CHAPTER 290 Congenital Abnormalities of the Thoracic and Lumbar Spine
Epidemiology and Associated Disorders
The overall incidence of congenital thoracolumbar abnormalities is low. Spinal dysraphism is seen in 20 of every 100,000 births in the United States and higher in offspring of affected parents.1 There is a greater risk in pregnancies with maternal diabetes mellitus.2 Congenital scoliosis has an overall incidence of 1 in 1000 to 2000.3 Known genetic abnormalities include linkage to chromosome 18 for multiple defects in segmentation and chromosome 17 for hemivertebrae.4,5
Abnormalities of the spine are often seen in association with disorders in other systems. In one series, up to 61% of patients with congenital spine disease had another system involved.6 The urologic system is the most commonly associated system seen in conjunction with congenital spine abnormalities. Oskovian and associates and Macewen and colleagues found 18% of their congenital scoliosis patients to have urologic abnormalities.7,8 Defects in the cardiac system have been found in 26% of patients with congenital spinal abnormalities as described by Basu and colleagues.9 There is a clear association with pulmonary insufficiency with severe thoracic vertebral deformities related to structural limitations imposed on the thorax by the deformity.10 There is also an association between congenital spine abnormalities and such syndromes as OEIS, VATER, and the Currarino triad (anorectal malformation, partial sacral agenesis, and presacral mass).11–13 The common association between congenital spinal anomalies and others mandates a thorough work-up of a patient with congenital spine abnormalities.
Imaging
Magnetic resonance imaging (MRI) of the entire neuraxis is indicated in the evaluation of congenital spinal abnormalities because 18% to 35% of patients with congenital scoliosis have abnormalities of the brain and spinal cord.14–18 Ultrasound can be useful in neonatal evaluation before the benefit of diagnosis outweighs the risk of the general anesthesia required for MRI.19 There is some debate that MRI should be reserved for patients with lower extremity, bowel, or bladder symptomatology, or signs of closed spinal dysraphism such as hairy patches. However, the utility, safety, and widespread availability of this imaging modality should be considered. The ability to assess cord tethering, asymptomatic mass lesions, and syringomyelia makes MRI pivotal in weighing the benefits, risks, and alternatives of management plans.
Embryology
Embryogenesis
The normal embryo transforms into the blastula after bilaminar induction by the second week of gestation. The epiblast is the origin of future ectoderm and the hypoblast is the origin of future mesoderm. The primitive streak is located on the dorsal embryonic midline and is composed of totipotent epiblastic cells. Human spine development occurs at the site of the primitive streak, which appears on day 15. Cells divide caudally to elongate the primitive streak, ereas the primitive knot or Hensen’s node is the rostral thickening. This elongation defines the rostrocaudal embryonic axis.20
The process of gastrulation involves the induction of the future mesoderm and this happens during week 3. The blastopore, an opening in the blastula, marks the embryo’s entry into gastrulation. Subsequently, the epiblastic cells migrate in a complex motion dictated by the central signaling apparatus, Hensen’s node, or the Spemann organizer, located on the axial side.21 The organizer crucially acts to allow migration of epiblastic cells into the primitive groove, an indentation within the primitive streak. These migrating epiblastic cells interpose in the epiblast-hypoblast transition to form mesoderm, making a trilaminar embryo composed of ectoderm, mesoderm, and endoderm. The midline migratory cells specifically form the basis of the notochord, axial skeleton, and neuroectoderm inductor. Abnormal gastrulation results in failed integration of neural structures into the axial midline and resultant split-cord malformations.22
Multiple genes have been implicated in abnormal gastrulation such as BMP and Pax. The homeobox and zinc finger class transcription factor gene families are necessary for proper induction of mesoderm formation, differentiation of neural crest, and development of the vertebral column and spinal cord.23
Neurogenesis
Cells migrate from the cranial end of the primitive streak from Hensen’s node to form the notochord on day 16. Organogenesis requires an intact notochord and primitive streak. During primary neurulation, the notochord interacts with the overlying ectoderm to form the neuroectoderm and neural plate. Subsequently, on day 18, the neural plate invaginates to form the neural groove in the midline and the neural folds laterally. By day 21, the neural folds fuse to form the precursor of the spinal cord, the neural tube. This portion of the developing spine and neuraxis extends only to the lumbosacral junction. During secondary neurulation, the neural tube distal to the second sacral vertebra forms from the caudal aspect of the primitive streak. These secondary neural elements cavitate with a terminal ventricle and eventually form the cauda equina and distal spinal cord. The conus medullaris contains a dilated central canal that is the remnant of the cavity with the secondary neural tube.24 The neural tube closes in a bidirectional, zipperlike fashion starting around the sixth to seventh cervical somite stage. The cranial neuropore closes around day 24 and the caudal neuropore closes around day 27. Failure of neuropore closure results in the spectrum of disorders known as spinal dysraphism.25
Skeletogenesis
The somite stage of development marks the formation of the spine around the notochord.26 The mesoderm bilaterally thickens and segments into paired cuboidal structures called somites at around 20 days of gestation. They form along a rostrocaudal axis. A total of 42 to 44 pairs of somites form, 4 occipital, 8 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 8 to 10 coccygeal. During development, there is regression of the first occipital and the last five to seven coccygeal somites, leaving a total of 38. The caudal eminence of the somites then becomes the hindgut, the terminal portion of the sacrum and coccyx and the terminal spinal cord. Failure of proper development of the caudal eminence, can lead to sacral agenesis, imperforate anus, cloacal exstrophy, and malformation/caudal regression.27–29
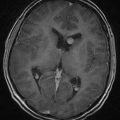
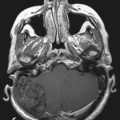
