[level-membership-for-neurology-category]
Chapter 44 Cognitive and Motor Regression
Epidemiology
Although the causes of PE are individually rare, the combined incidence of PE has been estimated to be as high as 1 in 2000 live births [Surtees, 2002]. Much of what has been published regarding these disorders has been retrospective and focused on individual conditions, providing little basis for a discussion of their collective epidemiology. A few studies have been notable exceptions.
An early paper examining the experience with PE at two large academic centers in the United States found that, of 1218 admissions to their child neurology services over the course of 10 years, 341 patients were diagnosed with 1 of more than 50 disorders causing neurological dysfunction [Dyken and Krawiecki, 1983]. Table 44-1 shows the results of their analysis of the relative frequency of the various diagnoses. Although 72 percent of the cases studied had a genetic or metabolic disorder causing PE, the study also included a significant number of children with pure lower motor neuron syndromes and acquired injuries due to infection, immunologic disorders, refractory epilepsy, chronic environmental insults, nutritional deficiencies, and iatrogenic factors. A study from the Children’s Hospital of Lahore, Pakistan [Sultan et al., 2006], found that, of the 1273 children admitted to the neurology service from 2004 to 2005, 66 were diagnosed with PE and most received a specific diagnosis. The most common diagnoses, in descending order of frequency, were metachromatic leukodystrophy (14 cases), adrenoleukodystrophy (11), subacute sclerosing panencephalitis (8), Wilson’s disease (6), Friedreich’s ataxia (5), liposis (4), Gaucher’s disease (3), Alexander’s disease (2), and pantothenate kinase-associated neurodegeneration (PKAN) (2). More than half of the patients underwent funduscopic examination, electroencephalography, and cerebrospinal fluid examination as part of their diagnostic work-up.
Table 44-1 Diagnosis in 340 Cases of Developmental Regression
| Diagnosis | Number of Cases |
|---|---|
| POLIODYSTROPHIES* | 129 |
| Lysosomal storage disorders | 39 |
| Hypoxic poliodystrophy | 29 |
| Idiopathic poliodystrophy | 24 |
| West’s syndrome | 17 |
| Lennox–Gastaut syndrome | 9 |
| Metabolic poliodystrophy | 4 |
| Toxoplasmosis | 3 |
| Post-vaccine poliodystrophy | 3 |
| Lowe’s syndrome | 1 |
| LEUKODYSTROPHIES† | 71 |
| SSPE | 26 |
| ADEM and MS | 17 |
| Adrenoleukodystrophy | 8 |
| Metachromatic leukodystrophy | 5 |
| Pelizaeus–Merzbacher disease | 4 |
| Krabbe’s disease | 4 |
| Phenylketonuria | 2 |
| Cockayne’s syndrome | 2 |
| Canavan’s disease | 1 |
| Alexander’s disease | 1 |
| Maple syrup urine disease | 1 |
| CORENCEPHALOPATHIES‡ | 26 |
| Idiopathic corencephalopathy | 8 |
| Huntington’s disease | 5 |
| Mitochondrial disorders | 4 |
| Dystonia musculorum deformans | 2 |
| Hallervorden–Spatz syndrome | 2 |
| Ataxia-telangiectasia | 1 |
| Congenital indifference to pain | 1 |
| Infantile neuroaxonal dystrophy | 1 |
| Riley–Day syndrome | 1 |
| Wilson’s disease | 1 |
| DIFFUSE ENCEPHALOPATHIES | 63 |
| Tuberous sclerosis | 19 |
| Idiopathic encephalopathy | 17 |
| Hyperammonemic disorders | 6 |
| Mitochondrial disorders | 4 |
| Neurofibromatosis | 4 |
| Achondroplasia | 2 |
| Organic acidurias | 2 |
| Letterer–Siwe disease | 2 |
| Sturge–Weber syndrome | 2 |
| Zellweger’s syndrome | 2 |
| Homocystinuria | 1 |
| Incontinentia pigmenti | 1 |
| Sjögren–Larsson syndrome | 1 |
| SPINOCEREBELLOPATHIES§ | 51 |
| Spinal muscular atrophy | 19 |
| Hereditary spastic paraplegia | 12 |
| Acute cerebellar ataxia | 8 |
| Infantile polymyoclonus | 4 |
| Charcot–Marie–Tooth disease | 2 |
| Friedreich’s ataxia | 2 |
| Marinesco–Sjögren syndrome | 1 |
| OPCA | 1 |
| Spinocerebellar degeneration | 1 |
| Refsum’s disease | 1 |
ADEM, acute disseminated encephalomyelitis; MS, multiple sclerosis; OPCA, olivopontocerebellar atrophy; SSPE, subacute sclerosing panencephalitis.
* Poliodystrophies = predominant cortical involvement.
† Leukodystrophies = predominant cerebral white-matter involvement.
‡ CORENCEPHALOPATHIES = predominant basal ganglia involvement.
§ Spinocerebellopathies = predominant spinal cord and cerebellar involvement.
(From Dyken P, Krawiecki N. Neurodegenerative diseases of infancy and childhood. Ann Neurol 1983;13:351–364.)
Following the initial description in 1996 of 10 cases of new variant Creutzfeldt–Jakob disease (nvCJD) affecting young adults in the United Kingdom [Will et al., 1996], several countries instituted prospective surveillance programs to collect data on patients with PE to better identify additional cases of nvCJD. Although these studies have relied on reports from pediatricians and have been unable to describe absolute incidence or prevalence figures, they have reported relative prevalences within their areas. The first report from the surveillance done in the UK [Devereux et al., 2004] collected and analyzed pediatric cases of progressive intellectual and neurological deterioration (PIND) over a 5-year span. The cases included children who had:
The study excluded children with intellectual and neurological deterioration after a nonprogressive insult, such as encephalitis, trauma, or global hypoxic-ischemic injury, but did include children with seizure disorders who otherwise met the case definition and children carrying diagnoses that could be expected to lead to progressive deterioration in the future. Of the 798 cases collected, 577 had a confirmed diagnosis, 6 had definite or probable nvCJD, and 211 had no clear etiologic diagnosis at the time of publication but did not have clinical features suggestive of nvCJD. There were nearly 100 different confirmed diagnoses, but more than one-quarter of the cases were explained by the five most common: mucopolysaccharidosis type III (Sanfilippo’s syndrome), adrenoleukodystrophy, late infantile neuronal ceroid-lipofuscinosis, mitochondrial diseases, and Rett’s syndrome. Higher rates of prevalence and of consanguinity were reported in families of South Asian origin. A follow-up of the UK study [Verity et al., 2010a] reported a confirmed etiologic diagnosis in 1047 of the 2493 cases of PIND that had been collected by 2008, with nearly one-quarter of cases again explained by the five most common diagnoses: neuronal ceroid-lipofuscinoses, mitochondrial diseases, mucopolysaccharidoses, GANGLIOSIDOSES, and Peroxisomal disorders. The most recent update of the study [Verity et al., 2010b] reported that, after 12 years, 147 different etiologies were found to explain 1114 of the 2636 cases of PIND collected. In total, only 6 children with confirmed or probable nvCJD had been identified. The 30 most common diagnoses identified in the study are presented in Table 44-2.
