[level-membership-for-neurology-category]
Chapter 28 Congenital Anomalies of the Skull
Introduction
Congenital anomalies of the skull can arise any time during gestation and must be distinguished from anomalies that arise after birth. During the first 4–6 weeks of development from conception, neural crest cells in the fetal head region migrate and differentiate into mesenchymal cells that form the bones of the face, while the cranial base and base of the skull are derived from the occipital somitomeres. Within the flat cranial bones, the mesenchyme differentiates directly into bone through membranous ossification. The neurocranium is the portion surrounding the brain, and it develops directly from mesenchyme derived from the occipital somitomeres. The viscerocranium is derived from neural crest and it forms the cartilaginous bones of the face [Sadler and Langman, 2009]. The neurocranium is divided into the membranous part, forming the flat bones of the cranial vault, and the chondrocranium, forming the cartilaginous bones of the base of the skull. The chondrocranium develops by fusion of a number of cartilaginous structures, which ossify by endochondral ossification to form the base of the skull. Posteriorly, the base of the occipital bone is formed from parachordal cartilage and three occipital sclerotomes. Anteriorly, the sphenoid and ethmoid bones are formed from the hypophyseal cartilages and trabeculae cranii. On either side of the medial plate, the ala orbicularis and ala temporalis form the sphenoid bones, and the periotic capsule forms the temporal bones. The membranous neurocranium (dura mater) ossifies to form the cranial vault through bone spicules, which progressively radiate from primary ossification centers near the center of each bony plate toward the periphery, where the sutures develop. Membranous bones enlarge during fetal and postnatal life by the apposition of new layers to the outer surface of the skull (ectocranial bone deposition), while endocranial osteoclastic bone resorption occurs on the inner surface. Growth of the cranial bones is directly related to brain growth, and premature fusion of cranial sutures can be related to the cessation of brain growth (e.g., primary microcephaly).
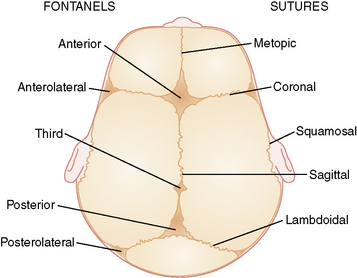
There are six fibrous areas where two or more cranial bones meet (fontanels). The five major sutures are the metopic, sagittal, coronal, squamosal, and lambdoid sutures; the six fontanels are the anterior (1), anteriolateral (sphenoidal) (2), posterolateral (2), and posterior (1) fontanels (Figure 28-1). The presence of the sutures and fontanels allows the bones of the skull to overlap each other (termed molding) during the birth process. Different sutures become ossified at different times, with the metopic suture being the first to ossify at 4–7 months and the remaining sutures not completely ossifying until adulthood.
Craniosynostosis versus Deformational Plagiocephaly
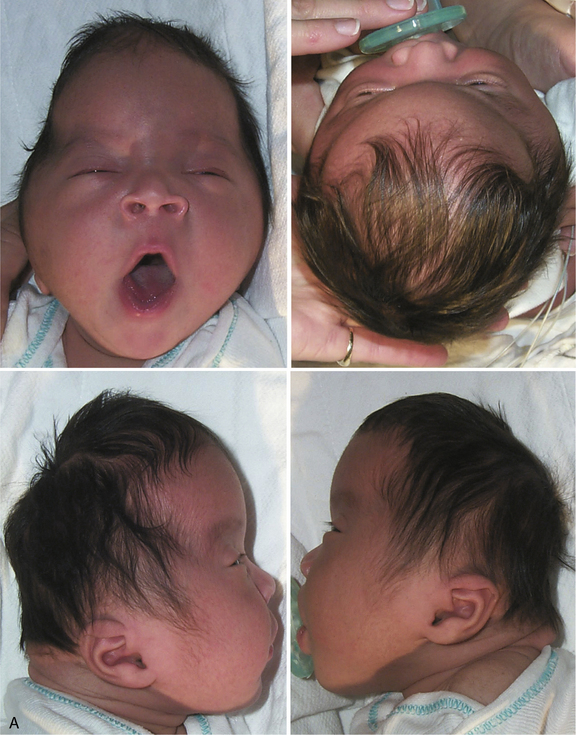
The term craniostenosis (literally “cranial narrowing”) is used to describe an abnormal head shape that results from premature fusion of one or more sutures, while craniosynostosis is the process of premature sutural fusion that results in craniostenosis. In clinical usage, the term craniosynostosis is used more widely, perhaps in an effort to distinguish deformational nonsynostotic head shapes from those caused by underlying sutural synostosis, but the two terms are often used interchangeably. Plagiocephaly is a nonspecific term used to describe an asymmetric head shape, which can result from either craniosynostosis or cranial deformation, and differentiation between these two causes is critical to determining the proper mode of treatment (i.e., surgery vs. physical or molding techniques). Craniosynostosis is usually treated with a neurosurgical procedure involving partial calvariectomy, while deformational plagiocephaly usually responds to early physical therapy, repositioning, and/or cranial orthotic therapy if those measures are unsuccessful [Graham and Smith, 2007; Graham et al., 2005].
In an otherwise normal fetus, prenatal relaxation of normal growth-stretch tensile forces in the underlying dura across a suture for a significant period of time during late fetal life can result in craniosynostosis [Graham et al., 1979, 1980; Higginbottom et al., 1980; Graham and Smith, 1980; Koskinen-Moffett et al., 1982]. This may also occur when the lack of growth stretch is caused by a deficit in brain growth, as in severe primary microcephaly. Experimental prolongation of gestation in pregnant mice, resulting in fetal crowding after installation of a cervical clip, has been shown to result in craniosynostosis [Koskinen-Moffett and Moffet, 1989]. The degree of craniosynostosis was greatest among those fetuses located more proximally in the uterine horns, where the crowding was most severe. The most common cause of craniosynostosis in otherwise normal infants is constraint of the fetal head in utero [Graham et al., 1979, 1980; Higginbottom et al., 1980; Graham and Smith, 1980; Koskinen-Moffett et al., 1982; Koskinen-Moffett and Moffet, 1989; Sanchez-Lara et al., 2010]. When external fetal head constraint limits growth stretch parallel to a cranial suture, it may lead to craniosynostosis of an intervening suture between the constraining points. Sagittal craniosynostosis (the most common type of craniosynostosis) usually is isolated and occurs in an otherwise normal child. The constrained suture tends to develop a bony ridge, especially at the point of maximal constraint between the biparietal eminences. Such ridging can easily be palpated or visualized on skull radiographs, and three-dimensional cranial computed tomography (3D-CT) allows the ridge to be seen even more clearly (Figure 28-2).
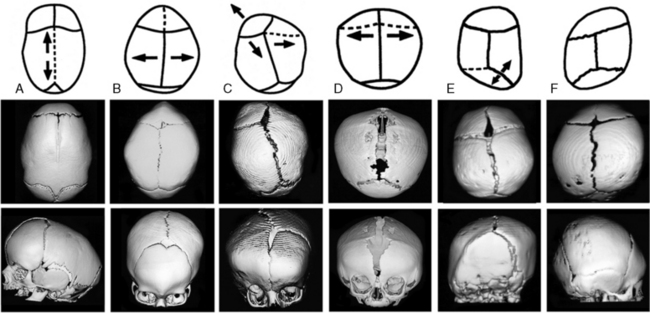
Craniosynostosis is usually recognized shortly after birth from the abnormal shape of the head and lack of molding resolving to normal. Early closure of a fontanel, head asymmetry, and/or palpable ridging along a closed suture can be presenting features. With synostosis, cranial radiographs may reveal sclerosis of the suture with no apparent intervening sutural ligament, but it may be difficult to distinguish an overlapping suture from synostosis. On cross-sectional images, dense ridging over the suture may be evident, particularly with sagittal and metopic synostosis. If there is uncertainty as to whether sutures are truly synostotic, 3D-CT can provide a more accurate appraisal (see Figure 28-2).
In general, craniosynostosis begins at one point and then spreads along a suture [Koskinen-Moffett and Moffet, 1989; Cohen and MacLean, 2000]. At the fused location, there is complete sutural obliteration, with nonlamellar bone extending completely across the sutural space, while further away from the initial site of fusion, the sutural margins are closely approximated with ossifying connective tissue. As age increases, there is a tendency for more of the suture to become synostotic, with synostosis usually beginning at only one location in most cases [Koskinen-Moffett and Moffet, 1989]. Synostosis prevents future expansion at that site, and the rapidly growing brain then distorts the calvarium into an aberrant shape, depending upon which sutures have become synostotic (see Figure 28-2). The earlier the synostosis takes place, the greater the effect on skull shape. Craniosynostosis may be caused by many different mechanisms, such as mutant genes, chromosome disorders, storage disorders, hyperthyroidism, or failure of normal brain growth The entire topic of craniosynostosis has been comprehensively reviewed by Cohen [Cohen and MacLean, 2000].
Sutural Anatomy and Head Shape
Different terms have been used to describe the different head shape alterations caused by craniosynostosis, with the resultant head shape dependent on the suture involved. A long, keel-shaped skull with prominent forehead and occiput is termed dolichocephaly or scaphocephaly. This head shape is usually associated with premature sagittal suture closure and a palpable ridge toward the posterior end of the suture (see Figure 28-2A). Sagittal synostosis must be distinguished from deformation of the infant cranium due to persistently sleeping on the side of the head (more common in prematurely born infants) or breech-head deformation sequence. Individuals with scaphocephaly and dolichocephaly have a decreased cephalic index (CI) of less than 76 percent (CI = head width/head length × 100 percent).
Premature fusion of both coronal sutures produces a high, wide forehead with a short skull, resulting in brachycephaly (see Figure 28-2), while fusion of one coronal suture produces an asymmetric head shape termed plagiocephaly. When coronal craniosynostosis occurs, it is important to examine the patient carefully for associated anomalies that might suggest a recognizable genetic syndrome. Evaluation of the limbs, ears, and cardiovascular system is quite helpful in diagnosing syndromes associated with coronal craniosynostosis. Limb defects, such as syndactyly, brachydactyly, carpal coalition, or broad, deviated thumbs and/or halluces, can suggest an associated genetic syndrome and indicate what types of molecular analysis to pursue. It is also important to examine both parents for similar anomalies, carpal coalition, and/or facial asymmetry, since these findings may represent variable expression of an altered gene in a parent.
Premature fusion of both the coronal and sagittal sutures generally leads to a tall, tower-like skull (turricephaly), with more severe synostosis of multiple sutures producing a tall, pointed skull (acrocephaly or oxycephaly). In this condition, the limitations of calvarial expansion are so extreme that there may be limited room for brain growth (Figure 28-3a). Synostosis of multiple cranial sutures is more likely to result in elevated intracranial pressure and to require shunting for hydrocephalus. In extreme cases, a cloverleaf head shape can result from multiple suture synostosis, usually with signs of increased intracranial pressure and a “beaten copper” radiographic appearance of the inner table of the skull (see Figure 28-3b). There may also be optic atrophy, proptosis, and loss of vision. Combinations of sutural synostosis, such as sagittal plus coronal, are also referred to as compound craniosynostosis, and multiple suture synostosis usually has a genetic basis.

A triangle-shaped skull (trigonocephaly) is caused by premature fusion of the metopic suture (see Figure 28-2). The similarity in epidemiological features between sagittal and metopic craniosynostosis suggests that prenatal lateral constraint of the frontal part of the head may be a frequent cause of metopic craniosynostosis. Examples of constraint-induced metopic synostosis have included a monozygotic triplet whose forehead was wedged between the buttocks of her two co-triplets, and an infant whose head was compressed within one horn of his mother’s bicornuate uterus [Graham and Smith, 1980]. Syndromic metopic synostosis can also occur, and trigonocephaly is seen in a variety of syndromes, some of which are associated with mental retardation or chromosome anomalies.
Unilateral lambdoid synostosis results in trapezoidal plagiocephaly, which differs from deformational posterior plagiocephaly due to supine positioning and torticollis, and from synostotic anterior plagiocephaly due to unicoronal synostosis (see Figure 28-2). Unlike coronal synostosis, facial structures and orbits are usually not affected by lambdoid synostosis. Radiographic signs include trapezoidal cranial asymmetry, small posterior fossa, and sutural sclerosis with ridging; however, sole reliance on skull radiographs and clinical signs can lead to misdiagnosis, so it is best to confirm the diagnosis of suspected lambdoid synostosis with a 3D-CT scan, which clearly images the involved suture(s) and permits secure diagnosis. Among 232 patients referred for either deformational posterior plagiocephaly or craniosynostosis, only 4 patients (3.1 percent) manifested clinical, imaging, and operative features of true unilambdoidal craniosynostosis [Huang et al., 1996]. These features included a thick bony ridge over the fused suture, with contralateral parietal and frontal bulging, and an ipsilateral occipitomastoid bulge, leading to tilting of the ipsilateral skull base and a downward/posterior displacement of the ear on the synostotic side. In contrast, infants with deformational, nonsynostotic posterior plagiocephaly had a parallelogram-shaped head, with forward displacement of the ear and frontal bossing on the side ipsilateral to the occipitoparietal flattening, accompanied by contralateral occipital bossing [Graham et al., 2005].
Plagiocephaly (which translates literally from the Greek term plagio kephale as “oblique head”) is a term used to describe asymmetry of the head shape, when viewed from the top [Graham and Smith, 2007]. The term deformational plagiocephaly should suffice to distinguish this type of defect and its proper type of management. The side of the plagiocephaly is usually indicated by the bone that has been most flattened by the deforming forces (usually the occiput for infants who sleep on their backs). Deformational plagiocephaly is usually not associated with premature closure of a cranial suture, but since craniosynostosis can also be caused by fetal head constraint, when both deformational plagiocephaly and craniosynostosis occur together, the diagnosis can be difficult, requiring complex management.
