Chapter 56 Coats Disease
History
Coats disease is an idiopathic condition characterized by telangiectatic and aneurysmal retinal vessels with intraretinal and subretinal exudation and fluid.1 Coats disease was first described by Scottish ophthalmologist George Coats in 1908.2 In his initial classification, Coats separated this new entity into three distinct groups. Group I included eyes with massive subretinal exudation and no demonstrable vascular abnormalities. Group II consisted of eyes with massive subretinal exudation and multiple retinal vascular abnormalities with intraretinal hemorrhage. Group III included eyes with massive subretinal exudation and frank retinal arteriovenous malformations.2 Eugen von Hippel later demonstrated that group III represented the distinctly separate entity of angiomatosis retinae (later retermed retinal capillary hemangioma or retinal hemangioblastoma), prompting Coats to drop this group from his classification. In 1912, and again in 1915, Theodor von Leber described a disease with similar telangiectatic and aneurysmal retinal vessels that lacked the massive subretinal exudation described by Coats.3,4 This condition was later named Leber multiple miliary aneurysms. In a 1916 paper, Leber concluded that what he had described was merely an earlier stage of the disease process identified by Coats.5,6 This conclusion was later reinforced by Reese, who described an eye with Leber miliary aneurysms that progressed into a classic case of Coats disease during long-term follow-up.7 Although some authors have disagreed, most authorities today classify Leber disease as an early or nonprogressive form of Coats disease.5,8,9
Histopathology, etiology, and pathogenesis
Early descriptions of Coats disease focused primarily on the morphology of the disease and attempted to explain the disease process solely on the basis of fundoscopic and histopathologic changes.10,11 The first observations published by Coats focused on histopathologic examination of six eyes enucleated for this condition that he received from various colleagues.2 From these histopathologic specimens, Coats described subretinal masses of fibrous tissue intimately adherent to the outer retina. Most of the eyes had associated retinal thickening, degeneration, and detachment, as well as diffuse vascular abnormalities. Four of the six eyes had associated retinal hemorrhages and most had cholesterol crystals. One eye had evidence of bone formation. Coats believed the vascular changes represented secondary manifestations of the underlying disease and that the exudation was secondary to organization and partial resorption of retinal hemorrhages, as he often found hemorrhages in association with the exudation.2 Coats also postulated that the primary process might be infectious in light of the mononuclear infiltrate found on histopathologic examination. This infectious theory of Coats disease was supported by a number of other reports, including that of Evens,12 Straub,13 and Müller,13 who suspected toxoplasmosis as the underlying cause. Subsequent investigations and the failure of anti-inflammatory adrenocorticotropic hormone and steroid therapies have refuted early theories of a primary infectious or inflammatory etiology.13,14
Pathologic samples of eyes with Coats disease were once abundant owing to enucleations for suspected intraocular tumor.15 Moreover, most histopathologic reports describe findings in eyes with advanced stages of the disease. Gross examination of enucleated eyes typically reveals bullous retinal detachment.16 Subretinal fluid has a viscous, lipid-rich, yellow appearance with glistening crystals.16 Microscopically, the subretinal exudation contains collections of foamy histiocytes and cholesterol clefts.16 The outer retina is thickened by exudation, with variable penetration of inner retinal layers.16 Areas of the inner retina also contain numerous, large, dilated, and telangiectatic vessels.16 Other vascular abnormalities include degenerated and hypocellular walls, narrowed lumen with or without thrombi, capillary dropout, neovascularization, as well as perivascular sheathing and/or pigmentation.1,9,17–23 Thickened vessel walls show characteristic heavy periodic acid–Schiff-positive deposits.1 Flat preparations prepared by trypsin digestion reveal vascular aneurysms measuring 50–350 µm, often forming large sausage-like outpouchings and located on shunt vessels.1 Advanced cases may have intraocular bone formation, with evidence of calcification on echography and computed tomography (CT) scanning that potentially confuses the diagnosis with retinoblastoma.24,25 Electron microscopy confirms diffuse structural abnormalities of the retinal vasculature. Tripathi and Ashton described prominent structural alterations of retinal vessels in areas of retinal thickening, with thickened walls mostly replaced by a laminated fibrous coating of basement membrane-like material.18 Most affected vessels were completely devoid of endothelium and pericytes, with red blood cells, plasma, and fibrin filling the lumina. There was patchy thinning or even absence of the vessel wall in some areas, with the vessel lumen extending to the basement membrane of adjacent glial cells.18 These findings support the widely held view that Coats disease is a disorder of vascular integrity.
Indeed, recent work in molecular genetics suggests that Coats disease may be part of a spectrum of related genetic disorders known as “retinal hypovasculopathies”26,27 which includes Norrie disease, familial exudative vitreoretinopathy (FEVR), fascioscapulohumeral muscular dystrophy (FSHD), and the osteoporosis pseudoglioma syndrome.27–35 These diseases share a similar ocular phenotype, characterized by a failure to vascularize fully the retinal periphery and associated telangiectatic, incompetent remnant vasculature. Each of these conditions can be related to abnormalities in the Wnt signalling pathway during retinal angiogenesis.26,27,36
Several reports have implicated a deficiency of Norrin, a retinal protein, in the pathogenesis of Coats disease.37,38 In one case report, a female with unilateral Coats disease gave birth to a son affected by Norrie disease.37 Both carried a missense mutation within the Norrie disease pseudoglioma (NDP) gene on chromosome Xp11.2. Further analysis on archival tissue from nine enucleated eyes from males with unilateral Coats disease revealed a mutation in the NDP gene in one subject. Knockout mouse models of Norrie disease model demonstrate abnormalities of the retinal vessels, including telangiectasia, bulb-like dilatations, and underdevelopment of the capillary bed.39,40 Elevated levels of hypoxia-inducible factor-1α and vascular endothelial growth factor (VEGF), as well as characteristic electroretinogram patterns in these mice, confirmed inner retinal hypoxia.40 Moreover, ectopic lens expression of Norrin in knockout mice induces the formation of normal deep retinal capillaries, and completely prevents abnormalities in retinal angiogenesis.41 These observations suggest a genetic basis for Coats disease and that a mutation in the NDP gene may be responsible.
Although FEVR and Coats disease share certain clinical features, they likely do not share a common genetic basis. Robataille and associates studied DNA expression of Frizzled-4 (FZD4) in 68 cases of FEVR and 16 cases of Coats disease.42 They found 11 FZD4 mutations in FEVR cases and no mutations in Coats disease cases. They concluded that germline mutations in FZD4 do not appear to be a common cause of Coats disease, implying that Coats disease does not likely represent asymmetric FEVR and is a distinct ocular condition.
Other genetic pathways have been explored. Cremers and associates found that 55% of eyes with retinitis pigmentosa and Coats-like secondary exudative vasculopathy contained a mutation in the CRB1 gene.43 This suggests that the CRB1 gene could be involved in primary Coats disease as well as other retinal diseases and dystrophies.
