[level-membership-for-anesthesiology-category]
Chapter 10 Clinical trials in critical care
Evidence based medicine is the conscientious, explicit, and judicious use of current best evidence in making decisions about the care of individual patients. The practice of evidence based medicine means integrating individual clinical expertise with the best available external clinical evidence from systematic research.1
RANDOMISED CLINICAL TRIALS
The result of any clinical trial may be due to three factors:
THE QUESTION TO BE ADDRESSED
Every trial should seek to answer a focused clinical question that can be clearly articulated at the outset. For example, ‘we sought to assess the influence of different volume replacement fluids on outcomes of intensive care patients’ is better expressed as the focused clinical question ‘we sought to address the hypothesis that when 4 percent albumin is compared with 0.9 percent sodium chloride (normal saline) for intravascular-fluid resuscitation in adult patients in the ICU, there is no difference in the rate of death from any cause at 28 days’.2 The focused clinical question defines the interventions to be compared, the population of patients to be studied and the primary outcome to be considered. This approach can be formalised using the PICO system. PICO stands for patient, intervention, comparison and outcome. In the example above:
Trials may be designed to answer two quite different questions about the same treatment and the design will be quite different depending on the questions to be answered. An efficacy trial seeks to determine whether a treatment will work under optimal conditions whereas an effectiveness trial seeks to determine the effects of the intervention when applied in normal clinical practice. For a detailed comparison of the features of efficacy and effectiveness trials, please see Hebert et al.3
POPULATION AND SAMPLE SIZE
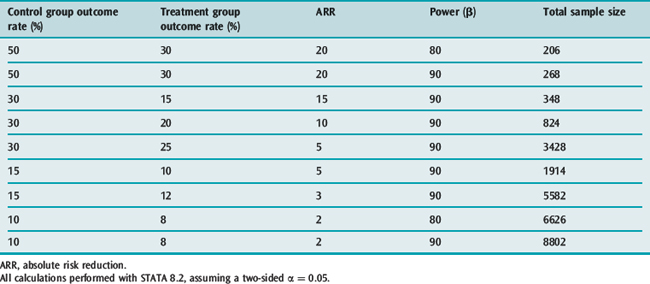
It is clear that many published trials addressing issues of importance in intensive care medicine are too small to detect clinically important treatment effects;4 fortunately this is now changing.2,5 This has almost certainly given rise to a significant number of false-negative results (type II errors). Type II errors result in potentially beneficial treatments being discarded. In order to avoid these errors, clinical trials have to include a surprisingly large numbers of participants. Examples of sample size calculations based on different baseline incidences, different treatment effects and different power are given in Table 10.1.
RANDOMISATION AND ALLOCATION CONCEALMENT
There are a number of benefits of using a random process to determine treatment allocation. Firstly, it eliminates the possibility of bias in treatment assignment (selection bias). In order for this to be ensured, both a truly random sequence of allocation must be produced and this sequence must not be known to the investigators prior to each participant entering the trial. Secondly, it reduces the chance that the trial results are affected by confounding. It is important that, prior to the intervention in a RCT being delivered, both groups have an equal chance of developing the outcome of interest. A clinical characteristic (such as advanced age, gender or disease severity, as measured by Acute Physiology, Age and Chronic Health Evaluation (APACHE) or Sequential Organ Failure Assessment (SOFA) scores) that is associated with the outcome is known as a confounding factor. Randomisation of a sufficient number of participants ensures that both known and unknown confounding factors (for example, genetic polymorphisms) are evenly distributed between the two treatment groups. The play of chance may result in uneven distribution of known confounding factors between the groups and this is particularly likely in trials with fewer than 200 participants.6 The third benefit of randomisation is that it allows the use of probability theory to quantify the role that chance could have played when differences are found between groups.7 Finally, randomisation with allocation concealment facilitates blinding, another important component in the minimisation of bias in clinical trials.8
Whatever method is used to produce a random allocation sequence, it is important that allocation concealment is maintained. Methods to ensure the concealment of allocation may be as simple as using sealed opaque envelopes,9 or as complex as the centralised automated telephone-based or web-based systems commonly used in large multicentre trials. Appropriate attention to this aspect of a clinical trial is essential as trials with poor allocation concealment produce estimates of treatment effects that may be exaggerated by up to 40%.10
THE INTERVENTIONS
The intervention being evaluated in any clinical trial should be described in sufficient detail that clinicians could implement the therapy if they so desired, or researchers could replicate the study to confirm the results. This may be a simple task if the intervention is a single drug given once at the beginning of an illness, or may be complex if the intervention being tested is the introduction of a process of care, such as the introduction of a medical emergency team.11 There are two additional areas with regard to the interventions delivered in clinical trials that require some thought by those conducting the trial and by clinicians evaluating the results, namely blinding and the control of concomitant interventions.
