Chapter 585 Congenital Anomalies of the Central Nervous System
585.1 Neural Tube Defects
Stephen L. Kinsman and Michael V. Johnston
The human nervous system originates from the primitive ectoderm that also develops into the epidermis. The ectoderm, endoderm, and mesoderm form the three primary germ layers that are developed by the 3rd wk. The endoderm, particularly the notochordal plate and the intraembryonic mesoderm, induces the overlying ectoderm to develop the neural plate in the 3rd wk of development (Fig. 585-1A). Failure of normal induction is responsible for most of the NTDs, as well as disorders of prosencephalic development. Rapid growth of cells within the neural plate causes further invagination of the neural groove and differentiation of a conglomerate of cells, the neural crest, which migrate laterally on the surface of the neural tube (see Fig. 585-1B). The notochordal plate becomes the centrally placed notochord, which acts as a foundation around which the vertebral column ultimately develops. With formation of the vertebral column, the notochord undergoes involution and becomes the nucleus pulposus of the intervertebral disks. The neural crest cells differentiate to form the peripheral nervous system, including the spinal and autonomic ganglia and the ganglia of cranial nerves V, VII, VIII, IX, and X. In addition, the neural crest forms the leptomeninges, as well as Schwann cells, which are responsible for myelination of the peripheral nervous system. The dura is thought to arise from the paraxial mesoderm. In the region of the embryo destined to become the head, similar patterns exist. In this region, the notocord is replaced by the precordal mesoderm.
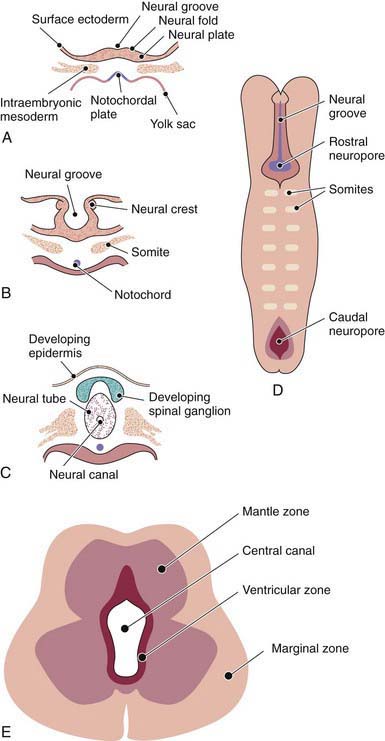
In the 3rd wk of embryonic development, invagination of the neural groove is completed and the neural tube is formed by separation from the overlying surface ectoderm (see Fig. 585-1C). Initial closure of the neural tube is accomplished in the area corresponding to the future junction of the spinal cord and medulla and moves rapidly both caudally and rostrally. For a brief period, the neural tube is open at both ends, and the neural canal communicates freely with the amniotic cavity (see Fig. 585-1D). Failure of closure of the neural tube allows excretion of fetal substances (α-fetoprotein [AFP], acetylcholinesterase) into the amniotic fluid, serving as biochemical markers for a NTD. Prenatal screening of maternal serum for AFP in the 16th-18th wk of gestation is an effective method for identifying pregnancies at risk for fetuses with NTDs in utero. Normally, the rostral end of the neural tube closes on the 23rd day and the caudal neuropore closes by a process of secondary neurulation by the 27th day of development, before the time that many women realize they are pregnant.
The embryonic neural tube consists of three zones: ventricular, mantle, and marginal (see Fig. 585-1E). The ependymal layer consists of pluripotential, pseudostratified, columnar neuroepithelial cells. Specific neuroepithelial cells differentiate into primitive neurons or neuroblasts that form the mantle layer. The marginal zone is formed from cells in the outer layer of the neuroepithelium, which ultimately becomes the white matter. Glioblasts, which act as the primitive supportive cells of the CNS, also arise from the neuroepithelial cells in the ependymal zone. They migrate to the mantle and marginal zones and become future astrocytes and oligodendrocytes. The importance of other pathways of progenitor cell generation and migration are also being elucidated. It is likely that microglia originate from mesenchymal cells at a later stage of fetal development when blood vessels begin to penetrate the developing nervous system.
