CHAPTER 15 Congenital dyserythropoietic anemias
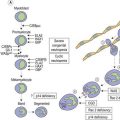
The congenital dyserythropoietic anemias (CDAs) are a heterogeneous group of rare inherited anemias, without additional cytopenias and with no tendency to neoplastic transformation.1,2 The cause of the peripheral anemia is a block in erythroid maturation, associated with a variable degree of erythroid dysplasia (dyserythropoiesis) in the bone marrow. The ineffectiveness of erythropoiesis results in a suboptimal reticulocyte response for the degree of anemia despite the presence of marked erythroid hyperplasia. Based largely on the nature of the dysplastic changes seen by light and electron microscopy, the CDAs have been divided into three major types, designated CDA types 1, 2 and 3 (Table 15.1), and other forms designated CDA groups IV–VII (see Table 15.2 below).3,4
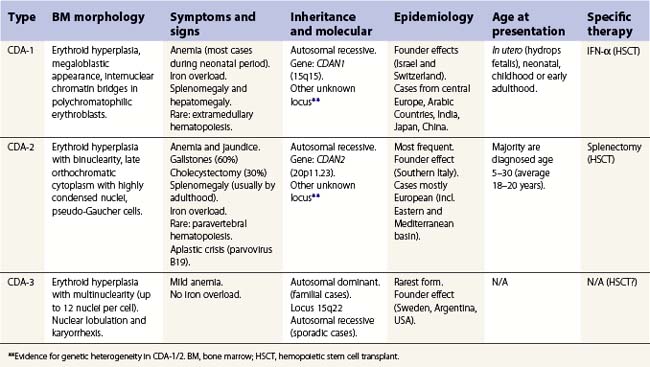
Table 15.2 Wickramasinghe’s CDA variants group classification
| For sake of clarity, groups of variants are indicated with Roman numerals, while major types are in Arabic numerals. To be considered a variant, at least three families must have been described with the features. First outlined by Sunitha Wickramasinghe (Wickramasinghe 2000).3 |
| Features | Inheritance | |
|---|---|---|
| Group CDA-IV | Severe transfusion-dependent normoblastic anemia with hypercellular marrow but absence of other features of CDA-1/2/3. If anemia is only moderate, then CDA-IVb. | Not clear |
| Group CDA-V | Mild normo/macrocytic anemia without medullary hyperplasia and erythroid dysplasia, but unconjugated hyperbilirubinemia. Has been reported in the literature as ‘primary shunt hyperbilirubinemia’ | Autosomal recessive/dominant |
| Group CDA-VI | Mild anemia with important macrocytosis (MCV 120–130 fl) and marked megaloblastoid hypercellular erythropoiesis. Orotic aciduria and thiamine-responsive anemia should be excluded. | Not clear |
| Group CDA-VII | As CDA-IV, but marked irregular nuclear shapes and with cytoplasmic globin-like inclusions (not reactive to α nor β antibodies). β thalassemia has to be excluded. | Not clear |
| Group CDA-VIII* | CDA with prominent post-splenectomy erythroblastemia (10–40 × 109 erythroblast/ml) but no resolution of a moderate anemia. Marrow showed irregular nuclei (clover-leaf forms) and karyorrhexis. | Not clear |
| Different variants | Reports of cases where classification and further delineation not possible. Many will possibly be classified within the spectrum of the major types when comprehensive molecular testing becomes available. |
* Previously identified as CDA with prominent post-splenectomy erythroblastemia.
Congenital dyserythropoietic anemia, type 1 (CDA-1)
CDA-1 has a variable presentation, as it can become apparent during the neonatal period or indeed at any time during the first five decades of life. It may also present in utero, with manifestations of fetal anemia requiring antenatal transfusions.5–7 However, patients usually have a mild to moderate anemia and may show jaundice, hepatosplenomegaly and cholelithiasis. A few are nevertheless severely anemic and transfusion-dependent. There is an increased tendency for some patients to develop marked iron overload despite a limited number of transfusions and lack of transfusion dependence (Fig. 15.1). Some cases have congenital abnormalities such as syndactyly, aberrations of the bones of the hands and feet (absence or hypoplasia of some phalanges, additional phalanges), dysplastic nails, short stature and abnormal pigmentation of areas of the skin (Fig. 15.2).
