[level-membership-for-neurosurgery-category]
CHAPTER 182 Craniosynostosis
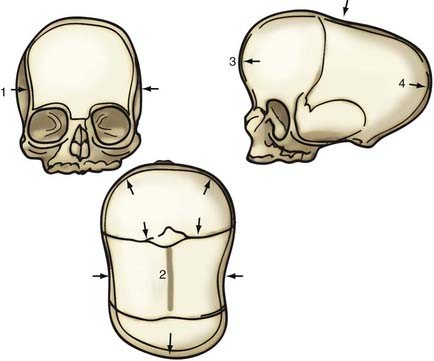
Although skull shape irregularity has been recognized since antiquity, the study of abnormal skull growth related to craniosynostosis had its scientific origin in the late 1700s. Sommerring1 noted that bone growth in the skull occurred primarily at suture lines and that when this growth site was prematurely bridged with bone, an abnormal skull shape developed. This was characterized by reconstruction of the skull’s growth in a plane perpendicular to the plane of the fused suture. Similar observations were made by Otto2 in 1830 and Virchow3 in 1821. It was Virchow’s widely publicized treatise on skull deformity that added impetus to the scientific study of abnormal skull form in craniosynostosis. He observed, as had earlier investigators, that skull growth occurred at suture lines in the skull and that when these suture lines were prematurely fused, skull deformity developed. Restriction of growth adjacent to the suture occurred, but compensatory growth occurred elsewhere in the skull to accommodate the growing brain. This understanding of normal and abnormal skull growth served as the basis for understanding skull abnormalities for the next 100 years (Fig. 182-1).
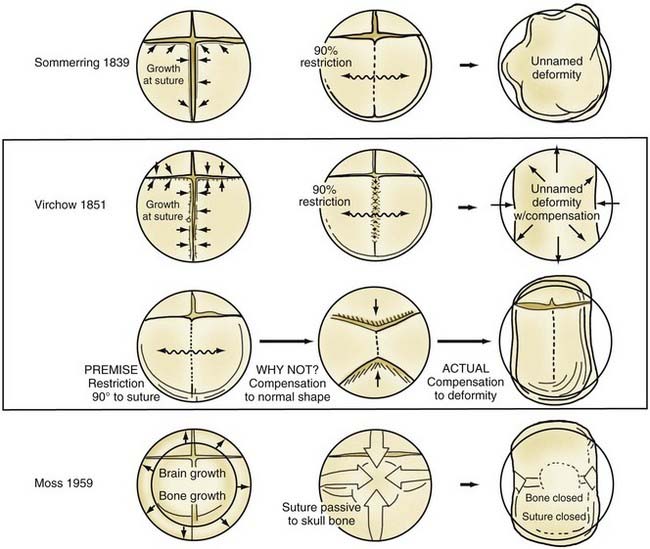
In the mid-20th century, the primary role of the cranial vault suture in the development of craniosynostosis skull deformities was questioned by Moss4 and van der Klaauw.5 Moss noted that surgeons operating on the skulls of children presumed to have craniosynostosis would occasionally find patent cranial vault sutures, despite what appeared to be typical craniosynostosis skull deformities (see Fig. 182-1).6 Also, he recognized that there were some characteristic cranial base deformities associated with individual forms of craniosynostosis, and because the cranial base matures embryologically before the remainder of the cranial vault, he suggested that the cranial base abnormality was the primary pathologic process and the cranial vault suture abnormality (fusion) was secondary.4 Testing this hypothesis in the laboratory, Moss7 removed a normal cranial vault suture and found that doing so did not affect overall skull length. This indicated that the cranial vault suture, unlike the epiphysis of a long bone, does not serve as a growth site that pushes the bone ends apart but is a passive recipient of growth influences.
The amount of bone deposited at the cranial vault suture is related to the strains that influence it. Brain enlargement, Moss8 believed, was the primary source of these tensile strains that caused the suture to deposit bone. This is known as the functional matrix theory, in which the functional enlargement or development of an organ system is the primary force in changing its overall shape and determining its final form. Even though brain enlargement is clearly the engine of skull remodeling, the precise role the vault suture plays in the development of the skull pathology associated with craniosynostosis must be determined by direct manipulation of growth at the suture.7
In 1979 Persson and colleagues9 reported that in animals, selective restriction of an individual cranial vault suture’s growth resulted in skull deformities that closely mimic the clinical condition of craniosynostosis involving the same cranial vault suture in humans. Moreover, cranial base and even facial deformities develop secondary to the cranial vault suture restrictions.10,11 This observation indicates that cranial vault suture pathology may be primary in the development of craniosynostosis skull deformities leading to cranial base and facial deformity. Subsequently, Mooney and coworkers12,13 studied an animal model of congenital craniosynostosis in which cranial vault suture, vault, and cranial base abnormalities closely resembled the findings of Babler and Persing10 on cranial suture restriction. In addition, the developmental studies of Opperman and colleagues14,15 demonstrated the significant influence of mesenchymal tissues, in particular the dura and the periosteum at the suture, on the maintenance of patency of cranial vault sutures during development.
Nonsyndromic and Syndromic Craniosynostosis
Researchers have found clinical evidence that cranial vault suture, not cranial base, pathology is of primary importance in the development of skull shape pathology in nonsyndromic craniosynostosis. Preoperative and postoperative computed tomography (CT) shows that surgical treatment of the cranial vault alone results in improvement of not only cranial vault but also cranial base pathology.16 This indicates that removing the abnormal influence in the cranial vault suture may improve cranial base shape. Further analysis of skull growth after simple suture closure has revealed a predictable pattern based on certain rules. The ability to predict and understand the stereotypical deformity from observation of the vault suture suggests its primary role.17,18 The dura clearly plays a role in this, as demonstrated by Drake and coworkers,19 who found that resected fused and nonfused cranial suture elements redevelop following the removal of cranial bone topographically connected to the fused and nonfused sutures. Distraction devices, such as those developed by Ilizarov for the distraction of extremity bones and by McCarthy for mandibular hypoplasia, are effective in elongating bone in animals. Using spring-like expanders, Persing and associates20 internalized the distraction of skull bone experimentally, and subsequently Maltese and colleagues21 employed this approach in the management of human craniosynostosis. The advantage of the distraction and spring techniques is that bone elongation is a gradual process, with minimal surgical intervention and operative time. One disadvantage is that if the cranial bone is already deformed, the irregularly shaped bone is advanced or expanded from the osteotomy site. In addition, the removal of spring and bone distraction devices requires an operative procedure. Similar to implanted metal fixation plates and screws in the skull, if left in place too long, these metal devices can migrate intracranially, with progressive resorption of the endocranial surface of the skull bone and positioning of bone on the ectocranial surface as the skull and brain enlarge. Concern about transcranial migration has been the main impetus for the avoidance of metallic fixation plates and screws in young children. Resorbable plates and screws, as well as suture material, have largely supplanted titanium devices in patients younger than 3 years.
Additional studies are being done to document skull growth changes by surgical manipulation, in particular by the use of mechanical devices to control or enhance growth influences.17,20 In the future, these devices may be used clinically to prolong corrective growth influences postoperatively.
Syndromic craniosynostosis is much less common and appears to be a more generalized disorder of mesenchymal development that may represent abnormalities in homeobox genes such as Runx2 or CBFA-δ. Crouzon’s syndrome occurs in 1 in 25,000 live births, and this bilateral coronal synostosis is frequently associated with exorbitism and midface hypoplasia. Apert’s syndrome occurs in 1 in 100,000 live births; the features of brachycephaly due to coronal synostosis and midface hypoplasia are evident, but extremity syndactylies are also characteristic. There are more than 64 known craniofacial syndromes associated with craniosynostosis22; however, the cause of craniosynostosis is still unknown, although in some cases, a genetic influence is undeniable. In the syndromic forms of craniosynostosis, autosomal dominant inheritance patterns are the general rule for the more common syndromes such as Apert’s, Crouzon’s, and Pfeiffer’s; autosomal recessive disorders (e.g., Carpenter’s syndrome, characterized by abnormal skull shape deformities and polydactyly) do not commonly occur.
Diagnosis
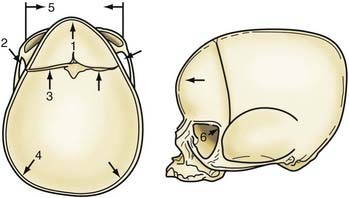
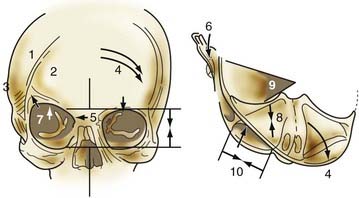
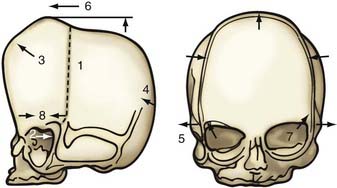
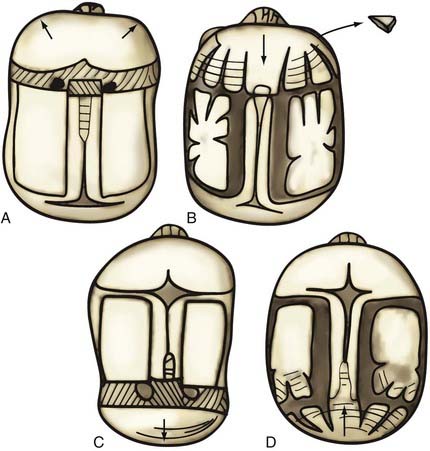
Nonsyndromic craniosynostosis involving a single vault suture is ordinarily diagnosed by the observation of a typically deformed skull shape, with radiographs serving as confirmatory evidence. We have modified the Virchow hypothesis of skull deformity to explain the skull shapes associated with individual forms of craniosynostosis.17,18 These patterns of skull abnormalities can be explained by invoking four tenets: (1) cranial vault bones directly adjacent to the prematurely fused vault suture act as a single bone plate, with reduced growth potential along all margins of that plate (Fig. 182-2A); (2) asymmetric bone deposition occurs at vault sutures along the perimeter of the bone plate, with increased bone deposition occurring at the suture margin located farther away from the plate (Fig. 182-2B); (3) the nonperimeter sutures in line with the fused suture deposit bone symmetrically at their sutural edges (Fig. 182-2C); and (4) perimeter and sutures (in line) abutting the prematurely fused suture compensate to a greater degree than distant sutures (Fig. 182-2D). The application of these principles to particular types of synostosis is as follows (Fig. 182-2E to H)  : Figure 182-2E represents sagittal synostosis, in which the coronal and lambdoid sutures show asymmetric growth, and the metopic suture shows symmetrical growth. Figure 182-2F depicts coronal synostosis, with asymmetric growth at the metopic, lambdoid, and sagittal sutures and symmetrical growth at the unfused coronal suture. Figure 182-2G illustrates metopic synostosis, with symmetrical growth at the sagittal suture and asymmetric growth at the coronal sutures. Figure 182-2H depicts lambdoid synostosis, with a fused lambdoid suture and bilateral growth at the lambdoid, unilateral at the sagittal, and unilateral at the squamosal sutures, displacing the ear downward rather than forward as in positional posterior plagiocephaly.
