Chapter 46 Diabetic Retinopathy
Genetics and Etiologic Mechanisms
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
Diabetic retinopathy is the leading cause of blindness among individuals between 25 and 74 years of age in the industrialized world. It affects three out of four diabetic patients after 15 years of disease duration. Chronic hyperglycemia is the primary factor leading to the development of diabetic retinopathy and other complications of the disease. The importance of long-term glycemic control has been conclusively established in the landmark clinical trials including the Diabetes Control and Complications Trial (DCCT),1 and the UK Prospective Diabetes Study (UKPDS).2,3 However, the mechanisms by which elevated blood sugar levels lead to the development of diabetic retinopathy and the anatomic changes visible histopathologically remain to be fully elucidated. This chapter will review the biochemical and molecular pathways believed to be responsible for the development of diabetic retinopathy and will highlight recent developments in genetics that provide insight into the influential role of genetic susceptibility.
Anatomic lesions
Loss of pericytes
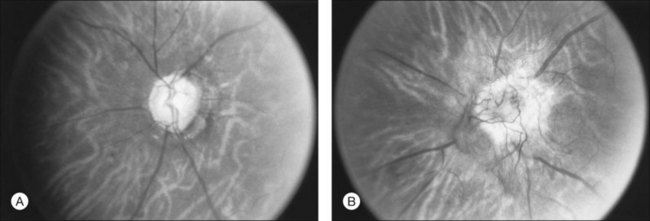
Loss of pericytes is one of the earliest and most specific signs of diabetic retinopathy. This finding was first described by Cogan, Kuwabara and coworkers after examining trypsin-digested retinal vasculature flat mounts from diabetic human subjects.4–6 Since their initial report, their findings have been confirmed by various investigators.2,7,8 In humans and canines, trypsin digestion of retinal vasculature flat mounts reveals the loss of pericytes as evidenced by the development of pericyte ghosts, empty aneurysmal spaces bulging from the capillary walls that lack the darkly staining nucleus of a viable pericyte (Fig. 46.1A). Pericytes are normally identifiable in these spaces by their nuclei which stain darkly and are spaced regularly along the capillary wall, producing the appearance of “bumps on a log” (Fig. 46.1B).
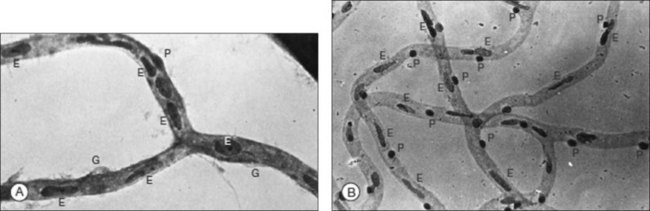
Pericytes are contractile cells that play an important role in microvascular autoregulation.9 Loss of pericytes leads to alterations of vascular intercellular contacts and impairment of the inner blood–retina barrier. These effects result in the venous dilation and beading that is visible clinically. Loss of intercellular contacts also appears to promote endothelial cell proliferation resulting in the development of microaneurysms.10 Loss of pericytes appears to be especially significant in the development of diabetic retinopathy, although pericyte loss has also been reported in diabetic peripheral neuropathy leading to neuronal ischemia.11 The mechanism by which hyperglycemia leads to pericyte degeneration remains largely unknown. The two leading hypotheses implicate the aldose reductase pathway and platelet-derived growth factor-beta (PDGF-β).
Akagi et al. reported the localization of the enzyme aldose reductase in retinal capillary pericytes but not in endothelial cells in human specimens using immunohistochemistry techniques.12 These findings would be consistent with the specific loss of capillary pericytes observed in diabetic retinopathy and other microvascular complications of diabetes.13 Two other groups, however, were unable to identify the presence of aldose reductase in rodent and canine retinal capillaries using immunohistochemistry techniques.14,15 In addition, a third group reported the presence of aldose reductase activity in cultured bovine retinal capillary pericytes as well as retinal capillary endothelial cells. They also found aldose reductase activity in cultured monkey retinal pericytes.16 These conflicting reports probably result from species-specific differences in aldose reductase expression and highlight the need to use caution when interpreting animal models of disease.
PDGF-β has been found to be critical in the recruitment of pericytes in the vasculature of various tissues and organs.17 It is well documented that endothelial cells express PDGF-β,18–21 and in vitro pericytes are known to express PDGF-β receptors and respond to PDGF-β.22,23 Lindahl et al. reported that in the PDGF-β-deficient mouse model, pericytes fail to develop in developing capillaries during angiogenesis.24 Subsequent studies using PDGF-β- and PDGF receptor-β (PDGFR-β)-deficient mice found that while PDGF-β/PDGFR-β-independent induction of pericyte precursors may occur, the expansion of pericytes is dependent on an intact PDGF-β/PDGFR-β paracrine signaling pathway.25 Ablation of either PDGF-β or PDGFR-β led to identical phenotypes in these mice.26 These studies suggest that endothelial cell-derived PDGF-β promotes the co-migration of PDGFR-β-expressing pericytes along sprouting new vessels, and the interruption of the PDGF-β/PDGFR-β results in the observed deficiency of pericytes in capillaries. Since PDGF-β has been found to be critical in the recruitment of pericytes during angiogenesis, it has been suggested that PDGF-β may play an important role in maintaining pericyte viability in mature vasculature, although no studies have confirmed this hypothesis.
Capillary basement membrane thickening
Thickening of capillary basement membranes is a well-documented lesion of diabetic retinopathy, visible on electron microscopy. Additional electron microscopic findings include deposition of fibrillar collagen and “Swiss cheese” vacuolization of the otherwise homogenous pattern of basement membrane collagen. The biochemical mechanism leading to basement membrane thickening remains unknown but studies suggest a role for the aldose reductase and the sorbitol pathway.27–30 Nondiabetic rats fed a galactose-rich diet for prolonged periods of time develop retinal capillary basement membrane thickening, fibrillar collagen deposition, and Swiss cheese vacuolization. By contrast, rats fed a control diet or a galactose-rich diet along with the aldose reductase inhibitor sorbinil do not develop basement membrane thickening.27,30 However, the observation that basement membrane thickening of renal glomeruli in diabetic and galactosemic rats is not inhibited by aldose reductase inhibitors casts doubt that the aldose reductase pathway is the primary pathway and suggests that basement membrane thickening may be a secondary nonspecific response.31
Glycation of basement membrane collagen by enzymatic32 and nonenzymatic processes33 may be another mechanism that plays a role in basement membrane thickening. Besides type IV collagen, the predominant type of collagen present in the basement membrane, other collagen types, and noncollagen macromolecules, such as laminin,34 entactin, heparan sulfate proteoglycan,35,36 and basement membrane-bound growth factors are also present.35 Glycation appears to alter the structure of the basement membrane by changing the chemical composition and relative amounts of these components. For example, Shimomura/Spiro and Spiro/Spiro reported decreased heparan sulfate proteoglycan in renal glomeruli from diabetic human subjects.37,38 Quantitative electron microscopic immunocytochemical studies have found that retinal and renal glomeruli basement membrane thickening in galactosemic rats is associated with a relative increase in the levels of type IV collagen and laminin, while the relative levels of heparan sulfate proteoglycan remain unchanged.29,31
Microaneurysms
Although pericyte loss is the earliest sign of diabetic retinopathy, it is only observable histologically. The earliest clinically visible sign of diabetic retinopathy is the microaneurysm.39 Microaneurysms appear as grape-like or spindle-shaped dilations of retinal capillaries on light microscopy.4 They can be either hypercellular or acellular. By ophthalmoscopic examination, microaneurysms appear as tiny, intraretinal red dots located in the inner retina. By fluorescein angiography, they appear as punctate hyperfluorescent dots with variable amounts of fluorescein leakage.
Pericyte cell death and loss of vascular intercellular contacts may lead to endothelial cell proliferation and microaneurysm development.24,40,41 Pericytes appear to exert an antiproliferative effect and pericyte loss may explain the development of hypercellular microaneurysms. However, this mechanism does not account for acellular microaneurysms. Acellular microaneurysms may develop from hypercellular microaneurysms that become acellular from endothelial cell and pericyte apoptosis.42
Pericyte loss may also result in weakening of the capillary wall, promoting the development of microaneurysms at the structural weak points. Pericytes contain myofibrils with contractile properties and may act as smooth muscle cells of larger vessels, exerting tone to the vessel wall in order to counteract the transmural pressure. Loss of pericyte tone may result in focal dilation of the vessel wall leading to the development of a microaneurysm. However, the transmural pressure of the capillary bed is low relative to the arterial circulation, and retinal capillary microaneurysms can develop in other diseases in which pericyte loss is not observed.43,44
Capillary acellularity
Complete loss of the cellular elements of the retinal capillary network can be seen as a more advanced microvascular lesion in diabetic retinopathy and other microvascular retinopathies. In clinicopathological correlations with fluorescein angiograms performed shortly before the eye was enucleated for therapeutic purposes or because the patient died and the eye was removed during autopsy, the retinal vascular digests of the enucleated eye showed that acellular capillaries are nonfunctional since they appeared as regions of nonperfusion on angiography.45 The mechanism by which capillaries become acellular is unknown and can be seen in diabetes, other retinal microvascular diseases, or in experimental diabetes or galactosemia in animal models. Since capillary acellularity is not unique to diabetes, a variety of pathogenic mechanisms may result in this nonspecific finding.