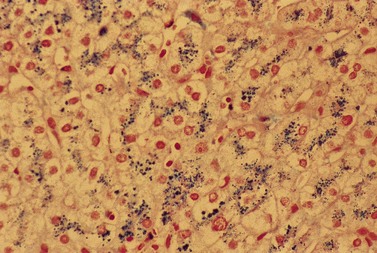
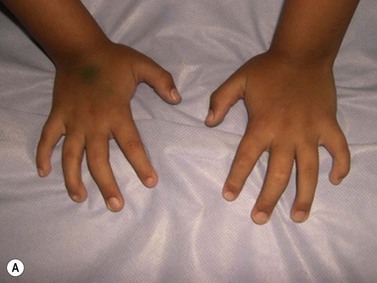
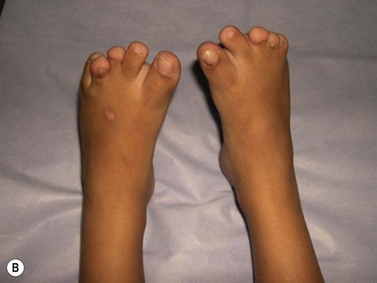
Fig. 15.2 (A, B) Dysmorphic features in the hands (A) and feet (B) of a child with CDA-1.
(Courtesy of Dr Aurora Feliu-Torres, Buenos Aires, Argentina.)
The disorder is inherited in an autosomal recessive manner. The disease gene was identified and localized to chromosome 15q15.1–15.3 in a highly inbred group of Israeli Bedouins.8 The CDAN1 gene has been highly conserved during evolution, has no known orthologues and the codanin-1 protein contains no recognizable motifs to provide clues as to its function. Over 20 mutations of CDAN1 have been described. The majority are missense mutations. Null mutations also occur, but to date no patients have been found who are either homozygous or compound heterzygous for these null mutations, suggesting such a combination is likely to be lethal. Incomplete sequencing may explain CDA-1 patients without CDAN1 mutations, but there is also likely to be genetic heterogeneity in this disorder.9
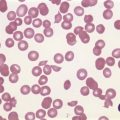
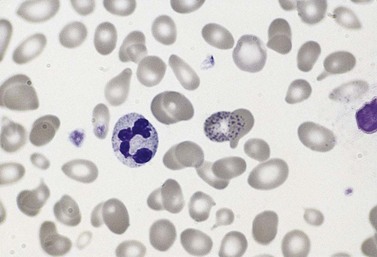
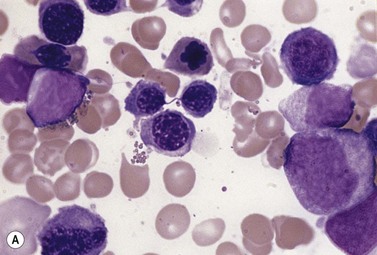
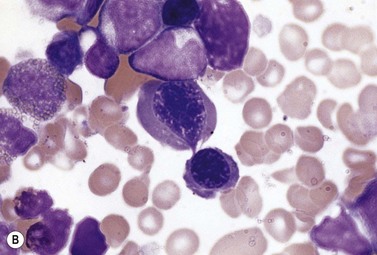
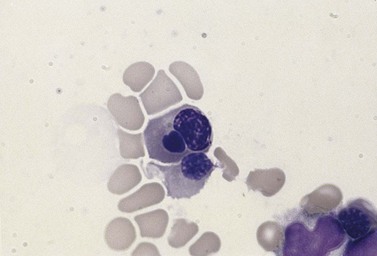
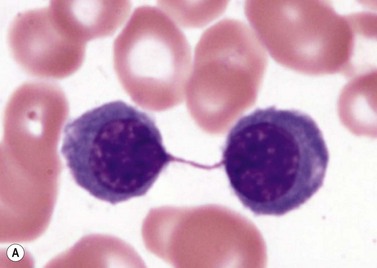
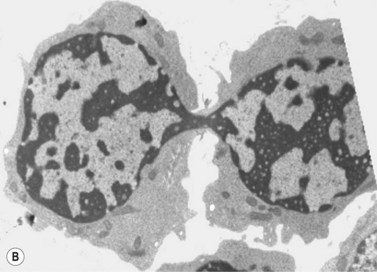
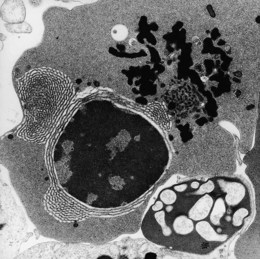
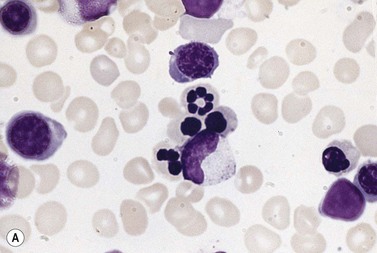
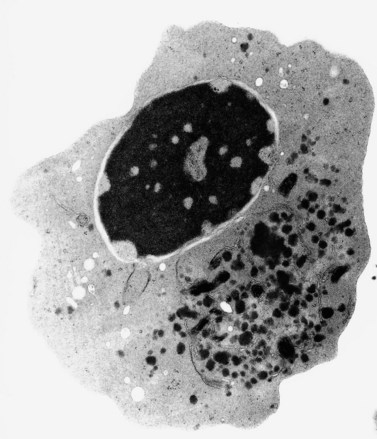
The average hemoglobin (Hb) is 9.0 g/dl (range 6.0–12.6 g/dl), and the mean cell volume (MCV) increased in 75% of cases. The peripheral blood film shows macrocytes and basophilic stippling (Fig. 15.3) and the reticulocyte count is normal or only minimally raised. There is megaloblastic erythroid hyperplasia in the bone marrow (Fig. 15.4). Some of the more mature basophilic erythroblasts and many of the early and late polychromatic erythroblasts show dyserythropoietic features. The characteristic morphological abnormality is internuclear bridging, where chromatin strands connect pairs of almost completely separated polychromatic erythroblasts (0.6–2.8 strands per 100 erythroblasts) (Fig.15.5A, B). There