BLINDING
Blinding, also known as masking, is the practice of keeping trial participants (and, in the case of critically ill patients, their relatives or other legal surrogate decision-makers), care-givers, data collectors, those adjudicating outcomes and sometimes those analysing the data and writing the study reports unaware of which treatment is being given to individual participants. Blinding serves to reduce bias by preventing clinicians from consciously or unconsciously treating patients differently on the basis of their treatment assignment within the trial. It prevents data collectors from introducing bias when recording parameters that require a subjective assessment, for example pain scores and sedation scores or the Glasgow Coma Score. Although many ICU trials cannot be blinded, for example, trials of intensive insulin therapy cannot blind treating staff who are responsible for monitoring blood glucose and adjusting insulin infusion rates, the successful blinding of the Saline versus Albumin Fluid Evaluation (SAFE) trial demonstrated the possibility of blinding even large complex trials if investigators are sufficiently committed and innovative.2 Blinded outcome assessment is also necessary when the chosen outcome measure requires a subjective judgement. In such cases the outcome measure is said to be subject to the potential for ascertainment bias. For example, a blinded outcome assessment committee should adjudicate the diagnosis of ventilator-associated pneumonia (VAP) and blinded assessors should be used when assessing functional neurological recovery using the extended Glasgow Outcome Scale; both the diagnosis of VAP and assessment of the Glasgow Outcome Scale require a degree of subjective assessment and are therefore said to be prone to ascertainment bias.
It has been traditional to describe trials as single-blinded, double-blinded or even triple-blinded. However these terms can be interpreted by clinicians to mean different things, and the terminology may be confusing.12 We recommend that reports of RCTs include a description of who was blinded and how this was achieved, rather than a simple statement that the trial was ‘single-blind’ or ‘double-blind’.13 Blinding is an important safeguard against bias in RCTs, and although not thought to be as essential as maintenance of allocation concealment, empirical studies have shown that unblinded studies may produce results that are biased by as much as 17%.10
CONCOMITANT TREATMENTS
Concomitant treatments are all treatments that are administered to patients during the course of a trial other than the study treatment. With the exception of the study treatment, patients assigned to the different treatment groups should be treated equally. When one group is treated in a way that is dependent on the treatment assignment, but not directly related to the treatment, there is the possibility that this third factor will influence the outcome. An example might be a trial of pulmonary artery catheters (PACs), compared to management without a PAC. If the group assigned to receive management based on the data from a PAC received an additional daily chest X-ray to confirm the position of the PAC, they could conceivably have other important complications noted earlier, such as pneumonia, pulmonary oedema or pneumothoraces, and this may affect outcome in a fashion unrelated to the data available from the PAC. Maintaining balance in concomitant treatments is facilitated by blinding. When trials cannot be blinded, use of concomitant treatments that may alter outcome should be recorded and reported, so that the potential impact of different concomitant treatments can be assessed.