Table 44-2 Common Diagnoses in 1114 Cases of Progressive Encephalopathy
| Diagnosis | Number of Cases |
|---|---|
| LEUKOENCEPHALOPATHIES | 183 |
| Metachromatic leukodystrophy | 59 |
| Krabbe’s disease | 33 |
| Pelizaeus–Merzbacher disease | 17 |
| Canavan’s disease | 13 |
| Vanishing white matter disease | 11 |
| Aicardi–Goutières syndrome | 10 |
| Alexander’s disease | 10 |
| Other | 31 |
| NEURONAL CEROID-LIPOFUSCINOSES | 141 |
| NCL late infantile | 73 |
| NCL juvenile | 44 |
| NCL infantile | 22 |
| Other | 2 |
| MITOCHONDRIAL | 122 |
| Leigh’s syndrome | 17 |
| NARP (including NARP/MILS) | 17 |
| Other | 88 |
| MUCOPOLYSACCHARIDOSES | 102 |
| Mucopolysaccharidosis IIIA (Sanfilippo’s syndrome) | 69 |
| Mucopolysaccharidosis IIA (Hunter’s disease) | 15 |
| Other | 18 |
| GANGLIOSIDOSES | 100 |
| GM2 gangliosidosis type 1 (Tay–Sachs disease) | 41 |
| GM2 gangliosidosis type 2 (Sandhoff’s disease) | 33 |
| GM1 gangliosidosis | 23 |
| Other | 3 |
| PEROXISOMAL | 69 |
| Adrenoleukodystrophy | 56 |
| Other | 13 |
| OTHER METABOLIC | 95 |
| Niemann–Pick disease type C | 38 |
| PKAN/NBIA | 21 |
| Menkes’ disease | 16 |
| Glutaric aciduria type 1 | 10 |
| Molybdenum co-factor deficiency | 10 |
| NONMETABOLIC | 135 |
| Rett’s syndrome | 60 |
| Huntington’s disease | 22 |
| Cockayne’s disease | 15 |
| Neuroaxonal dystrophy | 12 |
| Ataxia telangiectasia | 9 |
| Subacute sclerosing panencephalitis | 9 |
| Rasmussen’s syndrome | 8 |
MILS, maternally inherited Leigh’s syndrome; NARP, neuropathy, ataxia, and retinitis pigmentosa; NBIA, neurodegeneration with brain iron accumulation; NCL, neuronal ceroid-lipofuscinosis; PE, progressive encephalopathy; PKAN, pantothenate kinase-associated neurodegeneration (previously Hallervorden–Spatz disease).
(From Verity et al., The epidemiology of progressive intellectual and neurological deterioration in childhood. Arch Dis Child 2010b.)
A survey-based study conducted in Australia [Nunn et al., 2002] identified 230 cases of childhood PE in a 2-year period, with 134 patients having Rett’s syndrome, 20 having a lysosomal storage disorder, 16 having a leukodystrophy, and 15 having a mitochondrial disease. A study done in Oslo, Norway, gathered cases of pediatric PE over an 18-year period from the area’s one children’s hospital and from the national diagnostic laboratory for metabolic diseases [Strømme et al., 2007]. The authors excluded patients with diseases in which cognitive impairment was either atypical (e.g., spinocerebellar ataxia and spinal muscular atrophy) or typically seen only late in the course (multiple sclerosis). Also, unlike the studies already discussed, this study excluded disorders, such as regressive autism and Rett’s syndrome, in which intellectual deterioration may be seen early in the course but typically stabilizes. They reported a total of 84 cases of PE, of which they classified two-thirds as metabolic, one-third as neurodegenerative, and 2, both due to HIV/AIDS, as infectious. The metabolic and neurodegenerative cases were further subcategorized as shown in Table 44-3.
Table 44-3 Diagnoses in 84 Cases of Progressive Encephalopathy in Oslo, Norway
| Diagnosis | Number of Cases |
|---|---|
| METABOLIC | 55 |
| Subcellular organelles | 28 |
| Lysosomal | 23 |
| Mitochondrial | 3 |
| Peroxisomal | 2 |
| Intermediate metabolism | 27 |
| Organic aciduria | 11 |
| Fatty acid oxidation defect | 6 |
| Urea cycle disorder | 4 |
| Galactosemia | 4 |
| Unspecified | 2 |
| NEURODEGENERATIVE | 27 |
| Specified | 10 |
| Unspecified | 17 |
| INFECTIOUS | 2 |
(From Strømme P et al. Incidence rates of progressive childhood encephalopathy in Oslo, Norway: A population based study. BMC Pediatr 2007;7:25.)
There were 28 children with disorders of subcellular organelles (23 lysosomal, 3 mitochondrial, and 2 Peroxisomal) and 27 with disorders of intermediary metabolism (11 organic acidurias, 6 fatty acid oxidation disorders, 4 urea cycle disorders, 4 galactosemia, and 2 unspecified). The neurodegenerative cases included 10 children with a specific diagnosis (1 ataxia telangiectasia, 2 Cockayne’s syndrome, 1 megalencephalic leukoencephalopathy with subcortical cysts, 3 microphthalmia and brain atrophy, 1 pontocerebellar hypoplasia and infantile spinal muscular atrophy, and 2 Schinzel–Gideon syndrome) and 17 in which only the portion of the CNS most affected could be specified (8 cerebellum, 3 cerebral cortex, 3 cerebral white matter, 1 basal ganglia, 1 cerebellum and basal ganglia, and 1 cerebellum and brainstem). Analysis of the study data found that there was a 7-fold increase in risk of PE in children of Pakistani origin, due largely to the predominantly autosomal-recessive inheritance pattern for causes of PE and the much higher incidence of reported consanguinity in that community [Strømme et al., 2010]. It was estimated that 30 percent of all cases of PE, and at least 50 percent of the cases in children of Pakistani origin, would have been prevented if the practice of consanguinous marrage were avoided.
The same authors [Strømme et al., 2008] used local population data to calculate an overall incidence rate for PE of 6.43 per 100,000 person years (95 percent CI 5.15–7.97), with the age-specific rates being highest for infants <1 year old (79.9 per 100,000 person years) and lowest for children over 5 years (0.65 per 100,000 person years). They also found that the age at diagnosis averaged 0.5 years for patients with metabolic diseases and 4.5 years for patients with neurodegenerative diseases, and that children with neonatal onset and metabolic etiology had the highest risk of mortality.
Diagnostic Evaluation
These features are most readily observed when disorders have a later onset and more rapid deterioration, as is often seen in the cerebral forms of adrenoleukodystrophy [Moser et al., 2007]. PE may be difficult to recognize when disorders have a very early onset, develop very slowly, or prevent even initial development from being normal. A variety of metabolic and genetic conditions have been diagnosed in children initially thought to have cerebral palsy due to static encephalopathy [Gupta and Appleton, 2001], and those that have been reported in the literature are listed in Table 44-4. This experience suggests that children with a diagnosis of cerebral palsy should undergo further evaluation for neurodegenerative disorders when any of the following conditions is identified: no definite history of a preceding injury, a family history of neurologic symptoms, a history of parental consanguinity, or inadequately explained oculomotor abnormalities, involuntary movements, ataxia, muscle atrophy, or sensory loss. Children with recurrent, unexplained episodes of altered mental status, vomiting, or abnormal movements should be strongly suspected of having an IEM, such as a mitochondrial disease, aminoacidopathy, organic aciduria, or urea cycle enzyme defect.
Table 44-4 Progressive Encephalopathy Reported to Present as Cerebral Palsy
| Finding | Etiologic Diagnoses |
|---|---|
| Hypotonia | Duchenne muscular dystrophy Infantile neuroaxonal dystrophy Mitochondrial encephalopathy |
| Dystonia | 3-methylcrotonyl CoA carboxylase deficiency 3-methylglutaconic aciduria Dopa-responsive dystonia Glutaric aciduria type I Juvenile dystonic lipidosis Juvenile neuronal ceroid-lipofuscinosis Leigh’s disease Lesch–Nyhan syndrome Pelizaeus–Merzbacher disease Rett’s syndrome |
| Spasticity | Adrenoleukodystrophy Adrenomyeloneuropathy Arginase deficiency Hereditary progressive spastic paraplegia Holocarboxylase synthetase deficiency Metachromatic leukodystrophy |
| Ataxia | Angelman’s syndrome Ataxia telangiectasia GM1 gangliosidosis NARP Niemann–Pick disease type C Congenital disorder of glycosylation Posterior fossa tumor X-linked spinocerebellar ataxia |
NARP, neuropathy, ataxia, and retinitis pigmentosa.
The critical elements of the clinical evaluation are no different from those discussed in the evaluation of children with nonprogressive neurodevelopmental disorders, which is discussed in detail in Chapter 43. To establish the progressive nature of the child’s symptoms or clinical findings, however, it can be particularly helpful to review any records of the child’s appearance and abilities to which the caregivers can provide access, including photographs, videotapes, and examples of the child’s writing and drawing. Repeated examinations over months or even years may be necessary to uncover subtle regression in some children.