is also an increase in the percentage of binucleate polychromatic erythroblasts, which account for 3.5–7.0% of all erythroblasts. The latter may contain nuclei of different size and show partial fusion of the two nuclear masses; the two nuclei within the same cell may display different staining characteristics (Fig. 15.6). The most striking abnormality, seen on electron microscopy (EM), is the presence of nuclei with a spongy (or ‘Swiss-cheese’) appearance in a high proportion (up to 60%) of the mononucleate early and late polychromatic erythroblasts. These nuclei have multiple rounded electron-lucent areas within abnormally electron-dense heterochromatin (Fig. 15.7). Nuclei with a spongy appearance may also contain nuclear-membrane-lined cytoplasmic intrusions, sometimes with cytoplasmic organelles (Fig. 15.7B). An abnormally low percentage of mononucleate early polychromatic erythroblasts synthesize DNA; the non-DNA-synthesizing cells have DNA contents between 2c and 8c (1c = the haploid DNA content, i.e. the DNA content of a spermatozoon) and appear to be derived from the maturation of cells whose progress in the cell cycle has been arrested. These morphologically abnormal cells are destroyed in situ, resulting in ineffective erythropoiesis.10
All CDA-1 patients with confirmed mutations in CDAN1 have responded to α-interferon, with an increase in Hb, reduction in MCV, decrease in the proportion of erythroblasts with the ‘Swiss-cheese’ ultrastructural abnormality and a decrease in ineffective erythropoiesis.11,12 Currently, the first step of therapy for eligible CDA-1 patients (EM positive, awaiting/positive CDAN1 sequencing) should be with α-interferon. Some patients show a hematological response to splenectomy. The only available curative treatment for genetic conditions of hematopoiesis is hemopoietic stem cell transplantation (HSCT) which has been used with some success in patients with CDA-1, CDA-2 and a variant case.13–16 Transplantation has been limited to severe cases where splenectomy was ineffective, transfusion needs exceeded tolerance levels for iron overload, and a HLA-identical donor was available.
