[level-membership-for-pulmolory-and-respiratory-category]10
Diffuse Parenchymal Lung Diseases Associated with Known Etiologic Agents
This chapter focuses on several of the major categories of diffuse parenchymal (interstitial) lung disease for which an etiologic agent has been identified. The general principles discussed in Chapter 9 apply to most of these conditions, and the features emphasized here are those peculiar to or characteristic of each cause. Considering the vast number of diffuse parenchymal lung diseases, this chapter only scratches the surface of information available. When a physician is confronted with a patient having a particular type of diffuse parenchymal lung disease, it is best to relearn the details of the disease at that time.
Diseases Caused by Inhaled Inorganic Dusts
Silicosis
Although the pathogenesis of silicosis is not known with certainty, theories have focused on the potential toxicity of silica for macrophages. Silica particles in the lower respiratory tract are phagocytosed by alveolar macrophages. Freshly cut silica particles are more pathogenic than older particles. This property is thought to be due to the increased redox potential of the fresh surface, which is highly reactive. After engulfing the silica particle, the macrophage is activated and releases inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin-1, and arachidonic acid metabolites. Phagocytosis of silica particles leads to apoptotic cell death, and toxic silica particles are released that are capable of repeating the process after they are reingested by other macrophages. With their activation and destruction, the macrophages release chemical mediators that initiate or perpetuate an alveolitis, eventually leading to development of fibrosis. Pathologically, the inflammatory process initially is localized around the respiratory bronchioles but eventually becomes more diffuse throughout the parenchyma. The ongoing inflammatory process causes scarring and results in characteristic acellular nodules called silicotic nodules that are composed of connective tissue (Fig. 10-1). At first the nodules are small and discrete. With disease progression they become larger and may coalesce. Silicotic nodules are believed to be areas in which the cycle of macrophage ingestion, activation, destruction, and release of the toxic silica particles occurs.

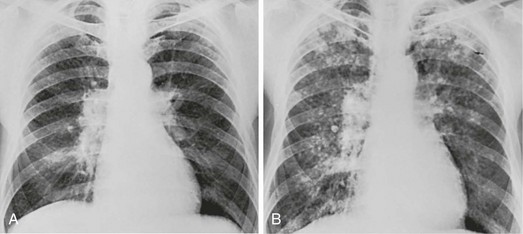
The most common radiographic appearance of silicosis is notable for small, rounded opacities or nodules. This pattern is described as simple chronic silicosis. Uncommonly, the nodules become larger and coalescent, in which case the pneumoconiosis is called complicated; the term progressive massive fibrosis has also been used (Fig. 10-2). As a general rule, in patients with silicosis the upper lung zones are affected more heavily than the lower zones. Enlargement of the hilar lymph nodes, which frequently calcify, may be seen.
In addition to the potential problem of progressive pulmonary involvement and eventual respiratory failure, abnormal immune regulation is associated with silicosis. Patients are at increased risk for certain autoimmune diseases including rheumatoid arthritis and systemic sclerosis. In addition, patients with silicosis are particularly susceptible to infections with mycobacteria, perhaps because of impaired macrophage function. The specific organisms may be either Mycobacterium tuberculosis, the etiologic agent for tuberculosis, or other species of mycobacteria, often called atypical or nontuberculous mycobacteria (see Chapter 24).
Coal Worker’s Pneumoconiosis
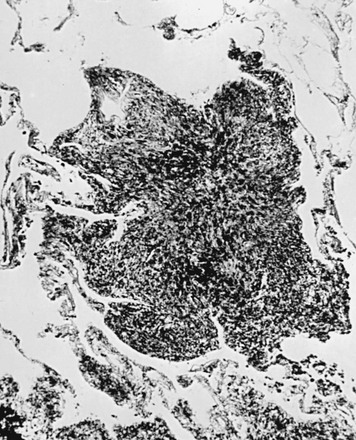
The pathologic hallmark of CWP is the coal macule, which is a focal collection of coal dust surrounded by relatively little cellular infiltration or fibrosis (Fig. 10-3). The initial lesions tend to be distributed primarily around respiratory bronchioles. Small associated regions of emphysema, termed focal emphysema, may be seen.
Asbestosis
Asbestos has been widely used because of its thermal and fire resistance. It is a fibrous derivative of silica, termed a fibrous silicate. It is a naturally occurring mineral that, because of its long narrow shape, can be woven into cloth. Among the health hazards it presents are the development of diffuse interstitial fibrosis, benign pleural plaques and effusions, and the potential for inducing several types of neoplasm, particularly bronchogenic carcinoma and mesothelioma. These latter problems are discussed in Chapters 15, 20, and 21. The term asbestosis should be reserved for the diffuse parenchymal lung disease that occurs as a result of asbestos exposure.