Clinical presentation
Coats disease is classically a painless ophthalmic condition. The disease affects males three times as often as females and has no reported racial or ethnic predilection.44 Coats disease is unilateral in 80–95% of cases.5,44,45 Some of the bilateral cases in older reports could represent secondary bilateral Coats-like retinopathy with systemic conditions and not true primary Coats disease. Any patient with bilateral presumed Coats disease should be evaluated for conditions that causes Coats-like exudative retinopathy such as retinitis pigmentosa, pars planitis, FSHD, FEVR, or other diseases.46 Based on a study of 150 consecutive cases, the mean age at diagnosis is 5 years with a range of 1 month to 63 years.44 Some speculate that the disease could be present at birth.13,47
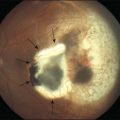
In an analysis of 150 consecutive cases of Coats disease, presenting symptoms included decreased visual acuity (43%), strabismus (23%: Fig. 56.1), leukocoria/xanthocoria (20%: Fig. 56.1), pain (3%), heterochromia (1%), nystagmus (1%), and no symptom (8%).44 Visual acuity was 20/20 to 20/50 in 12% of all eyes, 20/60 to 20/100 in 11%, 20/200 to counting fingers in 18%, and hand motions to no light perception in 58%. Nearly 90% of eyes had a normal anterior-segment examination. Those with findings included cataract (8%), iris neovascularization (8%), shallow anterior chamber (4%), corneal edema (3%), cholesterol in the anterior chamber (3%: Fig. 56.1),48,49 and megalocornea (2%). Retinal findings included telangiectasia (100%), intraretinal exudation (99%), exudative retinal detachment (81% with 42% demonstrating partial retinal detachment and 58% with total retinal detachment), retinal hemorrhage (13%), retinal macrocyst (11%), vasoproliferative tumor (6%), and optic disc neovascularization (1%). No eyes in this cohort presented with vitreous hemorrhage. Other studies have described macular fibrosis in up to 23% of patients, developing in an area of previous dense exudation, and involving the fovea in all cases.50
Adult cases of Coats disease are similar in clinical presentation and disease course, but generally have a smaller area of involvement, slower disease progression, more frequent hemorrhage near larger aneurysmal dilated vessels, and typically do not present with strabismus.51 Although the adult form of the disease has been described as frequently associated with hypercholesterolemia, such an association does not appear to occur in the juvenile form.14,52
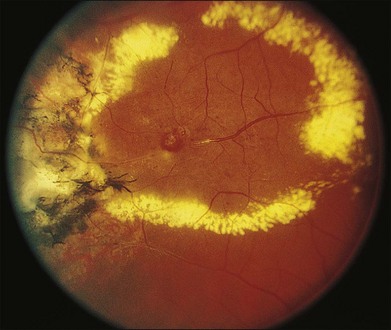
The typical ophthalmoscopic picture in Coats disease is that of localized, yellow, subretinal exudation associated with adjacent vascular anomalies, including sheathing, telangiectasia, tortuosity, aneurysmal dilation, zones of capillary dropout, and occasionally neovascularization.13,44 There is variability in this clinical picture.44 Exudation, hemorrhage, or a combination appears minimal during less active stages of the disease and can progress to massive findings, obscuring the retinal vasculature in more aggressive stages. The vascular abnormalities can be subtle and clinically undetectable or can be obvious as a dominant feature. The clinical course is also variable, but is generally slowly progressive. Spontaneous remission has been observed but is exceptional.53 Subretinal choroidal neovascularization rarely occurs in areas of lipid deposition. As subretinal fluid and exudation increase, the retinal detachment progresses and can reach a highly elevated state, visible behind the lens (Fig. 56.1). Hemorrhagic and nonhemorrhagic retinal macrocysts can occur as a result of chronic coalescent intraretinal cystoid edema.54 Secondary complications such as iridocyclitis, cataract, and secondary neovascular glaucoma can lead to phthisis bulbi in severe cases.2,15,55
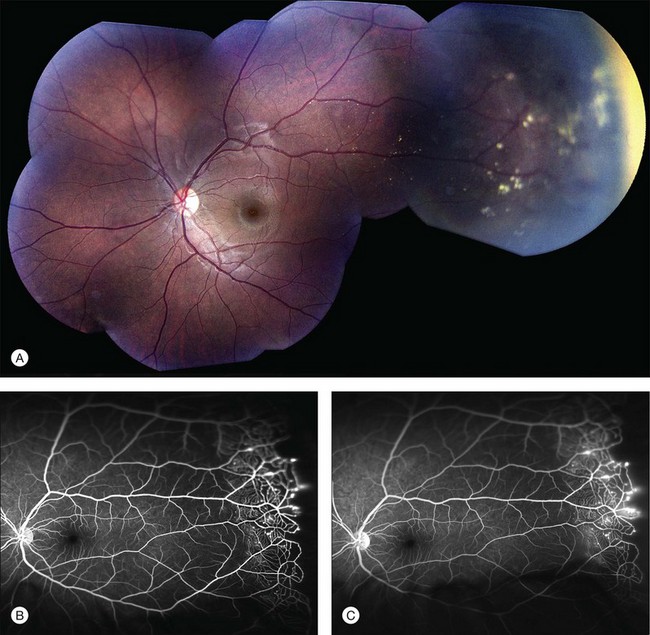
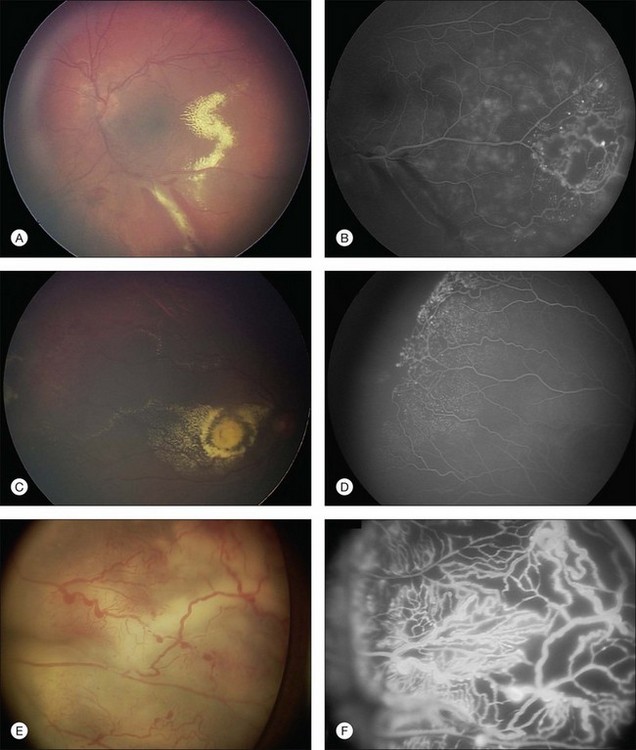
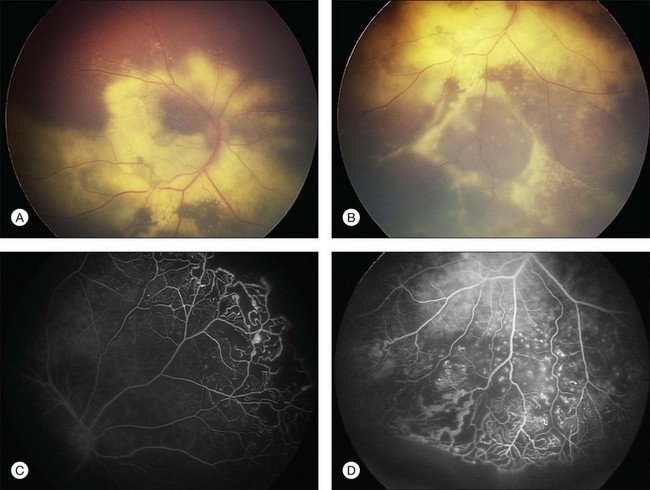
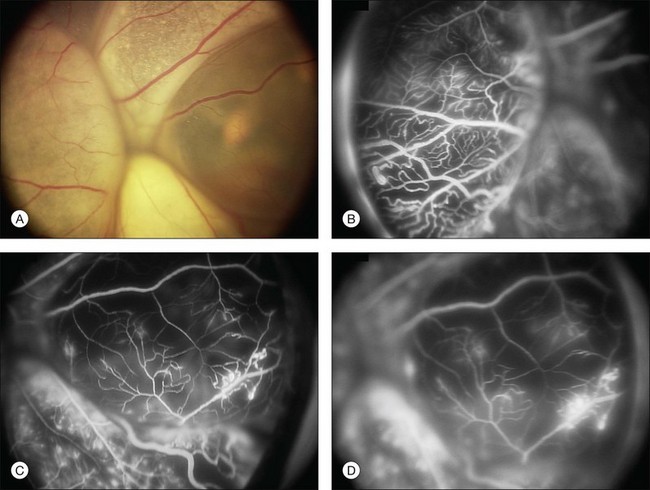
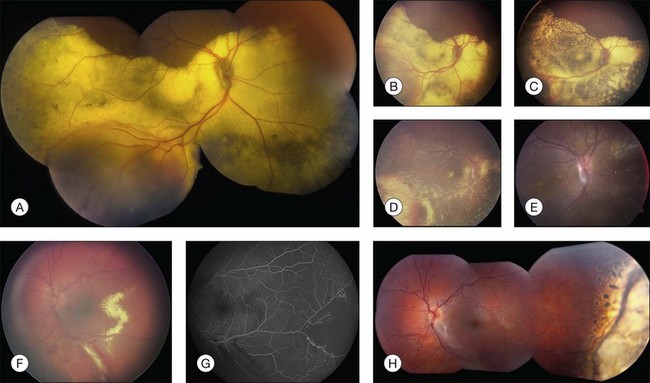