585.2 Spina Bifida Occulta (Occult Spinal Dysraphism)
Stephen L. Kinsman and Michael V. Johnston
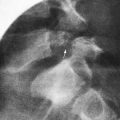
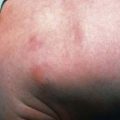
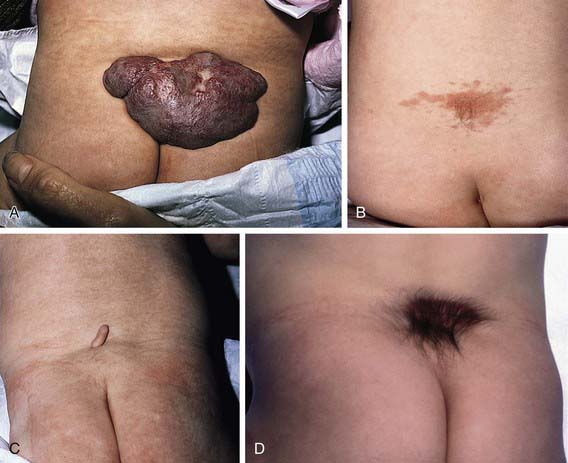
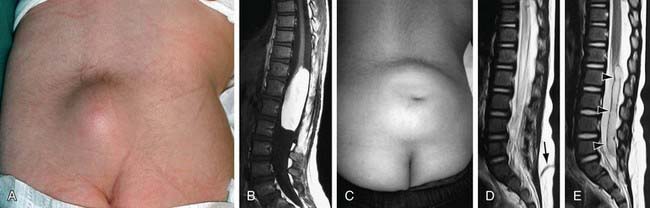
Spina bifida occulta is a common anomaly consisting of a midline defect of the vertebral bodies without protrusion of the spinal cord or meninges. Most patients are asymptomatic and lack neurologic signs, and the condition is usually of no consequence. Some consider the term spina bifida occulta to denote merely a posterior vertebral body fusion defect. This simple defect does not have an associated spinal cord malformation. Other clinically more significant forms of this closed spinal cord malformation are more correctly termed occult spinal dysraphism. In most of these cases, there are cutaneous manifestations such as a hemangioma, discoloration of the skin, pit, lump, dermal sinus, or hairy patch (Fig. 585-2). A spine roentgenogram in simple spina bifida occulta shows a defect in closure of the posterior vertebral arches and laminae, typically involving L5 and S1; there is no abnormality of the meninges, spinal cord, or nerve roots. Occult spinal dysraphism is often associated with more significant developmental abnormalities of the spinal cord, including syringomyelia, diastematomyelia, and/or a tethered cord. A spine roentgenogram in these cases might show bone defects or may be normal. All cases of occult spinal dysraphism are best investigated with MRI (Fig. 585-3). Initial screening in the neonate may include ultrasonography.
An approach to imaging of the spine in patients with cutaneous lesions is noted in Table 585-1.
Table 585-1 CUTANEOUS LESIONS ASSOCIATED WITH OCCULT SPINAL DYSRAPHISM
IMAGING INDICATED
IMAGING UNCERTAIN
IMAGING NOT REQUIRED
From Williams H: Spinal sinuses, dimples, pits and patches: what lies beneath? Arch Dis Child Educ Pract Ed 91:ep75–ep80, 2006.
585.4 Myelomeningocele
Clinical Manifestations
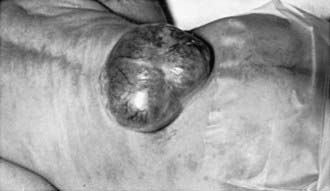
Myelomeningocele produces dysfunction of many organs and structures, including the skeleton, skin, and gastrointestinal and genitourinary tracts, in addition to the peripheral nervous system and the CNS. A myelomeningocele may be located anywhere along the neuraxis, but the lumbosacral region accounts for at least 75% of the cases. The extent and degree of the neurologic deficit depend on the location of the myelomeningocele and the associated lesions. A lesion in the low sacral region causes bowel and bladder incontinence associated with anesthesia in the perineal area but with no impairment of motor function. Newborns with a defect in the midlumbar or high lumbothoracic region typically have either a saclike cystic structure covered by a thin layer of partially epithelialized tissue (Fig. 585-4) or an exposed flat neural placode without overlying tissues. When a cyst or membrane is present, remnants of neural tissue are visible beneath the membrane, which occasionally ruptures and leaks CSF, whereas the placode is composed of neural tissue.
Treatment
Although incontinence of fecal matter is common and is socially unacceptable during the school years, it does not pose the same organ-damaging risks as urinary dysfunction, but occasionally fecal impaction and/or megacolon develop. Many children can be bowel-trained with a regimen of timed enemas or suppositories that allows evacuation at a predetermined time once or twice a day. Special attention to low anorectal tone and enema administration and retention is often required. Appendicostomy for antegrade enemas may also be helpful (Chapter 21.4).
In utero surgical closure of a spinal lesion has been successful in a few centers. Preliminary reports suggest a lower incidence of hindbrain abnormalities and hydrocephalus (fewer shunts) as well as improved motor outcomes. This suggests that the defects may be progressive in utero and that prenatal closure might prevent the development of further loss of function. In utero diagnosis is facilitated by maternal serum α-fetoprotein screening and by fetal ultrasonography (Chapter 90).
Adzick NS, Thom EA, Spong CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364:993-1004.
Bauer SB. Neurogenic bladder: etiology and assessment. Pediatr Nephrol. 2008;23:541-551.
Beeker T, Scheers M, Faber J, et al. Prediction of independence and intelligence at birth on meningomyelocele. Childs New Syst. 2006;22:33-37.
Bitsko RH, Reefhuis J, Romitti PA, et al. Periconceptional consumption of vitamins containing folic acid and risk for multiple congenital anomalies. Am J Med Genet. 2007;143A:2397-2405.
Bol KA, Collins JS, Kirby RS. Survival of infants with neural tube defects in the presence of folic acid fortification. Pediatrics. 2006;117:803-813.
Cameron M, Moran P. Prenatal screening and diagnosis of neural tube defects. Prenat Diagn. 2009;29:402-411.
Centers for Disease Control and Prevention. CDC Grand Rounds: additional opportunities to prevent neural tube defects with folic acid fortification. MMWR Morb Mortal Wkly Rep. 2010;59:980-984.