A characteristic finding of asbestos exposure is the ferruginous body, a rod-shaped body with clubbed ends (Fig. 10-4) that appears yellow-brown in stained tissue. Ferruginous bodies represent asbestos fibers that have been coated by macrophages with an iron-protein complex. Although large numbers of these structures are commonly seen by light microscopy in patients with asbestosis, not all such coated fibers are asbestos, and ferruginous bodies may be seen even in the absence of parenchymal lung disease. Uncoated asbestos fibers, which are long and narrow, cannot be seen by light microscopy and require electron microscopy for detection.
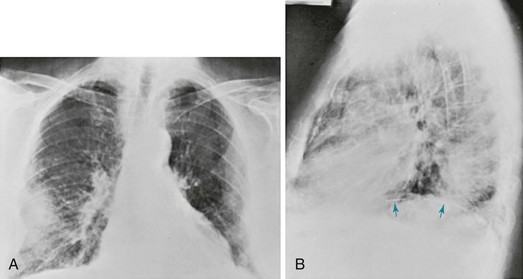
The chest radiograph in patients with asbestosis shows a pattern of linear streaking that is generally most prominent at the lung bases (Fig. 10-5). In advanced cases, the findings may be quite extensive and associated with cyst formation and honeycombing. Commonly there is evidence of associated pleural disease, either in the form of diffuse pleural thickening or localized plaques (which may be calcified) or, much less frequently, in the form of pleural effusions. Because asbestos is a predisposing factor in development of malignancies of the lung and pleura, either of these complications may be seen on the chest radiograph.
The clinical, pathophysiologic, and diagnostic features of asbestosis usually follow the general description of diffuse parenchymal lung disease discussed in Chapter 9. However, of the pneumoconioses already discussed, asbestosis is much more likely than either silicosis or CWP to be associated with clubbing of digits seen on physical examination.
Hypersensitivity Pneumonitis
Pathologic examination of the lung in patients with hypersensitivity pneumonitis reveals an alveolitis composed primarily of lymphocytes (especially cytotoxic/suppressor CD8+ cells) and macrophages, as well as the presence of granulomas. The granulomas often are loosely formed, unlike the well-defined granulomas characteristic of sarcoidosis (see Chapter 11). Often the pathologic changes have a peribronchiolar prominence, thus accounting for the frequent physiologic evidence for obstruction of small airways.
With an acute episode of hypersensitivity pneumonitis, the chest radiograph shows patchy or diffuse infiltrates. As the disease becomes chronic, the abnormality may take on a more nodular quality, eventually appearing as the reticulonodular pattern characteristic of the other chronic diffuse parenchymal lung diseases. In the chronic form of disease, an upper lobe predominance to the radiographic changes is often seen. High-resolution chest computed tomography (CT) scanning may be particularly helpful in suggesting the diagnosis, often demonstrating a mosaic ground-glass pattern (see Fig. 3-9).
Diseases Caused by Inhaled Inorganic Dusts
American Thoracic Society. Diagnosis and initial management of nonmalignant diseases related to asbestos. Am J Respir Crit Care Med. 2004;170:691–715.
Amicosante, M, Fontenot, AP. T cell recognition in chronic beryllium disease. Clin Immunol. 2006;121:134–143.
Antonescu-Turcu, AL, Schapira, RM. Parenchymal and airway diseases caused by asbestos. Curr Opin Pulm Med. 2010;16:155–161.
Cohen, R, Velho, V. Update on respiratory disease from coal mine and silica dust. Clin Chest Med. 2002;23:811–826.
Crosby, LM, Waters, CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;6:L715–L731.
Fontenot, AP, Amicosante, M. Metal-induced diffuse lung disease. Semin Respir Crit Care Med. 2008;29:662–669.
Glazer, CS, Newman, LS. Occupational interstitial lung disease. Clin Chest Med. 2004;25:467–478.
Hessel, PA, Gamble, JF, McDonald, JC. Asbestos, asbestosis, and lung cancer: a critical assessment of the epidemiological evidence. Thorax. 2005;60:433–436.
Jamrozik, E, de Klerk, N, Musk, AW. Asbestos-related disease. Intern Med J. 2011;41:372–380.
Kumagai-Takei, N, Maeda, M, Chen, Y, et al. Asbestos induces reduction of tumor immunity. Clin Dev Immunol. 481439, 2011.
