[level-membership-for-allergy-and-immunology-category]
Delayed-Type Hypersensitivity Reactions
Learning Objectives
• Identify the definition of delayed-type hypersensitivity (DTH)
• Compare and contrast immediate and cell-mediated hypersensitivities
• List the six Mycobacterium species that are human pathogens
• Recognize the components of an acid-fast cell wall
• Explain the relationship between interferon gamma (IFN-γ) and DTH
• Explain the relationship between vitamin D and leprosy
• Compare and contrast the three histologic stages of tuberculosis
• Recognize the cellular components in a granuloma
• Compare and contrast Mycobacterium tuberculosis and Bacille Calmette-Guérin (BCG)
• Explain the mechanism involved in the QuantiFERON-Gold test
• Recognize the four front-line drugs used to treat active tuberculosis and their mechanisms of action
• Recognize the second-line drugs used to treat active tuberculosis and their mechanisms of action
• Identify the definition of multidrug-resistant–tuberculosis (MDR–TB)
• Define extensively drug-resistant–tuberculosis (XDR–TB)
• Identify the dimorphic phases of histoplasmosis
• Compare the role of macroconidia and the yeast form in the pathogenesis of histoplasmosis
• Recognize regions of the United States where histoplasmosis is endemic
• Compare and contrast granuloma formation in histoplasmosis and tuberculosis
• Identify the groups at risk for progressive disseminated histoplasmosis
• Identify the dimorphic forms of Coccidioides immitis
• Identify regions of the United States where Coccidioides is endemic
• Recognize the role of arthroconidia in the pathogenesis of coccidioidomycosis
• Differentiate between spherules and endospores in the pathogenesis of coccidioidomycosis
• Compare and contrast contact dermatitis, irritant dermatitis, and photocontact dermatitis
• Recognize the drugs that are used to treat contact dermatitis
• Explain the immunologic skin response to nickel
• Identify the role(s) of Th17 cells in metal hypersensitivity
• Recognize the drugs that are used to treat nickel hypersensitivity
• List the three members of the Toxicodendron genus that cause skin reactions
• Identify the haptenic substance produced by the Toxicodendron species
• Explain the immunologic response to urushiol
• List the drugs that are used to treat poison ivy reaction
• Explain the immunologic and biologic responses active in celiac sprue
Key Terms
Allergic contact dermatitis
Arthroconidia
Bacille Calmette-Guérin (BCG)
Conidia
Cord factor
Extensively drug-resistant tuberculosis
Gliadin
Granuloma
Interferon gamma (IFN-γ)
Lysosomes
Mantoux test
Multidrug-resistant tuberculosis
Mycolic acids
Natural resistance–associated macrophage protein (NRAMP)
Photocontact dermatitis
T17 cells
Tumor necrosis factor
Urushiol
Vitamin D receptor
Introduction
Delayed-type hypersensitivity (DTH) is a unique type of cell-mediated immunity. The name originated from the skin test used in the diagnosis of tuberculosis and denotes cellular infiltrates causing induration and erythema at the skin test site within 24 to 72 hours. DTH was coined to describe the reaction to the tuberculosis skin test and to differentiate between delayed cellular skin test results and antibody-mediated immediate skin test results. The term has been expanded to include cell-mediated reactions to bacteria or fungi infecting lungs and skin responses to chemicals and plants. In the lung, cellular responses to tuberculosis, histoplasmosis, and coccidioidomycosis are considered DTH reactions. Skin reactions to nickel and poison ivy also are DTH reactions.
The principal effectors of DTH reactions are CD4Th1 lymphocytes, monocytes or macrophages, CD8Tc1 cells, and natural killer (NK) cells. The histopathology of DTH lesions varies with the nature of the antigen, the type of effector cells, and the anatomic location.
Delayed-Type Hypersensitivity Responses in the Lung
Tuberculosis
The family Mycobacteriaceae contains the Mycobacterium genus. Within the genus are six different human pathogens (Table 19-1). (1) M. tuberculosis causes most of the 9.1 million cases of pulmonary tuberculosis in the world. (2) M. leprae, which is also known as Hansen’s bacillus, is commonly found in tropical countries and is the etiological agent of leprosy. (3&4) Mycobacterium avium and M. intracellulare are found in soil, water systems, birds, and other domestic animals. Because the two species are difficult to identify in the laboratory, they are designated as the Mycobacterium avium complex (MAC). Patients with acquired immunodeficiency syndrome (AIDS) and therapeutically immunosuppressed individuals are frequently infected by MAC organisms. (5) M. bovis is a cattle-adapted species of tuberculosis that is also found in wild and domestic animals, but it causes less than 1% of all tuberculosis infections. Human infection is caused by inhalation of droplets containing live organisms or by ingestion of unpasteurized milk. Ingestion leads to extrapulmonary tuberculosis in lymph nodes and skeletal bones. (6) Mycobacterium africanum is commonly found in West Africa, where it accounts for 25% of tuberculosis cases.