Primary motor and sensory functions are readily assessed by the screening neurologic examination, even in uncooperative children, but higher cortical functions are far more difficult to evaluate. The collective observations of clinicians, parents, and teachers who suspect subtle cognitive decline should be supplemented by those of a child psychologist who is trained to administer age-specific tests and to give an appropriate assessment of the impact of potential confounders, such as primary sensory and motor deficits, inattentiveness, shyness, behavior problems, and cultural and language differences [Sparrow and Davis, 2000]. Children with PE generally have sufficiently impaired social and occupational functioning from loss of higher cortical function to be described as suffering dementia, although the term is rarely applied.
Disturbances of higher cortical function are well characterized and localized in adult patients with acquired and degenerative disorders, including amnesia (i.e., disturbed ability to form new memories or to recall previously learned information), aphasia (i.e., difficulty with the expression or comprehension of language), apraxia (i.e., difficulty carrying out learned motor tasks not caused by weakness or incoordination), agnosia (i.e., difficulty recognizing or identifying objects or sounds not caused by sensory loss), and disturbed executive functioning (i.e., poor planning, organizing, sequencing, and abstracting). The definitions and localizations of the most commonly encountered cognitive disturbances are listed in Table 44-5.
Table 44-5 Cortical Localization of Cognitive Impairments
| Cognitive Impairment | Clinical Features | Cortical Localization |
|---|---|---|
| APHASIA | ||
| Sensory (Wernicke) | Altered discrimination of sounds (phonetic errors) and substitution of sounds and syllables (phonemic errors), or words and phrases (verbal paraphasias) | Dominant posterosuperior temporal gyrus |
| Motor (Broca) | Disturbed speech production with loss of fluency and syntax | Dominant inferior frontal gyrus |
| Conduction | Poor repetition, despite normal fluency and comprehension | Dominant arcuate fasciculus |
| Alexia with agraphia | Isolated deficits in reading and writing language | Dominant supramarginal and angular gyri |
| Dysprosody (sensory, motor, and conduction) | Impaired discrimination, production, or repetition of the intonation, melody, or phrasing of language | Nondominant perisylvian cortex, with homology with the corresponding aphasia in the dominant hemisphere |
| AGNOSIA | ||
| Visual | Impaired recognition of colors (achromatopsia), object classes (visual object agnosia), or specific objects (prosopagnosia) | Bilateral occipitotemporal visual association cortex |
| Tactile | Decreased perception of object shape (astereognosis), weight, and texture | Contralateral parietal cortex |
| Auditory | Inability to recognize verbal (word deafness) or nonverbal (amusia) sounds | Bilateral temporoparietal cortex |
| Spatial | Inattention toward one side of the world or self that is partial (extinction) or complete (neglect) | Nondominant parietal cortex |
| APRAXIA | ||
| Ideational | Inability to perform or recognize learned motor skills, resulting from loss of mechanical knowledge | Dominant inferior parietal cortex |
| Ideomotor | Inability to copy gestures or use tools without errors in positioning, orientation, movement, and timing | Contralateral motor cortex |
| Limb-kinetic | Loss of finger dexterity | Contralateral and dominant premotor and supplementary motor cortex |
| Gerstmann’s syndrome | Impairments in writing (dysgraphia), performing arithmetic calculations (dyscalculia), distinguishing right from left, and identifying fingers (finger agnosia) | Dominant angular gyrus of the parietal lobe |
Laboratory Testing
One approach to the laboratory evaluation of children with nonspecific PE is outlined in Table 44-6, which lists screening tests that might be applicable to all such children, as well as basic studies for infectious, toxic, endocrinologic, genetic, neoplastic, metabolic, autoimmune, and nutritional disorders that may be suspected on the basis of the history or the results of the initial screening tests. The results of these tests will often suggest the need for:
Table 44-6 Screening Diagnostic Tests for Nonspecific Progressive Encephalopathy
| Type of Screening | Tests |
|---|---|
| General evaluation | Neuroimaging (MRI with and without contrast preferable to CT) Electroencephalogram (capturing wake and non-REM sleep) Comprehensive metabolic panel Complete blood count Ophthalmologic examination (by specialist if possible) Audiologic testing |
| ADDITIONAL SCREENING | |
| Autoimmune disorder | Serum sedimentation rate (Westergren), antinuclear antibody titer, complement levels |
| Autonomic disorder | Histamine skin test, sweat testing |
| Endocrinopathy | Serum T4, TSH, ACTH, and cortisol |
| Genetic disorder | Genomic microarray (preferable to karyotype) MECP2 mutation screening |
| Infection | CSF cell count, glucose, protein CSF bacterial, fungal, and viral cultures CSF HIV and HSV by polymerase chain reaction CSF fungal antigens CSF test for prion proteins CSF for viral antibodies (measles, mumps) |
| Intoxication | Serum lead and thin-layer chromatography Urine screen for drugs of abuse |
| Metabolic disorder | Serum amino acids, lactate, pyruvate, ammonia Serum carnitine (free and total) and acylcarnitines Serum cholesterol and lipid panel, very long-chain fatty acids Lymphocyte vacuolization, lysosomal enzyme analysis Urine organic acids, metabolic screen, porphyrins CSF lactate, amino acids, and neurotransmitter metabolites |
| Neoplastic disorder | CSF for cytologic analysis CSF and serum for paraneoplastic antibodies |
| Nutritional disorder | Serum niacin, thiamin, pyridoxine, cobalamine, vitamin E Serum homocysteine, methylmalonic acid |
ACTH, adrenocorticotropic hormone; CSF, cerebrospinal fluid; CT, computed tomography; HIV, human immunodeficiency virus, HSV, herpes simplex virus, MRI, magnetic resonance imaging; REM, rapid eye movement; TSH, thyroid stimulating hormone.
Another approach to the evaluation of a patient who may have a rare disease with which the clinician is unfamiliar is to use an interactive database to generate a broad differential diagnosis [Segal, 2007]. Simulconsult (www.simulconsult.com) is a web-based program of this kind that is freely available to clinicians and students. As each piece of clinical information about a patient is entered, including the age of onset of symptoms and pertinent negatives, the program re-orders the diagnoses on its suggested differential and updates the suggestions it makes for what additional pieces of clinical and laboratory data would most help in distinguishing between them. Currently, the program has information on more than 2300 predominantly metabolic and genetic neurological disorders, including more than 120 that are suggested when the finding of “regression (loss or deterioration of milestones)” is entered. A broad differential can help clinicians to avoid cognitive pitfalls that commonly contribute to diagnostic error and delay [Norman and Eva, 2010], including the biases of availability and representativeness (favoring familiar diagnoses over less well-known diagnoses or disease variants) and those of framing and premature closure (favoring findings that confirm rather than question a pre-existing diagnosis).
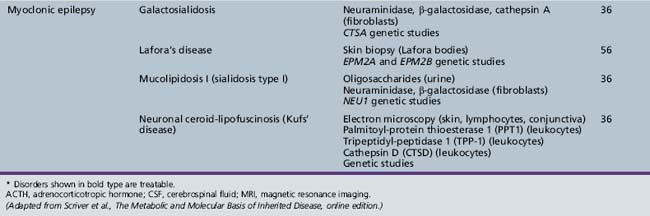
We begin with Table 44-7, which classifies the common metabolic disorders presenting in neonates based on clinical features, degree of acidosis and ketosis, and the results of tests for ammonia, lactate, glucose, calcium, and cell counts. For infants from 1–12 months old, disorders causing PE have been grouped into those associated with somatic abnormalities (Table 44-8), those with specific or suggestive neurological signs (Table 44-9), and those that typically cause slowly progressive developmental delays without more specific findings (Table 44-10). For children between the ages of 1 and 5 years, the disorders causing PE have been grouped into three different categories: those with somatic signs (Table 44-11), those with paraparesis (Table 44-12), and those with ataxia and incoordination (Table 44-13). The disorders causing PE in later childhood, between the ages of 5 and 15 years, have been divided into those that present with seizures and ataxia (Table 44-14), extrapyramidal signs, such as dystonia or choreoathetosis (Table 44-15), severe and diffuse CNS signs, including seizures and visual loss (Table 44-16), polymyoclonus (Table 44-17), cerebellar ataxia (Table 44-18), polyneuropathy (Table 44-19), and psychiatric symptoms (Table 44-20). Finally, disorders that may present with PE in late adolescence and adulthood are listed in Table 44-21.