OUTCOME MEASUREMENT
A clinically meaningful outcome is a measure of how patients feel, function or survive.14 Clinically meaningful outcomes are the most credible end-points for clinical trials that seek to change clinical practice. Phase III trials should always use clinically meaningful outcomes as the primary outcome. Examples of clinically meaningful outcomes include mortality and measures of health-related quality of life. In contrast, a surrogate outcome is a substitute for a clinically meaningful outcome; a reasonable surrogate outcome would be expected to predict clinical benefits based upon epidemiologic, therapeutic, pathophysiologic or other scientific evidence.14 Examples of surrogate end-points would include cytokine levels in sepsis trials, changes in oxygenation in ventilation trials or blood pressure and urine output in a fluid resuscitation trial.
Unless a surrogate outcome has been validated, it is unwise to rely on changes in surrogate outcomes to guide clinical practice. For example, it seemed intuitively sensible that after myocardial infarction the suppression of ventricular premature beats (a surrogate outcome) which were known to be linked to mortality (the clinically meaningful outcome) would be beneficial. Unfortunately the Cardiac Arrhythmia Suppression Trial (CAST) trial found increased mortality in participants assigned to receive antiarrhythmic therapy.15 The process for determining whether a surrogate outcome is a reliable indicator of clinically meaningful outcomes has been described.16
ANALYSIS
SUBGROUP ANALYSIS
Particular difficulties arise from the selection, analysis and reporting of subgroups. Subgroups should be predefined and kept to the minimum number possible. When many subgroups are examined, the likelihood of finding a subgroup where the treatment effect is different from that seen in the overall population increases. A well-known example of this was the analysis of the treatment effect of aspirin in patients with myocardial infarction in the large second International Study of Infarct Survival (ISIS-2) trial. Overall the trial indicated that aspirin reduced the relative risk of death at 1 month by 23%. To illustrate the unreliability of subgroup analyses, the participants were divided into subgroups according to their astrological birth signs; the analysis showed that patients born under Libra or Gemini did not benefit from treatment with aspirin.17 Although it is easy to identify this as a chance subgroup finding, this may be much harder when the choice of the subgroup appears rational and a theoretical explanation for the findings can be advanced. For example, in the Gruppo Italiano per lo Studio della Streptochinasi nell’infarto miocardico (GISSI) trial, subgroup analysis suggested that fibrinolytic therapy did not reduce mortality in patients who had suffered a previous myocardial infarct.18 Although this finding appears biologically plausible, subsequent trials have shown quite clearly that fibrinolytic therapy is just as effective in patients with prior infarction as in those without.19
TESTS OF INTERACTION VERSUS WITHIN-SUBGROUP COMPARISONS
REPORTING
The reporting of RCTs has been greatly improved by the work of the Consolidated Standards of Reporting Trials (CONSORT) group.13,20 The CONSORT statement provides a framework and checklist (Table 10.2) which can be followed by investigators and authors to provide a standardised high-quality report.20 An increasing number of journals require authors to follow the CONSORT recommendations when reporting the results of an RCT. The group also recommends the publication of a structured diagram which documents the flow of patients through four stages of the trial: enrolment, allocation, follow-up and analysis (Figure 10.1). It is likely that the use of the CONSORT statement to guide the reporting of RCTs does lead to improvements, at least in the quality of reporting of RCTs.21
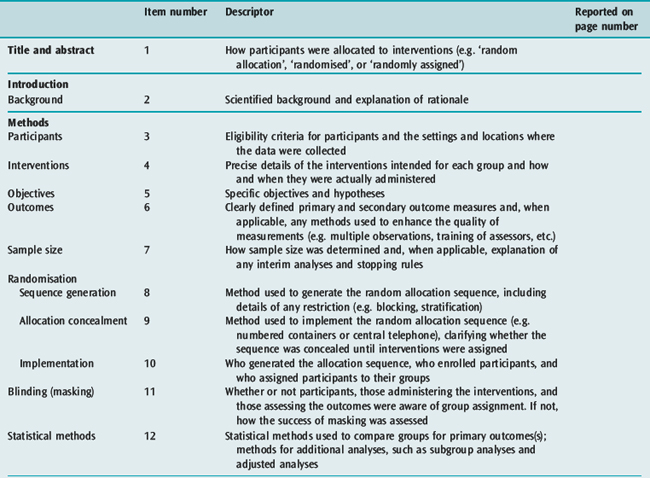
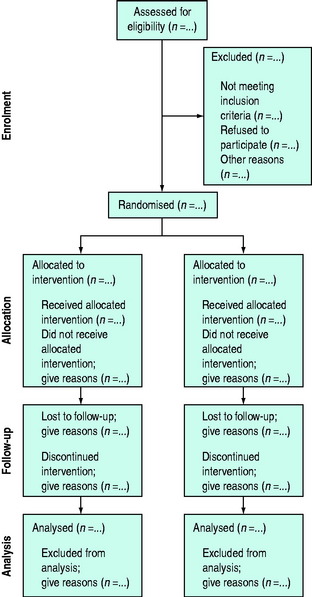
Figure 10.1 Flow diagram of the progress through the phases of a randomised trial.