Table 19-1
Species of Tuberculosis Infecting Humans
| Pathogen | Infection/Transmission |
| Mycobacterium tuberculosis | Human tuberculosis. Transmitted by inhalation route |
| Mycobacterium leprae | Hansen’s disease, or leprosy. Transmitted by inhalation route |
| Mycobacterium avium-intracellulare | Group of closely related opportunistic pathogens that infect immunosuppressed patients with human immunodeficiency virus (HIV). Transmitted by inhalation and oral routes |
| Mycobacterium bovis | Tuberculosis in cattle. Can be transmitted to humans via infected milk or aerosols |
| Mycobacterium africanum | Commonly found in West Africa. Spread by the airborne route |
Tuberculosis: Virulence Factors
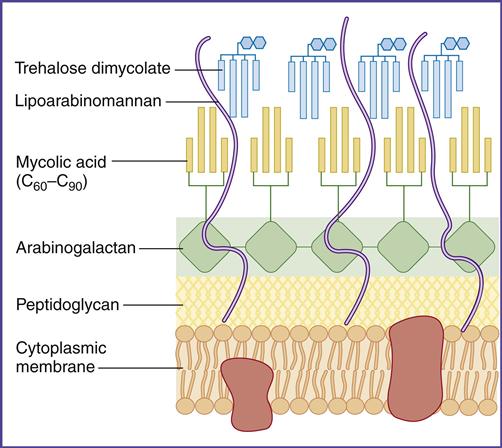
In the lung, survival of the tuberculosis bacterium is determined, in part, by cell-wall mycolic acids and a lipoarabinomannan known as the cord factor. Mycolic acids, which are 50% of the dry weight of the cell wall (Figure 19-1), form a hydrophobic barrier that prevents the entry of drugs, disinfectants, and other harsh chemicals. Cord factor inhibits the interferon (IFN)–induced activation of macrophages and stimulates the production of tumor necrosis factor alpha (TNF-α).
Laboratory Identification of Mycobacterium Species
Laboratory identification of mycobacteria is difficult because conventional stains do not penetrate the cell wall. However, mycobacteria can be identified by the Ziehl Neelsen stain, which uses a combination of heat and carbolfuchsin to drive the stain into the cell wall. Only the Mycobacterium and Nocardia species retain the dye following harsh treatment with an acid–alcohol mixture. Thus, these species are designated acid-fast organisms.
Pathophysiology of Tuberculosis
Exposure to tuberculosis occurs by inhalation of droplets containing live organisms. Because of their small size, the organisms are usually deposited in the distal areas of the lung. In the first stage of the disease, mycobacteria take up residence in phagosomes within alveolar macrophages and prevent intracellular killing by inhibiting maturation of the phagosome and fusion with endosomes and lysosomes which contain antimicrobial proteins and enzymes. Rapid logarithmic multiplication of mycobacteria in macrophages eventually causes the death of the cells and creates a small necrotic area in the lung. Free mycobacteria are ingested by circulating monocytes and transported to the mediastinal lymph node. Antigen stimulation in the lymph node generates CD4Th1, CD8, and long-lived memory cells.
In the second stage, which occurs 3 weeks after infection, CD4Th1 cells migrate from the lymph node to the infection site and undergo rapid proliferation. CD4Th1 and CD8 cells secrete TNF-α and IFN-γ. TNF-α is responsible for most of the lung damage noted in mycobacteria infections. IFN-γ– stimulated macrophages produce nitric oxide by oxidation of L-citrulline. Additional interactions between nitric oxide and singlet oxygen form peroxynitrite (ONOO–), which is microcidal. The role of CD8 cells in tuberculosis is unclear. They may contribute to the lesion by producing IFN-γ, by directly killing infected cells, or by both mechanisms (Figure 19-2).
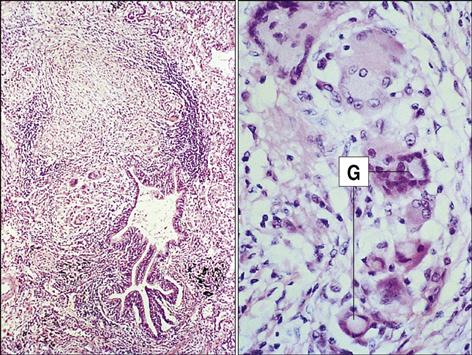
In the third phase of the disease, macrophage-derived reactive oxygen species cause necrosis of lung tissue. The production of chemotactic factors causes an influx of activated monocytes into the area. In an attempt to destroy the bacteria, activated monocytes surround the caseous center and release additional cytotoxic molecules. Some monocytes become flattened and are known as epithelioid cells (Figure 19-3). Fusion of macrophage membranes often creates giant cells with multiple nuclei.
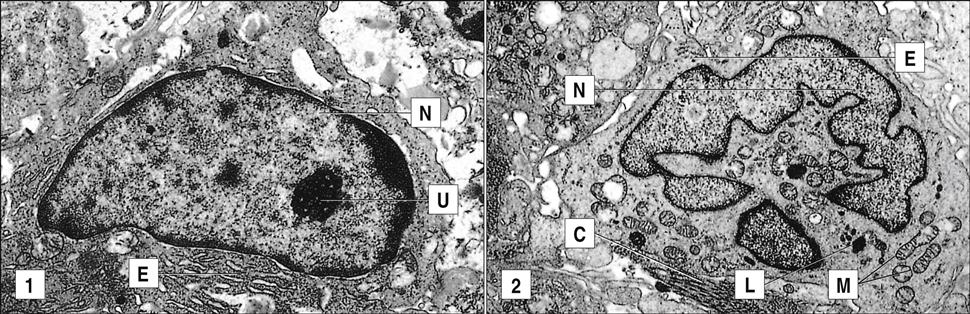
In a final attempt to contain the infection, fibroblasts are called into the area and produce collagen, which encapsulates the inflammatory cells surrounding the lesion. The accumulation of macrophages, lymphocytes, and fibroblasts is called a granuloma. Constant production of TNF and IFN-γ is required for the maintenance of the granuloma; if the granuloma is not maintained, it becomes unstable, liquefies, and creates a permissible environment for bacterial growth. As a consequence of rapid bacterial growth, the infection erodes into blood vessels and small airways. Dissemination in blood allows the organism to establish secondary infections in other organs and bone. This form of the disease is known as miliary tuberculosis. Mycobacteria in the airways also rapidly proliferate in the highly aerobic environment and are expelled from the lung by coughing or sneezing.