Epidemiology of Craniosynostosis
The incidence of craniosynostosis is 3.4 per 10,000 births, and it is usually an isolated, sporadic anomaly in an otherwise normal child. About 8 percent of all craniosynostosis cases are familial. Familial types of craniosynostosis are most frequent in coronal synostosis, accounting for 14.4 percent of coronal synostosis, 6 percent of sagittal synostosis, and 5.6 percent of metopic synostosis [Lajeunie et al., 1995, 1996, 1998], while lambdoidal synostosis is almost never familial. The frequency of associated twinning is increased, and most twin pairs are discordant, especially with sagittal and metopic synostosis, which would tend to support fetal crowding as a cause for these types of synostosis; concordance for coronal synostosis is much higher for monozygotic twins than for dizygotic twins, suggesting that many cases of coronal synostosis have a genetic basis [Lajeunie et al., 1995].
Sagittal synostosis is the most common type of craniosynostosis, accounting for 50–60 percent of cases and occurring in 1.9 per 10,000 births, with a 3.5:1 male:female sex ratio [Lajeunie et al., 1996]. Only 6 percent of cases are familial, with 72 percent of cases sporadic and no paternal or maternal age effects noted. Twinning occurred in 4.8 percent of 366 cases, with only one monozygotic twin pair being concordant [Lajeunie et al., 1996].
Coronal craniosynostosis is the second most frequent type of craniosynostosis (accounting for 20–30 percent of cases). Unilateral coronal craniosynostosis can be either genetic or due to fetal head constraint from an aberrant fetal lie, multiple gestation, or small uterine cavity [Graham et al., 1980; Higginbottom et al., 1980]. Approximately 71 percent of unilateral coronal craniosynostosis is right-sided, and 67 percent of vertex presentations are in the left occiput transverse position, possibly explaining the prevalence of right-sided, unilateral, coronal craniostenosis [Graham et al., 1980]. Nonsyndromic coronal craniosynostosis occurs 0.94 per 10,000 births, with 61 percent of cases sporadic, and 14.4 percent of 180 pedigrees familial. Bilateral cases occur much more frequently than unilateral cases, and coronal synostosis is more frequent in females (male to female ratio 1:2). The paternal age is statistically older than average (32.7 years), and these data have been interpreted as being consistent with fresh dominant mutation and autosomal-dominant inheritance with 60 percent penetrance, when the synostosis has a genetic basis [Lajeunie et al., 1995].
Metopic synostosis occurs in about 0.67 per 10,000 births, making it the third most frequent type of craniosynostosis (accounting for 10–20 percent of patients). Like sagittal synostosis, metopic synostosis is more frequent in males (3.3:1 male: female ratio) and seldom familial (5.6 percent of cases) [Lajeunie et al., 1998]. There is no maternal or paternal age effect, and the frequency of associated twinning was 7.8 percent of 179 pedigrees studied, with only 2 twin monozygotic pairs concordant [Lajeunie et al., 1998].
Familial craniosynostosis is usually transmitted as an autosomal-dominant trait with incomplete penetrance and variable expressivity. A wide variety of chromosomal anomalies have also been associated with craniosynostosis. This emphasizes the importance of chromosomal analysis for patients with syndromic craniosynostosis in whom a recognizable monogenic syndrome is not apparent, particularly when there is associated developmental delay and growth deficiency. As comparative genomic hybridization becomes more widely utilized, this will be even more useful than standard cytogenetics for many such cases. In addition, craniosynostosis can also occur as a component of numerous syndromes, many of which manifest phenotypic overlap and genetic heterogeneity [Cohen and MacLean, 2000] Craniosynostosis syndromes with a demonstrated mutational basis include Apert’s syndrome, Crouzon’s syndrome, Pfeiffer’s syndrome, Saethre–Chotzen syndrome, Jackson–Weiss syndrome, Boston craniosynostosis, Beare–Stevenson cutis gyrata syndrome, and fibroblast growth factor receptor 3 (FGFR3)-associated coronal synostosis; however, the efficiency of actually detecting a mutation for a given syndrome varies from about 60 percent for Crouzon’s syndrome to 98 percent for Apert’s syndrome [Cohen and MacLean, 2000].
Secondary craniosynostosis can occur with certain primary metabolic disorders (e.g., hyperthyroidism, rickets), storage disorders (e.g., mucopolysaccharidosis), hematological disorders (thalassemia, sickle cell anemia, polycythemia vera, congenital hemolytic icterus), brain malformations (e.g., holoprosencephaly, microcephaly, encephalocele, or overshunted hydrocephalus), and selected teratogenic exposures (e.g., diphenylhydantoin, retinoic acid, valproic acid, aminopterin, fluconazole, cyclophosphamide) [Cohen and MacLean, 2000].
Inability to demonstrate a mutation does not rule out a genetic basis for the craniosynostosis, and not every person with a pathogenic mutation manifests craniosynostosis. Bilateral coronal synostosis often lacks sutural ridging and usually has a genetic pathogenesis, suggesting that all such patients should be screened for mutations. Among 57 patients with bilateral coronal synostosis, mutations in FGFR genes were found for all 38 patients with a syndromic form of craniosynostosis. Among 19 patients with nonsyndromic bilateral craniosynostosis, mutations were found in or near exon 9 of FGFR2 in 4 patients, as well as the common Pro250Arg mutation being found in exon 7 of FGFR3 in 10 patients; only 5 patients (9 percent) lacked a detectable mutation in FGFR 1, 2, or 3 [Mulliken et al., 1999]. This study suggests that mutation analysis should be considered in most patients with bilateral coronal synostosis.
Kleeblattschadel (Cloverleaf Skull)
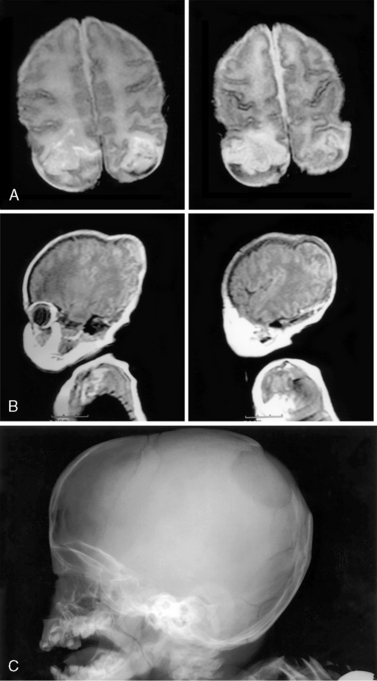
Kleeblattschadel is a term used to describe a cloverleaf skull configuration consisting of protrusion of each of the cranial bones, with broadening of the temporal region and face. These cranial protrusions are separated into focal bulges by furrows along the suture lines. The eyes often protrude, leading to corneal ulceration, scarring, and subsequent blindness, if the corneal surface remains unprotected [Stevenson, 1986]. Occipital encephaloceles can occur, and associated hydrocephalus is common. The palate is usually highly arched, but clefting is rare. The presence of Kleeblattschadel indicates that multiple sutural fusions occurred during early prenatal life. Increased thickening of the base of the occipital bone prevents lengthening of the skull, thus yielding the typical shape [Dambrain et al., 1987]. One child has also been described who had a cloverleaf skull without craniosynostosis (in which the primary defect was thought to be a cranial bone dysplasia that allowed for eventration of the brain and resultant cloverleaf configuration) [DR, 1988].
Multiple sutural synostosis is much more likely to result from genetic mutations in FGFR genes, TWIST, or MSX2, all of which result in syndromes which can present with cloverleaf skull [Cohen and MacLean, 2000]. The most common syndrome associated with cloverleaf skull is thanatophoric dysplasia type 2, which is due to mutations in FGFR3. Type 2 Pfeiffer’s syndrome, due to mutations in FGFR2, can result in cloverleaf skull, as can Crouzon’s syndrome and Apert’s syndrome, which are also due to mutations in FGFR2. Other syndromes, like FGFR3-associated coronal synostosis, rarely result in cloverleaf skull, but some rare syndromes like Boston craniosynostosis and Crouzon’s syndrome with acanthosis nigricans can sometimes manifest cloverleaf skull. In Boston craniosynostosis, the mutant MSX2 product has enhanced affinity for binding to its DNA target sequence, resulting in activated osteoblastic activity and aggressive cranial ossification [Warman et al., 1993; Jabs et al., 1993; Ma et al., 1996]. In Crouzon’s syndrome with acanthosis nigricans, a specific FGFR3 mutation (Ala391Glu), leads to early onset of acanthosis nigricans during childhood, often with associated choanal atresia and hydrocephalus [Schweitzer et al., 2001].
Eighty-five percent of children with Kleeblattschadel will have other anomalies, and the pattern is often consistent with a syndrome diagnosis. The prognosis is usually syndrome-dependent and can be quite poor, with early death due to respiratory difficulties or progressive brain damage from hydrocephalus being common [Frank et al., 1985]. However, subtotal craniectomy within the first 3 weeks of life in individuals with mild Kleeblattschadel may result in normal or near-normal development [Frank et al., 1985; Kroczek et al., 1986; Turner and Reynolds, 1980]. Early extensive calvariectomy is merited to preserve brain function and development, as well as to allow reformation of the craniofacial skeletal features. Many craniofacial surgeons prefer to begin with a posterior skull release in the early months of life (mean age 4 months), followed by fronto-orbital advancement around the end of the first year (mean age 14 months), with insertion of a ventriculoperitoneal shunt at the time of the first procedure if there is associated hydrocephalus [Sgouros et al., 1996]. The use of postoperative orthotic molding can help to channel brain growth into a more normal form, leading to improved postoperative results over those obtained via surgery alone [Schweitzer et al., 2001]. When lambdoid synostosis occurs as part of a syndrome with multiple suture involvement, there is often bilateral involvement, and early posterior release may alleviate some associated increased intracranial pressure. Patients with Kleeblattschadel need to be followed carefully for hydrocephalus, which may be part of the syndrome, rather than due to the multiple suture synostosis. Restricted growth of the posterior fossa is particularly common in severe craniofacial dysostosis syndromes. The purpose of surgery should be to decompress the brain, expand the bony orbits to accommodate the globes, and open airway passages [Kroczek et al., 1986].
Treatment and Outcomes of Craniosynostosis
Mild degrees of craniosynostosis may not always require surgery; however, in moderately severe cases, early surgery is usually warranted. The usual indication for surgery is to restore normal craniofacial shape and growth. When both the coronal and sagittal sutures are synostotic, impairing brain growth early in infancy, surgery is indicated to help prevent neurological and ophthalmologic complications associated with increased intracranial pressure and inadequate orbital volume. A variety of neurosurgical techniques have been developed for the treatment of craniosynostosis [Cohen and MacLean, 2000]. Most of these techniques involve removing the aberrant portion of the bony calvarium from its underlying dura, including the area surrounding the synostotic suture(s). If this is done within the first few months after birth, a new bony calvarium will usually develop within the remaining dura mater under the same principles that guide normal prenatal calvarial morphogenesis. As long as there is continued growth stretch from the expanding brain, the sites over the dural reflections remain unossified, thereby maintaining the sutures in a fibrous, open state. Thus, the calvarium and its sutures usually re-form normally after a partial calvariectomy for craniosynostosis. The new bony calvarium begins to develop within 2–3 weeks after surgery, and is usually firm by 5–8 weeks after surgery. If the procedure is done after 3–4 months of age, the approach is similar, with the exception that pieces of the calvarium are usually replaced in a mosaic pattern over the dura mater to act as niduses for the mineralization of new calvarium. Newer endoscopic repair techniques have been developed, followed by postoperative orthotic molding. Such procedures are most effective if done relatively early in infancy. These techniques are most effective in normal infants without a syndromic type of craniosynostosis.
Following early surgery for isolated craniosynostosis (primarily fronto-orbital advancement and/or calvarial vault remodeling at a mean age of 8 months), only 13 percent of 104 patients (10 bilateral coronal, 57 unilateral coronal, 29 metopic, and 8 sagittal) required a second cranial vault operation for residual defects at a mean age of 23 months. Perioperative complications were minimal (5 percent), with 87.5 percent of patients considered to have satisfactory craniofacial form, and low rates of hydrocephalus (3.8 percent), shunt placement (1 percent), and seizures (2.9 percent). Among such cases of isolated craniosynostosis, unilateral coronal synostosis was the most problematic type due to vertical orbital dystopia, nasal tip deviation, and altered craniofacial growth problems with residual craniofacial asymmetry [McCarthy et al., 1995]. In a second study of 167 children with both nonsyndromal (isolated) and syndromal craniosynostosis (12 bilateral coronal, 18 unilateral coronal, 39 metopic, and 46 sagittal), repeat operations were necessary in only 7 percent. Repeat operations were more common in syndromic cases (27.3 percent) than in nonsyndromic craniosynostosis (5.6 percent) [Williams et al., 1997].
Even though intracranial pressure can be elevated in patients with nonsyndromic craniosynostosis, they may not have decreased cranial volumes either before or after surgical repair, and as a group show slightly larger intracranial volumes when compared with normal controls. This could reflect the impact of fetal head constraint on fetuses with larger heads, or it might relate to the known association of macrocephaly with nonsyndromic coronal craniosynostosis due to the common Pro250Arg mutation in FGFR3 (which was not analyzed in these studies) [Polley et al., 1998]. Hydrocephalus occurs in 4–10 percent of patients with craniosynostosis and is more frequent with syndromic and multiple sutural craniosynostosis. In nonsyndromic patients, the rate of cerebral ventricular dilatation is the same as that observed in the general population, and it appears to be related to venous hypertension induced by jugular foramen stenosis. Such dilatation usually stabilizes spontaneously and rarely requires shunting. Some cases of progressive hydrocephalus in syndromic craniosynostosis cases were related to multiple sutural involvement, thereby constricting cranial volume, constricting the skull base, crowding the posterior fossa, and causing jugular foraminal stenosis [Cinalli et al., 1998]. These findings were most frequent among patients with Crouzon’s, Pfeiffer’s, and Apert’s syndrome, especially in association with cloverleaf skull abnormalities. A diffuse beaten copper pattern on skull radiographs, along with obliteration of anterior sulci or narrowing of basal cisterns in children under the age of 18 months, is predictive of increased intracranial pressure in over 95 percent of cases [Tuite and Lindquist, 1996].
