Complement
Learning Objectives
• Recognize the biologic functions of the complement cascade
• Identify the components of the classic complement pathway
• Discuss the roles of anaphylatoxin in an immune response
• Recognize the biologic function of C2b
• Name the complement components in classic pathway C3 convertase
• Relate complement activation to microbial insolubility, osponization, and phagocytosis
• Recognize the three different anaphylatoxins generated in complement pathways
• Identify the components and function of the membrane attack complex (MAC)
• Differentiate between the classic complement pathway and the alternative complement pathway
• Describe the unique structure and function of C9
• Identify the molecules involved in the control of the classic recognition complex
• Analyze the role of C3 in the activation of the alternative complement pathway
• Name the components of the C3 convertase in the alternative complement pathway
• Recognize the proteins that control the activation of the alternative C3 convertase
• List the seven molecules expressed on mammalian cell membranes that inhibit complement activation
• Identify the role of complement in B cell activation
• Identify the role of complement in the generation of memory cells
• Describe the immunologic defect in hereditary angioneurotic edema (HANE)
• Design a successful treatment regimen for HANE
• Describe the immunologic defects in paroxysmal nocturnal hemoglobulinuria (PNH)
Key Terms
Activation complex
Anaphylatoxin
Antigen recognition complex
Alternative complement pathway
C1 esterase inhibitor (C1INH)
C3 convertase
C4 binding protein (C4BP)
C5 convertase
CD59
Classic complement pathway
Complement receptor I
Decay accelerating factor (DAF)
Factor H
Factor I
Hereditary angioneurotic edema
Homologous restriction factor (HRF)
Membrane activation complex
Membrane cofactor protein (MCP)
Metastable C3
Paroxysmal nocturnal hemoglobulinuria
Properdin
S protein (vitronectin)
Introduction
Complement is a family of more than 20 plasma proteins that includes enzymes, proenzymes, enzyme inhibitors, and glycoproteins. Components interact in cascades and assist in the resolution of a microbial infection. For example, fragments indirectly render the microbe insoluble and draw phagocytic cells to the area of infection. Other fragments coat the microbe and act as opsonins, which promote phagocytosis. In a third function, complement components create lesions in the microbial cell wall that result in osmotic lysis. Complement fragments also provide the second signal necessary for B cell activation.
Three different complement cascades have been described—classical, alternative, and the mannose-binding lectin (MBL) pathways (see Chapter 2). Only the classic pathway requires an antibody for complement activation. In the MBL and alternative pathways, complement components are directly activated by highly conserved microbial antigens.
Classic Complement Pathway
The classic pathway was the first described using sheep red blood cells (SRBCs) coated with anti-SRBC immunoglobulin M (IgM). Over a number of years, nine complement proteins were found to be involved in the cascade. These proteins interact in the following sequence: Ab:C142356789. On the basis of function, the classic cascade is subdivided into recognition, activation, and membrane attack complexes.
Recognition Complex
The recognition complex consists of antibody (Ab) and C1. The C1 component is a complex of three proteins (C1q, r, and s). C1q is composed of polypeptide chains with a triple helix “collagen-like” amino terminus and a globular structure at the carboxyl terminus (Figure 11-1). C1r and C1s are inactive serine esterases.
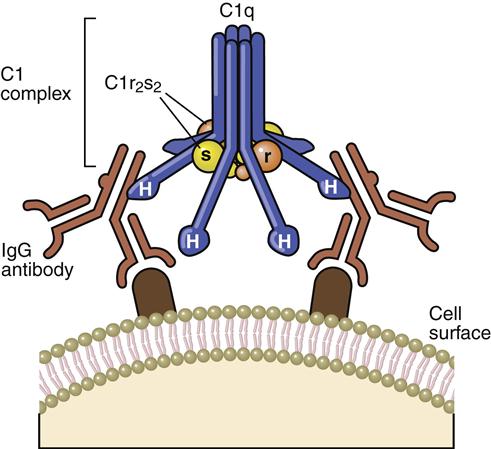
When the antibody binds to the antigen, a conformational change occurs in the hinge region, which exposes complement receptors. Although both IgG and IgM can activate the complement cascade, IgM is more efficient because of the proximity of multiple hinge regions. In the initial step of complement activation, C1q bridges two hinge regions. In the next step, C1r and C1s associate to form a tetrameric complex (two molecules of each protein), which binds to C1q. In the presence of calcium, C1r activates the serine esterase activity of C1s.