Several staging systems for Coats disease have been proposed.2,56 The most recent and widely used was introduced by Shields and associates in 2000 (Table 56.1).57 Stage 1 is characterized by retinal telangiectasia. Stage 2 has telangiectasia and intraretinal exudation, extrafoveal exudation defines stage 2A (Fig. 56.2), whereas foveal involvement defines stage 2B. Stage 3 is defined by exudative retinal detachment, 3A is a subtotal detachment, with 1 and 2 designating extrafoveal and foveal involvement, respectively (Fig. 56.3), and 3B is a total retinal detachment (Fig. 56.4). Stage 4 has total retinal detachment plus elevated intraocular pressure (Fig. 56.3), and stage 5 is endstage disease, occasionally with phthisis bulbi (Fig. 56.1). Using this classification for grouping of 124 cases, Shields and associates found stage 1 in 1%, stage 2 in 14% (2A in 8%, 2B in 6%), stage 3 in 69% (3A1 in 19%, 3A2 in 19%, 3B in 30%), stage 4 in 15%, and stage 5 in 2%.57 Based on classification and treatment, visual acuity strongly depended on stage at diagnosis.44,58 At the time of Coats disease control, poor visual acuity (20/200 or worse) was found in 0% of stage 1, 53% of stage 2, 74% of stage 3, and 100% of stage 4 and stage 5.57
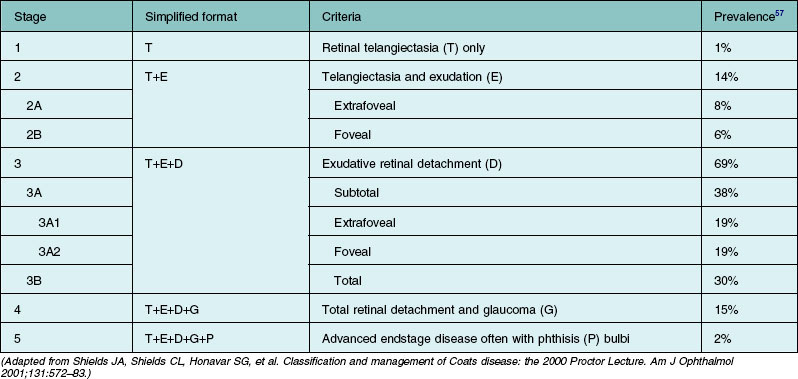
Diagnostic testing
Ancillary testing may be useful when the diagnosis of Coats disease is suspected, but other clinical entities must be ruled out, most notably retinoblastoma.59 Particularly when retinal detachment with subretinal exudation and dilated retinal vessels coexist, even an experienced clinician may have difficulty differentiating these entities ophthalmoscopically. Fluorescein angiography is critical to document classic findings to establish the diagnosis. Echography may enable differentiation between Coats disease and retinoblastoma on the basis of features such as the character of the retinal detachment and the presence or absence of subretinal calcifications. Echography is less useful when the retinoblastoma is poorly calcified and also has shortcomings in detecting optic nerve or extraocular extension of retinoblastoma when heavy calcification exists.60
Fluorescein angiography
The typical fluorescein angiographic picture of Coats disease is one of numerous localized anomalies of the retinal vasculature with peripheral retinal nonperfusion (Figs 56.2–56.6). Telangiectasia, aneurysms, beading of vessel walls, and various vascular communicating channels are found with larger vessels. These vessels show early and persistent leakage, which verifies their role as the source of exudation and hemorrhage. This leakage represents breakdown of the blood–retinal barrier, which can further be detected by vitreous fluorophotometry.61 Microvascular involvement is demonstrated with areas of diffuse loss of the capillary bed or areas of complete capillary nonperfusion on angiography. Geographic zones of microvascular involvement are typically surrounded by areas of arteriolar and venular anomalies. Although some early authors described cases of massive exudation or hemorrhage without areas of obvious vascular involvement, fluorescein angiography and histopathologic specimens invariably show unsuspected anomalous vessels. The ability to identify all anomalous vessels, especially those with the greatest degree of leakage, has made adequate treatment of Coats disease a possibility.5,57,62,63
Computed tomography
CT is valuable because of its ability to characterize intraocular morphology, quantify subretinal densities, identify vascularities within the subretinal space through the use of contrast enhancement, and detect other abnormalities that may be associated in the orbital or intracranial space. Spiral CT has the advantage of reducing anesthesia risk in small children and also decreases acquisition time and staff and equipment monitoring requirements.64
Magnetic resonance imaging
Magnetic resonance imaging (MRI) as an auxiliary test is useful because it permits multiplanar imaging and superior contrast resolution and yields biochemical insight into the structure and composition of tissues.65,66 It is less useful in detecting calcium than either ultrasound or CT scanning.60,67 An MRI study of 28 patients with leukocoria or intraocular mass, or both, found that retinoblastomas could be reliably distinguished from Coats disease, toxocariasis, and persistent hyperplastic primary vitreous.67 Calcification could not be reliably detected on MRI scanning.
Doppler ultrasonography
High-resolution Doppler ultrasound has been suggested as a diagnostic adjunct, providing unique information with real-time imaging and duplex pulse Doppler evaluation.68 This technique may delineate structural abnormalities not shown by CT or MRI.