The ventral portions of the somites are the sclerotomes that eventually become the skeletal system, including the vertebral bodies, the cartilaginous tissue, the disks, and the cells of the spinal meninges.25 The intermediate portions are the myotomes that become the striated musculature. The dorsal portions are the dermatomes that become the skin and subcutaneous tissues. As development progresses, the sclerotomes distinctly divide in two to form the vertebral bodies and the intervertebral disks. Within the disks, the notochord remnants form the nucleus pulposus as control by Pax gene expression.30 Chordomas arise from notochord remnants and occur primarily at the skull base and the sacrum (Fig. 290-1).31,32
The expression of Pax is also important in the overall patterning of the sclerotome and the development of the ventral vertebral body from the neural tube, as demonstrated in a knockout mouse model.30 The dorsal vertebral arch, however, requires the induction from the neural crest and this signaling is dependent on Zic, Gli, and Pax.33 Due to the separate inductive controls and temporal development of the ventral and dorsal elements of the vertebra, abnormalities can be independent of each other.7
Congenital Scoliosis
Congenital scoliosis is defined as an abnormality in the lateral curvature of the spine due to abnormal vertebrae present from birth that induce a coronal plane deformity.34 Specifically, congenital scoliosis differs from infantile idiopathic scoliosis, because the latter is not associated with abnormally formed vertebrae. Although difficult to estimate accurately due to undetected, benign cases, the overall incidence of congenital scoliosis is approximately 1 to 2 per 1000.3,16,34 Clinical presentation varies widely because of the myriad combinations of possible vertebral abnormalities. Approximately, 25% of patients with congenital scoliosis progress rapidly, 50% progress slowly, and the remaining 25% do not progress.7
The congenital scoliotic deformity is caused by some combination of vertebral dysgenesis and/or failure of segmentation.35 The most common vertebral anomaly is the hemivertebra. In contrast, a unilateral unsegmented bar is the most common segmentation failure and involves the facet and disk.16 Both segmentation and formation defects can result in severe progressive scoliosis, especially in the thoracolumbar spine.36 Hemivertebrae can be classified into three major types: fully segmented, semisegmented, and nonsegmented. Fully segmented hemivertebrae allow for full growth on one side and none on the other, leaving a significant potential for deformity based on the anatomic location. The scoliotic curves from fully segmented hemivertebrae may progress at the rate of one to two degrees annually. Semisegmented hemivertebrae are connected to one of the adjacent vertebrae, causing a wedge shape with differential side growths resulting in some scoliosis. Nonsegmented hemivertebrae are connected to both adjacent vertebrae and lack disk spaces, causing deformity but with limited progression due to the absence of growth plates.
The principle type of segmentation failure resulting in congenital scoliosis is the unilateral unsegmented bar. There are no growth plates on the affected side and the unaffected side of the spine continues to grow normally, resulting in a progressive deformity of approximately five degrees annually. This abnormality usually results in a substantial deformity and is usually treated surgically.34,37
From natural history data, the rate of scoliotic deterioration is determined by patient age, anomaly type, and anomalous level and curve pattern.3,34 It is generally agreed that progressive deformity ought to be treated surgically if orthotic therapy fails.7,16 Surgery remains the best treatment for progressive and severe deformity. The many surgical options for the treatment of congenital scoliosis are discussed in detail elsewhere in this text. Common procedures include hemivertebra excision, in situ fusion, instrumentation, and thoracoplasty with vertical expansion prosthetic titanium rib (VEPTR).7 Specific indications are evolving and are beyond the scope of this chapter. However, it is important to note that due to the 18% to 35% risk of associated abnormalities of the neural axis, surgical treatment carries the risk of neurological deterioration, particularly in cases of an untreated tethered spinal cord.15–1738
Congenital Lordosis and Kyphosis
Isolated congenital kyphosis and lordosis, both sagittal plane deformities, are considerably less prevalent than congenital scoliosis. They are both commonly seen in conjunction with coronal plane deformities and referred to as kyphoscoliosis and lordoscoliosis, respectively.7,39 Congenital kyphosis is divided into three classes under the Winter scheme: type I has failed vertebral body formation, type II has failed vertebral body segmentation, and type III has mixed features. There is also associated instability, likely from defects in the pars interarticularis, which predisposes to further neurological injury.39 Early posterior treatment for type I and type III kyphosis can help prevent paraplegia and allow for continued anterior growth to let the spine straighten with time.40 Treatment with posterior arthrodesis is advocated for types I and III kyphosis before it exceeds 60 degrees.41,42 Kyphosis exceeding 60 degrees requires an anterior release and subsequent posterior stabilization.42,43 A combined anterior and posterior approach using costotransversectomy has also been advocated for treatment of kyphosis and kyphoscoliosis.44 Type II congenital kyphosis is less common and not as commonly associated with neurologic injury as types I and III unless associated with kyphoscoliosis.41
Congenital lordosis is the rarest of curvature deformities but can be the most fatal due to pulmonary compromise in the case of thoracic location.45 It results from a failure of posterior segmentation.10 These cases require anterior correction due to excessive anterior growth and may be associated with preexisting diminished pulmonary function or pulmonary artery hypertension.43,45
Congenital Thoracolumbar Stenosis
There are three types of stenosis: congenital, developmental, and degenerative. A distinction is made for congenital stenosis because it results from a prenatal malformation that presents insidiously. Developmental stenosis implies chromosomal or spinal cord malformation such as that associated with a hereditary syndrome such as achondroplasia.46,47 The patients usually become symptomatic earlier in life and involve more spinal segments than their degenerative counterparts and have similar symptoms of claudication or radiculopathy that are relieved by resting or flexion.46,48,49 There are multiple classification schemes for congenital stenosis, including one from Verbiest and another delineated by Singh and colleagues. Neither scheme offers risk stratification for treatment options.7,46,48 Of fundamental importance in consideration of surgical treatment are the presence of neurological deficits, concomitant conditions such as spinal dysraphism, and other nonspinal comorbidities.