Activation Complex
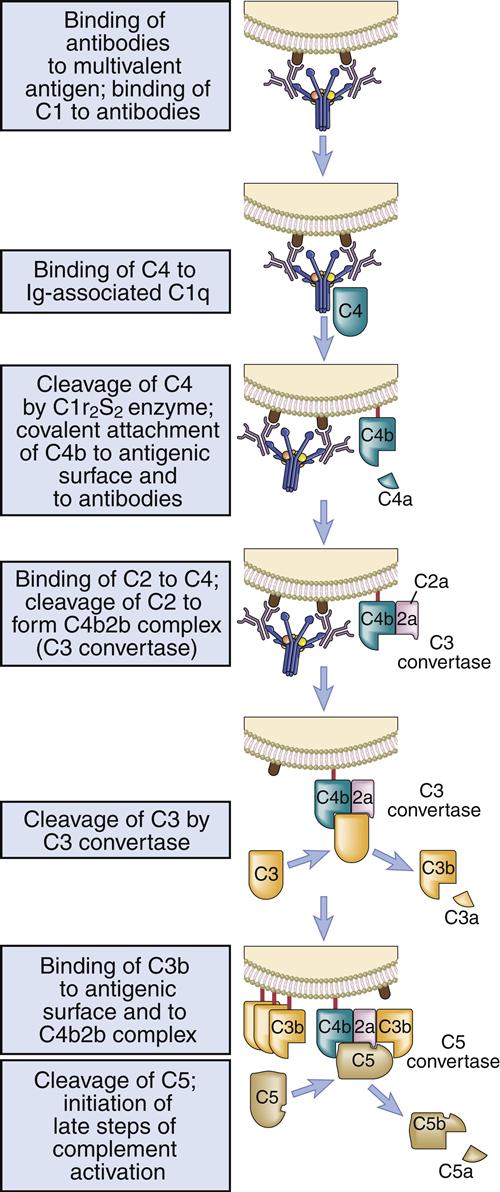
Activated C1s cleaves multiple C4 and C2 molecules. C4 is a large 210-kDal trimeric polypeptide, which contains an unusual thioester group. C4 is cleaved into C42a and C4b. C4a is a soluble protein that attracts phagocytic cells into the area where complement is being fixed. The C4b fragment is coupled to the microbial cell. Cleavage of C2 results in the production of C2a and C2b. The larger C2a (75-kDal) fragment remains in contact with C4b to create an AbC4b2a complex on the cell surface. Plasmin cleavage of the smaller, soluble C2b fragment creates a kinin, which causes leakage of fluid from vessels and tissue edema.
C3 Convertase
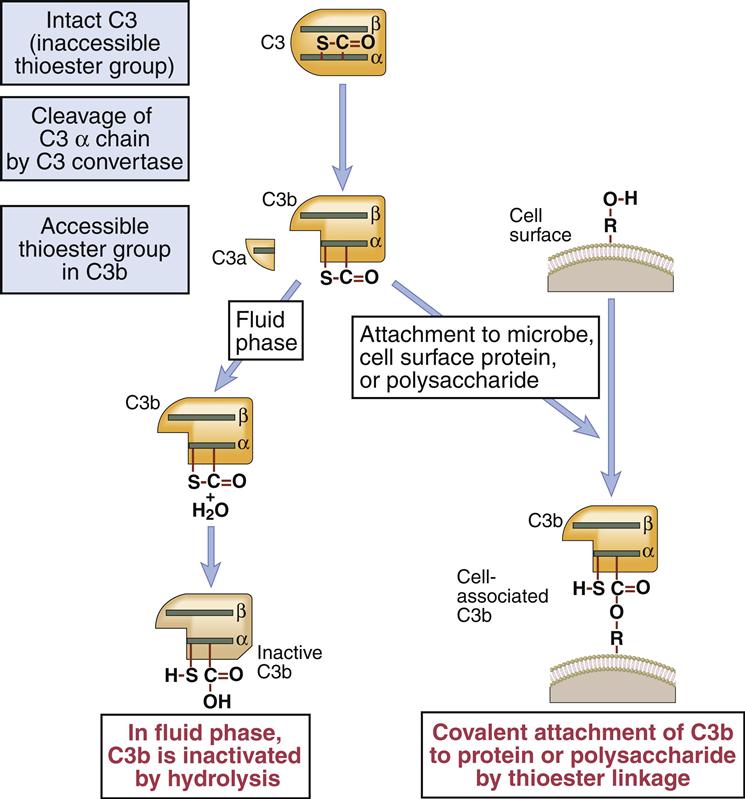
The C4b2a complex, or C3 convertase, catalyzes the cleavage of C3 into C3a and C3b. This is the most important step in the complement cascade and occurs in the classic, alternative, and MBL pathways. C3b is a highly unstable molecule that has a unique thioester that allows covalent binding to a microbial cell. Some C3b fragments bind to the target cell, where they act as opsonins. In the classic pathway, most C3b is found in an AbC4bC2aC3b complex on the cell surface, which is called the C5 convertase.
