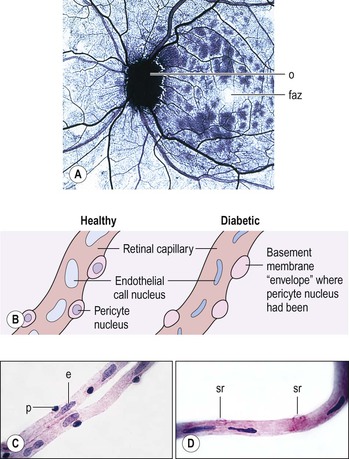
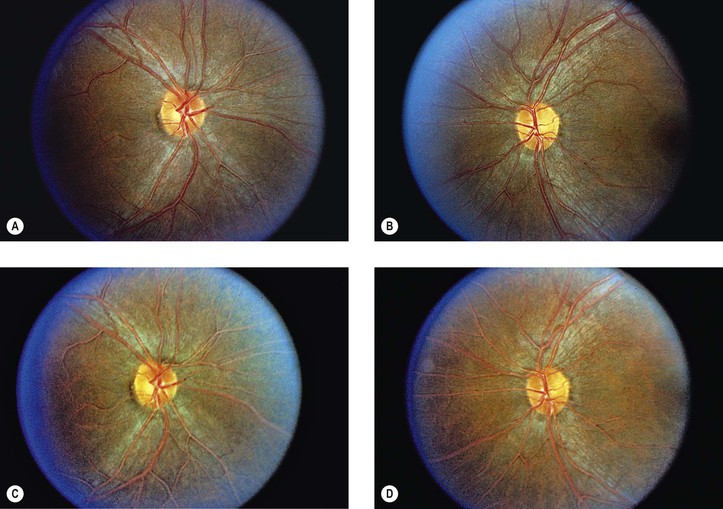
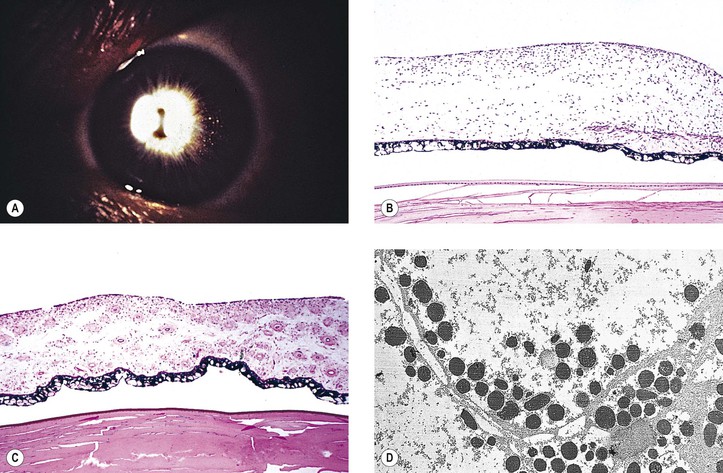
Bibliography
Natural History
Andresen AR, Christiansen JS, Jenson JK. Diagonal ear-lobe crease and diabetic retinal angiopathy [letter]. N Engl J Med. 1976;294:1182.
Canataroglu H, Varinli I, Ozcan AA, et al. Interleukin (IL)-6, interleukin (IL)-8 levels and cellular composition of the vitreous humor in proliferative diabetic retinopathy, proliferative vitreoretinopathy, and traumatic proliferative vitreoretinopathy. Ocul Immunol Inflamm. 2005;13:375.
Caprio S. Development of type 2 diabetes mellitus in the obese adolescent: A growing challenge. Endocr Pract. 2012;18:791.
Chase HP, Garg SK, Jackson WE, et al. Blood pressure and retinopathy in type 1 diabetes. Ophthalmology. 1990;97:155.
Chopra V, Varma R, Francis BA, et al. Type 2 diabetes mellitus and the risk of open-angle glaucoma: The Los Angeles Latino Eye Study. Ophthalmology. 2008;115:227.
Christen U, Bender C, von Herrath MG. Infection as a cause of type 1 diabetes? Curr Opin Rheumatol. 2012;24:417.
Davis MD, Hiller R, Magli YL, et al. Prognosis for life in patients with diabetes: Relation to severity of retinopathy. Trans Am Ophthalmol Soc. 1979;77:144.
Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther. 2008;88:1254.
Diabetes Control and Complications Trial Research Group. Early worsening of diabetic retinopathy in the Diabetes Control and Complications Trial. Ophthalmology. 1998;116:874.
Dielemans I, deJong PTVM, Stolk R, et al. Primary open-angle glaucoma, intraocular pressure, and diabetes mellitus in the general population: The Rotterdam Study. Ophthalmology. 1996;103:1271.
Efron N. The Glenn A. Fry award lecture 2010: Ophthalmic markers of diabetic neuropathy. Optom Vis Sci. 2011;88:661.
Forlenza GP, Rewers M. The epidemic of type 1 diabetes: What is it telling us? Curr Opin Endocrinol Diabetes Obes. 2011;18:248.
Frank RN. The Optic UK Lecture: Bench-to-bedside adventures of a diabetes researcher: Results past, results present. Eye (Lond). 2011;25:331.
Gale EA. Viruses and type 1 diabetes: Ignorance acquires a better vocabulary. Clin Exp Immunol. 2012;168:1.
Goldstein DE, Blinder KJ, Ide CH, et al. Glycemic control and development of retinopathy in youth-onset insulin-dependent diabetes mellitus. Ophthalmology. 1993;100:1125.
Grauslund J. Long-term mortality and retinopathy in type 1 diabetes. Acta Ophthalmol. 2010;88:1.
Grauslund J, Green A, Sjolie AK. Blindness in a 25-year follow-up of a population-based cohort of Danish type 1 diabetic patients. Ophthalmology. 2009;116:2170.
Grimsby JL, Porneala BC, Vassy JL, et al. Race–ethnic differences in the association of genetic loci with HbA1c levels and mortality in U.S. adults: The third National Health and Nutrition Examination Survey (NHANES III). BMC Med Genet. 2012;13:30.
Grosso A, Cheung N, Veglio F, et al. Similarities and differences in early retinal phenotypes in hypertension and diabetes. J Hypertens. 2011;29:1667.
Guery B, Choukroun G, Noel LH, et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol. 2003;14:2099.
Hober D, Sane F, Jaidane H, et al. Immunology in the clinic review series; focus on type 1 diabetes and viruses: Role of antibodies enhancing the infection with Coxsackievirus-B in the pathogenesis of type 1 diabetes. Clin Exp Immunol. 2012;168:47.
Ido Y, Vindigni A, Chang K, et al. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science. 1997;277:563.
Kaul K, Hodgkinson A, Tarr JM, et al. Is inflammation a common retinal–renal–nerve pathogenic link in diabetes? Curr Diabetes Rev. 2010;6:294.
Klein R, Klein BEK, Moss SE, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XIV. Ten-year incidence and progression of diabetic retinopathy. Arch Ophthalmol. 1994;112:1217.
Klein R, Klein BEK, Moss SE, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XV. The long-year incidence of macular edema. Ophthalmology. 1995;102:7.
Klein R, Klein BEK, Moss SE, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology. 1998;105:1801.
Klein R, Knudtson MD, Lee KE, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XXII. The twenty-five-year progression of retinopathy in persons with type 1 diabetes. Ophthalmology. 2008;115:1859.
Klein R, Meuer SM, Moss SE, et al. Retinal microaneurysm counts and progression of diabetic retinopathy. Arch Ophthalmol. 1995;113:1386.