Leung, CC, Yu, IT, Chen, W. Silicosis. Lancet. 2012;379:2008–2018.
Maeda, M, Nishimura, Y, Kumagai, N, et al. Dysregulation of the immune system caused by silica and asbestos. J Immunotoxicol. 2010;7:268–278.
McLeskey, TM, Buchner, V, Field, RW, et al. Recent advances in understanding the biomolecular basis of chronic beryllium disease: a review. Rev Environ Health. 2009;24:75–115.
Otsuki, T, Hayashi, H, Nishimura, Y, et al. Dysregulation of autoimmunity caused by silica exposure and alteration of Fas-mediated apoptosis in T lymphocyte derived from silicosis patients. Int J Immunopathol Pharmacol. 2011;24(1 Suppl):11S–16S.
Pipavath, SN, Godwin, JD, Kanne, JP. Occupational lung disease: a radiologic review. Semin Roentgenol. 2010;45:43–52.
Ross, MH, Murray, J. Occupational respiratory disease in mining. Occup Med (Lond). 2004;54:304–310.
Sawyer, RT, Maier, LA. Chronic beryllium disease: an updated model interaction between innate and acquired immunity. Biometals. 2011;24:1–17.
Yucesoy, B, Luster, MI. Genetic susceptibility in pneumoconiosis. Toxicol Lett. 2007;168:249–254.
Girard, M, Cormier, Y. Hypersensitivity pneumonitis. Curr Opin Allergy Clin Immunol. 2010;10:99–103.
Hirschmann, JV, Pipavath, SN, Godwin, JD. Hypersensitivity pneumonitis: a historical, clinical, and radiologic review. Radiographics. 2009;29:1921–1938.
Selman, M. Hypersensitivity pneumonitis: a multifaceted deceiving disorder. Clin Chest Med. 2004;25:531–547.
Selman, M, Lacasse, Y, Pardo, A, et al. Hypersensitivity pneumonitis caused by fungi. Proc Am Thorac Soc. 2010;7:229–236.
Selman, M, Pardo, A, King, TEJr. Hypersensitivity pneumonitis. Insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186:314–324.
Silva, CI, Churg, A, Müller, NL. Hypersensitivity pneumonitis: spectrum of high-resolution CT and pathologic findings. AJR Am J Roentgenol. 2007;188:334–344.
Drug-Induced Parenchymal Lung Disease
Camus, P, Bonniaud, P, Fanton, A, et al. Drug-induced and iatrogenic infiltrative lung disease. Clin Chest Med. 2004;25:479–519.
Camus, P, Martin, WJ, Rosenow, EC. Amiodarone pulmonary toxicity. Clin Chest Med. 2004;25:65–75.
Flieder, DB, Travis, WD. Pathologic characteristics of drug-induced lung disease. Clin Chest Med. 2004;25:37–45.
Limper, AH. Chemotherapy-induced lung disease. Clin Chest Med. 2004;25:53–64.
Lock, BJ, Eggert, M, Cooper, JA. Infiltrative lung disease due to noncytotoxic agents. Clin Chest Med. 2004;25:47–52.
Schwaiblmair, M, Berghaus, T, Haeckel, T, et al. Amiodarone-induced pulmonary toxicity: an under-recognized and severe adverse effect? Clin Res Cardiol. 2010;99:693–700.
Silva, CI, Müller, N. Drug-induced lung diseases: most common reaction patterns and corresponding high-resolution CT manifestations. Semin Ultrasound CT MR. 2006;27:111–116.
Sleijfer, S. Bleomycin-induced pneumonitis. Chest. 2001;120:617–624.
Radiation-Induced Lung Disease
Abratt, RP, Morgan, GW, Silvestri, G, et al. Pulmonary complications of radiation therapy. Clin Chest Med. 2004;25:167–177.
Cameron, EH, Crystal, RG. Radiation-induced lung injury. In: Crystal RG, West JB, Weibel ER, et al, eds. The lung: scientific foundations. ed 2. Philadelphia: Lippincott-Raven; 1997:2647–2651.
Ghafoori, P, Marks, LB, Vujaskovic, Z, et al. Radiation-induced lung injury. Assessment, management, and prevention, Oncology (Williston Park). 2008;22:37–47. discussion 52-3
Graves, PR, Siddiqui, F, Anscher, MS, et al. Radiation pulmonary toxicity: from mechanisms to management. Semin Radiat Oncol. 2010;20:201–207.
