[level-membership-for-pediatrics-category]
Chapter 80 Defects in Metabolism of Lipids
80.1 Disorders of Mitochondrial Fatty Acid β-Oxidation
Genetic defects have been identified in nearly all of the known steps in the fatty acid oxidation pathway; all are recessively inherited (Table 80-1).
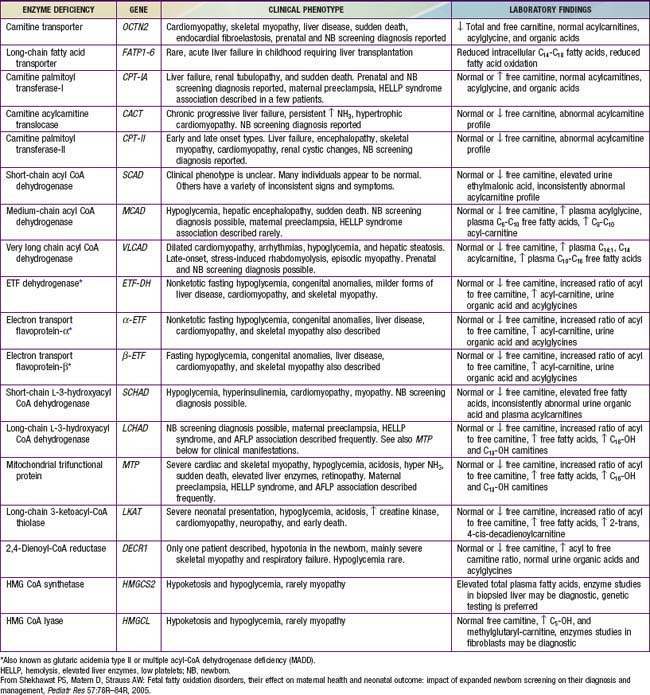
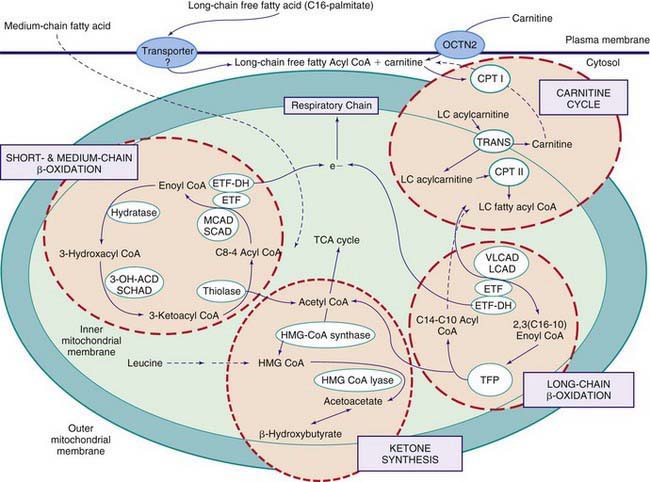
Figures 80-1 and 80-2 outline the steps involved in the oxidation of a typical long-chain fatty acid. In the carnitine cycle, fatty acids are transported across the barrier of the inner mitochondrial membrane as acylcarnitine esters. Within the mitochondria, successive turns of the 4-step β-oxidation cycle convert the coenzyme A (CoA)-activated fatty acid to acetyl CoA units. Two to three different chain-length specific isoenzymes are needed for each of these β-oxidation steps to accommodate the different-sized fatty acyl CoA species. The electron transfer pathway carries electrons generated in the 1st β-oxidation step (acyl CoA dehydrogenase) to the electron transport chain for adenosine triphosphate (ATP) production, while electrons generated from the third step (3-hydroxyacyl CoA dehydrogenase) enter the respiratory chain at the level of complex 1. Most of the acetyl CoA generated from hepatic β-oxidation flows through the pathway of ketogenesis to form β-hydroxybutyrate and acetoacetate.
Defects in the β-Oxidation Cycle
Medium-Chain Acyl CoA Dehydrogenase (MCAD) Deficiency
Treatment
Acute illnesses should be promptly treated with intravenous fluids containing 10% dextrose to treat or prevent hypoglycemia and to suppress lipolysis as rapidly as possible (Chapter 86). Chronic therapy consists of avoiding fasting. This usually requires simply adjusting the diet to ensure that overnight fasting periods are limited to <10-12 hr. Restricting dietary fat or treatment with carnitine is controversial. The necessity for active therapeutic intervention for individuals with the T199C variant has not yet been established.
Long-Chain 3-Hydroxyacyl CoA Dehydrogenase (LCHAD)/Mitochondrial Trifunctional Protein (TFP) Deficiency
Defects in the Carnitine Cycle
Defects in Electron Transfer Pathway
Defects in Ketone Synthesis Pathway
Defects in Ketone Utilization
De Leon DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57-68.
Gillingham MB, Weleber RG, Neuringer M, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005;86:124-133.
Greenberg CR, Dilling LA, Thompson GR, et al. The paradox of the carnitine palmitoyltransferase type 1a P479L variant in Canadian Aboriginal populations. Mol Genet Metab. 2009;96:201-207.
Hsu HW, Zytkovicz TH, Comeau AM, et al. Spectrum of medium-chain acyl-CoA dehydrogenase deficiency detected by newborn screening. Pediatrics. 2008;121:e1108-e1114.
Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol Genet Metab. 2008;95:195-200.
Longo N, di San Filippo CA, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77-85.
Loughrey C, Bennett MJ. Screening for MCAD deficiency in newborns. BMJ. 2009;338:843-846.
Molven A, Matre GE, Duran M, et al. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes. 2004;53:221-227.
Schulze A, Matern D, Hoffmann GF. Newborn Screening. In: Sarafoglou K, Hoffmann GF, Roth KS, editors. Pediatric endocrinology and inborn errors of metabolism. New York: McGraw-Hill; 2009:17-32.
Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57:78R-86R.
Strauss AW, Andresen BS, Bennett MJ. Mitochondrial fatty acid oxidation defects. In: Sarafoglou K, Hoffmann GF, Roth KS, editors. Pediatric endocrinology and inborn errors of metabolism. New York: McGraw-Hill; 2009:51-70.
van Maldegem BT, Duran M, Wanders RJA, et al. Fasting and fat-loading tests provide pathophysiological insight into short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 2010;156:121-127.
Wilcken B, Haas M, Joy P, et al. Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet. 2007;369:37-42.
80.2 Disorders of Very Long Chain Fatty Acids
Peroxisomal Disorders
Etiology
Peroxisomal disorders are subdivided into 2 major categories (Table 80-2).
Table 80-2 CLASSIFICATION OF PEROXISOMAL DISORDERS
A: DISORDERS OF PEROXISOME IMPORT
A1: Zellweger syndrome
A2: Neonatal adrenoleukodystrophy
A3: Infantile Refsum disease
A4: Rhizomelic chondrodysplasia punctata
B: DEFECTS OF SINGLE PEROXISOMAL ENZYME
B1: X-linked adrenoleukodystrophy
B2: Acyl CoA oxidase deficiency
B3: Bifunctional enzyme deficiency
B4: Peroxisomal thiolase deficiency
B5: Classic Refsum disease
B6: 2-Methylacyl CoA racemase deficiency
B7: DHAP acyltransferase deficiency
B8: Alkyl-DHAP synthase deficiency
B9: Mevalonic aciduria
B10: Glutaric aciduria type III
B11: Hyperoxaluria type I
B12: Acatalasemia
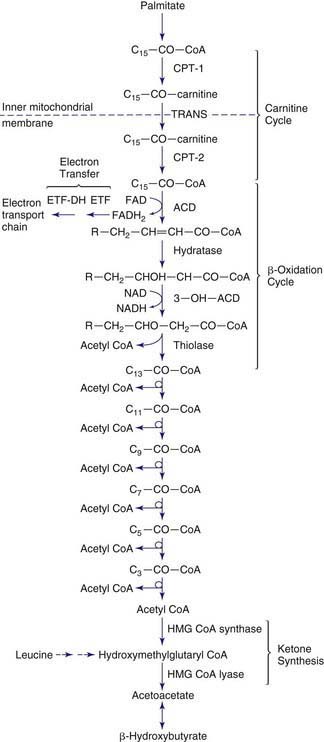
In category A, the peroxisomal biogenesis disorders (PBD), the basic defect is the failure to import one or more proteins into the organelle. In category B, defects affect a single peroxisomal protein. The peroxisome is present in all cells except mature erythrocytes and is a subcellular organelle surrounded by a single membrane; >50 peroxisomal enzymes are identified. Some enzymes are involved in the production and decomposition of hydrogen peroxide; others are concerned with lipid and amino acid metabolism. Most peroxisomal enzymes are first synthesized in their mature form on free polyribosomes and enter the cytoplasm. Proteins that are destined for the peroxisome contain specific peroxisome targeting sequences (PTS). Most peroxisomal matrix proteins contain PTS1, a 3-amino acid sequence at the carboxyl terminus. PTS2 is an amino-terminal sequence that is critical for the import of enzymes involved in plasmalogen and branched-chain fatty acid metabolism. Import of proteins involves a complex series of reactions that involves at least 23 distinct proteins. These proteins are referred to as peroxins encoded by PEX genes. Table 80-3 summarizes the PEX genes that are defective in human disease states.
Epidemiology
Except for X-linked adrenoleukodystrophy (X-ALD), all the peroxisomal disorders in Table 80-2 are autosomal recessive traits. X-ALD is the most common peroxisomal disorder, with an estimated incidence of 1/17,000. The combined incidence of the other peroxisomal disorders is estimated to be 1/50,000.
Pathogenesis
It is likely that all pathologic changes are secondary to the peroxisome defect. Multiple peroxisomal enzymes fail to function in the PBD (Table 80-4). The enzymes that are diminished or absent are synthesized but are degraded abnormally fast because they may be unprotected outside of the peroxisome. It is not clear how defective peroxisome functions lead to the widespread pathologic manifestations.
Table 80-4 ABNORMAL LABORATORY FINDINGS COMMON TO DISORDERS OF PEROXISOME BIOGENESIS
Peroxisomes absent to reduced in number
Catalase in cytosol
Deficient synthesis and reduced tissue levels of plasmalogens
Defective oxidation and abnormal accumulation of very long chain fatty acids
Deficient oxidation and age-dependent accumulation of phytanic acid
Defects in certain steps of bile acid formation and accumulation of bile acid intermediates
Defects in oxidation and accumulation of L-pipecolic acid
Increased urinary excretion of dicarboxylic acids
The PBD are associated with genetically determined import defects. The PBD have been subdivided into 12 complementation groups. The molecular defects have been defined in 10 of these groups (see Table 80-3). The pattern and severity of pathologic features vary with the nature of the import defects and the degree to which import is impaired. These gene defects lead to disorders that were named before their relationship to the peroxisome was recognized, namely, Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and rhizomelic chondrodysplasia punctata (RCDP). The first 3 disorders are considered to form a clinical continuum, with ZS the most severe, IRD the least severe, and NALD intermediate. They can be caused by 11 different gene defects, which involve mainly the import of proteins that contain the PTS1 targeting signal; the gene defects cannot be distinguished on the basis of clinical features. The clinical severity varies with the degree to which protein import is impaired. Mutations that abolish import completely are often associated with the ZS phenotype, whereas a missense mutation, in which some degree of import function is retained, leads to the somewhat milder phenotypes. A defect in PEX7, which involves the import of proteins that utilize PTS2, is associated with RCDP. PEX7 defects that leave import partially intact are associated with milder phenotypes, some of which resemble classic Refsum disease.
