessive primary microcephaly. BMC Med Genet. 2007;8:58.
Hayani A., Suarez C.R., Molnar Z., et al. Acute myeloid leukaemia in a patient with Seckel syndrome. J Med Genet. 1994;31:148-149.
Hazlett H.C., Poe M., Gerig G., et al. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry. 2005;62(12):1366-1376.
Hengst M., Tücke J., Zerres K., et al. Megalencephaly, mega corpus callosum, and complete lack of motor development: delineation of a rare syndrome. Am J Med Genet A. 2010;152A(9):2360-2364.
Herman G.E., Henninger N., Ratliff-Schaub K., et al. Genetic testing in autism: how much is enough? Genet Med. 2007;9(5):268-274. May
Hong S.E., Shugart Y.Y., Huang D.T., et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93-96.
Hood O.J., Rouse B.M., Lockhart L.H., et al. Proximal duplications of chromosome 15: clinical dilemmas. Clin Genet. 1986;29(3):234-240.
Hori A., Tamagawa K., Eber S.W., et al. Neuropathology of Seckel syndrome in fetal stage with evidence of intrauterine developmental retardation. Acta Neuropathol (Berl). 1987;74:397-401.
Inoki K., Li Y., Zhu T., et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648-657.
Jackson A.P., Eastwood H., Bell S.M., et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71:136-142.
Jacob F.D., Ramaswamy V., Andersen J., et al. Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet. 2009;17:1577-1581.
Jaworski J., Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol. 2006;34(3):205-219.
Jaworski J., Spangler S., Seeburg D.P., et al. Control of dendritic arborization by the phosphoinositide-3’-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25(49):11300-11312.
Jen Y.H., Musacchio M., Lander A.D. Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Dev. 2009;4:33.
Johnston J.J., Olivos-Glander I., Turner J., et al. Clinical and molecular delineation of the Greig cephalopolysyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndrome. Am J Med Genet A. 2003;123A(3):236-242.
Juric-Sekhar G., Kapur R.P., Glass I.A., et al. Neuronal migration disorders in microcephalic osteodysplastic primordial dwarfism type I/III. Acta Neuropathol. 2010.
Kalay E., Yigit G., Aslan Y., et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43(1):23-26.
Kalifa G.L., Chiron C., Sellier N., et al. Hemimegalencephaly: MR imaging in five children. Radiology. 1987;165(1):29-33.
Kannu P., Kelly P., Aftimos S. Microcephalic osteodysplastic primordial dwarfism type II: a child with cafe au lait lesions, cutis marmorata, and moyamoya disease. Am J Med Genet. 2004;128A:98-100.
Kantaputra P.N. Apparently new osteodysplastic and primordial short stature with severe microdontia, opalescent teeth, and rootless molars in two siblings. Am J Med Genet. 2002;111:420-428.
Kato M., Dobyns W.B. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12:R89-R96.
Kato M., Mizuguchi M., Sakuta R., et al. Hypertrophy of the cerebral white matter in hemimegalencephaly. Pediatr Neurol. 1996;14(4):335-338.
Kelley R.I., Robinson D., Puffenberger E.G., et al. Amish lethal microcephaly: a new metabolic disorder with severe congenital microcephaly and 2-ketoglutaric aciduria. Am J Med Genet. 2002;112:318-326.
Kemper T.L., Bauman M. Neuropathology of infantile autism. J Neuropathol Exp Neurol. 1998;57(7):645-652. Jul
Kimonis V.E., Goldstein A.M., Pastakia B., et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69(3):299-308.
Klinge L., Schaper J., Wieczorek D., et al. Microlissencephaly in microcephalic osteodysplastic primordial dwarfism: a case report and review of the literature. Neuropediatrics. 2002;33:309-313.
Knight S.J., Lese C.M., Precht K.S., et al. An optimized set of human telomere clones for studying telomere integrity and architecture. Am J Hum Genet. 2000;67:320-332.