Klein R, Meuer SM, Moss SE, et al. Incidence of retinopathy and associated risk factors from time of diagnosis of insulin-dependent diabetes. Arch Ophthalmol. 1997;115:351.
Klein R, Sharrett AR, Klein BE, et al. The association of atherosclerosis, vascular risk factors, and retinopathy in adults with diabetes: The Atherosclerosis Risk in Communities Study. Ophthalmology. 2002;109:1225.
Kyto JP, Harjutsalo V, Forsblom C, et al. Decline in the cumulative incidence of severe diabetic retinopathy in patients with type 1 diabetes. Diabetes Care. 2011;34:2005.
Leal EC, Santiago AR, Ambrosio AF. Old and new drug targets in diabetic retinopathy: From biochemical changes to inflammation and neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2005;4:421.
Lind K, Huhn MH, Flodstrom-Tullberg M. Immunology in the clinic review series—Focus on type 1 diabetes and viruses: The innate immune response to enteroviruses and its possible role in regulating type 1 diabetes. Clin Exp Immunol. 2012;168:30.
Lonneville YH, Ozdek SC, Onol M, et al. The effect of blood glucose regulation on retinal nerve fiber layer thickness in diabetic patients. Ophthalmologica. 2003;217:347.
Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2013;380:2095.
Marshall G, Garg SK, Jackson WE, et al. Factors influencing the onset and progression of diabetic retinopathy in subjects with insulin-dependent diabetes mellitus. Ophthalmology. 1993;100:1133.
McCollister KE, Zheng DD, Fernandez CA, et al. Racial disparities in quality-adjusted life-years associated with diabetes and visual impairment. Diabetes Care. 2012;35:1692.
Miceli MV, Newsome DA. Cultured retinal pigment cells from donors with type I diabetes show an altered insulin response. Invest Ophthalmol Vis Sci. 1991;32:2847.
Mitchell P, Smith W, Chey T, et al. Open-angle glaucoma and diabetes: The Blue Mountain Eye Study, Australia. Ophthalmology. 1997;104:712.
Mohan R, Rajendran B, Mohan V, et al. Retinopathy in tropical pancreatic diabetes. Arch Ophthalmol. 1985;103:1487.
Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981.
Moss SE, Klein R, Klein BEK. Ten-year incidence of visual loss in a diabetic population. Ophthalmology. 1994;101:1061.
Moss SE, Klein R, Meuer MB, et al. The association of iris color with eye disease in diabetes. Ophthalmology. 1987;94:1226.
Sacks DA, McDonald JM. The pathogenesis of type II diabetes mellitus: A polygenic disease. Am J Clin Pathol. 1996;105:149.
Sentell TL, He G, Gregg EW, Schillinger D. Racial/ethnic variation in prevalence estimates for United States prediabetes under alternative 2010 American Diabetes Association criteria: 1988–2008. Ethn Dis. 2012;22:451.
Sivaprasad S, Gupta B, Crosby-Nwaobi R, et al. Prevalence of diabetic retinopathy in various ethnic groups: A worldwide perspective. Surv Ophthalmol. 2012;57:347.
Takahashi H, Goto T, Shoji T, et al. Diabetes-associated retinal nerve fiber damage evaluated with scanning laser polarimetry. Am J Ophthalmol. 2006;142:88.
van de Enden M, Nyengaard JR, Ostrow E, et al. Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism: Implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 1995;36:1675.
Van Leiden HA, Dekker JM, Moll AC, et al. Risk factors for incident retinopathy in a diabetic and nondiabetic population. Arch Ophthalmol. 2003;121:245.
Wong TY, Cheung N, Islam FM, et al. Relation of retinopathy to coronary artery calcification: The multi-ethnic study of atherosclerosis. Am J Epidemiol. 2008;167:51.
Wong TY, Klein R, Islam FM, et al. Diabetic retinopathy in a multi-ethnic cohort in the United States. Am J Ophthalmol. 2006;141:446.
Yau JW, Rogers SL, Kawasaki R, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35:556.
Zhang X, Saaddine JB, Chou CF, et al. Prevalence of diabetic retinopathy in the United States, 2005–2008. JAMA. 2010;304:649.
Conjunctiva and Cornea
Ahmed A, Bril V, Orszag A, et al. Detection of diabetic sensorimotor polyneuropathy by corneal confocal microscopy in type 1 diabetes: A concurrent validity study. Diabetes Care. 2012;35:821.
Arnarsson A, Jonasson F, Sasaki H, et al. Risk factors for nuclear lens opacification: The Reykjavik Eye Study. Dev Ophthalmol. 2002;35:12.
Azar DT, Spurr-Michaud SJ, Tisdale AS, et al. Decreased penetration of anchoring fibrils into the diabetic stroma. Arch Ophthalmol. 1989;107:1520.
Brandt JD, Beiser JA, Kass MA, et al. Central corneal thickness in the Ocular Hypertension Treatment Study (OHTS). Ophthalmology. 2001;108:1779.
Chang PY, Carrel H, Huang JS, et al. Decreased density of corneal basal epithelium and subbasal corneal nerve bundle changes in patients with diabetic retinopathy. Am J Ophthalmol. 2006;142:488.
Cheung AT, Price AR, Duong PL, et al. Microvascular abnormalities in pediatric diabetic patients. Microvasc Res. 2002;63:252.
Cheung AT, Ramanujam S, Greer DA, et al. Microvascular abnormalities in the bulbar conjunctiva of patients with type 2 diabetes mellitus. Endocr Pract. 2001;7:358.
Dabbah MA, Graham J, Petropoulos I, et al. Dual-model automatic detection of nerve-fibres in corneal confocal microscopy images. Med Image Comput Comput Assist Interv. 2010;13:300.
Edwards K, Pritchard N, Vagenas D, et al. Utility of corneal confocal microscopy for assessing mild diabetic neuropathy: Baseline findings of the LANDMark study. Clin Exp Optom. 2012;95:348.
Efron N, Edwards K, Roper N, et al. Repeatability of measuring corneal subbasal nerve fiber length in individuals with type 2 diabetes. Eye Contact Lens. 2010;36:245.
Fujishima H, Tsubota K. Improvement of corneal fluorescein staining in post cataract surgery of diabetic patients by an oral aldose reductase inhibitor, ONO-2235. Br J Ophthalmol. 2002;86:860.
Gonzalez-Meijome JM, Jorge J, Queiros A, et al. Two single descriptors of endothelial polymegethism and pleomorphism. Graefes Arch Clin Exp Ophthalmol. 2010;248:1159.
He J, Bazan HE. Mapping the nerve architecture of diabetic human corneas. Ophthalmology. 2012;119:956.
Hertz P, Bril V, Orszag A, et al. Reproducibility of in vivo corneal confocal microscopy as a novel screening test for early diabetic sensorimotor polyneuropathy. Diabet Med. 2011;28:1253.
Hiraoka M, Amano S, Oshika T, et al. Factors contributing to corneal complications after vitrectomy in diabetic patients. Jpn J Ophthalmol. 2001;45:492.
Hugod M, Storr-Paulsen A, Norregaard JC, et al. Corneal endothelial cell changes associated with cataract surgery in patients with type 2 diabetes mellitus. Cornea. 2011;30:749.
Inoue K, Kato S, Ohara C, et al. Ocular and systemic factors relevant to diabetic keratoepitheliopathy. Cornea. 2001;20:798.
Jacques PF, Moeller SM, Hankinson SE, et al. Weight status, abdominal adiposity, diabetes, and early age-related lens opacities. Am J Clin Nutr. 2003;78:400.