PBD With Milder or Atypical Phenotypes
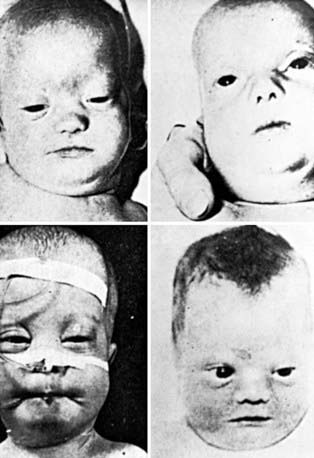
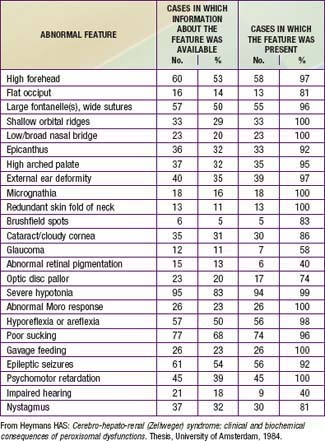
Newborn infants with Zellweger syndrome show striking and consistent, recognizable abnormalities. Of central diagnostic importance are the typical facial appearance (high forehead, unslanting palpebral fissures, hypoplastic supraorbital ridges, and epicanthal folds; Fig. 80-3), severe weakness and hypotonia, neonatal seizures, and eye abnormalities (cataracts, glaucoma, corneal clouding, Brushfield spots, pigmentary retinopathy, and nerve dysplasia). Because of the hypotonia and “mongoloid” appearance, Down syndrome may be suspected. Infants with Zellweger syndrome rarely live more than a few months. More than 90% show postnatal growth failure. Table 80-5 lists the main clinical abnormalities.
Rhizomelic Chondrodysplasia Punctata (Rcdp)
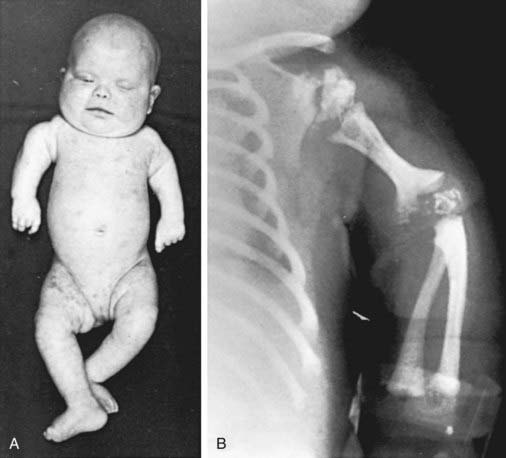
This disorder is characterized by the presence of stippled foci of calcification within the hyaline cartilage and is associated with dwarfing, cataracts (72%), and multiple malformations due to contractures. Vertebral bodies have a coronal cleft filled by cartilage that is a result of an embryonic arrest. Disproportionate short stature affects the proximal parts of the extremities (Fig. 80-4A). Radiologic abnormalities consist of shortening of the proximal limb bones, metaphyseal cupping, and disturbed ossification (Fig. 80-4B). Height, weight, and head circumference are less than the 3rd percentile, and these children are severely retarded mentally. Skin changes such as those observed in ichthyosiform erythroderma are present in about 25% of patients.
Isolated Defects of Peroxisomal Fatty Acid Oxidation
The disorders labeled B1 through B3 (see Table 80-2) each involve 1 of 3 enzymes involved in peroxisomal fatty acid oxidation. Their clinical manifestations resemble those of the Zellweger syndrome/neonatal ALD/infantile Refsum disease continuum; they can be distinguished from disorders of peroxisome biogenesis by laboratory tests. Defects of bifunctional enzyme are common and are found in about 15% of patients with the Zellweger syndrome/neonatal ALD/infantile Refsum disease phenotype. Patients with isolated acyl CoA oxidase deficiency have a somewhat milder phenotype that resembles that of neonatal ALD.
Isolated Defects of Plasmalogen Synthesis
Plasmalogens are lipids in which the 1st carbon of glycerol is linked to an alcohol rather than a fatty acid. They are synthesized through a complex series of reactions, the 1st two steps of which are catalyzed by the peroxisomal enzymes dihydroxyacetone phosphate alkyl transferase and synthase. Deficiency of either of these enzymes (B4 and B5 in Table 80-2) leads to a phenotype that is clinically indistinguishable from the peroxisomal import disorder RCDP. This latter disorder is caused by a defect in PEX7, the receptor for peroxisome targeting sequence 2. It shares the severe deficiency of plasmalogens with disorders B4 and B5 but, in addition, has defects of phytanic oxidation. The fact that disorders B4 and B5 are associated with the full phenotype of RCDP suggests that a deficiency of plasmalogens is sufficient to produce it.
Laboratory Findings
Laboratory tests for peroxisomal disorders can be viewed at three levels of complexity.
Level 1: Does the Patient have a Peroxisomal Disorder?
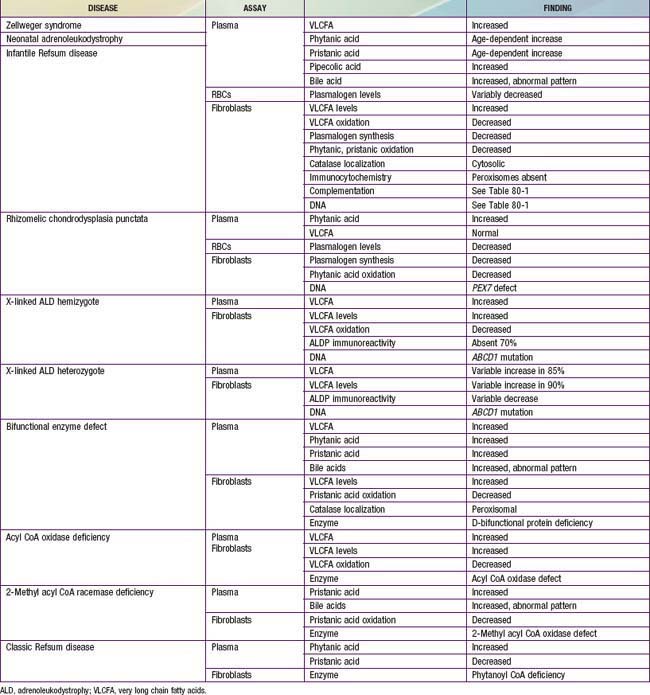
This can be resolved by noninvasive tests that are generally available (Table 80-6). Measurement of plasma VLCFA is the most commonly used assay. Whereas plasma VLCFA levels are elevated in many patients with peroxisomal disorders, this is not always the case. The most important exceptions are RCDP, in which VLCFA levels are normal, but plasma phytanic acid levels are increased and red blood cell plasmalogen levels are reduced. In some other peroxisomal disorders, the biochemical abnormalities are still more restricted. Therefore, a panel of tests is recommended and includes plasma levels of VLCFA and phytanic, pristanic, and pipecolic acids and red blood cell levels of plasmalogens. Tandem mass spectrometry techniques also permit convenient quantitation of bile acids in plasma and urine. This panel of tests can be performed on 2 mL samples of venous blood and permits detection of most peroxisomal disorders. Furthermore, normal results make the presence of a peroxisomal disorder unlikely.
Level 2: What is the Precise Nature of the Peroxisomal Disorder?
Table 80-6 lists the main biochemical abnormalities in the various peroxisomal disorders. When combined with the clinical presentation, the panel of level 1 tests (see earlier) is often sufficient to identify the precise nature of the defect. Elevated plasma VLCFA levels permit the precise diagnosis of X-ALD in male patients. Marked reduction of erythrocyte plasmalogen levels combined with elevated plasma phytanic acid permits precise diagnosis in a patient with the clinical features of RCDP. Classic Refsum disease can be diagnosed by demonstration of increased plasma phytanic acid combined with normal or reduced levels of pristanic acid levels, while in D-bifunctional enzyme deficiency and 2-methylacyl CoA racemase deficiency, the levels of pristanic and phytanic acid are both increased. Precise identification of some peroxisomal disorders may require more extensive studies in cultured skin fibroblasts. This may be required for the differentiation of PBD from defects in bifunctional enzyme. In PBD, the patient’s peroxisomes are absent and catalase is in the soluble fraction, whereas in bifunctional enzyme defect, peroxisomes are present and catalase is in the particulate fraction. Fibroblast studies are required to identify the nature of the molecular defect in PBD. Whether such specialized studies are clinically warranted depends on individual circumstances. Precise definition of the defect in a proband may improve the precision of prenatal diagnosis in at-risk pregnancies, and it is required for carrier detection. It is also of value in setting prognosis. Precise characterization is of prognostic value in patients with PEX1 defects. This defect is present in approximately 60% of PBD patients, and about half of the PEX1 defects have the G843D allele, which is associated with a significantly milder phenotype than is found in other mutations.
Diagnosis
There are several noninvasive laboratory tests that permit precise and early diagnosis of peroxisomal disorders (see Table 80-6). The challenge in PBD is to differentiate them from the large variety of other conditions that can cause hypotonia, seizures, failure to thrive, or dysmorphic features. Experienced clinicians can readily recognize classic Zellweger syndrome by its clinical manifestations. PBD patients often do not show the full clinical spectrum of disease and may be identifiable only by laboratory assays. Clinical features that may serve as indications for these diagnostic assays include severe psychomotor retardation; weakness and hypotonia; dysmorphic features; neonatal seizures; retinopathy, glaucoma, or cataracts; hearing deficits; enlarged liver and impaired liver function; and chondrodysplasia punctata. The presence of one or more of these abnormalities increases the likelihood of this diagnosis. Atypical milder forms presenting as peripheral neuropathy have also been described.
Genetic Counseling
All the peroxisomal disorders, except hyperoxaluria type 1, can be diagnosed prenatally in the 1st or 2nd trimester. The tests are similar to those described for postnatal diagnosis (see Table 80-6) and use chorionic villus sampling or amniocytes. More than 300 pregnancies have been monitored, and more than 60 affected fetuses have been identified without diagnostic error. Because of the 25% recurrence risk, couples with an affected child must be advised about the availability of prenatal diagnosis. Heterozygotes can be identified in X-ALD and in those disorders in which the molecular defect has been identified (see Table 80-3).
Adrenoleukodystrophy (X-Linked)
Etiology
The key biochemical abnormality is the tissue accumulation of unbranched saturated VLCFA, with a carbon chain length of 24 or more. Excess hexacosanoic acid (C26 : 0) is the most striking and characteristic feature. This accumulation of fatty acids is caused by genetically deficient peroxisomal degradation of fatty acid. The key biochemical defect involves the impaired function of peroxisomal lignoceroyl CoA ligase, the enzyme that catalyzes the formation of the CoA derivative of VLCFA. The gene that is defective (ABCD1) codes for a peroxisomal membrane (ALDP). More than 400 distinct mutations have been identified, and most families have a mutation that is “private” (unique to that kindred) and are updated on the website, www.x-ald.nl. The gene has been mapped to chromosome Xq28. The mechanism by which the ALDP defect leads to VLCFA accumulation and the pathology of X-ALD is unknown.
Laboratory and Radiographic Findings
CT And MRI
Patients with childhood cerebral or adolescent ALD show cerebral white matter lesions that are characteristic with respect to location and attenuation patterns on MRI. In 80% of patients, the lesions are symmetric and involve the periventricular white matter in the posterior parietal and occipital lobes. About 50% show location of a garland of accumulated contrast material adjacent and anterior to the posterior hypodense lesions (Fig. 80-5A). This zone corresponds to the zones of intense perivascular lymphocytic infiltration where the blood-brain barrier breaks down. In 12% of patients, the initial lesions are frontal. Unilateral lesions that produce a mass effect suggestive of a brain tumor may occur. MRI provides a clearer delineation of normal and abnormal white matter than does CT and may demonstrate abnormalities missed by CT (Fig. 80-5B).
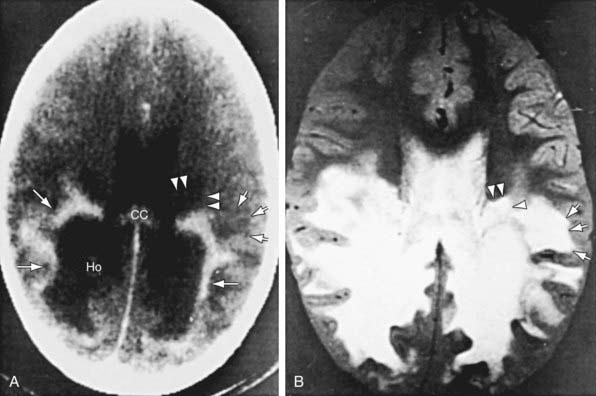
Diagnosis and Differential Diagnosis
The earliest manifestations of childhood cerebral ALD are difficult to distinguish from the more common attention-deficit disorders or learning disabilities. Rapid progression, signs of dementia, or difficulty in auditory discrimination suggest ALD. Even in early stages, CT or MRI may show strikingly abnormal changes. Other leukodystrophies (Chapters 592 and 605.10) or multiple sclerosis (Chapter 593.1) may mimic these radiographic findings. Definitive diagnosis depends on demonstration of VLCFA excess, which occurs only in X-ALD and the other peroxisomal disorders. The latter may be distinguished from X-ALD by their clinical presentation during the neonatal period.