Spondylolysis and Spondylolisthesis
Congenital defects in the pars interarticularis can present as breakage (spondylolysis) or slippage (spondylolisthesis). In their natural history study, Fredrickson and colleagues found the overall incidence of spondylolysis at age 6 as 4.4%, increasing to 5.8% by adulthood with a male-to-female ratio of 2 to 1. The maximum listhesis they found was 28% with rare symptoms aside from back pain and no progression past adolescence. There was a strong association with spina bifida.50 Wiltse and Jackson formulated the most widely used classification scheme of spondylolisthesis with five types: dysplastic (I), isthmic (II), degenerative (III), traumatic (IV), and pathologic (V). Types I and II have a congenital implication with dysplasia in the L5 or S1 neural arch (type I) or poorly formed pars interarticularis (type II). In type I, greater than 25% of listhesis can result in cauda equina injury.51 Some advocate decompression for symptomatic patients and fusion for patients with greater than 30% slippage.52 Low-grade spondylolisthesis Wiltse types I and II patients do better with combined anterior-posterior fusions.53 Molinari and colleagues suggest reduction and circumferential fusion for patients with high-grade spondylolisthesis.54
Sacral Agenesis and Caudal Regression Syndrome
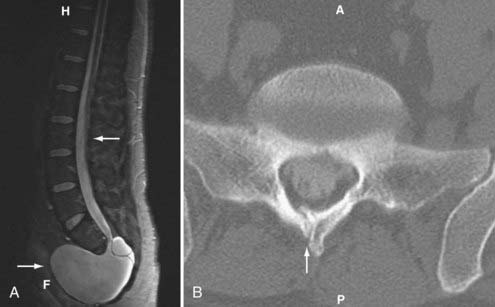
Caudal regression syndrome is a spectrum of disorders involving agenesis or absence of the lower thoracic, lumbar, sacral, or coccygeal spine. Agenesis above the 12th vertebral level is not compatible with life.55 Sacral agenesis is the most common of these conditions but still rare with an incidence of 1 in 25,000 (Fig. 290-2).7 There is a 1% prevalence of sacral agenesis in diabetic gestation, with diabetic mothers representing 16% of these.56 Caudal agenesis is mostly nonfamilial and sporadic, although genes have been identified for the autosomal dominant variant responsible for the Currarino triad, with associated anorectal abnormality and presacral mass lesion.57 There are also many associated abnormalities of the midline, including bladder exstrophy and imperforate anus. Clinical findings often include a stereotypical sitting described as the “Buddha” position with the arms resting on the hips, thereby providing support to the upper spine. Agenesis at the lumbosacral junction can result in knee flexion contractures and clubfeet. Agenesis above S2 results in neurogenic bladder. The common cause may be teratogenesis of the caudal eminence after the caudal neuropore closes. The Pang classification describes five types: type I—total sacral agenesis with missing lumbar vertebrae; type II—total sacral agenesis only; type III—subtotal sacral agenesis; type IV—hemisacrum; and type V—agenesis of the coccyx.27
Sacrococcygeal Teratoma
Sacrococcygeal teratoma is a relatively uncommon sacral tumor, whose overall incidence is approximately 1 in 35,000 births.58 There is a 4 : 1 female predominance.59 The approximate incidence in adults is 1 in 87,000.60 The tumors are usually benign and originate from the tissue around Hensen’s node or the primitive knot and mostly arise in the sacrococcygeal region. The most commonly used classification scheme comes from Altman and colleagues with four strata: type I—external; type II—mostly external; type III—mostly internal; and type IV—internal.61 Survival is reported at 95% at 1 to 5 years follow-up after operative resection without or with chemotherapy, depending upon malignant transformation.62
Spinal Dysraphism
Spinal dysraphism has a varied global incidence and is highest in Ireland.63 The United States incidence has greatly decreased due to folate supplementation. Since the 1998 mandate of cereal folate supplementation, the specific incidence of open neural tube defects has further decreased by 26%.1 There are many nutritional factors that have been implicated in preventing spinal dysraphism, including folate, vitamin C, calcium, zinc, and vitamin A. The risk of spinal dysraphism is increased in the first-degree relatives of afflicted patients.63 The historical differentiation of open and closed spinal dysraphism is based on presentation at birth. Open dysraphism involves open or almost open defects present at birth. Full layers of epidermis and dermis obfuscate the defects of closed dysraphism including lipomyelomeningocele, diastematomyelia, neurenteric cyst, dermoid cyst, terminal myelocystocele, “meningocele manqué,” fatty filum terminale, and dermal sinus tract. Such defects can be associated with cutaneous stigmata such as capillary hemangiomas, hypertrichosis, nevi, dimples, or sinus tracts.64