Blood testing
Aqueous lactic dehydrogenase and isoenzyme levels have not proved valuable in distinguishing between Coats disease and retinoblastoma. Examination of subretinal fluid, although rarely used, is accurate in confirming the diagnosis of Coats disease on the basis of cholesterol crystal and pigment-laden macrophages in the absence of tumor cells.60
Differential diagnosis
The differential diagnosis of Coats disease includes other conditions that produce leukocoria or strabismus,19 including retinoblastoma,15,69 retinal detachment, persistent hyperplastic primary vitreous, congenital cataract, Norrie disease, and FEVR,70 among other diagnoses. Shields and associates reviewed 150 cases of Coats disease and found that the diagnosis was correct in 64 cases (41%).44 The mistaken referring diagnoses included retinoblastoma in 43 cases (27%), retinal detachment in 12 (8%), retinal hemorrhage in 7 (4%), toxocariasis in 4 (3%), choroidal melanoma in 2 (1%), choroidal hemangioma in 2 (1%), coloboma in 2 (1%), endophthalmitis in 2 (1%), and single cases of cytomegalovirus retinitis, retinopathy of prematurity, traumatic retinopathy, and toxoplasmosis. In 18 cases (11%) there was no submitted diagnosis.
Numerous entities have been described as presenting with a Coats-like picture, including several systemic conditions, and Coats disease can also simulate other exudative retinal conditions such as FEVR, peripheral vasculitis, Eales disease, and FSHD (Table 56.2). FEVR is an important consideration and can present with peripheral avascularity with telangiectasia and exudation. FEVR can be differentiated based on the typical presence of retinal dragging, a positive family history, and bilaterality, all of which are rare in Coats disease. A Coats-like picture of bilateral retinal telangiectasia and exudation has been described in patients with deafness and FSHD.71–73 It is believed that the retinal vascular abnormalities, muscular dystrophy, and deafness in FSHD may all be a consequence of a common mechanism based on abnormal Wnt signaling.35
Table 56.2 Clinical conditions that can present with Coats-like retinal findings
| Systemic conditions | |
| Ocular conditions that can simulate juvenile Coats disease |
A Coats-like picture associated with varied skeletal defects, cerebellar and extrapyramidal movement disorder, epileptic seizures, leukodystrophic changes, and postnatal growth failure has been referred to as “Coats plus syndrome.”74 In order to avoid confusion, the term “Coats disease” should be reserved for cases of idiopathic retinal telangiectasia associated with intraretinal exudation with or without exudative retinal detachment, without evidence of vitreoretinal traction.7,44
Isolated case reports have described a number of other disorders occurring concurrently with Coats disease, including retinitis pigmentosa,75,76 Senior–Loken syndrome,77 the ichthyosis hystrix variant of epidermal nevus syndrome,78 Turner’s syndrome,79 diffuse central nervous system venous abnormality,80 and Hallermann–Streiff syndrome81 (Table 56.2). Small, in 1968, reported the combination of mental retardation, muscular dystrophy, and an exudative vasculopathy in four siblings.71 Egerer and associates noted histologic evidence of rosettes characteristic of retinal dysplasia in a series of nine enucleated eyes carrying the diagnosis of Coats disease.82 Fogle and colleagues noted an exudative vasculopathy with a clinical picture similar to that of Coats disease arising from abnormal choroidal vessels in both eyes of a patient with retinitis pigmentosa.83 Despite these reports, no definite connection has been made between other systemic or ocular conditions and Coats disease.
Treatment
Ablative therapies – laser photocoagulation and cryotherapy
In less severe cases of exudation due to Coats disease, with or without retinal detachment, argon or diode laser photocoagulation is the treatment of choice (Fig. 56.7).84–86 Fluorescein angiographic guidance assists directed treatment of vascular leakage. Most wavelengths of laser light are adequate for treatment, although those near the yellow portion of the spectrum have better absorption by blood in the target vascular channels and may be particularly useful if the vessels are in detached retina. Leaking lesions are treated directly with moderate to large (100–500 µm, depending on size and location of the target lesions) applications of moderate-intensity light. Scatter photocoagulation to areas of extensive nonperfusion is of unproven value but could decrease the risk of later neovascularization. If lesions are too peripheral to be reached with the slit-lamp delivery system, the indirect ophthalmoscope-mounted laser, transscleral laser, or cryotherapy can be used.87 These methods may be particularly useful in small children, who usually require general anesthesia for treatment. Of note, extensive laser photocoagulation can result in transient disruption of the blood–retinal barrier, paradoxically increasing retinal exudation. Several sessions of therapy, approximately every 3 months, are often necessary to produce complete resolution of exudation or detachment.57,84,88 Complications of photocoagulation in Coats disease include inflammation, choroidal detachment, progressive exudation, creation of chorioretinal and vitreochoroidal anastomoses, epiretinal membrane formation, sympathetic ophthalmia, rhegmatogenous retinal detachment, and hemorrhage.
Double freeze–thaw cryotherapy is also useful in ablating affected areas of the retina, and is particularly helpful in cases where laser is ineffective, such as extensive subretinal exudation or retinal detachment. In some cases it may be necessary to drain subretinal fluid to obtain sufficient retinal vascular freezing. As with laser photocoagulation, many more advanced cases require multiple sessions.57 To minimize side-effects in eyes with extensive vascular abnormalities, selective treatment with cryotherapy to two or fewer quadrants per session may be advised, as well as avoidance of the ciliary body. In some cases with advanced disease and imminent glaucoma, treatment of the entire four quadrants of the retina is necessary. Compared to laser, cryotherapy is associated with more inflammation and patient discomfort as well as potential complications, which include subcapsular cataract, proliferative vitreoretinopathy, and total retinal detachment.63
The use of photodynamic therapy in combination with intravitreal bevacizumab for adult Coats disease was reported in a single case report, with apparent success.89 Diathermy also can be used as an alternative to cryotherapy or laser.
Pharmacologic therapies
Intravitreal triamcinolone acetonide (IVTA) is effective in reducing macular edema and subretinal exudation.90,91Othman and colleagues recently described their experience in 15 eyes treated with 4 mg IVTA in combination with traditional treatment modalities and followed for at least 1 year. All patients experienced an improvement in visual acuity, although 40% required cataract surgery and one patient required intraocular pressure-lowering drops.90
Intravitreal anti-VEGF agents have been reported to be effective as adjunctive therapy to reduce subretinal fluid and macular exudation in children with Coats if given alone92–94 or in combination with IVTA,95,96 laser photocoagulation, or cryotherapy.94,95,97–99 Intraocular VEGF levels are known to be substantially elevated in eyes with Coats disease, with dramatic decline following intravitreal anti-VEGF injection.100,101 In a study of four eyes with Coats disease, the intraocular VEGF levels were nearly 2400 pg/mL, compared to 15 pg/mL in five eyes with rhegmatogenous retinal detachment.101 In one eye with stage 2B disease in that report, the intraocular VEGF level decreased from 1247 pg/mL to 20.4 pg/mL 1 month after a single injection of intravitreal bevacizumab, with a corresponding improvement in visual acuity. VEGF levels may be elevated secondary to retinal ischemia associated with the abnormal vasculature.
Although in most of the available reports anti-VEGF agents appeared to be well tolerated, the case numbers are small, with limited follow-up. In the largest published series to date of eight eyes, Ramasubramanian and Shields noted the development of vitreous fibrosis in four eyes after a mean of 1.75 injections and 5 months of follow-up. In three of these patients, this progressed to tractional retinal detachment.102 Furthermore, we have no data on the long-term consequences of repeated anti-VEGF injections in children. Larger studies with longer follow-up will be necessary to establish the safety of anti-VEGF in juvenile Coats disease.