Knisely A.S. Megalencephaly in thanatophoric dysplasia and in achondroplasia. J Pediatr. 1989;115(6):1026.
Krauss M.J., Morrissey A.E., Winn H.N., et al. Microcephaly: an epidemiologic analysis. Am J Obstet Gynecol. 2003;188:1484-1489.
Krishnan B.R., Ramesh A., Kumari M.P., et al. Genetic analysis of a group of mentally retarded children. Indian J Pediatr. 1989;56(2):249-258. Mar-Apr
Kroon A.A., Smit B.J., Barth P.G., et al. Lissencephaly with extreme cerebral and cerebellar hypoplasia. A magnetic resonance imaging study. Neuropediatrics. 1996;27:273-276.
Kuida K., Haydar T.F., Kuan C.Y., et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94(3):325-337.
Kuida K., Zheng T.S., Na S., et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384(6607):368-372.
Kumar A., Girimaji S.C., Duvvari M.R., et al. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. 2009;84:286-290.
Kumar R.A., Pilz D.T., Babatz T.D., et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010;19:2817-2827.
Kwan S.Y., Shyu H.Y., Lin J.H., et al. Corpus callosotomy in a patient of hemimegalencephaly and Lennox-Gastaut syndrome. Brain Dev. 2008;30(10):643-646.
Kwon C.H., Zhou J., Li Y., et al. Neuron-specific enolase-cre mouse line with cre activity in specific neuronal populations. Genesis. 2006;44(3):130-135.
Kwon C.H., Zhu X., Zhang J., et al. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA. 2003;100(22):12923-12928.
Kwon C.H., Zhu X., Zhang J., et al. Pten regulates neuronal soma size: a mouse model of Lhermitte–Duclos disease. Nat Genet. 2001;29:404-411.
Lacbawan F., Solomon B.D., Roessler E., et al. Clinical Spectrum of SIX3-Associated Mutations in Holoprosencephaly: Correlation between Genotype, Phenotype, and Function. J Med Genet. 2009;46:389-398.
Lainhart J.E., Bigler E.D., Bocian M., et al. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am J Med Genet A. 2006;140(21):2257-2274.
Lainhart J.E., Piven J., Wzorek M., et al. Macrocephaly in children and adults with autism. J Am Acad Child Adolesc Psychiatry. 1997;36(2):282-290.
Laisram N., Srivastava V.K., Srivastava R.K. Cerebral palsy – an etiological study. Indian J Pediatr. 1992;59:723-728.
Lalaguna-Mallada P., Alonso-del Val B., Abio-Albero S., et al. (Microcephalus as the reason for visiting a regional referral neuropaediatric service). Rev Neurol. 2004;38:106-110.
Lapunzina P., Gairí A., Delicado A., et al. Macrocephaly-cutis marmorata telangiectatica congenita: report of six new patients and a review. Am J Med Genet A. 2004;130A(1):45-51.
Laubscher B., Deonna T., Uske A., et al. Primitive megalencephaly in children: natural history, medium term prognosis with special reference to external hydrocephalus. Eur J Pediatr. 1990;149:502-507.
Lee da Y., Yeh T.H., Emnett R.J., et al. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 2010;24(20):2317-2329.
Lenski C., Abidi F., Meindl A., et al. Novel truncating mutations in the polyglutamine tract binding protein 1 gene (PQBP1) cause Renpenning syndrome and X-linked mental retardation in another family with microcephaly. Am J Hum Genet. 2004;74(4):777-780.
Leschot N.J., Lim K.S. “Complete” trisomy 5p; de novo translocation t(2;5)(q36;p11) with isochromosome 5p. Case report and review of the literature. Hum Genet. 1979;46(3):271-278.
Leviton A., Holmes L.B., Allred E.N., et al. Methodologic issues in epidemiologic studies of congenital microcephaly. Early Hum Dev. 2002;69:91-105.
Levy H.L., Lobbregt D., Barnes P.D., et al. Maternal phenylketonuria: magnetic resonance imaging of the brain in offspring. J Pediatr. 1996;128:770-775.