: Figure 182-2E represents sagittal synostosis, in which the coronal and lambdoid sutures show asymmetric growth, and the metopic suture shows symmetrical growth. Figure 182-2F depicts coronal synostosis, with asymmetric growth at the metopic, lambdoid, and sagittal sutures and symmetrical growth at the unfused coronal suture. Figure 182-2G illustrates metopic synostosis, with symmetrical growth at the sagittal suture and asymmetric growth at the coronal sutures. Figure 182-2H depicts lambdoid synostosis, with a fused lambdoid suture and bilateral growth at the lambdoid, unilateral at the sagittal, and unilateral at the squamosal sutures, displacing the ear downward rather than forward as in positional posterior plagiocephaly.
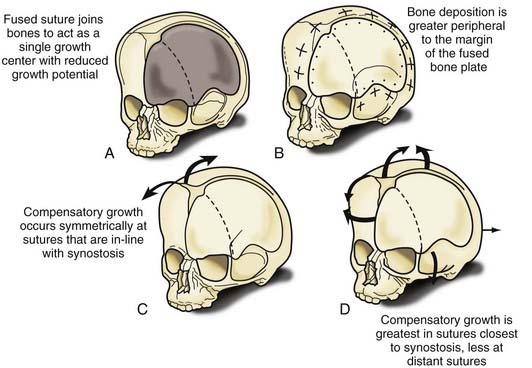
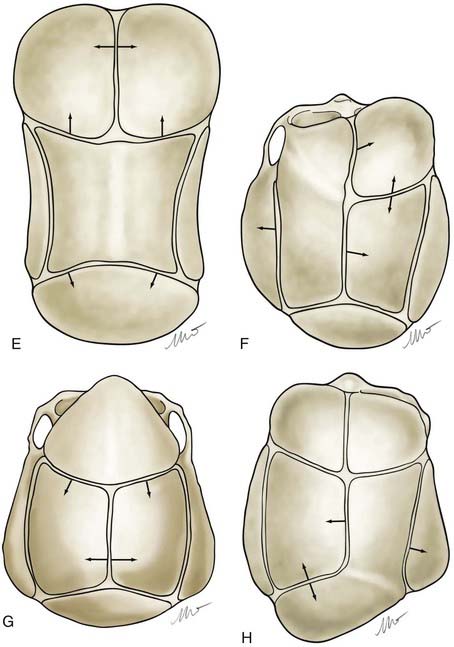
Figure 182-E2 contd E, Sagittal synostosis. F, Coronal synostosis. G, Metopic synostosis. H, Lambdoid synostosis.
(E-H, Courtesy of Dr. Michael McKisic.)
There is controversy regarding the advisability of CT scans in patients being evaluated for craniosynostosis. It has been reported that CT scans performed early in infancy are associated with an increased likelihood of malignancy later in life,23 although the concern for malignancy is greatest in children undergoing thoracoabdominal scans and in those scanned earlier in life.
Operative Timing and Approaches
A common question asked by parents is whether the condition is progressive—in other words, will it “look worse” with time? This is particularly pertinent in sagittal synostosis, where the answer appears to be affirmative. Serial CT imaging has shown that normal growth of the skull tends toward increasing roundness in the first year of life. Sagittal synostosis results in a progressively longer skull, but because the width also increases, the shape does not change as it should, resulting in a marked increase in the deformity.24
Experimental evidence demonstrates that improved skull form is achieved with an operation before the human equivalent of 6 months of age.25 In clinical studies, more significant degrees of improvement were noted in patients who underwent surgery when younger than 6 months compared with those having surgery after 6 months of age. Correction of the cranial base deformities established in unilateral coronal synostosis was also more successful if surgery was performed early.16
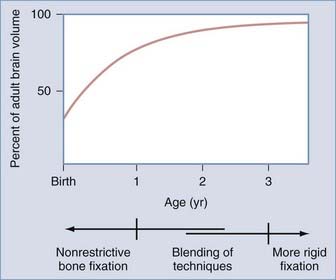
The influence of surgical methods is not yet clearly defined; however, limited craniectomy procedures of few sutures appear to be beneficial in very young patients with mild deformities, whereas more comprehensive approaches to established skull deformities are more effective in older children and in children of any age with more severe deformities. Objective documentation of these approaches is not yet widespread, but Marsh and coworkers25 compared preoperative and postoperative skull shape following a limited craniectomy procedure versus a whole-vault cranioplasty approach for sagittal synostosis; they found that skull deformity was reduced with both craniectomy and cranioplasty procedures. Heller and associates26 showed a normal cephalic index in 100% of patients undergoing whole-vault cranioplasty; however, the skull shape returned to normal only in patients who underwent the more extensive cranioplasty procedure. Additional data are required to confirm initial impressions, but they are consistent with the hypothesis that early rapid growth of the brain shapes the skull (Fig. 182-3).
Intracranial Pressure, Cognition, and Behavior
Treatment of brain deficiency by surgical attack on the deformed skull has an inglorious history dating to the second half of the 19th century. Many luminaries of the early years of neurosurgery were caught up in an international wave of unscientific enthusiasm for linear craniectomy as a therapy to “unlock” the brains of severely impaired, microcephalic children. The benefits were imaginary, and the mortality rates were appalling. A passionate campaign led by Abraham Jacobi, patriarch of the specialty of pediatrics and founder of the American Academy of Pediatrics, and supported by Harvey Cushing, among others, eventually drove the surgical treatment of mental retardation into deserved obscurity (see Feinsod and Davis27 for the full story). When craniosynostosis later emerged as a distinct diagnostic entity, and as surgical interventions became safe and effective from a cosmetic standpoint, the question of the relationship between skull deformity and brain development arose once again, and it has proved resistant to simple answers.
Elevated intracranial pressure (ICP) is a well-recognized feature of the syndromic forms of craniosynostosis, but it can be documented in a fraction of cases of single-suture synostosis as well.28–40 Generally it subsides after surgical treatment.36,39,41 ICP monitoring has been proposed as an adjunct to surgical decision-making in cases for which cosmetic indications are not compelling.49 The most extensive experience with ICP monitoring comes from the Hopital des Enfants Malades in Paris.30,32,38,40 In a 1989 report of 358 ICP recordings in children with craniosynostosis, Renier32 took ICP recorded during slow-wave sleep as “baseline.” In the absence of valid age-adjusted, normative ICP data, baseline measurements less than 10 torr were defined as normal. Measurements above 15 torr were considered abnormal, and measurements between 10 and 15 torr were considered borderline. Elevated ICP was recorded in 8% of cases of sagittal synostosis, 6% of metopic synostosis, and 12% of unilateral coronal synostosis. It was more prevalent in patients with multiple suture involvement and among syndromic cases. Patients who presented after 1 year of age had much higher rates of intracranial hypertension. Likewise, patients who presented after 1 year of age had lower developmental quotients (DQs) or intelligence quotients (IQs), and there was an inverse correlation between preoperative ICP and DQ or IQ.
The generalization of these data in support of early surgical treatment is difficult to accept. Patients were selected for ICP monitoring for unstated reasons, and the factors determining early and late presentation were unknown as well. It is possible that the parents of the older patients found their children’s deformities acceptable but brought them to medical attention because of developmental or behavioral issues—a selection bias that may have enriched the late presenting group with low DQs and IQs.40 Another possibility is that late presenting cases had experienced obstacles to access to care based on low socioeconomic status or low parental intelligence. This analysis has also been criticized for the interchangeable use of DQ and IQ data, because longitudinal studies have shown that DQ scores above the range of frank retardation are only weakly predictive of IQ later in life.40,42 The association of single-suture craniosynostosis with intracranial hypertension is further clouded by mechanistic questions. Perhaps contrary to expectations, measurements of cranial volume using CT have documented normal or increased volumes among infants with single-suture craniosynostosis compared with age-matched controls.43–45 The correlation between intracranial hypertension and lower than normal cranial volume seems to be quite poor, and the measurement of normal or even increased cranial volume does not eliminate the possibility of elevated ICP.29,30 As is true among patients with syndromic craniosynostosis, mechanisms other than craniocerebral disproportion, such as cerebral venous insufficiency, may be important.
A few studies have reported focal regions of hypoperfusion46,47 or hypometabolism48 subjacent to abnormal sutures among small numbers of selected infants with single-suture craniosynostosis. In most cases these abnormalities resolved after surgery. The prevalence of such focal physiologic disturbances cannot be estimated. The natural history and functional significance of such disturbances are unknown.
Recent investigations employing sophisticated methods of image analysis have begun to attempt to relate calvarial deformities to deformation of the underlying neuroanatomic structures and to development data.49–53 Efforts to demonstrate correlations between preoperative skull morphology and preoperative development testing have yielded contradictory findings.51–54 Distinctive alterations in cortical and subcortical brain morphology in association with sagittal and metopic synostosis have been demonstrated on magnetic resonance imaging in comparison with age-matched controls.49 Preoperative and postoperative studies of infants with sagittal synostosis indicate that these distortions of brain morphology do not revert toward normal even after successful surgical correction of the calvarial deformity.50
Since the beginning of the modern era of surgical treatment of craniosynostosis, there have been concerns about the intellectual and behavioral prognosis of affected children,55,56 leading to many detailed observational studies. Kapp-Simon and others have reviewed this literature lucidly.57–59 Points of observation fall into three time periods: at presentation in infancy, early in the postoperative period, and at school age or later. Observations in the first two periods are necessarily developmental. In the last period, intelligence can be tested, and behavioral disturbances and learning disabilities can be recognized. The standard instruments, typically the Bayley Scales of Infant Development and the Wechsler Intelligence Scales for Children—Revised, have age- and gender-adjusted norms, but some studies have employed matched community controls or sibling controls, with particular implications for the interpretation of results. In all three periods, the rates of true mental retardation (DQ or IQ more than 2 standard deviations below the mean) are probably no greater among children with single-suture synostosis than in the general population. Most investigators have found that in study groups of infants with single-suture involvement, the distributions of preoperative developmental test scores are normal or shifted downward to variable degrees, with variable statistical significance in comparison to normative data.52,59–66 In the early postoperative period, aggregate test scores are generally stable,61,62,66,67 although some studies have claimed improvements over preoperative scores that are not enjoyed by untreated patients.60 Later in childhood the differences between patients and unaffected children become more vivid: among patients, mean full-scale IQ scores are within the normal range but generally lower than among controls,68 and a growing fraction of patients are diagnosed with speech, language, or learning disabilities and behavioral disturbances. The literature suggests that between 35% and 50% of children with single-suture synostosis, regardless of treatment status, can be expected to exhibit some degree of cognitive or behavioral disability in the school years.54,59,69–74
Clinical research will never directly address the question of the effect of surgical treatment on the long-term cognitive and behavioral prognosis for children with single-suture craniosynostosis. A randomized trial cannot be conducted. Although there may be equipoise on the question of cognitive and behavioral outcomes, the contemporary standard of care dictates the treatment of almost all patients for cosmetic indications. Observational studies have drawn conflicting inferential conclusions based on the presence or absence of statistical correlations between treatment status or age at treatment and various outcomes of interest.32,40,54,65,72 A host of factors, known and unknown, determine whether and when a child gets surgical treatment, and many of these factors are potential determinants of cognitive and behavioral outcomes as well. Such sources of bias prohibit the confident interpretation of the existing literature.