In contrast, the safety profile of anti-VEGF injections in adults is well established, and several small studies have shown the efficacy of anti-VEGF agents in adult-onset Coats with macular involvement.103,104
Surgery
Less invasive measures are usually effective in treating exudative retinal detachment with Coats disease;105 however eyes with advanced retinal detachment abutting the crystalline lens or those with a rhegmatogenous component to the detachment may benefit from surgical repair.
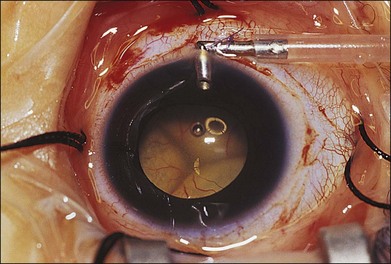
Machemer and Williams reported improvement in the clinical course of selected cases of Coats disease with vitreous surgery consisting of removal of vitreal and preretinal membranes to eliminate traction detachment, combined with destruction of leaking vessels.106 Other authors have advocated the use of vitrectomy with internal drainage of subretinal fluid to flatten the retina, before vasoablative procedures, and intraocular tamponade with gas or silicone oil.107–109 Silodor and associates have used intraocular infusion, drainage of subretinal fluid, and cryotherapy with success in eyes with advanced bullous retinal detachment.110 In their series of 13 children with blindness from Coats disease, six that were followed without surgery developed painful neovascular glaucoma necessitating enucleation and seven that were surgically repaired avoided glaucoma and the eyes remained cosmetically acceptable. When transscleral drainage of extensive subretinal fluid is necessary, a pediatric infusion cannula placed in the anterior chamber works well for maintaining the globe (Fig. 56.8).
Endstage cases associated with neovascular or angle closure glaucoma, or a blind painful eye may require enucleation.57,111 Alternatively, eyes with neovascular glaucoma may respond to transscleral diode laser cyclophotocoagulation.112
Outcomes
In a series of 103 eyes, published by Shields and associates in 2001, telangiectasia resolved completely (47%) or partially (53%) over a mean interval of 15 months following treatment.57 Following treatment, inactive telangiectasia and old exudation were found in 45% of cases at 17 months.57 Residual telangiectatic areas are often left untreated, especially if there is no fresh exudation or subretinal fluid. The goal of treatment is to close telangiectasia so that further leakage is halted. In 7% of cases by 10 years, recurrence of leakage from residual or new telangiectasia can occur.57
Anatomically, most cases (76%) stabilize or improve following treatment, with the minority (8%) progressively worsening.57 Approximately 20% require enucleation for neovascular glaucoma or painful phthisis bulbi.57 In the large series by Shields and colleagues on 150 patients, 117 individuals (124 eyes) were managed and followed for a median of 23 months and outcomes were determined. Management included observation in 22 eyes (18%), photocoagulation in 16 (13%), cryotherapy in 52 (42%), retinal detachment repair with drainage of subretinal fluid and cryotherapy or photocoagulation in 20 (17%), and enucleation in 14 (11%). Often multiple sessions were required. The median interval from initial treatment to complete resolution of telangiectasia was 10 months (range 2–123 months). Exudation resolved completely in 46 cases (45%) over a median of 12 months. Of the 88 eyes with retinal detachment prior to treatment, 50 (57%) had complete resolution of the detachment. Unfortunately, visual outcome with this condition is generally poor as foveal exudation and retinal detachment often destroy macular function. In that series, final visual acuity was 20/50 or better in 16%, 20/60 to 20/100 in 8%, 20/200 to finger counting in 29%, and hand motions to no light perception in 47%.57 Severe vision loss was due to persistent foveal exudation, retinal detachment, and/or subfoveal fibrosis. Risk factors predictive of poor visual outcome (20/200 or worse) included noncaucasian race, postequatorial (P = 0.01), diffuse (P = 0.01), or superior (P = 0.04) location of telangiectasia and exudation, failed resolution of subretinal fluid after treatment (P = 0.02), and presence of retinal macrocysts (P = 0.02). Significant risk factors for enucleation included elevated intraocular pressure (greater than 22 mm Hg; P < 0.001) and iris neovascularization (P < 0.001) at presentation.
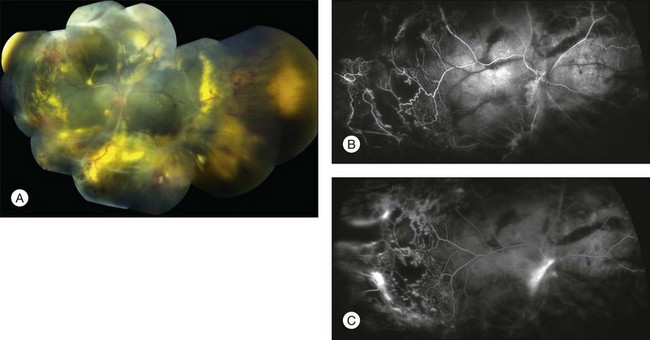
Following resolution of exudation, extensive subretinal fibrosis and pigmentation, particularly in the fovea, can limit visual recovery. Intraocular surgical intervention carries the added risks of endophthalmitis, retinal tear formation, rhegmatogenous retinal detachment, and proliferative vitreoretinopathy.5,79,113–116 Patients should be followed lifelong for recurrence (Fig. 56.9).57,117–119
1 Egbert PR, Chan CC, Winter FC. Flat preparations of the retinal vessels in Coats’ disease. J Pediatr Ophthalmol. 1976;13:336–339.
2 Coats G. Forms of retinal disease with massive exudation. R Lond Ophthalm Hosp Rep. 1908;525:440–525.
3 Leber TH. Über eine durch Vorkommen multipler Miliaraneurysmen charakterisierte Form von Retinaldegeneration. Graefes Arch Ophthalmol. 1912;81:1–14.
4 Leber TH. Retinitis exudativa (Coats), retinitis und chorioretinitis serofibrinosa degenerans. Graefe-Saemisch Augenheilk. 1915;7:1267–1319.
5 Egerer I, Tasman W, Tomer T. Coats disease. Arch Ophthalmol. 1974;92:109–112.
6 Leber T. Die Krankheiten der Netzhaut. In: Handbuch der gesamten Augenheilkunde. Leipzig: Engelmann, Graefe und Saemisch; 1916.
7 Reese AB. Telangiectasis of the retina and Coats’ disease. Am J Ophthalmol. 1956;42:1–8.
8 Bonnet M. Le syndrome de Coats. J Fr Ophthalmol. 1980;3:57–66.
9 Theodossiadis GP, Bairaktaris-Kouris E, Kouris T. Evolution of Leber’s miliary aneurysms: a clinicopathological study. J Pediatr Ophthalmol Strabismus. 1979;16:364–370.
10 Sugar HS. Coats’ disease: telangiectatic or multiple vascular origin. Am J Ophthalmol. 1958;45:508–517.
11 Wise GN. Coats’ disease. AMA Arch Ophthalmol. 1957;58:735–746.
12 Evens L, Francois J, Rabaey M, et al. [Title not available. ]. Ophthalmologica. 1956;132:1–12.
13 Imre G. Coats’ disease. Am J Ophthalmol. 1962;54:175.
14 Woods AC, Duke JR. Coats’s disease. I. Review of the literature, diagnostic criteria, clinical findings, and plasma lipid studies. Br J Ophthalmol. 1963;47:385–412.