Congenital dyserythropoietic anemia, type 2 (CDA-2)
CDA-2, previously referred to as hereditary erythroblastic multinuclearity with positive acidified serum test (HEMPAS), is the most common type of CDA. Many of the reported cases were of southern Italian ancestry, but cases have also been reported from northwestern Europe and North Africa. Clinical features include mild to moderate anemia, mild jaundice, splenomegaly in 65% of cases, cholelithiasis and a tendency to develop iron-overload. Patients usually present between the ages of 1 month and 25 years, but there is frequently a delay in establishing the diagnosis.17
The disorder is inherited in an autosomal recessive manner, and the disease gene localizes to chromosome 20p11. The gene, CDAN2, encodes for the SEC23B protein, a component of the COP-II coat protein complex, which plays a role in vesicular transport from the endoplasmic reticulum to the Golgi apparatus. More than 25 mutations have been described, both missense and null alleles. Two specific missense mutations constitute nearly half of all the mutant alleles.18 However, there is evidence that an additional locus could be involved in CDA-2.19
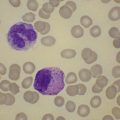
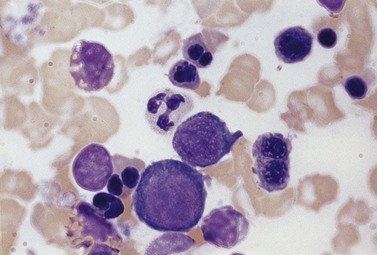
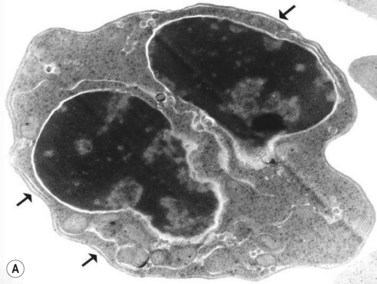
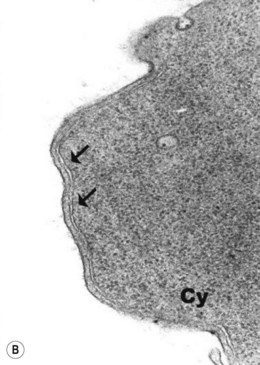
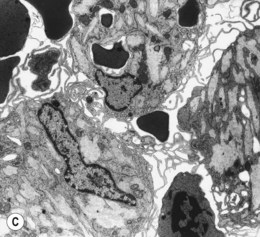
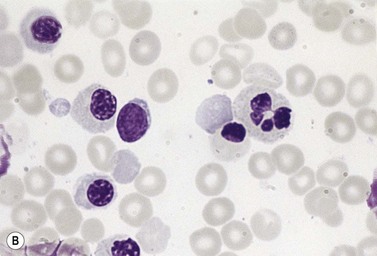
The average Hb is 8 g/dl. The red cells are normochromic and normocytic and show moderate anisocytosis, poikilocytosis and basophilic stippling. The reticulocyte count is either normal or minimally raised. Some cases present with minimal anemia, while occasional patients are transfusion-dependent, even without the co-inheritance of a β-thalassemia trait. The bone marrow shows normoblastic erythroid hyperplasia. A few of the basophilic erythropoietic cells and early polychromatic erythroblasts and 10–35% of the late erythroblasts are binucleate; the nuclei are usually of equal size (Fig. 15.8). In addition, a small proportion of the erythroblasts are trinucleate or multinucleate. Many of the mononucleate and binucleate late erythroblasts have orthochromatic cytoplasm and highly-condensed, apparently structureless nuclei. The anemia results from ineffective erythropoiesis. A high rate of phagocytosis of mononucleate and binucleate late erythroblasts by bone marrow macrophages is seen on bone marrow examination. Electron microscopy reveals a characteristic double membrane (consisting of smooth endoplasmic reticulum) aligned parallel to and at a distance of 40–60 nm from the cell membrane in a substantial proportion of the late erythroblasts (Fig. 15.9A, B). These double membranes are referred to as peripheral cisternae. Bone marrow macrophages are often laden with lipid and appear as pseudo-Gaucher cells (Fig. 15.9C).