The errors of closed dysraphism happen in the first 8 weeks of embryogenesis. The neural tube fails to develop normally and widens, hence the term spina bifida. When the neural tube does not completely separate from the ectoderm, the resultant malformations include cord tethering, dermal sinus tract, or diastematomyelia. Lipomas result when the ectoderm and tube prematurely separate because of incorporation of intervening mesenchymal elements. When the posterior spinal midline elements fail to form, myelomeningocele and other midline abnormalities occur.7
The clinical presentation of closed spinal dysraphism often involves tethered cord syndrome, first described clinically in 1891 by Jones.65 The clinical symptoms include back pain, loss of perineal sensation, lower extremity myelopathy, bowel incontinence, and neurogenic bladder. The associated findings can involve any of the described lesions and also a low-lying conus medullaris and necessitate total spine imaging. The symptoms are thought to result from hypoxic injury to the lower spinal cord from traction. Often these symptoms worsen with activity because there is thought to be more traction on the cord during activity.65
Lipomyelomeningocele
Intraspinal lipomas are uncommon and are usually found in the cervicothoracic spine.66 The entity of lipomyelomeningocele describes a lipoma fused to a dorsally split spinal cord. They comprise 20% to 56% of closed spinal dysraphism cases and 20% of epidermally closed caudal masses, but the overall prevalence is unknown because not all cases are symptomatic.67 Subcutaneous lipoma is the most common cutaneous sign.68,69 A defect in secondary neurulation, premature dysjunction of the ectodermal layer, is postulated to allow mesenchymal migration in the neural tube. Only fat can be induced to differentiate out of mesenchyme by the neural tube.70,71 The lipoma tethers to the superficial dermal elements and often presents with tethered cord syndrome. The clinical management of the lesion is predicated by the severity of the symptomatology. If the primary concern is back pain and paresthesia, surgical detethering can be postponed at the patient’s discretion. The presence of bladder or bowel compromise or myelopathy warrants more urgent care.
Diastematomyelia and Diplomyelia
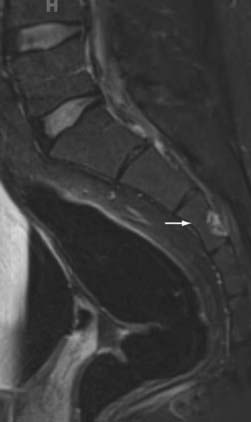
Pang72 provided a unified theory of split cord malformations (SCMs) that encompass both diastematomyelia and diplomyelia. The primary anomaly is an adhesion between ectoderm and endoderm during embryogenesis resulting from the accessory neurenteric canal forming through the midline of the embryonic disk. In those designated type I, there are two sagittally separate hemicords in individual dural sacs separated by a rigid osseocartilaginous septum, including diastematomyelia. Type II SCMs involve two separate hemicords within one dural sac separated by a flexible fibrous septum (Figs. 290-3 and 290-4). Medial nerve roots occur in 75% of cases and are almost always dorsal. Type I SCMs comprise 60% of cases.73,74 True diplomyelia is rare and describes duplicated cords.75
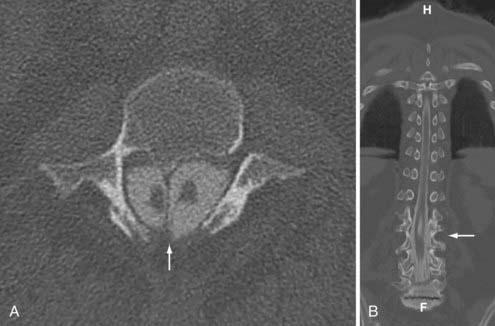
In regards to clinical findings, Keim and Greene reported that 70% of patients with diastematomyelia have associated scoliosis. They noted 50% of patients had cutaneous stigmata at the level of the malformation.76 The symptoms of SCMs can worsen with time as the patient ages, the spinal column grows, and the spinal cord elevates, but the osseous or cartilaginous septum remains fixed. Adults often have perineal anesthesia and urinary symptoms. The surgical management of these lesions aims at removing the septum and closing as much of the dural defect as possible. Ventral repairs can be challenging. In the setting of scoliosis or tethered cord, the SCM ought to be repaired first to prevent neurological injury from correction or detethering due to cord migration.
Neurenteric Cyst
Neurenteric cysts are rare lesions that result from endodermic communication of the developing foregut to the neural tube and usually occur in the caudal spinal cord. The remnant endoderm in the tract between the hemicords forms the cyst. Due to the rotation of the gut, there is a right-sided predominance of the cysts.77 The cysts can present insidiously with symptoms of myelopathy or meningitis.78,79 The surgical approach is intraspinal cyst resection and primary dural closure.
