Chapter 433 Diseases of the Myocardium
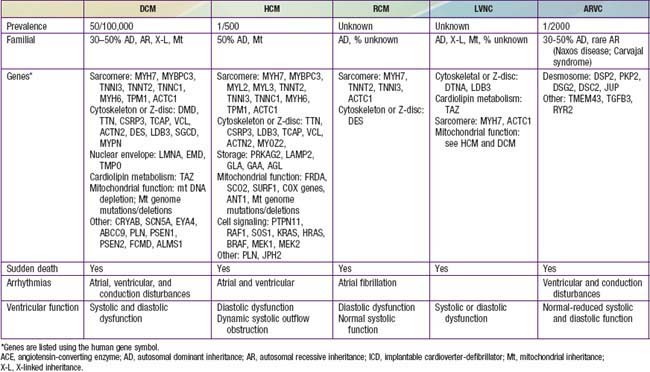
Table 433-1 classifies the cardiomyopathies based on their anatomic (ventricular morphology) and functional pathophysiology. Dilated cardiomyopathy, the most common form of cardiomyopathy, is characterized predominantly by left ventricular dilation and decreased left ventricular systolic function (Fig. 433-1). Hypertrophic cardiomyopathy demonstrates increased ventricular myocardial wall thickness, normal or increased systolic function, and often, diastolic (relaxation) abnormalities (Table 433-2). Restrictive cardiomyopathy is characterized by nearly normal ventricular chamber size and wall thickness with preserved systolic function, but dramatically impaired diastolic function leading to elevated filling pressures and atrial enlargement (see Fig. 433-3). Arrhythmogenic right ventricular cardiomyopathy and left ventricular non-compaction are characterized by specific morphologic abnormalities and heterogeneous functional disturbances.
Table 433-1 ETIOLOGY OF PEDIATRIC MYOCARDIAL DISEASE
| CARDIOMYOPATHY | |
| Dilated Cardiomyopathy (DCM) | |
| Neuromuscular diseases | Muscular dystrophies (Duchenne, Becker, limb girdle, Emery- Dreifuss, congenital muscular dystrophy, etc.), myotonic dystrophy, myofibrillar myopathy |
| Inborn errors of metabolism | Fatty acid oxidation disorders (trifunctional protein, VLCAD), carnitine abnormalities (carnitine transport, CPTI, CPTII), mitochondrial disorders (including Kearns-Sayre syndrome), organic acidemias (propionic acidemia) |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic DCM |
| Genetic syndromes | Alstrom syndrome, Barth syndrome (phospholipid disorders) |
| Ischemic | Most common in adults |
| Chronic tachyarrhythmias | |
| Hypertrophic Cardiomyopathy (HCM) | |
| Inborn errors of metabolism | Mitochondrial disorders (including Friedreich ataxia, mutations in nuclear or mitochondrial genome), storage disorders (glycogen storage disorders, especially Pompe; mucopolysaccharidoses; Fabry disease; sphingolipidoses; hemochromatosis; Danon disease) |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic HCM |
| Genetic syndromes | Noonan, Costello, cardio-faciocutaneous, Beckwith-Wiedemann syndrome |
| Infant of a diabetic mother | Transient hypertrophy |
| Restrictive Cardiomyopathy (RCM) | |
| Neuromuscular disease | Myofibrillar myopathies |
| Metabolic | Storage disorders |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic RCM |
| Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) | |
| Genetic mutations in cardiomyocyte structural apparatus | Familial or sporadic ARVC |
| Left Ventricular Noncompaction (LVNC) | X-linked (Barth syndrome), autosomal dominant, autosomal recessive, mitochondrial inheritance, or sporadic LVNC Sporadic LVNC |
| SECONDARY OR ACQUIRED MYOCARDIAL DISEASE | |
| Myocarditis |
Viral: parvovirus B19, adenovirus, coxsackievirus A and B, echovirus, rubella, varicella, influenza, mumps, Epstein-Barr virus, cytomegalovirus, measles, poliomyelitis, smallpox vaccine, hepatitis C virus, HIV virus, or opportunistic infections
Bacterial: diphtheria, mycoplasma, meningococcus, leptospirosis, Lyme disease, typhoid fever, tuberculosis, streptococcus, listeriosis
|