Operative Treatment
Because the objective of surgical treatment is to provide each patient with the best aesthetic outcome that can be achieved, the plan for correcting the deformity must be formulated according to the particular demands of each case. The technique of surgical reconstruction must be tailored to the type of synostosis present and the age of the patient. It is clear that reshaping bone in a child younger than 1 year is much more readily achieved without significant bone disruption than in an older child. The methods of fixation for reshaped skull segments should also be different for very young children to avoid abnormalities in brain development resulting from vault surgery and the subsequent restriction of brain growth. Patients who are younger than 3 years have a substantial amount of brain growth remaining, but because those older than 3 years have already achieved approximately 85% to 90% of their ultimate brain mass (see Fig. 182-3), the fixation techniques can be more rigid and inclusive in these older children. In the following sections, the treatment of patients younger than 1 year and older than 3 years is described separately; those between 1 and 3 years of age require a blending of techniques. Metallic fixation devices are generally avoided in patients younger than 3 years owing to concerns related to the inward “migration” of the metal with further skull development. Resorbable plates, screws, and sutures are used instead of metallic devices.
Metopic Synostosis
Metopic synostosis is characterized by metopic suture ridging, bilateral flattening of the frontal bones, anterior displacement of the coronal sutures, lateral flaring of the posterior parietal regions, hypotelorism, and flattening of the supraorbital ridges (Fig. 182-4). Viewed from above, the skull has a characteristic triangular shape known as trigonocephaly.
Operative Technique in Children Younger Than 1 Year
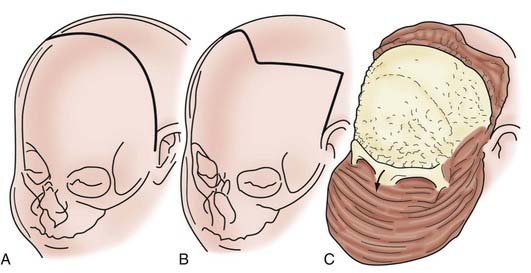
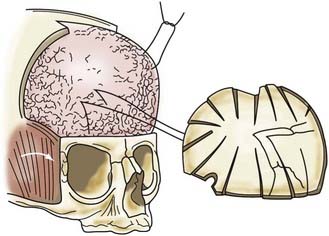
In children younger than 1 year, a zigzag variation on the coronal incision, sometimes called the stealth incision, can be used to minimize the visibility of incisional scalp alopecia (Fig. 182-5). Dissection of the anterior and posterior scalp flaps is conducted in the supraperiosteal plane; this is preferred because bleeding is reduced and the periosteal tissue left on the bone anchors individual bone fragments and maintains their alignment with subsequent remodeling.76
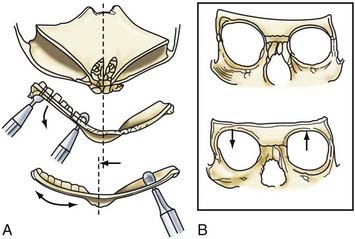
Incision of the periosteum approximately 1 cm above the supraorbital rim aids in subperiorbital dissection, allowing bilateral orbital rim osteotomy and advancement (Fig. 182-6A). A bifrontal craniotomy is performed, and the posterior extent is located just anterior to the coronal suture. The orbital rims are reshaped to assume a more convex configuration, particularly in the supralateral orbital rim. No attempt is made to correct the hypotelorism evident on preoperative examination; it is ordinarily relatively mild and does not constitute a significant long-term postoperative deformity.
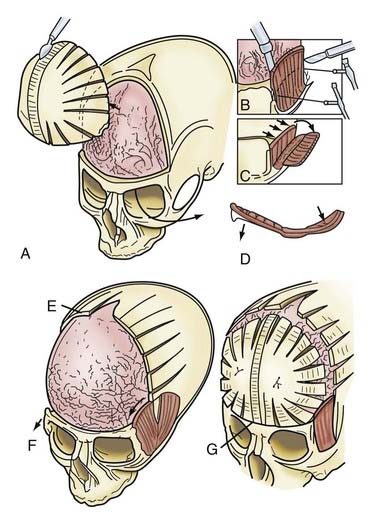
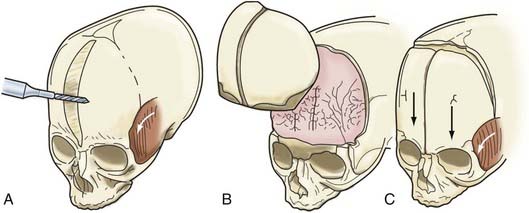
Additional reconstructive steps are taken in the temporal regions. Instead of dissecting the temporalis muscle from the infratemporal fossa, a composite resection of the squamous temporal bone and the overlying temporalis muscle is performed using a Gigli saw and a craniotome (Fig. 182-6B). The remaining basal bone in the temporal region is split into vertical “staves” and fractured laterally, increasing the flare in the temporal region to counteract the concavity typically found with metopic synostosis (Fig. 182-6C). The anterior parietal bone is divided with anteroposterior slats to increase the flare in the anterior parietal region. The bifrontal bone graft undergoes radial osteotomies, reshaping the bone to provide a less acutely angled midline shape and more prominence to the lateral superior frontal bone (see Fig. 182-6B). The midline frontal bone frequently requires shaping with a bur to reduce its prominence. The advanced supraorbital rim is affixed to the nasal bone. The orbital rim is angled forward at the frontozygomatic suture. The composite of temporalis muscle and squamous bone is attached laterally to the advanced supraorbital rims. At the coronal suture, the adjacent bone is removed, creating a neocoronal suture, to remove the bony restriction to dural expansion in this region after further anterior remodeling of the frontal bone. The scalp flap is then replaced.
A less radical procedure is used for less severe forms (Fig. 182-7).77 Occasionally, simple burring of a prominent metopic suture suffices. If the lateral orbital rims do not need advancement, the temporalis muscle can be detached and reinserted in a more anterior position. Also, it is acceptable to remove the metopic suture down to the nasal suture along with the thickened medial orbital rim or frontal bone and reinsert them on the frontal process of the zygoma, along with the temporalis muscle.
Operative Technique in Children Older Than 3 Years
Regardless of age, the same scalp and dissection plane is chosen, and a bifrontal bone graft is elevated. In patients older than 3 years, the graft may be split vertically into anteroposterior slats, in which case the intracranial surface of the bone undergoes “kerfing” (weakening of the bone) to allow remodeling (Fig. 182-8). The goal is to achieve a more round frontal form and less central V-shaped angularity. Supraorbital rims undergo the same form of elevation and advancement as in younger children. The temporalis muscle and squamous portion of the temporal bone are elevated as a composite flap, advanced, and attached to the advanced superior orbital rim. The individual bone slats are reattached to the advanced superior orbital rim medially and laterally, with care taken to avoid significant bony defects because osteogenesis is much less active in a more mature child. In some instances, split calvarial bone grafting may be necessary to fill in bone gaps.

Unilateral Coronal Synostosis
Unilateral coronal synostosis is characterized by ridging of the prematurely fused half of the coronal suture, flattening of the ipsilateral frontal and parietal bones, bulging of the ipsilateral squamous portion of the temporal bone, and bulging of the contralateral frontal and parietal bones (Fig. 182-9). The nasal radix is deviated to the side of the fused suture, and the ear ipsilateral to the fused suture is displaced anteriorly compared with the contralateral ear. Confirmatory radiographic findings include the “harlequin” orbit deformity, characterized by elevation of the greater and lesser wings of the sphenoid ipsilateral to the fused coronal suture. CT of the cranial base demonstrates a shortened anterior cranial fossa and a narrowed sphenopetrosal angle ipsilateral to the fused coronal suture.
Operative Technique in Children Younger Than 1 Year
A bifrontal craniotomy is performed, avoiding removal of the temporalis muscle from the underlying squamous temporal bone (Fig. 182-10). An orbital roof osteotomy, extending to the frontal zygomatic suture laterally to the midline, is carried out bilaterally to elevate and reshape the supraorbital rims. Care is taken to avoid detaching the orbital rim laterally from the zygoma at the frontozygomatic suture. Contouring of the inner portion of the zygomatic process of the frontal bone is performed to equal the mediolateral dimension of the orbit on the contralateral side (Fig. 182-11). The superior orbital rim contralateral to the fused suture is shaped with a bur to achieve symmetry. A composite flap (consisting of temporalis muscle and the squamous portion of the temporal bone) is elevated ipsilateral to the fused suture, and the basal coronal suture is resected to the frontal sphenoid suture.78 The orbital rims are contoured to achieve symmetry and to increase the projection of this supralateral orbital rim ipsilateral to the fused half of the coronal suture. The orbital rims are secured in the midline and angled forward at the fibrous union of the frontozygomatic suture. Deviation of the nasal radix is usually not corrected because it will be ameliorated with subsequent growth in the majority of patients. The composite flap is attached to the advanced superior orbital rim ipsilateral to the previously fused suture. The frontal craniotomy bone is reshaped by radial osteotomy to increase projection in the flattened bone segment ipsilateral to the fused suture and to reduce the projection of the bulging contralateral frontal bone. The bone is then affixed to the advanced supraorbital rims. For less severe conditions, particularly in younger children, simpler techniques can give good results (Fig. 182-12).79
FIGURE 182-10 Early unilateral coronal synostosis. A, Bicoronal takedown and bifrontal craniotomy are done with the bur hole placed superior to the temporal crest. B, Bilateral supraorbital rim advancement and contouring are associated with advancement of the composite myo-osseous flap (see Fig. 182-6C).