Tsoutsou, PG, Koukourakis, ML. Radiation pneumonitis and fibrosis: mechanisms underlying its pathogenesis and implications for future research. Int J Radiat Oncol Biol Phys. 2006;66:1281–1293.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]10
Diffuse Parenchymal Lung Diseases Associated with Known Etiologic Agents
This chapter focuses on several of the major categories of diffuse parenchymal (interstitial) lung disease for which an etiologic agent has been identified. The general principles discussed in Chapter 9 apply to most of these conditions, and the features emphasized here are those peculiar to or characteristic of each cause. Considering the vast number of diffuse parenchymal lung diseases, this chapter only scratches the surface of information available. When a physician is confronted with a patient having a particular type of diffuse parenchymal lung disease, it is best to relearn the details of the disease at that time.
Diseases Caused by Inhaled Inorganic Dusts
Silicosis
Although the pathogenesis of silicosis is not known with certainty, theories have focused on the potential toxicity of silica for macrophages. Silica particles in the lower respiratory tract are phagocytosed by alveolar macrophages. Freshly cut silica particles are more pathogenic than older particles. This property is thought to be due to the increased redox potential of the fresh surface, which is highly reactive. After engulfing the silica particle, the macrophage is activated and releases inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin-1, and arachidonic acid metabolites. Phagocytosis of silica particles leads to apoptotic cell death, and toxic silica particles are released that are capable of repeating the process after they are reingested by other macrophages. With their activation and destruction, the macrophages release chemical mediators that initiate or perpetuate an alveolitis, eventually leading to development of fibrosis. Pathologically, the inflammatory process initially is localized around the respiratory bronchioles but eventually becomes more diffuse throughout the parenchyma. The ongoing inflammatory process causes scarring and results in characteristic acellular nodules called silicotic nodules that are composed of connective tissue (Fig. 10-1). At first the nodules are small and discrete. With disease progression they become larger and may coalesce. Silicotic nodules are believed to be areas in which the cycle of macrophage ingestion, activation, destruction, and release of the toxic silica particles occurs.
The most common radiographic appearance of silicosis is notable for small, rounded opacities or nodules. This pattern is described as simple chronic silicosis. Uncommonly, the nodules become larger and coalescent, in which case the pneumoconiosis is called complicated; the term progressive massive fibrosis has also been used (Fig. 10-2). As a general rule, in patients with silicosis the upper lung zones are affected more heavily than the lower zones. Enlargement of the hilar lymph nodes, which frequently calcify, may be seen.
In addition to the potential problem of progressive pulmonary involvement and eventual respiratory failure, abnormal immune regulation is associated with silicosis. Patients are at increased risk for certain autoimmune diseases including rheumatoid arthritis and systemic sclerosis. In addition, patients with silicosis are particularly susceptible to infections with mycobacteria, perhaps because of impaired macrophage function. The specific organisms may be either Mycobacterium tuberculosis, the etiologic agent for tuberculosis, or other species of mycobacteria, often called atypical or nontuberculous mycobacteria (see Chapter 24).
Coal Worker’s Pneumoconiosis
The pathologic hallmark of CWP is the coal macule, which is a focal collection of coal dust surrounded by relatively little cellular infiltration or fibrosis (Fig. 10-3). The initial lesions tend to be distributed primarily around respiratory bronchioles. Small associated regions of emphysema, termed focal emphysema, may be seen.
Asbestosis
Asbestos has been widely used because of its thermal and fire resistance. It is a fibrous derivative of silica, termed a fibrous silicate. It is a naturally occurring mineral that, because of its long narrow shape, can be woven into cloth. Among the health hazards it presents are the development of diffuse interstitial fibrosis, benign pleural plaques and effusions, and the potential for inducing several types of neoplasm, particularly bronchogenic carcinoma and mesothelioma. These latter problems are discussed in Chapters 15, 20, and 21. The term asbestosis should be reserved for the diffuse parenchymal lung disease that occurs as a result of asbestos exposure.
A characteristic finding of asbestos exposure is the ferruginous body, a rod-shaped body with clubbed ends (Fig. 10-4) that appears yellow-brown in stained tissue. Ferruginous bodies represent asbestos fibers that have been coated by macrophages with an iron-protein complex. Although large numbers of these structures are commonly seen by light microscopy in patients with asbestosis, not all such coated fibers are asbestos, and ferruginous bodies may be seen even in the absence of parenchymal lung disease. Uncoated asbestos fibers, which are long and narrow, cannot be seen by light microscopy and require electron microscopy for detection.

[/not-level-membership-for-pulmolory-and-respiratory-category]