(Reproduced from Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001; 357: 1191–4.)
ETHICAL ISSUES SPECIFIC TO CLINICAL TRIALS IN CRITICAL CARE
The ethical principles guiding the conduct of research in human subjects are outlined in the International Ethical Guidelines for Biomedical Research Involving Human Subjects.22 In addition country-specific guidelines are provided by various national bodies. The ethical principles of integrity, respect for persons, beneficence and justice should be considered whenever research is conducted and an appropriately convened human research ethics committee or equivalent should assess all research to ensure adherence to these principles. As the potential participants in critical care research are particularly vulnerable due to the nature of their clinical conditions and the limitations to communication that exist, special consideration needs to be given to a number of areas, including informed consent.
INFORMED CONSENT
That all mentally competent participants in clinical research should give informed consent prior to entering a study is an important ethical principle. This is rarely possible for people suffering critical illness, where the disease process (e.g. traumatic brain injury, encephalitis, severe hypoxaemia) or the required treatment (e.g. intubation, use of sedative medications) may make it impossible to obtain informed consent. Even awake, alert patients may not be able to give fully informed consent when they are facing stressful and potentially life-threatening situations.23 This applies equally to surrogate decision-makers. However, the treatment of critically ill patients can only be improved through the conduct of research and in many jurisdictions this has been recognised by making special provisions for consent in emergency research, including research in the critically ill. In some circumstances, it may be ethical to allow a waiver of consent for research involving treatments that must be given in a time-dependent fashion (e.g. in the setting of cardiac arrest). A waiver of consent may well improve recruitment into clinical trials; it is unclear if this approach is universally acceptable. Another approach has been to allow delayed consent, where patients are included in the study and consent from the patient or the relevant surrogate decision-maker is sought as soon as practical. Neither approach is without problems.24
CRITICAL APPRAISAL
Clinicians reading reports of RCTs should use a structured framework to assess the methodological quality of the trial and the adequacy of the trial report. They should also address the magnitude and precision of reported treatment effects and ask themselves whether the results of the trial can be applied to their own clinical practice. There are a number of resources available to assist clinicians in this task, notably the Users’ Guides to Evidence-Based Practice, originally published in the Journal of the American Medical Association, and the Critical Appraisal Skills programme from Oxford, UK, both of which are freely available on the internet.25,26 These resources provide a structured framework which allows any reader to perform a systematic critical appraisal of almost any piece of medical literature. A checklist is provided for the appraisal of RCTs (Table 10.3).
Table 10.3 Critical appraisal checklist for randomised controlled trials
Rights were not granted to include this table in electronic media. Please refer to the printed book.
(Reproduced from Centre for Health Evidence. Users’ guides to evidence-based practice, 2007. Avilable online at: http://www.cche.net/usersguides/main.asp.)