Treatment
Corticosteroid replacement for adrenal insufficiency or adrenocortical hypofunction is effective (Chapter 569). It may be lifesaving and increase general strength and well-being, but it does not alter the course of the neurologic disability.
Ferdinandusse S, Ylianttila MS, Gloerich J, et al. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am J Hum Genet. 2006;78:112-124.
Fidaleo M. Peroxisomes and peroxisomal disorders: the main facts. Exp Toxicol Pathol. 2009 Sep 7. Epub ahead of print
Moser HW. Genotype-phenotype correlations in disorders of peroxisome biogenesis. Mol Genet Metab. 1999;68:316-327.
Motley AM, Brites P, Gerez L, et al. Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1. Am J Hum Genet. 2002;70:612-624.
Steinberg S, Chen L, Wei L, Moser A, et al. The PEX gene screen: Molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum. Mol Genet Metab. 2004;83:252-263.
Walter C, Gootjes J, Mooijer PA. Disorders of peroxisome biogenesis due to mutations in PEX1: phenotypes and PEX1 protein levels. Am J Hum Genet. 2001;69:35-48.
Wanders RJ, Jansen GA, Skjeldal OH. Refsum disease, peroxisomes and phytanic acid oxidation: a review. J Neuropathol Exp Neurol. 2001;60:1021-1031.
Wanders RJ, Komen JC. Peroxisomes, lipid metabolism, and peroxisomal disorders. Biochem Soc Trans. 2007;35:865-869.
Wanders RJ, Waterham HR. Peroxisomal disorders: the single peroxisomal enzyme deficiencies. Biochim Biophys Acta. 2006;1763:1707-1720.
Waterham HR, Koster J, van Roermund CWT, et al. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736-1740.
Adrenoleukodystrophy (X-Linked)
Kemp S, Pujol A, Waterham HR, et al. X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat. 2001;18:499-515.
Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3:150-151.
Moser HW, Moser AB, Hollandsworth K, et al. “Lorenzo’s oil” therapy for X-linked adrenoleukodystrophy: rationale and current assessment of efficacy. J Mol Neurosci. 2007;33:105-113.
Moser HW, Raymond GV, Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA. 2005;294:3131-3134.
Moser HW, Raymond GV, Lu SE, et al. Follow-up of 89 Lorenzo’s oil treated asymptomatic adrenoleukodystrophy patients. Arch Neurol. 2005;62:1073-1080.
Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881-888.
Stephenson DJ, Bezman L, Raymond GV. Acute presentation of childhood adrenoleukodystrophy. Neuropediatrics. 2000;31:293-297.
80.3 Disorders of Lipoprotein Metabolism and Transport
Blood Lipids and Atherogenesis
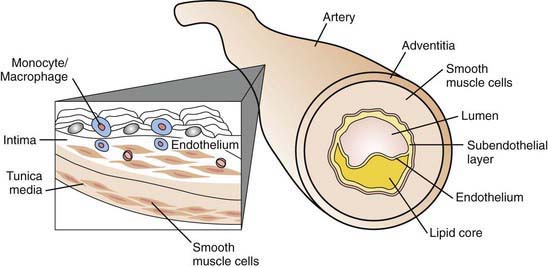
The early stage of development of atherosclerosis is thought to begin with vascular endothelial dysfunction and intima media thickness, which has been shown to occur in preadolescent children with risk factors such as obesity or familial hypercholesterolemia. The complex process of penetration of the intimal lining of the vessel may be due to a variety of insults, including the presence of highly toxic oxidized LDL particles. Lymphocytes and monocytes penetrate the damaged endothelial lining, where they become macrophages laden with LDL lipids and then become foam cells. Such accumulation is counterbalanced by HDL particles capable of removing lipid deposits from the vessel wall. Fundamental to plaque formation is an inflammatory process (elevated C-reactive protein) involving macrophages and the arterial wall. The deposition of lipid within the subendothelial lining of the arterial wall appears macroscopically as fatty streaks, which may to some degree be reversible. A later stage of plaque development involves disruption of arterial smooth muscle cells stimulated by the release of tissue cytokines and growth factors. The atheroma is composed of a core of fatty substance separated from the lumen by collagen and smooth muscle (Fig. 80-6). Growth of the atherosclerotic plaque may result in ischemia of the tissue supplied by the artery. Chronic inflammation within the atheroma, perhaps caused by infectious agents such as Chlamydia pneumoniae, results in plaque instability and subsequent rupture. Platelet adherence leads to clot formation at the site of rupture, resulting in myocardial infarction or a cerebrovascular event.
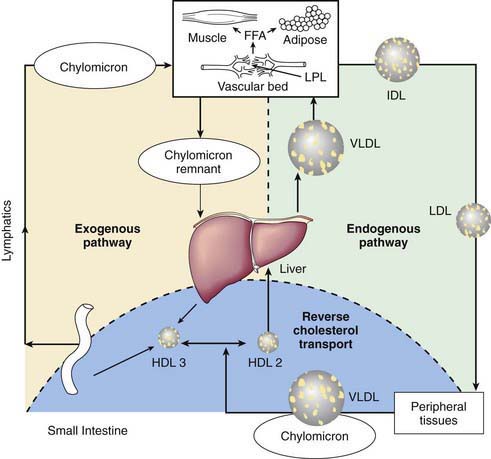
Plasma Lipoprotein Metabolism and Transport
Abnormalities of lipoprotein metabolism are associated with diabetes mellitus and premature atherosclerosis. Lipoproteins are soluble complexes of lipids and proteins that effect transport of fat absorbed from the diet, or synthesis by the liver and adipose tissues, for utilization and storage. Dietary fat is transported from the small intestine as chylomicrons. Lipids synthesized by the liver as very low density lipoproteins (VLDL) are catabolized to intermediate density lipoproteins (IDL) and low-density lipoproteins (LDL). High-density lipoproteins (HDL) are fundamentally involved in VLDL and chylomicron metabolism and cholesterol transport. Nonesterified free fatty acids (FFAs) are metabolically active lipids derived from lipolysis of triglycerides stored in adipose tissue bound to albumin for circulation in the plasma (Fig. 80-7).
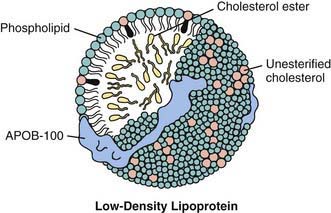
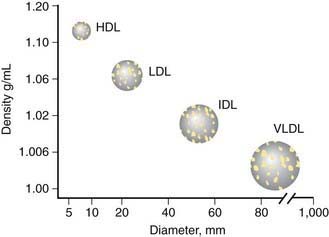
Lipoproteins consist of a central core of triglycerides and cholesteryl esters (CE) surrounded by phospholipids, cholesterol, and proteins (Fig. 80-8). The density of the several classes of lipoproteins is inversely proportional to the ratio of lipid to protein (Fig. 80-9).
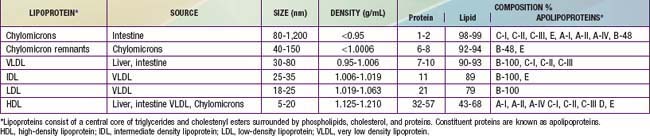
Constituent proteins are known as apolipoproteins (Table 80-7). They are responsible for a variety of metabolic functions in addition to their structural role, including cofactors or inhibitors of enzymatic pathways, and mediators of lipoprotein binding to cell surface receptors. ApoA is the major apoliprotein of HDL. ApoB is present in LDL, VLDL, IDL, and chylomicrons. ApoB-100 is derived from the liver, whereas apoB-48 comes from the small intestine. ApoC-I, C-II, and C-III are small peptides important in triglyceride metabolism. Likewise, apoE, which is present in VLDL, HDL, chylomicrons, and chylomicron remnants, plays an important role in the clearance of triglycerides.
Hyperlipoproteinemias
Hypercholesterolemia (Table 80-8)
Homozygous FH
The prognosis is poor regardless of the specific LDL receptor aberration. Severe atherosclerosis involving the aortic root and coronary arteries is present by early- to mid-childhood. These children usually present with xanthomas, which may cause thickening of the Achilles tendon or extensor tendons of the hands, or cutaneous lesions on the hands, elbows, knees, or buttocks (Figs. 80-10, 80-11, and 80-12). Corneal arcus may be present. Family history is informative because premature heart disease is strongly prevalent among relatives of both parents. The diagnosis may be confirmed by measuring LDL receptor activity in cultured skin fibroblasts. Phenotypic expression of the disease may also be assessed by measuring receptor activity on the surface of lymphocytes by using cell sorting techniques.
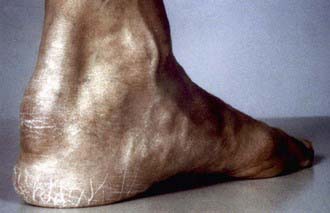
Figure 80-10 Achilles tendon xanthoma (heterozygous familial hypercholesterolemia).
(From Durrington P: Dyslipidaemia, Lancet 362:717–731, 2003.)
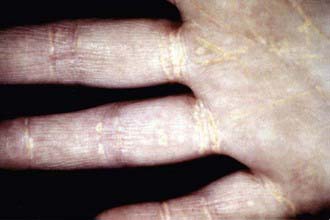
Figure 80-11 Striate palmar xanthomata.
(From Durrington P: Dyslipidaemia, Lancet 362:717–731, 2003.)
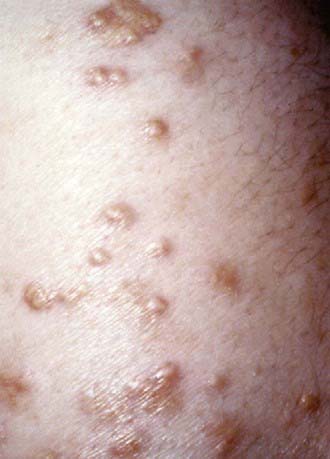
Heterozygous FH
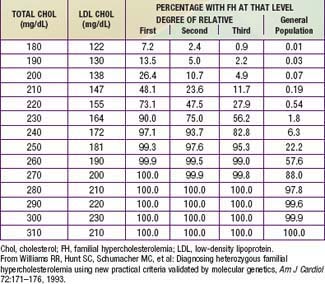
Very high cholesterol levels in children should prompt extensive screening of adult first- and second-degree relatives (“reverse” cholesterol screening). A child younger than 18 yr with total plasma cholesterol of 270 mg/dL and/or LDL-C of 200 mg/dL has an 88% chance of having FH. If there is a first-degree relative with proven FH, the diagnosis in the child is virtually certain (Table 80-9). Conversely, criteria for diagnosing probable FH in a child whose first-degree relative has known FH require only modest elevation of total cholesterol to 220 mg/dL (LDL-C 155 mg/dL).