Chescheir NC. Maternal-fetal surgery: where are we and how did we get here? Obstet Gynecol. 2009;113:717-731.
Cochrane DD. Cord untethering for lipomyelomeningocele: expectation after surgery. Neurosurg Focus. 2007;23:1-7.
de Jong TP, Chrzan R, Klijn AJ, et al. Treatment of the neurogenic bladder in spina bifida. Pediatr Nephrol. 2008;23:889-896.
Dicianno BE, Kurowski BG, Yang JM, et al. Rehabilitation and medical management of the adult with spina bifida. Am J Phys Med Rehabil. 2008;87:1027-1050.
Fichter MA, Dornseifer U, Henke J, et al. Fetal spina bifida repair—current trends and prospects of intrauterine neurosurgery. Fetal Diagn Ther. 2008;23:271-286.
Guggisberg D, Hadj-Rabia S, Viney C, et al. Skin markers of occult spinal dysraphism in children. Arch Dermatol. 2004;140:1109-1115.
Ickowicz V, Ewin D, Maugay-Laulom B, et al. Meckel-Gruber syndrome, sonography and pathology. Ultrasound Obstet Gynecol. 2006;27:296-300.
Joseph DB. Current approaches to the urologic care of children with spina bifida. Curr Urol Rep. 2008;9:151-157.
Kibar Z, Torban E, McDearmid JR, et al. Mutations in VANGLI associated with neural-tube defects. N Engl J Med. 2007;356:1432-1437.
Scales CD, Wiener JS. Evaluating outcomes of enterocystoplasty in patients with spina bifida: a review of the literature. J Urol. 2008;180:2323-2329.
Stevenson RE, Allen WP, Pai GS, et al. Decline in prevalence of neural tube defects in a high-risk region of the United States. Pediatrics. 2000;106:677-683.
Tubbs RS, Bui CJ, Loukas M, et al. The horizontal sacrum as an indicator of the tethered spinal cord in spina bifida aperta and occulta. Neurosurg Focus. 2007;23:1-4.
U.S. Preventive Services Task Force. Recommendation statement: folic acid for the prevention of neural tube defects. Ann Intern Med. 2009;150:626-631.
Williams H. Spinal sinuses, dimples, pits and patches: what lies beneath? Arch Dis Child. 2006;91:ep75-ep80.
585.7 Disorders of Neuronal Migration
Disorders of neuronal migration can result in minor abnormalities with little or no clinical consequence (small heterotopia of neurons) or devastating abnormalities of CNS structure and/or function (mental retardation, seizures, lissencephaly, and schizencephaly, particularly the open-lip form) (Fig. 585-5). One of the most important mechanisms in the control of neuronal migration is the radial glial fiber system that guides neurons to their proper site. Migrating neurons attach to the radial glial fiber and then disembark at predetermined sites to form, ultimately, the precisely designed six-layered cerebral cortex. Another important mechanism is the tangential migration of progenitor neurons destined to become cortical interneurons. The severity and the extent of the disorder are related to numerous factors, including the timing of a particular insult and a host of environmental and genetic contributors.
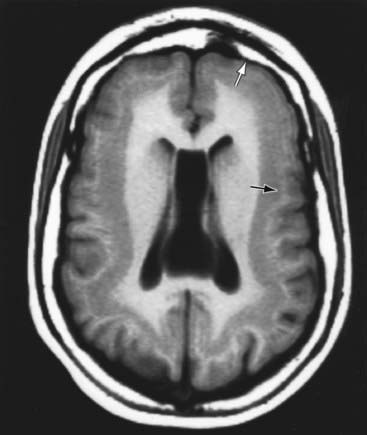
Lissencephaly
The gene LIS-1 (lissencephaly 1) that maps to chromosome region 17p13.3 is deleted in patients with MDS. CT and MRI scans typically show a smooth brain with an absence of sulci (Fig. 585-6). Doublecortin is an X chromosome gene that causes lissencephaly when mutated in males and subcortical band heterotopia when mutated in females. Other important forms of lissencephaly include the Walker-Warburg variant and other cobblestone cortical malformations.
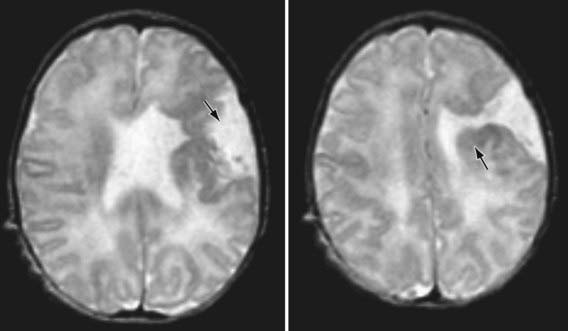
Schizencephaly
Schizencephaly is the presence of unilateral or bilateral clefts within the cerebral hemispheres owing to an abnormality of morphogenesis (Fig. 585-7). The cleft may be fused or unfused and, if unilateral and large, may be confused with a porencephalic cyst. Not infrequently, the borders of the cleft are surrounded by abnormal brain, particularly microgyria. MRI is the study of choice for elucidating schizencephaly and associated malformations.
Abdel Razek AAK, Kandell AY, Elsorogy LG, et al. Disorders of cortical formation: MR imaging features. AJNR Am J Neuroradiol. 2009;30:4-11.