Lewis B.A., Aram D.N., Horwitz S.J. Language and motor findings in benign megalencephaly. Percept Mot Skills. 1989;68:1051-1054.
Li J., Simpson L., Takahashi M., et al. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58(24):5667-5672.
Loebstein R., Koren G. Pregnancy outcome and neurodevelopment of children exposed in utero to psychoactive drugs: the Motherisk experience. J Psychiatry Neurosci. 1997;22:192-196.
Lorber J., Priestly B.L. Children with large heads: a practical approach to diagnosis in 557 children, with special reference to 109 children with megalencephaly. Dev Med Child Neurol. 1981;23:494-504.
Lubis M.U., Tjipta G.D., Marbun M.D., et al. Cerebral palsy. Paediatr Indones. 1990;30:65-70.
Lucio-Eterovic A.K., Singh M.M., Gardner J.E., et al. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci USA. 2010;107(39):16952-16957.
Majewski F., Goecke T. Microcephalic osteodysplastic primordial dwarfism type II: report of three cases and review. Am J Med Genet. 1998;80:25-31.
Majewski F., Goecke T. Studies of microcephalic primordial dwarfism I: approach to a delineation of the Seckel syndrome. Am J Med Genet. 1982;12:7-21.
Majewski F., Ranke M., Schinzel A. Studies of microcephalic primordial dwarfism II: the osteodysplastic type II of primordial dwarfism. Am J Med Genet. 1982;12:23-35.
Malan V., Chevallier S., Soler G., et al. Array-based comparative genomic hybridization identifies a high frequency of copy number variations in patients with syndromic overgrowth. Eur J Hum Genet. 2010;18(2):227-232.
Manning B.D., Tee A.R., Logsdon M.N., et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151-162.
Marks N., Berg M.J. Recent advances on neuronal caspases in development and neurodegeneration. Neurochem Int. 1999;35(3):195-220. Sep
Marsh D.J., Kum J.B., Lunetta K.L., et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461-1472. Aug
Martin H.P. Microcephaly and mental retardation. Am J Dis Child. 1970;119:128-131.
Matsubara O., Tanaka M., Ida T., et al. Hemimegalencephaly with hemihypertrophy (Klippel-Trénaunay-Weber syndrome). Virchows Arch A Pathol Anat Histopathol. 1983;400(2):155-162.
Mazzeu J.F., Pardono E., Vianna-Morgante A.M., et al. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am J Med Genet A. 2007;143(4):320-325.
McBride K.L., Varga E.A., Pastore M.T., et al. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3(3):137-141.
Meikle L., Pollizzi K., Egnor A., et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC 1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28(21):5422-5432.
Meikle L., Talos D.M., Onda H., et al. A mouse model of tuberoussclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myclination, seizure activity, and limited survival. J Neurosci. 2007;27:5546-5558.
Meinecke P., Schaefer E., Wiedemann H.R. Microcephalic osteodysplastic primordial dwarfism: further evidence for identity of the so-called types I and III. Am J Med Genet. 1991;39:232-236.
Mendicino A., Sabbadini G., Pergola M.S. Clark-Baraitser syndrome: report of a new case and review of the literature. Clin Dysmorphol. 2005;14(3):133-135.
Mignot C., Boespflug-Tanguy O., Gelof A., et al. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell Mol Life Sci. 2004;61:369-385.
Miles J.H., Hadden L.L., Takahashi T.N., et al. Head circumference is an independent clinical finding associated with autism. Am J Med Genet. 2000;95(4):339-350.
Mirzaa G., Dodge N.N., Glass I., et al. Megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus: a rare brain malformation syndrome associated with mental retardation and seizures. Neuropediatrics. 2004;35(6):353-359.
Moore C.A., Toriello H.V., Abuelo D.N., et al. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet. 1997;70(1):67-73.