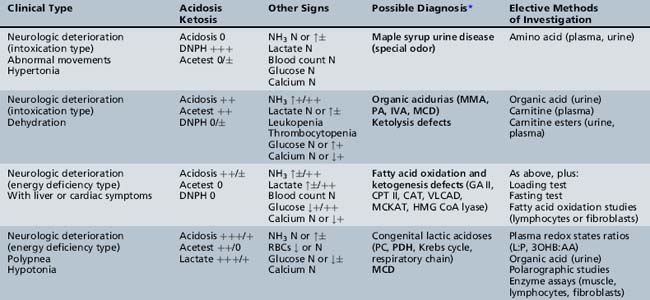
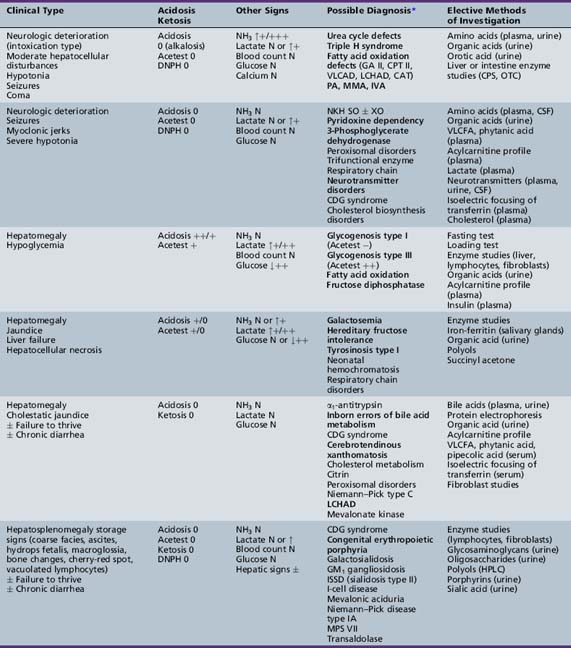

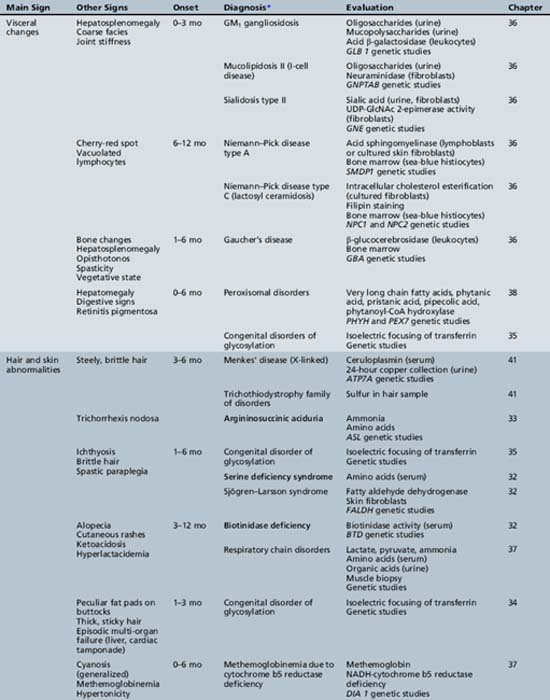
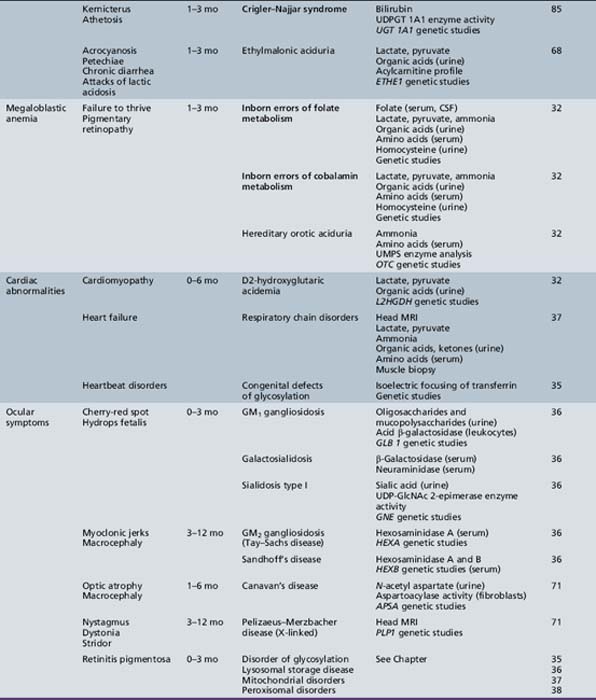
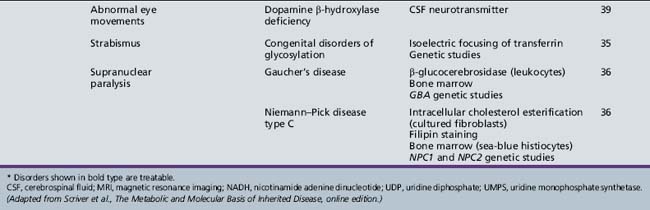
Table 44-9 Progressive Encephalopathy in Infancy (1–12 Months) with Specific or Suggestive Neurologic Signs
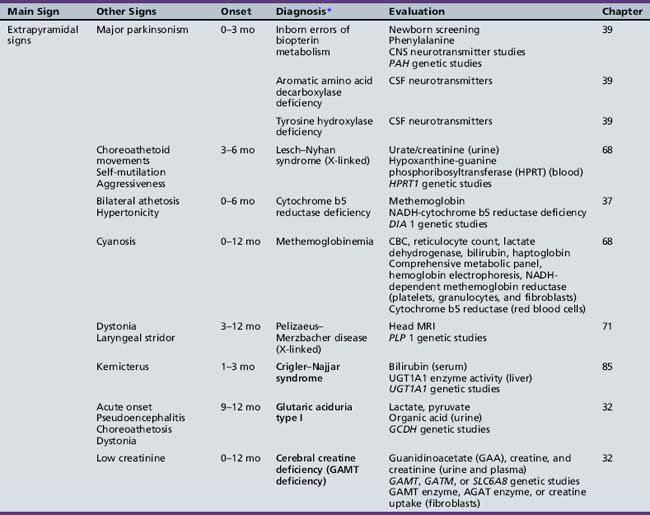
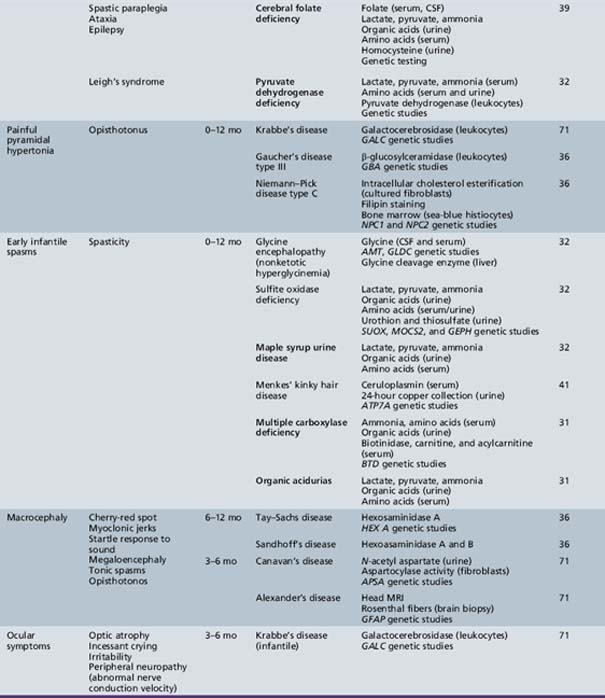
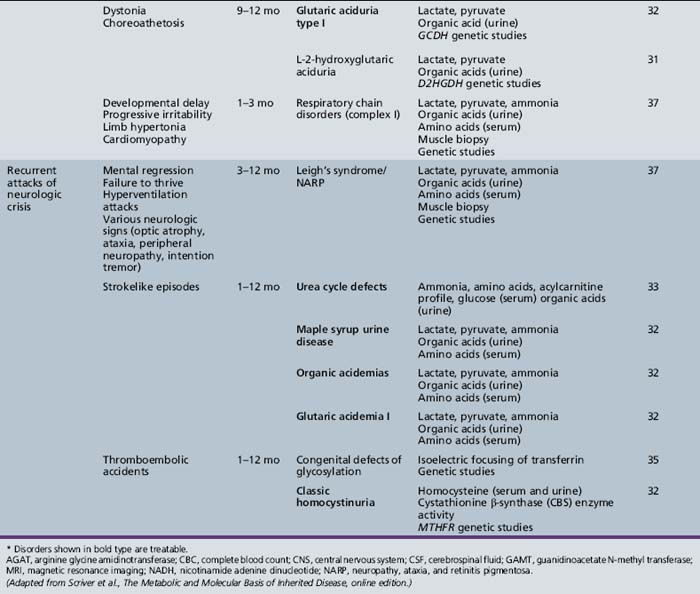
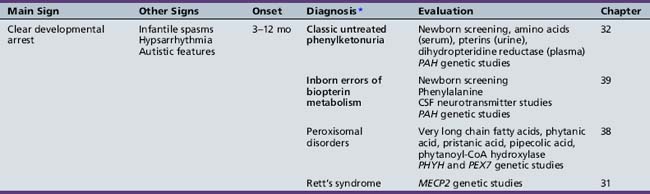

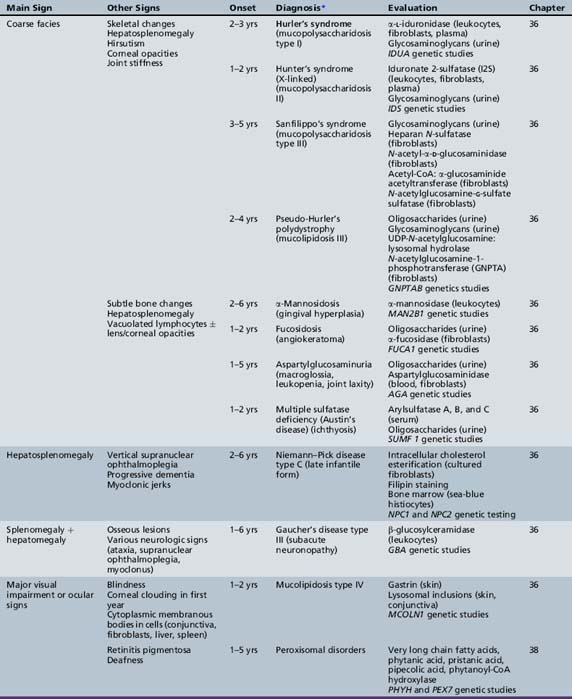

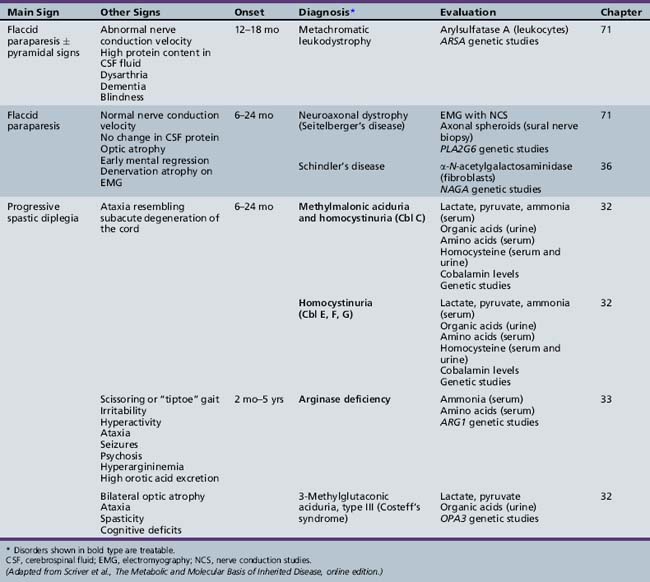
Table 44-13 Progressive Encephalopathy in Middle to Late Childhood (1–5 Years) with Ataxia and Incoordination


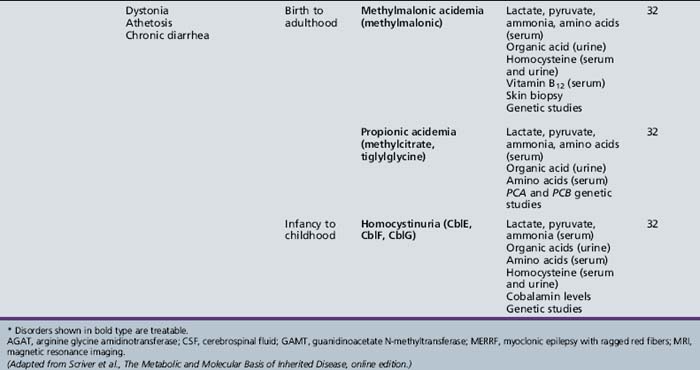
Table 44-14 Progressive Encephalopathy in Middle to Late Childhood (5–15 Years) with Seizures and Ataxia
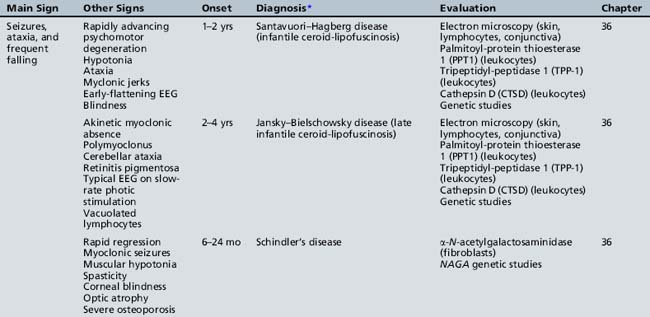
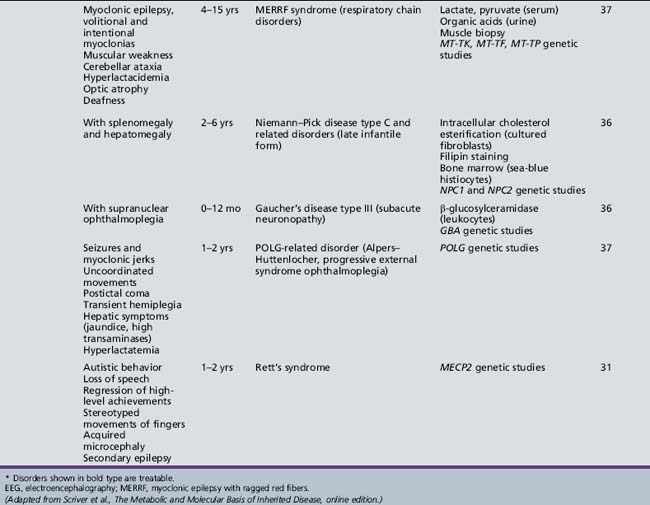
Table 44-15 Progressive Encephalopathy in Middle to Late Childhood (5–15 Years) with Predominant Extrapyramidal Signs
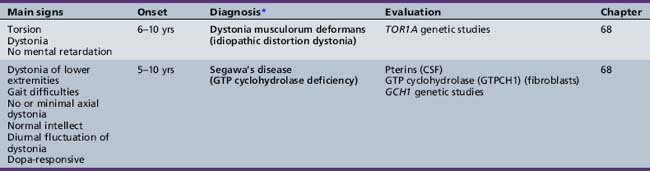
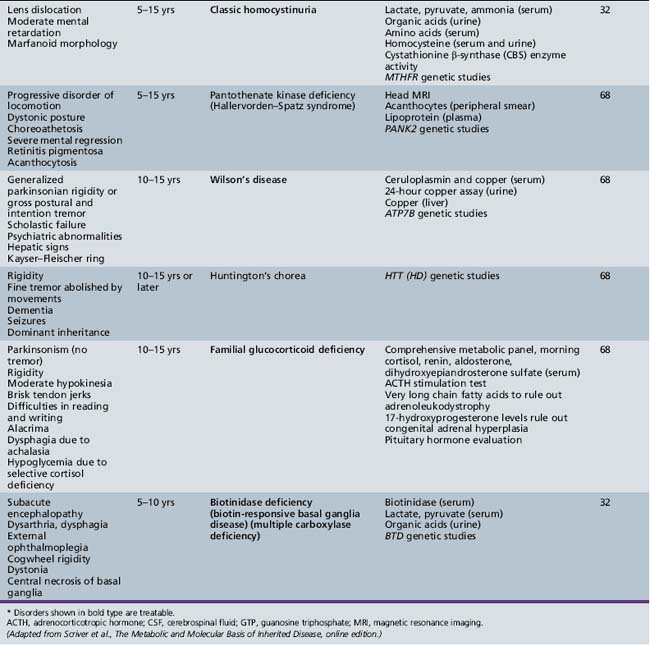
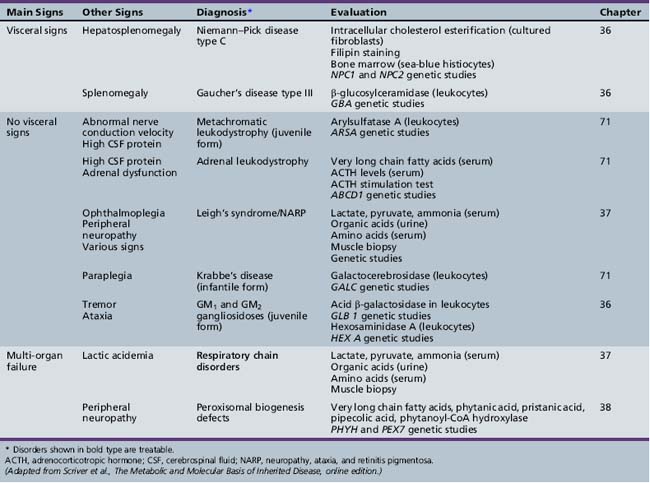
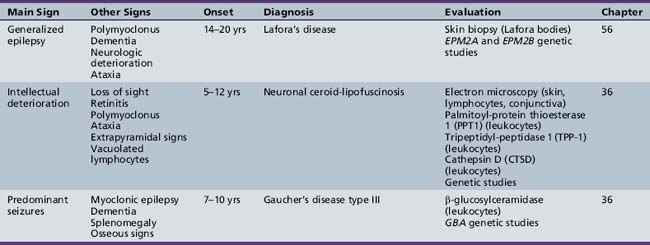
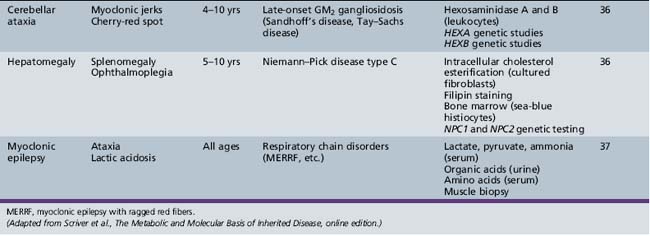
Table 44-18 Progressive Encephalopathy in Middle to Late Childhood (5–15 Years) with Predominant Cerebellar Ataxia
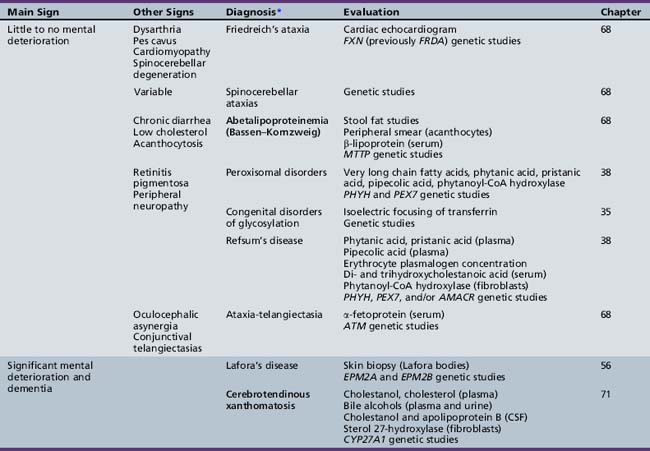
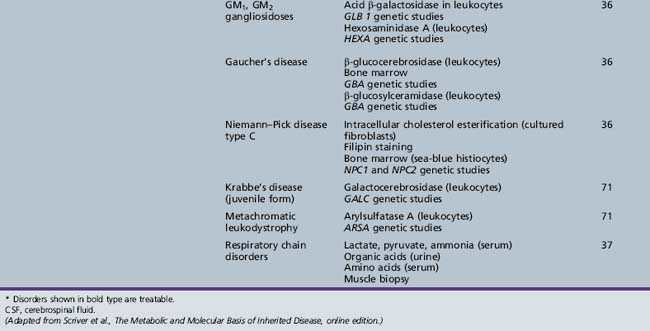
Table 44-19 Progressive Encephalopathy in Middle to Late Childhood (5–15 Yrs) with Predominant Polyneuropathy
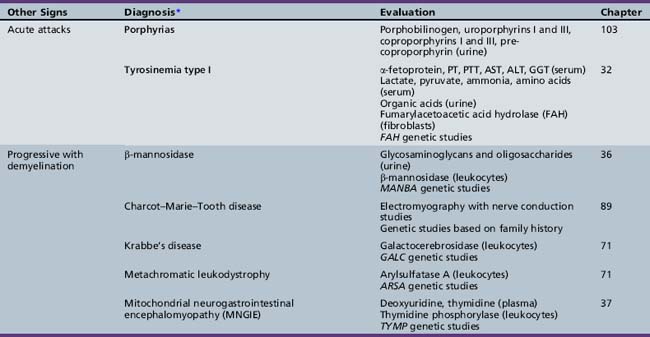
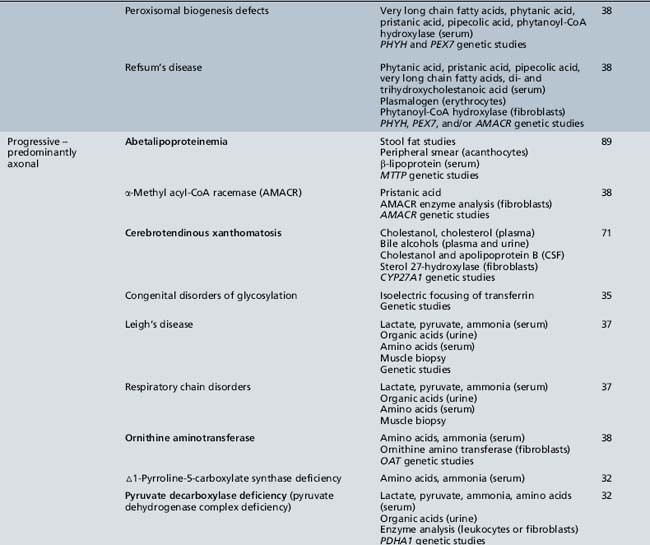
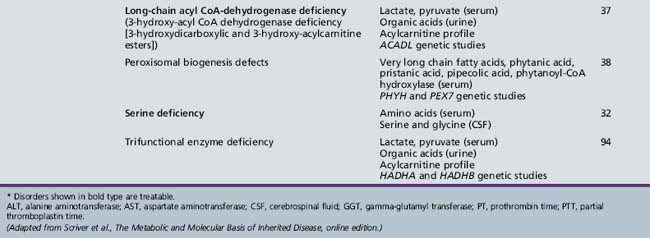
Table 44-20 Progressive Encephalopathy in Middle to Late Childhood (5–15 Years) with Predominantly Psychiatric Symptoms
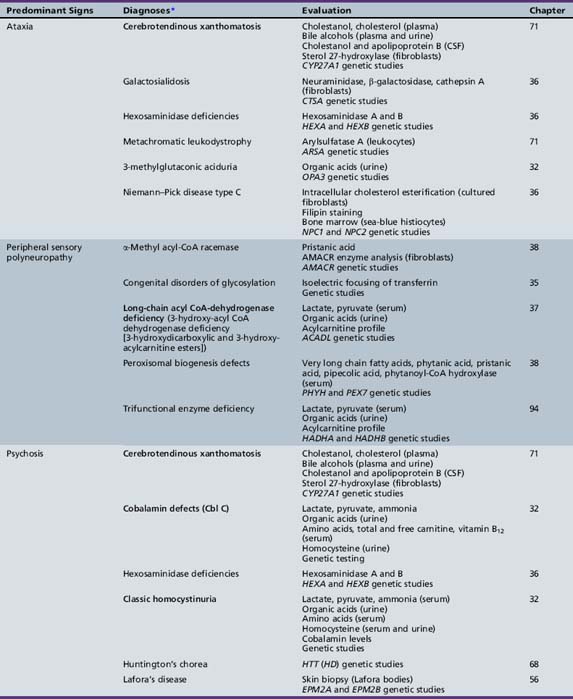
| Diagnosis* | Evaluation | Chapter |
|---|---|---|
| X-linked adrenoleukodystrophy | Very long chain fatty acids (serum) ACTH levels (serum) ACTH stimulation test ABCD1 genetic studies |