15 Naumann GD, Portwich E. [Etiology and final clinical cause for 1000 enucleations. (A clinico-pathologic study) (author’s transl.).]. Klin Monbl Augenheilkd. 1976;168:622–630.
16 Eagle RC, Jr. Eye pathology. Philadelphia: Lippincott Williams & Wilkins; 2011. pp. 221–2
17 Hogan M, Zimmerman L. Ophthalmic pathology, an atlas and textbook. Philadelphia: WB Saunders; 1962.
18 Tripathi R, Ashton N. Electron microscopical study of Coats’ disease. Br J Ophthalmol. 1971;55:289–301.
19 Chang MM, McLean IW, Merritt JC. Coats’ disease: a study of 62 histologically confirmed cases. J Pediatr Ophthalmol Strabismus. 1984;21:163–168.
20 Duke JR, Woods AC. Coats’s disease. II. Studies on the identity of the lipids concerned, and the probable role of mucopolysaccharides in its pathogenesis. Br J Ophthalmol. 1963;47:413–434.
21 Farkas TG, Potts AM, Boone C. Some pathologic and biochemical aspects of Coats’ disease. Am J Ophthalmol. 1973;75:289–301.
22 Takei Y. Origin of ghost cell in Coats’ disease. Invest Ophthalmol. 1976;15:677–681.
23 Yannuzzi L, Gitter K, Schatz H. The macula: a comprehensive text and atlas. Baltimore: Williams & Wilkins; 1979.
24 Senft SH, Hidayat AA, Cavender JC. Atypical presentation of Coats disease. Retina. 1994;14:36–38.
25 Pe’er J. Calcifications in Coats’ disease. Am J Ophthalmol. 1988;106:742–743.
26 Ye X, Wang Y, Cahill H, et al. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298.
27 Clevers H. Eyeing up new Wnt pathway players. Cell. 2009;139:227–229.
28 Lin P, Shankar SP, Duncan J, et al. Retinal vascular abnormalities and dragged maculae in a carrier with a new NDP mutation (c. 268delC) that caused severe Norrie disease in the proband. J AAPOS. 2010;14:93–96.
29 Dickinson JL, Sale MM, Passmore A, et al. Mutations in the NDP gene: contribution to Norrie disease, familial exudative vitreoretinopathy and retinopathy of prematurity. Clin Experiment Ophthalmol. 2006;34:682–688.
30 Warden SM, Andreoli CM, Mukai S. The Wnt signaling pathway in familial exudative vitreoretinopathy and Norrie disease. Semin Ophthalmol. 2007;22:211–217.
31 Qin M, Hayashi H, Oshima K, et al. Complexity of the genotype–phenotype correlation in familial exudative vitreoretinopathy with mutations in the LRP5 and/or FZD4 genes. Hum Mutat. 2005;26:104–112.
32 Jiao X, Ventruto V, Trese MT, et al. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am J Hum Genet. 2004;75:878–884.
33 Chen ZY, Battinelli EM, Fielder A, et al. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat Genet. 1993;5:180–183.
34 Chung BD, Kayserili H, Ai M, et al. A mutation in the signal sequence of LRP5 in a family with an osteoporosis-pseudoglioma syndrome (OPPG)-like phenotype indicates a novel disease mechanism for trinucleotide repeats. Hum Mutat. 2009;30:641–648.
35 Fitzsimons RB. Retinal vascular disease and the pathogenesis of facioscapulohumeral muscular dystrophy. A signalling message from Wnt? Neuromuscul Disord. 2011;214:263–271.
36 Xu Q, Wang Y, Dabdoub A, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895.
37 Black GC, Perveen R, Bonshek R, et al. Coats’ disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Mol Genet. 1999;8:2031–2035.
38 Shastry BS, Trese MT. Overproduction and partial purification of the Norrie disease gene product, norrin, from a recombinant baculovirus. Biochem Biophys Res Commun. 2003;312:229–234.
39 Berger W, van de Pol D, Bachner D, et al. An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum Mol Genet. 1996;5:51–59.
40 Luhmann UF, Lin J, Acar N, et al. Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest Ophthalmol Vis Sci. 2005;46:3372–3382.
41 Ohlmann A, Scholz M, Goldwich A, et al. Ectopic norrin induces growth of ocular capillaries and restores normal retinal angiogenesis in Norrie disease mutant mice. J Neurosci. 2005;25:1701–1710.
42 Robitaille JM, Zheng B, Wallace K, et al. The role of Frizzled-4 mutations in familial exudative vitreoretinopathy and Coats disease. Br J Ophthalmol. 2011;95:574–579.
43 Cremers FP, Maugeri A, den Hollander AI, et al. The expanding roles of ABCA4 and CRB1 in inherited blindness. Novartis Found Symp. 2004;255:68–79. discussion 79–84, 177–8
44 Shields JA, Shields CL, Honavar SG, et al. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol. 2001;131:561–571.
45 Shields JA, Shields CL. Differentiation of coats’ disease and retinoblastoma. J Pediatr Ophthalmol Strabismus. 2001;38:262–266. quiz 302–263
46 Vance SK, Wald KJ, Sherman J, et al. Subclinical facioscapulohumeral muscular dystrophy masquerading as bilateral Coats disease in a woman. Arch Ophthalmol. 2011;129:807–809.
47 Campbell FP. Coats’ disease and congenital vascular retinopathy. Trans Am Ophthalmol Soc. 1976;74:365–424.
48 Gupta N, Beri S, D’Souza P. Cholesterolosis bulbi of the anterior chamber in Coats disease. J Pediatr Ophthalmol Strabismus. 2009:1–3.
49 Shields JA, Eagle RC, Jr., Fammartino J, et al. Coats’ disease as a cause of anterior chamber cholesterolosis. Arch Ophthalmol. 1995;113:975–977.
50 Jumper JM, Pomerleau D, McDonald HR, et al. Macular fibrosis in Coats disease. Retina. 2010;30(Suppl):S9–14.
51 Smithen LM, Brown GC, Brucker AJ, et al. Coats’ disease diagnosed in adulthood. Ophthalmology. 2005;112:1072–1078.
52 Yeung J, Harris GS. Coats’ disease: A study of cholesterol transport in the eye. Can J Ophthalmol. 1976;11:61–68.
53 Deutsch TA, Rabb MF, Jampol LM. Spontaneous regression of retinal lesions in Coats’ disease. Can J Ophthalmol. 1982;17:169–172.
54 Goel SD, Augsburger JJ. Hemorrhagic retinal macrocysts in advanced Coats disease. Retina. 1991;11:437–440.
55 Friedenwald H, Friedenwald JS. Terminal stage in a case of retinitis with massive exudation. Trans Am Ophthalmol Soc. 1929;27:188–194.
56 Cahill M, O’Keefe M, Acheson R, et al. Classification of the spectrum of Coats’ disease as subtypes of idiopathic retinal telangiectasis with exudation. Acta Ophthalmol Scand. 2001;79:596–602.
57 Shields JA, Shields CL, Honavar SG, et al. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001;131:572–583.
58 Lai CH, Kuo HK, Wu PC, et al. Manifestation of Coats’ disease by age in Taiwan. Clin Experiment Ophthalmol. 2007;35:361–365.