OBSERVATIONAL STUDIES
DESCRIPTIVE STUDIES
Case reports, case series and cross-sectional studies are all examples of descriptive studies. These types of studies may be important in the initial identification of new diseases such as human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS)27–30 and severe acute respiratory syndrome (SARS).31 The purpose of these studies will be to describe the ‘who, when, where, what and why’ of the condition, and so further the understanding of the epidemiology of the disease. It is important that clear and standardised definitions of cases are utilised, so that the information gathered can be used by clinicians and researchers to identify similar cases. While there are some famous examples where data from simple observational studies have been used to solve particular problems,32 in general only very limited inferences can be drawn from descriptive data. In particular, it is dangerous to draw conclusions about ‘cause and effect’ using data from descriptive studies alone.33
ANALYTICAL OBSERVATIONAL STUDIES
Case-control studies are performed by identifying patients with a particular condition (the ‘cases’), and a group of people who do not have the condition (the ‘controls’). The researchers then look back in time to ascertain the exposure of the members of each group to the variables of interest.34 A case-control design may be appropriate when the disease has a long latency period and is rare. Cohort studies are performed by identifying a group of people who have been exposed to a certain risk factor and a group of people who are similar in most respects apart from their exposure to the risk factor. Both groups are then followed to ascertain whether they develop the outcome of interest. Cohort studies may be the appropriate design to determine the effects of a rare exposure, and have the advantage of being able to detect multiple outcomes that are associated with the same exposure.35
Both types of observational study are prone to bias. In particular, although it is possible to correct for known confounding factors using multivariate statistical techniques, it is not possible to control for unknown or unmeasured confounding factors. There are a number of other biases that may distort the results of observational studies; these include selection bias, information bias and differential loss to follow-up.35,36 Critical appraisal guides for observational studies are available to help readers assess the validity of these studies.37 These limitations and inherent biases mean that observational studies may not always provide reliable evidence to guide clinical practice, although it has been argued that this is not always the case.38,39
SYSTEMATIC REVIEWS AND META-ANALYSIS
Systematic reviews have been proposed as a solution to the problem of the ever expanding medical literature.40 A systematic review utilises specific methods to identify and critically appraise all the RCTs that address a particular clinical question, and, if appropriate, statistically combine the results of the primary RCTs in order to arrive at an overall estimate of the effect of the treatment. By systematically assembling all RCTs that address one specific topic, a methodologically sound systematic review can provide a valuable overview for the busy clinician. Systematic reviews play an important role in providing an objective appraisal of all available evidence and may reduce the possibility that treatments with moderate effects will be discarded due to false-negative results from small or underpowered studies.41 The use of meta-analysis could have resulted in the earlier introduction of life-saving therapies such as thrombolysis.42 By using systematic methods, meta-analyses can provide more accurate and unbiased overviews, drawing conclusions that are often at odds with those of ‘experts’ and narrative reviews.43,44
In spite of these advantages and benefits there are still problems with interpretation of meta-analyses. Like all clinical trials, they need to be performed with attention to methodological detail. There are guidelines for performing and reporting systematic reviews.45,46 It is clear that in the critical care literature these guidelines are not always followed.47 Clinicians should critically appraise the reports of all systematic reviews and meta-analyses regardless of the source of the review, using an appropriate guide.48,49 Problems with interpretation can arise when the results of a meta-analysis are at odds with the results of large RCTs which address the same issue;50,51 this is not uncommon and clinicians will have to compare the methodological quality of meta-analysis and the RCTs included in the meta-analysis to the validity of the large RCT in order to decide which provides the most reliable evidence.2,52
1 Sackett DL, Rosenberg WM, Gray JA, et al. Evidence based medicine: what it is and what it isn’t. Br Med J. 1996;312:71-72.
2 The SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247-2256.
3 Hebert PC, Cook DJ, Wells G, et al. The design of randomized clinical trials in critically ill patients. Chest. 2002;121:1290-1300.