Breakdown of blood–retina barrier
Breakdown of the blood–retina barrier is an important pathophysiologic feature of diabetic retinopathy that leads to the development of macular edema, the leading cause of vision loss in diabetic patients. One mechanism by which the function of this barrier becomes altered involves opening of the tight junctions between vascular endothelial cell processes.46,47 These tight junctions, also known as zonula occludens, appear as a pentalaminar structure on electron microscopy, consisting of two outer and one central electron-dense layer sandwiching two electron-lucent layers, giving the appearance of two “fused” plasma membranes. Using tracers such as lanthanum chloride or horseradish peroxidase, electron microscopy can be used to demonstrate that these molecules cannot pass in the presence of an intact tight junction. However, when the tight junctions are open, they become permeable to these tracer molecules.47 Several important proteins are involved with the formation and function of tight junctions, with ZO-1 (zonula occludens) and occludin being the best characterized. In the presence of histamine, the expression of ZO-1 in cultured retinal endothelial cells is reduced in a dose-dependent manner.48 Culturing in astrocyte-conditioned medium increases expression of ZO-1 while high glucose decreases expression of ZO-1.49 These in vitro results have also been supported by in vivo studies. Reduced expression and anatomic distribution of occludin was found in experimental diabetes.50 Likewise, in experimentally diabetic rats and in diabetic humans, antihistamines reduce leakage of fluorescein into the vitreous.51,52
Vascular endothelial growth factor (VEGF) has been found to be an important mediator leading to the breakdown of the inner blood–retina barrier. Before the discovery of its well-documented role in promoting neovascularization, VEGF was found to increase the permeability of vessels, leading to its alternative name “vascular permeability factor”.53 The mechanism by which VEGF leads to the breakdown of the inner blood–retina barrier appears to involve alteration of endothelial cell tight junctions. Intravitreal injection of VEGF in rats increased the production of the free radical nitric oxide and led to phosphorylation of ZO-154,55
Another important factor promoting retinal vascular permeability involves the kallikrein–kinin system. Proteonomic studies of the vitreous from patients with advanced diabetic retinopathy have identified components of the kallikrein kinin system, including plasma kallikrein, factor XII, and kininogen.56,57 In rodent models, activation of plasma kallikrein in the vitreous has been found to increase retinal vascular permeability.58 Likewise, inhibition of the kallikrein kinin system reduces retinal vascular leakage caused by diabetes and hypertension.58,59 The mechanism by which the kallikrein kinin system promotes vascular permeability probably involves bradykinin. Bradykinin, via nitric oxide, induces vasorelaxation of retinal arterioles.60 Intravenous infusion of bradykinin results in dilation of retinal arterioles and venules, an effect that is reduced by indomethacin and the cyclooxygenase-2 selective inhibitor nimesulide.61 Bradykinin is also a neuropeptide with a direct effect on glia and neurons, which can release vasoactive factors that affect blood flow and vascular permeability.62
Biochemical mechanisms in the pathogenesis of diabetic retinopathy
Although diabetic retinopathy does not manifest the classic “rubor, tumor, calor, and dolor” features of an inflammatory disease and lacks a prominent infiltration of inflammatory cells, there are characteristics that imply a chronic low-grade inflammatory component. In the retinas of diabetic rats, increased activation of leukocytes with increased amounts of inflammatory cytokines and adhesion molecules have been observed. The upregulation of these molecules enhances leukocyte adhesion to retinal capillary walls, leading to increased capillary stasis, occlusion, and ultimately hypoxia as seen in diabetic retinopathy.63–65 Clinically, it has been observed that intravitreal corticosteroid injections decrease diabetic macular edema, often with improvement of visual acuity.66,67 The mechanism by which intravitreal corticosteroids reduce macular edema in diabetes and other etiologies, like retinal vein occlusions, is unclear, but the anti-inflammatory effects of corticosteroids support an inflammatory component to the development of at least diabetic macular edema as well as macular edema from retinal vein occlusions. Likewise, there is growing evidence that the kinin–kallikrein system, which plays an important role in the inflammatory cascade by acting on phospholipase and promoting the release of arachidonic acid and the production of prostaglandins, contributes to the development of diabetic macular edema.68
The aldose reductase theory
Elevation of intracellular glucose levels can cause increased activation of the aldose reductase pathway. Also known as the polyol pathway or the sorbitol pathway, the aldose reductase pathway is a series of intracellular reactions that involves the enzymes aldose reductase and sorbitol dehydrogenase.69,70 Aldose reductase uses the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor to reduce many aldose sugars into their respective sugar alcohols. Glucose is reduced to sorbitol, which is then oxidized into fructose by sorbitol dehydrogenase. However, sorbitol may build up to high intracellular levels because the sorbitol dehydrogenase reaction is slow and the accumulating sorbitol does not easily cross the plasma membrane into the extracellular space. In normoglycemic conditions, the aldose reductase pathway is nonoperative because glucose is a poor substrate for aldose reductase due to its high binding constant (kM). However, in the setting of hyperglycemia as seen with uncontrolled diabetes, the aldose reductase pathway becomes activated once the other enzymatic pathways of glucose metabolism become saturated. Lens epithelium expresses high levels of aldose reductase and accumulation of sorbitol is believed to lead to the development of a cataract in diabetes.71 Osmotic stress has been proposed as the mechanism by which elevated intracellular sorbitol leads to the pathologic changes seen in diabetes.72 However, the levels of sorbitol in vascular cells are in the nanomolar range, which is orders of magnitude less than other glucose metabolites, which have ranges in the micromolar and millimolar range.73
A different mechanism that may account for the role of aldose reductase involves the cellular redox balance. Increases in the utilization of aldose reductase in the hyperglycemic state of diabetes will result in a decline in intracellular NADPH that alters the cellular redox balance. Reduction of intracellular NADPH may also decrease the production of nitric oxide in endothelial cells.74 Similarly, the increased use of sorbitol dehydrogenase can lead to an increase in the NADH/NAD+ ratio that alters the cellular redox balance and may lead to oxidative stress and cellular damage.75
Of interest to animal models of diabetes, galactose is reduced by aldose reductase into galactilol. However, sorbitol dehydrogenase cannot oxidize galactilol, resulting in the rapid intracellular accumulation of galactilol. In the setting of chronic galactosemia, diabetic-like vascular basement membrane changes,27,28 and pericyte loss, development of microaneurysms, and capillary acellularity76 have been reported. When these experiments were repeated and the animals were also treated with aldose reductase inhibitors, it was reported that the development of diabetic-like retinopathy changes was slowed, although most animals developed some degree of changes.30,77–79 While aldose reductase inhibitors were reported to slow or prevent some of the pathologic changes in animal models of diabetes, the aldose reductase inhibitor sorbinil was found not to be effective in humans in the Sorbinil Retinopathy trial.80 The lack of efficacy of aldose reductase inhibitors in human trials may, however, reflect dose-limiting side-effects of the drug that may have precluded it from achieving therapeutic levels in the tissues of interest.
Advanced glycation endproduct (AGE) theory
Accelerated aging by nonenzymatic glycation and crosslinking of proteins has been proposed as a mechanism to explain the complications of diabetes.81 Advanced glycation endproducts (AGEs) is the collective name given to proteins, lipids, and nucleic acids that undergo irreversible modification by reducing sugars or sugar-derived products. The series of chemical reactions that lead to the formation of AGEs is called the Maillard reaction. The Maillard reaction is responsible for the “browning” of tissue seen with aging as well as the “browning” of food during cooking. The initial chemical reaction is known as early glycation and involves reversible nonenzymatic binding of a sugar to amino acid groups on proteins, lipids, or nucleic acids. They form Schiff bases which can undergo rearrangement to form more stable Amadori products. Glycosylated hemoglobin (HbA1c) and fructosamine are well-known examples of Amadori products used clinically as markers of glycemic control. Although they are not AGEs, they can undergo further reactions to eventually lead to the formation of AGEs. Formation of AGEs may directly damage cells by impairing the function of a variety of proteins,82 including both extracellular proteins like collagen83 and intracellular proteins.84,85
The cellular effect of AGEs is also mediated by its binding to receptors, namely receptor for AGE (RAGE). RAGE is a multiligand transmembrane receptor that is part of the immunoglobulin superfamily of proteins.86,87 When bound to AGEs, it initiates a cascade of signal transduction involving at least p21ras, p44/p42 mitogen-activated protein kinase (MAPK), nuclear factor-kappa B (NF-κB), and protein kinase C (PKC).88–93 Activation of these intracellular kinases can subsequently lead to cell dysfunction.94 Other receptors have also been reported to bind to AGEs, such as the macrophage scavenger receptor, P60, P90, and galectin-3.94–96 Aminoguanidine is an inhibitor of AGE formation and has been reported to block the development of many of the microvascular complications of diabetes in animal models.97–100 The effects of aminoguanidine cannot be automatically attributed to the blockade of the AGE pathway, however, since aminoguanidine also has parallel action as an inhibitor of inducible nitric oxide synthase and oxidants.101 Owing to limits secondary to toxicity, clinical trials in humans have been inconclusive; in animal models, however, the use of soluble RAGE to block binding of AGEs to RAGE has been found to prevent many of the effects of hyperglycemia.102
Reactive oxygen intermediates (ROI) theory
One of the oldest theories proposes that chronic hyperglycemia leads to the complications of diabetes by increasing oxidative stress. The usual metabolic pathway of glucose is through glycolysis and the tricarboxylic acid cycle, which takes place in the mitochondria and yields reducing equivalents used to drive the synthesis of adenosine triphosphate via oxidative phosphorylation. However, byproducts of oxidative phosphorylation include free radicals, such as superoxide anion, whose production is increased by high levels of glucose.103 Free radicals can damage mitochondrial DNA104 as well as cellular proteins105 and are also produced by the autoxidation of glucose. Elevated oxidative stress also reduces nitric oxide levels,106,107 promotes leukocyte adhesion to the endothelium and decreases the barrier function of endothelial cells,108 and damages cellular proteins.109 In diabetic mice that overexpress Cu2+/Zn2+ superoxide dismutase, they develop less mesangial expansion compared to wild-type diabetic mice, suggesting that oxidative stress promotes at least some complications of diabetes.110 Oxidative stress can also activate PKC by increasing the formation of diacylglycerol (DAG).111
There is some evidence of increased oxidative stress in diabetic patients. It has been reported that diabetic patients have lower levels of antioxidants, such as vitamin C, vitamin E, and glutathione,112–114 although these results have not been unequivocally reproduced by other researchers.115 However, other markers of oxidative stress, such as oxidized low-density lipoprotein116 and urinary isoprostanes, are elevated in diabetic patients.117 In animal models of diabetes, the use of antioxidants has blocked the development of some of the microvascular complications of diabetes.118–122 One clinical trial reported that high doses of vitamin E (>1000 IU/day) and lipoic acid improved retinal blood flow and creatinine clearance in diabetic patients.123 However, most studies evaluating antioxidants to prevent the complications of diabetes in people have been unsuccessful.124
Protein kinase C (PKC) theory
PKC is a ubiquitous enzyme that appears to promote the development of many of the complications of diabetes without the involvement of the aldose reductase pathway. It has been observed that diabetes and galactosemia can produce elevation of DAG within cells of the retina and aorta in dogs despite treatment with the aldose reductase inhibitor sorbinil.125,126 Activation of PKC occurs through the activation of phospholipase C, which leads to an increase in intracellular Ca2+ and DAG, which in turn results in the activation of PKC.127 Hyperglycemia can result in pathological activation of PKC. Elevated glucose levels result in activation of the glycolytic pathway and lead to increased levels of intracellular glyceraldehyde-3-phosphate. Glyceraldehyde-3-phosphate can promote the de novo synthesis of DAG through glycerol-3-phosphate, which in turn activates PKC.128 Activation of PKC can also be mediated by AGE92 and ROI.111
Elevated levels of DAG and PKC activity has been detected in the tissues of animals with diabetes.129 The pathologic effects of PKC activation that cause vascular damage are mediated through increased vascular permeability,130 disruption of nitric oxide regulation,131,132 increased leukocyte adhesion to vessel walls,133 and changes in blood flow.134 In fact, the effects of PKC on retinal blood flow as measured by fluorescein video angiography, as well as glomerular filtration rate and albumin excretion rate, were improved in a dose-dependent fashion with ruboxistaurin (LY333531), a PKC-β inhibitor.135 VEGF136 and endothelin137 can also activate PKC, which in turn can promote the expression of growth factors like VEGF138 and transforming growth factor-beta (TGF-β).139 PKC activation can influence other signally pathways, such as MAPK or NF-κB.140
PKC inhibition by ruboxistaurin has been reported to block many of the vascular abnormalities in endothelial cells and contractile cells from the retina, arteries, and renal glomeruli.141 In animal models of diabetes, ruboxistaurin protected against or reversed many of the early vascular changes seen with retinopathy, nephropathy, and neuropathy.135,136,142,143 However, a prospective clinical trial of ruboxistaurin did not meet its primary outcome (progression to sight-threatening diabetic macular edema or application of focal/grid photocoagulation for diabetic macular edema) at 30 months although there was a significant reduction of progression to sight-threatening diabetic macular edema when considered alone.144 An open-label extension of the Protein Kinase C Diabetic Retinopathy Study 2 (PKCDRS-2) reported that over a 6-year study period patients with the greatest ruboxistaurin exposure (~5 years) had less sustained moderate vision loss (>15-letter decline) compared to those in the original placebo group (~2 years of ruboxistaurin use).145
Insulin receptors and glucose transporters
In certain types of cells, such as adipocytes and skeletal muscle cells, insulin is required to transport glucose from the extracellular fluid across the plasma membrane into the cytoplasm. This action requires a specific receptor for insulin on the plasma membrane. Although it has been commonly stated that the microvascular complications of diabetes do not occur in tissues in which insulin is required for the transport of glucose into cells, insulin receptors have been reported on the pericytes and endothelial cells of the retinal microvessels.146 There is no evidence, however, that the retinal microvascular insulin receptors are required for glucose transport, although insulin does enhance glycogen synthesis from radiolabeled glucose in retinal microvascular pericytes and endothelial cells and aortic smooth-muscle cells, but not in aortic endothelial cells.146 Insulin in physiologic concentrations (as low as 10 ng/ml) stimulated [3H]-thymidine incorporation into retinal microvascular pericytes and endothelial cells and aortic smooth-muscle cells but not aortic endothelial cells.146 It is noteworthy that, in these experiments, such low concentrations of insulin produced an effect, because unphysiologically high (e.g., 1 mg/ml) concentrations of insulin will stimulate proliferation of many types of cultured cells. However, since microvascular endothelial cells and pericytes do not normally proliferate in the mature retina,147 the importance of these results for normal retinal vascular physiology is unclear. Additionally, these results indicate that there are metabolic differences between microvascular endothelial cells and the endothelial cells of larger vessels, so that translation of results from one type of vascular endothelial cell to another must be done with great caution.