71 |
| Cerebrotendinous xanthomatosis | Cholestanol, cholesterol (plasma) Bile alcohols (plasma and urine) Cholestanol and apolipoprotein B (CSF) Sterol 27-hydroxylase (fibroblasts) CYP27A1 genetic studies |
71 |
| Cobalamin defects (Cbl C) | Lactate, pyruvate, ammonia Organic acids (urine) Amino acids (serum) Homocysteine (urine) Genetic testing |
32 |
| Classic homocystinuria | Lactate, pyruvate, ammonia (serum) Organic acids (urine) Amino acids (serum) Homocysteine (serum and urine) Cobalamin levels Genetic studies |
32 |
| Huntington’s chorea (juvenile form) | HTT (HD) genetic studies | 68 |
| Krabbe’s disease | Galactocerebrosidase (leukocytes) GALC genetic studies |
71 |
| Methylene tetrahydrofolate reductase deficiency | Homocysteine (urine) MTHFR genetic studies |
103 |
| Metachromatic leukodystrophy | Arylsulfatase A (leukocytes) ARSA genetic studies |
71 |
| Niemann–Pick disease type C | Intracellular cholesterol esterification (cultured fibroblasts) Filipin staining Bone marrow (sea-blue histiocytes) NPC1 and NPC2 genetic studies |
36 |
| Neuronal ceroid-lipofuscinosis | Electron microscopy (skin, lymphocytes, conjunctiva) Palmitoyl-protein thioesterase 1 (PPT1) (leukocytes) Tripeptidyl-peptidase 1 (TPP-1) (leukocytes) Cathepsin D (CTSD) (leukocytes) Genetic studies |
36 |
| Pantothenate kinase deficiency (Hallervorden–Spatz) |
Head MRI Acanthocytes (peripheral smear) Lipoprotein (plasma) PANK2 genetic studies |
68 |
| Pyruvate carboxylate deficiency (Leigh’s disease) | Lactate, pyruvate, ammonia (serum) Organic acids (urine) Amino acids (serum) Muscle biopsy Genetic studies |
37 |
| Sanfilippo’s syndrome | Heparan N-sulfatase (fibroblasts) N-acetyl-alpha-d-glucosaminidase (fibroblasts) Acetyl-CoA: alpha-glucosaminide acetyltransferase (fibroblasts) N-acetylglucosamine-g-sulfate sulfatase (fibroblasts) Glycosaminoglycans (urine) Genetic studies |
36 |