4 Roberts I, Schierhout G, Alderson P. Absence of evidence for the effectiveness of five interventions routinely used in the intensive care management of severe head injury: a systematic review. J Neurol Neurosurg Psychiatry. 1998;65:729-733.
5 Edwards P, Arango M, Balica L, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury – outcomes at 6 months. Lancet. 2005;365:1957-1959.
6 Lachin JM. Properties of simple randomization in clinical trials. Control Clin Trials. 1988;9:312-326.
7 Schulz KF, Grimes DA. Generation of allocation sequences in randomised trials: chance, not choice. Lancet. 2002;359:515-519.
8 Armitage P. The role of randomization in clinical trials. Stat Med. 1982;1:345-352.
9 Doig GS, Simpson F. Randomization and allocation concealment: a practical guide for researchers. J Crit Care. 2005;20:18-91. discussion 191–3
10 Schulz KF, Chalmers I, Hayes RJ, et al. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA. 1995;273:408-412.
11 Hillman K, Chen J, Cretikos M, et al. Introduction of the medical emergency team (MET) system: a cluster-randomised controlled trial. Lancet. 2005;365:2091-2097.
12 Montori VM, Bhandari M, Devereaux PJ, et al. In the dark: the reporting of blinding status in randomized controlled trials. J Clin Epidemiol. 2002;55:787-790.
13 Altman DG, Schulz KF, Moher D, et al. The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Ann Intern Med. 2001;134:66-94.
14 De Gruttola VG, Clax P, DeMets DL, et al. Considerations in the evaluation of surrogate endpoints in clinical trials. Summary of a National Institutes of Health workshop. Control Clin Trials. 2001;22:485-502.
15 Echt DS, Liebson PR, Mitchell LB, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781-788.
16 Bucher HC, Guyatt GH, Cook DJ, et al. Users’ guides to the medical literature: XIX. Applying clinical trial results. A. How to use an article measuring the effect of an intervention on surrogate end points. Evidence-Based Medicine Working Group. JAMA. 1999;282:771-778.
17 ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17 187 cases of suspected acute myocardial infarction: ISIS-2. Lancet. 1988;2:349-360.
18 Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI). Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet. 1986;1:397-402.
19 Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994;343:311-322.
20 Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet. 2001;357:1191-1194.
21 Plint AC, Moher D, Morrison A, et al. Does the CONSORT checklist improve the quality of reports of randomised controlled trials? A systematic review. Med J Aust. 2006;185:263-267.
22 Council for International Organizations of Medical Sciences. International ethical guidelines for biomedical research involving human subjects, 2002. Available online at: http://www.cioms.ch/frame_guidelines_nov_2002.htm
23 Wilets I, Schears RM, Gligorov N. Communicating with subjects: special challenges for resuscitation research. Acad Emerg Med. 2005;12:1060-1063.
24 Harvey SE, Elbourne D, Ashcroft J, et al. Informed consent in clinical trials in critical care: experience from the PAC-Man Study. Intens Care Med. 2006;32:2020-2025.
25 Centre for Health Evidence. Users’ guides to evidence-based practice, 2007. Avilable online at: http://www.cche.net/usersguides/main.asp
26 Learning and Development, Public Health Resource Unit. Critical Appraisal Skills Programme and Evidence-Based Practice. Oxford: Public Health Resouce Unit, 2005.
27 Masur H, Michelis MA, Greene JB, et al. An outbreak of community-acquired Pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction. N Engl J Med. 1981;305:1431-1438.
28 Gottlieb MS, Schroff R, Schanker HM, et al. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N Engl J Med. 1981;305:1425-1431.
29 Durack DT. Opportunistic infections and Kaposi’s sarcoma in homosexual men. N Engl J Med. 1981;305:1465-1467.
30 Siegal FP, Lopez C, Hammer GS, et al. Severe acquired immunodeficiency in male homosexuals, manifested by chronic perianal ulcerative herpes simplex lesions. N Engl J Med. 1981;305:1439-1444.