The red cells give a positive acidified serum lysis test (Ham test) with about 30% of fresh ABO-compatible normal sera but not with the patient’s own serum. The reactive sera contain an antibody that combines specifically with an antigen on the CDA-2 red cells. This IgM antibody can be removed by absorption with red cells of patients with CDA-2, but not with red cells of patients with paroxysmal nocturnal hemoglobinuria (PNH). Due to the problems in performing this test in routine laboratories because of the difficulty in obtaining appropriate control anti-sera, finding an alternative test has become a priority.20 The anion transport protein (band 3) of the red cell membrane is underglycosylated in CDA-2 and, consequently, is migrates faster than normal on SDS polyacrylamide gel electrophoresis (SDS-PAGE). This technique has progressively replaced the Ham test. If results are suggestive for CDA-2 in SDS-PAGE, genetic analysis of CDAN2 is warranted. Specific therapy for CDA-2 is not yet available, and supportive care constitutes the best standard practice, although splenectomy results in a substantial increase in Hb in some cases but without effect on iron overloading. Studies identifying the criteria for splenectomy in patients with CDA-2 need to be performed. HSCT has been attempted with some success in a very limited number of selected patients with CDA-2 (see discussion of HCST for CDA-1).
Congenital dyserythropoietic anemia, type 3 (CDA-3)
CDA-3 is the rarest of the three classical types of CDA. Familial cases have been described in Sweden, Argentina and the USA. The largest is a five-generation family from the Væsterbotten district of Sweden, whose ancestry can be traced back to the 19th century. It contains more than 30 cases inherited in an autosomal dominant manner.21 CDA-3 is not as severe as the other types and the anemia is mild. Patients present with ocular abnormalities (angioid streaks with macular degeneration), do not have iron overload (probably due to predominant intravascular hemolysis with hemosiderinuria) and do not have splenomegaly (except the Argentinian family). In the Swedish family there is an increased tendency to develop monoclonal gammopathy and multiple myeloma while sporadic cases have developed lymphoma.21,22 Sporadic cases do not have autosomal dominant inheritance as parents and relatives were entirely normal; they may represent de novo mutations or demonstrate genetic heterogeneity. The clinical features of the sporadic cases are also extremely variable.
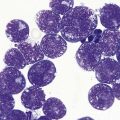
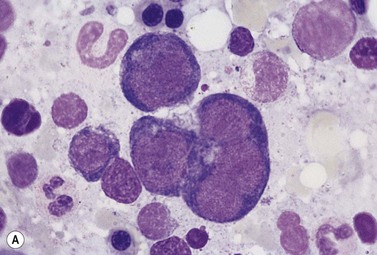
In CDA-3, Hb levels are usually between 7 and 14 g/dl, the MCV is usually raised or normal and the granulocyte and platelet counts are normal. The blood film shows macrocytes (and, sometimes, occasional giant erythrocytes), poikilocytosis, fragmented red cells and basophilic stippling. The reticulocyte count is normal or slightly raised. The acidified serum lysis test is negative. The bone marrow shows marked erythroid hyperplasia and megaloblastic erythropoiesis in the absence of vitamin B12 or folate deficiency. Large uninucleate erythroblasts with enlarged lobulated nuclei and many giant multinucleate erythroblasts with up to 12 nuclei per cell are commonly present. The tendency to multinuclearity begins in the basophilic erythropoietic cells but is most marked in the early and late polychromatic erythroblasts. Over 35% of all erythroblasts may be binucleate or multinucleate (Fig. 15.10) and the latter may have total DNA contents up to 40c. Sometimes the two nuclei of a binucleate cell or two or more of the nuclei within a multinucleate cell are joined together either by a narrow strand of chromatin or over a wide area of nuclear contact. The giant mononucleate erythroblasts have DNA contents of up to 20c. Other abnormalities affecting erythroblasts include coarse basophilic stippling of the cytoplasm and karyorrhexis. Electron microscopy studies show a variety of nonspecific abnormalities including differences in the ultrastructural appearances of different nuclei within the same multinucleate cell (Fig. 15.11). In addition, in some patients occasional erythroblast sections contain stellate or branching intracytoplasmic inclusions composed of precipitated β-globin chains (Fig. 15.12). The anemia results largely from ineffective erythropoiesis; both mononucleate and multinucleate cells may be seen within bone marrow macrophages.