Genetics and Mycobacterium Infections
The role of host genetics in Mycobacterium infection was suggested by an accidental administration of virulent M. tuberculosis to 249 babies in Lubeck, Germany, in 1926. Although 76 babies died of the disease, 123 had lung lesions that healed, and another 50 babies had no signs of infection. This suggested that genetics influences susceptibility to tuberculosis. Subsequent studies indicated that persons at risk for active tuberculosis have dysfunctional receptors for IFN-γ or vitamin D. Some individuals also have a defect in the production of natural resistance–associated macrophage proteins (NRAMPs).
Interferon Gamma Receptor Deficiency
IFN-γ is a CD4Th1-produced cytokine that activates IFN-γ receptors expressed by other T cells, NK cells, and macrophages or monocytes. IFN increases bacterial killing by monocytes and alveolar macrophages. Individuals with an IFN-γ receptor deficiency are at risk for infection with M. tuberculosis, M. africanum, and M. bovis.
Vitamin D Receptor Deficiency
Lymphocytes, dendritic cells, and macrophages have intracellular receptors for the active form to 1 alpha, 25-dihydroxyvitamin D3 [1,25(OH)2D3]. Association of vitamin D with the vitamin D receptor (VDR) initiates transcription and translation of gene products that activate macrophages and Th1 cells while suppressing the activity of Th2 cells.
Failure to express the VDR increases the risk for M. leprae infection and determines whether the patient develops the lepromatous form or the tuberculoid form of the disease. In individuals with VDR deficiency, a CD4Th2 response with antibody production occurs. The Th2 response results in lepromatous leprosy characterized by numerous intracellular bacterial growth and severe tissue damage. In contrast, patients with functional VDRs mount a vigorous Th1 inflammatory response, which isolates the bacteria in granuloma and minimizes tissue damage. This type of response is called tuberculoid leprosy.
Natural Resistance–Associated Macrophage Protein (NRAMP) Deficiency
Early in the infection, two NRAMPs play a role in the destruction of mycobacteria: (1) NRAMP1 is localized in endosomal vesicles that fuse with phagosomes. NRAMP1 reduces the pH of the phagosome to levels that are toxic to the bacteria. (2) NRAMP2 is an efflux pump that exports iron and other cations from the phagosome. Mutations in NRAMP1 and NRAMP2 genes influence the phagosomal function of alveolar macrophages. In patients with an NRAMP1 deficiency, the pH of the phagolysosomes is near pH 6.8, which fosters bacterial growth. As a consequence of defective NRAMP2 production, iron accumulates in the phagosome and is used in the synthesis of bacterial heme–containing cytochromes during bacterial replication.
In Vivo Tuberculin Skin Test
Exposure to tuberculosis generates a population of long-lived memory cells. The Mantoux, or PPD (purified protein derivative of tuberculosis), skin test is now the standard test to determine previous exposure to tuberculosis bacilli. In the Mantoux test, PPD is injected intradermally, and the reaction site is viewed at 48 and 72 hours. If memory cells are present, an influx of macrophages and lymphocytes around the injection site causes a hard, raised indurated area with clearly defined margins (Figure 19-4). The cellular development of a positive skin test response to tuberculosis is shown in Figure 19-5.
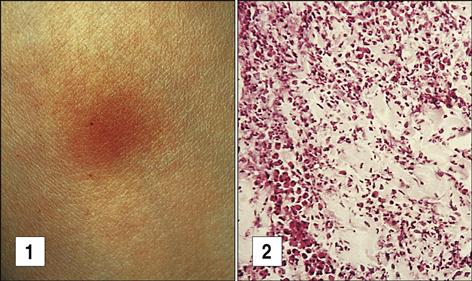
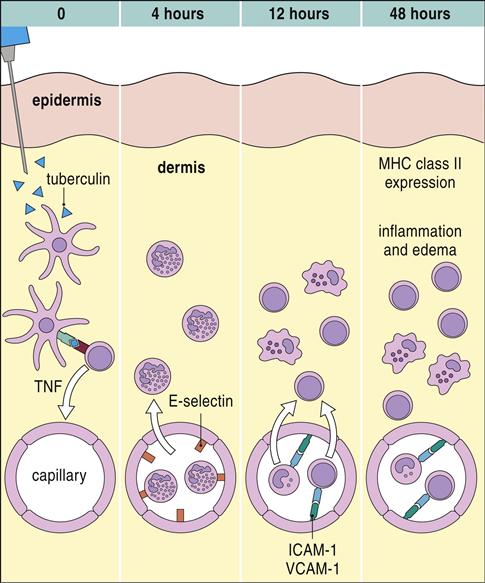
The interpretation of skin test reactions is difficult. A positive skin test only indicates previous exposure to tuberculosis or vaccination with an attenuated strain of M. bovis called Bacille Calmette-Guérin (BCG). However, a significant false-negative rate in the Mantoux test does occur. Between 20% and 25% of patients with active tuberculosis have a negative skin test. Clinical history, radiologic studies, and sputum cultures are used to support a tentative diagnosis of active tuberculosis.
In Vitro Tests for Exposure to Tuberculosis
The T-SPOT.TB and the QuantiFERON Gold test (GFT-G) are two in vitro tests that detect memory cells and identify tuberculosis or latent tuberculosis. The former is being evaluated by the U.S. Food and Drug Administration (FDA) for clinical use, and the latter test has been approved and is commercially available.
In the GFT-G test, blood lymphocytes are incubated with tuberculosis antigens not found in environmental tuberculosis species, PPD, or BCG vaccines. If an individual has memory cells to tuberculosis or a latent tuberculosis infection, T cells will produce INF-γ, which is measured in the laboratory using enzyme-linked immunosorbent assay (ELISA).