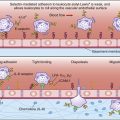
The soluble C3a fragment acts as an anaphylatoxin. By definition, an anaphylatoxin is a molecule that induces the movement of eosinophils and phagocytic cells (chemotaxis) toward increasing concentrations of C3a (Figure 11-2). Anaphylatoxins also release histamine and heparin from basophils and mast cells. Histamine and heparin cause vasoconstriction and increased vascular permeability, which reduce the solubility of microbes or antigen–antibody complexes.
Membrane Attack Complex
The C5 convertase cleaves C5 into C5a and C5b fragments. The C5b binds to the cell surface and serves as a platform for the membrane attack complex (MAC), which consists of C5bC6789 (Figure 11-3). C5a is the most potent anaphylatoxin (100–1000 times more potent than C3a) in the complement cascade.
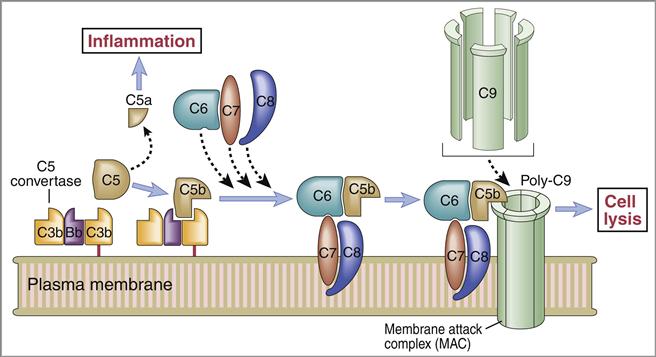
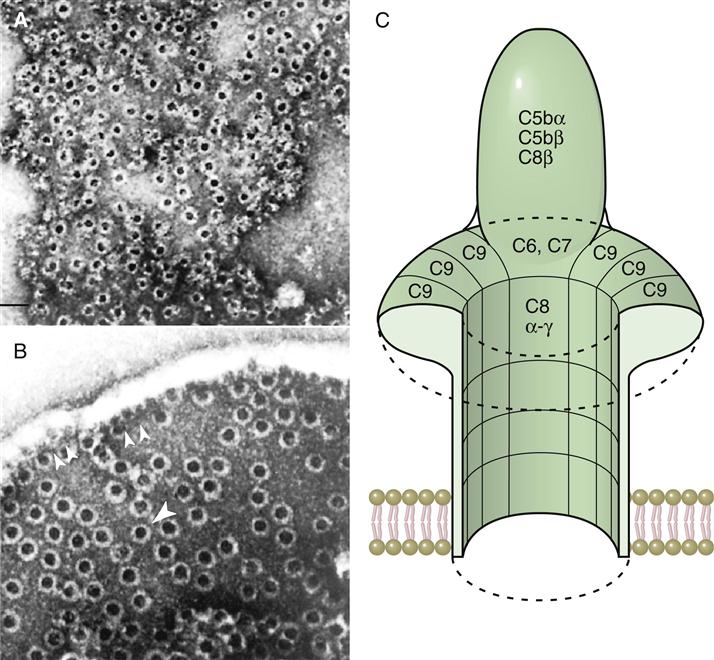
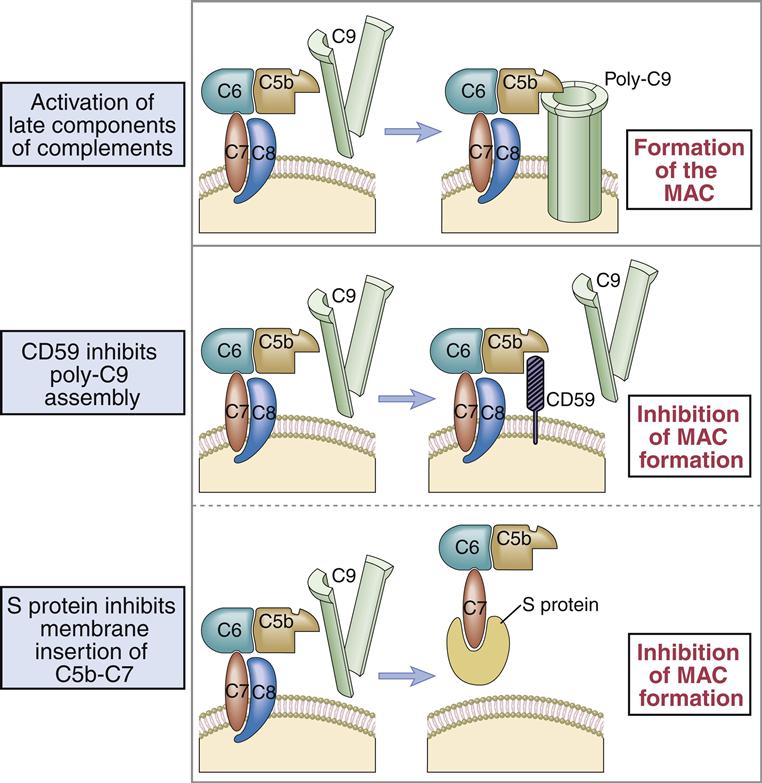