Kallinikos P, Berhanu M, O’Donnell C, et al. Corneal nerve tortuosity in diabetic patients with neuropathy. Invest Ophthalmol Vis Sci. 2004;45:418.
Keoleian GM, Pach JM, Hodge DO, et al. Structural and functional studies of the corneal endothelium in diabetes mellitus. Am J Ophthalmol. 1992;113:64.
Khalfaoui T, Lizard G, Ouertani-Meddeb A. Adhesion molecules (ICAM-1 and VCAM-1) and diabetic retinopathy in type 2 diabetes. J Mol Histol. 2008;39:243.
Kria L, Khalfaoui T, Mkannez G, et al. Immunohistochemical study of vascular endothelial growth factor (VEGF), tumor suppressor protein (p53) and intercellular adhesion molecule (ICAM-1) in the conjunctiva of diabetic patients. J Mol Histol. 2005;36:381.
Larson L-I, Bourne WM, Pach JM, et al. Structure and function of the corneal endothelium in diabetes mellitus type I and type II. Arch Ophthalmol. 1996;114:9.
Lee JS, Lee JE, Choi HY, et al. Corneal endothelial cell change after phacoemulsification relative to the severity of diabetic retinopathy. J Cataract Refract Surg. 2005;31:742.
Leem HS, Lee KJ, Shin KC. Central corneal thickness and corneal endothelial cell changes caused by contact lens use in diabetic patients. Yonsei Med J. 2011;52:322.
Leske MC, Wu SY, Nemesure B, et al. Risk factors for incident nuclear opacities. Ophthalmology. 2002;109:1303.
Malik RA, Kallinikos P, Abbott CA, et al. Corneal confocal microscopy: A non-invasive surrogate of nerve fibre damage and repair in diabetic patients. Diabetologia. 2003;46:683.
Mathew PT, David S, Thomas N. Endothelial cell loss and central corneal thickness in patients with and without diabetes after manual small incision cataract surgery. Cornea. 2011;30:424.
Messmer EM, Schmid-Tannwald C, Zapp D, et al. In vivo confocal microscopy of corneal small fiber damage in diabetes mellitus. Graefes Arch Clin Exp Ophthalmol. 2010;248:1307.
Midena E, Brugin E, Ghirlando A, et al. Corneal diabetic neuropathy: A confocal microscopy study. J Refract Surg. 2006;22:S1047.
Mimura T, Obata H, Usui T, et al. Pinguecula and diabetes mellitus. Cornea. 2012;31:264.
Mocan MC, Durukan I, Irkec M, et al. Morphologic alterations of both the stromal and subbasal nerves in the corneas of patients with diabetes. Cornea. 2006;25:769.
Modis L Jr, Szalai E, Kertesz K, et al. Evaluation of the corneal endothelium in patients with diabetes mellitus type I and II. Histol Histopathol. 2010;25:1531.
Morikubo S, Takamura Y, Kubo E, et al. Corneal changes after small-incision cataract surgery in patients with diabetes mellitus. Arch Ophthalmol. 2004;122:966.
Okamura N, Ito Y, Shibata MA, et al. Fas-mediated apoptosis in human lens epithelial cells of cataracts associated with diabetic retinopathy. Med Electron Microsc. 2002;35:234.
Price MO, Thompson RW Jr, Price FW Jr. Risk factors for various causes of failure in initial corneal grafts. Arch Ophthalmol. 2003;121:1087.
Saghizadeh M, Kramerov AA, Tajbakhsh J, et al. Proteinase and growth factor alterations revealed by gene microarray analysis of human diabetic corneas. Invest Ophthalmol Vis Sci. 2005;46:3604.
Sani JS, Mittal S. In vivo assessment of corneal endothelial function in diabetes mellitus. Arch Ophthalmol. 1996;114:649.
Schultz RO, van Horn DL, Peters MA, et al. Diabetic keratopathy. Trans Am Ophthalmol Soc. 1981;74:180.
Shenoy R, Khandekar R, Bialasiewicz A, et al. Corneal endothelium in patients with diabetes mellitus: A historical cohort study. Eur J Ophthalmol. 2009;19:369.
Shetlar DJ, Bourne WM, Campbell RJ. Morphologic evaluation of Descemet’s membrane and corneal endothelium in diabetes mellitus. Ophthalmology. 1989;96:247.
Siribunkum J, Kosrirukvongs P, Singalavanija A. Corneal abnormalities in diabetes. J Med Assoc Thai. 2001;84:1075.
Su DH, Wong TY, Foster PJ, et al. Central corneal thickness and its associations with ocular and systemic factors: The Singapore Malay Eye Study. Am J Ophthalmol. 2009;147:709.
Su DH, Wong TY, Wong WL, et al. Diabetes, hyperglycemia, and central corneal thickness: The Singapore Malay Eye Study. Ophthalmology. 2008;115:964.
Su F, Li B, Wang J, et al. Molecular regulation of vasculogenic mimicry in human uveal melanoma cells: Role of helix–loop–helix Id2 (inhibitor of DNA binding 2). Graefes Arch Clin Exp Ophthalmol. 2009;247:411.
Sudhir RR, Raman R, Sharma T. Changes in the corneal endothelial cell density and morphology in patients with type 2 diabetes mellitus: A population-based study, Sankara Nethralaya Diabetic Retinopathy and Molecular Genetics Study (SN-DREAMS, Report 23). Cornea. 2012;31:1119.
Tavakoli M, Boulton AJ, Efron N, et al. Increased Langerhan cell density and corneal nerve damage in diabetic patients: Role of immune mechanisms in human diabetic neuropathy. Cont Lens Anterior Eye. 2011;34:7.
Tavakoli M, Kallinikos P, Iqbal A, et al. Corneal confocal microscopy detects improvement in corneal nerve morphology with an improvement in risk factors for diabetic neuropathy. Diabet Med. 2011;28:1261.
Taylor HR, Kimsey RA. Corneal epithelial basement membrane changes in diabetes. Invest Ophthalmol. 1981;20:548.
To WJ, Telander DG, Lloyd ME, et al. Correlation of conjunctival microangiopathy with retinopathy in type-2 diabetes mellitus (T2DM) patients. Clin Hemorheol Microcirc. 2011;47:131.
Yoon KC, Im SK, Seo MS. Changes of tear film and ocular surface in diabetes mellitus. Korean J Ophthalmol. 2004;18:168.
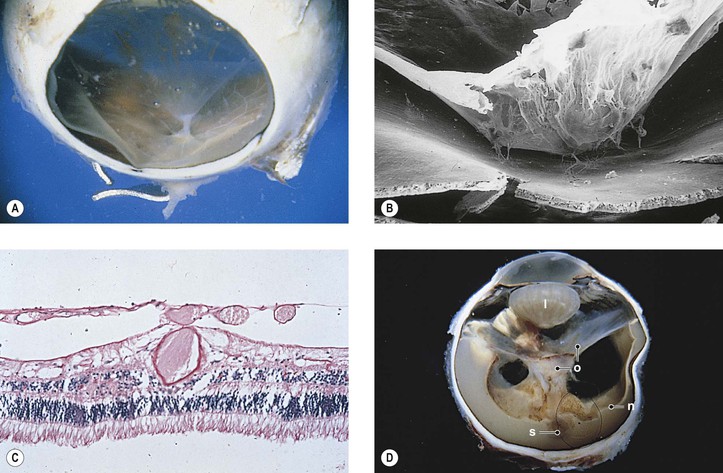
Lens
Agte VV, Tarwadi KV. Combination of diabetes and cataract worsens the oxidative stress and micronutrient status in Indians. Nutrition. 2008;24:617.