In vitro tests have many advantages. The test can be completed in 1 day with no need for a second patient visit. It is more accurate than the skin test, and the interpretation of results is not affected by a reader’s bias or capability. Moreover, previous BCG immunizations do not influence the results. In vitro testing has great usefulness for tuberculosis surveillance of transients or large congregate populations such as military personnel or prisoners.
Treatment for Tuberculosis
The diagnosis of tuberculosis is based on clinical signs and symptoms, radiologic studies, and sputum cultures. Patients with acid-fast bacilli in their sputum can be presumptively diagnosed and treated with anti-tuberculosis therapy, which also may be appropriate in patients with a negative sputum smear but who have clinical and radiographic findings consistent with pulmonary tuberculosis. Therapy is designed to treat tuberculosis caused by M. tuberculosis, M. bovis, and M. africanum. Treatment of active tuberculosis uses a four-drug combination, consisting of isoniazid, rifampin, pyrazinamide, and ethambutol. Isoniazid and ethambutol are the critical therapeutic agents. These agents inhibit the synthesis of mycolic acids and increase the permeability of mammalian cells. This allows rifampin and pyrazinamide to enter the cells. The mechanism of action of each drug is provided in Table 19-2.
Table 19-2
First-Line Drugs Used to Treat Tuberculosis
| Drug | Action | Type |
| Isoniazid | Inhibits mycolic acid synthesis | Bactericidal |
| Pyrazinamide | Analog of nicotinamide | Bactericidal or bacteriostatic, depending on concentration |
| Rifampin | Rifampin inhibits prokaryotic ribonucleic acid (RNA) polymerase. Inhibits the translation of proteins | Bactericidal |
| Ethambutol | Inhibits mycolic acid synthesis | Bacteriostatic |
Infections caused by genetically related organisms in the Mycobacterium avium complex (MAC) are more difficult to treat because of their resistance to most antibiotics and anti-tuberculosis drugs. Treatment usually involves second-generation macrolides (e.g., azithromycin), ethambutol, and rifabutin.
Multidrug-Resistant Tuberculosis Strains
Over the past 16 years, two forms of drug resistance have emerged. Multidrug-resistant tuberculosis (MDR-TB) is defined as a tuberculosis strain that is resistant to, at least, isoniazid and rifampin. Resistance is associated with random mutations that alter the structure or quantity of intracellular drug targets. In 2005, the overall rate of MDR-TB with resistance to isoniazid and rifampin was 1.2%, with the rates for individual states varying from 0 to 3.7%. Worldwide, the number of new MDR-TB cases in 2005 was estimated at 480,000. When second-line drugs for tuberculosis (Table 19-3) are mismanaged or misused, extensively drug-resistant tuberculosis (XDR-TB) strains develop. These strains are resistant to isoniazid and rifampin as well as any of the fluoroquinolones and any of the second-line drugs. An estimated 40,000 new cases of XDR-TB are reported yearly in the developing countries. Between 1993 and 2006, only 49 cases of XDR-TB were reported in the United States.
Table 19-3
Second-Line Drugs Used to Treat Tuberculosis
| Drug | Action | Type |
| Para-aminosalicylic acid (Sodium PAS) | Competitive inhibitor of folic acid | Bacteriostatic |
| Capreomycin | Believed to inhibit protein synthesis by binding to 70S ribosome | Bactericidal |
| Cycloserine | Structural analog of d-alanine in cell walls. Inhibits cell wall synthesis | Bacteriostatic |
| Avlosulfon | Competitive inhibitor of folic acid | Bacteriostatic (experimental) |
| Levofloxacin | Inhibits deoxyribonucleic acid (DNA) gyrase and causes DNA breakage | Bactericidal |
| Amikacin | Irreversibly binds to 30S subunit of bacterial ribosomes and blocks protein synthesis | Bactericidal |
| Rifabutin | Inhibits ribonucleic acid (RNA) polymerase | Bactericidal |
| Azithromycin | Inhibits transfer RNA (tRNA) peptidase | Bactericidal |
Histoplasmosis
Histoplasma capsulatum is a dimorphic saprophytic fungus that grows rapidly in damp, acidic soil contaminated with bird or bat guano. The fungus is endemic in the Ohio and Mississippi River valleys (Figure 19-6). Over 80% of persons living in these areas have been exposed to the organism and have a positive skin test to histoplasmin. Over 40 million people are currently infected with Histoplasma. In the majority of cases, the infection is asymptomatic and resolves in several months. Only 5% of the new infections cause clinical and pulmonary symptoms. Immunosuppressed patients, farmers, landscapers, cavers, and construction workers are at high risk for active infection.
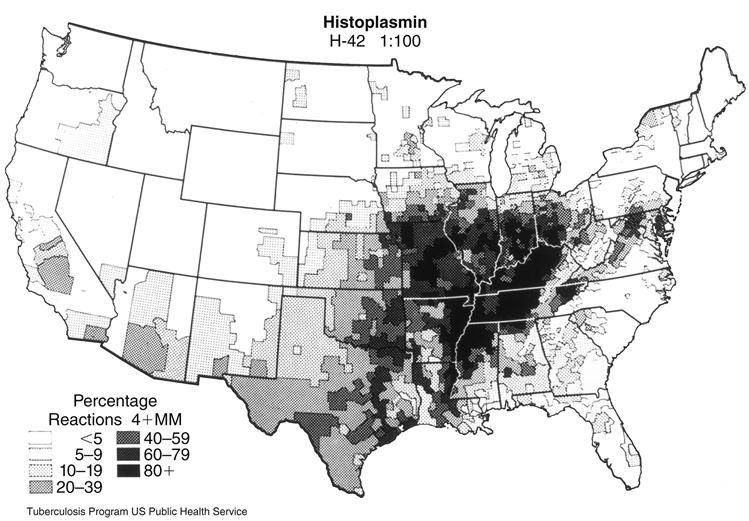
Pathophysiology of Histoplasmosis
At ambient temperatures in the soil, the fungus grows as a mycelium with hyphae that produce macroconidia and microconidia (Figure 19-7). When soil is disturbed by occupational or recreational activities, conidia are easily aerosolized and inhaled. Conidia are ingested by alveolar macrophages and germinate into a single-cell yeast form (Figure 19-8).