Moretti-Ferreira D., Koiffmann C.P., Listik M., et al. Macrosomia, obesity, macrocephaly and ocular abnormalities (MOMO syndrome) in two unrelated patients: delineation of a newly recognized overgrowth syndrome. Am J Med Genet. 1993;46(5):555-558.
Morrow J.D., Whitman B.Y., Accardo P.J. Autistic disorder in Sotos syndrome: a case report. Eur J Pediatr. 1990;149(8):567-569.
Nagamani S.C., Zhang F., Shchelochkov O.A., et al. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J Med Genet. 2009;46(12):825-833.
Najm J., Horn D., Wimplinger I., et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet. 2008;40:1065-1067.
Nelson K.B., Deutschberger J. Head size at one year as a predictor of four-year IQ. Dev Med Child Neurol. 1970;12:487-495.
Neri G., Gurrieri F., Zanni G., et al. Clinical and molecular aspects of the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1998;79(4):279-283. Oct 2
Nicholas A.K., Khurshid M., Desir J., et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet. 2010;42:1010-1014.
Nordlund M., Gu X., Shipley M.T., et al. Neurofibromin is enriched in the endoplasmic reticulum of CNS neurons. J Neurosci. 1993;13(4):1588-1600. Apr
Norman M., McGillivray B., Kalousek D., et al. Congenital malformations of the brain: pathological, embryological, clinical, radiological and genetic aspects. New York: Oxford University Press; 1995.
O’Connell E.J., Feldt R.H., Stickler G.B. Head circumference, mental retardation, and growth failure. Pediatrics. 1965;36:62-66.
O’Driscoll M., Jeggo P.A. Clinical impact of ATR checkpoint signalling failure in humans. Cell Cycle. 2003;2:194-195.
O’Driscoll M., Ruiz-Perez V.L., Woods C.G., et al. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497-501.
Ogawa S., Kwon C.H., Zhou J., et al. A seizure-prone phenotype is associated with altered free-running rhythm in Pten mutant mice. Brain Res. 2007;1168:112-123.
Okajima K., Ito T., Wakita A., et al. Male siblings with dyserythropoiesis, microcephaly and intrauterine growth retardation. Clin Dysmorphol. 2002;11:107-111.
Opitz J.M., Holt M.C. Microcephaly: general considerations and aids to nosology. J Craniofac Genet Dev Biol. 1990;10:175-204.
Opitz J.M., Smith J.F., Santoro L. The FG syndromes (Online Mendelian Inheritance in Man 305450): perspective in 2008. Adv Pediatr. 2008;55:123-170.
Ouellette E.M., Rosett H.L., Rosman N.P., et al. Adverse effects on offspring of maternal alcohol abuse during pregnancy. N Engl J Med. 1977;297:528-530.
Ozawa H., Takayama C., Nishida A., et al. Pachygyria in a girl with microcephalic osteodysplastic primordial short stature type II. Brain Dev. 2005;27:237-240.
Pavone L., Curatolo P., Rizzo R., et al. Epidermal nevus syndrome: a neurologic variant with hemimegalencephaly, gyral malformation, mental retardation, seizures, and facial hemihypertrophy. Neurology. 1991;41(2[pt1]):266-271.
Pavone P., Parano E., Polizzi A., et al. Colobomatous microphthalmia, microcephaly with cerebellar hypoplasia: association or new syndrome? Am J Med Genet. 2000;92:278-280.
Peiffer A., Singh N., Leppert M., et al. Microcephaly with simplified gyral pattern in six related children. Am J Med Genet. 1999;84:137-144.
Peippo M., Koivisto A.M., Särkämö T., et al. PAK3 related mental disability: further characterization of the phenotype. Am J Med Genet A. 2007;143A(20):2406-2416.
Perlman M., Levin M., Wittels B. Syndrome of fetal gigantism, renal hamartomas, and nephroblastomatosis with Wilms’ tumor. Cancer. 1975;35(4):1212-1217.
Petersson S., Sandberg Nordqvist A., Schalling M., et al. The megencephaly mouse has disturbances in the insulin-like growth factor (IGF) system. Brain Res Mol Brain Res. 1999;72(1):80-88.