Terminal Myelocystocele
Terminal myelocystoceles are cerebrospinal fluid–filled dilations of the distal spinal cord that are tethered to the skin and represent 5% of lumbosacral skin covered masses. These lesions can be silent at birth and insidiously have caudal cord symptoms due to terminal tethering. On imaging, there are two sacs, one with a proximally enlarged subarachnoid space and the other with a dilation of the terminal cord. The cause of this abnormality likely involves the idiopathic dilation of the terminal ventricle that disrupts the mesenchymal layer only and then adheres to the overlying skin and prevents spinal cord ascension. The operative management requires exposure of the sac and detethering along with adequate dural closure.80
Dermal Sinus Tract
Evaluation for a dermal sinus tract is part of the initial pediatric examination of the newborn, although patients often only become symptomatic later in life. The incidence of dermal sinus tracts is 1 in 2500 live births.81 They can present in a delayed fashion and should be suspected in cases of recurrent meningitis. The embryologic origin involves failed dysjunction of ectoderm from endoderm and results in a lined squamous epithelial tract extending from the skin to the subarachnoid space. They can occur anywhere in the spine but typically occur in the lumbosacral region. There is no role for observation because management demands prompt imaging and complete surgical excision of the tract to prevent infection of the central nervous system.82
Dermoids and Epidermoids
These masses represent 1% to 2% of all spinal tumors and occur with about 50% of dermal sinus tracts.83,84 They can either have acute chemical meningitis from rupture or compressive symptoms. The ruptured fat globules from dermoids are bright on T1-weighted sequences on MRI. Epidermoids restrict diffusion on diffusion-weighted T2 sequences on MRI and this is how they are distinguished from simple arachnoid cysts. There are numerous embryological explanations for intraspinal dermoids and epidermoids including isolated rests of pluripotent cells that differentiate into these tumors or failed dysjunction of ectoderm from endoderm during week 4 of development.7 Observation can be employed in the event of an incidental finding. Surgical management involves complete resection in the presence of symptomatology. Postoperative aseptic meningitis is not uncommon and the recurrence rate is low with total resection.85
Fatty Filum Terminale
This tethered cord syndrome is often attributed to the presence of a shortened, thickened, fat-filled filum terminale. Iskandar and colleagues observed this in 56% of their patients.86 The cord is anchored prematurely during development and the filum does not lengthen. As the spinal cord ascends during spinal column growth, the neurological deterioration can be profound. Usually, on MRI the conus medullaris lies below L1-2 in 82% to 86% of these patients, but the conus can also be at a normal height with similar symptoms. There was an overlying skin abnormality or vertebral anomaly in 13 of the 55 patients reviewed by Warder and Oakes.87 The surgical management involves a laminectomy at the level of the conus and sectioning of the abnormal filum. The dura is closed primarily. The incidence of cord “retethering” after filum sectioning is low. However, incorrect identification of the abnormal filum may occur, requiring re-exploration.88–89 Intraoperative nerve root stimulation may be helpful in these cases.
Meningocele Manqué
The term “meningocele manqué” refers to a dorsal dural band tethering the spinal cord. The band is composed of atretic or fibrotic neural tissue and can be remote from the location of obvious tethering like diastematomyelia.65 The dorsal bands can be detected on CT myelography or MRI.90 Surgical management involves lysis of the band.65
Ackerman LL, Menezes AH. Spinal congenital dermal sinuses: a 30-year experience. Pediatrics. 2003;112(Pt 1):641-647.
Banta JV, Nichols O. Sacral agenesis. J Bone Joint Surg Am. 1969;51:693-703.
Duhamel B. From the mermaid to anal imperforation: the syndrome of caudal regression. Arch Dis Child. 1961;186:152-155.
Fidas A, MacDonald HL, Elton RA, et al. Prevalence and patterns of spina bifida occulta in 2707 normal adults. Clin Radiol. 1987;38:537-542.
Fredrickson BE, Baker D, McHolick WJ, et al. The natural history of spondylolysis and spondylolisthesis. J Bone Joint Surg Am. 1984;66:699-707.
Guidetti B, Gagliardi FM. Epidermoid and dermoid cysts. Clinical evaluation and late surgical results. J Neurosurg. 1977;47:12-18.
Kaffenberger DA, Heinz ER, Oakes JW, Boyko O. Meningocele manque: radiologic findings with clinical correlation. AJNR Am J Neuroradiol. 1992;13:1083-1088.
Mathews TJ, Honein MA, Erickson JD. Spina bifida and anencephaly prevalence—United States, 1991-2001. MMWR Recomm Rep. 2002;51(RR-13):9-11.
McMaster MJ. Occult intraspinal anomalies and congenital scoliosis. J Bone Joint Surg Am. 1984;66:588-601.
McMaster MJ, Ohtsuka K. The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J Bone Joint Surg Am. 1982;64:1128-1147.
Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-778.
Pang D. Split cord malformation: part II: clinical syndrome. Neurosurgery. 1992;31:481-500.
Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
Schropp C, Sörensen N, Collmann H, Krauss J. Cutaneous lesions in occult spinal dysraphism—correlation with intraspinal findings. Childs Nerv Syst. 2006;22:125-131.