Nonsyndromic Craniosynostosis Neurocognitive Development
There is a growing body of evidence that single-suture craniosynostosis is associated with neurobehavioral problems [Kapp-Simon, 1998; Magge et al., 2002]. In the 2004 manuscript, Speltz et al. nicely summarized all of the published neurobehavioral studies conducted on children with single suture craniosynostosis [Speltz et al., 2004].
Few studies have documented actual rates of mental retardation (typically defined as standardized test scores below 70). Several older studies reported slightly increased rates of mental retardation (from 6.5 to 12 percent) in comparison to an expected rate of about 2.2 percent in the population [Kapp-Simon, 1998; Hunter and Rudd, 1976, 1977; Sidoti et al., 1996].
Syndromes with multiple-suture fusions have more commonly been associated with elevated rates of mental retardation and learning disabilities [Cohen, 1991]. Most studies have found adverse neurocognitive outcomes in about 35–40 percent of assessed cases [Bottero et al., 1998; Shimoji et al., 2002; Shipster et al., 2003], with an occasional study finding this number to be as high as 50 percent [Kapp-Simon, 1998; Magge et al., 2002]. A few studies have reported a 3–5 times higher than average risk of poor neurobehavioral outcome. In studies that directly measured the IQs of children with sagittal synostosis, a discrepancy of greater than 20 standard score points between language and nonverbal IQ scores was found [Magge et al., 2002; Shipster et al., 2003]. In 2002, Magge et al. reported a 50 percent incidence of learning disability and found that verbal IQ was significantly higher than nonverbal IQ.
Sagittal synostosis has not been found to affect the neurological function of the infant or toddler significantly [Kapp-Simon et al., 1993]. However, older children may demonstrate moderately severe speech and language difficulties in up to 37 percent of cases, and the tendency toward such problems is associated with a positive family history for such difficulties and later age of surgical correction [Shipster et al., 2003; Virtanen et al., 1999; Panchal et al., 1999]. In a study of 30 older untreated children (average age 9.25 years) with sagittal synostosis, almost all patients and parents were pleased with their decision and patients demonstrated normal cognitive and school performance, as well as normal behavior and psychological adjustment on standardized testing [Boltshauser et al., 2003].
In a long-term neurodevelopmental outcome study of nonsyndromic cases, a correlation was found between outcomes and timing of surgery, with 22.6 percent of those operated on before 12 months showing impaired mental development, versus 52.2 percent impaired mental development in those operated on after age 12 months. Overall, 31 percent of trigonocephaly cases had delayed development, and this appeared to be related to the severity of the problem and the presence of associated malformations [Bottero et al., 1998].
Wide Cranial Sutures
Cranial sutures are considered to be widened when the sutural separation is more than 2 SD above the mean sutural width for age. Diagnosis is confirmed radiographically, although it may also be appreciated on palpation of the skull. Criteria for determining suture width in infants up to 45 days old have been published by Erasmie and Ringertz [Erasmie and Ringertz, 1976]. Wide cranial sutures in themselves cause no impairment, but they can be an indication of increased intracranial pressure or caused by craniosynostosis in another part of the skull. Wide cranial sutures are a feature of numerous syndromes (e.g., cleidocranial dysostosis, Hajdu–Cheney syndrome, or pycnodysostosis) with several proposed causes including increased intracranial pressure, delayed maturation of bone, or resorption of bone. The younger the child, the earlier sutural diastasis will appear after acutely increased intracranial pressure. The prognosis depends on the underlying cause.
Anomalies of Fontanels
The anterior fontanel is the largest of the fontanels, is diamond-shaped, and normally closes by 18 months. The posterior fontanel is triangular in shape and is usually closed at birth. Fontanel size may be measured in several different ways. Measurement of the anterior fontanel may be expressed as width (measurement along the coronal suture), length (measurement along the sagittal suture), area (width × length), or diagonal diameter. Standard curves are available for measurements taken by each method [Hall, 2007]. The posterior fontanel is usually measured only in length (measurement along the sagittal suture), but may also be measured along the lambdoid suture (width) and the area calculated (length × width).
Depending on the cause, a small or absent anterior fontanel may be noted at birth by palpation and confirmed by measurement and comparison with normal values for length and width [Hall, 2007]. A small or absent anterior fontanel usually indicates some type of underlying pathology, with the most common etiologies including any cause of congenital microcephaly, craniosynostosis (particularly involving the metopic suture), or accelerated bone maturation such as occurs in hyperthyroidism [Popich and Smith, 1972].
Large fontanels may be noted at birth by palpation and confirmed by measurement. Causes of both large fontanels and delayed closure include increased intracranial pressure or delayed ossification of the cranium [Popich and Smith, 1972]. Cleidocranial dysplasia is a common genetic syndrome that results in delayed closure of the anterior fontanel with widened cranial sutures and hypoplastic clavicles.
Occasionally, the anterior fontanel will ossify into a bony plate, which may be slightly elevated in relation to the rest of the cranium. This may relate to decreased growth-stretch tensile forces across the anterior fontanel, and is sometimes seen in cases of craniosynostosis, but it can also occur in otherwise normal infants, in which case, it is considered a normal variant [Keats and Anderson, 2001]. Instead of the usual flat, uncalcified, diamond-shaped anterior fontanel, there is a slightly raised diamond-shaped plate of bone in its place.
A small or absent anterior fontanel may be secondary to microcephaly (because of decreased brain growth), craniosynostosis affecting the metopic, sagittal, and/or coronal suture, or accelerated osseous maturation. There have also been reports of normal infants with absent anterior fontanels at birth. The shape of the anterior fontanel bone often remains visible on skull radiographs throughout childhood and on into adulthood, with a characteristic appearance on Towne projection. The appearance of the fusing anterior fontanel bone can be confused with a depressed skull fracture in the lateral projection [Girdany and Blank, 1965]. Occasionally, children thought to have a small or absent anterior fontanel will be found to have an anterior intrafontanel bone, which is of no significance. Causes of large or late-closing fontanels include increased intracranial pressure or delayed ossification of the skull [Popich and Smith, 1972].
Anterior fontanel closure occurs in 1 percent of normal infants by age 3 months, in 38 percent by 12 months, in 70 percent by 18 months, and in 97 percent by 24 months [Duc and Largo, 1986]. Therefore, early or late closure is reasonably common, but care must be taken to rule out underlying pathology. In normal infants, there is no sex difference in the size and age of closure of the anterior fontanel; nor is there any correlation with gestational age at delivery, head circumference, or bone age. The size of the fontanel at birth does not predict time of closure [Duc and Largo, 1986].
Extra fontanels are inconsistently occurring bony defects situated along the suture lines or at the junction of major bone plates of the skull. One such extra fontanel occurs along the sagittal suture about 2 cm anterior to the posterior fontanel. This fontanel is called the obeliac, interparietal, or sagittal fontanel, as well as the fontanel of Gerdy. Being midline, it is distinct from the parietal foramen, but parietal foramina may involve the lateral extremes of a large sagittal fontanel. Glabellar, metopic, or cerebellar fontanels also exhibit extra fontanels [Sidoti et al., 1996]. Diagnosis of an extra fontanel is made by palpation or radiography. It is often an isolated malformation, but may sometimes be associated with rare syndromes. In all cases, prognosis is dependent on the underlying cause. If the anomaly is isolated, there is no ill effect.
A sagittal fontanel is found in 6.3 percent of all newborns and is caused by a lack of union at the medial edge of junction of the two parietal ossification centers. This union normally occurs by the seventh month of gestation, but may occur 2–3 months after birth [Cohen, 1991]. In one study of infants with a sagittal fontanel diameter of greater than 13 mm, 5 percent had major anomalies and 35 percent had minor anomalies. In those with a diameter less than 13 mm, 2 of 45 infants studied had Down’s syndrome and 1 had a major anomaly. The latter group accounted for two-thirds of all children with this third fontanel [Keats and Anderson, 2001]. Although it is unknown whether there is ethnic variability in the prevalence of sagittal fontanels, the majority of individuals with this feature are male [Cohen, 1991]. Individuals with Down syndrome commonly have large fontanels that close late, along with a sagittal fontanel and persistence of the metopic suture [Cohen, 1991].
Cranial Dermal Sinus
A cranial dermal sinus is a midline depression or tract lined by stratified squamous epithelium that extends from the skin toward the central nervous system or its coverings. Cranial dermal sinuses are associated with bony defects in 80 percent of cases [Shackelf et al., 1974]. Diagnosis is best achieved radiographically via CT scan, which reveals a low-density lesion that may be surrounded by an enhancing ring [Starinsky et al., 1988]. Cranial dermal sinuses are most common in the occipital region, but can be found anywhere. Size can vary from a very small defect to a large, expanding mass. Clinical presentation is often as a cutaneous localized swelling that presents an infection or cystic expansion of the sinus tract beneath the skin surface, occasionally with drainage. There is often an abnormal distribution of hair along the defect. Occasionally, cystic expansion occurs within the cranial cavity, which obstructs cerebrospinal fluid flow, compresses the adjacent neural structures, and/or ruptures to cause sterile meningitis, which can be recurrent [Shackelf et al., 1974; Starinsky et al., 1988]. Cranial dermal sinuses are thought to be the result of faulty separation of neuroectoderm from cutaneous ectoderm during early gestation [Shackelf et al., 1974]. The recommended treatment is surgery; prognosis is good if treatment is done prior to the development of complications.
In the facial region, the most frequent congenital midline mass is a nasal dermoid sinus cyst, which can have intracranial extension and be associated with other anomalies [Posnick et al., 1994; Wardinsky et al., 1991]. Nasal dermoid sinus cysts constitute 11–12 percent of dermoids found in the head and neck [Wardinsky et al., 1991], and usually arise sporadically, although reports of familial occurrence have been documented [Posnick et al., 1994; Wardinsky et al., 1991]. At 50–60 days gestation, the nasal and frontal bones develop through intramembranous ossification, remaining separated by a space termed the fonticulus nasofrontalis. During growth, the nasal process of the frontal bone separates the skin from the dura, which maintains a connection between the base of the skull and the nasal tip via a dural projection, which ends at an opening in the frontal bones termed the foramen cecum. The foramen cecum eventually fuses with the fonticulus nasofrontalis in the area of the future cribriform plate, thereby obliterating this previous neuroectodermal connection. If this process remains incomplete, dermal connections (termed nasal dermoid sinus cysts) may occur anywhere from the nasal tip to the intracranial space through the foramen cecum [Posnick et al., 1994]. Such dermoid sinus cysts contain both ectodermal and mesodermal derivatives, and are composed of a stratified squamous epithelial lining, hair follicles, pilosebaceous glands, and smooth muscle [Posnick et al., 1994]. Nasal pits are present in 50 percent of patients with nasal dermoids [Mccaffrey et al., 1979]. often with hairs protruding from the pit, and they can appear anywhere along the nose, with intracranial extension noted in 36–45 percent of cases [Posnick et al., 1994; Wardinsky et al., 1991]. Associated anomalies were present in 41 percent of one series examined by a multidisciplinary team, and they were associated with many different syndromes, including hemifacial microsomia, oral-facial-digital (OFD) syndrome (type 1), frontonasal dysplasia, VATER (vertebral anomalies, anal anomalies, tracheoesophageal fistulae, esophageal atresia, renal and/or radial anomalies) association, and chromosome anomalies [Wardinsky et al., 1991]. Complications can result from enlargement of the cyst, skeletal distortion, and recurrent infection, so surgical excision is recommended after CT scanning to identify intracranial extension and plan the surgical approach.
Parietal Foramina (Includes Cranium Bifidum)
Parietal foramina are small defects in the superoposterior angles of the parietal bones via which emissary veins may pass through the calvarium (Figure 28-4). Usually, parietal foramina present as symmetrical oval defects situated on each side of the sagittal suture, and their size diminishes with age. They are covered with normal scalp and hair, and are detected through palpation and radiography. Occasionally, brain covered by dura and intact scalp can bulge through extensive lesions, suggesting the possibility of an encephalocele, but the location of these lesions off the midline differentiates them from neural tube closure defects.
Parietal foramina are usually small, with 60 percent being less than 1 mm. Such small defects are only detectable radiographically; however, 10 percent are 5 mm or more and can be as large as 50 mm in diameter. Reported individuals have had defects as large as 57 mm in diameter, with seizures apparently secondary to venous obstruction [Epstein and Epstein, 1967]. Small parietal foramina are found in 60–70 percent of all adults, whereas large parietal foramina are present in less than 1 percent of adults [O’Rahilly and Twohig, 1952; Little et al., 1990; Currarino, 1976]. Small unilateral defects are more common than bilateral defects. When the defect is unilateral, it more often involves the right side, and males are more commonly affected than females, with a ratio of 5:3 [O’Rahilly and Twohig, 1952]. Parietal foramina themselves cause no impairment, usually manifest autosomal-dominant inheritance with variable expression, and can occur as part of the phenotype in a few syndromes [Little et al., 1990].
Wilkie et al. [2000] described heterozygous MSX2 mutations in three unrelated families with enlarged parietal foramina, suggesting that loss of MSX2 activity results in calvarial defects [Wilkie et al., 2000]. A second gene has been implicated in those families who do not link to 5q34–q35, where MSX2 is located, and mutations or deletions of ALX4 on 11p11.2 can also result in parietal foramina [Wu et al., 2000]. Parietal foramina can occur as an isolated trait due to mutations in or haploinsufficiency of MSX2 or ALX4, or as a component of a multiple congenital anomaly syndrome, such as Saethre–Chotzen syndrome, cleidocranial dysplasia, or Rubinstein–Taybi syndrome. The combination of parietal foramina with multiple exostoses is now known to be a contiguous gene deletion of ALX4 and EXT2 on chromosome 11p11–p12 (also termed DEFECT11 syndrome).