Hypercholesterolemia with Hypertriglyceridemia
Hypertriglyceridemias
Familial Chylomicronemia (Type I Hyperlipidemia)
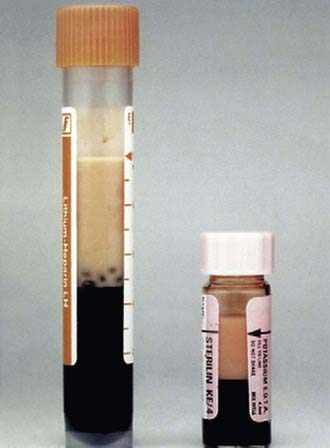
This rare single gene defect, like familial hypercholesterolemia, is due to mutations affecting clearance of apoB-containing lipoproteins. Deficiency or absence of lipoprotein lipase (LPL) or its cofactor apoC-II, which facilitates lipolysis by LPL, causes severe elevation of triglyceride rich plasma chylomicrons. HDL cholesterol levels are decreased. As clearance of these particles is markedly delayed, the plasma is noted to have a turbid appearance even after prolonged fasting (Fig. 80-13). Chylomicronemia caused by LPL deficiency is associated with modest elevation in triglycerides, whereas this is not the case when the cause is deficient or absent apoC-II. Both are autosomal recessive conditions with a frequency of approximately 1/1,000,000. The disease usually presents during childhood with acute pancreatitis. Eruptive xanthomas on the arms, knees, and buttocks may be present, and there may be hepatosplenomegaly. The diagnosis is established by assaying triglyceride lipolytic activity. Treatment of chylomicronemia is by vigorous dietary fat restriction supplemented by fat-soluble vitamins. Medium-chain triglycerides that are adsorbed into the portal venous system may augment total fat intake, and administration of fish oils may also be beneficial.
Familial Hypertriglyceridemia (FHTG, Type IV Hyperlipidemia)
Pediatric diseases associated with hyperlipidemia include hypothyroidism, nephrotic syndrome, biliary atresia, glycogen storage disease, Niemann-Pick disease, Tay-Sachs disease, systemic lupus erythematosus, hepatitis, and anorexia nervosa (Table 80-10). Certain medications exacerbate hyperlipidemia, including isotretinoin (Accutane), thiazide diuretics, oral contraceptives, steroids, β-blockers, immunosuppressants, and protease inhibitors used in the treatment of HIV.
Table 80-10 SECONDARY CAUSES OF HYPERLIPIDEMIA
HYPERCHOLESTEROLEMIA
Hypothyrodism
Nephrotic syndrome
Cholestasis
Anorexia nervosa
Drugs: progesterone, thiazides, tegretol, cyclosporine
HYPERTRIGLYCERIDEMIA
Obesity
Type II diabetes
Alcohol
Renal failure
Sepsis
Stress
Cushing syndrome
Pregnancy
Hepatitis
AIDS, protease inhibitions
Drugs: anabolic steroids, β blockers, estrogen, thiazides
REDUCED HIGH-DENSITY LIPOPROTEIN
Smoking
Obesity
Type II diabetes
Malnutrition
Drugs: β Blockers, anabolic steroids
Disorders of HDL Metabolism
Conditions Associated with Low Cholesterol
Smith-Lemli-Opitz Syndrome (SLOS)
Patients with SLOS often have multiple congenital anomalies and developmental delay caused by low plasma cholesterol and accumulated precursors (Tables 80-11 and 80-12). Family pedigree analysis has revealed its autosomal recessive inheritance pattern. Mutations in the DHCR7 (7-dehydrocholesterol-Δ7 reductase) gene result in deficiency of the microsomal enzyme DHCR7, which is necessary to complete the final step in cholesterol synthesis. It is not known why defects in cholesterol synthesis result in congenital malformations, but as cholesterol is a major component of myelin and a contributor to signal transduction in the developing nervous system, neurodevelopment is severely impaired. The incidence of SLOS is estimated to be 1/20,000-60,000 births among whites, with a somewhat higher frequency in Hispanics and lower incidence in individuals of African descent.
Table 80-11 MAJOR CLINICAL CHARACTERISTICS OF SMITH-LEMLI-OPITZ SYNDROME: FREQUENT ANOMALIES (>50% OF PATIENTS)
CRANIOFACIAL
Microcephaly
Blepharoptosis
Anteverted nares
Retromicrognathia
Low-set, posteriorly rotated ears
Midline cleft palate
Broad maxillary alveolar ridges
Cataracts (<50%)
SKELETAL ANOMALIES
Syndactyly of toes II/III
Postaxial polydactyly (<50%)
Equinovarus deformity (<50%)
GENITAL ANOMALIES
Hypospadias
Cryptorchidism
Sexual ambiguity (<50%)
DEVELOPMENT
Pre- and postnatal growth retardation
Feeding problems
Mental retardation
Behavioral abnormalities
From Haas D, Kelley RI, Hoffmann GF: Inherited disorders of cholesterol biosynthesis, Neuropediatrics 32:113–122, 2001.
Table 80-12 CHARACTERISTIC MALFORMATIONS OF INTERNAL ORGANS IN SEVERELY AFFECTED SMITH-LEMLI-OPITZ PATIENTS
CENTRAL NERVOUS SYSTEM
Frontal lobe hypoplasia
Enlarged ventricles
Agenesis of corpus callosum
Cerebellar hypoplasia
Holoprosencephaly
CARDIOVASCULAR
Atrioventricular canal
Secundum atrial septal defect
Patent ductus arteriosus
Membranous ventricular septal defect
URINARY TRACT
Renal hypoplasia or aplasia
Renal cortical cysts
Hydronephrosis
Ureteral duplication
GASTROINTESTINAL
Hirschsprung disease
Pyloric stenosis
Refractory dysmotility
Cholestatic and noncholestatic progressive liver disease
PULMONARY
Pulmonary hypoplasia
Abnormal lobation
ENDOCRINE
Adrenal insufficiency
From Haas D, Kelley RI, Hoffmann GF: Inherited disorders of cholesterol biosynthesis, Neuropediatrics 32:113–122, 2001.
Disorders of Intracellular Cholesterol Metabolism
Lipoprotein Patterns in Children and Adolescents
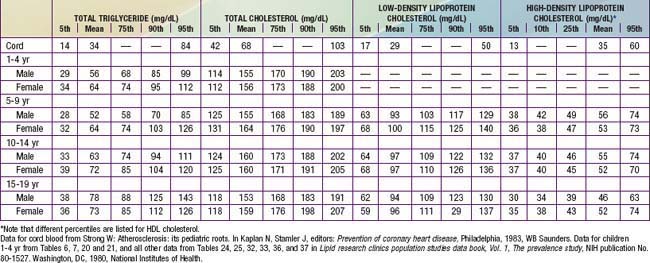
Table 80-13, derived primarily from the Lipid Research Clinics Population Studies, shows the distribution of lipoprotein levels in American youth at various ages. Total plasma cholesterol rises rapidly from a mean of 68 mg/dL at birth to a level approximately twice that by the end of the neonatal period. A very gradual rise in total cholesterol level occurs until puberty, at which time the mean level reaches 160 mg/dL. Total cholesterol falls transiently during puberty, in males due to a small decrease in HDL cholesterol, and in females secondary to a slight fall in LDL cholesterol. Blood cholesterol levels track reasonably well as individuals age. High blood cholesterol tends to aggregate in families, a reflection of genetic and environmental influences.
Blood Cholesterol Screening
Being overweight confers special risk of CVD because of the strong association with the insulin resistance syndrome (metabolic syndrome). Though there is no single definition of metabolic syndrome defined for youth, it is likely that half of all severely obese children are insulin resistant. Among a large cohort of 5th grade school children who had comprehensive screening of CVD risk factors conducted by the Coronary Artery Risk Detection in Appalachian Communities (CARDIAC) Project, 49% of those with the hyperpigmented rash, acanthosis nigricans, had 3 or more risk factors for the metabolic syndrome, including insulin resistance, hypertension, and abnormal lipid levels (triglycerides >150 mg/dL; HDL-C <40 mg/dL) (Chapter 44).
Risk Assessment and Treatment of Hyperlipidemia
The National Cholesterol Education Program (NCEP) recommends a population-based approach toward healthy lifestyle applicable to all children, and an individualized approach directed at those children at high risk (Fig. 80-14). The important focus on maintenance of a healthy lifestyle rather than aggressive weight reduction is recommended by the AAP.
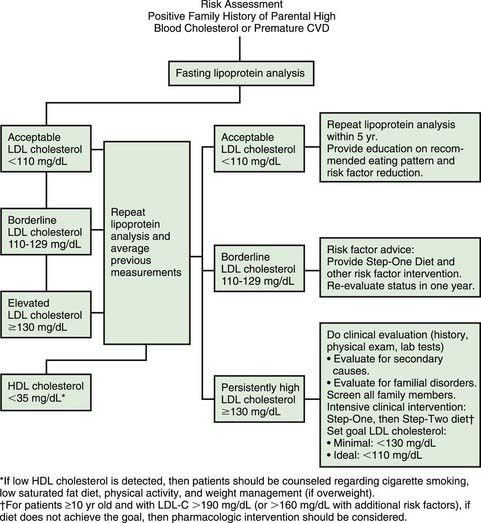
Persistence of an elevated LDL cholesterol >130 mg/dL indicates the need for more comprehensive evaluation and lifestyle modification. History and physical examination and additional laboratory tests aimed at ruling out secondary causes of hyperlipidemia (see Table 80-10) should be performed. Other family members should have blood cholesterol screening. If the LDL cholesterol level does not achieve the minimal goal of <130 mg/dl the AHA Step II diet should be recommended. This diet allows the same average fat consumption of no more than 30% of total calories, but restricts saturated fats to <7-8% of total calories and cholesterol intake to <200 mg per day. Follow-up lab tests, measurement of height and weight for the calculation of body mass index (BMI), and dietary history should be scheduled at 3-6 mo intervals.
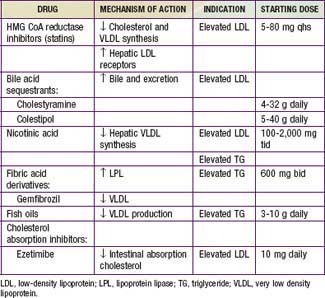
Drug Therapy (Tables 80-14 And 80-15)
| DRUG AND SITE OR TYPE OF EFFECT | EFFECT |
|---|---|
| STATINS | |
| Skin | Rash |
| Nervous system | Loss of concentration, sleep disturbance, headache, peripheral neuropathy |
| Liver | Hepatitis, loss of appetite, weight loss, and increases in serum aminotransferases to two to three times the upper limit of the normal range |
| Gastrointestinal tract | Abdominal pain, nausea, diarrhea |
| Muscles | Muscle pain or weakness, myositis (usually with serum creatine kinase >1,000U/L), rhabdomyolysis with renal failure |
| Immune system | Lupus-like syndrome (lovastatin, simvastatin, or fluvastatin) |
| Protein binding | Diminished binding of warfarin (lovastatin, simvastatin, fluvastatin) |
| BILE ACID-BINDING RESINS | |
| Gastrointestinal tract | Abdominal fullness, nausea, gas, constipation, hemorrhoids, anal fissure, activation of diverticulitis, diminished absorption of vitamin D in children |
| Liver | Mild serum aminotransferase elevations, which can be exacerbated by concomitant treatment with a statin |
| Metabolic system | Increases in serum triglycerides of ≈10% (greater increases in patients with hypertriglyceridemia) |
| Electrolytes | Hyperchloremic acidosis in children and patients with renal failure (cholestyramine) |
| Drug interactions | Binding of warfarin, digoxin, thiazide diuretics, thyroxine, statins |
| NICOTINIC ACID | |
| Skin | Flushing, dry skin, pruritus, ichthyosis, acanthosis nigricans |
| Eyes | Conjunctivitis, cystoid macular edema, retinal detachment |
| Respiratory tract | Nasal stuffiness |
| Heart | Supraventricular arrhythmias |
| Gastrointestinal tract | Heartburn, loose bowel movements or diarrhea |
| Liver | Mild increase in serum aminotransferases, hepatitis with nausea and fatigue |
| Muscles | Myositis |
| Metabolic system | Hyperglycemia (incidence, ≈5% higher in patients with diabetes), increase of 10% in serum uric acid |
| FIBRATES | |
| Skin | Rash |
| Gastrointestinal tract | Stomach upset, abdominal pain (mainly gemfibrozil), cholesterol-saturated bile, increase of 1-2% in gallstone incidence |
| Genitourinary tract | Erectile dysfunction (mainly clofibrate) |
| Muscles | Myositis with impaired renal function |
| Plasma proteins | Interference with binding of warfarin, requiring reduction in the dose of warfarin by ≈30% |
| Liver | Increased serum aminotransferases |
From Knopp RH: Drug treatment of lipid disorders, N Engl J Med 341:498–512, 1999.