| Urea cycle defects | Lactate, pyruvate, ammonia, amino acids (serum) Organic acids (urine) Genetic studies |
33 |
| Wilson’s disease | Ceruloplasmin and copper (serum) ATP7B genetic studies |
68 |
ACTH, adrenocorticotropic hormone; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging.
* Disorders shown in bold type are treatable.
(Adapted from Scriver et al., The Metabolic and Molecular Basis of Inherited Disease, online edition.)

Up-to-date information about the sensitivity and availability of tests for specific genetic disorders is available through the Gene Tests website (www.genetests.org), maintained by the University of Washington at Seattle through funding from the National Institutes of Health. Some genetic tests can be performed on a research basis through direct communication and cooperation between the clinician and research laboratory.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Devereux G., Stellitano L., Verity C.M., et al. Variations in neurodegenerative disease across the UK: Findings from the national study of Progressive Intellectual and Neurological Deterioration (PIND). Arch Dis Child. 2004;89:8-12.
Dyken P., Krawiecki N. Neurodegenerative diseases of infancy and childhood. Ann Neurol. 1983;13:351-364.
Gupta R., Appleton R.E. Cerebral palsy: Not always what it seems. Arch Dis Child. 2001;85:356-360.
Moser H.W., Mahmood A., Raymond G.V. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3(3):140-151.
Norman G.R., Eva K.W. Diagnostic error and clinical reasoning. Med Educ. 2010;44(1):94-100.
Nunn K., Williams K., Ouvrier R. The Australian Childhood Dementia Study. Eur Child Adolesc Psychiatry. 2002;11:63-70.
Segal M. How doctors think, and how software can help avoid cognitive errors in diagnosis. Acta Paediatr. 2007;96:1720-1722.
Sparrow S.S., Davis S.M. Recent advances in the assessment of intelligence and cognition. J Child Psychol Psychiatry. 2000;41:117-131.