Petrij F., Giles R.H., Dauwerse H.G., et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376(6538):348-351.
Pharoah P.O. Prevalence and pathogenesis of congenital anomalies in cerebral palsy. Arch Dis Child Fetal Neonatal Ed. 2007;92:F489-F493.
Phelan M.C., Thomas G.R., Saul R.A., et al. Cytogenetic, biochemical, and molecular analyses of a 22q13 deletion. Am J Med Genet. 1992;43(5):872-876.
Pierre-Kahn A., Hirsch J.F., Renier D., et al. Hydrocephalus and achondroplasia. A study of 25 observations. Childs Brain. 1980;7(4):205-219.
Pilarski R., Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41(5):323-326.
Pisano T., Meloni M., Cianchetti C., et al. Megalencephaly, polymicrogyria, and hydrocephalus (MPPH) syndrome: a new case with syndactyly. J Child Neurol. 2008;23(8):916-918.
Platt M., Nash A. Benign familial megalencephaly. Pediatr Res. 1972;6:426.
Potter C.J., Pedraza L.G., Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658-665.
Priestley B.L., Lorber J. Ventricular size and intelligence in achondroplasia. Z Kinderchir. 1981;34:320-326.
Proud V.K., Braddock S.R., Cook L., et al. Weaver syndrome: autosomal dominant inheritance of the disorder. Am J Med Genet. 1998;79(4):305-310.
Pryor H.B., Thelander H. Abnormally small head size and intellect in children. J Pediatr. 1968;73:593-598.
Puffenberger E.G., Strauss K.A., Ramsey K.E., et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130(Pt 7):1929-1941.
Qazi Q.H., Reed T.E. A problem in diagnosis of primary versus secondary microcephaly. Clin Genet. 1973;4:46-52.
Quezada E., Gripp K.W. Costello syndrome and related disorders. Curr Opin Pediatr. 2007;19(6):636-644.
Rauch A., Trautmann U., Pfeiffer R.A. Clinical and molecular cytogenetic observations in three cases of “trisomy 12p syndrome”. Am J Med Genet. 1996;63(1):243-249.
Reichenbach H., Holland H., Dalitz E., et al. De novo complete trisomy 5p: clinical report and FISH studies. Am J Med Genet. 1999;85(5):447-451.
Rio M., Clech L., Amiel J., et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet. 2003;40(6):436-440.
Robain O., Lyon G. Familial micrencephalies due to cerebral malformation: clinical and anatomical review. Act Neuropathol (Berlin). 1972;20:96-109.
Roberts A., Allanson J., Jadico S.K., et al. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43(11):833-842.
Robertson S.P., Gattas M., Rogers M., et al. Macrocephaly–cutis marmorata telangiectatica congenita: report of five patients and a review of the literature. Clin Dysmorphol. 2000;9(1):1-9.
Roboz P. Microcephaly. Aust J Ment Retard. 1973;2:173-179.
Roche A.F., Mukherjee D., Guo S.M., et al. Head circumference reference data: birth to 18 years. Pediatrics. 1987;79:706-712.
Roessler E., Belloni E., Gaudenz K., et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357-360.
Roggenbuck J.A., Mendelsohn N.J., Tenenholz B., et al. Duplication of the distal long arm of chromosome 15: report of three new patients and review of the literature. Am J Med Genet A. 2004;126A(4):398-402.
Rosenberg M.J., Agarwala R., Bouffard G., et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat Genet. 2002;32:175-179.
Rouse B., Azen C., Koch R., et al. Maternal phenylketonuria collaborative study (MPKUCS) offspring: facial anomalies, malformations, and early neurological sequelae. Am J Med Genet. 1997;69:89-95.
Rutter M. Developmental catch-up, and deficit, following adoption after severe global early privation. English and Romanian Adoptees (ERA) Study Team. J Child Psychol Psychiatry. 1998;39:465-476.