Operative Technique in Children Older Than 3 Years
A coronal skin flap and periorbital dissection are performed, similar to that in younger children (Fig. 182-13). A bifrontal bone graft is elevated, and a unilateral (rather than bilateral) orbital rim elevation is performed ipsilateral to the fused coronal sutures. In older children, the abnormal mediolateral dimension of the orbital rim (i.e., mediolateral shortening) ipsilateral to the fused suture is corrected by contouring the inner aspect of the zygomatic process of the frontal bone. The temporalis muscle composite flap is advanced, attaching the squamous portion of the temporal bone to the advanced orbital rim laterally. The bifrontal bone graft is split into slats approximately 1.5 cm wide, weakened on their cranial undersurface by transversely oriented kerf channels, and then greenstick fractured to achieve the desired form. The individual slats are attached with resorbable plates, cork screws, or sutures to the superior orbital rim and to one another to achieve the final desired symmetry.
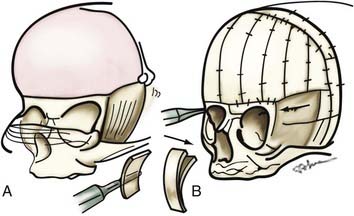
Bilateral Coronal Synostosis
Bilateral coronal synostosis is characterized by ridging of the coronal suture, flattening of the caudal portion of the frontal bones and supraorbital ridges, and bulging of the cephalad portion of the frontal bones (Fig. 182-14). The occiput is flattened, and the squamous portion of the temporal bones is unusually prominent. The vertex of the skull is more anteriorly situated than normal radiographically, bilateral harlequin abnormalities are present in the orbits, and basal CT demonstrates shortened anterior cranial fossae and bilaterally narrowed sphenoid petrosal angles.
Operative Technique in Children Younger Than 1 Year
Because the abnormalities of coronal synostosis are situated both anteriorly and posteriorly within the skull, a different approach to patient positioning is elected. The modified prone position, in which the anterior and posterior portions of the skull are exposed simultaneously, is optimal; however, use of this position requires certain precautions.80 The presence of craniovertebral anomalies and the patient’s ability to tolerate this position must be assessed preoperatively. Cervical spine radiographs with flexion and extension lateral views and basal CT are routinely performed to identify any patient who might be at risk for neurological injury. If anomalies such as the Arnold-Chiari malformation and instability of the cervical spine are identified, a two-stage approach, with the patient alternately in a supine and then a prone position, should be elected.
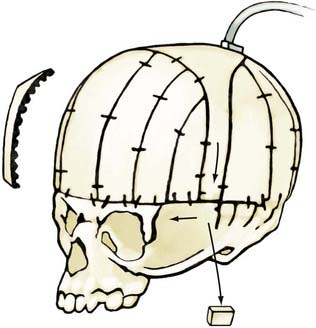
If no vertebral abnormalities exist, the patient’s head is placed on a well-padded chin support (usually a combination of a beanbag and a well-cushioned anterior portion of a Philadelphia collar for stabilization).81 A coronal incision is performed, and anterior and posterior supraperiosteal dissection is carried out (Fig. 182-15). The posterior limit is the foramen magnum, and the anterior limit is the lateral portion of the orbital rims at the level of the frontal zygomatic suture bilaterally. Frontal and parieto-occipital bone grafts are elevated, leaving bone over the vertex of the skull and two lateral struts of bone extending from the vertex to the base of the skull. Subperiosteal dissection is performed posteriorly in the occiput. “Barrel stave” osteotomies are performed in the occipital region,82 with the individual bone segments fractured posteriorly to increase the potential space in the posterior skull for the repositioned brain and dural envelope. The superior orbital rims are elevated bilaterally to the level of the frontozygomatic suture, contoured, angled forward (at the frontozygomatic suture), and fixed in the advanced position. The squamous portion of the temporal bone is elevated with the overlying temporalis muscle and advanced and attached to the superior orbital rims. The more basal temporal bone undergoes osteotomy and infracture to reduce the abnormal convexity in the region. An ICP monitor is placed in the right parietal bone, off the midline. The struts extending from the vertex of the skull in the parietal region to the basal temporal region are then divided, shifted posteriorly, and reduced in height (approximately 1 cm) to change the position of the vertex of the skull, flatten the frontal contour, and reduce the height of the skull.
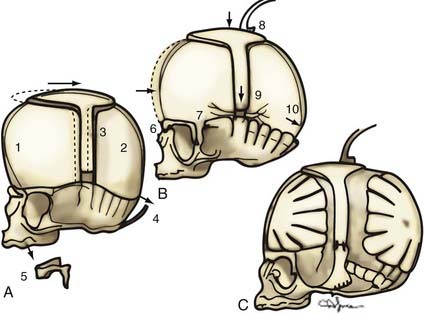
Operative Technique in Children Older Than 3 Years
A zigzag coronal incision and a supraperiosteal dissection plane are carried out, similar to the technique used for younger children. After elevation of the bifrontal and parietal occipital bone grafts, barrel stave osteotomies are performed in the occipital region (Fig. 182-16). The orbital rims are elevated and advanced as a unit, extending into the frontal process of the zygoma. The two C-shaped rings of bone are advanced and stabilized.83–85 The squamous temporal bone and temporalis muscle are elevated as a composite flap and advanced to attach to the posterior border of the advanced orbital rim. The height of the skull is again reduced, and the ICP is monitored as in younger children; however, patients older than 3 years require a much more prolonged period of accommodation than younger children do, and less height reduction is ordinarily achieved. Correction greater than 1 cm is unusual in children older than 3 years, whereas 1 to 1.2 cm of correction is usually well tolerated in children younger than 1 year.
Sagittal Synostosis
Sagittal synostosis results in a skull shape characterized by biparietal narrowing, ridging of the sagittal suture, and bilateral bulging of the frontal or occipital region or both (Fig. 182-17). Radiographically, fusion of the sagittal suture is evident. An individually tailored surgical technique is necessary to address the most salient deformities. Usually, one of three varieties of sagittal synostosis predominates: anterior compensation, posterior compensation, and “golf tee” deformities. The anterior type can be treated with the standard π (pi) procedure, with optional supplementary frontal craniotomy for reshaping and dura plication.86 The posterior variety needs a posterior π procedure, but this often requires elevation of the parietal region as well. This is a variety of the golf tee deformity, which may also involve frontal bossing and requires surgery in a modified prone position.
Operative Technique in Children Younger Than 1 Year
As in children with bilateral coronal synostosis, patients with complete sagittal synostosis frequently have simultaneous evidence of deformity in the anterior and posterior skull. The most complete correction of these abnormalities requires that the patient be placed in the modified prone position80; however, basal CT and cervical spine radiography should be performed beforehand to rule out cranial vertebral anomalies such as Arnold-Chiari type I malformation, cervical spine instability, or both. If anomalies are evident, a two-stage approach is more prudent.
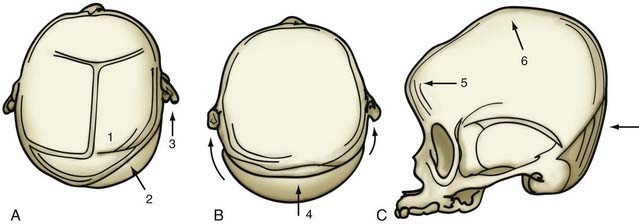
If no cranial vertebral abnormalities exist, the patient’s head is placed on a well-padded chin support, and a zigzag coronal incision is carried out. The currently used procedure is a modification of the previously described π procedure (Fig. 182-18).86,87 Anterior and posterior scalp flaps are dissected in the supraperiosteal plane, and bifrontal, individual parietal, and occipital bone grafts are elevated. Because there are no significant orbital abnormalities in sagittal synostosis, no orbital dissection is performed. However, because of the characteristic hollowing seen in this area, lateral barrel stave osteotomies are carried out temporally through the squamous portion of the temporal bone and temporalis muscle. These bone segments are then outfractured to increase projection in this region. The skull is shortened anteriorly to posteriorly by first reshaping the occipital bone to a flatter plane and then attaching the reshaped bone posteriorly to the occiput. The paramedian parietal bone adjacent to the sagittal sinuses is elevated, the ridge contoured, and the bone attached posteriorly to the occipital bone. The bifrontal bone graft is ordinarily reshaped by radial osteotomy to correct abnormal convexity bilaterally in the supralateral frontal bones. To angulate the forehead posteriorly and resolve abnormal frontal prominence, a segment of the median parietal bone is removed, usually approximately 1 cm long. When this is accomplished, an overlap of the lateral frontal bone is seen over the superolateral orbital rim. A triangular wedge of bone is removed over this area to allow precise alignment with the superior orbital rim. The parietal bones are then reshaped with radial osteotomies to give them a more convex form. Two paramedian neosagittal sutures are created by tailoring the remaining bone grafts so that there is a gap of approximately 3 to 10 cm between the midline bone and the parietal bone grafts. To allow relatively free lateral movement of the brain and bone in this region, the parietal bone grafts are sutured only to the underlying dura and not to adjacent bone, resulting in a more rounded skull form.
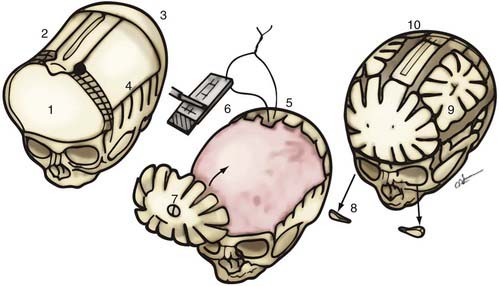
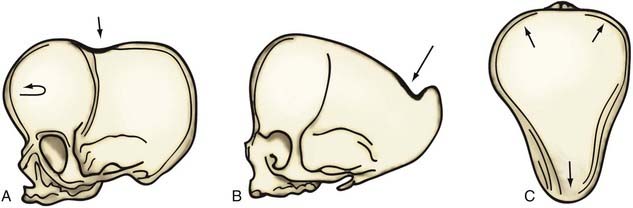
An alternative mode of treatment addresses the most salient deformity—that is, the variety of sagittal synostosis that predominates: anterior compensation (Fig. 182-19A), posterior compensation (Fig. 182-19B), or golf tee deformity (Fig. 182-19C). The anterior type can be treated by the standard π procedure, with an optional supplementary frontal craniotomy with reshaping and dural plication (Fig. 182-20A and B).87 The posterior variety needs a posterior π procedure (Fig. 182-20C and D).
Operative Technique in Children Older Than 3 Years
The child is placed in the modified prone position, and anterior and posterior supraperiosteal scalp dissection, similar to Marchac’s original procedure, is carried out as discussed for younger children.32 Serial bifrontal, bifrontoparietal, biparietal, and right occipital bone grafts are elevated (Fig. 182-21). The occiput convexity is flattened by radial osteotomy and greenstick fracture. However, the parietal bone is reshaped to a more convex form using kerfs on the endocranial surface of the bone and a series of controlled greenstick fractures. Bone grafts harvested from the occiput and anterior parietal region are inserted into the gaps created by lateral remodeling of the parietal bone in the temporal regions. The frontal bone graft is angulated posteriorly to achieve a normal forehead contour. The bone overlapping the anterior parietal paramedian bone is resected in the form of a triangle. Barrel stave osteotomies are performed in the temporal region, creating multiple composite flaps, and are fractured outward. The remodeled bone grafts are secured anterior to posterior and medial to lateral.