Cranium bifidum literally means “cleft skull,” and presents as a wide opening between the frontal and parietal bones, which normally begin their process of intramembranous ossification in the center of each bone and then spread towards the sutures. During mid-childhood, these areas ossify, leaving only symmetric openings in the frontal and parietal bones. One reported family included individuals with both cranium bifidum and parietal foramina, confirming the fact that cranium bifidum in infancy and early childhood can evolve into large parietal foramina in later childhood and adulthood [Little et al., 1990].
Wormian Bones
Wormian bones were named after Dr. Worm, who initially described them as accessory bones that occur within cranial suture lines or fontanels [Gooding, 1971; Pryles and Khan, 1979]. They can occur singly, or in large numbers, and are diagnosed radiographically. Although they can occur within any suture, they are rare in coronal or sagittal sutures (Figure 28-5). Although they do not cause any impairment themselves, their significance is variable. In one study, the majority of children with an “excessive” number of Wormian bones had some abnormality of the central nervous system [Pryles and Khan, 1979]. These abnormalities ranged from gross malformations to minimal brain dysfunction, though this study may have been biased since it arose from a hospital-based population. Thus, some individuals with many Wormian bones may have other anomalies and/or central nervous system dysfunction.
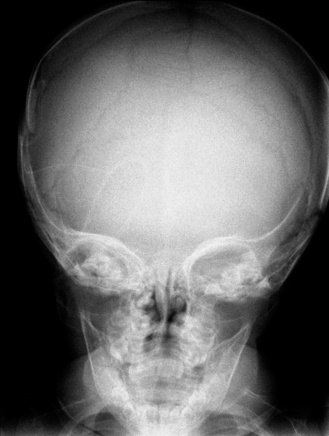
Fig. 28-5 Osteogenesis type IV with marked osteopenia of the skull and spine, and multiple Wormian bones.
The patient also had a history of multiple fractures with minimal trauma.
The pathogenesis of Wormian bones is thought to be related to intracranial strain along with open sutures causing ossification defects [Sanchez-Lara et al., 2007]. Such sutural bones persist and are not incorporated into the adjacent bone during mineralization and maturation [Gooding, 1971]. Although the prevalence of Wormian bones in the general population is 17 percent, the prevalence varies with age. Males are more often affected than females, and differences between ethnic groups have been noted. Wormian bones are commonly seen in osteogenesis imperfecta and other disorders resulting in defective cranial bone mineralization. The more severe the defective mineralization, the more numerous the Wormian bones, and such infants can also become quite brachycephalic as a consequence of postnatal supine positioning with soft cranial bones [Graham et al., 2005]. The increased frequency of Wormian bones in Chinese infants might relate to traditional supine sleep positioning practices in this population, and their resultant brachycephaly. If so, an increased frequency of Wormian bones may soon be noted in other cultures that now follow recommendations for supine sleep positioning to prevent sudden infant death syndrome (SIDS).
Scalp Vertex Aplasia
Scalp vertex aplasia, or aplasia cutis congenita (ACC), is a relatively common congenital defect resulting in localized absence of skin, usually occurring on the scalp as an isolated finding not associated with other abnormalities. Scalp vertex aplasia begins as multiple or solitary, sharply marginated, raw areas with absence of skin. These lesions mature into atrophic scars devoid of adnexal structures, usually in the vertex area or midline superior occipital region (Figure 28-6) [Tann and Tay, 1997]. The cause of these lesions is heterogeneous and includes vascular disruption, trauma, teratogens, and genetic factors [Evers et al., 1995]. Because vascular disruption and placental infarcts are seen in antiphospholipid antibody syndrome, some cases of extensive scalp vertex aplasia may be related to the effects of this maternal disease state or some other type of thrombophilia during pregnancy [Evers et al., 1995; Roll et al., 1999]. The frequency is 1 per 3000 live births, and a classification system of subtypes for ACC has been suggested by Frieden.
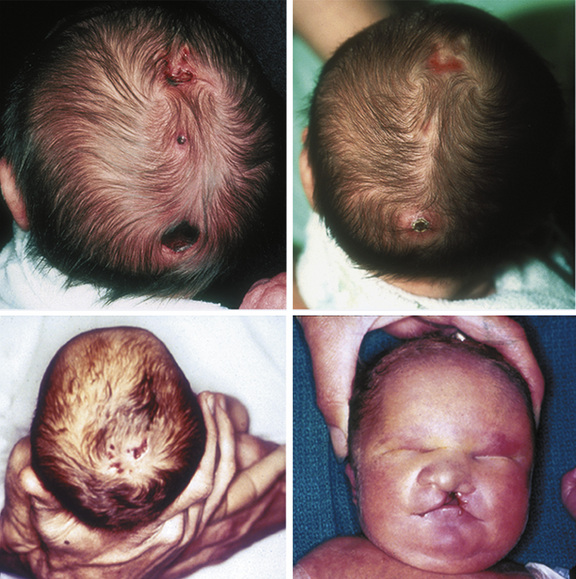
Lesions may be ulcerated, bullous, cicatricial, or covered with a tough, translucent membrane, and they occasionally extend to the bone or dura [Tann and Tay, 1997]. They may be circular, elongated, stellate, or triangular in shape, and of variable depth [Rudolph et al., 1974], with 86 percent of the solitary lesions occurring on the scalp, most often near the parietal hair whorl [Demmel, 1975; Stephan et al., 1982]. Type 1 ACC manifests scalp involvement without other abnormalities, and when familial, it manifests autosomal-dominant inheritance [Fimiani et al., 1999; Itin and Pletscher, 1988]. Less frequently, other parts of the body may be involved, with or without associated defects. When the lesions are midline and overlying the spine or midcranium, they can be associated with occult spinal dysraphism or tiny encephaloceles (ACC type 4). When there are multiple areas of ACC involving primarily the lower extremities, particularly the flank, thighs, and knees, a careful examination of the placenta may reveal a fetus papyraceus, which occurs 1 in 12,000 live births and affects 1 in 200 twin pregnancies [Leaute-Labreze et al., 1998; Daw, 1983; Mannino et al., 1977].
Larger defects can be complicated by hemorrhage, venous thrombosis, and rarely meningitis. With extensive ACC and other vasculodisruptive defects, survival during the neonatal period can be severely compromised [Lane and Zanol, 2000]. ACC can be associated with teratogenic or genetic disorders, and it is important to search for associated malformations in order to counsel parents concerning prognosis and recurrence risks in relation to the underlying disorder. ACC associated with epidermal nevus or nevus sebaceus syndrome (or ACC type 3) is the association of a sebaceous nevus (a linear yellow verrucous nevus) on the head and neck with variable ocular, cerebral, neurological, skeletal, cardiac, and other abnormalities, usually on a sporadic basis [Shields et al., 1997; Hogler et al., 1999]. Epidermolysis bullosa (EB) is a term applied to a group of hereditary skin disorders that result in the formation of blisters after minor skin trauma. EB can be associated with pyloric atresia and/or ACC; it manifests autosomal-recessive inheritance and histopathology is usually of the junctional type [Maman et al., 1998]. These findings suggest that when ACC occurs with EB, it is most likely to be autosomal-recessive, and some cases of junctional EB with pyloric atresia have demonstrated mutations in integrin beta 4 [Maman et al., 1998]. Finally, ACC has been associated with numerous cytogenetic and genetic malformation syndromes as ACC type 9 [Evers et al., 1995; Fimiani et al., 1999; Zvulunov et al., 1998; Edwards et al., 1994].
Wound treatment in cases of superficial ulceration is conservative, with antibacterial dressings, but extensive or deep lesions may require reconstruction of the scalp. Small hairless areas can be excised and covered with a neighboring flap from the scalp [Kruk-Jeromin et al., 1998]. This approach works for the most common scalp ACC lesions, which are frequently round, punched-out lesions in the vertex region, or less frequently triangular lesions in the temporal region (termed temporal triangular alopecia) [Kruk-Jeromin et al., 1998; Trakimas et al., 1994]. With extensive scalp lesions (over 6 cm in diameter), it is especially important to avoid eschar formation immediately after birth by covering exposed dura with split-thickness skin grafts from adjacent healthy scalp and moist dressings. Prompt closure is important because of the high risk of fatal hemorrhage from the sagittal sinus when the eschar becomes dry and separated, causing the underlying dura to become damaged and to tear [Yang and Yang, 2000]. Once the superficial defect is completely healed, the subsequent scar alopecia may be treated by tissue-expanded local flaps, pericranial flaps, or free vascularized flaps when the child is older. With prompt closure and a healthy underlying dura, cranial bone growth will occur after prompt early wound closure, and the risk of fatal hemorrhage or meningitis is greatly lessened [Yang and Yang, 2000].
Thin Cranial Bones
Thin cranial bones are those that appear thinner than average and have little or no diploe. The diagnosis of thin cranial bones is made radiographically and is usually subjective [Ethier, 1971]. Thin calvarial bones can be secondary to craniosynostosis (particularly adjacent to the ridging in sagittal synostosis) and hydrocephalus, or can occur as part of several syndromes in which undermineralization is a feature [Hodges, 1989]. Areas of radiolucency are called craniolacunae (luckenschadel) and are often secondary to spinal dysraphism. These generally disappear by age 1 year [McRae, 1971]. Regionally thinned areas can also occur in association with porencephaly, subdural hygroma, arachnoid cyst, and some tumors [Hodges, 1989]. Generalized thinning of cranial bones also results from increased dural distension, when expansion of the brain outpaces the growth of the skull. In other instances, undermineralization is the cause. As with thickened cranial bones, it is unknown whether thin cranial bones can occur as an isolated trait. Craniolacunae probably represent defective membranous bone formation, particularly along the inner periosteum of the cranial vault [McRae, 1971]. Craniolacunae probably do not occur as isolated traits. Prognosis is dependent on the underlying cause of this condition, and lacunar skull defects themselves have no direct effects on the infant.
Undermineralization of the Skull
Undermineralization of the skull results in increased radiolucency of the cranial bones and is attributable to decreased calcium deposition. Congenital undermineralization occurs in a number of syndromes, particularly osteogenesis imperfecta and hypophosphatasia (see Figure 28-5). Hypophosphatasia occurs in at least three forms, including infantile, childhood, and adult forms. Undermineralization is most pronounced in the infantile form and least evident in the adult form. The infantile form can usually be diagnosed by fetal ultrasound, whereas the other forms are often diagnosed after birth by radiographs and measurement of serum alkaline phosphatase levels [Goodman and Gorlin, 1983; Wynne-Davies et al., 1985]. Fluorosis and vitamin D-dependent rickets can also produce postnatal undermineralization of the skull, but areas of sclerosis are also present in fluorosis. The incidence of undermineralization is low, and prognosis is dependent on cause, varying widely from stillbirth or death during infancy to little effect at all.
Craniotabes
Prolonged forceful pressure on the fetal vertex may result in diminished cranial mineralization affecting the superior portions of the parietal bones. Such craniotabes is more likely to occur in first-born infants, especially with early fetal head descent into a vertex presentation for a prolonged period of time. Mild degrees of craniotabes occur in about 2 percent of newborn babies, and more extensive degrees of craniotabes are less common [Fox and Maier, 1984]. Craniotabes was first described in congenital syphilis, and it can also be seen with subclinical rickets due to vitamin D deficiency [Kokkonen et al., 1983]. Rickets should be considered in any infant with a nonvertex presentation, whose mother might be at risk for nutritional deficiency, and such infants usually manifest generalized craniotabes with osteomalacia.
With compression-related craniotabes, the superior parieto-occipital region tends to be soft to palpation, and often indents upon finger compression. In extreme cases, the entire top of the head can be involved. The presence of normally firm bone along the sides of the calvarium and in the mastoid regions readily differentiates this benign form of craniotabes from more generalized problems of decreased mineralization, such as hypophosphatasia, osteogenesis imperfecta, or infantile rickets. Within the affected region of the calvarium, the sutures and fontanels may also feel wider than usual. Accentuated vertex molding can be an associated feature in a fetus with prolonged vertex engagement. Benign vertex craniotabes has not been reported in babies in breech presentation, and radiolucency of the parietal bones in the vertex of the skull is considered to be a normal anatomic variant on neonatal head CT scans [Pastakia and Herdt, 1984].
With compression-related craniotabes, the prognosis is excellent, and the calvarium usually mineralizes in a normal fashion within 1–2 months after birth [Graham and Smith, 2007]. If the mother has vitamin D-deficient rickets and there is more generalized craniotabes and osteomalacia, this condition usually manifests a prompt response to vitamin D therapy over the next few months. Vitamin D-deficient rickets generally is accompanied by metaphyseal changes at the wrist and low 25-hydroxyvitamin D concentrations (less than 12 ng/mL), with a variably elevated alkaline phosphatase level. As in other defects of skeletal mineralization, such as osteogenesis imperfecta and hypophosphatasia, initial care must be taken to avoid fractures. Infants with osteogenesis imperfecta or hypophosphatasia usually show generalized osteomalacia, brittle bones, and Wormian bones.
Thick Cranial Bones
Numerous syndromes with thick calvarial bones have been described. It is unknown whether thick cranial bones can occur as an isolated trait, and the prognosis depends on the underlying condition. Thickening of the middle table is usually a manifestation of overproliferation of bone marrow in hemolytic disease or bone diseases. In hemolytic diseases, such as thalassemia, vertical striations (“hair-on-end” appearance) occur, whereas in bone diseases such as osteopetrosis, sclerosis occurs [Hodges, 1989]. Overgrowth of the middle table can also occur in microcephaly. In situations in which a shunt has been placed to relieve hydrocephalus, thickening of both inner and middle tables can occur.