Said S.M., Yeh T.L., Greenwood R.S., et al. MRI morphometric analysis and neuropsychological function in patients with neurofibromatosis. Neuroreport. 1996;7(12):1941-1944. Aug 12
Saillour Y., Zanni G., Des Portes V., et al. Mutations in the AP1S2 gene encoding the sigma 2 subunit of the adaptor protein 1 complex are associated with syndromic X-linked mental retardation with hydrocephalus and calcifications in basal ganglia. J Med Genet. 2007;44(11):739-744.
Salen G., Shefer S., Batta A.K., et al. Abnormal cholesterol biosynthesis in the Smith-Lemli-Opitz syndrome. J Lipid Res. 1996;37(6):1169-1180.
Schaefer G.B., Bodensteiner J.B., Buehler B.A., et al. The neuroimaging findings in Sotos syndrome. Am J Med Genet. 1997;68(4):462-465.
Schinzel A. Postaxial polydactyly, hallux duplication, absence of the corpus callosum, macrencephaly and severe mental retardation: a new syndrome? Helv Paediatr Acta. 1979;34(2):141-146. May
Schinzel A., Schmid W. Hallux duplication; postaxial polydactyly, absence of the corpus callosum, severe mental retardation, and additional anomalies in two unrelated patients: a new syndrome. Am J Med Genet. 1980;6(3):241-249.
Schreier H., Rapin I., Davis J. Familial megalencephaly or hydrocephalus? Neurology. 1974;24(3):232-236.
Schwartz C.E., Tarpey P.S., Lubs H.A., et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet. 2007;44(7):472-477.
Scott R.E. Ataxia-telangiectasia. Its association with megalencephaly and mesenteric lymphangiectasia. Arch Pathol. 1969;88(1):78-84.
Seckel H. Bird-headed dwarfs: studies in developmental anthropology including human proportions. Springfield, IL: Charles C Thomas; 1960.
Sells C.J. Microcephaly in a normal school population. Pediatrics. 1977;59:262-265.
Shanske A., Caride D.G., Menasse-Palmer L., et al. Central nervous system anomalies in Seckel syndrome: report of a new family and review of the literature. Am J Med Genet. 1997;70:155-158.
Sharma S., Sankhyan N., Kabra M., et al. Hypomelanosis of Ito with hemimegalencephaly. Dermatol Online J. 2009;15(11):12.
Sheen V.L., Ganesh V.S., Topcu M., et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36(1):69-76.
Shen J., Gilmore E.C., Marshall C.A., et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. 2010;42(3):245-249.
Shevell M.I., Majnemer A., Morin I. Etiologic yield of cerebral palsy: a contemporary case series. Pediatr Neurol. 2003;28:352-359.
Sigaudy S., Toutain A., Moncla A., et al. Microcephalic osteodysplastic primordial dwarfism Taybi-Linder type: report of four cases and review of the literature. Am J Med Genet. 1998;80:16-24.
Simpson J.L., Landey S., New M., et al. A previously unrecognized X-linked syndrome of dysmorphia. Birth Defects Orig Artic Ser. 1975;11(2):18-24.
Simpson L., Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264(1):29-41.
Singhal B.S., Gorospe J.R., Naidu S. Megalencephalic lenkoencephalopathy with subcortical cysts. J Child Neurol. 2003;18:646-652.
Smigiel R., Pilch J., Makowska I., et al. The Pallister-Killian syndrome in a child with rare karyotype – a diagnostic problem. Eur J Pediatr. 2008;167(9):1063-1065.
Smith R.D. Abnormal head circumference in learning-disabled children. Develop Med Child Neurol. 1981;23:626-632.
Solomon B.D., Lacbawan F., Mercier S., et al. Mutations in ZIC2 in human holoprosencephaly: description of a Novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J Med Genet. 2010;47:513-524.
Sparks B.F., Friedman S.D., Shaw D.W., et al. Brain structural abnormalities in young children with autism spectrum disorder. Neurology. 2002;59(2):184-192.