Barber AJ. A new view of diabetic retinopathy: A neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:283.
Biswas S, Harris F, Singh J, et al. Role of calpains in diabetes mellitus-induced cataractogenesis: A mini review. Mol Cell Biochem. 2004;261:151.
Chung SS, Chung SK. Genetic analysis of aldose reductase in diabetic complications. Curr Med Chem. 2003;10:1375.
Cunliffe IA, Flanagan DW, George NDL, et al. Extracapsular cataract surgery with lens implantation in diabetics with and without proliferative retinopathy. Br J Ophthalmol. 1991;75:9.
Datiles MB, Kador PF. Type I diabetic cataract. Arch Ophthalmol. 1999;117:284.
Degenring RF, Vey S, Kamppeter B, et al. Effect of uncomplicated phacoemulsification on the central retina in diabetic and non-diabetic subjects. Graefes Arch Clin Exp Ophthalmol. 2007;245:18.
Fan H, Suzuki T, Ogata M, et al. Expression of PCNA, ICAM-1, and vimentin in lens epithelial cells of cataract patients with and without type 2 diabetes. Tokai J Exp Clin Med. 2012;37:51.
Freel CD, al-Ghoul KJ, Kuszak JR, et al. Analysis of nuclear fiber cell compaction in transparent and cataractous diabetic human lenses by scanning electron microscopy. BMC Ophthalmol. 2003;3:1.
Gardner TW, Antonetti DA, Barber AJ, et al. Diabetic retinopathy: More than meets the eye. Surv Ophthalmol. 2002;47(Suppl 2):S253.
Grauslund J. Eye complications and markers of morbidity and mortality in long-term type 1 diabetes. Acta Ophthalmol. 2011;89:1.
Gul A, Rahman MA, Hasnain SN, et al. Could oxidative stress associate with age products in cataractogenesis? Curr Eye Res. 2008;33:669.
Gul A, Rahman MA, Salim A, et al. Advanced glycation end products in senile diabetic and nondiabetic patients with cataract. J Diabetes Complications. 2009;23:343.
Hayashi K, Hayashi H, Nakao F, et al. Posterior capsule opacification after cataract surgery in patients with diabetes mellitus. Am J Ophthalmol. 2002;134:10.
Jaffe GJ, Burton TC, Kuhn E, et al. Progression of nonproliferative diabetic retinopathy and visual outcome after extracapsular cataract extraction and intraocular lens implantation. Am J Ophthalmol. 1992;114:448.
Kim B, Kim SY, Chung SK. Changes in apoptosis factors in lens epithelial cells of cataract patients with diabetes mellitus. J Cataract Refract Surg. 2012;38:1376.
Klein BEK, Klein R, Moss SE. Prevalence of cataracts in a population-based study of persons with diabetes mellitus. Ophthalmology. 1985;92:1191.
Kumamoto Y, Takamura Y, Kubo E, et al. Epithelial cell density in cataractous lenses of patients with diabetes: Association with erythrocyte aldose reductase. Exp Eye Res. 2007;85:393.
Lopes de Faria JM, Katsumi O, Cagliero E, et al. Neurovisual abnormalities preceding the retinopathy in patients with long-term type 1 diabetes mellitus. Graefes Arch Clin Exp Ophthalmol. 2001;239:643.
Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44:791.
Palsamy P, Ayaki M, Elanchezhian R, et al. Promoter demethylation of Keap1 gene in human diabetic cataractous lenses. Biochem Biophys Res Commun. 2012;423:542.
Patel JI, Hykin PG, Cree IA. Diabetic cataract removal: Postoperative progression of maculopathy—Growth factor and clinical analysis. Br J Ophthalmol. 2006;90:697.
Pollack A, Dotan S, Oliver M. Course of diabetic retinopathy following cataract surgery. Br J Ophthalmol. 1991;75:2.
Rahim A, Iqbal K. To assess the levels of zinc in serum and changes in the lens of diabetic and senile cataract patients. J Pak Med Assoc. 2011;61:853.
Richter GM, Choudhury F, Torres M, et al. Risk factors for incident cortical, nuclear, posterior subcapsular, and mixed lens opacities: The Los Angeles Latino eye study. Ophthalmology. 2012;119:2040.
Santiago AP, Rosenbaum AL, Masket S. Insulin-dependent diabetes mellitus appearing as bilateral mature diabetic cataracts in a child. Arch Ophthalmol. 1997;115:422.
Schatz H, Atienza D, McDonald HR, et al. Severe diabetic retinopathy after cataract surgery. Am J Ophthalmol. 1994;117:314.
Smith R. Diabetic retinopathy and cataract surgery. Br J Ophthalmol. 1991;75:1.
Tkachov SI, Lautenschlager C, Ehrich D, et al. Changes in the lens epithelium with respect to cataractogenesis: Light microscopic and Scheimpflug densitometric analysis of the cataractous and the clear lens of diabetics and non-diabetics. Graefes Arch Clin Exp Ophthalmol. 2006;244:596.
Tsilimbaris M, Diakonis VF, Kymionis GD, et al. Prospective study of foveal thickness alterations after cataract surgery assessed by optical coherence tomography. Ophthalmologica. 2012;228:53.
Yanoff M. Ocular pathology of diabetes mellitus. Am J Ophthalmol. 1969;67:21.
Neurosensory Retina
Bandello F, Berchicci L, La SC, et al. Evidence for anti-VEGF treatment of diabetic macular edema. Ophthalmic Res. 2012;48(Suppl 1):16.
Baskin DE. Optical coherence tomography in diabetic macular edema. Curr Opin Ophthalmol. 2010;21:172.
Beutel J, Peters S, Luke M, et al. Bevacizumab as adjuvant for neovascular glaucoma. Acta Ophthalmol. 2010;88:103.
Bhagat N, Grigorian RA, Tutela A, et al. Diabetic macular edema: Pathogenesis and treatment. Surv Ophthalmol. 2009;54:1.
Biro Z, Balla Z. Foveal and perifoveal retinal thickness measured by OCT in diabetic patients after phacoemulsification cataract surgery. Oftalmologia. 2009;53(2):54.
Biro Z, Balla Z. OCT measurements on the foveal and perifoveal retinal thickness on diabetic patients after phacoemulsification and IOL implantation. Eye (Lond). 2010;24:639.
Ebihara Y, Kato S, Oshika T, et al. Posterior capsule opacification after cataract surgery in patients with diabetes mellitus. J Cataract Refract Surg. 2006;32:1184.
Feng Y, Busch S, Gretz N, et al. Crosstalk in the retinal neurovascular unit: Lessons for the diabetic retina. Exp Clin Endocrinol Diabetes. 2012;120:199.
Feng Y, vom Hagen F, Pfister F, et al. Impaired pericyte recruitment and abnormal retinal angiogenesis as a result of angiopoietin-2 overexpression. Thromb Haemost. 2007;97:99.
Fletcher EL, Phipps JA, Ward MM, et al. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr Pharm Des. 2007;13:2699.
Fletcher EL, Phipps JA, Wilkinson-Berka JL. Dysfunction of retinal neurons and glia during diabetes. Clin Exp Optom. 2005;88:132.
Forst T, Weber MM, Mitry M, et al. Pilot study for the evaluation of morphological and functional changes in retinal blood flow in patients with insulin resistance and/or type 2 diabetes mellitus. J Diabetes Sci Technol. 2012;6:163.
Gambhir D, Ananth S, Veeranan-Karmegam R, et al. GPR109A as an anti-inflammatory receptor in retinal pigment epithelial cells and its relevance to diabetic retinopathy. Invest Ophthalmol Vis Sci. 2012;53:2208.