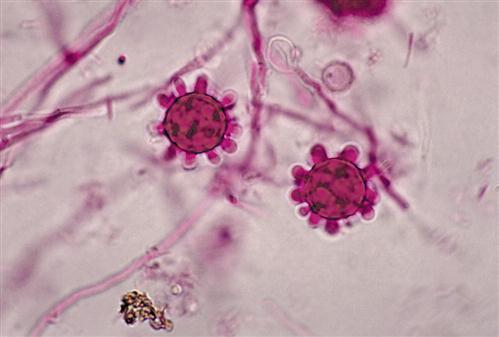
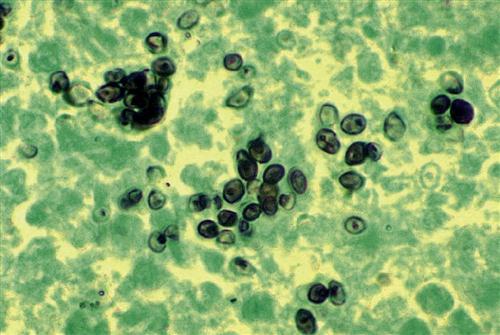
The pathophysiology of the disease is similar to that of tuberculosis. The yeast form survives in alveolar macrophages by preventing NRAMP1 acidification of phagosomes. Some of the infected macrophages migrate to the hilar or mediastinal lymph, where they stimulate a cell-mediated response.
A cell-mediated response is mounted 10 to 14 days after infection. IFN-γ and TNF-α produced by CD4Th1 memory cells activate the fungistatic activities of the resident alveolar macrophages and peripheral blood monocytes. To contain the infection, lesions are ultimately encapsulated with collagen. Over a period of several years, calcium is deposited around the capsule, and the lesion becomes totally calcified.
A progressive form of histoplasmosis is an emerging problem in patients with secondary or therapy-induced immunodeficiencies. Individuals with AIDS have a high risk of developing progressive disease, in which the fungus or its toxic products disseminate throughout the body and cause septic shock and kidney and liver failure.
Coccidioidomycosis or Valley Fever
Coccidioides immitis is a saprophytic, dimorphic fungus that is restricted to the San Joaquin valley in California, southern Arizona, New Mexico, west Texas, and the Sonoran desert regions of Mexico. In the soil, the fungus grows as a mold with septate hyphae or arthroconidia. Alternate arthroconidia undergo autolysis creating a fragile, segmented hyphal structure.
Pathophysiology of Coccidioidomycosis
When the soil is disturbed, segmented hyphae rupture and release arthroconidia into the atmosphere. Arthroconidia are easily inhaled and localize in the alveoli, where they transform into multi-nucleated spherules that contain hundreds of endospores (Figure 19-9).
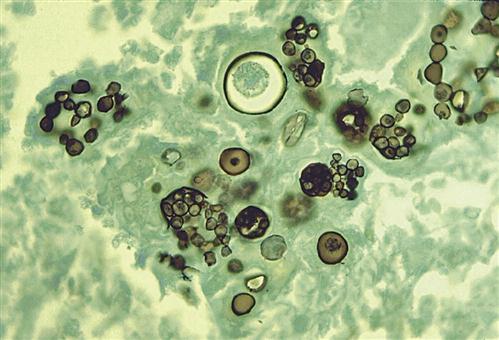
Rupture of the spherule releases the endospores into the lung alveoli. Some of the endospores are ingested by alveolar macrophages and monocytes, which migrate to local lymph nodes and stimulate a cell-mediated response. This is followed by an influx of monocytes, neutrophils, and CD4Th1 lymphocytes into the infected area. The evolution of the granuloma is similar to that described for tuberculosis. The granuloma, however, is unique in that B lymphocytes are found in the lesion, and antibodies to coccidioidal antigens are generated. Complement activation may also play a role in the immune response by generating neutrophil chemotactic factors.
Over 60% of infections are asymptomatic. Most active infections are confined to the lung and lung-associated lymph nodes. Less than 1% of individuals with clinical symptoms develop disseminated disease that spreads to the lungs, liver, brain, skin, heart, and pericardium. African Americans, Filipino Americans, and Hispanic Americans have a 10-fold higher risk for disseminated coccidioidomycosis compared with the general population. Disseminated disease is also found in individuals with secondary immunodeficiencies as a result of disease or therapeutic immunosuppression.
Treatment for Histoplasmosis and Coccidioidomycosis
Therapy is designed to inhibit sterol synthesis in the fungal membranes. The plasma membranes of Histoplasma and Coccidioides contain ergosterol, a nonpolar sterol. Sterols control membrane fluidity, passage of material in and out of the fungi, and some physiologic functions. Inhibition of ergosterol synthesis causes membrane failure, and osmotic lyses of the fungi. Therapeutic agents used in the treatment of histoplasmosis and coccidioidomycosis and their mechanisms of action are provided in Table 19-4.