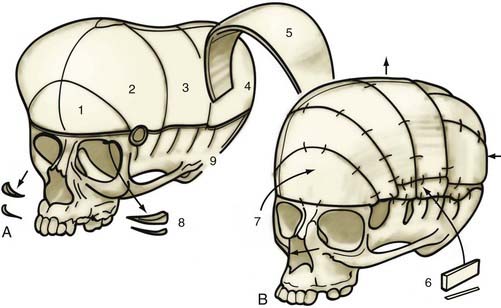
Lambdoid Synostosis
Children with lambdoid synostosis typically have flattening of the parietal and occipital regions ipsilateral to the fused portion of the suture. If synostosis has occurred unilaterally, an asymmetric occiput is seen, characterized by flattening of the occiput ipsilateral to the fused suture and contralateral bulging evident in the parietal occiput region (Fig. 182-22). If bilateral synostosis has occurred, symmetrical flattening of the occiput is seen. If the synostosis is severe, skull deformities in the frontal region, which may include elevation of the vertex of the skull, are evident.
Operative Technique in Children Younger Than 1 Year
Individualization of surgical technique is required; however, most deformities can be satisfactorily corrected with the patient in the prone position and surgical manipulations in the occipital bone.88 If, however, vertex and frontal abnormalities exist, the patient should be placed in the modified prone position, similar to that used for the correction of bilateral coronal synostosis. This discussion is restricted to patients having deformities in the parietal occiput.
A patient with an occipital deformity is placed in the prone position, and a posteriorly “displaced” coronal incision is carried out (Fig. 182-23A). Dissection is performed in the supraperiosteal plane. A biparietal occipital bone graft is elevated in patients with unilateral synostosis or bilateral lambdoid synostosis, with care taken to avoid injuring the transverse sinus. Barrel stave osteotomies are performed in both unilateral and bilateral synostosis; in bilateral lambdoid synostosis with bilateral posterior flattening, barrel stave osteotomies are performed to increase bilateral occipital projection. In patients with unilateral lambdoid synostosis, the occiput is fractured posteriorly, ipsilateral to the fused suture unilaterally, and inwardly on the contralateral bulging side. The biparietal occipital bone graft undergoes radial osteotomy and contouring to achieve symmetry (Fig. 182-23B). In unilateral synostosis, the convex side is made flatter, and the flatter side is made more convex with the use of greenstick fractures. In patients with bilateral lambdoid synostosis, a bilaterally convex occiput is achieved by similar means. The bone is then reattached to the dura and not to adjacent bone.
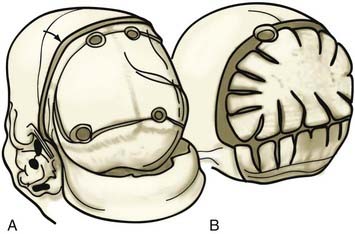
Posterior Synostosis
Posterior deformities include unilateral and bilateral lambdoid synostosis with or without posterior sagittal synostosis or squamosal synostosis. These are rare conditions, and the principal surgical difficulty lies in not being able to maintain the corrected posterior projection. Jane has been using a single microplate attached just above the foramen magnum to correct this projection (Fig. 182-24). The dead space obtained is so large that it is unlikely that significant inward migration could occur.
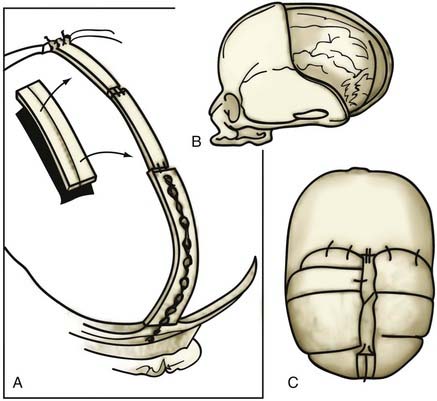
Operative Complications
Although surgical procedures for craniosynostosis are quite safe despite their extensive nature, complications may arise. Problems associated with cranioplasty procedures are blood loss and air embolus.89 Patients undergoing these operative procedures lose blood at a slow rate from the cancellous portion of the bone, particularly if basal skull osteotomies are performed. Adequate preparation for blood replacement and monitoring for blood loss are necessary. Because of low circulating blood volumes in very young patients, it is imperative that attention be paid to achieving good hemostasis intraoperatively and monitoring blood loss postoperatively. The most appropriate place to do so is a highly skilled intensive care unit.
Philosophy of Treatment
For many years, we have advocated a more radical approach to the treatment of craniosynostosis. Early on, it was recognized that simple synostectomy was insufficient and that more extensive procedures produced better results.86 This led to a plea for bilateral procedures for unilateral synostosis.79 The theoretical basis for this clinical approach is the observation that single-suture closure gives rise to a progressive series of compensations that often represent the principal deformity needing treatment.18 Simple synostectomy may suffice for an early sagittal synostosis before frontal bossing or an occipital knob has occurred. An early unilateral coronal synostosis may not (but usually does) need a contralateral frontal correction to achieve an ideal result, but a bilateral coronal synostosis has a brachycephalic deformity that, in our experience, cannot be corrected without extensive cranial remodeling. However, an individualized approach to each deformity is necessary. The surgical techniques discussed in this chapter are not meant to be rigidly adhered to, but they should serve as a framework for treatment in particular patients.
Alden TD, Link Y, Jane JA. Mechanism of premature closure of cranial suture. Childs Nerv Syst. 1999;15:670-675.
Arnaud E, Renier D, Marchac D. Prognosis for mental function in scaphocephaly. J Neurosurg. 1995;83:476-479.
Babler WJ, Persing JA. Experimental alteration of cranial suture growth: effects on the neurocranium, basicranium and midface. In: Dixon AP, Samat BG, editors. Factors and Mechanisms Influencing Bone Growth. New York: Alan R. Liss; 1982:333-345.
Delashaw JB, Persing JA, Broaddus WC, et al. Rules for cranial vault growth. J Neurosurg. 1989;70:159-165.
Gault DT, Renier D, Marchac D, Jones BM. Intracranial pressure and intracranial volume in children with craniosynostosis. Plast Reconstr Surg. 1992;90:377-381.
Jane JA, Persing JA. Neurosurgical treatment of craniosynostosis. In: Cohen MM, editor. Craniosynostosis: Diagnosis and Evaluation of Management. New York: Raven Press; 1986:249-320.
Kapp-Simon KA, Speltz ML, Cunningham ML, et al. Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst. 2007;23:269-281.
Maltese G, Tarnow P, Lauritzen Claes G. Spring-assisted correction of hypotelorism in metopic synostosis. Plast Reconstr Surg. 2007;119:977-984.
Marsh J, Jenny A, Galic M, et al. Surgical management of sagittal synostosis. In: Persing J, Edgerton M, Jane J, editors. Scientific Foundations and Surgical Treatment of Craniosynostosis. Baltimore: Williams & Wilkins; 1989:263-269.
Marsh J, Vannier MW. Cranial base changes following surgical treatment of craniosynostosis. J Neurosurg. 1981;54:601-606.
Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat. 1959;37:351-370.
Moss ML. Growth of the calvaria in the rat: the determination of osseous morphology. Am J Anat. 1954;94:333-362.
Otto AW. Lehrbuch der Pathologischen des Menschen and der Thiere. Berlin: Rucker; 1830.
Persing JA, Babler WD, Winn HR, et al. Age as a critical factor in the success of surgical correction of craniosynostosis. J Neurosurg. 1981;54:601-606.
Persing JA, Babler WF, Nagorsky MJ, et al. Skull expansion in experimental craniosynostosis. Plast Reconstr Surg. 1986;78:594-603.
Posnick JC, Armstrong D, Bite U. Metopic and sagittal synostosis: intracranial volume measurements prior to and after cranio-orbital reshaping in childhood. Plast Reconstr Surg. 1995;96:299-309.
Renier D. Intracranial pressure in craniosynostosis: pre- and postoperative recordings—correlation with functional results. In: Persing JA, Edgerton MT, Jane JA, editors. Scientific Foundations and Surgical Treatment of Craniosynostosis. Baltimore: Williams & Wilkins; 1989:263-269.
Renier D, Sainte-Rose C, Marchac D, Hirsch JF. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57:370-377.
Shillito JJr, Matson DD. Craniosynostosis: a review of 519 surgical patients. Pediatrics. 1968;41:829-853.
Sommerring SF. Vom Baue des Menschlichen, 2nd ed. Leipzig: Voss; 1839.
Speltz ML, Kapp-Simon KA, Cunningham M, et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol. 2004;29:651-668.
Tuite GF, Evanson J, Chong WK, et al. The beaten copper cranium: a correlation between intracranial pressure, cranial radiographs, and computed tomographic scans in children with craniosynostosis. Neurosurgery. 1996;39:691-699.
Virchow R. Ueber den cretinismus, namentlich in Franken: Und euber pathologische Schadelformen. Verh Phys Med Gesane Wurzburg. 1851;2:230-271.
Whittle IR, Johnston IH, Besser M. Intracranial pressure changes in craniostenosis. Surg Neurol. 1984;21:367-372.
1 Sommerring SF. Vom Baue des Menschlichen, 2nd ed. Leipzig: Voss; 1839.
2 Otto AW. Lehrbuch der Pathologischen des Menschen and der Thiere. Berlin: Rucker; 1830.
3 Virchow R. Ueber den cretinismus, namentlich in Franken: Und euber pathologische Schadelformen. Verh Phys Med Gesane Wurzburg. 1851;2:230-271.
4 Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat. 1959;37:351-370.
5 van der Klaauw CJ. Cerebral skull and facial skull. Arch Neerl Zool. 1946;7:1-37.
6 Bolk L. On the premature obliteration of sutures in the human skull. Am J Anat. 1915;17:495-523.
7 Moss ML. Growth of the calvaria in the rat: the determination of osseous morphology. Am J Anat. 1954;94:333-362.
8 Moss ML. Functional anatomy of cranial synostosis. Childs Brain. 1975;1:22-33.
9 Persson KM, Roy WA, Persing JA, et al. Craniofacial growth following experimental craniosynostosis and craniectomy in rabbits. J Neurosurg. 1979;50:187-197.
10 Babler WJ, Persing JA. Experimental alteration of cranial suture growth: effects on the neurocranium, basicranium and midface. In: Dixon AP, Samat BG, editors. Factors and Mechanisms Influencing Bone Growth. New York: Alan R. Liss; 1982:333-345.