There are at least five different types of facilitated cell membrane glucose transporters, designated GLUT1, GLUT2, GLUT3, GLUT4, and GLUT5, that appear to be most important for the intracellular transport of glucose in tissues like the retina that do not require insulin. Of these, GLUT1 appears to be the most prevalent in the retina,148–150 occurring in microvascular and macrovascular endothelial cells and on RPE cells, as well as in the Müller cells. GLUT2 localization has been reported by immunocytochemistry at the apical ends of the Müller cells of the rat retina, facing the interphotoreceptor matrix,151 while GLUT3 has been reported by similar techniques to be localized to the plexiform layers of the rat152 and human150 retina. An initial report using light microscopic immunocytochemistry in human eyes148 also reported GLUT1 in the nerve fiber layer of the retina and in photoreceptor cell bodies, but GLUT1 was absent from retinal neovascular proliferations in the eyes of diabetic subjects. Subsequently, these investigators used quantitative immunogold electron microscopic immunocytochemistry to examine GLUT1 localization in the eyes of two nondiabetic subjects and three diabetic subjects with little or no retinopathy.149 In approximately half of the retinal microvessels from the diabetic subjects there was no quantitative difference in GLUT1 immunoreactivity by comparison with microvessels from the two normal individuals. However, in the other half of the retinal microvessels studied from diabetic individuals, these investigators reported an increase of GLUT1 immunoreactivity of about 18-fold over normal on the luminal plasma membranes. If these findings can be confirmed in a much larger number of eyes, such upregulation could be a mechanism that initiates glucose-mediated cellular damage by permitting a much greater influx of glucose into cells.
Whether galactose also enters cells by one or more of these facilitated glucose transporters has not been directly tested. However, the fact that rats fed a 50% galactose diet – which also contains the normal amount of glucose – double their food intake by comparison with normal rats, but nevertheless gain weight at only 60–70% of the rate of the normal animals,153 suggests that the excessive amount of galactose competes with glucose for the transport sites, thereby limiting the entry of glucose into cells and diminishing glucose-requiring cellular energy metabolism. However, galactose can participate in other cellular pathways along with glucose, including protein glycation/advanced glycation endproduct formation and synthesis of DAG to activate PKC.126 Whether glucose or galactose can upregulate the mRNAs governing synthesis of any of the GLUT proteins in a fashion such that this upregulation persists long after cessation of the hyperglycemic or galactosemic state has not yet been explored.
Genetic factors in the pathogenesis of diabetic retinopathy
There is good evidence that diabetic retinopathy has a genetic predisposition.154 Diabetic Retinopathy Study (DRS) data indicated that only 50% of nonproliferative diabetic retinopathy (NPDR) patients developed PDR, and many diabetic patients never developed diabetic retinopathy (DR). Twin studies of DR also lend support to this notion.155 Some investigators have also suggested that aldose reductase gene polymorphisms may be associated with risk of DR.156–162 There appears to be considerable value in further investigation of genetic factors related to the pathogenesis of the more severe forms of diabetic retinopathy: severe nonproliferative and proliferative retinopathy, as well as macular edema. Nearly all individuals with type 1 diabetes, and most with type 2 disease, will demonstrate some of the lesions of early retinopathy with sufficient disease duration, but only 50% or less will develop proliferative disease.163,164 Like clinically evident diabetic nephropathy, which similarly affects fewer than 50% of all diabetic subjects regardless of the duration of their diabetes, this suggests genetic factors, in addition to chronic hyperglycemia, are likely involved in the development of these severe forms of retinopathy.
Several studies have explored the relationship between human leukocyte antigen (HLA) antigens, expressed on cell surfaces (and customarily tested with leukocytes withdrawn by venipuncture), and the presence, or severity, of diabetic retinopathy. Rand et al.,165 used a case–control design and found a strong association (relative risk, 3.74) between proliferative retinopathy and the presence of HLA-DR phenotypes 4/0, 3/0, and X/X (neither 3 nor 4). Subjects who had HLA-DR phenotypes 3/4, 3/X, and 4/X had no increased risk of proliferative retinopathy as compared with “control” diabetic subjects, matched for age, sex, and diabetes duration, but without retinopathy. This question was also investigated in a group of 425 subjects with insulin-dependent diabetes who were randomly selected from a much larger population-based study.166 After adjustments were made for duration of diabetes, glycemic control, hypertension, and nephropathy, these authors also found a significantly increased risk of proliferative diabetic retinopathy in subjects with the HLA DR4+ DR3 phenotype.
More recent studies have investigated other aspects of the genetics of diabetic retinopathy. Of particular note is a report from the DCCT research group,167 which examined familial clustering of severe diabetic retinopathy (Early Treatment Diabetic Retinopathy Study (ETDRS) score >47, i.e., severe preproliferative disease) among families of DCCT subjects with multiple diabetic members. Significant associations were found when the correlation of retinopathy severity among family members was investigated in several different ways. However, a less strong familial clustering of diabetic nephropathy was found. This is surprising since evidence from other studies has demonstrated considerable familial clustering of diabetic nephropathy, a complication of diabetes that is now considered to have a strong genetic component.168–171 With increased sophistication in molecular biology, a number of investigators have examined several genetic loci for abnormalities that might be related to a hereditary susceptibility to complications of diabetes. Although these studies are not definitive, one of these claimed that a mutation in the aldose reductase gene conferred increased susceptibility to early-onset diabetic retinopathy in patients with type 2 diabetes.172
Although many genes and proteins of vascular growth have been studied in association with PDR, few if any definitive predisposing genes for PDR have been identified. Below is a brief summary of several studies performed on candidate genes. One of the best known and most well-studied genes is the VEGF gene.173–175 VEGF refers to 2 families of proteins created by alternate splicing of exon 8 in the VEGF gene176 and is an important mediator of ischemia-induced vascularization and neovascularization. Current research of VEGF has focused on the role that certain single nucleotide polymorphisms (SNP) may play in contributing either a risk or protective effect in patients.176–178 Recently, three SNPs in the promoter and 5’UTR regions of the gene (C(-7)T, C(-634)G, T(-1498), and G(-1190)A) were studied for frequency differences between patients with and without PDR.177 These studies have looked at different genetic populations, focusing on Japanese and Indian patients. The results of these genetic studies are at times confusing, as a disease-associated SNP in one population may not confer a risk in another population. For example, in the Japanese population, the CC genotype at the C(-634)G region was significantly associated with PDR, whereas CG genotype at the same site was found to be risk-associated in the Indian population. In addition, disease duration, age, and sex must also be taken into account.
Ramprasad and coworkers in 2007 described work evaluating the role of SNPs within the receptor for advanced glycation end products (RAGE) in PDR.179 At least 20 different polymorphisms have been studied within the RAGE gene. Interaction between the receptor RAGE and its associated glycation ligands plays a role in initiating a proinflammatory cascade, and this interaction has been studied in many disorders of chronic inflammation, including peripheral vascular disease and PDR. Research in these SNPs has shown disease association with the NPDR disease phenotype, yet this association needs to be replicated in other independent cohorts. More recently, Balasubbu et al.180 analyzed the association of nine candidate genes (RAGE, PEDF, AKR1B1, EPO, HTRA1, ICAM, HFE, CFH, and ARMS2) but only found a significant association with diabetic retinopathy in one locus (rs2070600 in RAGE → reduces risk).