Gustavsson C, Agardh CD, Zetterqvist AV, et al. Vascular cellular adhesion molecule-1 (VCAM-1) expression in mice retinal vessels is affected by both hyperglycemia and hyperlipidemia. PLoS One. 2010;5:e12699.
Hayashi Y, Kato S, Maeda T, et al. Immunohistologic study of interleukin-1, transforming growth factor-beta, and alpha-smooth muscle actin in lens epithelial cells in diabetic eyes. J Cataract Refract Surg. 2005;31:2187.
Ho AC, Scott IU, Kim SJ, et al. Anti-vascular endothelial growth factor pharmacotherapy for diabetic macular edema: A report by the American Academy of Ophthalmology. Ophthalmology. 2012;119:2179.
Kakehashi A, Inoda S, Mameuda C, et al. Relationship among VEGF, VEGF receptor, AGEs, and macrophages in proliferative diabetic retinopathy. Diabetes Res Clin Pract. 2008;79:438.
Koleva-Georgieva DN, Sivkova NP. Optical coherence tomography for the detection of early macular edema in diabetic patients with retinopathy. Folia Med (Plovdiv). 2010;52:40.
Lang GE. Diabetic macular edema. Ophthalmologica. 2012;227(Suppl 1):21.
Meleth AD, Agron E, Chan CC, et al. Serum inflammatory markers in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2005;46:4295.
Miller JW, Le Couter J, Strauss EC, et al. Vascular endothelial growth factor A in intraocular vascular disease. Ophthalmology. 2013;120:106.
Patel JI, Saleh GM, Hykin PG, et al. Concentration of haemodynamic and inflammatory related cytokines in diabetic retinopathy. Eye (Lond). 2008;22:223.
Sun C, Wang JJ, Mackey DA, et al. Retinal vascular caliber: Systemic, environmental, and genetic associations. Surv Ophthalmol. 2009;54:74.
Verma A, Raman R, Vaitheeswaran K, et al. Does neuronal damage precede vascular damage in subjects with type 2 diabetes mellitus and having no clinical diabetic retinopathy? Ophthalmic Res. 2012;47:202.
Verma A, Rani PK, Raman R, et al. Is neuronal dysfunction an early sign of diabetic retinopathy? Microperimetry and spectral domain optical coherence tomography (SD-OCT) study in individuals with diabetes, but no diabetic retinopathy. Eye (Lond). 2009;23:1824.
Wiemer NG, Dubbelman M, Hermans EA, et al. Changes in the internal structure of the human crystalline lens with diabetes mellitus type 1 and type 2. Ophthalmology. 2008;115:2017.
Zeng HY, Green WR, Tso MO. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. 2008;126:227.
Zong H, Ward M, Madden A, et al. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia. 2010;53:2656.
Iris
Aiello LM, Wand M, Liang G. Neovascular glaucoma and vitreous hemorrhage following cataract surgery in patients with diabetes mellitus. Ophthalmology. 1983;90:814.
Fine BS, Berkow JW, Helfgott JA. Diabetic lacy vacuolation of iris pigment epithelium. Am J Ophthalmol. 1970;69:197.
Smith ME, Glickman P. Diabetic vacuolation of the iris pigment epithelium. Am J Ophthalmol. 1975;79:875.
Yanoff M. Ocular pathology of diabetes mellitus. Am J Ophthalmol. 1969;67:21.
Yanoff M, Fine BS, Berkow JW. Diabetic lacy vacuolation of iris pigment epithelium. Am J Ophthalmol. 1970;69:201.
Ciliary Body and Choroid
Cao J, McCeod DS, Merges CA, et al. Choriocapillaris degeneration and related pathologic changes in human diabetic eyes. Arch Ophthalmol. 1998;116:589.
Fryczkowski AW, Sato SE, Hodes BL. Changes in the diabetic choroidal vasculature: Scanning electron microscopy findings. Ann Ophthalmol. 1988;20:299.
Hidayat AA, Fine BS. Diabetic choroidopathy: Light and electron microscopic observations of seven cases. Ophthalmology. 1985;92:512.
Rothova A, Meenken C, Michels RPJ, et al. Uveitis and diabetes mellitus. Am J Ophthalmol. 1988;106:17.
Yanoff M. Ocular pathology of diabetes mellitus. Am J Ophthalmol. 1969;67:21.
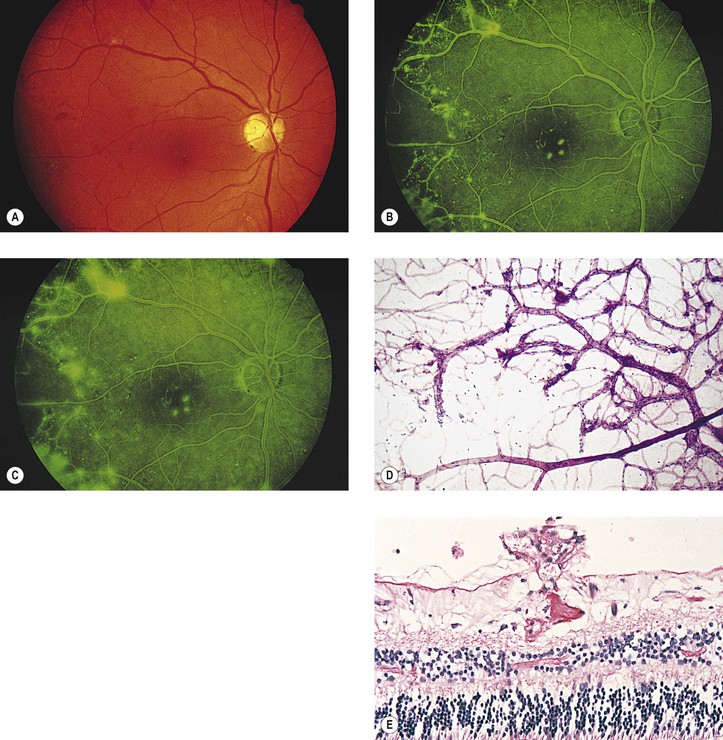
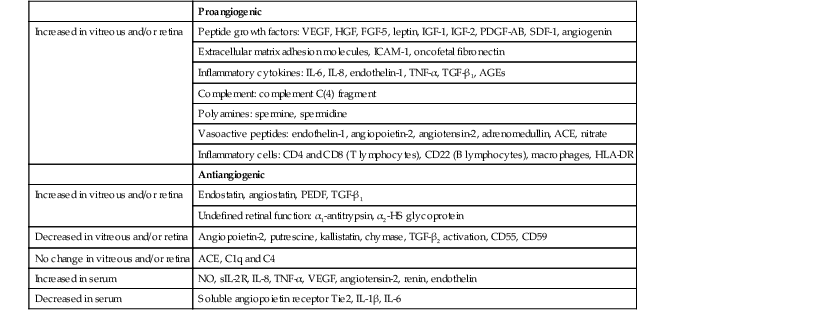
Retina
Adamis AP, Miller JW, Bernal M-T, et al. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol. 1994;118:445.
Aguilar E, Friedlander MF, Gariano RF, et al. Endothelial proliferation in diabetic retinal aneurysms. Arch Ophthalmol. 2003;121:740.
Aiello LP, Northrup JM, Keyt BA, et al. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol. 1995;113:1538.
Alzaid AA, Dinneen SF, Melton LJ III, et al. The role of growth hormone in the development of diabetic retinopathy. Diabetes Care. 1994;17:531.