Spemann H, Mangold H. Induction of embryonic primordia by implantation of organizers from a different species. 1923. Int J Dev Biol. 2001;45:13-38.
Suh SW, Sarwark JF, Vora A, Huang BK. Evaluating congenital spine deformities for intraspinal anomalies with magnetic resonance imaging. J Pediatr Orthop. 2001;21:525-531.
Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115-1123.
Verbiest H. Results of surgical treatment of idiopathic developmental stenosis of the lumbar vertebral canal. A review of twenty-seven years’ experience. J Bone Joint Surg Br. 1977;59:181-188.
Winter RB, Moe JH, Bradford DS. Congenital thoracic lordosis. J Bone Joint Surg Am. 1978;60:806-810.
Winter RB, Moe JH, Wang JF. Congenital kyphosis. Its natural history and treatment as observed in a study of one hundred and thirty patients. J Bone Joint Surg Am. 1973;55:223-256.
1 Mathews TJ, Honein MA, Erickson JD. Spina bifida and anencephaly prevalence—United States, 1991-2001. MMWR Recomm Rep. 2002;51(RR-13):9-11.
2 Aberg A, Westbom L, Kallen B. Congenital malformations among infants whose mothers had gestational diabetes or preexisting diabetes. Early Hum Dev. 2001;61:85-95.
3 Shands ARJr, Eisberg HB. The incidence of scoliosis in the state of Delaware: a study of 50,000 minifilms of the chest made during a survey for tuberculosis. J Bone Joint Surg Am. 1955;37:1243-1249.
4 Gottlieb MI, Hirschhorn K, Cooper HL, et al. Trisomy-17 syndrome. Report of three cases and review of the literature. Am J Med. 1962;33:763-773.
5 Dowton SB, Hing AV, Sheen-Kaniecki V, Watson MS. Chromosome 18q22.2—>qter deletion and a congenital anomaly syndrome with multiple vertebral segmentation defects. J Med Genet. 1997;34:414-417.
6 Beals RK, Robbins JR, Rolfe B. Anomalies associated with vertebral malformations. Spine. 1993;18:1329-1332.
7 Oskouian RJJr, Sansur CA, Shaffrey CI. Congenital abnormalities of the thoracic and lumbar spine. Neurosurg Clin N Am. 2007;18:479-498.
8 Macewen GD, Winter RB, Hardy JH. Evaluation of kidney anomalies in congenital scoliosis. J Bone Joint Surg Am. 1972;54:1451-1454.
9 Basu PS, Elsebaie H, Noordeen MH. Congenital spinal deformity: a comprehensive assessment at presentation. Spine. 2002;27:2255-2259.
10 Winter RB, Moe JH, Bradford DS. Congenital thoracic lordosis. J Bone Joint Surg Am. 1978;60:806-810.
11 Currarino G, Coln D, Votteler T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981;137:395-398.
12 Carey JC, Greenbaum B, Hall BD. The OEIS complex (omphalocele, exstrophy, imperforate anus, spinal defects). Birth Defects Orig Artic Ser. 1978;14:253-263.
13 Quan L, Smith DW. The VATER association. Vertebral defects, anal atresia, T-E fistula with esophageal atresia, radial and renal dysplasia: a spectrum of associated defects. J Pediatr. 1973;82:104-107.
14 Belmont PJJr, Kuklo TR, Taylor KF, et al. Intraspinal anomalies associated with isolated congenital hemivertebra: the role of routine magnetic resonance imaging. J Bone Joint Surg Am. 2004;86:1704-1710.
15 McMaster MJ. Occult intraspinal anomalies and congenital scoliosis. J Bone Joint Surg Am. 1984;66:588-601.
16 Hedequist D, Emans J. Congenital scoliosis: a review and update. J Pediatr Orthop. 2007;27:106-116.
17 Prahinski JR, Polly DWJr, McHale KA, Ellenbogen RG. Occult intraspinal anomalies in congenital scoliosis. J Pediatr Orthop. 2000;20:59-63.
18 Suh SW, Sarwark JF, Vora A, Huang BK. Evaluating congenital spine deformities for intraspinal anomalies with magnetic resonance imaging. J Pediatr Orthop. 2001;21:525-531.
19 Hughes JA, De Bruyn R, Patel K, Thomspon D. Evaluation of spinal ultrasound in spinal dysraphism. Clin Radiol. 2003;58:227-233.
20 Selleck MA, Stern CD. Fate mapping and cell lineage analysis of Hensen’s node in the chick embryo. Development. 1991;112:615-626.
21 Spemann H, Mangold H. Induction of embryonic primordia by implantation of organizers from a different species. 1923. Int J Dev Biol. 2001;45:13-38.
22 Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: part i: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
23 Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115-1123.
24 Nievelstein RA, Hartwig NG, Vermeij-Keers C, Valk J. Embryonic development of the mammalian caudal neural tube. Teratology. 1993;48:21-31.
25 Rossi A, Gandolfo C, Morana G, et al. Current classification and imaging of congenital spinal abnormalities. Semin Roentgenol. 2006;41:250-273.
26 Christ B, Wilting J. From somites to vertebral column. Ann Anat. 1992;174:23-32.
27 Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-778.