11 Persing JA, Morgan EP, Cronin AJ, et al. Skull base expansion: craniofacial effects. Plast Reconstr Surg. 1991;87:1028-1033.
12 Mooney MP, Losken HW, Siegel ML, et al. Development of a strain of rabbits with congenital simple non-syndromic coronal suture synostosis: I. Breeding demographics, inheritance pattern, and craniofacial anomalies. Cleft Palate Craniofac J. 1994;31:1-7.
13 Mooney MP, Losken HW, Siegel ML, et al. Development of a strain of rabbits with congenital simple non-syndromic coronal suture synostosis: II. Somatic and craniofacial growth patterns. Cleft Palate Craniofac J. 1994;31:8-16.
14 Opperman LA, Sheen R, Persing JA, et al. In the absence of periosteum, transplanted fetal and neonatal rat coronal sutures resist osseous obliteration. J Craniofac Surg. 1994;5:327-332.
15 Opperman LA, Sweeney TM, Redmon J, et al. Tissue interactions with underlying dura mater inhibit osseous obliteration of developing cranial sutures. J Dev Dynam. 1993;198:312-322.
16 Marsh J, Vannier MW. Cranial base changes following surgical treatment of craniosynostosis. J Neurosurg. 1981;54:601-606.
17 Delashaw JB, Persing JA, Broaddus WC, et al. Rules for cranial vault growth. J Neurosurg. 1989;70:159-165.
18 Jane JA, Persing JA. Neurosurgical treatment of craniosynostosis. In: Cohen MM, editor. Craniosynostosis: Diagnosis and Evaluation of Management. New York: Raven Press; 1986:249-320.
19 Drake DB, Persing JA, Berman DE, Ogle RC. Calvarial deformity regeneration following subtotal craniectomy for craniosynostosis: a case report and theoretical implications. J Craniofac Surg. 1993;4:85-90.
20 Persing JA, Babler WF, Nagorsky MJ, et al. Skull expansion in experimental craniosynostosis. Plast Reconstr Surg. 1986;78:594-603.
21 Maltese G, Tarnow P, Lauritzen Claes G. Spring-assisted correction of hypotelorism in metopic synostosis. Plast Reconstr Surg. 2007;119:977-984.
22 Cohen MMJr. Craniosynostosis: Diagnosis, Evaluation, and Management. New York: Raven Press; 1986. 1-606
23 Brenner DJ, Hall EJ, Phil HD. Computed tomography—an increasing source of radiation exposure. N Engl J Med. 2007;357:2277-2284.
24 Persing JA, Babler WD, Winn HR, et al. Age as a critical factor in the success of surgical correction of craniosynostosis. J Neurosurg. 1981;54:601-606.
25 Marsh J, Jenny A, Galic M, et al. Surgical management of sagittal synostosis. In: Persing J, Edgerton M, Jane J, editors. Scientific Foundations and Surgical Treatment of Craniosynostosis. Baltimore: Williams & Wilkins; 1989:263-269.
26 Heller J, Knoll B, Gabbey J, et al. Intracranial volume and cephalic index outcomes for total calvarial reconstruction among non-syndromic sagittal synostosis patients. Plast Reconstr Surg. 2008;121:187-195.
27 Feinsod M, Davis NL. Unlocking the brain: attempts to improve mental function of microcephalic retarded children by “craniotomy. ”. Neurosurgery. 2003;53;:723-730.
28 Eide PK, Helseth E, Due-Tonnessen B, Lundar T. Assessment of continuous intracranial pressure recordings in childhood craniosynostosis. Pediatr Neurosurg. 2002;37:310-320.
29 Fok H, Jones BM, Gault DG, et al. Relationship between intracranial pressure and intracranial volume in craniosynostosis. Br J Plast Surg. 1992;45:394-397.
30 Gault DT, Renier D, Marchac D, Jones BM. Intracranial pressure and intracranial volume in children with craniosynostosis. Plast Reconstr Surg. 1992;90:377-381.
31 Martinez-Lage JF, Almao L, Poza M. Raised intracranial pressure in minimal forms of craniosynostosis. Childs Nerv Syst. 1999;15:11-15.
32 Renier D. Intracranial pressure in craniosynostosis: pre- and postoperative recordings—correlation with functional results. In: Persing JA, Edgerton MT, Jane JA, editors. Scientific Foundations and Surgical Treatment of Craniosynostosis. Baltimore: Williams & Wilkins; 1989:263-269.
33 Stavrou P, Sgouros S, Willshaw HE, et al. Visual failure caused by raised intracranial pressure in craniosynostosis. Childs Nerv Syst. 1997;13:64-67.
34 Tamburrini G, Caldarelli M, Massimi L, et al. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Childs Nerv Syst. 2005;21:913-921.
35 Thompson DN, Harkness W, Jones B, et al. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst. 1995;11:269-275.
36 Thompson DN, Malcolm GP, Jones BM, et al. Intracranial pressure in single-suture craniosynostosis. Pediatr Neurosurg. 1995;22:235-240.
37 Tuite GF, Chong WK, Evanson J, et al. The effectiveness of papilledema as an indicator of raised intracranial pressure in children with craniosynostosis. Neurosurgery. 1996;38:272-278.
38 Tuite GF, Evanson J, Chong WK, et al. The beaten copper cranium: a correlation between intracranial pressure, cranial radiographs, and computed tomographic scans in children with craniosynostosis. Neurosurgery. 1996;39:691-699.
39 Whittle IR, Johnston IH, Besser M. Intracranial pressure changes in craniostenosis. Surg Neurol. 1984;21:367-372.
40 Arnaud E, Renier D, Marchac D. Prognosis for mental function in scaphocephaly. J Neurosurg. 1995;83:476-479.
41 Renier D, Sainte-Rose C, Marchac D, Hirsch JF. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57:370-377.
42 Lekovic GP, Bristol RE, Rekate HL. Cognitive impact of craniosynostosis. Semin Pediatr Neurol. 2004;11:305-310.
43 Netherway DJ, Abbott AH, Anderson PJ, David DJ. Intracranial volume in patients with nonsyndromal craniosynostosis. J Neurosurg. 2005;103:137-141.
44 Posnick JC, Armstrong D, Bite U. Metopic and sagittal synostosis: intracranial volume measurements prior to and after cranio-orbital reshaping in childhood. Plast Reconstr Surg. 1995;96:299-309.
45 Sgouros S, Hockley AD, Goldin JH, et al. Intracranial volume change in craniosynostosis. J Neurosurg. 1999;91:617-625.
46 Sen A, Dougal P, Padhy AK, et al. Technetium-99m-HMPAO spect cerebral blood flow study in children with craniosynostosis. J Nucl Med. 1995;36:394-398.
47 Shimoji T, Tomiyama N. Mild trigonocephaly and intracranial pressure: report of 56 patients. Childs Nerv Syst. 2004;20:749-756.
48 David LR, Genecov DG, Camastra AA, et al. Positron emission tomography studies confirm the need for early surgical intervention in patiens with single-suture craniosynostosis. J Craniofac Surg. 1999;10:38-42.
49 Aldridge K, Marsh JL, Govier D, Richtsmeier JT. Central nervous system phenotypes in craniosynostosis. J Anat. 2002;201:31-39.
50 Aldridge K, Kane AA, Marsh JL, et al. Relationship of brain and skull in pre- and postoperative sagittal synostosis. J Anat. 2005;206:373-385.
51 Lin HJ, Ruiz-Correa S, Shapiro LG, et al. Predicting neuropsychological development from skull imaging. Conf Proc IEEE Eng Med Biol Soc. 2006;1:3450-3455.
52 Warschausky S, Angobaldo J, Kewman D, et al. Early development of infants with untreated metopic craniosynostosis. Plast Reconstr Surg. 2005;115:1518-1523.
53 Ruiz-Correa S, Starr JR, Lin JH, et al. Severity of skull malformation is unrelated to presurgery neurobehavioral status of infants with sagittal synostosis. Cleft Palate Craniofac J. 2007;44:548-554.
54 Bottero L, Lajeunie E, Arnaud E, et al. Functional outcome after surgery for trigonocephaly. Plast Reconstr Surg. 1998;102:952-958.
55 Ingraham FD, Matson DD. Craniosynostosis. In: Neurosurgery of Infancy and Childhood. Springfield, IL: Charles C. Thomas; 1954. 83-104
56 Shillito JJr, Matson DD. Craniosynostosis: a review of 519 surgical patients. Pediatrics. 1968;41:829-853.
57 Kapp-Simon KA, Speltz ML, Cunningham ML, et al. Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst. 2007;23:269-281.
58 Speltz ML, Kapp-Simon KA, Cunningham M, et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol. 2004;29:651-668.
59 Becker DB, Petersen JD, Kane AA, et al. Speech, cognitive, and behavioral outcomes in nonsyndromic craniosynostosis. Plast Reconstr Surg. 2005;116:400-407.
60 Bellew M, Chumas P, Mueller R, et al. Pre- and postoperative developmental attainment in sagittal synostosis. Arch Dis Child. 2005;90:346-350.
61 Gewalli F, Guimaraes-Ferreira JP, Sahlin P, et al. Mental development after modified pi procedure: dynamic cranioplasty for sagittal synostosis. Ann Plast Surg. 2001;46:415-420.
62 Kapp-Simon KA, Figueroa A, Jocher CA, Schafer M. Longitudinal assessment of mental development in infants with nonsyndromic craniosynostosis with and without cranial release and reconstruction. Plast Reconstr Surg. 1993;92:831-839.
63 Kapp-Simon KA, Lerous B, Cunningham M, Speltz ML. Multisite study of infants with single-suture craniosynostosis: preliminary report of presurgery development. Cleft Palate Craniofac J. 2005;42:377-384.
64 Panchal J, Amirsheybani H, Gurwitch R, et al. Neurodevelopment in children with single-suture craniosynostosis and plagiocephaly without synostosis. Plast Reconstr Surg. 2001;108:1492-1498.
65 Speltz ML, Kapp-Simon K, Collett B, et al. Neurodevelopment of infants with single-suture craniosynostosis: presurgery comparisons with case-matched controls. Plast Reconstr Surg. 2007;119:1874-1881.
66 Starr JR, Kapp-Simon KA, Cloona YK, et al. Presurgical and postoperative assessment of the neurodevelopment of infants with single-suture craniosynostosis: comparison with controls. J Neurosurg. 2007;107(2 suppl):103-110.
67 Toth K, Collett B, Kapp-Simon KA, et al. Memory and response inhibition in young children with single-suture craniosynostosis. Child Neuropsychol. 2007;26(Sep):1-14. [Epub ahead of print]
68 Da Costa AC, Walters I, Savarirayan R, et al. Intellectual outcomes in children and adolescents with syndromic and nonsyndromic craniosynostosis. Plast Reconstr Surg. 2006;118:175-181.