Ambati J, Chalam KV, Chawla DK, et al. Elevated γ-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115:1161.
Ashton N. Vascular basement membrane changes in diabetic retinopathy. Br J Ophthalmol. 1974;58:344.
Ballantyne AJ, Lowenstein A. Diseases of the retina: I. The pathology of diabetic retinopathy. Trans Ophthalmol Soc UK. 1943;63:95.
Baudouin C, Gordon WC, Fredj-Reygrobellet D, et al. Class II antigen expression in diabetic preretinal membranes. Am J Ophthalmol. 1990;109:70.
Bek T. A clinicopathological study of venous loops and reduplications in diabetic retinopathy. Acta Ophthalmol Scand. 2002;80:69.
Bengtsson B, Heijl A, Agardh E. Visual fields correlate better than visual acuity to severity of diabetic retinopathy. Diabetologia. 2005;48:2494.
Bloodworth JMB Jr. Diabetic microangiopathy. Diabetes. 1963;12:99.
Boehm BO, Lang G, Feldmann B, et al. Proliferative diabetic retinopathy is associated with a low level of the natural ocular anti-angiogenic agent pigment epithelium-derived factor (PEDF) in aqueous humor: A pilot study. Horm Metab Res. 2003;35:382.
Boehm BO, Lang G, Volpert O, et al. Low content of the natural ocular anti-angiogenic agent pigment epithelium-derived factor (PEDF) in aqueous humor predicts progression of diabetic retinopathy. Diabetologia. 2003;46:394.
Bronson SK, Reiter CE, Gardner TW. An eye on insulin. J Clin Invest. 2003;111:1817.
Brooks PC, Clark RAF, Cheresh DA. Requirement of vascular integrin for angiogenesis. Science. 1994;264:569.
bu-El-Asrar AM, Dralands L, Missotten L, et al. Expression of apoptosis markers in the retinas of human subjects with diabetes. Invest Ophthalmol Vis Sci. 2004;45:2760.
Chen YJ, Kuo HK, Huang HW. Retinal outcomes in proliferative diabetic retinopathy presenting during and after pregnancy. Chang Gung Med J. 2004;27:678.
Chew EY, Klein ML, Ferris FL, et al. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy: Early Treatment Diabetic Retinopathy Study (ETDRS) report 22. Arch Ophthalmol. 1996;114:1079.
Clermont AC, Aiello LP, Mori F, et al. Vascular endothelial growth factor and severity of nonproliferative diabetic retinopathy mediate retinal hemodynamics in vivo: A potential role for vascular endothelial growth factor in the progression of nonproliferative diabetic retinopathy. Am J Ophthalmol. 1997;124:433.
Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns: IV. Diabetic retinopathy. Arch Ophthalmol. 1961;66:366.
Cussick M, Chew EY, Chan C-C, et al. Histopathology and regression of retinal hard exudates in diabetic retinopathy after reduction of elevated serum lipid levels. Ophthalmology. 2003;110:2126.
Daria B, Maiello M, Lorenzi M. Increased incidence of micro thrombosis in retinal capillaries of diabetic individuals. Diabetes. 2002;50:1432.
Diabetes Control and Complications Trial Research Group. Early worsening of diabetic retinopathy in the diabetes control and complications trial. Ophthalmology. 1998;116:874.
Economopoulou M, Bdeir K, Cines DB, et al. Inhibition of pathologic retinal neovascularization by alpha-defensins. Blood. 2005;106:3831.
Frank RN. On the pathogenesis of diabetic retinopathy: A 1990 update. Ophthalmology. 1991;98:586.
Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48.
Fritsche P, van der HR, Suttorp-Schulten MS, et al. Retinal thickness analysis (RTA): An objective method to assess and quantify the retinal thickness in healthy controls and in diabetics without diabetic retinopathy. Retina. 2002;22:768.
Funatsu H, Yamashita H, Noma H, et al. Stimulation and inhibition of angiogenesis in diabetic retinopathy. Jpn J Ophthalmol. 2001;45:577.
Gargiulo P, Giusti C, Pietrobono D, et al. Diabetes mellitus and retinopathy. Dig Liver Dis. 2004;36(Suppl 1):S101.
Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438:960.
Giebel SJ, Menicucci G, McGuire PG, et al. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood–retinal barrier. Lab Invest. 2005;85:597.
Goebel W, Kretzchmar-Gross T. Retinal thickness in diabetic retinopathy: A study using optical coherence tomography (OCT). Retina. 2002;22:759.
Güven D, Ozdemir H, Hasanreisoglu B. Hemodynamic alterations in diabetic retinopathy. Ophthalmology. 1996;103:1245.
Hammes HP, Du X, Edelstein D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9:294.
Hanneken A, de Juan E Jr, Lutty GA, et al. Altered distribution of basic fibroblast growth factor in diabetic retinopathy. Arch Ophthalmol. 1991;109:1005.
Helbig H, Kornacker S, Berweck S, et al. Membrane potentials in retinal capillary pericytes: Excitability and effect of vasoactive substances. Invest Ophthalmol Vis Sci. 1992;33:2105.
Hellstedt T, Immonen I. Disappearance and formation rates of microaneurysms in early diabetic retinopathy. Br J Ophthalmol. 1996;80:135.
Hersh PS, Green WR, Thomas JV. Tractional venous loops in diabetic retinopathy. Am J Ophthalmol. 1981;92:661.
Hinton DR, Spee C, He S, et al. Accumulation of NH2-terminal fragment of connective tissue growth factor in the vitreous of patients with proliferative diabetic retinopathy. Diabetes Care. 2004;27:758.
Hussain A, Hussain N, Nutheti R. Comparison of mean macular thickness using optical coherence tomography and visual acuity in diabetic retinopathy. Clin Exp Ophthalmol. 2005;33:240.
Ioachim E, Stefaniotou M, Gorezis S, et al. Immunohistochemical study of extracellular matrix components in epiretinal membranes of vitreoproliferative retinopathy and proliferative diabetic retinopathy. Eur J Ophthalmol. 2005;15:384.
Kador PF, Akagi Y, Takahashi Y, et al. Prevention of retinal vessel changes associated with diabetic retinopathy in galactose-fed dogs by aldose reductase inhibitors. Arch Ophthalmol. 1990;108:1301.
Karacorlu M, Ozdemir H, Karacorlu S, et al. Intravitreal triamcinolone as a primary therapy in diabetic macular oedema. Eye. 2005;19:382.
Klein R, Klein BEK, Moss SE, et al. Retinal vascular abnormalities in persons with type 1 diabetes: The Wisconsin Epidemiologic Study of diabetic retinopathy: XVIII. Ophthalmology. 2003;110:2118.
Klein R, Meuer SM, Moss SE, et al. The relationship of retinal microaneurysm counts to the 4-year progression of diabetic retinopathy. Arch Ophthalmol. 1989;107:1780.
Klein R, Meuer SM, Moss SE, et al. Retinal microaneurysm counts and progression of diabetic retinopathy. Arch Ophthalmol. 1995;113:1386.
Klein R, Moss SE, Klein BEK, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XI. The incidence of macular edema. Ophthalmology. 1989;96:1501.
Knudsen LL, Lervang HH. Can a cilio-retinal artery influence diabetic maculopathy? Br J Ophthalmol. 2002;86:1252.
Kuhn F, Kiss G, Mester V, et al. Vitrectomy with internal limiting membrane removal for clinically significant macular oedema. Graefes Arch Clin Exp Ophthalmol. 2004;242:402.
Lahdenranta J, Pasqualini R, Schlingemann RO, et al. An anti-angiogenic state in mice and humans with retinal photoreceptor cell degeneration. Proc Natl Acad Sci USA. 2001;98:10368.