Proliferative diabetic retinopathy (PDR) and endstage renal disease (ESRD) are two of the most common and severe microvascular complications of diabetes. There is a high concordance in the development of PDR and ESRD in diabetic patients, as well as strong familial aggregation of these complications, suggesting a common underlying genetic mechanism. However, the precise gene(s) and genetic variant(s) involved remain largely unknown. Erythropoietin (EPO) is a potent angiogenic factor observed in the diabetic human and mouse eye. By a combination of case–control association and functional studies, Tong et al.181 demonstrated that the T allele of SNP rs1617640 in the promoter of the EPO gene is significantly associated with PDR and ESRD. The study was performed in three European-American cohorts (Utah: P = 1.91×10-3; GoKinD: P = 2.66×10-8; Boston: P = 2.1×10-2). The EPO concentration in human vitreous was 7.5-fold higher in normal subjects with the TT risk genotype than in those with the GG genotype. Computational analysis suggests that the risk allele (T) of rs1617640 creates a matrix match with the EVI1/MEL1 or AP1 binding site, accounting for an observed 25-fold enhancement of luciferase reporter expression as compared to the G allele. These results suggest that rs1617640 in the EPO promoter is significantly associated with PDR and ESRD and suggest EPO as a potential pathway mediating severe diabetic microvascular complications.181
Genome-wide association studies (GWAS) of diabetic retinopathy have also been pursued. In a Taiwanese population, Huang et al. found genetic associations for susceptibility for development of diabetic retinopathy in five loci, including PLXDC2 and ARHGAP22, which are genes implicated in endothelial cell proliferation and capillary permeability.182 In addition, one group recently published their preliminary results of a GWAS on a small (286 total) Mexican-American diabetic cohort. They identified 32 SNPs (in 11 regions) with a nominal association for severe diabetic retinopathy, though none was located in traditional candidate genes for diabetes or diabetic retinopathy.183 These findings have yet to be replicated, and as noted above, the cohort was small. In two large type I diabetes cohorts, several novel genetic loci associated with sight-threatening complications due to diabetic retinopathy, were identified, including rs10521145 in the intron of CCDC101, a histone acetyltransferase.184
Catalyzed by the rapid advances in whole-genome SNP genotyping technologies and the construction of reference haplotype maps, genetic variants associated with ~300 traits have successfully been mapped by GWAS in the past few years, using a P-value threshold of 10-5. GWAS is based on the hypothesis that common diseases are mainly caused by common genetic variants in the population (Common Disease Common Variants, CD-CV), each having relatively weak effects.185 This assumption appears to be generally true for many genetic traits such as age-related macular degeneration.186–188 Another hypothesis is Common Disease Rare Variants (CD-RV), or that common diseases are due to the presence of many rare variants in the same genes or pathways, each having relative strong effects. There is mounting evidence suggesting that CD-RV is also true for many diseases.189–192 In diseases such as cancer, coronary atherosclerosis, and Parkinson disease, nonsynonymous variants in the disease genes were found more frequently in the disease samples compared to the controls. In addition, a higher proportion of these are predicted to be damaging.189,193,194 For example, Cohen et al. sequenced genes in individuals at risk for coronary disease.189 They found that 16% of the individuals in the disease group had novel nonsynonymous variants in candidate genes, in contrast to only 2% for individuals not at risk. Causal variants under the CD-RV hypothesis are difficult to detect in GWAS, because they are in very weak linkage disequilibrium with the tagging SNPs included in genotyping assays. Deep resequencing in the case and control population is required to uncover such variants.195 Due to the high cost of large-scale resequencing, only a very limited number of candidate genes have been screened.
Other ocular factors
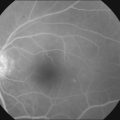
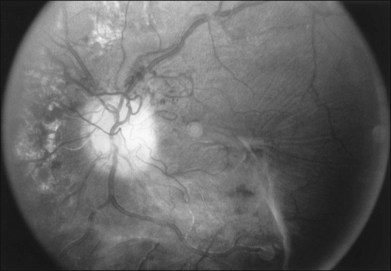
Becker196 reported in 1967 that glaucoma was associated with a decreased prevalence and severity of diabetic retinopathy in affected eyes. Other studies have reported similar results. This has never been confirmed in a methodologically precise epidemiologic study, although Becker’s claim seems to be correct based on other clinical observations (Fig. 46.2). This is an important point that should be evaluated in a proper case–control study. If it is true, the explanation is unclear. If the effect is observable only in true glaucoma and not in ocular hypertension, in which the intraocular pressure is chronically elevated without damage to the retinal ganglion cell or optic nerve fiber layers, then it may be related in some way to loss of metabolic activity in the retina with degeneration of ganglion cells. If the effect is related simply to elevated intraocular pressure, the explanation for this observation would be less obvious.
It has also been reported that myopia is associated with a decreased prevalence and severity of diabetic retinopathy.197 This effect of myopia on the prevalence of proliferative diabetic retinopathy has been confirmed by Rand et al.165 who found an interesting interaction between myopia of greater than 2 diopters and HLA-D-group antigens. Fewer subjects in their “case” group with proliferative retinopathy had myopia of this degree than did subjects in the control group, who had diabetes of 15 years’ or more duration and minimal or no retinopathy. Subjects with 2 D or more of myopia and HLA-D group phenotypes 3/0, 4/0, or X/X had a relative risk for proliferative disease of 1.0 as compared with the control group (i.e., their risk was no different from that of the controls), while the overall risk for all subjects, regardless of refractive error, with these HLA-D group phenotypes was 3.74.
The initial observations leading to the development of panretinal photocoagulation (or scatter) treatment for proliferative diabetic retinopathy were made by Aiello and colleagues at the Symposium on the Treatment of Diabetic Retinopathy,198 who noted that eyes with a great deal of retinochoroidal scarring from trauma, inflammatory disease, etc. had markedly reduced prevalence and severity of diabetic retinopathy. The effect is unexplained, but the most widespread current hypothesis is that it results from decreased retinal metabolism – in particular, a decreased need for oxygen, with a resultant diminished production of a vasoproliferative (angiogenic) factor.199,200 The immediate practical application of this observation was the attempt, through extensive photocoagulation of the mid-peripheral retina (panretinal or scatter photocoagulation), to produce the same effect iatrogenically, a technique that significantly reduces the rate of progression to severe visual loss in PDR.201
Retinopathy in different forms of diabetes
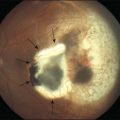
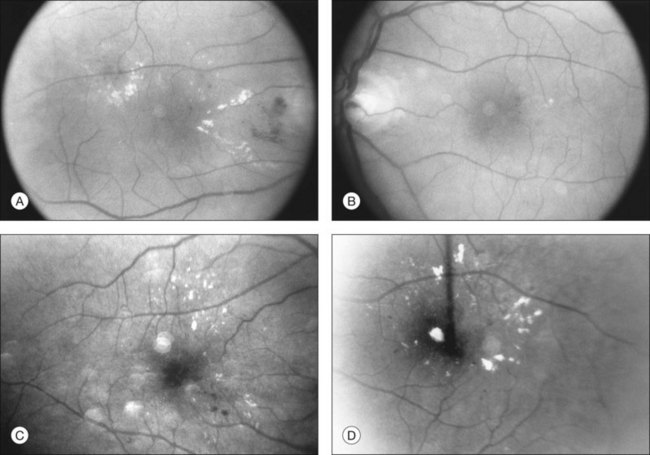
There is no evidence that retinopathy differs in different forms of diabetes. Proliferative retinopathy is more prevalent at any given duration of the systemic disease in type 1 than in type 2 diabetes,163,164 but, as noted earlier, it is not clear whether this is due to different metabolic factors in the two types of diabetes, to differences in the ages of the patients (type 1 patients are, on average, much younger), or to the higher mean blood glucose levels in type 1 patients. Macular edema probably occurs with equal prevalence as a function of disease duration in both type 1 and type 2 diabetes.202 Type 2 diabetes is more common after the age of 30164 (although “type 2 diabetes of youth” is becoming increasingly frequently recognized203), but, despite the apparently mild metabolic defect, proliferative retinopathy can occur as well in this form of the disease (Fig. 46.3). Similarly, vision-threatening retinopathy can occur in patients with “secondary” diabetes – that which occurs, for example, following pancreatitis, hemochromatosis, or acromegaly. The patient whose retinal photographs are shown in Fig. 46.4A and B had acromegaly secondary to a pituitary adenoma and also developed background diabetic retinopathy with macular edema. Since the acromegaly had been treated and the patient had normal levels of circulating growth hormone, the retinopathy may have been related to diabetes secondary to the acromegaly, or to a type 2 diabetes that would have developed regardless of the presence of acromegaly.
Animal models in the study of diabetic retinopathy
Studies of the pathogenesis and treatment of human disease can be facilitated by the development of models of the disease in animals. There have been numerous attempts to reproduce the lesions of diabetic retinopathy in animals. Although several authors have claimed positive results, there are only a few animal models in which one or more of the lesions of diabetic retinopathy have been produced with unquestioned validity. Foremost among these are dogs, with spontaneous204 or induced205 diabetes of 3–5 years’ duration. These animals develop loss of capillary pericytes and ultimately also of endothelial cells with nonfunctional, acellular capillaries; capillary basement membrane thickening, and microaneurysm formation. Early intraretinal neovascularization has also been observed. However, more advanced lesions, including retinal edema (dogs do not have a macula, so true macular edema cannot develop) and neovascularization into the vitreous, have not been reported.
Because of the ease of working with small animals, there have been many attempts to develop diabetic retinopathy in rodents, including mice and, in particular, rats. Claims of producing lesions such as microaneurysms97 and pericyte dropout97 in rats with experimental diabetes of less than 1 year have not been validated by Engerman et al.205 or Tilton et al.206 However, rats with diabetes or galactosemia27,28 develop retinal capillary basement membrane thickening, a lesion that has been widely observed in many microvascular systems in the body in human and animal diabetes.207 In addition, rats that have been galactosemic for 18 months or more develop pericyte loss and, eventually, capillary acellularity,79,153,208,209 and some rats that have been fed a 30–50% galactose diet for up to 24 months develop a halo of dilated, hypercellular vessels surrounding the optic nerve.79,153,208,209 Whether these represent intraretinal neovascularization or simply dilated pre-existing vascular channels is uncertain. Of particular interest is a report that pericyte and endothelial cell nuclei in short-term diabetic or galactosemic rats, and in retinal capillaries from donor eyes of humans with diabetes, undergo apoptosis as demonstrated by appropriate nuclear labeling techniques.42 It is unknown what stimulus is induced by prolonged hyperglycemia, or galactosemia, that causes programmed death of retinal capillary cells to a greater extent than capillary cells elsewhere in the body.
Because of the similarity of their retinal anatomy to that of humans, one might expect that nonhuman primates with diabetes of sufficient duration would be good models for human diabetic retinopathy. However, studies of rhesus monkeys with diabetes for as long as 10 years have revealed occasional microaneurysms but no other lesions.205,210
Doubtless a major reason for the difficulty in producing lesions of diabetic retinopathy in animals with diabetes is the factor of disease duration. In diabetic dogs a minimum disease duration of 3–5 years is necessary for the development of the earliest lesions of retinopathy, and this is identical to the duration required for retinopathy to develop in humans with type 1 diabetes.164,211,212 Rats and mice normally have a lifespan of under 3 years, and after the onset of diabetes it is difficult to maintain these animals for much more than 1 year. Several investigators have reported the development of lesions resembling diabetic retinopathy in rats fed a 50% galactose diet for 28 months.79,153,208 These lesions include pericyte “ghosts,” acellular capillaries, and vascular dilation and tortuosity. Whether microaneurysms occur in galactosemic rats is controversial.42,79,153,208–210 The use of a high-galactose diet to produce a model of diabetic retinopathy originated from the hypothesis that the enzyme sequence known as the “sorbitol pathway” is responsible for the earliest lesions of diabetic retinopathy. Since galactose, along with glucose, is a substrate for this pathway, its use in producing models of retinopathy is a good test of the hypothesis. Previously, Kern and Engerman,100 as well as Kador and associates,77,78 have produced a diabetic-like retinopathy in dogs fed a 50% galactose diet for 3–4 years. Kern and Engerman have also reported a diabetic-like retinopathy in mice fed a 30% galactose diet for 21–26 months.213 They reported that, unlike galactosemic rats, galactosemic mice developed true microaneurysms. These findings raise two important questions. First, why might mice, dogs, and humans develop microaneurysms after chronic diabetes, or galactosemia, and rats do not? Second, how might the presence of retinopathy in chronically galactosemic mice be relevant to the hypothesis that the “sorbitol pathway” is an important causal mechanism for diabetic retinopathy? Both these questions will be considered in more detail in later sections of this chapter.