69 Boltshauser E, Ludwig S, Dietrich F, Landolt MA. Sagittal craniosynostosis: cognitive development, behavior, and quality of life in unoperated children. Neuropediatrics. 2003;34:293-300.
70 Kapp-Simon KA. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac J. 1998;35:197-2003.
71 Magge SN, Westerveld M, Pruzinsky T, Persing JA. Long-term neuropsychological effects of sagittal craniosynostosis on child development. J Craniofac Surg. 2002;13:99-104.
72 Shipster C, Hearst D, Somerville A, et al. Speech, language and cognitive development in children with isolated sagittal synostosis. Dev Med Child Neurol. 2003;45:34-43.
73 Sidoti EJJr, Marsh JL, Marty-Grames L, Noetzel MJ. Long-term studies of metopic synostosis: frequency of cognitive impairment and behavioral disturbances. Plast Reconstr Surg. 1996;97:276-281.
74 Virtane R, Korhonen T, Fagerholm J, Viljanto J. Neurocognitive sequelae of scaphocephaly. Pediatrics. 1999;103:791-795.
75 Alden TD, Link Y, Jane JA. Mechanism of premature closure of cranial suture. Childs Nerv Syst. 1999;15:670-675.
76 Persing JA, Luce C. Remodeling techniques for immature and mature cranial vault bone: technical note. J Craniofac Surg. 1990;1:145-149.
77 Delashaw JB, Persing JA, Parks TS, et al. Surgical approach for the correction of metopic synostosis. Neurosurgery. 1986;19:228-234.
78 Persing JA, Mayer P, Spinelli H, et al. Prevention of temporal hollowing following fronto-orbital advancement for craniosynostosis. J Craniofac Surg. 1994;5:271-274.
79 Jane JA, Park TS, Zide BA, et al. Alternative techniques in the treatment of unilateral coronal synostosis. J Neurosurg. 1984;61:550-556.
80 Park TS, Haworth CS, Jane JA, et al. Modified prone position for cranial remodeling procedures in children with craniofacial dysmorphism: a technical note. Neurosurgery. 1985;16:212-214.
81 Park TS, Broaddus WC, Harris M, et al. Vacuum-stiffened bean bag for cranial remodeling procedures in the modified prone position. J Neurosurg. 1989;71:623-625.
82 Persing JA, Edgerton MT, Park TS, et al. Barrel stave osteotomy for correction of turribrachycephaly craniosynostosis deformity. Ann Plast Surg. 1987;18:488-493.
83 Marchac D, Renier D. Craniofacial Surgery for Craniosynostosis. Boston: Little Brown; 1982.
84 Persing JA, Jane JA, Park TS, et al. Floating C-shaped orbital osteotomy for orbital rim advancement in craniosynostosis: preliminary report. J Neurosurg. 1990;72:22-26.
85 Whitaker LA, Schut L, Kerr LP. Early surgery for isolated craniofacial dysostosis: improvement and possible prevention of increasing deformity. Plast Reconstr Surg. 1977;60:575-581.
86 Jane JA, Edgerton MT, Futrell JW, et al. Immediate correction of sagittal synostosis. J Neurosurg. 1978;49:705-710.
87 Vollmer DG, Jane JA, Park TS, et al. Variants of sagittal synostosis: strategies for correction. J Neurosurg. 1984;61:557-562.
88 Persing JA, Delashaw JB, Jane JA, et al. Lambdoid synostosis: surgical consideration. Plast Reconstr Surg. 1988;81:852-860.
89 Harris MM, Stratford MA, Rowe RW, et al. Venous air embolism and cardiac arrest during craniectomy in a supine infant. Anesthesiology. 1986;65:547-550.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 182 Craniosynostosis
Although skull shape irregularity has been recognized since antiquity, the study of abnormal skull growth related to craniosynostosis had its scientific origin in the late 1700s. Sommerring1 noted that bone growth in the skull occurred primarily at suture lines and that when this growth site was prematurely bridged with bone, an abnormal skull shape developed. This was characterized by reconstruction of the skull’s growth in a plane perpendicular to the plane of the fused suture. Similar observations were made by Otto2 in 1830 and Virchow3 in 1821. It was Virchow’s widely publicized treatise on skull deformity that added impetus to the scientific study of abnormal skull form in craniosynostosis. He observed, as had earlier investigators, that skull growth occurred at suture lines in the skull and that when these suture lines were prematurely fused, skull deformity developed. Restriction of growth adjacent to the suture occurred, but compensatory growth occurred elsewhere in the skull to accommodate the growing brain. This understanding of normal and abnormal skull growth served as the basis for understanding skull abnormalities for the next 100 years (Fig. 182-1).
In the mid-20th century, the primary role of the cranial vault suture in the development of craniosynostosis skull deformities was questioned by Moss4 and van der Klaauw.5 Moss noted that surgeons operating on the skulls of children presumed to have craniosynostosis would occasionally find patent cranial vault sutures, despite what appeared to be typical craniosynostosis skull deformities (see Fig. 182-1).6 Also, he recognized that there were some characteristic cranial base deformities associated with individual forms of craniosynostosis, and because the cranial base matures embryologically before the remainder of the cranial vault, he suggested that the cranial base abnormality was the primary pathologic process and the cranial vault suture abnormality (fusion) was secondary.4 Testing this hypothesis in the laboratory, Moss7 removed a normal cranial vault suture and found that doing so did not affect overall skull length. This indicated that the cranial vault suture, unlike the epiphysis of a long bone, does not serve as a growth site that pushes the bone ends apart but is a passive recipient of growth influences.
The amount of bone deposited at the cranial vault suture is related to the strains that influence it. Brain enlargement, Moss8 believed, was the primary source of these tensile strains that caused the suture to deposit bone. This is known as the functional matrix theory, in which the functional enlargement or development of an organ system is the primary force in changing its overall shape and determining its final form. Even though brain enlargement is clearly the engine of skull remodeling, the precise role the vault suture plays in the development of the skull pathology associated with craniosynostosis must be determined by direct manipulation of growth at the suture.7
In 1979 Persson and colleagues9 reported that in animals, selective restriction of an individual cranial vault suture’s growth resulted in skull deformities that closely mimic the clinical condition of craniosynostosis involving the same cranial vault suture in humans. Moreover, cranial base and even facial deformities develop secondary to the cranial vault suture restrictions.10,11 This observation indicates that cranial vault suture pathology may be primary in the development of craniosynostosis skull deformities leading to cranial base and facial deformity. Subsequently, Mooney and coworkers12,13 studied an animal model of congenital craniosynostosis in which cranial vault suture, vault, and cranial base abnormalities closely resembled the findings of Babler and Persing10 on cranial suture restriction. In addition, the developmental studies of Opperman and colleagues14,15 demonstrated the significant influence of mesenchymal tissues, in particular the dura and the periosteum at the suture, on the maintenance of patency of cranial vault sutures during development.
Nonsyndromic and Syndromic Craniosynostosis
Researchers have found clinical evidence that cranial vault suture, not cranial base, pathology is of primary importance in the development of skull shape pathology in nonsyndromic craniosynostosis. Preoperative and postoperative computed tomography (CT) shows that surgical treatment of the cranial vault alone results in improvement of not only cranial vault but also cranial base pathology.16 This indicates that removing the abnormal influence in the cranial vault suture may improve cranial base shape. Further analysis of skull growth after simple suture closure has revealed a predictable pattern based on certain rules. The ability to predict and understand the stereotypical deformity from observation of the vault suture suggests its primary role.17,18 The dura clearly plays a role in this, as demonstrated by Drake and coworkers,19 who found that resected fused and nonfused cranial suture elements redevelop following the removal of cranial bone topographically connected to the fused and nonfused sutures. Distraction devices, such as those developed by Ilizarov for the distraction of extremity bones and by McCarthy for mandibular hypoplasia, are effective in elongating bone in animals. Using spring-like expanders, Persing and associates20 internalized the distraction of skull bone experimentally, and subsequently Maltese and colleagues21 employed this approach in the management of human craniosynostosis. The advantage of the distraction and spring techniques is that bone elongation is a gradual process, with minimal surgical intervention and operative time. One disadvantage is that if the cranial bone is already deformed, the irregularly shaped bone is advanced or expanded from the osteotomy site. In addition, the removal of spring and bone distraction devices requires an operative procedure. Similar to implanted metal fixation plates and screws in the skull, if left in place too long, these metal devices can migrate intracranially, with progressive resorption of the endocranial surface of the skull bone and positioning of bone on the ectocranial surface as the skull and brain enlarge. Concern about transcranial migration has been the main impetus for the avoidance of metallic fixation plates and screws in young children. Resorbable plates and screws, as well as suture material, have largely supplanted titanium devices in patients younger than 3 years.
Additional studies are being done to document skull growth changes by surgical manipulation, in particular by the use of mechanical devices to control or enhance growth influences.17,20 In the future, these devices may be used clinically to prolong corrective growth influences postoperatively.
Syndromic craniosynostosis is much less common and appears to be a more generalized disorder of mesenchymal development that may represent abnormalities in homeobox genes such as Runx2 or CBFA-δ. Crouzon’s syndrome occurs in 1 in 25,000 live births, and this bilateral coronal synostosis is frequently associated with exorbitism and midface hypoplasia. Apert’s syndrome occurs in 1 in 100,000 live births; the features of brachycephaly due to coronal synostosis and midface hypoplasia are evident, but extremity syndactylies are also characteristic. There are more than 64 known craniofacial syndromes associated with craniosynostosis22; however, the cause of craniosynostosis is still unknown, although in some cases, a genetic influence is undeniable. In the syndromic forms of craniosynostosis, autosomal dominant inheritance patterns are the general rule for the more common syndromes such as Apert’s, Crouzon’s, and Pfeiffer’s; autosomal recessive disorders (e.g., Carpenter’s syndrome, characterized by abnormal skull shape deformities and polydactyly) do not commonly occur.