28 Muller F, O’Rahilly R. The development of the human brain, the closure of the caudal neuropore, and the beginning of secondary neurulation at stage 12. Anat Embryol (Berl). 1987;176:413-430.
29 Duhamel B. From the mermaid to anal imperforation: the syndrome of caudal regression. Arch Dis Child. 1961;186:152-155.
30 Wallin J, Wilting J, Koseki H, et al. The role of Pax-1 in axial skeleton development. Development. 1994;120:1109-1121.
31 Firooznia H, Pinto RS, Lin JP, et al. Chordoma: radiologic evaluation of 20 cases. AJR Am J Roentgenol. 1976;127:797-805.
32 Horwitz T. Chordal ectopia and its possible relation to chordoma. Arch Pathol. 1941;31:354.
33 Aruga J, Mizugishi K, Koseki H, et al. Zic1 regulates the patterning of vertebral arches in cooperation with Gli3. Mech Dev. 1999;89:141-150.
34 McMaster MJ, Ohtsuka K. The natural history of congenital scoliosis. A study of two hundred and fifty-one patients. J Bone Joint Surg Am. 1982;64:1128-1147.
35 Hedequist D, Emans J. Congenital scoliosis. J Am Acad Orthop Surg. 2004;12:266-275.
36 McMaster MJ. Congenital scoliosis caused by a unilateral failure of vertebral segmentation with contralateral hemivertebrae. Spine. 1998;23:998-1005.
37 McMaster MJ. Spinal growth and congenital deformity of the spine. Spine. 2006;31:2284-2287.
38 MacEwen GD, Bunnell WP, Sriram K. Acute neurological complications in the treatment of scoliosis. A report of the Scoliosis Research Society. J Bone Joint Surg Am. 1975;57:404-448.
39 Winter RB, Moe JH, Wang JF. Congenital kyphosis. Its natural history and treatment as observed in a study of one hundred and thirty patients. J Bone Joint Surg Am. 1973;55:223-256.
40 Winter RB. Congenital spine deformity: “What’s the latest and what’s the best”? Spine. 1989;14:1406-1409.
41 McMaster MJ, Singh H. Natural history of congenital kyphosis and kyphoscoliosis. A study of one hundred and twelve patients. J Bone Joint Surg Am. 1999;81:1367-1383.
42 McMaster MJ, Singh H. The surgical management of congenital kyphosis and kyphoscoliosis. Spine. 2001;26:2146-2154.
43 Lonstein JE. Congenital spine deformities: scoliosis, kyphosis, and lordosis. Orthop Clin North Am. 1999;30:387-405. viii
44 Smith JT, Gollogly S, Dunn HK. Simultaneous anterior-posterior approach through a costotransversectomy for the treatment of congenital kyphosis and acquired kyphoscoliotic deformities. J Bone Joint Surg Am. 2005;87:2281-2289.
45 Winter RB, Lovell WW, Moe JH. Excessive thoracic lordosis and loss of pulmonary function in patients with idiopathic scoliosis. J Bone Joint Surg Am. 1975;57:972-977.
46 Verbiest H. Results of surgical treatment of idiopathic developmental stenosis of the lumbar vertebral canal. A review of twenty-seven years’ experience. J Bone Joint Surg Br. 1977;59:181-188.
47 Grabias S. Current concepts review. The treatment of spinal stenosis. J Bone Joint Surg Am. 1980;62:308-313.
48 Singh K, Samartzis D, Vaccaro AR, et al. Congenital lumbar spinal stenosis: a prospective, control-matched, cohort radiographic analysis. Spine J. 2005;5:615-622.
49 Verbiest H. A radicular syndrome from developmental narrowing of the lumbar vertebral canal. J Bone Joint Surg Br. 1954;36:230-237.
50 Fredrickson BE, Baker D, McHolick WJ, et al. The natural history of spondylolysis and spondylolisthesis. J Bone Joint Surg Am. 1984;66:699-707.
51 Wiltse LL, Jackson DW. Treatment of spondylolisthesis and spondylolysis in children. Clin Orthop Relat Res. 117. 1976:92-100.
52 Blackburne JS, Velikas EP. Spondylolisthesis in children and adolescents. J Bone Joint Surg Br. 1977;59:490-494.
53 Swan J, Hurwitz E, Malek F, et al. Surgical treatment for unstable low-grade isthmic spondylolisthesis in adults: a prospective controlled study of posterior instrumented fusion compared with combined anterior-posterior fusion. Spine J. 2006;6:606-614.
54 Molinari RW, Bridwell KH, Lenke LG, et al. Complications in the surgical treatment of pediatric high-grade, isthmic dysplastic spondylolisthesis. A comparison of three surgical approaches. Spine. 1999;24:1701-1711.
55 Banta JV, Nichols O. Sacral agenesis. J Bone Joint Surg Am. 1969;51:693-703.
56 Passarge E, Lenz W. Syndrome of caudal regression in infants of diabetic mothers: observations of further cases. Pediatrics. 1966;37:672-675.
57 Lynch SA, Bond PM, Copp AJ, et al. A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36. Nat Genet. 1995;11:93-95.
58 Riker W, Potts WJ. Sacrococcygeal teratoma in infancy: a report of six cases. Ann Surg. 1948;128:89-100.
59 Ein SH, Adeyemi SD, Mancer K. Benign sacrococcygeal teratomas in infants and children: a 25 year review. Ann Surg. 1980;191:382-384.