Species- and organ-specific variations in anatomy and in metabolic pathways may be important considerations in the development of microvascular lesions in the retina or other organs. Pericyte dropout and microaneurysm formation have not been found either in the brains of diabetic human subjects,214 or in those of diabetic or galactosemic dogs,215 even though these lesions are common in the retina and were present in the retinas of the same human or animal subjects in whose cerebral cortexes they could not be found. It might be argued that, in these studies, retinas were examined by the trypsin digest procedure in which the entire, intact retinal vasculature can be spread on a microscope slide and examined in detail. This cannot be done with cerebral cortical vasculature, which must be examined histologically following homogenization and sieving through a nylon mesh that retains only vascular fragments. Capillaries that have lost cells or are otherwise abnormal may be sufficiently fragile that they are broken into smaller pieces by this technique and are lost in the sieving process. Although the retina is derived embryologically from the brain, and both retina and brain have a microvasculature featuring thick endothelial cell cytoplasm and tight junctions between endothelial cells that produce a blood–tissue barrier to many molecules, pericyte coverage of the endothelial cell tube of capillaries of the retina is substantially greater in the two species that have been studied (rat and monkey) than it is in the brains of these species.216,217 The same is probably true in humans, where the data from retina closely resemble data from the retinas of monkeys, but it has not been possible to obtain retinal and brain tissue from the same human donors adequate to perform these morphometric studies. Although galactosemic rats develop a diabetic-like retinopathy, rats have a much smaller ratio of pericytes to endothelial cells in their retinal microcirculations than do humans.206,216,218 Comparisons of the retinal and cerebral pericyte : endothelial cell ratios in retinas and brains of rats with those of dogs, mice, and humans have not been carried out, but would be of interest because dogs, humans, and (at least according to one group of investigators) mice develop true capillary microaneurysms with long-term diabetes (dogs and humans) and galactosemia (dogs and mice), while rats do not.
Two useful animal models of neovascularization exist, both in nondiabetic animals. The first, originally described by Ashton196 and by Patz,219 is produced by exposing neonatal kittens or puppies to an atmosphere high in oxygen for up to a few days just after birth. This produces at least a peripheral, or in more severe cases a generalized,220 vasoconstriction, followed by the development of retinal new vessels. These vessels are transient, however, and regress spontaneously after a few weeks. This model was initially developed to simulate retinopathy of prematurity, a condition that may develop following exposure of premature human infants to a high-oxygen atmosphere. The development of the new vessels, according to the hypothesis that has been favored for a number of years,200 presumably resulted from production by the hypoxic retina cells (hypoxic because of the profound vasoconstriction that followed the hyperoxia) of an “angiogenesis factor.”221 More recently, hyperoxygenation during the neonatal period has been applied to mice and rats, producing a model of peripheral retinal neovascularization similar to that of retinopathy of prematurity in human infants (Fig. 46.5), that has been exploited for studies of the production, and inhibition, of angiogenesis factors.222–224A second model of intraretinal and subretinal (beneath the neural retina, but arising from the retinal and not the choroidal circulation) neovascularization has recently been described, using transgenic mice that overexpress the gene for VEGF.225 This model is additional evidence that VEGF is capable of producing retinal neovascularization, although in a quite artificial situation that may not be relevant to human ocular diseases associated with neovascularization. The new vessels in these transgenic mice extend from the retinal circulation to beneath the photoreceptor layer of the neural retina but do not enter the RPE. The reason for this outward growth, rather than inward into the vitreous, is that the promoter used to carry the VEGF gene into the retina is the rhodopsin gene, which is localized to the photoreceptor cells.
Cell culture studies
Since the mid-1970s, techniques have been developed to isolate and grow in culture the component cells of both large and small blood vessels, including those from the retina. In 1975, Buzney et al. described the culture of retinal microvascular pericytes from bovine and monkey eyes.226 Subsequently Frank et al.227 and several others228–230 described the culture of retinal microvascular endothelial cells. More recent evidence indicates that glial cells of the retina and optic nerve may also be important in the development, at least in the later stages of diabetic retinopathy, of neovascularization and, perhaps, macular edema. Methods are available for retinal glial cell culture. These techniques add an extra tool for research in diabetic retinopathy, but one that must be used with caution. One can use cell culture studies to investigate certain biochemical processes, but with great caution that the processes being studied have not been greatly modified in culture from those that occur in vivo. The same can be said for the investigation of physiologic function, e.g., phagocytosis, cell contraction, or cell motility. Diabetic retinopathy requires years to develop in the intact human or experimental animal. It has not yet been possible to maintain cultures of retinal cells for that duration, and there is no way of ascertaining that alterations produced in cells by exposure to high glucose or galactose over a short time in culture are truly related to the development of the anatomic and functional lesions of diabetic retinopathy over a very long time in the intact retina. Finally, although capillary tube-like structures composed of micro- and macrovascular endothelial cells have been produced under certain conditions in culture,231–233 these may not be truly analogous either to normal vessels or to abnormal new vessels in the intact retina.
Despite these caveats, there are several results using cultured retinal microvascular cells that appear to be relevant to the physiology of the intact retinal microcirculation. These include the elegant studies of D’Amore and associates,40,41 in which microvascular endothelial cells were co-cultured with a variety of other cell types that had been growth-arrested with an antibiotic and were then plated together with endothelial cells in different proportions. Most of the cells used for the co-cultures, including bovine RPE cells, human skin fibroblasts, mouse 3T3 fibroblasts, and Madin–Darby canine kidney cells, greatly stimulated endothelial cell proliferation. By contrast, pericytes and vascular smooth-muscle cells dramatically inhibited endothelial cell proliferation, even when the pericytes or smooth-muscle cells were added in ratios as low as 1 : 10 with the endothelial cells.40 For the co-culture to be effective in retarding endothelial cell proliferation, pericyte or smooth-muscle cell processes had to make contact with the endothelial cells. Transfer of “conditioned media” from pericyte or smooth-muscle cell cultures was ineffective in producing growth inhibition. In studying the mechanism of the inhibition, these authors found that it was produced by the release, and activation, of transforming growth factor-beta (TGF-β).41 Pericytes or smooth-muscle cells alone produce this polypeptide in an inactive form. The co-cultured cells, in physical contact with one another, both produce and activate TGF-beta. This finding suggests that one function of pericytes in the retina is to inhibit the proliferation of endothelial cells. Thus, the loss of pericytes that occurs early in the course of diabetic retinopathy may facilitate the later development of microaneurysms (clusters of newly formed endothelial cells) and of frank neovascularization. In fact, just this result has been reported in an experiment using mice with a targeted disruption (“knockout”) of the gene for the B-chain of platelet-derived growth factor (PDGF-β).24 This genetic defect is lethal, but histopathologic examination of the fetal PDGF-β-deficient animals shows an absence of capillary pericytes – whose antenatal development is evidently controlled by this growth factor – and the frequent appearance of microaneurysms throughout the microcirculation of the retina and brain.
Pericytes are considered to be contractile cells, regulating flow through the capillaries analogous to the function of the smooth-muscle cells of the larger vessels. Cultured pericytes are immunocytochemically positive for smooth-muscle actin,234 and in culture they contract either spontaneously235 or in response to a variety of agents.236–238 Thus, loss of pericytes from the retinal microcirculation may produce alterations in retinal blood flow. However, direct evidence for pericyte contraction in the circulation of the intact retina has never been obtained. Tilton et al.239 performed a morphometric study of capillaries in rats following infusion of various vasoconstrictor agents and found evidence of contraction of pericytes in skeletal muscle capillaries but not of those in cardiac muscle. Butryn and coworkers conducted a similar study in the retinal vessels of rats following intravitreal infusion of ET-1, an extremely powerful vasoconstrictor.240 Although they found evidence of contraction of arteriolar smooth muscle, they could not demonstrate contraction of retinal capillary pericytes.
 For online acknowledgments visit
For online acknowledgments visit 1 The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986.
2 UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352:837–853.
3 UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes (UKPDS 38). BMJ. 1998;317:703–713.
4 Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol. 1961;66:366–378.
5 Kuwabara T, Cogan DG. Studies of retinal vascular patterns. I. Normal architecture. Arch Ophthalmol. 1960;64:904–911.
6 Kuwabara T, Cogan DG. Retinal vascular patterns. VI. Mural cells of the retinal capillaries. Arch Ophthalmol. 1963;69:492–502.
7 Speiser P, Gittelsohn AM, Patz A. Studies on diabetic retinopathy. 3. Influence of diabetes on intramural pericytes. Arch Ophthalmol. 1968;80:332–337.
8 Yanoff M. Diabetic retinopathy. N Engl J Med. 1966;274:1344–1349.
9 Shepro D, Morel NM. Pericyte physiology. FASEB J. 1993;7:1031–1038.
10 Aiello LP, Cavallerano J, Bursell SE. Diabetic eye disease. Endocrinol Metab Clin North Am. 1996;25:271–291.
11 Cameron NE, Eaton SE, Cotter MA, et al. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001;44:1973–1988.
12 Akagi Y, Kador PF, Kuwabara T, et al. Aldose reductase localization in human retinal mural cells. Invest Ophthalmol Vis Sci. 1983;24:1516–1519.
13 Tilton RG, Hoffmann PL, Kilo C, et al. Pericyte degeneration and basement membrane thickening in skeletal muscle capillaries of human diabetics. Diabetes. 1981;30:326–334.
14 Kern TS, Engerman RL. Distribution of aldose reductase in ocular tissues. Exp Eye Res. 1981;33:175–182.
15 Ludvigson MA, Sorenson RL. Immunohistochemical localization of aldose reductase. II. Rat eye and kidney. Diabetes. 1980;29:450–459.
16 Buzney SM, Frank RN, Varma SD, et al. Aldose reductase in retinal mural cells. Invest Ophthalmol Vis Sci. 1977;16:392–396.
17 Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820.
18 Barrett TB, Gajdusek CM, Schwartz SM, et al. Expression of the sis gene by endothelial cells in culture and in vivo. Proc Natl Acad Sci U S A. 1984;81:6772–6774.