Diagnosis
Nonsyndromic craniosynostosis involving a single vault suture is ordinarily diagnosed by the observation of a typically deformed skull shape, with radiographs serving as confirmatory evidence. We have modified the Virchow hypothesis of skull deformity to explain the skull shapes associated with individual forms of craniosynostosis.17,18 These patterns of skull abnormalities can be explained by invoking four tenets: (1) cranial vault bones directly adjacent to the prematurely fused vault suture act as a single bone plate, with reduced growth potential along all margins of that plate (Fig. 182-2A); (2) asymmetric bone deposition occurs at vault sutures along the perimeter of the bone plate, with increased bone deposition occurring at the suture margin located farther away from the plate (Fig. 182-2B); (3) the nonperimeter sutures in line with the fused suture deposit bone symmetrically at their sutural edges (Fig. 182-2C); and (4) perimeter and sutures (in line) abutting the prematurely fused suture compensate to a greater degree than distant sutures (Fig. 182-2D). The application of these principles to particular types of synostosis is as follows (Fig. 182-2E to H) : Figure 182-2E represents sagittal synostosis, in which the coronal and lambdoid sutures show asymmetric growth, and the metopic suture shows symmetrical growth. Figure 182-2F depicts coronal synostosis, with asymmetric growth at the metopic, lambdoid, and sagittal sutures and symmetrical growth at the unfused coronal suture. Figure 182-2G illustrates metopic synostosis, with symmetrical growth at the sagittal suture and asymmetric growth at the coronal sutures. Figure 182-2H depicts lambdoid synostosis, with a fused lambdoid suture and bilateral growth at the lambdoid, unilateral at the sagittal, and unilateral at the squamosal sutures, displacing the ear downward rather than forward as in positional posterior plagiocephaly.
Figure 182-E2 contd E, Sagittal synostosis. F, Coronal synostosis. G, Metopic synostosis. H, Lambdoid synostosis.
(E-H, Courtesy of Dr. Michael McKisic.)
There is controversy regarding the advisability of CT scans in patients being evaluated for craniosynostosis. It has been reported that CT scans performed early in infancy are associated with an increased likelihood of malignancy later in life,23 although the concern for malignancy is greatest in children undergoing thoracoabdominal scans and in those scanned earlier in life.
Operative Timing and Approaches
A common question asked by parents is whether the condition is progressive—in other words, will it “look worse” with time? This is particularly pertinent in sagittal synostosis, where the answer appears to be affirmative. Serial CT imaging has shown that normal growth of the skull tends toward increasing roundness in the first year of life. Sagittal synostosis results in a progressively longer skull, but because the width also increases, the shape does not change as it should, resulting in a marked increase in the deformity.24
Experimental evidence demonstrates that improved skull form is achieved with an operation before the human equivalent of 6 months of age.25 In clinical studies, more significant degrees of improvement were noted in patients who underwent surgery when younger than 6 months compared with those having surgery after 6 months of age. Correction of the cranial base deformities established in unilateral coronal synostosis was also more successful if surgery was performed early.16
The influence of surgical methods is not yet clearly defined; however, limited craniectomy procedures of few sutures appear to be beneficial in very young patients with mild deformities, whereas more comprehensive approaches to established skull deformities are more effective in older children and in children of any age with more severe deformities. Objective documentation of these approaches is not yet widespread, but Marsh and coworkers25 compared preoperative and postoperative skull shape following a limited craniectomy procedure versus a whole-vault cranioplasty approach for sagittal synostosis; they found that skull deformity was reduced with both craniectomy and cranioplasty procedures. Heller and associates26 showed a normal cephalic index in 100% of patients undergoing whole-vault cranioplasty; however, the skull shape returned to normal only in patients who underwent the more extensive cranioplasty procedure. Additional data are required to confirm initial impressions, but they are consistent with the hypothesis that early rapid growth of the brain shapes the skull (Fig. 182-3).
Intracranial Pressure, Cognition, and Behavior
Treatment of brain deficiency by surgical attack on the deformed skull has an inglorious history dating to the second half of the 19th century. Many luminaries of the early years of neurosurgery were caught up in an international wave of unscientific enthusiasm for linear craniectomy as a therapy to “unlock” the brains of severely impaired, microcephalic children. The benefits were imaginary, and the mortality rates were appalling. A passionate campaign led by Abraham Jacobi, patriarch of the specialty of pediatrics and founder of the American Academy of Pediatrics, and supported by Harvey Cushing, among others, eventually drove the surgical treatment of mental retardation into deserved obscurity (see Feinsod and Davis27 for the full story). When craniosynostosis later emerged as a distinct diagnostic entity, and as surgical interventions became safe and effective from a cosmetic standpoint, the question of the relationship between skull deformity and brain development arose once again, and it has proved resistant to simple answers.
Elevated intracranial pressure (ICP) is a well-recognized feature of the syndromic forms of craniosynostosis, but it can be documented in a fraction of cases of single-suture synostosis as well.28–40 Generally it subsides after surgical treatment.36,39,41 ICP monitoring has been proposed as an adjunct to surgical decision-making in cases for which cosmetic indications are not compelling.49 The most extensive experience with ICP monitoring comes from the Hopital des Enfants Malades in Paris.30,32,38,40 In a 1989 report of 358 ICP recordings in children with craniosynostosis, Renier32 took ICP recorded during slow-wave sleep as “baseline.” In the absence of valid age-adjusted, normative ICP data, baseline measurements less than 10 torr were defined as normal. Measurements above 15 torr were considered abnormal, and measurements between 10 and 15 torr were considered borderline. Elevated ICP was recorded in 8% of cases of sagittal synostosis, 6% of metopic synostosis, and 12% of unilateral coronal synostosis. It was more prevalent in patients with multiple suture involvement and among syndromic cases. Patients who presented after 1 year of age had much higher rates of intracranial hypertension. Likewise, patients who presented after 1 year of age had lower developmental quotients (DQs) or intelligence quotients (IQs), and there was an inverse correlation between preoperative ICP and DQ or IQ.
The generalization of these data in support of early surgical treatment is difficult to accept. Patients were selected for ICP monitoring for unstated reasons, and the factors determining early and late presentation were unknown as well. It is possible that the parents of the older patients found their children’s deformities acceptable but brought them to medical attention because of developmental or behavioral issues—a selection bias that may have enriched the late presenting group with low DQs and IQs.40 Another possibility is that late presenting cases had experienced obstacles to access to care based on low socioeconomic status or low parental intelligence. This analysis has also been criticized for the interchangeable use of DQ and IQ data, because longitudinal studies have shown that DQ scores above the range of frank retardation are only weakly predictive of IQ later in life.40,42 The association of single-suture craniosynostosis with intracranial hypertension is further clouded by mechanistic questions. Perhaps contrary to expectations, measurements of cranial volume using CT have documented normal or increased volumes among infants with single-suture craniosynostosis compared with age-matched controls.43–45 The correlation between intracranial hypertension and lower than normal cranial volume seems to be quite poor, and the measurement of normal or even increased cranial volume does not eliminate the possibility of elevated ICP.29,30 As is true among patients with syndromic craniosynostosis, mechanisms other than craniocerebral disproportion, such as cerebral venous insufficiency, may be important.
A few studies have reported focal regions of hypoperfusion46,47 or hypometabolism48 subjacent to abnormal sutures among small numbers of selected infants with single-suture craniosynostosis. In most cases these abnormalities resolved after surgery. The prevalence of such focal physiologic disturbances cannot be estimated. The natural history and functional significance of such disturbances are unknown.
Recent investigations employing sophisticated methods of image analysis have begun to attempt to relate calvarial deformities to deformation of the underlying neuroanatomic structures and to development data.49–53 Efforts to demonstrate correlations between preoperative skull morphology and preoperative development testing have yielded contradictory findings.51–54 Distinctive alterations in cortical and subcortical brain morphology in association with sagittal and metopic synostosis have been demonstrated on magnetic resonance imaging in comparison with age-matched controls.49 Preoperative and postoperative studies of infants with sagittal synostosis indicate that these distortions of brain morphology do not revert toward normal even after successful surgical correction of the calvarial deformity.50
Since the beginning of the modern era of surgical treatment of craniosynostosis, there have been concerns about the intellectual and behavioral prognosis of affected children,55,56 leading to many detailed observational studies. Kapp-Simon and others have reviewed this literature lucidly.57–59 Points of observation fall into three time periods: at presentation in infancy, early in the postoperative period, and at school age or later. Observations in the first two periods are necessarily developmental. In the last period, intelligence can be tested, and behavioral disturbances and learning disabilities can be recognized. The standard instruments, typically the Bayley Scales of Infant Development and the Wechsler Intelligence Scales for Children—Revised, have age- and gender-adjusted norms, but some studies have employed matched community controls or sibling controls, with particular implications for the interpretation of results. In all three periods, the rates of true mental retardation (DQ or IQ more than 2 standard deviations below the mean) are probably no greater among children with single-suture synostosis than in the general population. Most investigators have found that in study groups of infants with single-suture involvement, the distributions of preoperative developmental test scores are normal or shifted downward to variable degrees, with variable statistical significance in comparison to normative data.52,59–66 In the early postoperative period, aggregate test scores are generally stable,61,62,66,67 although some studies have claimed improvements over preoperative scores that are not enjoyed by untreated patients.60 Later in childhood the differences between patients and unaffected children become more vivid: among patients, mean full-scale IQ scores are within the normal range but generally lower than among controls,68 and a growing fraction of patients are diagnosed with speech, language, or learning disabilities and behavioral disturbances. The literature suggests that between 35% and 50% of children with single-suture synostosis, regardless of treatment status, can be expected to exhibit some degree of cognitive or behavioral disability in the school years.54,59,69–74
Clinical research will never directly address the question of the effect of surgical treatment on the long-term cognitive and behavioral prognosis for children with single-suture craniosynostosis. A randomized trial cannot be conducted. Although there may be equipoise on the question of cognitive and behavioral outcomes, the contemporary standard of care dictates the treatment of almost all patients for cosmetic indications. Observational studies have drawn conflicting inferential conclusions based on the presence or absence of statistical correlations between treatment status or age at treatment and various outcomes of interest.32,40,54,65,72 A host of factors, known and unknown, determine whether and when a child gets surgical treatment, and many of these factors are potential determinants of cognitive and behavioral outcomes as well. Such sources of bias prohibit the confident interpretation of the existing literature.
Operative Treatment
Because the objective of surgical treatment is to provide each patient with the best aesthetic outcome that can be achieved, the plan for correcting the deformity must be formulated according to the particular demands of each case. The technique of surgical reconstruction must be tailored to the type of synostosis present and the age of the patient. It is clear that reshaping bone in a child younger than 1 year is much more readily achieved without significant bone disruption than in an older child. The methods of fixation for reshaped skull segments should also be different for very young children to avoid abnormalities in brain development resulting from vault surgery and the subsequent restriction of brain growth. Patients who are younger than 3 years have a substantial amount of brain growth remaining, but because those older than 3 years have already achieved approximately 85% to 90% of their ultimate brain mass (see Fig. 182-3), the fixation techniques can be more rigid and inclusive in these older children. In the following sections, the treatment of patients younger than 1 year and older than 3 years is described separately; those between 1 and 3 years of age require a blending of techniques. Metallic fixation devices are generally avoided in patients younger than 3 years owing to concerns related to the inward “migration” of the metal with further skull development. Resorbable plates, screws, and sutures are used instead of metallic devices.