Binding of the C6C7 component creates a trimolecular, lipophilic complex, which is inserted into cell membranes. C8 also is inserted into the microbial cell wall or membrane, further increasing the stability of the C5b678 complex (Figure 11-3). Polymerized C9 is inserted into the cell membrane and creates pores in the cell membrane that cause osmotic lysis (Figure 11-4).
Control of the Classic Complement Cascade
Inadvertent or aberrant complement activation is controlled by soluble proteins that inhibit the antigen recognition, activation, and membrane attack complexes.
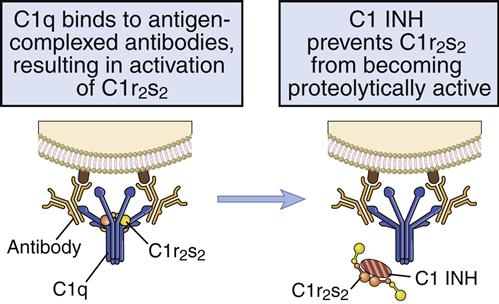
Control of the Recognition Complex
C1 esterase inhibitor (C1INH) is a soluble protein that inhibits C1 activation by disassociating C1r and C1s and preventing the activation of C4 (Figure 11-5).
Control of the Activation Complex
The C4 binding protein (C4BP) is a plasma protein that inhibits the classic and MBL complement pathways by blocking the interaction between C4b and C2a (Table 11-1).