19 Collins T, Ginsburg D, Boss JM, et al. Cultured human endothelial cells express platelet-derived growth factor B chain: cDNA cloning and structural analysis. Nature. 1985;316:748–750.
20 Collins T, Pober JS, Gimbrone MA, Jr., et al. Cultured human endothelial cells express platelet-derived growth factor A chain. Am J Pathol. 1987;126:7–12.
21 DiCorleto PE, Bowen-Pope DF. Cultured endothelial cells produce a platelet-derived growth factor-like protein. Proc Natl Acad Sci U S A. 1983;80:1919–1923.
22 Bernstein LR, Antoniades H, Zetter BR. Migration of cultured vascular cells in response to plasma and platelet-derived factors. J Cell Sci. 1982;56:71–82.
23 D’Amore PA, Smith SR. Growth factor effects on cells of the vascular wall: a survey. Growth Factors. 1993;8:61–75.
24 Lindahl P, Johansson BR, Leveen P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245.
25 Hellstrom M, Kalen M, Lindahl P, et al. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055.
26 Leveen P, Pekny M, Gebre-Medhin S, et al. Mice deficient for PDGF B show renal, cardiovascular, hematological abnormalities. Genes Dev. 1994;8:1875–1887.
27 Robison WG, Jr., Kador PF, Kinoshita JH. Retinal capillaries: basement membrane thickening by galactosemia prevented with aldose reductase inhibitor. Science. 1983;221:1177–1179.
28 Frank RN, Keirn RJ, Kennedy A, et al. Galactose-induced retinal capillary basement membrane thickening: prevention by Sorbinil. Invest Ophthalmol Vis Sci. 1983;24:1519–1524.
29 Das A, Frank RN, Zhang NL, et al. Increases in collagen type IV and laminin in galactose-induced retinal capillary basement membrane thickening – prevention by an aldose reductase inhibitor. Exp Eye Res. 1990;50:269–280.
30 Robison WG, Jr., Kador PF, Akagi Y, et al. Prevention of basement membrane thickening in retinal capillaries by a novel inhibitor of aldose reductase, tolrestat. Diabetes. 1986;35:295–299.
31 Das A, Frank RN, Zhang NL. Sorbinil does not prevent galactose-induced glomerular capillary basement membrane thickening in the rat. Diabetologia. 1990;33:515–521.
32 Nishio Y, Warren CE, Buczek-Thomas JA, et al. Identification and characterization of a gene regulating enzymatic glycosylation which is induced by diabetes and hyperglycemia specifically in rat cardiac tissue. J Clin Invest. 1995;96:1759–1767.
33 Brownlee M, Cerami A. The biochemistry of the complications of diabetes mellitus. Annu Rev Biochem. 1981;50:385–432.
34 Timpl R, Rohde H, Robey PG, et al. Laminin – a glycoprotein from basement membranes. J Biol Chem. 1979;254:9933–9937.
35 Grant DS, Kleinman HK. Regulation of capillary formation by laminin and other components of the extracellular matrix. EXS. 1997;79:317–333.
36 Kennedy A, Frank RN, Mancini MA. In vitro production of glycosaminoglycans by retinal microvessel cells and lens epithelium. Invest Ophthalmol Vis Sci. 1986;27:746–754.
37 Shimomura H, Spiro RG. Studies on macromolecular components of human glomerular basement membrane and alterations in diabetes. Decreased levels of heparan sulfate proteoglycan and laminin. Diabetes. 1987;36:374–381.
38 Spiro RG, Spiro MJ. Effect of diabetes on the biosynthesis of the renal glomerular basement membrane. Studies on the glucosyltransferase. Diabetes. 1971;20:641–648.
39 Friedenwald JS. Diabetic retinopathy. Am J Ophthalmol. 1950;33:1187–1199.
40 Orlidge A, D’Amore PA. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol. 1987;105:1455–1462.
41 Antonelli-Orlidge A, Saunders KB, Smith SR, et al. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A. 1989;86:4544–4548.
42 Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890.
43 Ashton N, Kok DA, Foulds WS. Ocular pathology in macroglobulinaemia. J Pathol Bacteriol. 1963;86:453–461.
44 Duke JR, Wilkinson CP, Sigelman S. Retinal microaneurysms in leukaemia. Br J Ophthalmol. 1968;52:368–374.
45 Kohner EM, Henkind P. Correlation of fluorescein angiogram and retinal digest in diabetic retinopathy. Am J Ophthalmol. 1970;69:403–414.
46 Daneman D, Drash AL, Lobes LA, et al. Progressive retinopathy with improved control in diabetic dwarfism (Mauriac’s syndrome). Diabetes Care. 1981;4:360–365.
47 Wallow IH, Engerman RL. Permeability and patency of retinal blood vessels in experimental diabetes. Invest Ophthalmol Vis Sci. 1977;16:447–461.
48 Gardner TW, Lesher T, Khin S, et al. Histamine reduces ZO-1 tight-junction protein expression in cultured retinal microvascular endothelial cells. Biochem J. 1996;320(Pt 3):717–721.
49 Gardner TW. Histamine, ZO-1 and increased blood-retinal barrier permeability in diabetic retinopathy. Trans Am Ophthalmol Soc. 1995;93:583–621.
50 Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–3568.
51 Enea NA, Hollis TM, Kern JA, et al. Histamine H1 receptors mediate increased blood–retinal barrier permeability in experimental diabetes. Arch Ophthalmol. 1989;107:270–274.
52 Gardner TW, Eller AW, Friberg TR, et al. Antihistamines reduce blood–retinal barrier permeability in type I (insulin-dependent) diabetic patients with nonproliferative retinopathy. A pilot study. Retina. 1995;15:134–140.
53 Keck PJ, Hauser SD, Krivi G, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312.
54 Antonetti DA, Barber AJ, Hollinger LA, et al. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467.
55 Lakshminarayanan S, Antonetti DA, Gardner TW, et al. Effect of VEGF on retinal microvascular endothelial hydraulic conductivity: the role of NO. Invest Ophthalmol Vis Sci. 2000;41:4256–4261.
56 Gao BB, Clermont A, Rook S, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. 2007;13:181–188.
57 Gao BB, Chen X, Timothy N, et al. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res. 2008;7:2516–2525.
58 Phipps JA, Clermont AC, Sinha S, et al. Plasma kallikrein mediates angiotensin II type 1 receptor-stimulated retinal vascular permeability. Hypertension. 2009;53:175–181.
59 Abdouh M, Talbot S, Couture R, et al. Retinal plasma extravasation in streptozotocin-diabetic rats mediated by kinin B(1) and B(2) receptors. Br J Pharmacol. 2008;154:136–143.
60 Jeppesen P, Aalkjaer C, Bek T. Bradykinin relaxation in small porcine retinal arterioles. Invest Ophthalmol Vis Sci. 2002;43:1891–1896.
61 Kojima N, Saito M, Mori A, et al. Role of cyclooxygenase in vasodilation of retinal blood vessels induced by bradykinin in Brown Norway rats. Vascul Pharmacol. 2009;51:119–124.
62 Parpura V, Basarsky TA, Liu F, et al. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747.
63 Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–365.
64 Joussen AM, Murata T, Tsujikawa A, et al. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152.
65 Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440.
66 Martidis A, Duker JS, Greenberg PB, et al. Intravitreal triamcinolone for refractory diabetic macular edema. Ophthalmology. 2002;109:920–927.
67 Yilmaz T, Weaver CD, Gallagher MJ, et al. Intravitreal triamcinolone acetonide injection for treatment of refractory diabetic macular edema: a systematic review. Ophthalmology. 2009;116:902–911. quiz 912–903
68 Feener EP. Plasma kallikrein and diabetic macular edema. Curr Diab Rep. 2010;10:270–275.
69 Gabbay KH. The sorbitol pathway and the complications of diabetes. N Engl J Med. 1973;288:831–836.
70 Kinoshita JH. Cataracts in galactosemia. The Jonas S. Friedenwald Memorial Lecture. Invest Ophthalmol. 1965;4:786–799.
71 Kinoshita JH. Mechanisms initiating cataract formation. Proctor Lecture. Invest Ophthalmol. 1974;13:713–724.
72 Gabbay KH. Hyperglycemia, polyol metabolism, complications of diabetes mellitus. Annu Rev Med. 1975;26:521–536.
73 Van den Enden MK, Nyengaard JR, Ostrow E, et al. Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism. Implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 1995;36:1675–1685.
74 Tesfamariam B. Free radicals in diabetic endothelial cell dysfunction. Free Radic Biol Med. 1994;16:383–391.
75 Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813.
76 Engerman RL, Kern TS. Experimental galactosemia produces diabetic-like retinopathy. Diabetes. 1984;33:97–100.
77 Kador PF, Akagi Y, Takahashi Y, et al. Prevention of retinal vessel changes associated with diabetic retinopathy in galactose-fed dogs by aldose reductase inhibitors. Arch Ophthalmol. 1990;108:1301–1309.
78 Kador PF, Akagi Y, Terubayashi H, et al. Prevention of pericyte ghost formation in retinal capillaries of galactose-fed dogs by aldose reductase inhibitors. Arch Ophthalmol. 1988;106:1099–1102.
79 Robinson WG, Jr., Laver NM, Jacot JL, et al. Diabetic-like retinopathy ameliorated with the aldose reductase inhibitor WAY-121,509. Invest Ophthalmol Vis Sci. 1996;37:1149–1156.
80 Sorbinil Retinopathy Trial Research Group. A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Arch Ophthalmol. 1990;108:1234–1244.
81 Monnier VM, Kohn RR, Cerami A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci U S A. 1984;81:583–587.
82 Brownlee M, Vlassara H, Cerami A. Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann Intern Med. 1984;101:527–537.
83 Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234.
84 Chibber R, Molinatti PA, Kohner EM. Intracellular protein glycation in cultured retinal capillary pericytes and endothelial cells exposed to high-glucose concentration. Cell Mol Biol (Noisy-le-grand). 1999;45:47–57.
85 Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J Clin Invest. 1994;94:110–117.
86 Neeper M, Schmidt AM, Brett J, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004.
87 Schmidt AM, Vianna M, Gerlach M, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997.
88 Lander HM, Tauras JM, Ogiste JS, et al. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–17814.
89 Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901.
90 Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274:19919–19924.
91 Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphotericin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360.
92 Beauchamp MC, Michaud SE, Li L, et al. Advanced glycation end products potentiate the stimulatory effect of glucose on macrophage lipoprotein lipase expression. J Lipid Res. 2004;45:1749–1757.
93 Yan SD, Schmidt AM, Anderson GM, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–9897.
94 Schmidt AM, Stern DM. RAGE: a new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol Metab. 2000;11:368–375.
95 el Khoury J, Thomas CA, Loike JD, et al. Macrophages adhere to glucose-modified basement membrane collagen IV via their scavenger receptors. J Biol Chem. 1994;269:10197–10200.