Sclerosis and Hyperostosis of the Skull
Increased density or overmineralization of the cranial bones can be generalized or localized, and this is termed sclerosis or hyperostosis of the skull. Sclerosis generally refers to an increase in bone density without an alteration in width, while hyperostosis is caused by bone overgrowth that leads to an increase in density and width, though not all cases fit cleanly into one category or the other [Kozlowski and Beighton, 1995]. Hyperostosis is distinct from thick cranial bones, although hyperostosis and occasionally sclerosis can also cause thick cranial bones [Ethier, 1971]. Most of the sclerosing bone dysplasias manifest generalized changes, which are classified on the basis of the distribution and configuration of these abnormalities. One subclassification divides these disorders into osteosclerosis, craniotubular dysplasis, and craniotubular hyperostoses. In such conditions, basal sclerosis may be present without significant calvarial involvement, but the converse rarely occurs [Kozlowski and Beighton, 1995].
The presence of sclerosis or hyperostosis can be diagnosed radiographically or by CT, and scintigraphy may provide information on disease progression (Figure 28-7) [Kumar et al., 1981]. Radiologic changes are age-related, and definitive diagnosis may be difficult in early childhood [Kozlowski and Beighton, 1995]. All conditions that cause generalized osteosclerosis affect the skull [Kozlowski and Beighton, 1995]. Localized sclerosis of the base of the skull can occur in fibrous dysplasia, Jansen-type metaphyseal dysplasia, severe anemia, hypercalciuria, and Paget’s disease. It may also be seen with a meningioma or inflammation [Kozlowski and Beighton, 1995]. Symptoms include narrowing of cranial nerve foramina, which in turn can cause nerve palsy, deafness, or vision defects [Beighton et al., 1976]. Increased intracranial pressure is not uncommon, and papilledema can also occur as a complication. Craniosynostosis also occurs in some cases, perhaps related to overstimulation of bone growth along suture edges. Several syndromes have sclerosis as a feature.
Fig. 28-7 Craniometaphyseal dysplasia presenting with progressive craniofacial changes and hearing loss.
Two primary pathologic processes can lead to sclerosis: overproduction of bone and/or failure of osteoclastic absorption of bone [Ethier, 1971]. The prognosis depends on the underlying cause and varies from individuals being asymptomatic to sudden death from medullary compression. In addition, facial palsy, as well as hearing and vision loss, may occur due to cranial nerve compression within stenotic foramina, which may require surgical decompression [Kumar et al., 1981; Beighton et al., 1976]. Craniotomy to relieve increased intracranial pressure may also be indicated.
Anomalies of the Sella Turcica
Anomalies include abnormal size and/or shape of the sella turcica, which is the central depression within the sphenoid bone that contains the pituitary gland. Assessment of the sella turcica can best be done radiographically or by CT [Pribam, 1971]. Measurements of normal sella turcica size have been published, with considerable overlap between normal and abnormal ranges [Fisher and Dichiro, 1964; Oon, 1963]. Small sellas have been described in patients with hypopituitarism and myotonic dystrophy, whereas large sellas occur in patients with storage disorders, pituitary tumors, empty sella syndrome, craniopharyngioma, intrasellar aneurysm, untreated hypogonadism, and hypothyroidism.
A J-shaped sella describes the lateral profile of the sella turcica, in which the sella resembles a “J” lying on its side. A J-shaped sella can occur as a normal variant, but may also occur in individuals with calvarial enlargement or optic nerve gliomas [Swischuk, 1972]. Bridged sella is caused by bony bridging between anterior and posterior clinoids and can be a normal variant. It can also be seen in nevoid basal cell carcinoma syndrome (Gorlin’s syndrome).
The sphenoid bone consists of two main cartilaginous parts (hypophyseal cartilage) until the seventh or eighth month of gestation. The presphenoid will contribute to the anterior part of the sella turcica, while the postsphenoid forms the remainder. At birth, the sella is only a small depression; it begins to ossify soon after birth. Since 80 percent of the sella is occupied by the pituitary gland, it is not unusual for pituitary anomalies to cause abnormalities in the sella. In individuals with optic nerve gliomas, the chiasmatic groove will appear scalloped, whereas in cases of calvarial enlargement it will appear elongated [Swischuk, 1972]. Sellar abnormalities do not themselves require treatment. However, they are usually indicative of an underlying pathologic process that may require treatment. Prognosis, therefore, is dependent on the underlying cause.
Anomalies of Foramen Magnum
The foramen magnum is normally an oval-shaped opening in the occipital bone bounded anteriorly by the basiocciput, laterally by the occipital condyles, and posteriorly by the supraocciput [McRae, 1971]. These bones are separated by two anterior and two posterior synchondroses that begin to fuse at 12 months and completely fuse by 3–4 years and 7 years, respectively [Hecht et al., 1985]. If enchondral ossification is abnormal, or suture fusion premature, or both, a small foramen magnum is the result. Anomalies include either small or large size, or a keyhole shape. MRI best achieves diagnostic assessment of foramen magnum size or shape, although radiography or CT may also be used. Tables have been published indicating normal foramen magnum size [Hecht et al., 1985]. Effects of a small foramen magnum vary from producing no symptoms to being associated with weakness, apneic spells, hyperreflexia, hydrocephalus, and abnormal somatosensory-evoked potentials and/or polysomnograms [Hecht et al., 1985]. Achondroplasia is the most common syndrome in which a small foramen magnum occurs, but other skeletal dysplasias and disorders associated with sclerosis of the skull can also lead to a small foramen magnum. Patients with achondroplasia usually do not experience neurological complications until the foramen magnum is 4 SD or more below the mean [Hecht et al., 1985], and 96 percent of achondroplastic patients with neurological manifestations have foramen magnum sizes more than 3 SD below the mean [Wang et al., 1987]. A small foramen magnum can be accompanied by a short cranial base, and anterior herniation of the brain through an open metopic suture has been known to occur in such cases [McRae, 1971]. Premature synostosis of one or two sutures may cause asymmetry [Coin, 1971]. A large foramen magnum usually results from chronic increased intracranial pressure or from direct effects of an expanding process within the foramen magnum (syringomyelia, Arnold–Chiari malformation) [Wang et al., 1987; Coin, 1971; Salonen et al., 1981]. Asymmetry of the foramen magnum occurs with craniovertebral anomalies or premature synostosis of one or more of the occipital synchondroses [Coin, 1971]. Children with the latter may tend to hold their heads obliquely. A keyhole-shaped foramen magnum has been described in the hydrolethalus syndrome [Salonen et al., 1981].
Prognosis for a small foramen magnum is variable, but the most serious outcome of brainstem compression may be sudden death. Recommended treatment is suboccipital craniectomy [Hecht et al., 1985]. The prognosis for a large or abnormally shaped foramen magnum is dependent on the underlying cause.
Basilar Impression
Basilar impression is a malformation or deformation of the cranial base consisting of indentation of the base of the skull at the craniospinal junction. Primary basilar impression is a malformation in which the base of the skull and the upper two cervical vertebrae fail to segment and exist as a bony mass within which the posterior fossa, brainstem, and upper cervical spinal cord may become compressed. Some degree of basilar impression may occur with platybasia, defined as a craniocervical angle of greater than 140°. Basilar impression may be suspected when there is limited movement and shortening of the neck, but definitive diagnosis requires radiography, CT scan, or MRI scan [Sondheimer, 1971]. In basilar impression, the odontoid moves cephalad and can protrude into the foramen magnum, thus compromising function of the spinal cord, brainstem, and cerebellum, as well as impeding the flow of cerebrospinal fluid. Symptoms include pain, limitation of movement, increased intracranial pressure, hydrocephalus, and cranial nerve symptoms [Teodori and Painter, 1984; Roger et al., 1948]. Symptoms may appear suddenly or develop over several months.
Primary basilar impression is caused by a congenital defect of osseous structures in the cervico-occipital region, and can occur as an autosomal-dominant trait [Bull et al., 1955]. Secondary basilar impression is related to disease of the skull. It has been reported in association with Paget’s disease, histiocytosis X, rheumatoid arthritis, rickets, and hypoparathyroidism. It also occurs in several skeletal dysplasias and malformation syndromes. The incidence in the general population is 1 in 3300, although it may be more common in Eskimos, and in cultures where carrying heavy loads on the top of the head is practiced [Teodori and Painter, 1984].
The prognosis is quite variable. Affected individuals may be asymptomatic, develop sudden or progressive symptoms, or die suddenly. Most patients present with symptoms in late childhood or early adulthood, which corresponds to the time of closure of the anterior synchondroses (6 years) and spheno-occipital synchondrosis (25 years) of the occipital bone [Adam, 1987]. Treatment consists of immobilization or, in severe cases, decompression of the foramen magnum, laminectomy of the first and second cervical vertebrae, and cervico-occipital fusion [Rush et al., 1989]. Shunting for hydrocephalus may also be indicated.
Miscellaneous Anomalies of the Skull
Paracondylar Process
A paracondylar process is an asymptomatic anatomic variant that is visible only on CT; it consists of a process of bone that arises from the lateral aspect of the condyloid process and extends toward the transverse process of the atlas. It is generally of no significance, but has been reported in children with hemifacial microsomia. It is present in 7–8 percent of all human skulls [Silverman et al., 1993].
Bathrocephaly
Bathrocephaly is a skull deformation that appears as a steplike deformity at the back of the skull, and it is also termed an occipital shelf. It is not associated with craniosynostosis, but can occur secondary to breech position in utero. When associated with breech presentation, the head is usually also dolichocephalic in shape. When associated with other abnormal fetal positions, such as a transverse lie, face presentation, or brow presentation, the forehead may be compressed. Hence, any constraining fetal position that retroflexes the head against the posterior neck and shoulders can result in bathrocephaly. Other anomalies related to breech presentation are often present, and these include dislocated hips, congenital muscular torticollis, clubfoot, or uplifted earlobes. In general, this anomaly is of no significance, and spontaneous improvement usually occurs. Prognosis is poor only if breech position is secondary to malformation or neurological dysfunction [Silverman et al., 1993].
Occipital Horns
Occipital horns are bony protuberances situated on both sides of the foramen magnum and pointing caudad. They have only been described in individuals with an X-linked syndrome that also includes obstructive uropathy, joint laxity, and other features of Ehlers–Danlos type IX [Lazoff et al., 1975].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adam A.M. Skull radiograph measurements of normals and patients with basilar impression; use of Landzert’s angle. Surg Radiol Anat. 1987;9:225-229.
Beighton P., Durr L., Hamersma H. The clinical features of sclerosteosis. A review of the manifestations in twenty-five affected individuals. Ann Intern Med. 1976;84:393-397.
Boltshauser E., Ludwig S., Dietrich F., et al. Sagittal craniosynostosis: cognitive development, behaviour, and quality of life in unoperated children. Neuropediatrics. 2003;34:293-300.
Bottero L., Lajeunie E., Arnaud E., et al. Functional outcome after surgery for trigonocephaly. Plast Reconstr Surg. 1998;102:952-958. discussion 9–60
Bull J.W., Nixon W.L., Pratt R.T. The radiological criteria and familial occurrence of primary basilar impression. Brain. 1955;78:229-247.
Cinalli G., Sainte-Rose C., Kollar E.M., et al. Hydrocephalus and craniosynostosis. J Neurosurg. 1998;88:209-214.
Cohen M.M.Jr. Etiopathogenesis of craniosynostosis. Neurosurg Clin N Am. 1991;2:507-513.
Cohen M.M., MacLean R.E. Craniosynostosis: diagnosis, evaluation, and management, ed 2. New York: Oxford University Press; 2000.
Coin C.G.M.D. Foramen magnum. In: Newton T.H.P.D., editor. Radiology of the Skull and Brain. Great Neck, NY: Medibooks; 1971:275.
Currarino G. Normal variants and congenital anomalies in the region of the obelion. Am J Roentgenol. 1976;127:487-494.
Dambrain R., Freund M., Verellen G., et al. Considerations about the cloverleaf skull. J Craniofac Genet Dev Biol. 1987;7:387-401.
Daw E. Fetus papyraceus–11 cases. Postgrad Med J. 1983;59:598-600.
Demmel U. Clinical aspects of congenital skin defects. I. Congenital skin defects on the head of the newborn. Eur J Pediatr. 1975;121:21-50.
Duc G., Largo R.H. Anterior Fontanel – Size and Closure in Term and Preterm Infants. Pediatrics. 1986;78:904-908.
Edwards M.J., McDonald D., Moore P., et al. Scalp-ear-nipple syndrome: additional manifestations. Am J Med Genet. 1994;50:247-250.
Epstein J.A., Epstein B.S. Deformities of the skull surfaces in infancy and childhood. J Pediatr. 1967;70:636-647.
Erasmie U., Ringertz H. Normal width of cranial sutures in the neonate and infant. An objective method of assessment. Acta Radiol Diagn (Stockh). 1976;17:565-572.
Ethier R. Thickness and texture. In: Newton T., Potts D.G., editors. Radiology of the Skull and Brain. Great Neck, NY: Medibooks; 1971:154.
Ethier R. Thickness and texture. In: Newton T.P., Potts D.G., editors. Radiology of the Skull and Brain. Great Neck, NY: Medibooks, 1971.
Evers M.E., Steijlen P.M., Hamel B.C. Aplasia cutis congenita and associated disorders: an update. Clin Genet. 1995;47:295-301.
Fimiani M., Seri M., Rubegni P., et al. Autosomal dominant aplasia cutis congenita: report of a large Italian family and no hint for candidate chromosomal regions. Arch Dermatol Res. 1999;291:637-642.
Fisher R.L., Dichiro G. the small sella turcica. Am J Roentgenol Radium Ther Nucl Med. 1964;91:996-1008.
Fox G.N., Maier M.K. Neonatal craniotabes. Am Fam Physician. 1984;30:149-151.
Frank L.M., Mason M.A., Magee W.P.Jr, et al. The kleeblattschadel deformity: neurologic outcome with early treatment. Pediatr Neurol. 1985;1:379-381.
Girdany B.R., Blank E. Anterior fontanel bones. Am J Roentgenol Radium Ther Nucl Med. 1965;95:148-153.