Almgren M, Schalling M, Lavebratt C. Idiopathic megalencephaly—possible cause and treatment opportunities: from patient to lab. Eur J Paediat Neurol. 2008;12:438-445.
Breedveld G, de Coo IF, Lequin MH, et al. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J Med Genet. 2006;43:490-495.
Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167-1171.
Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008;31:154-162.
Hayashi N, Tsutsumi Y, Barkovich AJ. Polymicrogyria without porencephaly/schizencephaly. MRI analysis of the spectrum and the prevalence of macroscopic findings in the clinical population. Neuroradiology. 2002;44:647-655.
Kerjan G, Gleeson JG. Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 2007;23:623-630.
Sharif U, Kuban K. Prenatal intracranial hemorrhage and neurologic complications in alloimmune thrombocytopenia. J Child Neurol. 2001;16:838-842.
Sherlock RL, Synnes AR, Grunau RE, et al. Long-term outcome after neonatal intraparenchymal echodensities with porencephaly. Arch Dis Child Fetal Neonatal Ed. 2008;93:F127-F131.
Spalice A, Parisi P, Nicita F, et al. Neuronal migration disorders: clinical, neuroradiologic and genetics aspects. Acta Paediatrica. 2009;98:421-433.
van der Knaap MS, Smit LME, Barkhof F, et al. Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol. 2006;59:504-511.
Wynshaw-Boris A. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72:296-304.
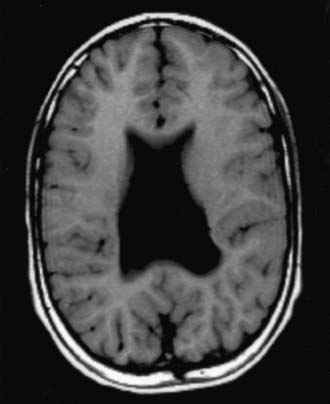
585.8 Agenesis of the Corpus Callosum
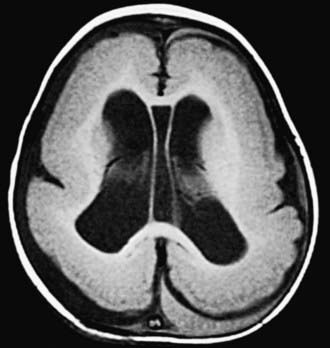
Agenesis of the corpus callosum consists of a heterogeneous group of disorders that vary in expression from severe intellectual and neurologic abnormalities to the asymptomatic and normally intelligent patient (Fig. 585-8). The corpus callosum develops from the commissural plate that lies in proximity to the anterior neuropore. Either a direct insult to the commissural plate or disruption of the genetic signaling that specifies and organizes this area during early embryogenesis causes agenesis of the corpus callosum.
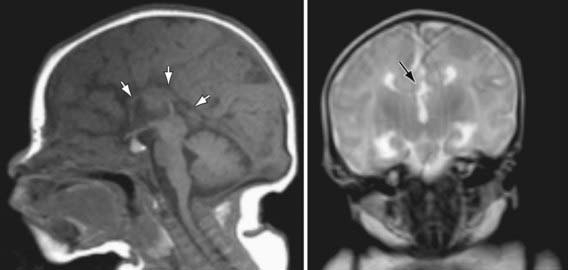
Holoprosencephaly
Holoprosencephaly is a developmental disorder of the brain that results from defective formation of the prosencephalon and inadequate induction of forebrain structures. The abnormality, which represents a spectrum of severity, is classified into 3 groups: alobar, semilobar, and lobar, depending on the degree of the cleavage abnormality (Fig. 585-9). A fourth type, the middle interhemispheric fusion (MIHF) variant or syntelencephaly, involves a segmental area of noncleavage, actually a nonseparation, of the posterior frontal and parietal lobes. Facial abnormalities including cyclopia, synophthalmia, cebocephaly, single nostril, solitary central incisor tooth, and premaxillary agenesis are common in severe cases, because the prechordal mesoderm that induces the ventral prosencephalon is also responsible for induction of the median facial structures. Alobar holoprosencephaly is characterized by a single ventricle, an absent falx, and nonseparated deep cerebral nuclei. Care must be taken not to overdiagnose holoprosencephaly based on ventricular abnormalities alone. Evidence of nonseparated midline deep brain structures such as caudate, putamen, globus pallidus, and hypothalamus is the critical element for diagnosis.
Bedeschi MF, Bonaglia MC, Grasso R, et al. Agenesis of the corpus callosum: clinical and genetic study in 63 young patients. Pediatr Neurol. 2006;34:186-193.
Bendavid C, Dubourg C, Gicquel I, et al. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet. 2006;119:1-8.
Cohen MM. Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res Part A Clin Mol Teratol. 2006;76:658-673.
Dubourg C, Lazaro L, Pasquier L, et al. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: mutation review and genotype-phenotype correlations. Hum Mutat. 2004;24:43-51.
Glass HC, Shaw GM, Ma C, et al. Agenesis of the corpus callosum in California 1983–2003: a population-based study. Am J Med Genet. 2008;146A:2495-2500.
Goetzinger KR, Stamilio DM, Dicke JM, et al. Evaluating the incidence and likelihood ratios for chromosomal abnormalities in fetuses with common central nervous system malformations. Am J Obstet Gynecol. 2008;199(285):e1-e6.