Table 19-4
Drugs Used to Treat Histoplasmosis and Coccidioidomycosis
| Drug | Used to Treat | Action |
| Amphotericin B | Histoplasmosis and coccidioidomycosis | Binds to sterols, and ergosterol in the cell membrane causing leakage of cytoplasmic fluids |
| Itraconazole | Histoplasmosis and coccidioidomycosis | Inhibits the synthesis of ergosterol |
| Ketoconazole | Histoplasmosis and coccidioidomycosis | Inhibits the synthesis of ergosterol |
| Fluconazole | Histoplasmosis and coccidioidomycosis | Prevents the conversion of lanosterol to ergosterol |
| Posaconazole | Histoplasmosis | Blocks ergosterol synthesis in membrane |
| Naproxen | Histoplasmosis | Decreases leukotriene synthesis, lysosomal enzyme release, neutrophil aggregation |
| Ibuprofen | Histoplasmosis | Decreased prostaglandin synthesis |
Delayed-Type Hypersensitivity Reactions in the Skin
Dermatitis can be caused by both nonimmunologic and immunologic mechanisms. Over 80% of skin eruptions are an irritant dermatitis caused by exposure to acids, alkalis, solvents, soaps, and metals. These skin eruptions have no immunologic basis. Only photocontact dermatitis and allergic contact dermatitis are believed to be immunologically mediated.
Photocontact Dermatitis
Quinolones, sulfonamides, tetracyclines, and trimethoprim are the major agents of photocontact dermatitis. Drug reactions usually begin 7 to 10 days after the initiation of therapy and usually occur in the sun-exposed areas of the skin. Lesions are usually pruritic, erythematous, and vesicular and resemble severe sunburn. Symptoms subside rapidly following cessation of the drug therapy.
Photocontact dermatitis is a cell-mediated response to drugs or drug metabolites that act as haptens. Photohaptenic drugs bind to class II molecules on Langerhans cells. Exposure to sunlight induces structural changes in the bound hapten and the class II molecule creating an antigen that is recognized by CD4Th1 cells. Cytokines released from the T cells induce changes in skin cells.
Contact Dermatitis
Twenty-five chemicals and one plant species cause the majority of DTH reactions in the skin. The agents include nickel, paraphenylenediamine (permanent hair dyes), formaldehyde releasers (quaternium-15 and isothiazolinones), neomycin, latex manufacturing process contaminants, and poison ivy. With the exception of nickel and poison ivy, the nature of the cell-mediated responses to the other irritants is unclear.
Nickel Hypersensitivity
Nickel is a major component of studded earrings, costume jewelry, watches, belt buckles, dental appliances, and orthopedic prosthetics. Usually, the nickel is covered by an outer layer of gold or silver alloy. With normal wear, the outer coat erodes, liberating nickel ions. Animal models suggest that nickel sensitivity differs from classic DTH reactions. In the nickel reaction, Langerhans cells transport nickel to the lymph node, where it stimulates a specialized T cell population known as Th17 cells. These T cells produce a specialized cytokine known as interleukin 17A (IL-17A). Activated Th17 cells migrate to the skin, where they release IL-17A, which upregulates cellular adhesion factors on keratinocytes and promotes localization of the inflammatory cells in the skin.
A genetic component associated with nickel sensitivity does exist. When compared with the normal population, a significant increase is seen in the frequency of HLA-DRw6 in individuals with nickel contact dermatitis.
Treatment of Contact Dermatitis
Topical and systemic corticosteroids are the mainstays of contact dermatitis treatment modalities. However, other topical immunomodulators (TIMs) and immunosuppressive agents are used to treat dermatitis as well. The full spectrum of available drugs are listed in Table 19-5.
Table 19-5
Drugs Used to Treat Contact Dermatitis
| Drug | Action |
| Prednisone | Anti-inflammatory drug that prevents diapedesis and suppresses polymorphonuclear (PMN) functions |
| Pimecrolimus | Binds to cytostolic immunophilin receptor 2 and inhibits production of proinflammatory cytokines |
| Tacrolimus | Inhibits transcription of interleukin 3 (IL-3), IL-4, IL-5, and tumor necrosis factor alpha (TNF-α) |
| Triamcinolone | Inhibits migration of PMNs and decreases capillary permeability |
Plants and Hypersensitivity Reactions
Members of the plant genus Toxicodendron cause contact dermatitis. This genus includes poison ivy, poison oak, and poison sumac. Poison ivy usually grows east of the Rocky Mountains and in Canada. Poison oak usually grows in the western and southeastern parts of the United States, in Canada, and in Mexico. Sumac is also found in the eastern parts of the United States and in southern Canada.
Sap from these Toxicodendron species contains a mixture of saturated and unsaturated catechols known as urushiol. The immunologic response to haptenic urushiol is unique. Langerhans cells ingest free urushiol and use cross-priming to load haptens onto class I molecules. Unlike classic DTH reactions, urushiol-specific cytotoxic CD8 T cells proliferate in the lymph node and enter the peripheral circulation. The CD8 cells attack any cell that has urushiol on its surface. Lysis of skin cells allows fluid to enter the skin, creating the characteristic weeping poison ivy lesions.
Treatment for Poison Ivy Dermatitis
Drugs used to treat poison ivy dermatitis are similar to those used for treating contact dermatitis. Nonprescription topical products such as hydrocortisone, antihistamines, astringents, and anesthetics are useful in alleviating the pruritus and inflammation associated with poison ivy skin reactions. A list of other drugs used to treat poison ivy and their mechanisms of action is provided in Table 19-6.