| Systemic Inflammatory Disease | Systemic lupus erythematosus (SLE), infant of mother with SLE, scleroderma, Churg-Strauss vasculitis, rheumatoid arthritis, rheumatic fever, sarcoidosis, dermatomyositis, periarteritis nodosa, hypereosinophilic syndrome (Löffler syndrome), acute eosinophilic necrotizing myocarditis, giant cell myocarditis |
| Nutritional Deficiency | Beriberi (thiamine deficiency), kwashiorkor, Keshan disease (selenium deficiency) |
| Drugs, Toxins | Doxorubicin (Adriamycin), cyclophosphamide, chloroquine, ipecac (emetine), sulfonamides, mesalazine, chloramphenicol, alcohol, hypersensitivity reaction, envenomations, irradiation, herbal remedy (blue cohosh) |
| Coronary Artery Disease | Kawasaki disease, medial necrosis, anomalous left coronary artery from the pulmonary artery (ALCAPA), other congenital coronary anomalies (anomalous right coronary, coronary ostial stenosis), familial hypercholesterolemia |
| Hematology-Oncology | Anemia, sickle cell disease, leukemia |
| Endocrine-Neuroendocrine | Hyperthyroidism, carcinoid tumor, pheochromocytoma |
Bibliography
Judge DP: Use of genetics in the clinical evaluation of cardiomyopathy, JAMA 302:2471–2476, 2009.
433.1 Dilated Cardiomyopathy
Etiology and Epidemiology
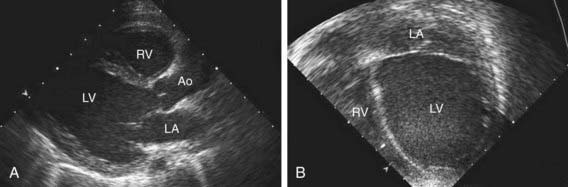
Dilated cardiomyopathy (DCM), the most common form of cardiomyopathy in children, is the cause of significant morbidity and mortality as well as a common indication for cardiac transplantation. The etiologies are diverse. Unlike adult patients with dilated cardiomyopathy, ischemic etiologies are rare in children, although these include anomalous origin of the left coronary artery from the pulmonary artery, premature coronary atherosclerosis (homozygous type II hypercholesterolemia), and coronary inflammatory diseases, such as Kawasaki disease. It is estimated that up to 50% of cases are genetic, including some with metabolic causes (see Table 433-1). Although the most common etiology of dilated cardiomyopathy remains idiopathic, it is likely that undiagnosed familial/genetic conditions and myocarditis predominate. The annual incidence of DCM in children younger than 18 yr is 0.57 cases per 100,000 per year. Incidence is higher in males, African Americans, and in infants less than 1 yr old.
Pathogenesis
In 20-50% of cases, the DCM is familial with autosomal dominant inheritance most common (see Table 433-2). Duchenne and Becker muscular dystrophies (Chapter 601.1) are X-linked cardiomyopathies that account for 5-10% of familial dilated cardiomyopathy cases. These dystrophinopathies result in an abnormal sarcomere-cytoskeleton connection, causing impaired myocardial force generation, myocyte damage/scarring, chamber enlargement, and altered function.
Laboratory Findings
Electrocardiographic screening reveals atrial or ventricular hypertrophy, nonspecific T-wave abnormalities, and occasionally atrial or ventricular arrhythmias. The chest x-ray demonstrates cardiomegaly and may reveal pulmonary vascular prominence or pleural effusions. The echocardiogram is often diagnostic, demonstrating the characteristic findings of left ventricular enlargement, decreased ventricular contractility, and occasionally a globular (remodeled) left ventricular contour (see Fig. 433-1). Right ventricular enlargement and depressed function are occasionally noted. Echo Doppler studies can reveal evidence of pulmonary hypertension, mitral regurgitation, or other structural cardiac or coronary abnormalities.