Austin MA, Hutter CH, Zimmern RL, et al. Familial hypercholesterolemia and coronary heart disease: a huge association review. Am J Epidemiol. 2004;160:421-429.
Bhatnagar D, Soran H, Durrington PN. Hypercholesterolaemia and its management. BMJ. 2008;337:503-508.
Brunzell JD. Hypertriglyceridemia. N Engl J Med. 2007;357:1009-1016.
Centers for Disease Control and Prevention. Prevalence of abnormal lipid levels among youths—United States, 1999–2006. MMWR. 2010;59:29-33.
Chan YM, Merkens LS, Connor WB, et al. Effects of dietary cholesterol and simvastatin on cholesterol synthesis in Smith-Lemli-Opitz syndrome. Pediatr Res. 2009;65:681-685.
Daniels SR, Greer FR, et al. Lipid screening and cardiovascular health in childhood. Pediatrics. 2008;122:198-208.
De Jongh S, Ose L, Szamosi T, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia. Circulation. 2002;106:2231-2237.
Ford ES, Li C, Zhao G, Mokdad AH. Concentrations of low-density lipoprotein cholesterol and total cholesterol among children and adolescents in the United States. Circulation. 2009;119:1108-1115.
Grundy SM, Hansen B, Smith SC, et al. Clinical management of metabolic syndrome: report of the American Heart Association/National Heart, Lung, Blood Institute/American Diabetes Association Conference on Scientific Issues Related to Management. Circulation. 2004;109:551-556.
Kumanyika SK, Obarzanek E. Population-based prevention of obesity. The need for comprehensive practices of healthful eating, physical activity and energy balance. A scientific statement from American Heart Association Council on Epidemiology and Prevention. Circulation. 2008;118:428-464.
Lebenthal Y, Horvath A, Dziechciarz P, et al. Are treatment targets for hypercholesterolemia evidence-based? Systematic review and meta-analysis of randomized controlled trials. Arch Dis Child. 2010;95:673-680.
Magnussen CG, Raitakari OT, Thomson R, et al. Utility of currently recommended pediatric dyslipidemia classifications in predicting dyslipidemia in adulthood. Circulation. 2008;117:32-42.
Manlhiot C, Larsson P, Gurofsky R, et al. Spectrum and management of hypertriglyceridemia among children in clinical practice. Pediatrics. 2009;123:458-465.
The Medical Letter. Fenofibric acid (trilipix). Med Lett. 2009;51:33-34.
Merkens LS, Connor WE, Linck LM, et al. Effects of dietary cholesterol on plasma lipoproteins in Smith-Lemli-Opitz syndrome. Pediatr Res. 2004;56:726-732.
Raitakari OT. Arterial abnormalities in children with familial hypercholesteremia. Lancet. 2004;363:342-343.
The SEARCH Collaborative Group. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med. 2008;359:789-799.
Silverstein J, Haller M. Coronary artery disease in youth: present markers, future hope? J Pediatr. 2010;157(4):523-524.
Singhal A, Cole TJ, Fewtrell M, et al. Breast milk feeding and lipoprotein profile in adolescents born preterm: follow-up of a prospective randomized study. Lancet. 2004;363:1571-1578.
Wald DS, Bestwick JP, Wald NJ. Child-parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis. BMJ. 2007;335:599-603.
Wiegman A, Hutten BA, de Groot E, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia. JAMA. 2004;292:331-337.
80.4 Lipidoses (Lysosomal Storage Disorders)
The lysosomal lipid storage diseases are diverse disorders each due to an inherited deficiency of a lysosomal hydrolase leading to the intralysosomal accumulation of the enzyme’s particular substrate (Tables 80-16 and 80-17). With the exception of Wolman disease and cholesterol ester storage disease, the lipid substrates share a common structure that includes a ceramide backbone (2-N-acylsphingosine) from which the various sphingolipids are derived by substitution of hexoses, phosphorylcholine, or one or more sialic acid residues on the terminal hydroxyl group of the ceramide molecule. The pathway of sphingolipid metabolism in nervous tissue (Fig. 80-15) and in visceral organs (Fig. 80-16) is known; each catabolic step, with the exception of the catabolism of lactosylceramide, has a genetically determined metabolic defect and a resultant disease. Because sphingolipids are essential components of all cell membranes, the inability to degrade these substances and their subsequent accumulation results in the physiologic and morphologic alterations and characteristic clinical manifestations of the lipid storage disorders (see Table 80-16). Progressive lysosomal accumulation of glycosphingolipids in the central nervous system leads to neurodegeneration, whereas storage in visceral cells can lead to organomegaly, skeletal abnormalities, pulmonary infiltration, and other manifestations. The storage of a substrate in a specific tissue is dependent on its normal distribution in the body.
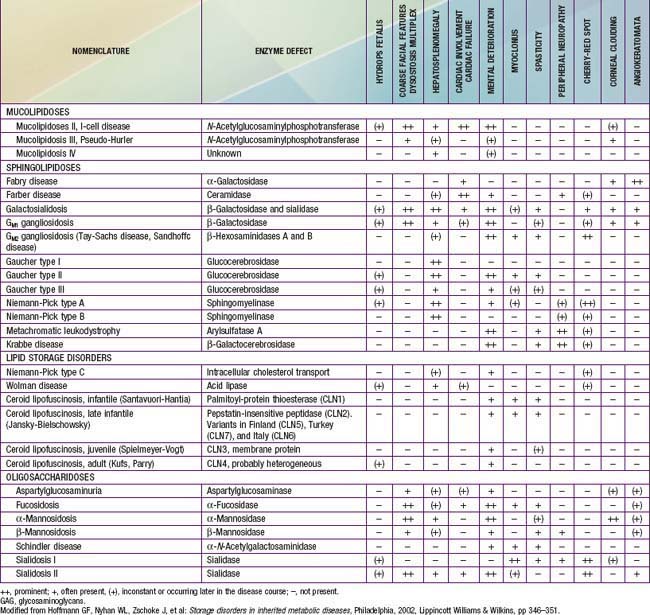
Table 80-17 SYMPTOMS ENCOUNTERED IN PATIENTS WITH LYSOSOMAL STORAGE DISORDERS
| System | Manifestations |
| Neurologic | Hypotonia |
| Floppy-infant syndrome | |
| Trismus | |
| Strabismus | |
| Opisthotonus | |
| Spasticity | |
| Seizures | |
| Peripheral neuropathy | |
| Developmental delay | |
| Irritability | |
| Extrapyramidal movement disorder | |
| Hydrocephalus | |
| Respiratory | Congenital lobar emphysema |
| Impaired cough | |
| Recurrent respiratory infections | |
| Hoarseness | |
| Endocrine | Osteopenia |
| Metabolic bone disease | |
| Secondary hyperparathyroidism | |
| Congenital adrenal hyperplasia | |
| Cardiovascular | Cardiomegaly |
| Congenital heart failure | |
| Arrhythmias | |
| Wolff-Parkinson-White syndrome | |
| Cardiomyopathy | |
| Dysmorphology | |
| Head and neck | Microcephaly |
| Enlarged nuchal translucency | |
| Microstomia | |
| Micrognathia/microretrognathia | |
| Long philtrums | |
| Limbs | Bilateral broad thumbs and toes |
| Bilateral club feet | |
| Eversed lips | |
| Flattened nasal bridge | |
| Short nasal columella | |
| Oral | Macroglossia |
| Molar hypoplasia | |
| Hypertrophic gums | |
| Absent nasal septum | |
| Bilateral epicanthal inferior orbital creases | |
| Palpebral edema | |
| Hypertelorism | |
| Facial | Coarse facies |
| Low-set ears | |
| Gastrointestinal | Hepatosplenomegaly |
| Neonatal cholestasis | |
| Bones and joints | Lytic bone lesions |
| Joint contractures | |
| Dysostosis multiplex | |
| Hyperphosphatasemia | |
| Vertebral breaking | |
| Broadening of tubular bones | |
| Punctuate epiphysis | |
| Craniosynostosis | |
| Painful joint swelling | |
| Skin | Congenital ichthyosis |
| Collodion infant | |
| Hypopigmentation | |
| Telangiectasias | |
| Extended Mongolian spots | |
| Ocular | Corneal clouding |
| Megalocornea | |
| Glaucoma | |
| Cherry-red spots | |
| Fundi hypopigmentation | |
| Bilateral cataracts | |
| Hematologic | Anemia |
| Thrombocytopenia | |
| Hydrops fetalis | NIHF |
| Congenital ascites | |
| Recurrent fetal losses |
From Staretz-Chacham O, Lang TC, LaMarca ME, et al: Lysosomal storage disorders in the newborn, Pediatrics 123:1191–1207, 2009.
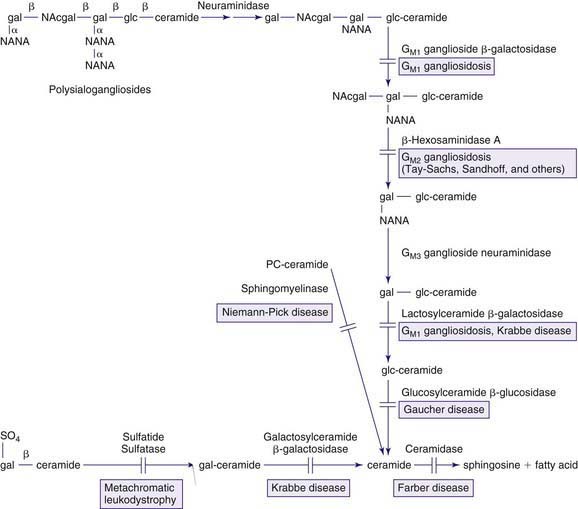
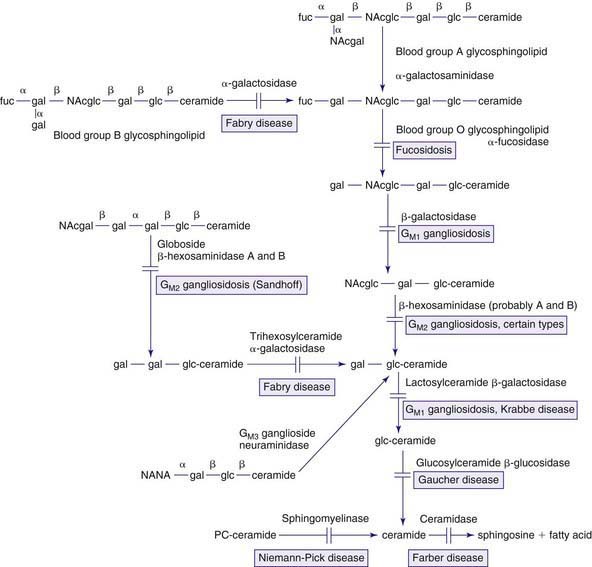
Figure 80-16 Pathways in the degradation of sphingolipids found in visceral organs and red or white blood cells. See also the legend for Figure 80-15. Fuc, fucose; NAcglc, N-acetylglucosamine.
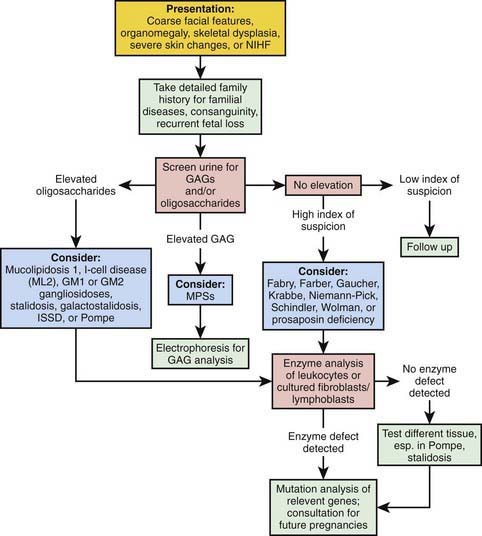
Diagnostic assays for the identification of affected individuals rely on the measurement of the specific enzymatic activity in isolated leukocytes or cultured fibroblasts or lymphoblasts. An approach to differentiating these disorders is noted in Figure 80-17. For most disorders, carrier identification and prenatal diagnosis are available; a specific diagnosis is essential to permit genetic counseling. The characterization of the genes that encode the specific enzymes required for sphingolipid metabolism permit the development of therapeutic options, such as recombinant enzyme replacement therapy, as well as the potential of cell or gene therapy. Identification of specific disease-causing mutations improves diagnosis, prenatal detection, and carrier identification. For several disorders (Gaucher, Fabry, and Niemann-Pick disease), it has been possible to make genotype-phenotype correlations that predict disease severity and allow more precise genetic counseling. Inheritance is autosomal recessive except for X-linked Fabry disease.