96 Vlassara H, Li YM, Imani F, et al. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med. 1995;1:634–646.
97 Hammes HP, Martin S, Federlin K, et al. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A. 1991;88:11555–11558.
98 Brownlee M, Vlassara H, Kooney A, et al. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232:1629–1632.
99 Friedman EA. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care. 1999;22(Suppl 2):B65–B71.
100 Kern TS, Engerman RL. Pharmacological inhibition of diabetic retinopathy: aminoguanidine and aspirin. Diabetes. 2001;50:1636–1642.
101 Nilsson BO. Biological effects of aminoguanidine: an update. Inflamm Res. 1999;48:509–515.
102 Wautier JL, Zoukourian C, Chappey O, et al. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J Clin Invest. 1996;97:238–243.
103 Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790.
104 Suzuki S, Hinokio Y, Komatu K, et al. Oxidative damage to mitochondrial DNA and its relationship to diabetic complications. Diabetes Res Clin Pract. 1999;45:161–168.
105 Hunt JV, Dean RT, Wolff SP. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J. 1988;256:205–212.
106 Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267.
107 Tesfamariam B. Selective impairment of endothelium-dependent relaxations by prostaglandin endoperoxide. J Hypertens. 1994;12:41–47.
108 Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–C741.
109 Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412.
110 Craven PA, Melhem MF, Phillips SL, et al. Overexpression of Cu2+/Zn2+ superoxide dismutase protects against early diabetic glomerular injury in transgenic mice. Diabetes. 2001;50:2114–2125.
111 Taher MM, Garcia JG, Natarajan V. Hydroperoxide-induced diacylglycerol formation and protein kinase C activation in vascular endothelial cells. Arch Biochem Biophys. 1993;303:260–266.
112 Jain SK, McVie R. Effect of glycemic control, race (white versus black), duration of diabetes on reduced glutathione content in erythrocytes of diabetic patients. Metabolism. 1994;43:306–309.
113 Jennings PE, Chirico S, Jones AF, et al. Vitamin C metabolites and microangiopathy in diabetes mellitus. Diabetes Res. 1987;6:151–154.
114 Karpen CW, Cataland S, O’Dorisio TM, et al. Production of 12-hydroxyeicosatetraenoic acid and vitamin E status in platelets from type I human diabetic subjects. Diabetes. 1985;34:526–531.
115 Oberley LW. Free radicals and diabetes. Free Radic Biol Med. 1988;5:113–124.
116 Hayden JM, Reaven PD. Cardiovascular disease in diabetes mellitus type 2: a potential role for novel cardiovascular risk factors. Curr Opin Lipidol. 2000;11:519–528.
117 Devaraj S, Hirany SV, Burk RF, et al. Divergence between LDL oxidative susceptibility and urinary F(2)-isoprostanes as measures of oxidative stress in type 2 diabetes. Clin Chem. 2001;47:1974–1979.
118 Cameron NE, Cotter MA. Neurovascular dysfunction in diabetic rats. Potential contribution of autoxidation and free radicals examined using transition metal chelating agents. J Clin Invest. 1995;96:1159–1163.
119 Cameron NE, Cotter MA, Archibald V, et al. Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic and streptozotocin-diabetic rats. Diabetologia. 1994;37:449–459.
120 Nagamatsu M, Nickander KK, Schmelzer JD, et al. Lipoic acid improves nerve blood flow, reduces oxidative stress, improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167.
121 Lal MA, Korner A, Matsuo Y, et al. Combined antioxidant and COMT inhibitor treatment reverses renal abnormalities in diabetic rats. Diabetes. 2000;49:1381–1389.
122 Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–1942.
123 Bursell SE, Clermont AC, Aiello LP, et al. High-dose vitamin E supplementation normalizes retinal blood flow and creatinine clearance in patients with type 1 diabetes. Diabetes Care. 1999;22:1245–1251.
124 Yusuf S, Dagenais G, Pogue J, et al. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:154–160.
125 Xia P, Aiello LP, Ishii H, et al. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, endothelial cell growth. J Clin Invest. 1996;98:2018–2026.
126 Xia P, Inoguchi T, Kern TS, et al. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43:1122–1129.
127 Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614.
128 Inoguchi T, Battan R, Handler E, et al. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–11063.
129 Ishii H, Koya D, King GL. Protein kinase C activation and its role in the development of vascular complications in diabetes mellitus. J Mol Med. 1998;76:21–31.
130 Nagpala PG, Malik AB, Vuong PT, et al. Protein kinase C beta 1 overexpression augments phorbol ester-induced increase in endothelial permeability. J Cell Physiol. 1996;166:249–255.
131 Kuboki K, Jiang ZY, Takahara N, et al. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation. 2000;101:676–681.
132 Bohlen HG, Nase GP. Arteriolar nitric oxide concentration is decreased during hyperglycemia-induced betaII PKC activation. Am J Physiol Heart Circ Physiol. 2001;280:H621–H627.
133 Nonaka A, Kiryu J, Tsujikawa A, et al. PKC-beta inhibitor (LY333531) attenuates leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 2000;41:2702–2706.
134 Shiba T, Inoguchi T, Sportsman JR, et al. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Am J Physiol. 1993;265:E783–E793.
135 Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–731.
136 Aiello LP, Bursell SE, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480.
137 Schiffrin EL, Touyz RM. Vascular biology of endothelin. J Cardiovasc Pharmacol. 1998;32(Suppl 3):S2–13.
138 Williams B, Gallacher B, Patel H, et al. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes. 1997;46:1497–1503.
139 Koya D, Jirousek MR, Lin YW, et al. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–126.
140 Tomlinson DR. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia. 1999;42:1271–1281.
141 Meier M, King GL. Protein kinase C activation and its pharmacological inhibition in vascular disease. Vasc Med. 2000;5:173–185.
142 Danis RP, Bingaman DP, Jirousek M, et al. Inhibition of intraocular neovascularization caused by retinal ischemia in pigs by PKCbeta inhibition with LY333531. Invest Ophthalmol Vis Sci. 1998;39:171–179.
143 Nakamura J, Kato K, Hamada Y, et al. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48:2090–2095.
144 Effect of ruboxistaurin in patients with diabetic macular edema: thirty-month results of the randomized PKC-DMES clinical trial. Arch Ophthalmol. 2007;125:318–324.
145 Sheetz MJ, Aiello LP, Shahri N. Effect of ruboxistaurin (RBX) on visual acuity decline over a 6-year period with cessation and reinstitution of therapy: results of an open-label extension of the Protein Kinase C Diabetic Retinopathy Study 2 (PKC-DRS2). Retina. 2011;31:1053–1059.
146 King GL, Buzney SM, Kahn CR, et al. Differential responsiveness to insulin of endothelial and support cells from micro- and macrovessels. J Clin Invest. 1983;71:974–979.
147 Engerman RL, Pfaffenb D, Davis MD. Cell turnover of capillaries. Lab Invest. 1967;17:738–743.
148 Kumagai AK, Glasgow BJ, Pardridge WM. GLUT1 glucose transporter expression in the diabetic and nondiabetic human eye. Invest Ophthalmol Vis Sci. 1994;35:2887–2894.
149 Kumagai AK, Vinores SA, Pardridge WM. Pathological upregulation of inner blood-retinal barrier Glut1 glucose transporter expression in diabetes mellitus. Brain Res. 1996;706:313–317.
150 Mantych GJ, Hageman GS, Devaskar SU. Characterization of glucose transporter isoforms in the adult and developing human eye. Endocrinology. 1993;133:600–607.
151 Watanabe T, Mio Y, Hoshino FB, et al. GLUT2 expression in the rat retina: localization at the apical ends of Muller cells. Brain Res. 1994;655:128–134.
152 Watanabe T, Matsushima S, Okazaki M, et al. Localization and ontogeny of GLUT3 expression in the rat retina. Brain Res Dev Brain Res. 1996;94:60–66.
153 Frank RN, Amin R, Kennedy A, et al. An aldose reductase inhibitor and aminoguanidine prevent vascular endothelial growth factor expression in rats with long-term galactosemia. Arch Ophthalmol. 1997;115:1036–1047.
154 Warpeha KM, Chakravarthy U. Molecular genetics of microvascular disease in diabetic retinopathy. Eye (Lond). 2003;17:305–311.
155 Field LL. Genetic linkage and association studies of Type I diabetes: challenges and rewards. Diabetologia. 2002;45:21–35.
156 Kao YL, Donaghue K, Chan A, et al. An aldose reductase intragenic polymorphism associated with diabetic retinopathy. Diabetes Res Clin Pract. 1999;46:155–160.
157 Demaine A, Cross D, Millward A. Polymorphisms of the aldose reductase gene and susceptibility to retinopathy in type 1 diabetes mellitus. Invest Ophthalmol Vis Sci. 2000;41:4064–4068.
158 Demaine AG. Polymorphisms of the aldose reductase gene and susceptibility to diabetic microvascular complications. Curr Med Chem. 2003;10:1389–1398.
159 Wang Y, Ng MC, Lee SC, et al. Phenotypic heterogeneity and associations of two aldose reductase gene polymorphisms with nephropathy and retinopathy in type 2 diabetes. Diabetes Care. 2003;26:2410–2415.
160 Sivenius K, Niskanen L, Voutilainen-Kaunisto R, et al. Aldose reductase gene polymorphisms and susceptibility to microvascular complications in Type 2 diabetes. Diabet Med. 2004;21:1325–1333.
161 Petrovic MG, Peterlin B, Hawlina M, et al. Aldose reductase (AC)n gene polymorphism and susceptibility to diabetic retinopathy in Type 2 diabetes in Caucasians. J Diabetes Complications. 2005;19:70–73.
162 Richeti F, Noronha RM, Waetge RT, et al. Evaluation of AC(n) and C(-106)T polymorphisms of the aldose reductase gene in Brazilian patients with DM1 and susceptibility to diabetic retinopathy. Mol Vis. 2007;13:740–745.
163 Klein R, Klein BE, Moss SE, et al. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol. 1984;102:527–532.
164 Klein R, Klein BE, Moss SE, et al. The Wisconsin epidemiologic study of diabetic retinopathy. II. Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch Ophthalmol. 1984;102:520–526.
165 Rand LI, Krolewski AS, Aiello LM, et al. Multiple factors in the prediction of risk of proliferative diabetic retinopathy. N Engl J Med. 1985;313:1433–1438.
166 Cruickshanks KJ, Vadheim CM, Moss SE, et al. Genetic marker associations with proliferative retinopathy in persons diagnosed with diabetes before 30 yr of age. Diabetes. 1992;41:879–885.
167 The Diabetes Control and Complications Trial Research Group. Clustering of long-term complications in families with diabetes in the diabetes control and complications trial. Diabetes. 1997;46:1829–1839.