59 Smirniotopoulos JG, Bargallo N, Mafee MF. Differential diagnosis of leukokoria: radiologic-pathologic correlation. Radiographics. 1994;14:1059–1079. quiz 1052–81
60 Haik BG. Advanced Coats’ disease. Trans Am Ophthalmol Soc. 1991;89:371–476.
61 Cunha-Vaz JG. The blood–retinal barriers. Doc Ophthalmol. 1976;41:287–327.
62 Ridley ME, Shields JA, Brown GC, et al. Coats’ disease. Evaluation of management. Ophthalmology. 1982;89:1381–1387.
63 Tarkkanen A, Laatikainen L. Coat’s disease: clinical, angiographic, histopathological findings and clinical management. Br J Ophthalmol. 1983;67:766–776.
64 O’Brien JM, Char DH, Tucker N, et al. Efficacy of unanesthetized spiral computed tomography scanning in initial evaluation of childhood leukocoria. Ophthalmology. 1995;102:1345–1350.
65 Beets-Tan RG, Hendriks MJ, Ramos LM, et al. Retinoblastoma: CT and MRI. Neuroradiology. 1994;36:59–62.
66 Lai WW, Edward DP, Weiss RA, et al. Magnetic resonance imaging findings in a case of advanced Coats’ disease. Ophthalmic Surg Lasers. 1996;27:234–238.
67 Mafee MF, Goldberg MF, Cohen SB, et al. Magnetic resonance imaging versus computed tomography of leukocoric eyes and use of in vitro proton magnetic resonance spectroscopy of retinoblastoma. Ophthalmology. 1989;96:965–975. discussion 966–75
68 Glasier CM, Brodsky MC, Leithiser RE, Jr., et al. High resolution ultrasound with Doppler: a diagnostic adjunct in orbital and ocular lesions in children. Pediatr Radiol. 1992;22:174–178.
69 Lam HD, Samuel MA, Rao NA, et al. Retinoblastoma presenting as Coats’ disease. Eye (Lond). 2008;22:1196–1197.
70 Plager DA, Orgel IK, Ellis FD, et al. X-linked recessive familial exudative vitreoretinopathy. Am J Ophthalmol. 1992;114:145–148.
71 Small RG. Coats’ disease and muscular dystrophy. Trans Am Acad Ophthalmol Otolaryngol. 1968;72:225–231.
72 Padberg GW, Brouwer OF, de Keizer RJ, et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995;2:S73–S80.
73 Shields CL, Zahler J, Falk N, et al. Neovascular glaucoma from advanced Coats disease as the initial manifestation of facioscapulohumeral dystrophy in a 2-year-old child. Arch Ophthalmol. 2007;125:840–842.
74 Crow YJ, McMenamin J, Haenggeli CA, et al. Coats’ plus: a progressive familial syndrome of bilateral Coats’ disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics. 2004;35:10–19.
75 Khan JA, Ide CH, Strickland MP. Coats’-type retinitis pigmentosa. Surv Ophthalmol. 1988;32:317–332.
76 Pruett RC. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc. 1983;81:693–735.
77 Schuman JS, Lieberman KV, Friedman AH, et al. Senior–Loken syndrome (familial renal-retinal dystrophy) and Coats’ disease. Am J Ophthalmol. 1985;100:822–827.
78 Burch JV, Leveille AS, Morse PH. Ichthyosis hystrix (epidermal nevus syndrome) and Coats’ disease. Am J Ophthalmol. 1980;89:25–30.
79 Asdourian G. Vascular anomalies of the retina. In: Peyman G, Sanders D, Goldberg M. Principles and practices of ophthalmology. Philadelphia: Saunders, 1980.
80 Robitaille JM, Monsein L, Traboulsi EI. Coats’ disease and central nervous system venous malformation. Ophthalmic Genet. 1996;17:215–218.
81 Newell SW, Hall BD, Anderson CW, et al. Hallermann–Streiff syndrome with Coats disease. J Pediatr Ophthalmol Strabismus. 1994;31:123–125.
82 Egerer I, Rodrigues MM, Tasman WS. Retinal dysplasia in Coat’s disease. Can J Ophthalmol. 1975;10:79–85.
83 Fogle JA, Welch RB, Green WR. Retinitis pigmentosa and exudative vasculopathy. Arch Ophthalmol. 1978;96:696–702.
84 Schefler AC, Berrocal AM, Murray TG. Advanced Coats’ disease. Management with repetitive aggressive laser ablation therapy. Retina. 2008;28(Suppl):S38–S41.
85 Spitznas M, Joussen F, Wessing A. Treatment of Coats’ disease with photocoagulation. Graefes Arch Klin Exp Ophthalmol. 1976;199:31–37.
86 Shapiro MJ, Chow CC, Karth PA, et al. Effects of green diode laser in the treatment of pediatric Coats disease. Am J Ophthalmol. 2011;151:725–731. e722
87 Sneed SR, Blodi CF, Pulido JS. Treatment of Coats’ disease with the binocular indirect argon laser photocoagulator. Arch Ophthalmol. 1989;107:789–790.
88 Couvillion SS, Margolis R, Mavrofjides E, et al. Laser treatment of Coats’ disease. J Pediatr Ophthalmol Strabismus. 2005;42:367–368.
89 Kim J, Park KH, Woo SJ. Combined photodynamic therapy and intravitreal bevacizumab injection for the treatment of adult Coats’ disease: a case report. Korean J Ophthalmol. 2010;24:374–376.
90 Othman IS, Moussa M, Bouhaimed M. Management of lipid exudates in Coats disease by adjuvant intravitreal triamcinolone: effects and complications. Br J Ophthalmol. 2010;94:606–610.
91 Jarin RR, Teoh SC, Lim TH. Resolution of severe macular oedema in adult Coat’s syndrome with high-dose intravitreal triamcinolone acetonide. Eye (Lond). 2006;20:163–165.
92 Entezari M, Ramezani A, Safavizadeh L, et al. Resolution of macular edema in Coats’ disease with intravitreal bevacizumab. Indian J Ophthalmol. 2010;58:80–82.
93 Alvarez-Rivera LG, Abraham-Marin ML, Flores-Orta HJ, et al. [Coat’s disease treated with bevacizumab (Avastin).]. Arch Soc Esp Oftalmol. 2008;83:329–331.
94 Lin CJ, Hwang JF, Chen YT, et al. The effect of intravitreal bevacizumab in the treatment of Coats disease in children. Retina. 2010;30:617–622.
95 Cakir M, Cekic O, Yilmaz OF. Combined intravitreal bevacizumab and triamcinolone injection in a child with Coats disease. J AAPOS. 2008;12:309–311.
96 Bergstrom CS, Hubbard GB, 3rd. Combination intravitreal triamcinolone injection and cryotherapy for exudative retinal detachments in severe Coats disease. Retina. 2008;28(Suppl):S33–S37.
97 Venkatesh P, Mandal S, Garg S. Management of Coats disease with bevacizumab in 2 patients. Can J Ophthalmol. 2008;43:245–246.
98 Cackett P, Wong D, Cheung CM. Combined intravitreal bevacizumab and argon laser treatment for Coats’ disease. Acta Ophthalmol. 2010;88:e48–e49.
99 Kaul S, Uparkar M, Mody K, et al. Intravitreal anti-vascular endothelial growth factor agents as an adjunct in the management of Coats’ disease in children. Indian J Ophthalmol. 2010;58:76–78.