The GM2 Gangliosidoses
Patients with the infantile form of Tay-Sachs disease have clinical manifestations in infancy including loss of motor skills, increased startle reaction, and macular pallor and retinal cherry red spots (see Table 80-16). Affected infants usually develop normally until 4-5 mo of age when decreased eye contact and an exaggerated startle response to noise (hyperacusis) are noted. Macrocephaly, not associated with hydrocephalus, may develop. In the 2nd yr of life, seizures develop which may be refractory to anticonvulsant therapy. Neurodegeneration is relentless, with death occurring by the age of 4 or 5 yr. The juvenile and later-onset forms initially present with ataxia and dysarthria and may not be associated with a macular cherry red spot.
Gaucher Disease
This disease is a multisystemic lipidosis characterized by hematologic abnormalities, organomegaly, and skeletal involvement, the latter usually manifesting as bone pain and pathologic fractures (see Table 80-16). It is the most common lysosomal storage disease and the most prevalent genetic defect among Ashkenazi Jews. There are 3 clinical subtypes delineated by the absence or presence and progression of neurologic manifestations: type 1 or the adult, non-neuronopathic form; type 2, the infantile or acute neuronopathic form; and type 3, the juvenile or subacute neuronopathic form. All are autosomal recessive traits. Type 1, which accounts for 99% of cases, has a striking predilection for Ashkenazi Jews, with an incidence of about 1/1,000 and a carrier frequency of about 1/18.
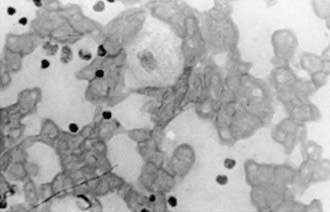
The pathologic hallmark of Gaucher disease is the Gaucher cell in the reticuloendothelial system, particularly in the bone marrow (Fig. 80-18). These cells, which are 20-100 µm in diameter, have a characteristic wrinkled paper appearance resulting from the presence of intracytoplasmic substrate inclusions. The cytoplasm of the Gaucher cell reacts strongly positive with the periodic acid–Schiff stain. The presence of this cell in bone marrow and tissue specimens is highly suggestive of Gaucher disease, although it also may be found in patients with granulocytic leukemia and myeloma.
Neimann-Pick Disease (NPD)
The original description of NPD was what is now known as type A NPD, a fatal disorder of infancy characterized by failure to thrive, hepatosplenomegaly, and a rapidly progressive neurodegenerative course that leads to death by 2-3 yr of age. Type B disease is a non-neuronopathic form observed in children and adults. Type C disease is a neuronopathic form that results from defective cholesterol transport. All subtypes are inherited as autosomal recessive traits and display variable clinical features (see Table 80-16).
Type C NPD patients often present with prolonged neonatal jaundice, appear normal for 1-2 yr, and then experience a slowly progressive and variable neurodegenerative course. Their hepatosplenomegaly is less severe than that of patients with types A or B NPD, and they may survive into adulthood. The underlying biochemical defect in type C patients is an abnormality in cholesterol transport, leading to the accumulation of sphingomyelin and cholesterol in their lysosomes and a secondary partial reduction in acid sphingomyelinase activity (Chapter 80.3).
Fabry Disease
This condition is an X-linked inborn error of glycosphingolipid metabolism characterized by angiokeratomas (telangiectatic skin lesions), hypohidrosis, corneal and lenticular opacities, acroparesthesias, and vascular disease of the kidney, heart, and/or brain (see Table 80-16). The classic phenotype is due to the deficient activity of the enzyme α-galactosidase A and has an estimated prevalence of approximately 1/50,000 males. Later-onset affected males with residual α-galactosidase A activity may present with cardiac and/or renal disease including hypertrophic cardiomyopathy and renal failure and is more prevalent than the classic phenotype. Heterozygous females for the classic phenotype can be asymptomatic or as severely affected as the males, the variability due to random X-inactivation. The disease results from mutations in the α-galactosidase A gene located on the long arm of the X chromosome (Xq22). The enzymatic defect leads to the systemic accumulation of neutral glycosphingolipids, primarily globotriaosylceramide, particularly in the plasma and lysosomes of vascular endothelial and smooth muscle cells. The progressive vascular glycosphingolipid deposition in classically affected males results in ischemia and infarction, leading to the major disease manifestations. The cDNA and genomic sequences encoding α-galactosidase A have identified more than 500 different mutations in the α-galactosidase A gene that are responsible for this lysosomal storage disease, including amino acid substitutions, gene rearrangements, and mRNA splicing defects.
Affected males with the classic phenotype have the skin lesions, acroparesthesias, hypohidrosis, and ocular changes, whereas males with the later onset phenotypes lack these findings and present with cardiac and/or renal disease in adulthood. The classic angiokeratomas usually occur in childhood and may lead to early diagnosis (Fig. 80-19). They increase in size and number with age and range from barely visible to several mm in diameter. The lesions are punctate, dark red to blue-black, and flat or slightly raised. They do not blanch with pressure, and the larger ones may show slight hyperkeratosis. Characteristically, the lesions are most dense between the umbilicus and knees, in the “bathing trunk area,” but may occur anywhere, including the oral mucosa. The hips, thighs, buttocks, umbilicus, lower abdomen, scrotum, and glans penis are common sites, and there is a tendency toward symmetry. Variants without skin lesions have been described. Sweating is usually decreased or absent. Corneal opacities and characteristic lenticular lesions, observed under slit-lamp examination, are present in affected males as well as in about 90% of heterozygotes. Conjunctival and retinal vascular tortuosity is common and results from the systemic vascular involvement.
Fucosidosis
This is a rare autosomal recessive disorder caused by the deficient activity of α-fucosidase and the accumulation of fucose-containing glycosphingolipids, glycoproteins, and oligosaccharides in the lysosomes of the liver, brain, and other organs (see Table 80-16). The α-fucosidase gene is on chromosome 1 (1p24), and specific mutations are known. Although the disorder is panethnic, most affected patients are from Italy and the USA. There is wide variability in the clinical phenotype, with the most severely affected patients presenting in the first year of life with developmental delay and somatic features similar to those of the mucopolysaccharidoses. These features include frontal bossing, hepatosplenomegaly, coarse facial features, and macroglossia. The central nervous system storage results in a relentless neurodegenerative course, with death in childhood. Patients with milder disease have angiokeratomas and longer survival. No specific therapy exists for the disorder, which can be diagnosed by the demonstration of deficient α-fucosidase activity in peripheral leukocytes or cultured fibroblasts. Carrier identification studies and prenatal diagnosis are possible by determination of the enzymatic activity or the specific family mutations.
Schindler Disease
This is an autosomal recessive neurodegenerative disorder that results from the deficient activity of α-N-acetylgalactosaminidase and the accumulation of sialylated and asialoglycopeptides and oligosaccharides (see Table 80-16). The gene for the enzyme is located on chromosome 22 (22q11). The disease is clinically heterogeneous, and two major phenotypes have been identified. Type I disease is an infantile-onset neuroaxonal dystrophy. Affected infants have normal development for the 1st 9-15 mo of life followed by a rapid neurodegenerative course that results in severe psychomotor retardation, cortical blindness, and frequent myoclonic seizures. Type II disease is characterized by a variable age at onset, mild retardation, and angiokeratomas. There is no specific therapy for either form of the disorder. The diagnosis is by demonstration of the enzymatic deficiency in leukocytes or cultured skin fibroblasts or specific gene mutations.
Metachromatic Leukodystrophy (MLD)
The clinical manifestations of the late infantile form of MLD, which is most common, usually presents between 12 and 18 mo of age as irritability, inability to walk, and hyperextension of the knee, causing genu recurvatum. The clinical progression of the disease relates to the pathological involvement of both central and peripheral nervous system, giving a mixture of upper and lower motor neuron and cognitive and psychiatric signs. Deep tendon reflexes are diminished or absent. Gradual muscle wasting, weakness, and hypotonia become evident and lead to a debilitated state. As the disease progresses, nystagmus, myoclonic seizures, optic atrophy, and quadriparesis appear, with death in the 1st decade of life (see Table 80-16). The juvenile form of the disorder has a more indolent course with onset that may occur as late as 20 yr of age. This form of the disease presents with gait disturbances, mental deterioration, urinary incontinence, and emotional difficulties. The adult form, which presents after the 2nd decade, is similar to the juvenile form in its clinical manifestations, although emotional difficulties and psychosis are more prominent features. Dementia, seizures, diminished reflexes, and optic atrophy also occur in both the juvenile and adult forms. The pathologic hallmark of MLD is the deposition of metachromatic bodies, which stain strongly positive with periodic acid–Schiff and Alcian blue, in the white matter of the brain. Neuronal inclusions may be seen in the midbrain, pons, medulla, retina, and spinal cord; demyelination occurs in the peripheral nervous system. Bone marrow transplantation has resulted in normal enzymatic levels in peripheral blood, but no clear evidence for clinical efficacy in terms of the neurologic course; supportive care remains the primary intervention.
Krabbe Disease
This condition, also called globoid cell leukodystrophy, is an autosomal recessive fatal disorder of infancy. It results from the deficiency of the enzymatic activity of galactocerebrosidase (GALC) and the white matter accumulation of galactosylceramide, which is normally found almost exclusively in the myelin sheath. Both peripheral and central myelin are affected, resulting in spasticity and cognitive impairment coupled with deceptively normal or even absent deep tendon reflexes. The galactocerebrosidase gene is on chromosome 14 (14q31), and specific disease-causing mutations are known. The infantile form of Krabbe disease is rapidly progressive and patients present in early infancy with irritability, seizures, and hypertonia (see Table 80-16). Optic atrophy is evident in the 1st yr of life, and mental development is severely impaired. As the disease progresses, optic atrophy and severe developmental delay become apparent; affected children exhibit opisthotonos and die before 3 yr of age. A 2nd, late infantile form of Krabbe disease also exists and patients present after the age of 2 yr. Affected individuals have a disease course similar to that of the early infantile form.
Farber Disease
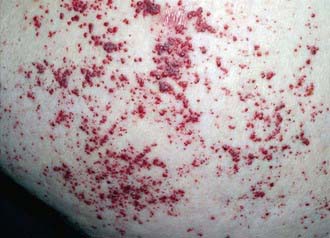
This is a rare autosomal recessive disorder that results from the deficiency of the lysosomal enzyme acid ceramidase and the accumulation of ceramide in various tissues, especially the joints. Symptoms can begin as early as the 1st year of life with painful joint swelling and nodule formation (Fig. 80-20), which is sometimes diagnosed as rheumatoid arthritis. As the disease progresses, nodule or granulomatous formation on the vocal cords can lead to hoarseness and breathing difficulties; failure to thrive is common. In some patients, moderate central nervous system dysfunction is present (see Table 80-16). Patients may die of recurrent pneumonias in their teens; there is currently no specific therapy. The diagnosis of this disorder should be suspected in patients who have nodule formation over the joints but no other findings of rheumatoid arthritis. In such patients, ceramidase activity should be determined in cultured skin fibroblasts or peripheral leukocytes. Various disease causing mutations have been identified in the acid ceramidase gene. Carrier detection and prenatal diagnosis are available.