Table 19-6
Drugs Used to Treat Poison Ivy
| Drug | Action |
| Prednisone | Anti-inflammatory that prevents diapedesis and suppresses polymorphonuclear (PMN) functions |
| Diphenhydramine | Prevents the release of mediators from basophils and mast cells |
| Hydroxyzine | Antagonizes H1 receptors in the skin |
| Cimetidine | Antagonizes H2 receptors in the skin |
Delayed-Type Hypersensitivity Reactions in the Intestine
Celiac Sprue or Gluten-Sensitive Enteropathy
Celiac sprue is a chronic autoimmune inflammatory response which damages the intestinal mucosa and impedes normal digestion and absorption of foods. Patients with the classic form of celiac sprue present with moderate to severe diarrhea and failure to thrive.
The etiologic agent of the disease is gliadin, a component of gluten found in wheat, rye, barley, and oats. In the intestine, gliadin is deaminated by a transaminase to create two negatively charged proteins. A large protein fragment remains complexed with catalytic transaminase and is processed and presented to T cells in the context of class II molecules. Within the lamina propria, antigen presentation stimulates proliferation of CD4Th1 cells and IFN-γ production. IFN activates macrophages that release enzymes and cytokines, which increase mucosal permeability and damage the mucosa.
The second protein, a small negatively charged molecule, stimulates the enteric epithelial cells to secrete IL-15, which upregulates the expression of antigenic proteins on mucosal cells. These proteins are the target of both CD8 and NK cells.
A genetic component to celiac sprue does exist. Approximately 95% of individuals with celiac sprue have a DQ2 heterodimer composed of DQB1∗02 and DQA1∗05. These class II molecules avidly and selectively bind negatively charged proteins.
Treatment for Celiac Sprue
The primary treatment is the removal of gluten from the diet. In patients undergoing an acute clinical episode, a combination of corticosteroids and gluten elimination bring the disease into remission. At this point, diet control is sufficient to control the disease.
Summary
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Delayed-Type Hypersensitivity Reactions
Learning Objectives
• Identify the definition of delayed-type hypersensitivity (DTH)
• Compare and contrast immediate and cell-mediated hypersensitivities
• List the six Mycobacterium species that are human pathogens
• Recognize the components of an acid-fast cell wall
• Explain the relationship between interferon gamma (IFN-γ) and DTH
• Explain the relationship between vitamin D and leprosy
• Compare and contrast the three histologic stages of tuberculosis
• Recognize the cellular components in a granuloma
• Compare and contrast Mycobacterium tuberculosis and Bacille Calmette-Guérin (BCG)
• Explain the mechanism involved in the QuantiFERON-Gold test
• Recognize the four front-line drugs used to treat active tuberculosis and their mechanisms of action
• Recognize the second-line drugs used to treat active tuberculosis and their mechanisms of action
• Identify the definition of multidrug-resistant–tuberculosis (MDR–TB)
• Define extensively drug-resistant–tuberculosis (XDR–TB)
• Identify the dimorphic phases of histoplasmosis
• Compare the role of macroconidia and the yeast form in the pathogenesis of histoplasmosis
• Recognize regions of the United States where histoplasmosis is endemic
• Compare and contrast granuloma formation in histoplasmosis and tuberculosis
• Identify the groups at risk for progressive disseminated histoplasmosis
• Identify the dimorphic forms of Coccidioides immitis
• Identify regions of the United States where Coccidioides is endemic
• Recognize the role of arthroconidia in the pathogenesis of coccidioidomycosis
• Differentiate between spherules and endospores in the pathogenesis of coccidioidomycosis
• Compare and contrast contact dermatitis, irritant dermatitis, and photocontact dermatitis
• Recognize the drugs that are used to treat contact dermatitis
• Explain the immunologic skin response to nickel
• Identify the role(s) of Th17 cells in metal hypersensitivity
• Recognize the drugs that are used to treat nickel hypersensitivity
• List the three members of the Toxicodendron genus that cause skin reactions
• Identify the haptenic substance produced by the Toxicodendron species
• Explain the immunologic response to urushiol
• List the drugs that are used to treat poison ivy reaction
• Explain the immunologic and biologic responses active in celiac sprue
Key Terms
Allergic contact dermatitis
Arthroconidia
Bacille Calmette-Guérin (BCG)
Conidia
Cord factor
Extensively drug-resistant tuberculosis
Gliadin
Granuloma
Interferon gamma (IFN-γ)
Lysosomes
Mantoux test
Multidrug-resistant tuberculosis
Mycolic acids
Natural resistance–associated macrophage protein (NRAMP)
Photocontact dermatitis
T17 cells
Tumor necrosis factor
Urushiol
Vitamin D receptor
Introduction
Delayed-type hypersensitivity (DTH) is a unique type of cell-mediated immunity. The name originated from the skin test used in the diagnosis of tuberculosis and denotes cellular infiltrates causing induration and erythema at the skin test site within 24 to 72 hours. DTH was coined to describe the reaction to the tuberculosis skin test and to differentiate between delayed cellular skin test results and antibody-mediated immediate skin test results. The term has been expanded to include cell-mediated reactions to bacteria or fungi infecting lungs and skin responses to chemicals and plants. In the lung, cellular responses to tuberculosis, histoplasmosis, and coccidioidomycosis are considered DTH reactions. Skin reactions to nickel and poison ivy also are DTH reactions.
The principal effectors of DTH reactions are CD4Th1 lymphocytes, monocytes or macrophages, CD8Tc1 cells, and natural killer (NK) cells. The histopathology of DTH lesions varies with the nature of the antigen, the type of effector cells, and the anatomic location.