Layton CJ, Becker S, Osborne NN. The effect of insulin and glucose levels on retinal glial cell activation and pigment epithelium-derived fibroblast growth factor-2. Mol Vis. 2006;12:43.
Lee VS, Kingsley RM, Lee ET, et al. The diagnosis of diabetic retinopathy. Ophthalmology. 1993;100:1504.
Leto G, Pricci F, Amadio L, et al. Increased retinal endothelial cell monolayer permeability induced by the diabetic milieu: role of advanced non-enzymatic glycation and polyol pathway activation. Diabetes Metab Res Rev. 2001;17:448.
Li W, Liu X, Yanoff M, et al. Cultured retinal capillary pericytes die by apoptosis after an abrupt fluctuation from high to low glucose levels: A comparative study with retinal capillary endothelial cells. Diabetologia. 1996;39:537.
Li W, Tao L, Yanoff M. Agonist-induced phosphatidylinositide breakdown and mitogenesis in retinal capillary pericytes. Ophthalmic Res. 1994;26:36.
Lindahl P, Johansson BR, Levéen P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242.
Ljubimov AV, Caballero S, Aoki A, et al. Involvement of protein kinase CK2 in angiogenesis and retinal neovascularization. Invest Ophthalmol Vis Sci. 2004;45:4583.
Lobo CL, Bernardes RC, de A Jr, et al. One-year follow-up of blood–retinal barrier and retinal thickness alterations in patients with type 2 diabetes mellitus and mild nonproliferative retinopathy. Arch Ophthalmol. 2001;119:1469.
Lyons TJ, Jenkins AJ, Zhen D, et al. Diabetic retinopathy and serum lipoprotein subclasses in the DCCT/EDIC cohort. Invest Ophthalmol Vis Sci. 2004;45:910.
Mansour AM, Schachat A, Bodiford G, et al. Foveal avascular zone in diabetes mellitus. Retina. 1993;13:125.
Massin P, Audren F, Haouchine B, et al. Intravitreal triamcinolone acetonide for diabetic diffuse macular edema: Preliminary results of a prospective controlled trial. Ophthalmology. 2004;111:218.
Matsuoka M, Ogata N, Minamino K, et al. Expression of pigment epithelium-derived factor and vascular endothelial growth factor in fibrovascular membranes from patients with proliferative diabetic retinopathy. Jpn J Ophthalmol. 2006;50:116.
McFarland TJ, Zhang Y, Appukuttan B, et al. Gene therapy for proliferative ocular diseases. Expert Opin Biol Ther. 2004;4:1053.
Nagaoka T, Kitaya N, Sugawara R, et al. Alteration of choroidal circulation in the foveal region in patients with type 2 diabetes. Br J Ophthalmol. 2004;88:1060.
Nguyen QD, Shah SM, Van AE, et al. Supplemental oxygen improves diabetic macular edema: A pilot study. Invest Ophthalmol Vis Sci. 2004;45:617.
Nicoletti VG, Nicoletti R, Ferrara N, et al. Diabetic patients and retinal proliferation: An evaluation of the role of vascular endothelial growth factor (VEGF). Exp Clin Endocrinol Diabetes. 2003;111:209.
Nishiwaki H, Shahidi M, Vitale S, et al. Relation between retinal thickening and clinically visible fundus pathologies in mild nonproliferative diabetic retinopathy. Ophthalmic Surg Lasers. 2002;33:127.
Noma H, Funatsu H, Yamashita H, et al. Regulation of angiogenesis in diabetic retinopathy: Possible balance between vascular endothelial growth factor and endostatin. Arch Ophthalmol. 2002;120:1075.
North PE, Anthony DC, Young TL, et al. Retinal neovascular markers in retinopathy of prematurity: Aetiological implications. Br J Ophthalmol. 2003;87:275.
Perrin RM, Konopatskaya O, Qiu Y, et al. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia. 2005;48:2422.
Poulaki V, Joussen AM, Mitsiades N, et al. Insulin-like growth factor-I plays a pathogenetic role in diabetic retinopathy. Am J Pathol. 2004;165:457.
Romeo G, Liu WH, Asnaghi V, et al. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241.
Roy S, Cagliero E, Lorenzi M. Fibronectin overexpression in retinal microvessels of patients with diabetes. Invest Ophthalmol Vis Sci. 1996;37:258.
Savage HI, Hendrix JW, Peterson DC, et al. Differences in pulsatile ocular blood flow among three classifications of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2004;45:4504.
Schneeberger SA, Hjelmeland LM, Tucker RP, et al. Vascular endothelial growth factor and fibroblastic growth factor 5 are colonized in vascular and avascular epiretinal membranes. Am J Ophthalmol. 1997;124:433.
Schröder S, Palinski W, Schmid-Schönbein GW. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol. 1991;139:81.
Sennlaub F, Valamanesh F, Vazquez-Tello A, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198.
Sinclair SH. Macular retinal capillary hemodynamics in diabetic patients. Ophthalmology. 1991;98:1580.
Siren V, Immonen I. uPA, tPA and PAI-1 mRNA expression in periretinal membranes. Curr Eye Res. 2003;27:261.
Sonkin PL, Sinclair SH, Hatchell DL. The effect of pentoxifylline on retinal capillary blood flow velocity and whole blood velocity. Am J Ophthalmol. 1993;115:775.
Stitt AW, Frizzell N, Thorpe SR. Advanced glycation and advanced lipoxidation: Possible role in initiation and progression of diabetic retinopathy. Curr Pharm Des. 2004;10:3349.
Sugimoto M, Sasoh M, Ido M, et al. Detection of early diabetic change with optical coherence tomography in type 2 diabetes mellitus patients without retinopathy. Ophthalmologica. 2005;219:379.
Tang J, Mohr S, Du YD, et al. Non-uniform distribution of lesions and biochemical abnormalities within the retina of diabetic humans. Curr Eye Res. 2003;27:7.
Vinores SA, Gadegbeku C, Compochiaro PA, et al. Immunohistochemic localization of blood–retinal barrier breakdown in human diabetes. Am J Pathol. 1989;134:231.
Vitale S, Maguire MG, Murphy RP, et al. Clinically significant macular edema in type I diabetes. Ophthalmology. 1995;102:1170.
Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87.
Watanabe D, Suzuma K, Matsui S, et al. Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med. 2005;353:782.
Wilkinson CP, Ferris FL, Klein RE, et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003;110:1677.
Wilkinson-Berka JL. Angiotensin and diabetic retinopathy. Int J Biochem Cell Biol. 2006;38:752.
Witmer AN, Blaauwgeers HG, Weich HA, et al. Altered expression patterns of VEGF receptors in human diabetic retina and in experimental VEGF-induced retinopathy in monkey. Invest Ophthalmol Vis Sci. 2002;43:849.
Yamagishi S, Matsui T, Nakamura K, et al. Pigment epithelium-derived factor is a pericyte mitogen secreted by microvascular endothelial cells: Possible participation of angiotensin II-elicited PEDF downregulation in diabetic retinopathy. Int J Tissue React. 2005;27:197.
Yanoff M. Diabetic retinopathy. N Engl J Med. 1966;274:1344.
Yanoff M. Ocular pathology of diabetes mellitus. Am J Ophthalmol. 1969;67:21.
Yanoff M. Histopathogenesis of diabetic retinopathy. Acta Diabetol Lat. 1972;9:527.