Additional testing should include CBC, renal and liver function tests, CPK, cardiac troponin I, lactate, plasma amino acids, urine organic acids, and an acylcarnitine profile. Additional genetic and enzymatic testing may be useful (see Table 433-2). Cardiac catheterization and endomyocardial biopsy are not routine but may be useful in patients with acute dilated cardiomyopathy. Biopsy samples can also be assessed for the presence of mononuclear cell infiltrates, myocardial damage, storage abnormalities, and viral infection or genomes. It is important to consider screening of 1st-degree family members utilizing echocardiography and ECG.
Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969-981.
Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752-760.
Mordente A, Meucci E, Silvestrini A, et al. New developments in anthracycline-induced cardiotoxicity. Curr Med Chem. 2009;16:1656-1672.
Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867-1876.
Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57:78R-86R.
433.2 Hypertrophic Cardiomyopathy
Etiology and Epidemiology
Hypertrophic cardiomyopathy (HCM) is a heterogeneous, relatively common, and potentially severe form of cardiomyopathy. The causes of hypertrophic cardiomyopathy are heterogeneous and include inborn errors of metabolism, neuromuscular disorders, syndromic conditions, and genetic abnormalities of the structural components of the cardiomyocyte (see Table 433-1). Both the age of onset and associated features are helpful in identifying the underlying etiology.
HCM is a genetic disorder and frequently occurs as a result of mutations in sarcomere or cytoskeletal components of the cardiomyocyte. Mutations of the genes encoding cardiac β-myosin heavy-chain (MYH7) and myosin-binding protein C (MYBPC3) are the most common (see Table 433-2). Mutations are inherited in an autosomal dominant pattern with widely variable penetrance; many cases represent de novo mutations. Some patients have mutations in more than 1 gene; this may result in early onset and more severe symptoms. Additional genetic causes for HCM include nonsarcomeric protein mutations, such as the γ-2-regulatory subunit of AMP-activated protein kinase (PRKAG2) and the lysosome-associated membrane protein 2α-galactosidase (Danon disease, a form of glycogen storage disease). Syndromic conditions, such as Noonan syndrome, may manifest with hypertrophic cardiomyopathy at birth and recognition of extracardiac manifestations is important in making the diagnosis.
Diagnosis
The electrocardiogram typically demonstrates left ventricular hypertrophy with ST segment and T-wave abnormalities. Intraventricular conduction delays and signs of ventricular preexcitation (Wolff-Parkinson-White syndrome) may be present and should raise the possibility of Danon disease or Pompe disease. Chest radiography demonstrates normal or mildly increased heart size with a prominence of the left ventricle. Echocardiography is diagnostic in identifying, localizing, and quantifying the degree of myocardial hypertrophy (Fig. 433-2). Doppler interrogation defines, localizes, and quantifies the degree of ventricular outflow tract obstruction and also demonstrates and quantifies the degree of mitral insufficiency. Diastolic dysfunction can be confirmed by M-mode, flow, and tissue Doppler techniques.
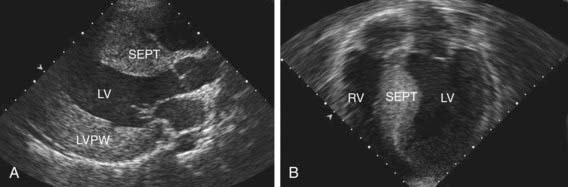
Additional diagnostic studies include metabolic testing, genetic testing for specific syndromic conditions, or genetic testing for mutations in genes known to cause isolated HCM (see Table 433-2). The clinical availability of these tests is expanding. In adults, where isolated HCM is a common genetic diagnosis, it has been possible to identify a subset of mutations that confer an increased risk for arrhythmia or sudden death. As identification of the molecular basis of disease in children increases, similar correlations are expected to emerge. In addition, genetic diagnosis is useful to identify at risk family members that require ongoing surveillance.
Colan SD, Lipshultz SE, Lowe AM, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115:773-781.
Ho CY, Lopez B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363(6):552-562.
Kaski JP, Esteban MTT, Lowe M, et al. Outcomes after implantable cardioverter-defibrillator treatment in children with hypertrophic cardiomyopathy. Heart. 2007;93:372-374.