Table 11-1
Molecules and Receptors That Inhibit Complement Activation
| Receptor | Interacts with | Function |
| C1 esterase inhibitor | C1r, C1s | Binds to C1r and C1s and disassociates them from C1q |
| C4 binding protein | C4b | Binds C4b and displaces C2 |
| Membrane cofactor protein (MCP, CD46) | C3b, C4b | Cofactor for factor I mediated cleavage of C4b |
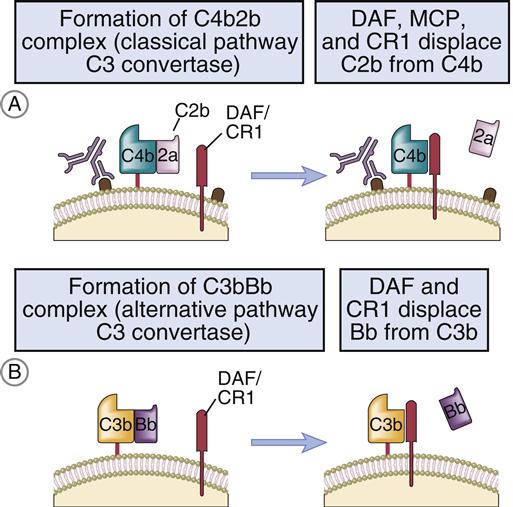
| Decay accelerating factor (DAF) | C4b, C2a | Displaces C2a from C4b |
| Factor I | C4b, C3b | Cleaves C3b and C4b |
| Factor H | C3b | Binds C3b |
| Complement receptor type 1 (CR1, CD35) | C3b, C4b, iC3B | Promotes dissociation of C3 convertase |
Control of the Membrane Attack Complex
S Protein (vitronectin) is a plasma protein that associates C5bC6C7 with C8.
Alternative Pathway of Complement Activation
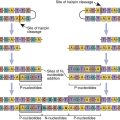
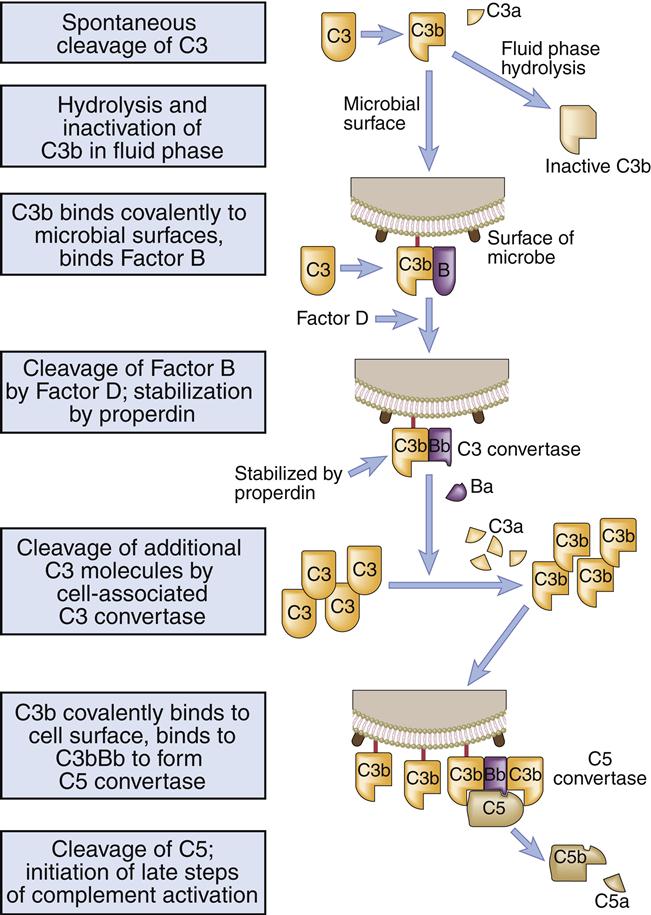
Because it is inherently unstable, serum C3 spontaneously fragments into C3a and C3b. Metastable C3b covalently binds to highly conserved microbial polysaccharides (e.g., inulin, zymosan) and lipopolysaccharides (Figure 11-6).
Activation Complex
Binding of C3b causes a conformational change that exposes a binding site for factor B. On the cell surface, factor B complexes with C3b. Factor D, a serine protease, cleaves factor B into a soluble Ba and a Bb fragment. The C3bBb fragment becomes a C3 convertase, which is stabilized by another molecule called properdin.
Some C3b is liberated by the novel convertase and binds to the complex itself, creating a C3bBbC3b. The new complex acts as a C5 convertase that hydrolyzes the alpha chain, liberating C5a and C5b (Figure 11-7). C5b catalyzes the formation of the MAC.
Control Mechanisms for the Alternative Pathway
Soluble factor H downregulates the alternative pathway by inactivating soluble or bound C3b and amplifying the decay of the C3bBb complex. Factor I is a serine protease that cleaves C3b and C4b and prevents further activation of complement.