Strømme P., Kanavin O.J., Abdelnoor M., et al. Incidence rates of progressive childhood encephalopathy in Oslo, Norway: a population based study. BMC Pediatr. 2007;7:25.
Strømme P., Magnus P., Kanavin ØJ, et al. Mortality in childhood progressive encephalopathy from 1985 to 2004 in Oslo, Norway: a population-based study. Acta Paediatr. 2008;97:35-40.
Strømme P., Suren P., Kanavin O.J., et al. Parental consanguinity is associated with a seven-fold increased risk of progressive encephalopathy: a cohort study from Oslo, Norway. Eur J Paediatr Neurol. 2010;14(2):138-145.
Sultan T., Qureshi A.A., Rehman M., et al. The spectrum of neurodegeneration in children. J Coll Physicians Surg Pak. 2006;16(11):721-724.
Surtees R. Understanding neurodegenerative disorders. Curr Paediatr. 2002;12:191-198.
Valle D, Beaudet AL, Vogelstein B, et al: The Online Metabolic and Molecular Bases of Inherited Disease. http://omimbid.com/
Verity C.M., Winstone A.M., Stellitano L., et al. The clinical presentation of mitochondrial diseases in children with progressive intellectual and neurological deterioration: a national, prospective, population-based study. Dev Med Child Neurol. 2010;52(5):434-440.
Verity C., Winstone A.M., Stellitano L., et al. The epidemiology of progressive intellectual and neurological deterioration in childhood. Arch Dis Child. 2010;95(5):361-364.
Will R.G., Ironside J.W., Zeidler M., et al. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet. 1996;347:921-925.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 44 Cognitive and Motor Regression
Epidemiology
Although the causes of PE are individually rare, the combined incidence of PE has been estimated to be as high as 1 in 2000 live births [Surtees, 2002]. Much of what has been published regarding these disorders has been retrospective and focused on individual conditions, providing little basis for a discussion of their collective epidemiology. A few studies have been notable exceptions.