Griffiths PD, Batty R, Connolly DA, et al. Effects of failed commissuration on the septum pellucidum and fornix: implications for fetal imaging. Neuroradiology. 2009;51:347-356.
Herman-Sucharska I, Bekiesinska-Figatowska M, Urbanik A. Fetal central nervous system malformations on MR images. Brain Dev. 2009;31:185-199.
Kerjan G, Gleeson JG. Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 2007;23:623-630.
Miller SP, Shevell MI, Patenaude Y, et al. Septo-optic dysplasia plus: a spectrum of malformations of cortical development. Neurology. 2000;54:1701-1703.
Moes P, Schilmoeller K, Schilmoeller G. Physical, motor, sensory and developmental features associated with agenesis of the corpus callosum. Child Care Health Dev. 2009;35:656-672.
Nerdich JA, Nussbaum RL, Packer TC, et al. Heterogeneity of clinical severity and molecular lesions in Aicardi syndrome. J Paediatr. 1990;116:911-917.
Passos-Bueno MR, et al. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol. 2008;12:107-143.
Polizzi A, Pavone P, Iannetti P, et al. Septo-optic dysplasia complex: a heterogeneous malformation syndrome. Pediatr Neurol. 2006;34:66-71.
Schell-Apacik CC, Wagner K, Bihler M, et al. Agenesis and dysgenesis of the corpus callosum: clinical, genetic and neuroimaging findings in a series of 41 patients. Am J Med Genet. 2008;146A:2501-2511.
Smith T, Tekes A, Boltshauser E, et al. Commissural malformations: beyond the corpus callosum. J Neuroradiol. 2008;35:301-303.
Tang PH, Bartha AI, Norton ME, et al. Agenesis of the corpus callosum: an MR imaging analysis of associated abnormalities in the fetus. AJNR Am J Neuroradiol. 2009;30:257-263.
Volpe P, Campobasso G, De Robertis V, et al. Disorders of prosencephalic development. Prenat Diagn. 2009;29:340-354.
585.9 Agenesis of the Cranial Nerves and Dysgenesis of the Posterior Fossa
Bolduc M, Limperopoulos C. Neurodevelopmental outcomes in children with cerebellar malformations: a systematic review. Dev Med Child Neurol. 2009;51:256-267.
Huang H, Hwang CW, Lai PH, et al. Möbius syndrome as a syndrome of rhombencephalic maldevelopment: a case report. Pediatr Neonatol. 2009;50:36-38.
Jaspan T. New concepts on posterior fossa malformations. Pediatr Radiol. 2008;38:S409-S414.
Long A, Moran P, Robson S. Outcome of fetal cerebral posterior fossa anomalies. Prenat Diagn. 2006;26:707-710.
Sasaki-Adams D, Elbabaa SK, Jewells V, et al. The Dandy-Walker variant: a case series of 24 pediatric patients and evaluation of associated anomalies, incidence of hydrocephalus, and developmental outcomes. J Neurosurg Pediatr. 2008;2:194-199.
Traboulsi EI. Congenital abnormalities of cranial nerve development: overview, molecular mechanisms, and further evidence of heterogeneity and complexity of syndromes with congenital limitation of eye movements. Trans Am Ophthalmol Soc. 2004;102:373-389.
585.10 Microcephaly
Etiology
Primary microcephaly refers to a group of conditions that usually have no associated malformations and follow a mendelian pattern of inheritance or are associated with a specific genetic syndrome. Affected infants are usually identified at birth because of a small head circumference. The more common types include familial and autosomal dominant microcephaly and a series of chromosomal syndromes that are summarized in Table 585-2. Primary microcephaly is also associated with at least 7 loci, and 4 single etiologic genes have been identified. It is known as autosomal recessive primary microcephaly (MCPH) and has autosomal inheritance. Many X-linked causes of microcephaly are caused by gene mutations that lead to severe structural brain malformations such as lissencephaly, and these should be sought on MRI. Secondary microcephaly results from a large number of noxious agents that can affect a fetus in utero or an infant during periods of rapid brain growth, particularly the 1st 2 yr of life.