Gooding C.A. Cranial sutures and fontanelles. Great Neck, NY: Medibooks; 1971.
Goodman R.M., Gorlin R.J. The malformed infant and child: an illustrated guide. New York: Oxford University Press; 1983.
Graham J.M.Jr, Smith D.W. Metopic craniostenosis as a consequence of fetal head constraint: two interesting experiments of nature. Pediatrics. 1980;65:1000-1002.
Graham J.M., Smith D.W. Smith’s recognizable patterns of human deformation, ed 3. Philadelphia: Saunders/Elsevier; 2007.
Graham J.M.Jr, deSaxe M., Smith D.W. Sagittal craniostenosis: fetal head constraint as one possible cause. J Pediatr. 1979;95:747-750.
Graham J.M.Jr, Badura R.J., Smith D.W. Coronal craniostenosis: fetal head constraint as one possible cause. Pediatrics. 1980;65:995-999.
Graham J.M.Jr, Gomez M., Halberg A., et al. Management of deformational plagiocephaly: repositioning versus orthotic therapy. J Pediatr. 2005;146:258-262.
Graham J.M.Jr, Kreutzman J., Earl D., et al. Deformational brachycephaly in supine-sleeping infants. J Pediatr. 2005;146:253-257.
Hall J.G. Handbook of physical measurements, ed 2. Oxford; New York: Oxford University Press; 2007.
Hecht J.T., Nelson F.W., Butler I.J., et al. Computerized tomography of the foramen magnum: achondroplastic values compared to normal standards. Am J Med Genet. 1985;20:355-360.
Higginbottom M.C., Jones K.L., James H.E. Intrauterine constraint and craniosynostosis. Neurosurgery. 1980;6:39-44.
Hodges F.I. Pathology of the skull. In: Taveras J., editor. Neuroradiology of the Head and Neck. Philadelphia: JB Lippincott Co, 1989.
Hogler W., Sidoroff A., Weber F., et al. Aplasia cutis congenita, uvula bifida and bilateral retinal dystrophy in a girl with Naevus sebaceous syndrome. Br J Dermatol. 1999;140:542-543.
Huang M.H., Gruss J.S., Clarren S.K., et al. The differential diagnosis of posterior plagiocephaly: true lambdoid synostosis versus positional molding. Plast Reconstr Surg. 1996;98:765-774. discussion 75–6
Hunter A.G., Rudd N.L. Craniosynostosis. I. Sagittal synostosis: its genetics and associated clinical findings in 214 patients who lacked involvement of the coronal suture(s). Teratology. 1976;14:185-193.
Hunter A.G., Rudd N.L. Craniosynostosis. II. Coronal synostosis: its familial characteristics and associated clinical findings in 109 patients lacking bilateral polysyndactyly or syndactyly. Teratology. 1977;15:301-309.
Itin P., Pletscher M. Familial aplasia cutis congenita of the scalp without other defects in 6 members of three successive generations. Dermatologica. 1988;177:123-125.
Jabs E.W., Muller U., Li X., et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75:443-450.
Kapp-Simon K.A. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac J. 1998;35:197-203.
Kapp-Simon K.A., Figueroa A., Jocher C.A., et al. Longitudinal assessment of mental development in infants with nonsyndromic craniosynostosis with and without cranial release and reconstruction. Plast Reconstr Surg. 1993;92:831-839. discussion 40–1
Keats T.E., Anderson M. Atlas of normal roentgen variants that may simulate disease, ed 7. St. Louis, Mo: Mosby; 2001.
Kokkonen J., Koivisto M., Lautala P., et al. Serum calcium and 25-OH-D3 in mothers of newborns with craniotabes. J Perinat Med. 1983;11:127-131.
Koskinen-Moffett L.K., Moffett B.C.Jr, Graham J.M.Jr. Cranial synostosis and intra-uterine compression: a developmental study of human sutures. Prog Clin Biol Res. 1982;101:365-378.
Koskinen-Moffett L., Moffet B.C. Sutures and intrauterine deformation. In: Pershing J.A., MTEaJAJ. Scientific Foundations and Surgical Treatment of Craniosynostosis. Williams & Wilkins; 1989:96-106.
Kozlowski K., Beighton P. Gamut index of skeletal dysplasias: an aid to radiodiagnosis, ed 2. London; New York: Springer; 1995.
Kroczek R.A., Muhlbauer W., Zimmermann I. Cloverleaf skull associated with Pfeiffer syndrome: pathology and management. Eur J Pediatr. 1986;145:442-445.
Kruk-Jeromin J., Janik J., Rykala J. Aplasia cutis congenita of the scalp. Report of 16 cases. Dermatol Surg. 1998;24:549-553.
Kumar B., Murphy W.A., Whyte M.P. Progressive diaphyseal dysplasia (Engelmann disease): scintigraphic-radiographic-clinical correlations. Radiology. 1981;140:87-92.
Lajeunie E., Le Merrer M., Bonaiti-Pellie C., et al. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet. 1995;55:500-504.
Lajeunie E., Le Merrer M., Bonaiti-Pellie C., et al. Genetic study of scaphocephaly. Am J Med Genet. 1996;62:282-285.
Lajeunie E., Le Merrer M., Marchac D., et al. Syndromal and nonsyndromal primary trigonocephaly: analysis of a series of 237 patients. Am J Med Genet. 1998;75:211-215.
Lane W., Zanol K. Duodenal atresia, biliary atresia, and intestinal infarct in truncal aplasia cutis congenita. Pediatr Dermatol. 2000;17:290-292.
Lazoff S.G., Rybak J.J., Parker B.R., et al. Skeletal dysplasia, occipital horns, diarrhea and obstructive uropathy – a new hereditary syndrome. Birth Defects Orig Artic Ser. 1975;11:71-74.
Leaute-Labreze C., Depaire-Duclos F., Sarlangue J., et al. Congenital cutaneous defects as complications in surviving co-twins. Aplasia cutis congenita and neonatal volkmann ischemic contracture of the forearm. Arch Dermatol. 1998;134:1121-1124.
Little B.B., Knoll K.A., Klein V.R., et al. Hereditary cranium bifidum and symmetric parietal foramina are the same entity. Am J Med Genet. 1990;35:453-458.
Magge S.N., Westerveld M., Pruzinsky T., et al. Long-term neuropsychological effects of sagittal craniosynostosis on child development. J Craniofac Surg. 2002;13:99-104.
Ma L., Golden S., Wu L., et al. The molecular basis of Boston-type craniosynostosis: the Pro148His mutation in the N-terminal arm of the MSX2 homeodomain stabilizes DNA binding without altering nucleotide sequence preferences. Hum Mol Genet. 1996;5:1915-1920.
Maman E., Maor E., Kachko L., et al. Epidermolysis bullosa, pyloric atresia, aplasia cutis congenita: histopathological delineation of an autosomal recessive disease. Am J Med Genet. 1998;78:127-133.
Mannino F.L., Jones K.L., Benirschke K. Congenital skin defects and fetus papyraceus. J Pediatr. 1977;91:559-564.
Mccaffrey T.V., Mcdonald T.J., Gorenstein A. Dermoid Cysts of the Nose – Review of 21 Cases. Otolaryng Head Neck Surg. 1979;87:52-59.
McCarthy J.G., Glasberg S.B., Cutting C.B., et al. Twenty-year experience with early surgery for craniosynostosis: I. Isolated craniofacial synostosis–results and unsolved problems. Plast Reconstr Surg. 1995;96:272-283.
McRae D. Craniovertebral junction. In: Newton T.H.P.D., editor. Radiology of the Skull and Brain. Great Neck, NY: Medibooks; 1971:260.
McRae D. Lacunar skull, luckenschadel. In: Newton T., Potts D.G., editors. Radiology of the Skull and Brain. Great Neck, NY: Medibooks; 1971:648.
Mulliken J.B., Steinberger D., Kunze S., et al. Molecular diagnosis of bilateral coronal synostosis. Plast Reconstr Surg. 1999;104:1603-1615.
Oon C.L. The size of the pituitary fossa in adults. Br J Radiol. 1963;36:294-299.
O’Rahilly R., Twohig M.J. Foramina parietalia permagna. Am J Roentgenol Radium Ther Nucl Med. 1952;67:551-561.
Panchal J., Marsh J.L., Park T.S., et al. Sagittal craniosynostosis outcome assessment for two methods and timings of intervention. Plast Reconstr Surg. 1999;103:1574-1584.
Pastakia B., Herdt J.R. Radiolucent “zones” in parietal bones seen on computed tomography: a normal anatomic variant. J Comput Assist Tomogr. 1984;8:108-109.
Polley J.W., Charbel F.T., Kim D., et al. Nonsyndromal craniosynostosis: longitudinal outcome following cranio-orbital reconstruction in infancy. Plast Reconstr Surg. 1998;102:619-628. discussion 29–32
Popich G.A., Smith D.W. Fontanels – Range of Normal Size. J Pediatr. 1972;80:749-752.
Posnick J.C., Bortoluzzi P., Armstrong D.C., et al. Intracranial Nasal Dermoid Sinus Cysts – Computed Tomographic Scan Findings and Surgical Results. Plast Reconstr Surg. 1994;93:745-754.
dGPribam H.W. Sella turcica. In: Newton T.H.P.D., editor. Radiology of the Skull and Brain. Great Neck, NY: Medibooks; 1971:357.
Pryles C.V., Khan A.J. Wormian bones. A marker of CNS abnormality? Am J Dis Child. 1979;133:380-382.
Roger H., Alliez J., Payan H. Bull Mem Soc Med Hop Paris. 1948;64:524-529.
Roll C., Hanssler L., Voit T., et al. Aplasia cutis congenita–etiological relationship to antiphospholipid syndrome? Clin Dysmorphol. 1999;8:215-217.
Rudolph R.I., Schwartz W., Leyden J.J. Bitemporal aplasia cutis congenita. Occurrence with other cutaneous abnormalities. Arch Dermatol. 1974;110:615-618.
Rush P.J., Berbrayer D., Reilly B.J. Basilar impression and osteogenesis imperfecta in a three-year-old girl: CT and MRI. Pediatr Radiol. 1989;19:142-143.
Sadler T.W., Langman J. Langman’s medical embryology, ed 11. Baltimore, Md: Lippincott William & Wilkins; 2009.
Salonen R., Herva R., Norio R. The hydrolethalus syndrome: delineation of a “new”, lethal malformation syndrome based on 28 patients. Clin Genet. 1981;19:321-330.
Sanchez-Lara P.A., Graham J.M.Jr, Hing A.V., et al. The morphogenesis of wormian bones: a study of craniosynostosis and purposeful cranial deformation. Am J Med Genet A. 2007;143A:3243-3251.
Sanchez-Lara P.A., Carmichael S.L., Graham J.M.Jr, et al. Fetal constraint as a potential risk factor for craniosynostosis. Am J Med Genet A. 2010;152A:394-400.
Schweitzer D.N., Graham J.M.Jr, Lachman R.S., et al. Subtle radiographic findings of achondroplasia in patients with Crouzon syndrome with acanthosis nigricans due to an Ala391Glu substitution in FGFR3. Am J Med Genet. 2001;98:75-91.
Sgouros S., Goldin J.H., Hockley A.D., et al. Posterior skull surgery in craniosynostosis. Childs Nerv Syst. 1996;12:727-733.
Shackelf G.D., Shackelf P.G., Schwetsc P.R., et al. congenital occipital dermal sinus. Radiology. 1974;111:161-166.
Shields J.A., Shields C.L., Eagle R.C.Jr, et al. Ocular manifestations of the organoid nevus syndrome. Ophthalmology. 1997;104:549-557.
Shimoji T., Shimabukuro S., Sugama S., et al. Mild trigonocephaly with clinical symptoms: analysis of surgical results in 65 patients. Childs Nerv Syst. 2002;18:215-224.
Shipster C., Hearst D., Somerville A., et al. Speech, language, and cognitive development in children with isolated sagittal synostosis. Dev Med Child Neurol. 2003;45:34-43.
Sidoti E.J.Jr, Marsh J.L., Marty-Grames L., et al. Long-term studies of metopic synostosis: frequency of cognitive impairment and behavioral disturbances. Plast Reconstr Surg. 1996;97:276-281.
Silverman F.N., Kuhn J.P., Caffey J. Caffey’s pediatric X-ray diagnosis: an integrated imaging approach, ed 9. St. Louis: Mosby Year Book; 1993.
Sondheimer F. Basal foramina and canals. Radiology of the Skull and Brain. In: Newton T.H.P.D., editor. Radiology of the Skull and Brain. Great Neck, NY: Medibooks; 1971:287.
Speltz M.L., Kapp-Simon K.A., Cunningham M., et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol. 2004;29:651-668.
Starinsky R., Wald U., Michowitz S.D., et al. Dermoids of the Posterior-Fossa – Case-Reports and Review. Clin Pediatr. 1988;27:579-582.
Stephan M.J., Smith D.W., Ponzi J.W., et al. Origin of scalp vertex aplasia cutis. J Pediatr. 1982;101:850-853.
Stevenson R.E.S.R. The significance of the kleeblattschadel malformation. Proc Greenwood Genet Center. 5, 1986.
Swischuk L.E. The normal pediatric skull. Variations and artefacts. Radiol Clin North Am. 1972;10:277-290.
Tann H.H., Tay Y.K. Familial aplasia cutis congenita of the scalp; a case report and review. Ann Acad Med Singapore. 1997;26:500-502.
Teodori J.B., Painter M.J. Basilar impression in children. Pediatrics. 1984;74:1097-1099.
Trakimas C., Sperling L.C., Skelton H.G.3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
Tuite M.F., Lindquist S.L. Maintenance and inheritance of yeast prions. Trends Genet. 1996;12:467-471.
Turner P.T., Reynolds A.F. Generous craniectomy for Kleeblattschadel anomaly. Neurosurgery. 1980;6:555-558.