Spohr H.L., Willms J., Steinhausen H.C. Prenatal alcohol exposure and long-term developmental consequences. Lancet. 1993;341:907-910.
Steen R.G., Taylor J.S., Langston J.W., et al. Prospective evaluation of the brain in asymptomatic children with neurofibromatosis type 1: relationship of macrocephaly to T1 relaxation changes and structural brain abnormalities. Am J Neuroradiol. 2001;22(5):810-817.
Stevenson R.E., Schroer R.J., Skinner C., et al. Autism and macrocephaly. Lancet. 1997;349(9067):1744-1745.
Stoll C., Alembik Y., Dott B., et al. Congenital eye malformations in 212,479 consecutive births. Ann Genet. 1997;40:122-128.
Strømme P., Andersen W. Developmental aspects in apple peel intestinal atresia-ocular anomalies-microcephaly syndrome. Clin Genet. 1997;52(2):133.
Sugimoto T., Yasuhara A., Nishida N., et al. MRI of the head in the evaluation of microcephaly. Neuropediatrics. 1993;24:4-7.
Sugio Y., Tsukahara M., Kajii T. Two Japanese cases with microcephalic primordial dwarfism: classical Seckel syndrome and osteodysplastic primordial dwarfism type II. Jpn J Hum Genet. 1993;38:209-217.
Suzuki J., Ito M., Tomiwa K., et al. A clinical study of cerebral palsy in Shiga; 1977–1986–II. Severity of the disability and complications in various types of cerebral palsy. No To Hattatsu. 1999;31:336-342.
Sztriha L., Al-Gazali L.I., Varady E., et al. Autosomal recessive micrencephaly with simplified gyral pattern, abnormal myelination and arthrogryposis. Neuropediatrics. 1999;30:141-145.
Sztriha L., Al-Gazali L., Varady E., et al. microlissencephaly. Pediatr Neurol. 1998;18:362-365.
Tabolacci E., Zollino M., Lecce R., et al. Two brothers with 22q13 deletion syndrome and features suggestive of the Clark-Baraitser syndrome. Clin Dysmorphol. 2005;14(3):127-132.
Takashima S., Chan F., Becker L.E., et al. Aberrant neuronal development in hemimegalencephaly: immunohistochemical and Golgi studies. Pediatr Neurol. 1991;7(4):275-280.
Tatton-Brown K., Douglas J., Coleman K., et al. Childhood Overgrowth Collaboration. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005;77(2):193-204.
Tavazoie S.F., Alvarez V.A., Ridenour D.A., et al. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8(12):1727-1734.
Taybi H. Microcephalic osteodysplastic primordial dwarfism and cephalo-skeletal dysplasia (Taybi-Linder syndrome). Am J Med Genet. 1992;43:628-629.
ten Donkelaar H.J., Wesseling P., Semmekrot B.A., et al. Severe, non X-linked congenital microcephaly with absence of the pyramidal tracts in two siblings. Acta Neuropathol (Berl). 1999;98:203-211.
Thong M.K., Thompson E., Keenan R., et al. A child with hemimegalencephaly, hemihypertrophy, macrocephaly, cutaneous vascular malformation, psychomotor retardation intestinal lymphangiectasia – a diagnostic dilemma. Clin Dysmorphol. 1999;8(4):283-286.
Thurel R., Gruner J.E. (Monakow-type microcephaly). Rev Neurol (Paris). 1960;102:98-99.
Tohyama J., Akasaka N., Saito N., et al. Megalencephaly and polymicrogyria with polydactyly syndrome. Pediatr Neurol. 2007;37(2):148-151.
Tolmie J., McNay M., Stephenson J., et al. Microcephaly: genetic counseling and antenatal diagnosis after the birth of an affected child. Am J Med Genet. 1987;27:583-594.
Tonsgard J.H. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol. 2006;13(1):2-7.