| CAUSES | CHARACTERISTIC FINDINGS |
|---|---|
| PRIMARY (GENETIC) | |
| Familial (autosomal recessive) | Incidence 1/40,000 births Typical appearance with slanted forehead, prominent nose and ears; severe mental retardation and prominent seizures; surface convolutional markings of the brain, poorly differentiated and disorganized cytoarchitecture |
| Autosomal dominant | Nondistinctive facies, upslanting palpebral fissures, mild forehead slanting, and prominent ears Normal linear growth, seizures readily controlled, and mild or borderline mental retardation |
| Syndromes | |
| Down (trisomy 21) | Incidence 1/800 Abnormal rounding of occipital and frontal lobes and a small cerebellum; narrow superior temporal gyrus, propensity for Alzheimer neurofibrillary alterations, ultrastructure abnormalities of cerebral cortex |
| Edward (trisomy 18) | Incidence 1/6,500 Low birthweight, microstomia, micrognathia, low-set malformed ears, prominent occiput, rocker-bottom feet, flexion deformities of fingers, congenital heart disease, increased gyri, heterotopias of neurons |
| Cri-du-chat (5 p-) | Incidence 1/50,000 Round facies, prominent epicanthic folds, low-set ears, hypertelorism, characteristic cry No specific neuropathology |
| Cornelia de Lange | Prenatal and postnatal growth delay, synophrys, thin downturning upper lip Proximally placed thumb |
| Rubinstein-Taybi | Beaked nose, downward slanting of palpebral fissures, epicanthic folds, short stature, broad thumbs and toes |
| Smith-Lemli-Opitz | Ptosis, scaphocephaly, inner epicanthic folds, anteverted nostrils Low birthweight, marked feeding problems |
| SECONDARY (NONGENETIC) | |
| Congenital Infections | |
| Cytomegalovirus | Small for dates, petechial rash, hepatosplenomegaly, chorioretinitis, deafness, mental retardation, seizures Central nervous system calcification and microgyria |
| Rubella | Growth retardation, purpura, thrombocytopenia, hepatosplenomegaly, congenital heart disease, chorioretinitis, cataracts, deafness Perivascular necrotic areas, polymicrogyria, heterotopias, subependymal cavitations |
| Toxoplasmosis | Purpura, hepatosplenomegaly, jaundice, convulsions, hydrocephalus, chorioretinitis, cerebral calcification |
| Drugs | |
| Fetal alcohol | Growth retardation, ptosis, absent philtrum and hypoplastic upper lip, congenital heart disease, feeding problems, neuroglial heterotopia, disorganization of neurons |
| Fetal hydantoin | Growth delay, hypoplasia of distal phalanges, inner epicanthic folds, broad nasal ridge, anteverted nostrils |
| Other Causes | |
| Radiation | Microcephaly and mental retardation most severe with exposure before 15th wk of gestation |
| Meningitis/encephalitis | Cerebral infarcts, cystic cavitation, diffuse loss of neurons |
| Malnutrition | Controversial cause of microcephaly |
| Metabolic | Maternal diabetes mellitus and maternal hyperphenylalaninemia |
| Hyperthermia | Significant fever during 1st 4-6 wk has been reported to cause microcephaly, seizures, and facial anomalies Pathologic studies show neuronal heterotopias Further studies showed no abnormalities with maternal fever |
| Hypoxic-ischemic encephalopathy | Initially diffuse cerebral edema; late stages characterized by cerebral atrophy and abnormal signals on MR imaging |
Treatment
Once the cause of microcephaly has been established, the physician must provide accurate and supportive genetic and family counseling. Because many children with microcephaly are also mentally retarded, the physician must assist with placement in an appropriate program that will provide for maximal development of the child (Chapter 33).
Abuelo D. Microcephaly syndromes. Semin Pediatr Neurol. 2007;14:118-127.
Barkovich AJ, Kuzniecky RI, Jackson MD, et al. Classification system for malformations of cortical development. Neurology. 2001;57:2168-2178.
Clark GD. The classification of cortical dysplasias through molecular genetics. Brain Dev. 2004;26:351-362.
Denis D, Chateil JF, Brun M, et al. Schizencephaly: clinical and imaging features in 30 infantile cases. Brain Dev. 2000;22:475-483.
d’Orsi G, Tinuper P, Bisulli F, et al. Clinical features and long term outcome of epilepsy in periventricular nodular heterotopia. Simple compared with plus forms. J Neurol Neurosurg Psychiatry. 2004;75:873-878.
Guerrini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci. 2008;31:154-162.
Hayashi N, Tsutsumi Y, Barkovich AJ. Morphological features and associated anomalies of schizencephaly in the clinical population: detailed analysis of MR images. Neuroradiology. 2002;44:418-427.
Kinsman SL, Plawner LL, Hahn JS. Holoprosencephaly: recent advances and insights. Curr Opin Neurol. 2000;13:127-132.
Parrish ML, Roessmann U, Levinsohn MW. Agenesis of the corpus callosum: a study of the frequency of associated malformations. Ann Neurol. 1979;6:349-354.
Richards LJ, Plachez C, Ren T. Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet. 2004;66:276-289.
Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genetics. 2005;76:717-728.
Wynshaw-Boris A. Lissencephaly and LIS1: insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72:296-304.
585.11 Hydrocephalus
Hydrocephalus is not a specific disease; it represents a diverse group of conditions that result from impaired circulation and absorption of CSF or, in rare circumstances, from increased production of CSF by a choroid plexus papilloma (Table 585-3). Because megalencephaly is often discovered as part of an evaluation for hydrocephalus in children with macrocephaly, it is included in this section.
Table 585-3 CAUSES OF HYDROCEPHALUS
COMMUNICATING
NONCOMMUNICATING
HYDRANENCEPHALY
* Toxoplasmosis, neurocysticercosis mumps.
From Fenichel GM: Clinical pediatric neurology, ed 5, Philadelphia, 2005, Elsevier, p 354.
Clinical Manifestations
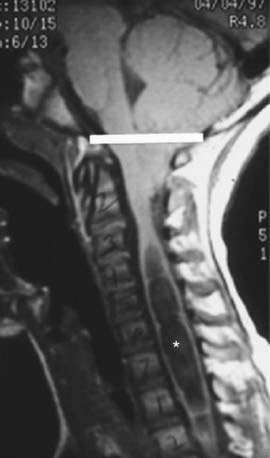
Chiari malformation consists of two major subgroups. Type I typically produces symptoms during adolescence or adult life and is usually not associated with hydrocephalus. Patients complain of recurrent headache, neck pain, urinary frequency, and progressive lower extremity spasticity. The deformity consists of displacement of the cerebellar tonsils into the cervical canal (Fig. 585-10). Although the pathogenesis is unknown, a prevailing theory suggests that obstruction of the caudal portion of the 4th ventricle during fetal development is responsible. Other theories include tethering of the cord or additional anomalies (syrinx).