Virtanen R., Korhonen T., Fagerholm J., et al. Neurocognitive sequelae of scaphocephaly. Pediatrics. 1999;103:791-795.
Wang H., Rosenbaum A.E., Reid C.S., et al. Pediatric patients with achondroplasia: CT evaluation of the craniocervical junction. Radiology. 1987;164:515-519.
Wardinsky T.D., Pagon R.A., Kropp R.J., et al. Nasal Dermoid Sinus Cysts – Association with Intracranial Extension and Multiple Malformations. Cleft Palate Craniofac J. 1991;28:87-95.
Warman M.L., Mulliken J.B., Hayward P.G., et al. Newly recognized autosomal dominant disorder with craniosynostosis. Am J Med Genet. 1993;46:444-449.
Wilkie A.O., Tang Z., Elanko N., et al. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet. 2000;24:387-390.
Williams J.K., Cohen S.R., Burstein F.D., et al. A longitudinal, statistical study of reoperation rates in craniosynostosis. Plast Reconstr Surg. 1997;100:305-310.
Witt DR. Rare kleeblattschadel anomaly without craniosynostosis. Proc Greenwood Genet Center. 7, 1988.
Wu Y.Q., Badano J.L., McCaskill C., et al. Haploinsufficiency of ALX4 as a potential cause of parietal foramina in the 11p11.2 contiguous gene-deletion syndrome. Am J Hum Genet. 2000;67:1327-1332.
Wynne-Davies R., Hall C.M., Apley A.G. Atlas of skeletal dysplasias. Edinburgh; New York: Churchill Livingstone; 1985.
Yang J.Y., Yang W.G. Large scalp and skull defect in aplasia cutis congenita. Br J Plast Surg. 2000;53:619-622.
Zvulunov A., Kachko L., Manor E., et al. Reticulolinear aplasia cutis congenita of the face and neck: a distinctive cutaneous manifestation in several syndromes linked to Xp22. Br J Dermatol. 1998;138:1046-1052.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 28 Congenital Anomalies of the Skull
Introduction
Congenital anomalies of the skull can arise any time during gestation and must be distinguished from anomalies that arise after birth. During the first 4–6 weeks of development from conception, neural crest cells in the fetal head region migrate and differentiate into mesenchymal cells that form the bones of the face, while the cranial base and base of the skull are derived from the occipital somitomeres. Within the flat cranial bones, the mesenchyme differentiates directly into bone through membranous ossification. The neurocranium is the portion surrounding the brain, and it develops directly from mesenchyme derived from the occipital somitomeres. The viscerocranium is derived from neural crest and it forms the cartilaginous bones of the face [Sadler and Langman, 2009]. The neurocranium is divided into the membranous part, forming the flat bones of the cranial vault, and the chondrocranium, forming the cartilaginous bones of the base of the skull. The chondrocranium develops by fusion of a number of cartilaginous structures, which ossify by endochondral ossification to form the base of the skull. Posteriorly, the base of the occipital bone is formed from parachordal cartilage and three occipital sclerotomes. Anteriorly, the sphenoid and ethmoid bones are formed from the hypophyseal cartilages and trabeculae cranii. On either side of the medial plate, the ala orbicularis and ala temporalis form the sphenoid bones, and the periotic capsule forms the temporal bones. The membranous neurocranium (dura mater) ossifies to form the cranial vault through bone spicules, which progressively radiate from primary ossification centers near the center of each bony plate toward the periphery, where the sutures develop. Membranous bones enlarge during fetal and postnatal life by the apposition of new layers to the outer surface of the skull (ectocranial bone deposition), while endocranial osteoclastic bone resorption occurs on the inner surface. Growth of the cranial bones is directly related to brain growth, and premature fusion of cranial sutures can be related to the cessation of brain growth (e.g., primary microcephaly).
There are six fibrous areas where two or more cranial bones meet (fontanels). The five major sutures are the metopic, sagittal, coronal, squamosal, and lambdoid sutures; the six fontanels are the anterior (1), anteriolateral (sphenoidal) (2), posterolateral (2), and posterior (1) fontanels (Figure 28-1). The presence of the sutures and fontanels allows the bones of the skull to overlap each other (termed molding) during the birth process. Different sutures become ossified at different times, with the metopic suture being the first to ossify at 4–7 months and the remaining sutures not completely ossifying until adulthood.
Craniosynostosis versus Deformational Plagiocephaly
The term craniostenosis (literally “cranial narrowing”) is used to describe an abnormal head shape that results from premature fusion of one or more sutures, while craniosynostosis is the process of premature sutural fusion that results in craniostenosis. In clinical usage, the term craniosynostosis is used more widely, perhaps in an effort to distinguish deformational nonsynostotic head shapes from those caused by underlying sutural synostosis, but the two terms are often used interchangeably. Plagiocephaly is a nonspecific term used to describe an asymmetric head shape, which can result from either craniosynostosis or cranial deformation, and differentiation between these two causes is critical to determining the proper mode of treatment (i.e., surgery vs. physical or molding techniques). Craniosynostosis is usually treated with a neurosurgical procedure involving partial calvariectomy, while deformational plagiocephaly usually responds to early physical therapy, repositioning, and/or cranial orthotic therapy if those measures are unsuccessful [Graham and Smith, 2007; Graham et al., 2005].
In an otherwise normal fetus, prenatal relaxation of normal growth-stretch tensile forces in the underlying dura across a suture for a significant period of time during late fetal life can result in craniosynostosis [Graham et al., 1979, 1980; Higginbottom et al., 1980; Graham and Smith, 1980; Koskinen-Moffett et al., 1982]. This may also occur when the lack of growth stretch is caused by a deficit in brain growth, as in severe primary microcephaly. Experimental prolongation of gestation in pregnant mice, resulting in fetal crowding after installation of a cervical clip, has been shown to result in craniosynostosis [Koskinen-Moffett and Moffet, 1989]. The degree of craniosynostosis was greatest among those fetuses located more proximally in the uterine horns, where the crowding was most severe. The most common cause of craniosynostosis in otherwise normal infants is constraint of the fetal head in utero [Graham et al., 1979, 1980; Higginbottom et al., 1980; Graham and Smith, 1980; Koskinen-Moffett et al., 1982; Koskinen-Moffett and Moffet, 1989; Sanchez-Lara et al., 2010]. When external fetal head constraint limits growth stretch parallel to a cranial suture, it may lead to craniosynostosis of an intervening suture between the constraining points. Sagittal craniosynostosis (the most common type of craniosynostosis) usually is isolated and occurs in an otherwise normal child. The constrained suture tends to develop a bony ridge, especially at the point of maximal constraint between the biparietal eminences. Such ridging can easily be palpated or visualized on skull radiographs, and three-dimensional cranial computed tomography (3D-CT) allows the ridge to be seen even more clearly (Figure 28-2).
Craniosynostosis is usually recognized shortly after birth from the abnormal shape of the head and lack of molding resolving to normal. Early closure of a fontanel, head asymmetry, and/or palpable ridging along a closed suture can be presenting features. With synostosis, cranial radiographs may reveal sclerosis of the suture with no apparent intervening sutural ligament, but it may be difficult to distinguish an overlapping suture from synostosis. On cross-sectional images, dense ridging over the suture may be evident, particularly with sagittal and metopic synostosis. If there is uncertainty as to whether sutures are truly synostotic, 3D-CT can provide a more accurate appraisal (see Figure 28-2).
In general, craniosynostosis begins at one point and then spreads along a suture [Koskinen-Moffett and Moffet, 1989; Cohen and MacLean, 2000]. At the fused location, there is complete sutural obliteration, with nonlamellar bone extending completely across the sutural space, while further away from the initial site of fusion, the sutural margins are closely approximated with ossifying connective tissue. As age increases, there is a tendency for more of the suture to become synostotic, with synostosis usually beginning at only one location in most cases [Koskinen-Moffett and Moffet, 1989]. Synostosis prevents future expansion at that site, and the rapidly growing brain then distorts the calvarium into an aberrant shape, depending upon which sutures have become synostotic (see Figure 28-2). The earlier the synostosis takes place, the greater the effect on skull shape. Craniosynostosis may be caused by many different mechanisms, such as mutant genes, chromosome disorders, storage disorders, hyperthyroidism, or failure of normal brain growth The entire topic of craniosynostosis has been comprehensively reviewed by Cohen [Cohen and MacLean, 2000].
Sutural Anatomy and Head Shape
Different terms have been used to describe the different head shape alterations caused by craniosynostosis, with the resultant head shape dependent on the suture involved. A long, keel-shaped skull with prominent forehead and occiput is termed dolichocephaly or scaphocephaly. This head shape is usually associated with premature sagittal suture closure and a palpable ridge toward the posterior end of the suture (see Figure 28-2A). Sagittal synostosis must be distinguished from deformation of the infant cranium due to persistently sleeping on the side of the head (more common in prematurely born infants) or breech-head deformation sequence. Individuals with scaphocephaly and dolichocephaly have a decreased cephalic index (CI) of less than 76 percent (CI = head width/head length × 100 percent).
Premature fusion of both coronal sutures produces a high, wide forehead with a short skull, resulting in brachycephaly (see Figure 28-2), while fusion of one coronal suture produces an asymmetric head shape termed plagiocephaly. When coronal craniosynostosis occurs, it is important to examine the patient carefully for associated anomalies that might suggest a recognizable genetic syndrome. Evaluation of the limbs, ears, and cardiovascular system is quite helpful in diagnosing syndromes associated with coronal craniosynostosis. Limb defects, such as syndactyly, brachydactyly, carpal coalition, or broad, deviated thumbs and/or halluces, can suggest an associated genetic syndrome and indicate what types of molecular analysis to pursue. It is also important to examine both parents for similar anomalies, carpal coalition, and/or facial asymmetry, since these findings may represent variable expression of an altered gene in a parent.
Premature fusion of both the coronal and sagittal sutures generally leads to a tall, tower-like skull (turricephaly), with more severe synostosis of multiple sutures producing a tall, pointed skull (acrocephaly or oxycephaly). In this condition, the limitations of calvarial expansion are so extreme that there may be limited room for brain growth (Figure 28-3a). Synostosis of multiple cranial sutures is more likely to result in elevated intracranial pressure and to require shunting for hydrocephalus. In extreme cases, a cloverleaf head shape can result from multiple suture synostosis, usually with signs of increased intracranial pressure and a “beaten copper” radiographic appearance of the inner table of the skull (see Figure 28-3b). There may also be optic atrophy, proptosis, and loss of vision. Combinations of sutural synostosis, such as sagittal plus coronal, are also referred to as compound craniosynostosis, and multiple suture synostosis usually has a genetic basis.
A triangle-shaped skull (trigonocephaly) is caused by premature fusion of the metopic suture (see Figure 28-2). The similarity in epidemiological features between sagittal and metopic craniosynostosis suggests that prenatal lateral constraint of the frontal part of the head may be a frequent cause of metopic craniosynostosis. Examples of constraint-induced metopic synostosis have included a monozygotic triplet whose forehead was wedged between the buttocks of her two co-triplets, and an infant whose head was compressed within one horn of his mother’s bicornuate uterus [Graham and Smith, 1980]. Syndromic metopic synostosis can also occur, and trigonocephaly is seen in a variety of syndromes, some of which are associated with mental retardation or chromosome anomalies.
Unilateral lambdoid synostosis results in trapezoidal plagiocephaly, which differs from deformational posterior plagiocephaly due to supine positioning and torticollis, and from synostotic anterior plagiocephaly due to unicoronal synostosis (see Figure 28-2). Unlike coronal synostosis, facial structures and orbits are usually not affected by lambdoid synostosis. Radiographic signs include trapezoidal cranial asymmetry, small posterior fossa, and sutural sclerosis with ridging; however, sole reliance on skull radiographs and clinical signs can lead to misdiagnosis, so it is best to confirm the diagnosis of suspected lambdoid synostosis with a 3D-CT scan, which clearly images the involved suture(s) and permits secure diagnosis. Among 232 patients referred for either deformational posterior plagiocephaly or craniosynostosis, only 4 patients (3.1 percent) manifested clinical, imaging, and operative features of true unilambdoidal craniosynostosis [Huang et al., 1996]. These features included a thick bony ridge over the fused suture, with contralateral parietal and frontal bulging, and an ipsilateral occipitomastoid bulge, leading to tilting of the ipsilateral skull base and a downward/posterior displacement of the ear on the synostotic side. In contrast, infants with deformational, nonsynostotic posterior plagiocephaly had a parallelogram-shaped head, with forward displacement of the ear and frontal bossing on the side ipsilateral to the occipitoparietal flattening, accompanied by contralateral occipital bossing [Graham et al., 2005].
Plagiocephaly (which translates literally from the Greek term plagio kephale as “oblique head”) is a term used to describe asymmetry of the head shape, when viewed from the top [Graham and Smith, 2007]. The term deformational plagiocephaly should suffice to distinguish this type of defect and its proper type of management. The side of the plagiocephaly is usually indicated by the bone that has been most flattened by the deforming forces (usually the occiput for infants who sleep on their backs). Deformational plagiocephaly is usually not associated with premature closure of a cranial suture, but since craniosynostosis can also be caused by fetal head constraint, when both deformational plagiocephaly and craniosynostosis occur together, the diagnosis can be difficult, requiring complex management.
Epidemiology of Craniosynostosis
The incidence of craniosynostosis is 3.4 per 10,000 births, and it is usually an isolated, sporadic anomaly in an otherwise normal child. About 8 percent of all craniosynostosis cases are familial. Familial types of craniosynostosis are most frequent in coronal synostosis, accounting for 14.4 percent of coronal synostosis, 6 percent of sagittal synostosis, and 5.6 percent of metopic synostosis [Lajeunie et al., 1995, 1996, 1998], while lambdoidal synostosis is almost never familial. The frequency of associated twinning is increased, and most twin pairs are discordant, especially with sagittal and metopic synostosis, which would tend to support fetal crowding as a cause for these types of synostosis; concordance for coronal synostosis is much higher for monozygotic twins than for dizygotic twins, suggesting that many cases of coronal synostosis have a genetic basis [Lajeunie et al., 1995].