Lupsa BC, Sachdev V, Lungu AO, et al. Cardiomyopathy in congenital and acquired generalized lipodystrophy. Medicine. 2010;89(4):245-250.
Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
Morita H, Rehm HL, Menesses A, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899-1908.
Nugent AW, Daubeney PE, Chondros P, et al. National Australian Childhood Cardiomyopathy Study. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation. 2005;112:1332-1338.
Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007-1012.
433.3 Restrictive Cardiomyopathy
Diagnosis
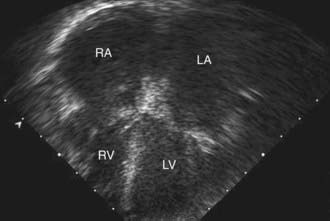
The characteristic electrocardiographic finding of prominent P waves is usually associated with normal QRS voltages and nonspecific ST and T-wave changes. Right ventricular hypertrophy occurs in patients with pulmonary hypertension. The chest x-ray may be normal or demonstrate a prominent atrial shadow and pulmonary vascular redistribution. The echocardiogram is often diagnostic, demonstrating normal-sized ventricles with preserved systolic function and dramatic enlargement of the atria (Fig. 433-3). Flow and tissue Doppler interrogation reveal abnormal filling parameters. Differential diagnosis from constrictive pericarditis is critical, as the latter can be treated surgically. Magnetic resonance imaging may be necessary to demonstrate the thickened or calcified pericardium often present in constrictive pericardial disease.
Kaski JP, Syrris P, Burch M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478-1484.
Rivenes SM, Kearney DL, Smith EO, et al. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation. 2000;102:876-882.
433.4 Left Ventricular Noncompaction, Arrhythmogenic Right Ventricular Cardiomyopathy, and Endocardial Fibroelastosis
Left ventricular noncompaction (LVNC) was initially believed to be a rare disorder found only in children, but is now known to affect individuals of all ages. LVNC is characterized by a distinctive trabeculated or spongy appearing left ventricle (Fig. 433-4) commonly associated with left ventricular hypertrophy and/or dilation, and at times, systolic or diastolic dysfunction. LVNC may be isolated or associated with structural congenital cardiac defects. Patients may present with signs of heart failure, arrhythmias, syncope, sudden death, or as an asymptomatic finding during screening of family members.
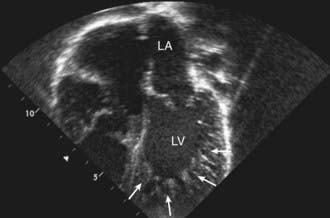
Figure 433-4 Echocardiogram of a patient with left ventricular noncompaction cardiomyopathy. Apical view showing the abnormal trabeculations of the left ventricle at the apex (arrows). For comparison, see the smooth walled LV in Fig. 433-1. LA, left atrium; LV, left ventricle.
Left Ventricular Noncompaction
Fazio G, Pipitone S, Iacona MA, et al. The noncompaction of the left ventricular myocardium: our paediatric experience. J Cardiovasc Med. 2007;8:904-908.
Klaassen S, Probst S, Oechslin E, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893-2901.
Murphy RT, Thaman R, Blanes JG, et al. Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J. 2005;26:187-192.
Pantazis AA, Elliott PM. Left ventricular noncompaction. Curr Opin Cardiol. 2009;24:209-213.
Pignatelli RH, McMahon CJ, Dreyer WJ, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108:2672-2678.
Arrhythmogenic Right Ventricular Cardiomyopathy
Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075-1084.
Basso C, Corrado D, Marcus FI, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289-1300.
Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823-3832.
Hulot JS, Jouven X, Empana JP, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;11:1879-1884.