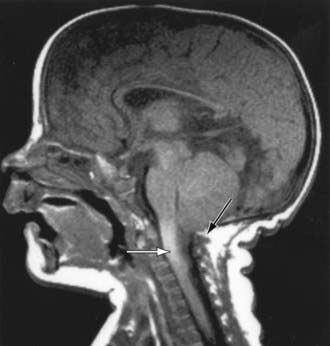
The type II Chiari malformation is characterized by progressive hydrocephalus with a myelomeningocele. This lesion represents an anomaly of the hindbrain, probably owing to a failure of pontine flexure during embryogenesis, and results in elongation of the 4th ventricle and kinking of the brainstem, with displacement of the inferior vermis, pons, and medulla into the cervical canal (Fig. 585-11). Approximately 10% of type II malformations produce symptoms during infancy, consisting of stridor, weak cry, and apnea, which may be relieved by shunting or by decompression of the posterior fossa. A more indolent form consists of abnormalities of gait, spasticity, and increasing incoordination during childhood.
The Dandy-Walker malformation consists of a cystic expansion of the 4th ventricle in the posterior fossa and midline cerebellar hypoplasia, which results from a developmental failure of the roof of the 4th ventricle during embryogenesis (Fig. 585-12). Approximately 90% of patients have hydrocephalus, and a significant number of children have associated anomalies, including agenesis of the posterior cerebellar vermis and corpus callosum. Infants present with a rapid increase in head size and a prominent occiput. Transillumination of the skull may be positive. Most children have evidence of long-tract signs, cerebellar ataxia, and delayed motor and cognitive milestones, probably due to the associated structural anomalies. The Dandy-Walker malformation is managed by shunting the cystic cavity (and on occasion the ventricles as well) in the presence of hydrocephalus.
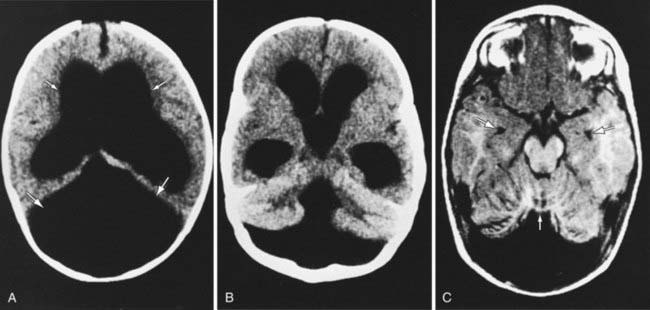
Hydranencephaly
Hydranencephaly may be confused with hydrocephalus. The cerebral hemispheres are absent or represented by membranous sacs with remnants of frontal, temporal, or occipital cortex dispersed over the membrane. The midbrain and brainstem are relatively intact (Fig. 585-13). The cause of hydranencephaly is unknown, but bilateral occlusion of the internal carotid arteries during early fetal development would explain most of the pathologic abnormalities. Affected infants can have a normal or enlarged head circumference at birth that grows at an excessive rate postnatally. Transillumination shows an absence of the cerebral hemispheres. The child is irritable, feeds poorly, develops seizures and spastic quadriparesis, and has little or no cognitive development. A ventriculoperitoneal shunt prevents massive enlargement of the cranium.
Acakpo-Satchivi L, Shannon CN, Tubbs RS, et al. Death in shunted hydrocephalic children: a follow-up study. Childs Nerv Syst. 2008;24:197-201.
Chumas P, Tyagi A, Livingston J. Hydrocephalus—what’s new? Arch Dis Child Fetal Neonatal Ed. 2001;85:F149-F154.
Cochrane DD, Myles ST, Nimrod C, et al. Intrauterine hydrocephalus and ventriculomegaly: associated abnormalities and fetal outcome. Can J Neurol Sci. 1985;12:51-59.
Drake JM. The surgical management of pediatric hydrocephalus. Neurosurgery. 2008;62(Suppl 2):633-640.
Greene M, Benacerraf B, Crawford J. Hydranencephaly: US appearance during in utero evolution. Radiology. 1985;156:779-780.
Hirsch JF, Pierre-Kahn A, Renier D, et al. The Dandy-Walker malformation. J Neurosurg. 1984;61:515-522.
Long A, Moran P, Robson S. Outcome of fetal cerebral posterior fossa anomalies. Prenat Diag. 2006;26:707-710.
Novegno F, Caldarelli M, Massa A, et al. The natural history of the Chiari type I anomaly. J Neurosurg Pediatr. 2008;2:179-187.
Olney AH. Macrocephaly syndromes. Semin Pediatr Neurol. 2007;14:128-135.
Parisi MA, Dobyns WB. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab. 2003;80:36-53.
Persson E, Anderson S, Wiklund L, et al. Hydrocephalus in children born in 1999–2002: epidemiology, outcome and ophthalmological findings. Childs Nerv Syst. 2007;23:1111-1118.