An early paper examining the experience with PE at two large academic centers in the United States found that, of 1218 admissions to their child neurology services over the course of 10 years, 341 patients were diagnosed with 1 of more than 50 disorders causing neurological dysfunction [Dyken and Krawiecki, 1983]. Table 44-1 shows the results of their analysis of the relative frequency of the various diagnoses. Although 72 percent of the cases studied had a genetic or metabolic disorder causing PE, the study also included a significant number of children with pure lower motor neuron syndromes and acquired injuries due to infection, immunologic disorders, refractory epilepsy, chronic environmental insults, nutritional deficiencies, and iatrogenic factors. A study from the Children’s Hospital of Lahore, Pakistan [Sultan et al., 2006], found that, of the 1273 children admitted to the neurology service from 2004 to 2005, 66 were diagnosed with PE and most received a specific diagnosis. The most common diagnoses, in descending order of frequency, were metachromatic leukodystrophy (14 cases), adrenoleukodystrophy (11), subacute sclerosing panencephalitis (8), Wilson’s disease (6), Friedreich’s ataxia (5), liposis (4), Gaucher’s disease (3), Alexander’s disease (2), and pantothenate kinase-associated neurodegeneration (PKAN) (2). More than half of the patients underwent funduscopic examination, electroencephalography, and cerebrospinal fluid examination as part of their diagnostic work-up.
Table 44-1 Diagnosis in 340 Cases of Developmental Regression
| Diagnosis | Number of Cases |
|---|---|
| POLIODYSTROPHIES* | 129 |
| Lysosomal storage disorders | 39 |
| Hypoxic poliodystrophy | 29 |
| Idiopathic poliodystrophy | 24 |
| West’s syndrome | 17 |
| Lennox–Gastaut syndrome | 9 |
| Metabolic poliodystrophy | 4 |
| Toxoplasmosis | 3 |
| Post-vaccine poliodystrophy | 3 |
| Lowe’s syndrome | 1 |
| LEUKODYSTROPHIES† | 71 |
| SSPE | 26 |
| ADEM and MS | 17 |
| Adrenoleukodystrophy | 8 |
| Metachromatic leukodystrophy | 5 |
| Pelizaeus–Merzbacher disease | 4 |
| Krabbe’s disease | 4 |
| Phenylketonuria | 2 |
| Cockayne’s syndrome | 2 |
| Canavan’s disease | 1 |
| Alexander’s disease | 1 |
| Maple syrup urine disease | 1 |
| CORENCEPHALOPATHIES‡ | 26 |
| Idiopathic corencephalopathy | 8 |
| Huntington’s disease | 5 |
| Mitochondrial disorders | 4 |
| Dystonia musculorum deformans | 2 |
| Hallervorden–Spatz syndrome | 2 |
| Ataxia-telangiectasia | 1 |
| Congenital indifference to pain | 1 |
| Infantile neuroaxonal dystrophy | 1 |
| Riley–Day syndrome | 1 |
| Wilson’s disease | 1 |
| DIFFUSE ENCEPHALOPATHIES | 63 |
| Tuberous sclerosis | 19 |
| Idiopathic encephalopathy | 17 |
| Hyperammonemic disorders | 6 |
| Mitochondrial disorders | 4 |
| Neurofibromatosis | 4 |
| Achondroplasia | 2 |
| Organic acidurias | 2 |
| Letterer–Siwe disease | 2 |
| Sturge–Weber syndrome | 2 |
| Zellweger’s syndrome | 2 |
| Homocystinuria | 1 |
| Incontinentia pigmenti | 1 |
| Sjögren–Larsson syndrome | 1 |
| SPINOCEREBELLOPATHIES§ | 51 |
| Spinal muscular atrophy | 19 |
| Hereditary spastic paraplegia | 12 |
| Acute cerebellar ataxia | 8 |
| Infantile polymyoclonus | 4 |
| Charcot–Marie–Tooth disease | 2 |
| Friedreich’s ataxia | 2 |
| Marinesco–Sjögren syndrome | 1 |
| OPCA | 1 |
| Spinocerebellar degeneration | 1 |
| Refsum’s disease | 1 |
ADEM, acute disseminated encephalomyelitis; MS, multiple sclerosis; OPCA, olivopontocerebellar atrophy; SSPE, subacute sclerosing panencephalitis.
* Poliodystrophies = predominant cortical involvement.
† Leukodystrophies = predominant cerebral white-matter involvement.
‡ CORENCEPHALOPATHIES = predominant basal ganglia involvement.
§ Spinocerebellopathies = predominant spinal cord and cerebellar involvement.
(From Dyken P, Krawiecki N. Neurodegenerative diseases of infancy and childhood. Ann Neurol 1983;13:351–364.)
Following the initial description in 1996 of 10 cases of new variant Creutzfeldt–Jakob disease (nvCJD) affecting young adults in the United Kingdom [Will et al., 1996], several countries instituted prospective surveillance programs to collect data on patients with PE to better identify additional cases of nvCJD. Although these studies have relied on reports from pediatricians and have been unable to describe absolute incidence or prevalence figures, they have reported relative prevalences within their areas. The first report from the surveillance done in the UK [Devereux et al., 2004] collected and analyzed pediatric cases of progressive intellectual and neurological deterioration (PIND) over a 5-year span. The cases included children who had:
The study excluded children with intellectual and neurological deterioration after a nonprogressive insult, such as encephalitis, trauma, or global hypoxic-ischemic injury, but did include children with seizure disorders who otherwise met the case definition and children carrying diagnoses that could be expected to lead to progressive deterioration in the future. Of the 798 cases collected, 577 had a confirmed diagnosis, 6 had definite or probable nvCJD, and 211 had no clear etiologic diagnosis at the time of publication but did not have clinical features suggestive of nvCJD. There were nearly 100 different confirmed diagnoses, but more than one-quarter of the cases were explained by the five most common: mucopolysaccharidosis type III (Sanfilippo’s syndrome), adrenoleukodystrophy, late infantile neuronal ceroid-lipofuscinosis, mitochondrial diseases, and Rett’s syndrome. Higher rates of prevalence and of consanguinity were reported in families of South Asian origin. A follow-up of the UK study [Verity et al., 2010a] reported a confirmed etiologic diagnosis in 1047 of the 2493 cases of PIND that had been collected by 2008, with nearly one-quarter of cases again explained by the five most common diagnoses: neuronal ceroid-lipofuscinoses, mitochondrial diseases, mucopolysaccharidoses, GANGLIOSIDOSES, and Peroxisomal disorders. The most recent update of the study [Verity et al., 2010b] reported that, after 12 years, 147 different etiologies were found to explain 1114 of the 2636 cases of PIND collected. In total, only 6 children with confirmed or probable nvCJD had been identified. The 30 most common diagnoses identified in the study are presented in Table 44-2.
Table 44-2 Common Diagnoses in 1114 Cases of Progressive Encephalopathy
| Diagnosis | Number of Cases |
|---|---|
| LEUKOENCEPHALOPATHIES | 183 |
| Metachromatic leukodystrophy | 59 |
| Krabbe’s disease | 33 |
| Pelizaeus–Merzbacher disease | 17 |
| Canavan’s disease | 13 |
| Vanishing white matter disease | 11 |
| Aicardi–Goutières syndrome | 10 |
| Alexander’s disease | 10 |
| Other | 31 |
| NEURONAL CEROID-LIPOFUSCINOSES | 141 |
| NCL late infantile | 73 |
| NCL juvenile | 44 |
| NCL infantile | 22 |
| Other | 2 |
| MITOCHONDRIAL | 122 |
| Leigh’s syndrome | 17 |
| NARP (including NARP/MILS) | 17 |
| Other | 88 |
| MUCOPOLYSACCHARIDOSES | 102 |
| Mucopolysaccharidosis IIIA (Sanfilippo’s syndrome) | 69 |
| Mucopolysaccharidosis IIA (Hunter’s disease) | 15 |
| Other | 18 |
| GANGLIOSIDOSES | 100 |
| GM2 gangliosidosis type 1 (Tay–Sachs disease) | 41 |
| GM2 gangliosidosis type 2 (Sandhoff’s disease) | 33 |
| GM1 gangliosidosis | 23 |
| Other | 3 |
| PEROXISOMAL | 69 |
| Adrenoleukodystrophy | 56 |
| Other | 13 |
| OTHER METABOLIC | 95 |
| Niemann–Pick disease type C | 38 |
| PKAN/NBIA | 21 |
| Menkes’ disease | 16 |
| Glutaric aciduria type 1 | 10 |
| Molybdenum co-factor deficiency |