433.5 Myocarditis
Acute or chronic inflammation of the myocardium is characterized by inflammatory cell infiltrates, myocyte necrosis, or myocyte degeneration and may be caused by infectious, connective tissue, granulomatous, toxic, or idiopathic processes. There may be associated systemic manifestations of the disease and on occasion the endocardium or pericardium is involved, though coronary pathology is uniformly absent. Patients may be asymptomatic, have nonspecific prodromal symptoms, or present with overt congestive heart failure, compromising arrhythmias, or sudden death. It is thought that viral infections are the most common etiology though myocardial toxins, drug exposures, hypersensitivity reactions, and immune disorders may also lead to myocarditis (Table 433-3).
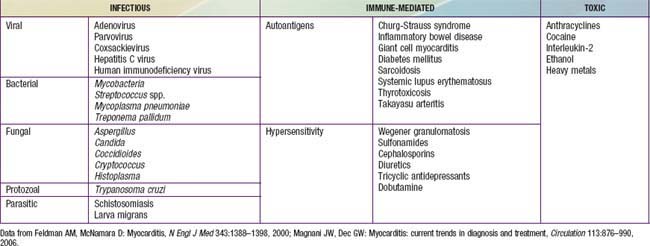
Etiology and Epidemiology
Bacterial Infections
Bacterial myocarditis has become far less common with the advent of advanced public health measures, which have minimized infectious causes such as diphtheria. Diphtheritic myocarditis (Chapter 180) is unique as bacterial toxin may produce circulatory collapse and toxic myocarditis characterized by atrioventricular block, bundle branch block, or ventricular ectopy. Any overwhelming systemic bacterial infection can manifest with circulatory collapse and shock with evidence of myocardial dysfunction characterized by tachycardia, gallop rhythm, and low cardiac output. Additional nonviral infectious causes of myocarditis include rickettsia, protozoa, parasitic infections, and fungal disease.
Differential Diagnosis
The predominant diseases mimicking acute myocarditis include carnitine deficiency, other metabolic disorders of energy generation, hereditary mitochondrial defects, idiopathic dilated cardiomyopathy, pericarditis, EFE, and anomalies of the coronary arteries (see Table 433-1).
Treatment
Primary therapy for acute myocarditis is supportive (Chapter 436). Acutely, the use of inotropic agents, preferably milrinone, should be entertained but used with caution because of their proarrhythmic potential. Diuretics are often required as well. If in extremis, mechanical ventilatory support and mechanical circulatory support with ventricular assist device implantation or ECMO may be needed to stabilize the patient’s hemodynamic status and act as a bridge to recovery or cardiac transplantation. Diuretics, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers are of use in patients with compensated congestive heart failure in the outpatient setting, but may be contraindicated in those presenting with fulminant heart failure and cardiovascular collapse. In patients manifesting with significant atrial or ventricular arrhythmias, specific antiarrhythmic agents (for example, amiodarone) should be administered and ICD placement considered.
Bowles NE, Ni J, Kearney DL, et al. Detection of viruses in myocardial tissues by polymerase chain reaction: evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42:466-472.
Cooper LT. Myocarditis. New Engl J Med. 2009;360:1526-1538.
Costello JM, Alexander ME, Greco KM, et al. Lyme carditis in children: presentation, predictive factors, and clinical course. Pediatrics. 2009;123:e835-e841.
Freedman SB, Haladyn JK, Floh A, et al. Pediatric myocarditis: emergency department clinical findings and diagnostic evaluation. Pediatrics. 2007;120:1278-1285.
Freund MW, Kleinveld G, Krediet TG, et al. Prognosis for neonates with enterovirus myocarditis. Arch Dis Child Fetal Neonatal Ed. 2010;95:F206-F212.
Hrobon P, Kuntz KM, Hare JM. Should endomyocardial biopsy be performed for detection of myocarditis? A decision analytic approach. J Heart Lung Transplant. 1998;17:479-486.
Pinkert S, Westermann D, Wang X, et al. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation. 2009;120:2358-2366.
Thanjan MT, Ramaswamy P, Lai WW, et al. Acute myopericarditis after multiple vaccinations in an adolescent: case report and review of the literature. Pediatrics. 2007;119:e1400-1403.
Weber MA, Ashworth MT, Risdon RA, et al. Clinicopathological features of paediatric deaths due to myocarditis: an autopsy series. Arch Dis Child. 2008;93:594-598.