60 Miles RM, Stewart GSJr. Sacrococcygeal teratomas in adult. Ann Surg. 1974;179:676-683.
61 Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics surgical section survey—1973. J Pediatr Surg. 1974;9:389-398.
62 Huddart SN, Mann JR, Robinson K, et alChildren’s Cancer Study Group. Sacrococcygeal teratomas: the UK Children’s cancer study group’s experience. I. Neonatal. Pediatr Surg Int. 2003;19:47-51.
63 Mitchell LE, Adzick NS, Melchionne J, et al. Spina bifida. Lancet. 2004;364:1885-1895.
64 Fidas A, MacDonald HL, Elton RA, et al. Prevalence and patterns of spina bifida occulta in 2707 normal adults. Clin Radiol. 1987;38:537-542.
65 Warder DE. Tethered cord syndrome and occult spinal dysraphism. Neurosurg Focus. 2001;10:E1.
66 Caram PC, Carton CA, Scarcella G. Intradural lipomas of the spinal cord: with particular emphasis on the intramedullary lipomas. J Neurosurg. 1957;14:28-42.
67 Swanson HS, Barnett JCJr. Intradural lipomas in children. Pediatrics. 1962;29:911-926.
68 Pierre-Kahn A, Zerah M, Renier D, et al. Congenital lumbosacral lipomas. Childs Nerv Syst. 1997;13:298-334.
69 Schropp C, Sörensen N, Collmann H, Krauss J. Cutaneous lesions in occult spinal dysraphism—correlation with intraspinal findings. Childs Nerv Syst. 2006;22:125-131.
70 Li YC, Shin SH, Cho BK, et al. Pathogenesis of lumbosacral lipoma: a test of the “premature dysjunction” theory. Pediatr Neurosurg. 2001;34:124-130.
71 McLone DG, Mutluer S, Naidich D. Lipomeningoceles of the conus medullaris. Concepts Pediatr Neurosurg. 1983;3:170-177.
72 Pang D. Split cord malformation: part ii: clinical syndrome. Neurosurgery. 1992;31:481-500.
73 Naidich TP, Harwood-Nash DC. Diastematomyelia: hemicord and meningeal sheaths: single and double arachnoid and dural tubes. AJNR Am J Neuroradiol. 1983;4:633-636.
74 Mahapatra AK, Gupta DK. Split cord malformations: a clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg. 2005;103(6 suppl):531-536.
75 Hori A, Fischer G, Dietrich-Schott B, Ikeda K. Dimyelia, diplomyelia, and diastematomyelia. Clin Neuropathol. 1982;1:23-30.
76 Keim HA, Greene AF. Diastematomyelia and scoliosis. J Bone Joint Surg Am. 1973;55:1425-1435.
77 Reed JC, Sobonya RE. Morphologic analysis of foregut cysts in the thorax. Am J Roentgenol Radium Ther Nucl Med. 1974;120:851-860.
78 Chang IC. Thoracic neurenteric cyst in a middle-aged adult presenting with Brown-Sequard syndrome. Spine. 2003;28:E515-E518.
79 Lieb G, Krauss J, Collmann H, et al. Recurrent bacterial meningitis. Eur J Pediatr. 1996;155:26-30.
80 McLone DG, Naidich TP. Terminal myelocystocele. Neurosurgery. 1985;16:36-43.
81 Powell KR, Cherry JD, Hougen TJ, et al. A prospective search for congenital dermal abnormalities of the craniospinal axis. J Pediatr. 1975;87:744-750.
82 Ackerman LL, Menezes AH. Spinal congenital dermal sinuses: a 30-year experience. Pediatrics. 2003;112(pt 1):641-647.
83 Guille JT, Sarwark JF, Sherk HH, Kumar SJ. Congenital and developmental deformities of the spine in children with myelomeningocele. J Am Acad Orthop Surg. 2006;14:294-302.
84 Byrd SE, Darling CF, McLone DG, Tomita T. MR imaging of the pediatric spine. Magn Reson Imaging Clin N Am. 1996;4:797-833.
85 Guidetti B, Gagliardi FM. Epidermoid and dermoid cysts. Clinical evaluation and late surgical results. J Neurosurg. 1977;47:12-18.
86 Iskandar BJ, Fulmer BB, Hadley MN, Oakes WJ. Congenital tethered spinal cord syndrome in adults. Neurosurg Focus. 2001;10:E7.
87 Warder DE, Oakes WJ. Tethered cord syndrome: the low-lying and normally positioned conus. Neurosurgery. 1994;34:597-600.
88 Herman JM, McLone DG, Storrs BB, Dauser RC. Analysis of 153 patients with myelomeningocele or spinal lipoma reoperated upon for a tethered cord. Presentation, management and outcome. Pediatr Neurosurg. 1993;19:243-249.
89 Souweidane MM, Drake JM. Retethering of sectioned fibrolipomatous filum terminales: report of two cases. Neurosurgery. 1998;42:1390-1393.
90 Kaffenberger DA, Heinz ER, Oakes JW, Boyko O. Meningocele manque: radiologic findings with clinical correlation. AJNR Am J Neuroradiol. 1992;13:1083-1088.