Mammalian Cell Inactivation of Complement
Activated C3b from either the classic pathway or the alternative pathway can bind to mammalian cell membranes and activate the complement MAC. To prevent lysis, mammalian cells express molecules that inactivate individual complement components or accelerate the destruction of C3 convertases formed by the classic and alternative pathways. The following are known membrane-bound complement inhibitors:
• Factor H binds to sialic residues on cell membrane and inactivates C3b inadvertently bound to cells.
• Membrane cofactor protein (MCP; CD46), in combination with other proteins, inactivates C4b.
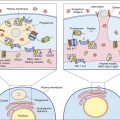
• Decay accelerating factor (DAF; CD55) is a glycoprotein that is anchored in the membrane by a covalent linkage to glycosylphosphatidylinositol (GPI). It functions to disassociate the C3 convertases by releasing C2a from the AbC4bC2a complex in the classic pathway and C3b and Bb in the alternative pathway (Figure 11-8).
• CD59 is a GPI-linked protein that binds to C8 and C9 and inhibits complement-mediated lysis of red blood cells, platelets, and leukocytes (Figure 11-9).
Complement, B Cell Activation, and Immunologic Memory
Assuming a multi-valent antigenic molecule, two signals are necessary to activate B cells. The second signal is provided by a B cell co-receptor complex that consists of CR2, CD19, and CD81 (TAPA-1). The complement C3d decay fragment binds to the B cell CD2 molecule. At the same time, other epitopes bind to the B cell receptor (BCR). Complement binding activates kinases, which phosphorylate the CD19 cytoplasmic tail and activation of PI-3 signaling pathway.
Immunologic memory is also potentiated by the complement decay products iC3b and C3dg. Antigens coated with these fragments bind to iccosomes on follicular dendritic cells. Iccosome-bound antigens continually stimulate B cells which differentiate into plasma and memory cells.
Complement Deficiencies
Hereditary Angioneurotic Edema
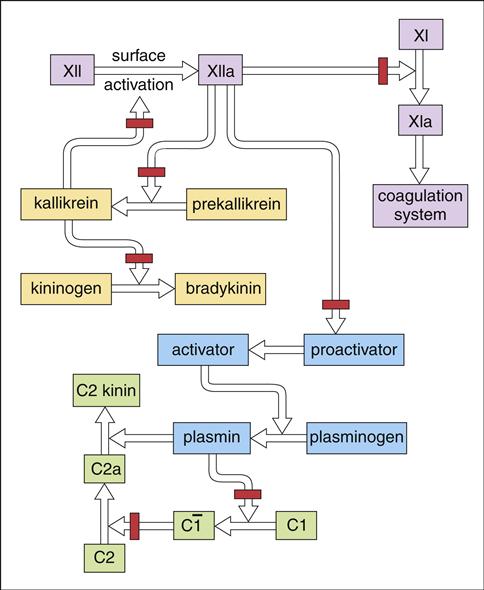
In addition to its role in complement activation, C1 INH inhibits the activation of the clotting, kinin, and plasmin pathways (Figure 11-10). In the absence of C1 INH, precursors are converted to plasmin, bradykinin, and coagulation pathways. Activated C2 kinins allow fluid to escape into tissue, causing edema.
Hereditary angioneurotic edema (HANE) is caused by C1 INH deficiency. During a clinical episode, well-circumscribed edema is localized to the face, tongue, and larynx (Figure 11-11). Edema in the larynx is often severe and restricts normal breathing.
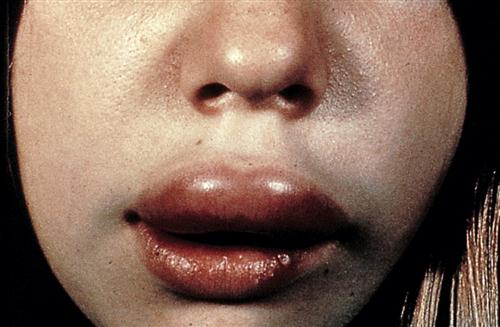
Over 66% of patients with HANE require emergency medical intervention during a clinical episode of laryngeal edema. Without adequate medical treatment, 33% of these patients die during a clinical episode.