Wolman Disease and Cholesterol Ester Storage Disease (CESD)
These are autosomal recessive lysosomal storage diseases that result from the deficiency of acid lipase and the accumulation of cholesterol esters and triglycerides in histiocytic foam cells of most visceral organs. The gene for lysosomal acid lipase is on chromosome 10 (10q24-q25). Wolman disease is the more severe clinical phenotype and is a fatal disorder of infancy. Clinical features become apparent in the 1st wk of life and include failure to thrive, relentless vomiting, abdominal distention, steatorrhea, and hepatosplenomegaly (see Table 80-16). There usually is hyperlipidemia. Hepatic dysfunction and fibrosis may occur. Calcification of the adrenal glands is pathognomonic for the disorder. Death usually occurs within 6 mo.
Diagnosis and carrier identification are based on measuring acid lipase activity in peripheral leukocytes or cultured skin fibroblasts. Disease causing mutations have been identified in the acid ceramide gene. Prenatal diagnosis depends on measuring decreased enzyme levels or identifying specific mutations in cultured chorionic villi or amniocytes. There is no specific therapy available for either disorder, although pharmacologic agents to suppress cholesterol synthesis, in combination with cholestyramine and diet modification, have been used in patients with cholesterol ester storage disease (see Chapter 80.3).
Andersson H, Kaplan P, Kacena K, Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics. 2008;122:1182-1190.
Clark JTR. Narrative review: Fabry disease. Ann Intern Med. 2007;146:425-433.
Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;20:2069-2080.
Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372:1263-1271.
Itzchaki M, Lebel E, Dweck A, et al. Orthopedic considerations in Gaucher disease since the advent of enzyme replacement therapy. Acta Orthop Scand. 2004;75:641-653.
Kaplan P, Anderson HC, Kacena KA, et al. The clinical and demographic characteristics of nonneuronpathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160:603-608.
Martins AM, D’Almeida VD, Kyosen SO, et al. Guidelines to diagnosis and monitoring of Fabry disease and review of treatment experiences. J Pediatr. 2009;155:S19-S30.
McGovern MM, Wasserstein MP, Giugliani R, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics. 2008;122:e341-e349.
Mehta A, Beck M, Elliott P, et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374:1986-1996.
Meikle PJ, Ranieri E, Simonsen H, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004;114:909-916.
Park NJ, Morgan C, Sharma R, et al. Improving accuracy of Tay Sachs carrier screening of the non-Jewish population: analysis of 34 carriers and six late-onset patients with HEXA enzyme and DNA sequence analysis. Pediatr Res. 2010;67:217-220.
Staretz-Chachem O, Lang TC, LaMarca ME, et al. Lysosomal storage disorders in the newborn. Pediatrics. 2009;123:1191-1207.
West M, Nicholls K, Mehta A, et al. Agalsidase alfa and kidney dysfunction in Fabry disease. J Ann Soc Nephrol. 2009;20:1132-1139.
Wraith JE, Tylki-Szymanska A, Guffon N, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr. 2008;152:563-570.
Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427-1435.
80.5 Mucolipidoses
I-cell disease (mucolipidosis II [ML-II]) and pseudo-Hurler polydystrophy (mucolipidosis III [ML-III]) are rare autosomal recessive disorders that share some clinical features with Hurler syndrome (Chapter 82). These diseases result from the abnormal transport of newly synthesized lysosomal enzymes that are normally targeted to the lysosome by the presence of mannose-6-phosphate residues and recognized by specific lysosomal membrane receptors. These mannose-6-phosphate recognition markers are synthesized in a 2-step reaction that occurs in the Golgi apparatus and is mediated by 2 enzymatic activities. The enzyme that catalyzes the 1st step, the UDP-N-acetylglucosamine : lysosomal enzyme N-acetylglucosamine-1-phosphotransferase, is defective in both ML-II and ML-III, which are allelic disorders resulting from mutations in the GlcNAc-phosphotransferase alpha/beta-subunits precursor gene (GNPTAB). This enzyme deficiency results in abnormal targeting of the lysosomal enzymes that are consequently secreted into the extracellular matrix. Because the lysosomal enzymes require the acidic medium of the lysosome to function, patients with this defect accumulate a variety of different substrates due to the intracellular deficiency of all lysosomal enzymes. The diagnosis of ML-II and ML-III can be made by the determination of the serum lysosomal enzymatic activities, which are elevated, or by the demonstration of reduced enzymatic activity levels in cultured skin fibroblasts. Direct measurement of the phosphotransferase activity is possible as well. Prenatal diagnosis is available for both disorders by measurement of lysosomal enzymatic activities in amniocytes or chorionic villus cells; carrier identification studies are available for both disorders using cultured skin fibroblasts. Neonatal screening by tandem mass spectroscopy may detect I-cell disease.
I-Cell Disease
This disorder shares many of the clinical manifestations of Hurler syndrome (Chapter 82), although there is no mucopolysacchariduria and the presentation is earlier (see Table 80-16). Some patients have clinical features evident at birth, including coarse facial features, craniofacial abnormalities, restricted joint movement, and hypotonia. Nonimmune hydrops may be present in the fetus. The remainder of patients present in the first year with severe psychomotor retardation, coarse facial features, and skeletal manifestations that include kyphoscoliosis and a lumbar gibbus. Patients may also have congenital dislocation of the hips, inguinal hernias, and gingival hypertrophy. Progressive, severe psychomotor retardation leads to death in early childhood. No treatment is available.
Pseudo-Hurler Polydystrophy
Pseudo-Hurler polydystrophy is a less severe disorder than I-cell disease, with later onset and survival to adulthood reported. Affected children may present around the age of 4 or 5 yr with joint stiffness and short stature. Progressive destruction of the hip joints and moderate dysostosis multiplex are evident. Radiographic evidence of low iliac wings, flattening of the proximal femoral epiphyses with valgus deformity of the femoral head, and hypoplasia of the anterior third of the lumbar vertebrae are characteristic findings. Ophthalmic findings include corneal clouding, retinopathy, and astigmatism; visual complaints are uncommon (see Table 80-16). Some patients have learning disabilities or mental retardation. Treatment, which should include orthopedic care, is symptomatic.
* This chapter has been extensively evaluated by Drs. Charles Stanley and Michael Bennett and the reference list has been updated to reflect more recent articles. However, Drs. Stanley and Bennett find that there have been no substantive diagnostic pathophysiological or therapeutic advances in this field since the previous version was written and, with deep respect to the late Dr. Moser, consider that no textual modifications are required for this present edition of the text book.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 80 Defects in Metabolism of Lipids
80.1 Disorders of Mitochondrial Fatty Acid β-Oxidation
Genetic defects have been identified in nearly all of the known steps in the fatty acid oxidation pathway; all are recessively inherited (Table 80-1).
Figures 80-1 and 80-2 outline the steps involved in the oxidation of a typical long-chain fatty acid. In the carnitine cycle, fatty acids are transported across the barrier of the inner mitochondrial membrane as acylcarnitine esters. Within the mitochondria, successive turns of the 4-step β-oxidation cycle convert the coenzyme A (CoA)-activated fatty acid to acetyl CoA units. Two to three different chain-length specific isoenzymes are needed for each of these β-oxidation steps to accommodate the different-sized fatty acyl CoA species. The electron transfer pathway carries electrons generated in the 1st β-oxidation step (acyl CoA dehydrogenase) to the electron transport chain for adenosine triphosphate (ATP) production, while electrons generated from the third step (3-hydroxyacyl CoA dehydrogenase) enter the respiratory chain at the level of complex 1. Most of the acetyl CoA generated from hepatic β-oxidation flows through the pathway of ketogenesis to form β-hydroxybutyrate and acetoacetate.
Defects in the β-Oxidation Cycle
Medium-Chain Acyl CoA Dehydrogenase (MCAD) Deficiency
Treatment
Acute illnesses should be promptly treated with intravenous fluids containing 10% dextrose to treat or prevent hypoglycemia and to suppress lipolysis as rapidly as possible (Chapter 86). Chronic therapy consists of avoiding fasting. This usually requires simply adjusting the diet to ensure that overnight fasting periods are limited to <10-12 hr. Restricting dietary fat or treatment with carnitine is controversial. The necessity for active therapeutic intervention for individuals with the T199C variant has not yet been established.
Long-Chain 3-Hydroxyacyl CoA Dehydrogenase (LCHAD)/Mitochondrial Trifunctional Protein (TFP) Deficiency
Defects in the Carnitine Cycle
Defects in Electron Transfer Pathway
Defects in Ketone Synthesis Pathway
Defects in Ketone Utilization
De Leon DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57-68.
Gillingham MB, Weleber RG, Neuringer M, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005;86:124-133.
Greenberg CR, Dilling LA, Thompson GR, et al. The paradox of the carnitine palmitoyltransferase type 1a P479L variant in Canadian Aboriginal populations. Mol Genet Metab. 2009;96:201-207.
Hsu HW, Zytkovicz TH, Comeau AM, et al. Spectrum of medium-chain acyl-CoA dehydrogenase deficiency detected by newborn screening. Pediatrics. 2008;121:e1108-e1114.
Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol Genet Metab. 2008;95:195-200.
Longo N, di San Filippo CA, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77-85.
Loughrey C, Bennett MJ. Screening for MCAD deficiency in newborns. BMJ. 2009;338:843-846.
Molven A, Matre GE, Duran M, et al. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes. 2004;53:221-227.
Schulze A, Matern D, Hoffmann GF. Newborn Screening. In: Sarafoglou K, Hoffmann GF, Roth KS, editors. Pediatric endocrinology and inborn errors of metabolism. New York: McGraw-Hill; 2009:17-32.
Shekhawat PS, Matern D, Strauss AW. Fetal fatty acid oxidation disorders, their effect on maternal health and neonatal outcome: impact of expanded newborn screening on their diagnosis and management. Pediatr Res. 2005;57:78R-86R.
Strauss AW, Andresen BS, Bennett MJ. Mitochondrial fatty acid oxidation defects. In: Sarafoglou K, Hoffmann GF, Roth KS, editors. Pediatric endocrinology and inborn errors of metabolism. New York: McGraw-Hill; 2009:51-70.
van Maldegem BT, Duran M, Wanders RJA, et al. Fasting and fat-loading tests provide pathophysiological insight into short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 2010;156:121-127.
Wilcken B, Haas M, Joy P, et al. Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet. 2007;369:37-42.
80.2 Disorders of Very Long Chain Fatty Acids
Peroxisomal Disorders
Etiology
Peroxisomal disorders are subdivided into 2 major categories (Table 80-2).
Table 80-2 CLASSIFICATION OF PEROXISOMAL DISORDERS
A: DISORDERS OF PEROXISOME IMPORT
A1: Zellweger syndrome
A2: Neonatal adrenoleukodystrophy
A3: Infantile Refsum disease
A4: Rhizomelic chondrodysplasia punctata
B: DEFECTS OF SINGLE PEROXISOMAL ENZYME
B1: X-linked adrenoleukodystrophy
B2: Acyl CoA oxidase deficiency
B3: Bifunctional enzyme deficiency
B4: Peroxisomal thiolase deficiency
B5: Classic Refsum disease
B6: 2-Methylacyl CoA racemase deficiency
B7: DHAP acyltransferase deficiency
B8: Alkyl-DHAP synthase deficiency
B9: Mevalonic aciduria
B10: Glutaric aciduria type III
B11: Hyperoxaluria type I
B12: Acatalasemia
In category A, the peroxisomal biogenesis disorders (PBD), the basic defect is the failure to import one or more proteins into the organelle. In category B, defects affect a single peroxisomal protein. The peroxisome is present in all cells except mature erythrocytes and is a subcellular organelle surrounded by a single membrane; >50 peroxisomal enzymes are identified. Some enzymes are involved in the production and decomposition of hydrogen peroxide; others are concerned with lipid and amino acid metabolism. Most peroxisomal enzymes are first synthesized in their mature form on free polyribosomes and enter the cytoplasm. Proteins that are destined for the peroxisome contain specific peroxisome targeting sequences (PTS). Most peroxisomal matrix proteins contain PTS1, a 3-amino acid sequence at the carboxyl terminus. PTS2 is an amino-terminal sequence that is critical for the import of enzymes involved in plasmalogen and branched-chain fatty acid metabolism. Import of proteins involves a complex series of reactions that involves at least 23 distinct proteins. These proteins are referred to as peroxins encoded by PEX genes. Table 80-3 summarizes the PEX genes that are defective in human disease states.