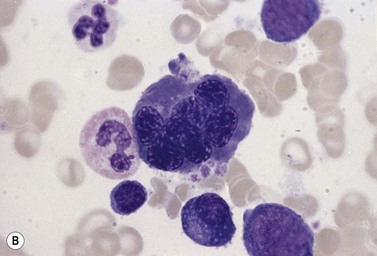
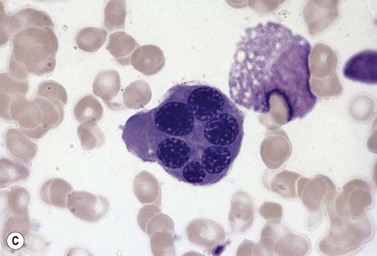
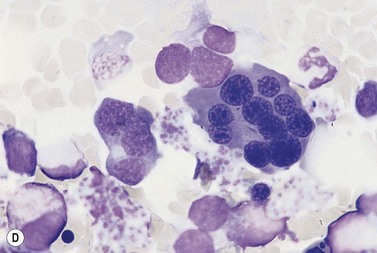
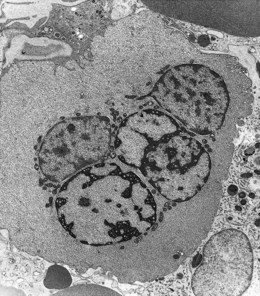
Other types of congenital dyserythropoietic anemia
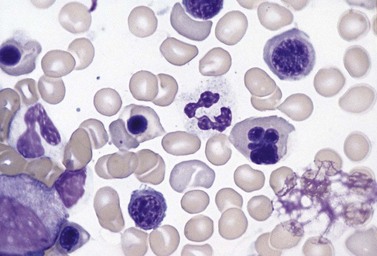
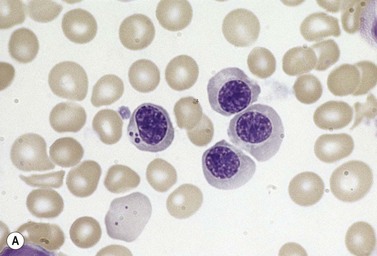
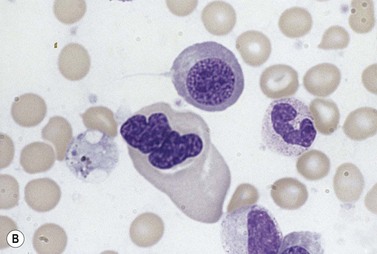
Approximately 40% of cases of CDA diagnosed in the UK are of types other than types 1, 2 and 3. A tentative classification of other forms of CDA has been attempted (Table 15.2).3 Examples of these atypical cases are illustrated in Figs 15.13–15.16. It is not clear how many of these are distinct clinical or hematological entities. Sequencing of CDAN1 and CDAN2 genes will determine whether atypical cases are allele specific phenotypes of the known genes.
Diagnosis of congenital dyserythropoietic anemia
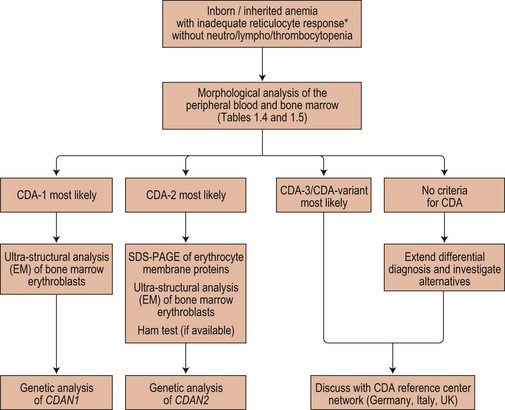
The possibility of CDA should be considered whenever there is a suboptimal absolute reticulocyte count for the degree of anemia, unexplained hyperbilirubinemia or unexplained iron overload. Acquired dyserythropoiesis and causes of congenital dyserythropoiesis of known etiology (conventionally not included under the heading CDA) must be excluded. Causes of acquired dyserythropoiesis are given in Chapters 5, 14 and 20. The diagnosis of specific types or groups of CDA depends on the demonstration of their characteristic features and especially on the light and electron microscope appearances of the bone marrow erythroblasts, and should follow Fig. 15.17.