Sacko O, Boetto S, Lauwers-Cances V, et al. Endoscopic third ventriculostomy: overcome analysis in 368 procedures. J Neurosurg Pediatr. 2010;5:68-74.
Stevenson KL. Chiari type II malformation: past, present, and future. Neurosurg Focus. 2004;16:E5.
Van Landingham M, Nguyen TV, Roberts A, et al. Risk factors of congenital hydrocephalus: a 10 year retrospective study. J Neurol Neurosurg Psychiatry. 2009;80:213-217.
585.12 Craniosynostosis
Development and Etiology
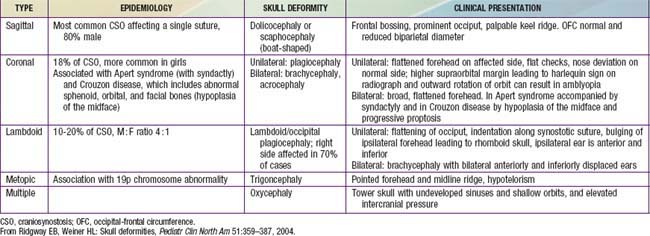
The bones of the cranium are well developed by the 5th mo of gestation (frontal, parietal, temporal, and occipital) and are separated by sutures and fontanels. The brain grows rapidly in the 1st several years of life and is normally not impeded because of equivalent growth along the suture lines. The cause of craniosynostosis is unknown, but the prevailing hypothesis suggests that abnormal development of the base of the skull creates exaggerated forces on the dura that act to disrupt normal cranial suture development. Genetic factors have been identified for some isolated and for many syndromic causes of craniosynostosis (Table 585-4).
Table 585-4 COMMONLY USED CLINICAL GENETIC CLASSIFICATIONS OF CRANIOSYNOSTOSES
| DISORDER | CAUSE |
|---|---|
| ISOLATED CRANIOSYNOSTOSIS | |
| Morphologically described | Unknown, uterine constraint, or FGFR3 mutation |
| SYNDROMIC CRANIOSYNOSTOSIS | |
| Antler-Bixler syndrome | Unknown |
| Apert syndrome | Usually one of two mutations in FGFR2 |
| Baere-Stevenson syndrome | Mutation in GFGR2 or FGFR3 |
| Bailler-Gerold syndrome | Mutation in TWIST heterogenous |
| Carpenter syndrome | Unknown |
| Craniofrontonasal dysplasia | Unknown gene at Xp22 |
| Crouzon syndrome | Numerous different mutations at FGFR2 |
| Crouzonomesodermoskeletal syndrome | Mutation in FGFR3 |
| Jackson-Weiss syndrome | Mutation in FGFR2 |
| Muenke syndrome | Mutation in FGFR3 |
| Pfeiffer syndrome | Mutation in FGFR1 or numerous mutation in FGFR2 |
| Saethre-Chotzen syndrome | Mutation in TWIST |
| Shprintzen-Goldberg syndrome | Mutation in FBEN1 |
From Ridgway EB, Weiner HL: Skull deformaties, Pediatr Clin North Am 51:359–387, 2004.
Clinical Manifestations and Treatment
Most cases of craniosynostosis are evident at birth and are characterized by a skull deformity that is a direct result of premature suture fusion. Palpation of the suture reveals a prominent bony ridge, and fusion of the suture may be confirmed by plain skull roentgenograms, CT scan or bone scan in ambiguous cases (Table 585-5).
Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989–2003. Am J Med Genet. 2008;146A:984-991.
Di Rocco F, Arnaud E, Meyer P, et al. Focus session on the changing “epidemiology” of craniosynostosis (comparing two quinquennia: 1985–1989 and 2003–2007) and its impact on the daily clinical practice: a review from Necker Enfants Malades. Childs Nerv Syst. 2009;25:807-811.
Losee JE, Mason AC. Deformational plagiocephaly: diagnosis, prevention, and treatment. Clin Plast Surg. 2005;32:53-64.
McKinney CM, Cunningham ML, Holt VL, et al. Characteristics of 2733 cases diagnosed with deformational plagiocephaly and changes in risk factors over time. Cleft Palate Craniofac J. 2008;45:208-216.
Passos-Bueno MR, Serti Eacute AE, Jehee FS, et al. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol. 2008;12:107-143.
Rasmussen SA, Yazdy MM, Frías JL, et al. Priorities for public health research on craniosynostosis: summary and recommendations from a Centers for Disease Control and Prevention–sponsored meeting. Am J Med Genet. 2008;146A:149-158.
Ridgway EB, Weiner HL. Skull deformities. Pediatr Clin North Am. 2004;51:359-387.
Speltz ML, Kapp-Simon KA, Cunningham M, et al. Single-suture craniosynostosis: a review of neurobehavioral research and theory. J Pediatr Psychol. 2004;29:651-668.
van der Meulen J, van der Hulst R, van Adrichem L, et al. The increase of metopic synostosis: a pan-European observation. J Craniofac Surg. 2009;20:283-286.
van Vlimmeren LA, van der Graaf Y, Boere-Boonekamp MM, et al. Risk factors for deformational plagiocephaly at birth and at 7 weeks of age: a prospective cohort study. Pediatrics. 2007;119:e408-e418.