Epidemiology
Except for X-linked adrenoleukodystrophy (X-ALD), all the peroxisomal disorders in Table 80-2 are autosomal recessive traits. X-ALD is the most common peroxisomal disorder, with an estimated incidence of 1/17,000. The combined incidence of the other peroxisomal disorders is estimated to be 1/50,000.
Pathogenesis
It is likely that all pathologic changes are secondary to the peroxisome defect. Multiple peroxisomal enzymes fail to function in the PBD (Table 80-4). The enzymes that are diminished or absent are synthesized but are degraded abnormally fast because they may be unprotected outside of the peroxisome. It is not clear how defective peroxisome functions lead to the widespread pathologic manifestations.
Table 80-4 ABNORMAL LABORATORY FINDINGS COMMON TO DISORDERS OF PEROXISOME BIOGENESIS
Peroxisomes absent to reduced in number
Catalase in cytosol
Deficient synthesis and reduced tissue levels of plasmalogens
Defective oxidation and abnormal accumulation of very long chain fatty acids
Deficient oxidation and age-dependent accumulation of phytanic acid
Defects in certain steps of bile acid formation and accumulation of bile acid intermediates
Defects in oxidation and accumulation of L-pipecolic acid
Increased urinary excretion of dicarboxylic acids
The PBD are associated with genetically determined import defects. The PBD have been subdivided into 12 complementation groups. The molecular defects have been defined in 10 of these groups (see Table 80-3). The pattern and severity of pathologic features vary with the nature of the import defects and the degree to which import is impaired. These gene defects lead to disorders that were named before their relationship to the peroxisome was recognized, namely, Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and rhizomelic chondrodysplasia punctata (RCDP). The first 3 disorders are considered to form a clinical continuum, with ZS the most severe, IRD the least severe, and NALD intermediate. They can be caused by 11 different gene defects, which involve mainly the import of proteins that contain the PTS1 targeting signal; the gene defects cannot be distinguished on the basis of clinical features. The clinical severity varies with the degree to which protein import is impaired. Mutations that abolish import completely are often associated with the ZS phenotype, whereas a missense mutation, in which some degree of import function is retained, leads to the somewhat milder phenotypes. A defect in PEX7, which involves the import of proteins that utilize PTS2, is associated with RCDP. PEX7 defects that leave import partially intact are associated with milder phenotypes, some of which resemble classic Refsum disease.
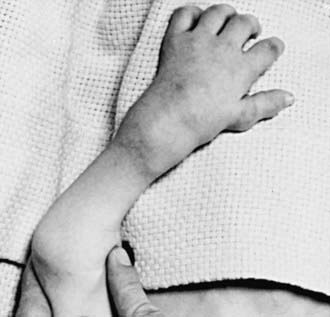
PBD With Milder or Atypical Phenotypes
Newborn infants with Zellweger syndrome show striking and consistent, recognizable abnormalities. Of central diagnostic importance are the typical facial appearance (high forehead, unslanting palpebral fissures, hypoplastic supraorbital ridges, and epicanthal folds; Fig. 80-3), severe weakness and hypotonia, neonatal seizures, and eye abnormalities (cataracts, glaucoma, corneal clouding, Brushfield spots, pigmentary retinopathy, and nerve dysplasia). Because of the hypotonia and “mongoloid” appearance, Down syndrome may be suspected. Infants with Zellweger syndrome rarely live more than a few months. More than 90% show postnatal growth failure. Table 80-5 lists the main clinical abnormalities.
Rhizomelic Chondrodysplasia Punctata (Rcdp)
This disorder is characterized by the presence of stippled foci of calcification within the hyaline cartilage and is associated with dwarfing, cataracts (72%), and multiple malformations due to contractures. Vertebral bodies have a coronal cleft filled by cartilage that is a result of an embryonic arrest. Disproportionate short stature affects the proximal parts of the extremities (Fig. 80-4A). Radiologic abnormalities consist of shortening of the proximal limb bones, metaphyseal cupping, and disturbed ossification (Fig. 80-4B). Height, weight, and head circumference are less than the 3rd percentile, and these children are severely retarded mentally. Skin changes such as those observed in ichthyosiform erythroderma are present in about 25% of patients.
Isolated Defects of Peroxisomal Fatty Acid Oxidation
The disorders labeled B1 through B3 (see Table 80-2) each involve 1 of 3 enzymes involved in peroxisomal fatty acid oxidation. Their clinical manifestations resemble those of the Zellweger syndrome/neonatal ALD/infantile Refsum disease continuum; they can be distinguished from disorders of peroxisome biogenesis by laboratory tests. Defects of bifunctional enzyme are common and are found in about 15% of patients with the Zellweger syndrome/neonatal ALD/infantile Refsum disease phenotype. Patients with isolated acyl CoA oxidase deficiency have a somewhat milder phenotype that resembles that of neonatal ALD.
Isolated Defects of Plasmalogen Synthesis
Plasmalogens are lipids in which the 1st carbon of glycerol is linked to an alcohol rather than a fatty acid. They are synthesized through a complex series of reactions, the 1st two steps of which are catalyzed by the peroxisomal enzymes dihydroxyacetone phosphate alkyl transferase and synthase. Deficiency of either of these enzymes (B4 and B5 in Table 80-2) leads to a phenotype that is clinically indistinguishable from the peroxisomal import disorder RCDP. This latter disorder is caused by a defect in PEX7, the receptor for peroxisome targeting sequence 2. It shares the severe deficiency of plasmalogens with disorders B4 and B5 but, in addition, has defects of phytanic oxidation. The fact that disorders B4 and B5 are associated with the full phenotype of RCDP suggests that a deficiency of plasmalogens is sufficient to produce it.
Laboratory Findings
Laboratory tests for peroxisomal disorders can be viewed at three levels of complexity.
Level 1: Does the Patient have a Peroxisomal Disorder?
This can be resolved by noninvasive tests that are generally available (Table 80-6). Measurement of plasma VLCFA is the most commonly used assay. Whereas plasma VLCFA levels are elevated in many patients with peroxisomal disorders, this is not always the case. The most important exceptions are RCDP, in which VLCFA levels are normal, but plasma phytanic acid levels are increased and red blood cell plasmalogen levels are reduced. In some other peroxisomal disorders, the biochemical abnormalities are still more restricted. Therefore, a panel of tests is recommended and includes plasma levels of VLCFA and phytanic, pristanic, and pipecolic acids and red blood cell levels of plasmalogens. Tandem mass spectrometry techniques also permit convenient quantitation of bile acids in plasma and urine. This panel of tests can be performed on 2 mL samples of venous blood and permits detection of most peroxisomal disorders. Furthermore, normal results make the presence of a peroxisomal disorder unlikely.
Level 2: What is the Precise Nature of the Peroxisomal Disorder?
Table 80-6 lists the main biochemical abnormalities in the various peroxisomal disorders. When combined with the clinical presentation, the panel of level 1 tests (see earlier) is often sufficient to identify the precise nature of the defect. Elevated plasma VLCFA levels permit the precise diagnosis of X-ALD in male patients. Marked reduction of erythrocyte plasmalogen levels combined with elevated plasma phytanic acid permits precise diagnosis in a patient with the clinical features of RCDP. Classic Refsum disease can be diagnosed by demonstration of increased plasma phytanic acid combined with normal or reduced levels of pristanic acid levels, while in D-bifunctional enzyme deficiency and 2-methylacyl CoA racemase deficiency, the levels of pristanic and phytanic acid are both increased. Precise identification of some peroxisomal disorders may require more extensive studies in cultured skin fibroblasts. This may be required for the differentiation of PBD from defects in bifunctional enzyme. In PBD, the patient’s peroxisomes are absent and catalase is in the soluble fraction, whereas in bifunctional enzyme defect, peroxisomes are present and catalase is in the particulate fraction. Fibroblast studies are required to identify the nature of the molecular defect in PBD. Whether such specialized studies are clinically warranted depends on individual circumstances. Precise definition of the defect in a proband may improve the precision of prenatal diagnosis in at-risk pregnancies, and it is required for carrier detection. It is also of value in setting prognosis. Precise characterization is of prognostic value in patients with PEX1 defects. This defect is present in approximately 60% of PBD patients, and about half of the PEX1 defects have the G843D allele, which is associated with a significantly milder phenotype than is found in other mutations.
Diagnosis
There are several noninvasive laboratory tests that permit precise and early diagnosis of peroxisomal disorders (see Table 80-6). The challenge in PBD is to differentiate them from the large variety of other conditions that can cause hypotonia, seizures, failure to thrive, or dysmorphic features. Experienced clinicians can readily recognize classic Zellweger syndrome by its clinical manifestations. PBD patients often do not show the full clinical spectrum of disease and may be identifiable only by laboratory assays. Clinical features that may serve as indications for these diagnostic assays include severe psychomotor retardation; weakness and hypotonia; dysmorphic features; neonatal seizures; retinopathy, glaucoma, or cataracts; hearing deficits; enlarged liver and impaired liver function; and chondrodysplasia punctata. The presence of one or more of these abnormalities increases the likelihood of this diagnosis. Atypical milder forms presenting as peripheral neuropathy have also been described.
Genetic Counseling
All the peroxisomal disorders, except hyperoxaluria type 1, can be diagnosed prenatally in the 1st or 2nd trimester. The tests are similar to those described for postnatal diagnosis (see Table 80-6) and use chorionic villus sampling or amniocytes. More than 300 pregnancies have been monitored, and more than 60 affected fetuses have been identified without diagnostic error. Because of the 25% recurrence risk, couples with an affected child must be advised about the availability of prenatal diagnosis. Heterozygotes can be identified in X-ALD and in those disorders in which the molecular defect has been identified (see Table 80-3).
Adrenoleukodystrophy (X-Linked)
Etiology
The key biochemical abnormality is the tissue accumulation of unbranched saturated VLCFA, with a carbon chain length of 24 or more. Excess hexacosanoic acid (C26 : 0) is the most striking and characteristic feature. This accumulation of fatty acids is caused by genetically deficient peroxisomal degradation of fatty acid. The key biochemical defect involves the impaired function of peroxisomal lignoceroyl CoA ligase, the enzyme that catalyzes the formation of the CoA derivative of VLCFA. The gene that is defective (ABCD1) codes for a peroxisomal membrane (ALDP). More than 400 distinct mutations have been identified, and most families have a mutation that is “private” (unique to that kindred) and are updated on the website, www.x-ald.nl. The gene has been mapped to chromosome Xq28. The mechanism by which the ALDP defect leads to VLCFA accumulation and the pathology of X-ALD is unknown.
Laboratory and Radiographic Findings
CT And MRI
Patients with childhood cerebral or adolescent ALD show cerebral white matter lesions that are characteristic with respect to location and attenuation patterns on MRI. In 80% of patients, the lesions are symmetric and involve the periventricular white matter in the posterior parietal and occipital lobes. About 50% show location of a garland of accumulated contrast material adjacent and anterior to the posterior hypodense lesions (Fig. 80-5A). This zone corresponds to the zones of intense perivascular lymphocytic infiltration where the blood-brain barrier breaks down. In 12% of patients, the initial lesions are frontal. Unilateral lesions that produce a mass effect suggestive of a brain tumor may occur. MRI provides a clearer delineation of normal and abnormal white matter than does CT and may demonstrate abnormalities missed by CT (Fig. 80-5B).
Diagnosis and Differential Diagnosis
The earliest manifestations of childhood cerebral ALD are difficult to distinguish from the more common attention-deficit disorders or learning disabilities. Rapid progression, signs of dementia, or difficulty in auditory discrimination suggest ALD. Even in early stages, CT or MRI may show strikingly abnormal changes. Other leukodystrophies (Chapters 592 and 605.10) or multiple sclerosis (Chapter 593.1) may mimic these radiographic findings. Definitive diagnosis depends on demonstration of VLCFA excess, which occurs only in X-ALD and the other peroxisomal disorders. The latter may be distinguished from X-ALD by their clinical presentation during the neonatal period.
Treatment
Corticosteroid replacement for adrenal insufficiency or adrenocortical hypofunction is effective (Chapter 569). It may be lifesaving and increase general strength and well-being, but it does not alter the course of the neurologic disability.