1 Renella R, Wood WG. The congenital dyserythropoietic anemias. Hematol Oncol Clin North Am. 2009;23:283-306.
2 Wickramasinghe SN. Congenital dyserythropoietic anaemias: clinical features, haematological morphology and new biochemical data. Blood Rev. 1998;12:178-200.
3 Wickramasinghe SN. Congenital dyserythropoietic anemias. Curr Opin Hematol. 2000;7:71-78.
4 Heimpel H, Wendt F. Congenital dyserythropoietic anemia with karyorrhexis and multinuclearity of erythroblasts. Helv Med Acta. 1968;34:103-115.
5 Heimpel H, Schwarz K, Ebnother M, et al. Congenital dyserythropoietic anemia type I (CDA I): molecular genetics, clinical appearance, and prognosis based on long-term observation. Blood. 2006;107:334-340.
6 Shalev H, Kapelushnik J, Moser A, et al. A comprehensive study of the neonatal manifestations of congenital dyserythropoietic anemia type I. J Pediatr Hematol Oncol. 2004;26:746-748.
7 Shalev H, Kapleushnik Y, Haeskelzon L, et al. Clinical and laboratory manifestations of congenital dyserythropoietic anemia type I in young adults. Eur J Haematol. 2002;68:170-174.
8 Dgany O, Avidan N, Delaunay J, et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet. 2002;71:1467-1474.
9 Ahmed MR, Chehal A, Zahed L, et al. Linkage and mutational analysis of the CDAN1 gene reveals genetic heterogeneity in congenital dyserythropoietic anemia type I. Blood. 2006;107:4968-4969.
10 Wickramasinghe SN, Pippard MJ. Studies of erythroblast function in congenital dyserythropoietic anaemia, type I: evidence of impaired DNA, RNA, and protein synthesis and unbalanced globin chain synthesis in ultrastructurally abnormal cells. J Clin Pathol. 1986;39:881-890.
11 Lavabre-Bertrand T, Blanc P, Navarro R, et al. alpha-Interferon therapy for congenital dyserythropoiesis type I. Br J Haematol. 1995;89:929-932.
12 Lavabre-Bertrand T, Ramos J, Delfour C, et al. Long-term alpha interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. Eur J Haematol. 2004;73:380-383.
13 Remacha AF, Badell I, Pujol-Moix N, et al. Hydrops fetalis-associated congenital dyserythropoietic anemia treated with intrauterine transfusions and bone marrow transplantation. Blood. 2002;100:356-358.
14 Ayas M, al-Jefri A, Baothman A, et al. Transfusion-dependent congenital dyserythropoietic anemia type I successfully treated with allogeneic stem cell transplantation. Bone Marrow Transplant. 2002;29:681-682.
15 Iolascon A, Sabato V, de Mattia D, Locatelli F. Bone marrow transplantation in a case of severe, type II congenital dyserythropoietic anaemia (CDA II). Bone Marrow Transplant. 2001;27:213-215.
16 Ariffin WA, Karnaneedi S, Choo KE, Normah J. Congenital dyserythropoietic anaemia: report of three cases. J Paediatr Child Health. 1996;32:191-193.
17 Heimpel H, Anselstetter V, Chrobak L, et al. Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood. 2003;102:4576-4581.
18 Schwarz K, Iolascon A, Verissimo F, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41:936-940.
19 Iolascon A, De Mattia D, Perrotta S, et al. Genetic heterogeneity of congenital dyserythropoietic anemia type II. Blood. 1998;92:2593-2594.
20 Denecke J, Marquardt T. Congenital dyserythropoietic anemia type II (CDAII/HEMPAS): Where are we now? Biochim Biophys Acta.. 2008.
21 Sandstrom H, Wahlin A. Congenital dyserythropoietic anemia type III. Haematologica. 2000;85:753-757.
22 McCluggage WG, Hull D, Mayne E, et al. Malignant lymphoma in congenital dyserythropoietic anaemia type III. J Clin Pathol. 1996;49:599-602.