Inherited and acquired forms of HANE exist. The inherited form is an autosomal dominant trait and appears during the first decade of life. It is characterized by normal levels of nonfunctional C1 INH. Acquired HANE is often associated with lymphoproliferative disorders or the presence of specific autoantibodies directed toward the C1 INH molecule. Most patients with acquired HANE have increased catabolism of C1 INH, which is reflected in the low serum levels of C1 INH.
Treatment of Hereditary Angioneurotic Edema
Oral androgens, anabolic steroids, and antifibrinolytic agents are commonly used to treat HANE. In cases of acquired C1 INH deficiency, glucocorticosteroids are effective as an emergency treatment and are usually tapered off when the patient begins taking androgens. Long-term treatment usually involves androgens and aminocaproic acid. Although the mechanism of action is unclear, it is presumed that synthetic androgens (e.g., danazol) increase the production of C1 INH and C4 by the liver. Aminocaproic acids function as an antifibrolytic agent that prevents plasmin activation.
Pooled human C1 INH concentrate and recombinant C1 INH are also currently undergoing investigational studies in the United States and Europe and may be highly effective in emergency treatment.
Paroxysmal Nocturnal Hemoglobulinuria
Paroxysmal nocturnal hemoglobulinuria (PNH) is associated with short, rapid episodes (paroxysmal) of red blood cell lysis occurring only at night (nocturnal). However, the terms “nocturnal” and “paroxysmal” are incorrect when describing the disease. Lysis of red blood cells is not exclusively nocturnal and occurs continuously throughout the day.
PNH is the result of a somatic mutation in the pig-A gene that synthesizes the GPI protein. Mutant GPI cannot covalently bind the complement regulatory proteins DAF (CD55), HRF, and CD59. Individuals with PNH are missing both DAF and CD59 and have increased sensitivity to complement lysis. This results in intravascular red cell hemolysis and hematoglobinuria (loss of iron) that is characteristic of the disease.
The absence of CD59 on platelets also contributes to recurrent thrombosis that is often fatal in individuals with PNH. MAC localization on platelets creates membrane pores that allow exovesiculation of platelet vesicles. These vesicles, which localize on the platelet surface, contain a membrane receptor for coagulation factor VI. Activation of the clotting sequence creates thrombi, which localize in the mesenteric, hepatic, and cerebral veins.
Treatment of Paroxysmal Nocturnal Hemoglobulinuria
In the treatment of PNH, clinicians must deal with five separate problems: (1) the lack of red blood cells, (2) deficient erythropoiesis, (3) increased thrombosis, (4) loss of iron (hematoglobinuria), and (5) persistent complement activation. Transfusion of packed red blood cells can replace lost cells. Androgens and recombinant erythropoietin are commonly used to accelerate erythropoiesis. Thrombotic complications are reduced by standard therapy, which includes heparin and maintenance doses of common anticoagulants. Iron loss is corrected by the administration of supplemental iron. To reduce complement activation, eculizumab can be used to block the activation of C5 and the generation of the MAC.
Defects in Complement Components
Genetic defects are present in the synthesis of complement components. A defect in C2 synthesis is the most commonly identified deficiency and increases the risk for neisserial infections. Conversely, C2 and C4 defects are not associated with an increased risk of fatal bacterial infections. This may be due to the alternative complement pathway and Fc receptors on phagocytic cells which compensate for the loss of C2.
Deficiencies in complement components that form or stabilize the classic or alternative C3 convertase increase the risk of pyogenic (pus-forming) infections with streptococcal and staphylococcal species. Individuals with a C3 deficiency usually have recurrent infections of the respiratory tract, gut, and skin.
Life-threatening meningitis is associated with defects in properdin, factor H, factor I, and the complement MAC. Properdin deficiencies are usually X linked, and meningococcal infections are usually fatal.
Treatment of Complement Deficiencies
Individuals with C2, properdin, factor H, factor I, or complement MAC deficiency have a significant risk of developing meningococcal infections. Vaccination is indicated to create high levels of protective antibodies. In the choice of vaccines, the age of the individual and the meningococcal serotype should be considered.