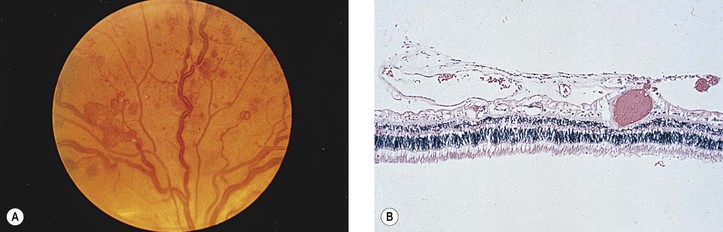
Vitreous
Anderson B Jr. Activity and diabetic vitreous hemorrhages. Ophthalmology. 1980;87:173.
Bergren RL, Brown GC, Duker JS. Prevalence and association of asteroid hyalosis with systemic disease. Am J Ophthalmol. 1991;111:289.
Citirik M, Kabatas EU, Batman C, et al. Vitreous vascular endothelial growth factor concentrations in proliferative diabetic retinopathy versus proliferative vitreoretinopathy. Ophthalmic Res. 2012;47:7.
Fawzi AA, Vo B, Kriwanek R, et al. Asteroid hyalosis in an autopsy population: The University of California at Los Angeles (UCLA) experience. Arch Ophthalmol. 2005;123:486.
Feke GT, Zuckerman R, Green GJ, et al. Response of human retinal blood flow to light and dark. Invest Ophthalmol Vis Sci. 1983;24:136.
Foos RY, Kreiger AE, Forsythe AB, et al. Posterior vitreous detachment in diabetic subjects. Ophthalmology. 1980;87:122.
Foos RY, Kreiger AE, Nofsinger K. Pathologic study following vitrectomy for proliferative diabetic retinopathy. Retina. 1985;5:101.
Gandorfer A, Rohleder M, Grosselfinger S, et al. Epiretinal pathology of diffuse diabetic macular edema associated with vitreomacular traction. Am J Ophthalmol. 2005;139:638.
Hernandez C, Ortega F, Garcia-Ramirez M, et al. Lipopolysaccharide-binding protein and soluble CD14 in the vitreous fluid of patients with proliferative diabetic retinopathy. Retina. 2010;30:345.
Hixson A, Reynolds S. Peripapillary vitreoretinal traction. Optometry. 2011;82:602.
Jerdan JA, Michels RG, Glaser BM. Diabetic preretinal membranes: An immunohistochemical study. Arch Ophthalmol. 1986;104:286.
Kim T, Kim SJ, Kim K, et al. Profiling of vitreous proteomes from proliferative diabetic retinopathy and nondiabetic patients. Proteomics. 2007;7:4203.
Kuiper EJ, de Smet MD, van Meurs JC, et al. Association of connective tissue growth factor with fibrosis in vitreoretinal disorders in the human eye. Arch Ophthalmol. 2006;124:1457.
Luxenberg M, Sime D. Relationship of asteroid hyalosis to diabetes mellitus and plasma lipid levels. Am J Ophthalmol. 1969;67:406.
Nasrallah FP, Jalkh AE, Van Coppenolle F, et al. The role of the vitreous in diabetic macular edema. Ophthalmology. 1988;95:1335.
Ophir A, Trevino A, Fatum S. Extrafoveal vitreous traction associated with diabetic diffuse macular oedema. Eye (Lond). 2010;24:347.
Roy MS, Podgor MJ, Bungay P, et al. Posterior vitreous fluorophotometry in diabetic patients with minimal or no retinopathy. Retina. 1987;7:170.
Schwartzman ML, Iserovich P, Gotlinger K, et al. Profile of lipid and protein autacoids in diabetic vitreous correlates with the progression of diabetic retinopathy. Diabetes. 2010;59:1780.
Yamakiri K, Yamashita T, Miyazaki M, et al. Fibrous proliferation of the pre-papillary canal in proliferative diabetic retinopathy: Cloquet’s canal as a scaffold for proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 2005;243:204.
Yanoff M. Ocular pathology of diabetes mellitus. Am J Ophthalmol. 1969;67:21.
Yoshimura T, Sonoda KH, Sugahara M, et al. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS One. 2009;4:e8158.
Optic Nerve
Appen RE, Chandra SR, Klein R, et al. Diabetic papillopathy. Am J Ophthalmol. 1980;90:203.
Barr CC, Glaser JS, Blankenship G. Acute disc swelling in juvenile diabetes: Clinical profile and natural history of 12 cases. Arch Ophthalmol. 1980;98:2185.
Bayraktar Z, Alacali N, Bayraktar S. Diabetic papillopathy in type II diabetic patients. Retina. 2002;22:752.
Cankaya AB, Ozdamar Y, Ozalp S, et al. Impact of panretinal photocoagulation on optic nerve head parameters. Ophthalmologica. 2011;225:193.
Giuliari GP, Sadaka A, Chang PY, et al. Diabetic papillopathy: Current and new treatment options. Curr Diabetes Rev. 2011;7:171.
Ho AC, Maguire AM, Fisher YL, et al. Rapidly progressive optic disc neovascularization after diabetic papillopathy. Am J Ophthalmol. 1995;120:673.
Isaacs TW, Barry C. Rapid resolution of severe disc new vessels in proliferative diabetic retinopathy following a single intravitreal injection of bevacizumab (Avastin). Clin Experiment Ophthalmol. 2006;34:802.
Kim HY, Cho HK. Peripapillary retinal nerve fiber layer thickness change after panretinal photocoagulation in patients with diabetic retinopathy. Korean J Ophthalmol. 2009;23:23.
Lopes de Faria JM, Russ H, Costa VP. Retinal nerve fibre layer loss in patients with type 1 diabetes mellitus without retinopathy. Br J Ophthalmol. 2002;86:725.
Mallika PS, Aziz S, Asok T, et al. Severe diabetic papillopathy mimicking non-arteritic anterior ischemic optic neuropathy (NAION) in a young patient. Med J Malaysia. 2012;67:228.
Mathews MK. Nonarteritic anterior ischemic optic neuropathy. Curr Opin Ophthalmol. 2005;16:341.
Nakamura M, Kanamori A, Negi A. Diabetes mellitus as a risk factor for glaucomatous optic neuropathy. Ophthalmologica. 2005;219:1.
Nelson K, Singh G, Boyer S, et al. Two presentations of nonarteritic ischemic optic neuropathy. Optometry. 2010;81:587.
Ozdek S, Lonneville YH, Onol M, et al. Assessment of nerve fiber layer in diabetic patients with scanning laser polarimetry. Eye. 2002;16:761.
Pavan PR, Aiello LM, Wafai Z, et al. Optic disc edema in juvenile-onset diabetes. Arch Ophthalmol. 1980;98:2193.
Regillo CD, Brown GC, Savino PJ, et al. Diabetic papillopathy: Patient characteristics and fundus findings. Arch Ophthalmol. 1995;113:889.
Saito Y, Ueki N, Hamanaka N, et al. Transient optic disc edema by vitreous traction in a quiescent eye with proliferative diabetic retinopathy mimicking diabetic papillopathy. Retina. 2005;25:83.
Sato T, Fujikado T, Hosohata J, et al. Development of bilateral, nonarteritic anterior ischemic optic neuropathy in an eye with diabetic papillopathy. Jpn J Ophthalmol. 2004;48:158.
Soares AS, Artes PH, Andreou P, et al. Factors associated with optic disc hemorrhages in glaucoma. Ophthalmology. 2004;111:1653.
Yassur Y, Pickle LW, Fine SL, et al. Optic disc neovascularisation in diabetic retinopathy: II. Natural history and results of photocoagulation treatment. Br J Ophthalmol. 1980;64:77.