Delayed-Type Hypersensitivity Responses in the Lung
Tuberculosis
The family Mycobacteriaceae contains the Mycobacterium genus. Within the genus are six different human pathogens (Table 19-1). (1) M. tuberculosis causes most of the 9.1 million cases of pulmonary tuberculosis in the world. (2) M. leprae, which is also known as Hansen’s bacillus, is commonly found in tropical countries and is the etiological agent of leprosy. (3&4) Mycobacterium avium and M. intracellulare are found in soil, water systems, birds, and other domestic animals. Because the two species are difficult to identify in the laboratory, they are designated as the Mycobacterium avium complex (MAC). Patients with acquired immunodeficiency syndrome (AIDS) and therapeutically immunosuppressed individuals are frequently infected by MAC organisms. (5) M. bovis is a cattle-adapted species of tuberculosis that is also found in wild and domestic animals, but it causes less than 1% of all tuberculosis infections. Human infection is caused by inhalation of droplets containing live organisms or by ingestion of unpasteurized milk. Ingestion leads to extrapulmonary tuberculosis in lymph nodes and skeletal bones. (6) Mycobacterium africanum is commonly found in West Africa, where it accounts for 25% of tuberculosis cases.
Table 19-1
Species of Tuberculosis Infecting Humans
| Pathogen | Infection/Transmission |
| Mycobacterium tuberculosis | Human tuberculosis. Transmitted by inhalation route |
| Mycobacterium leprae | Hansen’s disease, or leprosy. Transmitted by inhalation route |
| Mycobacterium avium-intracellulare | Group of closely related opportunistic pathogens that infect immunosuppressed patients with human immunodeficiency virus (HIV). Transmitted by inhalation and oral routes |
| Mycobacterium bovis | Tuberculosis in cattle. Can be transmitted to humans via infected milk or aerosols |
| Mycobacterium africanum | Commonly found in West Africa. Spread by the airborne route |
Tuberculosis: Virulence Factors
In the lung, survival of the tuberculosis bacterium is determined, in part, by cell-wall mycolic acids and a lipoarabinomannan known as the cord factor. Mycolic acids, which are 50% of the dry weight of the cell wall (Figure 19-1), form a hydrophobic barrier that prevents the entry of drugs, disinfectants, and other harsh chemicals. Cord factor inhibits the interferon (IFN)–induced activation of macrophages and stimulates the production of tumor necrosis factor alpha (TNF-α).
Laboratory Identification of Mycobacterium Species
Laboratory identification of mycobacteria is difficult because conventional stains do not penetrate the cell wall. However, mycobacteria can be identified by the Ziehl Neelsen stain, which uses a combination of heat and carbolfuchsin to drive the stain into the cell wall. Only the Mycobacterium and Nocardia species retain the dye following harsh treatment with an acid–alcohol mixture. Thus, these species are designated acid-fast organisms.
Pathophysiology of Tuberculosis
Exposure to tuberculosis occurs by inhalation of droplets containing live organisms. Because of their small size, the organisms are usually deposited in the distal areas of the lung. In the first stage of the disease, mycobacteria take up residence in phagosomes within alveolar macrophages and prevent intracellular killing by inhibiting maturation of the phagosome and fusion with endosomes and lysosomes which contain antimicrobial proteins and enzymes. Rapid logarithmic multiplication of mycobacteria in macrophages eventually causes the death of the cells and creates a small necrotic area in the lung. Free mycobacteria are ingested by circulating monocytes and transported to the mediastinal lymph node. Antigen stimulation in the lymph node generates CD4Th1, CD8, and long-lived memory cells.
In the second stage, which occurs 3 weeks after infection, CD4Th1 cells migrate from the lymph node to the infection site and undergo rapid proliferation. CD4Th1 and CD8 cells secrete TNF-α and IFN-γ. TNF-α is responsible for most of the lung damage noted in mycobacteria infections. IFN-γ– stimulated macrophages produce nitric oxide by oxidation of L-citrulline. Additional interactions between nitric oxide and singlet oxygen form peroxynitrite (ONOO–), which is microcidal. The role of CD8 cells in tuberculosis is unclear. They may contribute to the lesion by producing IFN-γ, by directly killing infected cells, or by both mechanisms (Figure 19-2).
In the third phase of the disease, macrophage-derived reactive oxygen species cause necrosis of lung tissue. The production of chemotactic factors causes an influx of activated monocytes into the area. In an attempt to destroy the bacteria, activated monocytes surround the caseous center and release additional cytotoxic molecules. Some monocytes become flattened and are known as epithelioid cells (Figure 19-3). Fusion of macrophage membranes often creates giant cells with multiple nuclei.
In a final attempt to contain the infection, fibroblasts are called into the area and produce collagen, which encapsulates the inflammatory cells surrounding the lesion. The accumulation of macrophages, lymphocytes, and fibroblasts is called a granuloma. Constant production of TNF and IFN-γ is required for the maintenance of the granuloma; if the granuloma is not maintained, it becomes unstable, liquefies, and creates a permissible environment for bacterial growth. As a consequence of rapid bacterial growth, the infection erodes into blood vessels and small airways. Dissemination in blood allows the organism to establish secondary infections in other organs and bone. This form of the disease is known as miliary tuberculosis. Mycobacteria in the airways also rapidly proliferate in the highly aerobic environment and are expelled from the lung by coughing or sneezing.
Genetics and Mycobacterium Infections
The role of host genetics in Mycobacterium infection was suggested by an accidental administration of virulent M. tuberculosis to 249 babies in Lubeck, Germany, in 1926. Although 76 babies died of the disease, 123 had lung lesions that healed, and another 50 babies had no signs of infection. This suggested that genetics influences susceptibility to tuberculosis. Subsequent studies indicated that persons at risk for active tuberculosis have dysfunctional receptors for IFN-γ or vitamin D. Some individuals also have a defect in the production of natural resistance–associated macrophage proteins (NRAMPs).
Interferon Gamma Receptor Deficiency
[/not-level-membership-for-allergy-and-immunology-category]