Torregrosa A., Martí-Bonmatí L., Higueras V., et al. Klippel-Trenaunay syndrome: frequency of cerebral and cerebellar hemihypertrophy on MRI. Neuroradiology. 2000;42(6):420-423.
Trimborn M., Richter R., Sternberg N., et al. The first missense alteration in the MCPH1 gene causes autosomal recessive microcephaly with an extremely mild cellular and clinical phenotype. Hum Mutat. 2005;26(5):496.
Varga E.A., Pastore M., Prior T., et al. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11(2):111-117.
Vargas J.E., Allred E.N., Leviton A., et al. Congenital microcephaly: phenotypic features in a consecutive sample of newborn infants. J Pediatr. 2001;139:210-214.
Vigevano F., Bertini E., Boldrini R., et al. Hemimegalencephaly and intractable epilepsy: benefits of hemispherectomy. Epilepsia. 1989;30(6):833-843.
Vogels A., Devriendt K., Legius E., et al. The macrocephaly-cutis marmorata telangiectatica congenita syndrome. Long-term follow-up data in 4 children and adolescents. Genet Couns. 1998;9(4):245-253.
Volpe J.J. Overview: normal and abnormal human brain development. Ment Retard Dev Disabil Res Rev. 2000;6(1):1-5.
Walenkamp M.J., Wit J.M. Genetic disorders in the GH IGF-I axis in mouse and man. Eur J Endocrinol. 2007;157(Suppl 1):S15-S26.
Watemberg N., Silver S., Harel S., et al. Significance of microcephaly among children with developmental disabilities. J Child Neurol. 2002;17:117-122.
Weaver D.D., Graham C.B., Thomas I.T., et al. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr. 1974;84(4):547-552.
Weng L., Brown J., Eng C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum Mol Genet. 2001;10(3):237-242.
Weng L.P., Brown J.L., Baker K.M., et al. PTEN blocks insulin-mediated ETS-2 phosphorylation through MAP kinase, independently of the phosphoinositide 3-kinase pathway. Hum Mol Genet. 2002;11(15):1687-1696.
Weng L.P., Smith W.M., Dahia P.L., et al. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59(22):5808-5814.
Wiedemann H.R., Burgio G.R., Aldenhoff P., et al. The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr. 1983;140(1):5-12.
Williams C.A., Driscoll D.J., Dagli A.I. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385-395.
Wisniewski K., Dambska M., Sher J.H., et al. A clinical neuropathological study of the fetal alcohol syndrome. Neuropediatrics. 1983;14:197-201.
Wiznitzer M., Rapin I., Van de Water T.R. Neurologic findings in children with ear malformations. Int J Pediatr Otorhinolaryngol. 1987;13:41-55.
Woodhouse W., Bailey A., Rutter M., et al. Head circumference in autism and other pervasive developmental disorders. J Child Psychol Psychiatry. 1996;37(6):665-671.
Wood J.W., Johnson K.G., Omori Y. In utero exposure to the Hiroshima atomic bomb. An evaluation of head size and mental retardation: twenty years later. Pediatrics. 1967;39:385-392.
Woods C.G., Bond J., Enard W. Autosomal Recessive Primary Microcephaly (MCPH): A Review of Clinical, Molecular, and Evolutionary Findings. Am J Hum Genet. 2005;76:717-728.
Yoshida H., Kong Y.Y., Yoshida R., et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94(6):739-750.
Young I.D., Barrow M., Hall C.M. Microcephalic osteodysplastic primordial short stature type II with cafe-au-lait spots and moyamoya disease: another patient. Am J Med Genet. 2004;127A:218-220.
Yu T.W., Mochida G.H., Tischfield D.J., et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet. 2010;42:1015-1020.
Zhang G., Assadi A.H., McNeil R.S., et al. The Pafah1b complex interacts with the reelin receptor VLDLR. PLoS ONE. 2007;2(2):e252.
Zhou J., Blundell J., Ogawa S., et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29(6):1773-1783.
Zweier C., de Jong E.K., Zweier M., et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85:655-666.