168 Krolewski AS, Canessa M, Warram JH, et al. Predisposition to hypertension and susceptibility to renal disease in insulin-dependent diabetes mellitus. N Engl J Med. 1988;318:140–145.
169 Krolewski AS, Doria A, Magre J, et al. Molecular genetic approaches to the identification of genes involved in the development of nephropathy in insulin-dependent diabetes mellitus. J Am Soc Nephrol. 1992;3:S9–17.
170 Pettitt DJ, Saad MF, Bennett PH, et al. Familial predisposition to renal disease in two generations of Pima Indians with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1990;33:438–443.
171 Seaquist ER, Goetz FC, Rich S, et al. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320:1161–1165.
172 Ko BC, Lam KS, Wat NM, et al. An (A-C)n dinucleotide repeat polymorphic marker at the 5’ end of the aldose reductase gene is associated with early-onset diabetic retinopathy in NIDDM patients. Diabetes. 1995;44:727–732.
173 Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487.
174 Aiello LP. Angiogenic pathways in diabetic retinopathy. N Engl J Med. 2005;353:839–841.
175 Miller JW, Adamis AP, Aiello LP. Vascular endothelial growth factor in ocular neovascularization and proliferative diabetic retinopathy. Diabetes Metab Rev. 1997;13:37–50.
176 Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865.
177 Suganthalakshmi B, Anand R, Kim R, et al. Association of VEGF and eNOS gene polymorphisms in type 2 diabetic retinopathy. Mol Vis. 2006;12:336–341.
178 Buraczynska M, Ksiazek P, Baranowicz-Gaszczyk I, et al. Association of the VEGF gene polymorphism with diabetic retinopathy in type 2 diabetes patients. Nephrol Dial Transplant. 2007;22:827–832.
179 Ramprasad S, Radha V, Mathias RA, et al. Rage gene promoter polymorphisms and diabetic retinopathy in a clinic-based population from South India. Eye (Lond). 2007;21:395–401.
180 Balasubbu S, Sundaresan P, Rajendran A, et al. Association analysis of nine candidate gene polymorphisms in Indian patients with type 2 diabetic retinopathy. BMC Med Genet. 2010;11:158.
181 Tong Z, Yang Z, Patel S, et al. Promoter polymorphism of the erythropoietin gene in severe diabetic eye and kidney complications. Proc Natl Acad Sci U S A. 2008;105:6998–7003.
182 Huang YC, Lin JM, Lin HJ, et al. Genome-wide association study of diabetic retinopathy in a Taiwanese population. Ophthalmology. 2011;118:642–648.
183 Fu YP, Hallman DM, Gonzalez VH, et al. Identification of Diabetic Retinopathy Genes through a Genome-Wide Association Study among Mexican-Americans from Starr County, Texas. J Ophthalmol. 2010. 2010
184 Grassi MA, Tikhomirov A, Ramalingam S, et al. Genome-wide meta-analysis for severe diabetic retinopathy. Hum Mol Genet. 2011;20:2472–2481.
185 Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510.
186 Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389.
187 Dewan A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992.
188 Yang Z, Camp NJ, Sun H, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–993.
189 Cohen JC, Kiss RS, Pertsemlidis A, et al. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872.
190 Kotowski IK, Pertsemlidis A, Luke A, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78:410–422.
191 Ahituv N, Kavaslar N, Schackwitz W, et al. Medical sequencing at the extremes of human body mass. Am J Hum Genet. 2007;80:779–791.
192 Romeo S, Pennacchio LA, Fu Y, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–516.
193 Smigrodzki R, Parks J, Parker WD. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol Aging. 2004;25:1273–1281.
194 Bielas JH, Loeb KR, Rubin BP, et al. Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A. 2006;103:18238–18242.
195 Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888.
196 Becker B. Diabetes and glaucoma; and Ashton N. Oxygen and the growth and development of retinal vessels: in vivo and in vitro studies. In: Kimura SJ, Caygill WM. Vascular complications of diabetes mellitus, with special emphasis on microangiopathy of the eye. St Louis: Mosby, 1967.
197 Jain IS, Luthra CL, Das T. Diabetic retinopathy and its relation to errors of refraction. Arch Ophthalmol. 1967;77:59–60.
198 Goldberg MF, Fine SL, United States Public Health Service. Symposium on the Treatment of Diabetic Retinopathy. Arlington, VA: US Neurological and Sensory Disease Control Program; for sale by the Superintendent of Documents, US Government Printing Office, Washington, DC; 1969.
199 Patz A. Studies on retinal neovascularization. Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1980;19:1133–1138.
200 Weiter JJ, Zuckerman R. The influence of the photoreceptor-RPE complex on the inner retina. An explanation for the beneficial effects of photocoagulation. Ophthalmology. 1980;87:1133–1139.
201 The Diabetic Retinopathy Study Research Group. Photocoagulation treatment of proliferative diabetic retinopathy. Clinical application of Diabetic Retinopathy Study (DRS) findings, DRS Report number 8. Ophthalmology. 1981;88:583–600.
202 Klein R, Klein BE, Moss SE, et al. The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology. 1984;91:1464–1474.
203 Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24:44–53.
204 Patz A, Maumenee AE. Studies on diabetic retinopathy. I. Retinopathy in a dog with spontaneous diabetes mellitus. Am J Ophthalmol. 1962;54:532–541.
205 Engerman R, Finkelstein D, Aguirre G, et al. Ocular complications. Diabetes. 1982;31:82–88.
206 Tilton RG, LaRose LS, Kilo C, et al. Absence of degenerative changes in retinal and uveal capillary pericytes in diabetic rats. Invest Ophthalmol Vis Sci. 1986;27:716–721.
207 Engerman RL, Colquhoun PJ. Epithelial and mesothelial basement membranes in diabetic patients and dogs. Diabetologia. 1982;23:521–524.
208 Kern TS, Engerman RL. Galactose-induced retinal microangiopathy in rats. Invest Ophthalmol Vis Sci. 1995;36:490–496.
209 Robison WG, Jr., McCaleb ML, Feld LG, et al. Degenerated intramural pericytes (‘ghost cells’) in the retinal capillaries of diabetic rats. Curr Eye Res. 1991;10:339–350.
210 Bloodworth JM, Jr., Engerman RL, Anderson PJ. Microangiopathy in the experimentally diabetic animal. Adv Metab Disord. 1973;2(Suppl 2):245–250.
211 Frank RN, Hoffman WH, Podgor MJ, et al. Retinopathy in juvenile-onset diabetes of short duration. Ophthalmology. 1980;87:1–9.
212 Palmberg P, Smith M, Waltman S, et al. The natural history of retinopathy in insulin-dependent juvenile-onset diabetes. Ophthalmology. 1981;88:613–618.
213 Kern TS, Engerman RL. A mouse model of diabetic retinopathy. Arch Ophthalmol. 1996;114:986–990.
214 de Oliveira F. Pericytes in diabetic retinopathy. Br J Ophthalmol. 1966;50:134–143.
215 Kern TS, Engerman RL. Capillary lesions develop in retina rather than cerebral cortex in diabetes and experimental galactosemia. Arch Ophthalmol. 1996;114:306–310.
216 Frank RN, Dutta S, Mancini MA. Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Invest Ophthalmol Vis Sci. 1987;28:1086–1091.
217 Frank RN, Turczyn TJ, Das A. Pericyte coverage of retinal and cerebral capillaries. Invest Ophthalmol Vis Sci. 1990;31:999–1007.
218 Tilton RG, Miller EJ, Kilo C, et al. Pericyte form and distribution in rat retinal and uveal capillaries. Invest Ophthalmol Vis Sci. 1985;26:68–73.
219 Patz A. The role of oxygen in retrolental fibroplasia. Trans Am Ophthalmol Soc. 1968;66:940–985.
220 Kremer I, Kissun R, Nissenkorn I, et al. Oxygen-induced retinopathy in newborn kittens. A model for ischemic vasoproliferative retinopathy. Invest Ophthalmol Vis Sci. 1987;28:126–130.
221 Wise GN, Dollery CT, Henkind P. The retinal circulation. New York: Harper & Row; 1971.
222 Smith LE, Kopchick JJ, Chen W, et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science. 1997;276:1706–1709.
223 Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A. 1995;92:10457–10461.
224 Penn JS, Rajaratnam VS, Collier RJ, et al. The effect of an angiostatic steroid on neovascularization in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2001;42:283–290.
225 Okamoto N, Tobe T, Hackett SF, et al. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol. 1997;151:281–291.
226 Buzney SM, Frank RN, Robison WG, Jr. Retinal capillaries: proliferation of mural cells in vitro. Science. 1975;190:985–986.
227 Frank RN, Kinsey VE, Frank KW, et al. In vitro proliferation of endothelial cells from kitten retinal capillaries. Invest Ophthalmol Vis Sci. 1979;18:1195–1200.
228 Bowman PD, Betz AL, Goldstein GW. Primary culture of microvascular endothelial cells from bovine retina: selective growth using fibronectin coated substrate and plasma derived serum. In Vitro. 1982;18:626–632.
229 Buzney SM, Massicotte SJ. Retinal vessels: proliferation of endothelium in vitro. Invest Ophthalmol Vis Sci. 1979;18:1191–1195.
230 Gitlin JD, D’Amore PA. Culture of retinal capillary cells using selective growth media. Microvasc Res. 1983;26:74–80.
231 Madri JA, Williams SK. Capillary endothelial cell cultures: phenotypic modulation by matrix components. J Cell Biol. 1983;97:153–165.
232 Montesano R, Orci L, Vassalli P. In vitro rapid organization of endothelial cells into capillary-like networks is promoted by collagen matrices. J Cell Biol. 1983;97:1648–1652.
233 Goto F, Goto K, Weindel K, et al. Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab Invest. 1993;69:508–517.
234 Herman IM, D’Amore PA. Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol. 1985;101:43–52.
235 Kelley C, D’Amore P, Hechtman HB, et al. Microvascular pericyte contractility in vitro: comparison with other cells of the vascular wall. J Cell Biol. 1987;104:483–490.
236 Das A, Frank RN, Weber ML, et al. ATP causes retinal pericytes to contract in vitro. Exp Eye Res. 1988;46:349–362.
237 Kelley C, D’Amore P, Hechtman HB, et al. Vasoactive hormones and cAMP affect pericyte contraction and stress fibres in vitro. J Muscle Res Cell Motil. 1988;9:184–194.
238 Chakravarthy U, Gardiner TA, Anderson P, et al. The effect of endothelin 1 on the retinal microvascular pericyte. Microvasc Res. 1992;43:241–254.
239 Tilton RG, Kilo C, Williamson JR, et al. Differences in pericyte contractile function in rat cardiac and skeletal muscle microvasculatures. Microvasc Res. 1979;18:336–352.
240 Butryn RK, Ruan H, Hull CM, et al. Vasoactive agonists do not change the caliber of retinal capillaries of the rat. Microvasc Res. 1995;50:80–93.