Sagittal synostosis is the most common type of craniosynostosis, accounting for 50–60 percent of cases and occurring in 1.9 per 10,000 births, with a 3.5:1 male:female sex ratio [Lajeunie et al., 1996]. Only 6 percent of cases are familial, with 72 percent of cases sporadic and no paternal or maternal age effects noted. Twinning occurred in 4.8 percent of 366 cases, with only one monozygotic twin pair being concordant [Lajeunie et al., 1996].
Coronal craniosynostosis is the second most frequent type of craniosynostosis (accounting for 20–30 percent of cases). Unilateral coronal craniosynostosis can be either genetic or due to fetal head constraint from an aberrant fetal lie, multiple gestation, or small uterine cavity [Graham et al., 1980; Higginbottom et al., 1980]. Approximately 71 percent of unilateral coronal craniosynostosis is right-sided, and 67 percent of vertex presentations are in the left occiput transverse position, possibly explaining the prevalence of right-sided, unilateral, coronal craniostenosis [Graham et al., 1980]. Nonsyndromic coronal craniosynostosis occurs 0.94 per 10,000 births, with 61 percent of cases sporadic, and 14.4 percent of 180 pedigrees familial. Bilateral cases occur much more frequently than unilateral cases, and coronal synostosis is more frequent in females (male to female ratio 1:2). The paternal age is statistically older than average (32.7 years), and these data have been interpreted as being consistent with fresh dominant mutation and autosomal-dominant inheritance with 60 percent penetrance, when the synostosis has a genetic basis [Lajeunie et al., 1995].
Metopic synostosis occurs in about 0.67 per 10,000 births, making it the third most frequent type of craniosynostosis (accounting for 10–20 percent of patients). Like sagittal synostosis, metopic synostosis is more frequent in males (3.3:1 male: female ratio) and seldom familial (5.6 percent of cases) [Lajeunie et al., 1998]. There is no maternal or paternal age effect, and the frequency of associated twinning was 7.8 percent of 179 pedigrees studied, with only 2 twin monozygotic pairs concordant [Lajeunie et al., 1998].
Familial craniosynostosis is usually transmitted as an autosomal-dominant trait with incomplete penetrance and variable expressivity. A wide variety of chromosomal anomalies have also been associated with craniosynostosis. This emphasizes the importance of chromosomal analysis for patients with syndromic craniosynostosis in whom a recognizable monogenic syndrome is not apparent, particularly when there is associated developmental delay and growth deficiency. As comparative genomic hybridization becomes more widely utilized, this will be even more useful than standard cytogenetics for many such cases. In addition, craniosynostosis can also occur as a component of numerous syndromes, many of which manifest phenotypic overlap and genetic heterogeneity [Cohen and MacLean, 2000] Craniosynostosis syndromes with a demonstrated mutational basis include Apert’s syndrome, Crouzon’s syndrome, Pfeiffer’s syndrome, Saethre–Chotzen syndrome, Jackson–Weiss syndrome, Boston craniosynostosis, Beare–Stevenson cutis gyrata syndrome, and fibroblast growth factor receptor 3 (FGFR3)-associated coronal synostosis; however, the efficiency of actually detecting a mutation for a given syndrome varies from about 60 percent for Crouzon’s syndrome to 98 percent for Apert’s syndrome [Cohen and MacLean, 2000].
Secondary craniosynostosis can occur with certain primary metabolic disorders (e.g., hyperthyroidism, rickets), storage disorders (e.g., mucopolysaccharidosis), hematological disorders (thalassemia, sickle cell anemia, polycythemia vera, congenital hemolytic icterus), brain malformations (e.g., holoprosencephaly, microcephaly, encephalocele, or overshunted hydrocephalus), and selected teratogenic exposures (e.g., diphenylhydantoin, retinoic acid, valproic acid, aminopterin, fluconazole, cyclophosphamide) [Cohen and MacLean, 2000].
Inability to demonstrate a mutation does not rule out a genetic basis for the craniosynostosis, and not every person with a pathogenic mutation manifests craniosynostosis. Bilateral coronal synostosis often lacks sutural ridging and usually has a genetic pathogenesis, suggesting that all such patients should be screened for mutations. Among 57 patients with bilateral coronal synostosis, mutations in FGFR genes were found for all 38 patients with a syndromic form of craniosynostosis. Among 19 patients with nonsyndromic bilateral craniosynostosis, mutations were found in or near exon 9 of FGFR2 in 4 patients, as well as the common Pro250Arg mutation being found in exon 7 of FGFR3 in 10 patients; only 5 patients (9 percent) lacked a detectable mutation in FGFR 1, 2, or 3 [Mulliken et al., 1999]. This study suggests that mutation analysis should be considered in most patients with bilateral coronal synostosis.
Kleeblattschadel (Cloverleaf Skull)
Kleeblattschadel is a term used to describe a cloverleaf skull configuration consisting of protrusion of each of the cranial bones, with broadening of the temporal region and face. These cranial protrusions are separated into focal bulges by furrows along the suture lines. The eyes often protrude, leading to corneal ulceration, scarring, and subsequent blindness, if the corneal surface remains unprotected [Stevenson, 1986]. Occipital encephaloceles can occur, and associated hydrocephalus is common. The palate is usually highly arched, but clefting is rare. The presence of Kleeblattschadel indicates that multiple sutural fusions occurred during early prenatal life. Increased thickening of the base of the occipital bone prevents lengthening of the skull, thus yielding the typical shape [Dambrain et al., 1987]. One child has also been described who had a cloverleaf skull without craniosynostosis (in which the primary defect was thought to be a cranial bone dysplasia that allowed for eventration of the brain and resultant cloverleaf configuration) [DR, 1988].
Multiple sutural synostosis is much more likely to result from genetic mutations in FGFR genes, TWIST, or MSX2, all of which result in syndromes which can present with cloverleaf skull [Cohen and MacLean, 2000]. The most common syndrome associated with cloverleaf skull is thanatophoric dysplasia type 2, which is due to mutations in FGFR3. Type 2 Pfeiffer’s syndrome, due to mutations in FGFR2, can result in cloverleaf skull, as can Crouzon’s syndrome and Apert’s syndrome, which are also due to mutations in FGFR2. Other syndromes, like FGFR3-associated coronal synostosis, rarely result in cloverleaf skull, but some rare syndromes like Boston craniosynostosis and Crouzon’s syndrome with acanthosis nigricans can sometimes manifest cloverleaf skull. In Boston craniosynostosis, the mutant MSX2 product has enhanced affinity for binding to its DNA target sequence, resulting in activated osteoblastic activity and aggressive cranial ossification [Warman et al., 1993; Jabs et al., 1993; Ma et al., 1996]. In Crouzon’s syndrome with acanthosis nigricans, a specific FGFR3 mutation (Ala391Glu), leads to early onset of acanthosis nigricans during childhood, often with associated choanal atresia and hydrocephalus [Schweitzer et al., 2001].
Eighty-five percent of children with Kleeblattschadel will have other anomalies, and the pattern is often consistent with a syndrome diagnosis. The prognosis is usually syndrome-dependent and can be quite poor, with early death due to respiratory difficulties or progressive brain damage from hydrocephalus being common [Frank et al., 1985]. However, subtotal craniectomy within the first 3 weeks of life in individuals with mild Kleeblattschadel may result in normal or near-normal development [Frank et al., 1985; Kroczek et al., 1986; Turner and Reynolds, 1980]. Early extensive calvariectomy is merited to preserve brain function and development, as well as to allow reformation of the craniofacial skeletal features. Many craniofacial surgeons prefer to begin with a posterior skull release in the early months of life (mean age 4 months), followed by fronto-orbital advancement around the end of the first year (mean age 14 months), with insertion of a ventriculoperitoneal shunt at the time of the first procedure if there is associated hydrocephalus [Sgouros et al., 1996]. The use of postoperative orthotic molding can help to channel brain growth into a more normal form, leading to improved postoperative results over those obtained via surgery alone [Schweitzer et al., 2001]. When lambdoid synostosis occurs as part of a syndrome with multiple suture involvement, there is often bilateral involvement, and early posterior release may alleviate some associated increased intracranial pressure. Patients with Kleeblattschadel need to be followed carefully for hydrocephalus, which may be part of the syndrome, rather than due to the multiple suture synostosis. Restricted growth of the posterior fossa is particularly common in severe craniofacial dysostosis syndromes. The purpose of surgery should be to decompress the brain, expand the bony orbits to accommodate the globes, and open airway passages [Kroczek et al., 1986].
Treatment and Outcomes of Craniosynostosis
Mild degrees of craniosynostosis may not always require surgery; however, in moderately severe cases, early surgery is usually warranted. The usual indication for surgery is to restore normal craniofacial shape and growth. When both the coronal and sagittal sutures are synostotic, impairing brain growth early in infancy, surgery is indicated to help prevent neurological and ophthalmologic complications associated with increased intracranial pressure and inadequate orbital volume. A variety of neurosurgical techniques have been developed for the treatment of craniosynostosis [Cohen and MacLean, 2000]. Most of these techniques involve removing the aberrant portion of the bony calvarium from its underlying dura, including the area surrounding the synostotic suture(s). If this is done within the first few months after birth, a new bony calvarium will usually develop within the remaining dura mater under the same principles that guide normal prenatal calvarial morphogenesis. As long as there is continued growth stretch from the expanding brain, the sites over the dural reflections remain unossified, thereby maintaining the sutures in a fibrous, open state. Thus, the calvarium and its sutures usually re-form normally after a partial calvariectomy for craniosynostosis. The new bony calvarium begins to develop within 2–3 weeks after surgery, and is usually firm by 5–8 weeks after surgery. If the procedure is done after 3–4 months of age, the approach is similar, with the exception that pieces of the calvarium are usually replaced in a mosaic pattern over the dura mater to act as niduses for the mineralization of new calvarium. Newer endoscopic repair techniques have been developed, followed by postoperative orthotic molding. Such procedures are most effective if done relatively early in infancy. These techniques are most effective in normal infants without a syndromic type of craniosynostosis.
Following early surgery for isolated craniosynostosis (primarily fronto-orbital advancement and/or calvarial vault remodeling at a mean age of 8 months), only 13 percent of 104 patients (10 bilateral coronal, 57 unilateral coronal, 29 metopic, and 8 sagittal) required a second cranial vault operation for residual defects at a mean age of 23 months. Perioperative complications were minimal (5 percent), with 87.5 percent of patients considered to have satisfactory craniofacial form, and low rates of hydrocephalus (3.8 percent), shunt placement (1 percent), and seizures (2.9 percent). Among such cases of isolated craniosynostosis, unilateral coronal synostosis was the most problematic type due to vertical orbital dystopia, nasal tip deviation, and altered craniofacial growth problems with residual craniofacial asymmetry [McCarthy et al., 1995]. In a second study of 167 children with both nonsyndromal (isolated) and syndromal craniosynostosis (12 bilateral coronal, 18 unilateral coronal, 39 metopic, and 46 sagittal), repeat operations were necessary in only 7 percent. Repeat operations were more common in syndromic cases (27.3 percent) than in nonsyndromic craniosynostosis (5.6 percent) [Williams et al., 1997].
Even though intracranial pressure can be elevated in patients with nonsyndromic craniosynostosis, they may not have decreased cranial volumes either before or after surgical repair, and as a group show slightly larger intracranial volumes when compared with normal controls. This could reflect the impact of fetal head constraint on fetuses with larger heads, or it might relate to the known association of macrocephaly with nonsyndromic coronal craniosynostosis due to the common Pro250Arg mutation in FGFR3 (which was not analyzed in these studies) [Polley et al., 1998]. Hydrocephalus occurs in 4–10 percent of patients with craniosynostosis and is more frequent with syndromic and multiple sutural craniosynostosis. In nonsyndromic patients, the rate of cerebral ventricular dilatation is the same as that observed in the general population, and it appears to be related to venous hypertension induced by jugular foramen stenosis. Such dilatation usually stabilizes spontaneously and rarely requires shunting. Some cases of progressive hydrocephalus in syndromic craniosynostosis cases were related to multiple sutural involvement, thereby constricting cranial volume, constricting the skull base, crowding the posterior fossa, and causing jugular foraminal stenosis [Cinalli et al., 1998]. These findings were most frequent among patients with Crouzon’s, Pfeiffer’s, and Apert’s syndrome, especially in association with cloverleaf skull abnormalities. A diffuse beaten copper pattern on skull radiographs, along with obliteration of anterior sulci or narrowing of basal cisterns in children under the age of 18 months, is predictive of increased intracranial pressure in over 95 percent of cases [Tuite and Lindquist, 1996].
Nonsyndromic Craniosynostosis Neurocognitive Development
There is a growing body of evidence that single-suture craniosynostosis is associated with neurobehavioral problems [Kapp-Simon, 1998; Magge et al., 2002]. In the 2004 manuscript, Speltz et al. nicely summarized all of the published neurobehavioral studies conducted on children with single suture craniosynostosis [Speltz et al., 2004].
Few studies have documented actual rates of mental retardation (typically defined as standardized test scores below 70). Several older studies reported slightly increased rates of mental retardation (from 6.5 to 12 percent) in comparison to an expected rate of about 2.2 percent in the population [Kapp-Simon, 1998; Hunter and Rudd, 1976, 1977; Sidoti et al., 1996].
Syndromes with multiple-suture fusions have more commonly been associated with elevated rates of mental retardation and learning disabilities [Cohen, 1991]. Most studies have found adverse neurocognitive outcomes in about 35–40 percent of assessed cases [Bottero et al., 1998; Shimoji et al., 2002; Shipster et al., 2003], with an occasional study finding this number to be as high as 50 percent [Kapp-Simon, 1998; Magge et al., 2002
[/not-level-membership-for-neurology-category]