100 Sun Y, Jain A, Moshfeghi DM. Elevated vascular endothelial growth factor levels in Coats disease: rapid response to pegaptanib sodium. Graefes Arch Clin Exp Ophthalmol. 2007;45:1387–1388.
101 He YG, Wang H, Zhao B, et al. Elevated vascular endothelial growth factor level in Coats’ disease and possible therapeutic role of bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2010;248:1519–1521.
102 Ramasubramanian A, Shields CL. Bevacizumab for Coats’ disease with exudative retinal detachment and risk of vitreoretinal traction. Br J Ophthalmol. 2012;96:356–359.
103 Goel N, Kumar V, Seth A, et al. Role of intravitreal bevacizumab in adult onset Coats’ disease. Int Ophthalmol. 2011;31:183–190.
104 Jun JH, Kim YC, Kim KS. Resolution of severe macular edema in adult coats’ disease with intravitreal triamcinolone and bevacizumab injection. Korean J Ophthalmol. 2008;22:190–193.
105 Zhao T, Wang K, Ma Y, et al. Resolution of total retinal detachment in Coats’ disease with intravitreal injection of bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2011;249:1745–1746.
106 Machemer R, Williams JM, Sr. Pathogenesis and therapy of traction detachment in various retinal vascular diseases. Am J Ophthalmol. 1988;105:170–181.
107 Yoshizumi MO, Kreiger AE, Lewis H, et al. Vitrectomy techniques in late-stage Coats’-like exudative retinal detachment. Doc Ophthalmol. 1995;90:387–394.
108 Muftuoglu G, Gulkilik G. Pars plana vitrectomy in advanced coats’ disease. Case Report Ophthalmol. 2011;2:15–22.
109 Peyman GA, Dellacroce JT, Ebrahim SA. Removal of submacular exudates in a patient with coats disease: a case report. Retina. 2006;26:836–839.
110 Silodor SW, Augsburger JJ, Shields JA, et al. Natural history and management of advanced Coats’ disease. Ophthalmic Surg. 1988;19:89–93.
111 Shields JA, Shields CL. Review: coats disease: the 2001 LuEsther T. Mertz lecture. Retina. 2002;22:80–91.
112 de Silva DJ, Brookes JL. Cyclodiode treatment of neovascular glaucoma secondary to Coats’ disease. Br J Ophthalmol. 2007;91:690–691.
113 Harris GS. Coats’ disease, diagnosis and treatment. Can J Ophthalmol. 1970;5:311–320.
114 Schatz H, Burton T, Yanuzzi L, et al. Abnormal retinal and disc vessels and retinal leak. In: Schatz H, ed. Interpretation of fundus fluorescein angiography. St. Louis: Mosby, 1978.
115 Mondon H, Hamard H, Girard P, et al. [Retinal gliosis associated with Coats’ disease.]. Bull Soc Ophtalmol Fr. 1970;70:881–883.
116 Theodossiadis GP. Some clinical, fluorescein-angiographic, and therapeutic-aspects of Coats’ disease. J Pediatr Ophthalmol Strabismus. 1979;16:257–262.
117 Shienbaum G, Tasman WS. Coats disease: a lifetime disease. Retina. 2006;26:422–424.
118 Tasman W. Coats’ disease. Am Fam Physician. 1977;15:107.
119 Egerer I, Tasman W, Tomer TT. Coats disease. Arch Ophthalmol. 1974;92:109–112.
120 Matsuzaka T, Sakuragawa N, Terasawa K, et al. Facioscapulohumeral dystrophy associated with mental retardation, hearing loss, and tortuosity of retinal arterioles. J Child Neurol. 1986;1:218–223.
121 Desai UR, Sabates FN. Long-term follow-up of facioscapulohumeral muscular dystrophy and Coats’ disease. Am J Ophthalmol. 1990;110:568–569.
122 Gurwin EB, Fitzsimons RB, Sehmi KS, et al. Retinal telangiectasis in facioscapulohumeral muscular dystrophy with deafness. Arch Ophthalmol. 1985;103:1695–1700.
123 Cameron JD, Yanoff M, Frayer WC. Coats’ disease and turner’s syndrome. Am J Ophthalmol. 1974;78:852–854.
124 Folk JC, Genovese FN, Biglan AW. Coats’ disease in a patient with Cornelia de Lange syndrome. Am J Ophthalmol. 1981;91:607–610.
125 Kondra L, Cangemi FE, Pitta CG. Alport’s syndrome and retinal telangiectasia. Ann Ophthalmol. 1983;15:550–551.
126 Genkova P, Toncheva D, Tzoneva M, et al. Deletion of 13q12. 1 in a child with Coats disease. Acta Paediatr Hung. 1986;27:141–143.
127 Berger M, Lieberman KV, Schoeneman MJ, et al. Coats’ disease in a renal transplant recipient. Nephrol Dial Transplant. 1987;2:120–123.
128 Skuta GL, France TD, Stevens TS, et al. Apparent Coats’ disease and pericentric inversion of chromosome 3. Am J Ophthalmol. 1987;104:84–86.
129 Kajtar P, Mehes K. Bilateral coats retinopathy associated with aplastic anaemia and mild dyskeratotic signs. Am J Med Genet. 1994;49:374–377.
130 Bhushan M, Kumar S, Griffiths CE. Multiple glomus tumours, Coats’ disease and basic fibroblast growth factor. Br J Dermatol. 1997;137:454–456.
131 Gärtner J, Draf W. Leber’s miliary aneurysms associated with telangiectasia of the nasal mucosa. Am J Ophthalmol. 1975;79:56–58.
132 Frontali M, Stomeo C, Dallapiccola B. Osteoporosis-pseudoglioma syndrome: report of three affected sibs and an overview. Am J Med Genet. 1985;22:35–47.
133 Reynolds BC, Lemmers RJ, Tolmie J, et al. Focal segmental glomerulosclerosis, Coats’-like retinopathy, sensorineural deafness and chromosome 4 duplication: a new association. Pediatr Nephrol. 2010;25:1551–1554.
134 Luckie AP, Hamilton AM. Adult Coats’ disease in branch retinal vein occlusion. Aust N Z J Ophthalmol. 1994;22:203–206.
135 Frezzotti R, Berengo A, Guerra R, et al. Toxoplasmic Coats’ retinitis. A parasitologically proved case. Am J Ophthalmol. 1965;59:1099–1102.
136 Kremer I, Cohen S, Izhak RB, et al. An unusual case of congenital unilateral Coats’s disease associated with morning glory optic disc anomaly. Br J Ophthalmol. 1985;69:32–37.
137 Green WR. Bilateral Coats’ disease. Massive gliosis of the retina. Arch Ophthalmol. 1967;77:378–383.
138 Lanier JD, McCrary JA, 3rd., Justice J. Autosomal recessive retinitis pigmentosa and Coats disease: a presumed familial incidence. Arch Ophthalmol. 1976;94:1737–1742.
139 Spallone A, Carlevaro G, Ridling P. Autosomal dominant retinitis pigmentosa and Coats’-like disease. Int Ophthalmol. 1985;8:147–151.
140 Arrigg PG, Lahav M, Hutchins RK, et al. Pigmentary retinal degeneration and Coats’ disease: a case study. Ophthalmic Surg. 1988;19:432–436.
141 Kim RY, Kearney JJ. Coats-type retinitis pigmentosa in a 4-year-old child. Am J Ophthalmol. 1997;124:846–848.