31 Zhong NS, Zheng BJ, Li YM, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet. 2003;362:1353-1358.
32 150th anniversary of John Snow and the pump handle. MMWR Morb Mortal Wkly Rep. 2004;53:783.
33 Grimes DA, Schulz KF. Descriptive studies: what they can and cannot do. Lancet. 2002;359:145-149.
34 Schulz KF, Grimes DA. Case-control studies: research in reverse. Lancet. 2002;359:431-434.
35 Grimes DA, Schulz KF. Cohort studies: marching towards outcomes. Lancet. 2002;359:341-345.
36 MacMahon S, Collins R. Reliable assessment of the effects of treatment on mortality and major morbidity, II: observational studies. Lancet. 2001;357:455-462.
37 Levine M, Walter S, Lee H, et al. Users’ guides to the medical literature. IV. How to use an article about harm. Evidence-Based Medicine Working Group. JAMA. 1994;271:1615-1619.
38 Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med. 2000;342:1878-1886.
39 Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med. 2000;342:1887-1892.
40 Cook DJ, Meade MO, Fink MP. How to keep up with the critical care literature and avoid being buried alive. Crit Care Med. 1996;24:1757-1768.
41 Egger M, Smith GD. Meta-analysis. Potentials and promise. Br Med J. 1997;315:1371-1374.
42 Lau J, Antman EM, Jimenez-Silva J, et al. Cumulative meta-analysis of therapeutic trials for myocardial infarction. N Engl J Med. 1992;327:248-254.
43 Antman EM, Lau J, Kupelnick B, et al. A comparison of results of meta-analyses of randomized control trials and recommendations of clinical experts. Treatments for myocardial infarction. JAMA. 1992;268:240-248.
44 Mulrow CD. The medical review article: state of the science. Ann Intern Med. 1987;106:485-488.
45 Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions. 2008 version 5.0.0 (updated February 2008). The Cochrane Collaboration Available from www.cochrane-handbook.org
46 Moher D, Cook DJ, Eastwood S, et al. Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. Quality of Reporting of Meta-analyses. Lancet. 1999;354:1896-1900.
47 Delaney A, Bagshaw SM, Ferland A, et al. A systematic evaluation of the quality of meta-analyses in the critical care literature. Crit Care. 2005;9:R5-R82.
48 Delaney A, Bagshaw SM, Ferland A, et al. The quality of reports of critical care meta-analyses in the Cochrane Database of Systematic Reviews: an independent appraisal. Crit Care Med. 2007;35:589-594.
49 Oxman AD, Cook DJ, Guyatt GH. Users’ guides to the medical literature. VI. How to use an overview. Evidence-Based Medicine Working Group. JAMA. 1994;272:1367-1371.
50 LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536-542.
51 Villar J, Carroli G, Belizan JM. Predictive ability of meta-analyses of randomised controlled trials. Lancet. 1995;345:772-776.
52 Cochrane Injuries Group Albumin Reviewers. Human albumin administration in critically ill patients: systematic review of randomised controlled trials. Br Med J. 1998;317:235-240.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 10 Clinical trials in critical care
Evidence based medicine is the conscientious, explicit, and judicious use of current best evidence in making decisions about the care of individual patients. The practice of evidence based medicine means integrating individual clinical expertise with the best available external clinical evidence from systematic research.1
RANDOMISED CLINICAL TRIALS
The result of any clinical trial may be due to three factors:
THE QUESTION TO BE ADDRESSED
Every trial should seek to answer a focused clinical question that can be clearly articulated at the outset. For example, ‘we sought to assess the influence of different volume replacement fluids on outcomes of intensive care patients’ is better expressed as the focused clinical question ‘we sought to address the hypothesis that when 4 percent albumin is compared with 0.9 percent sodium chloride (normal saline) for intravascular-fluid resuscitation in adult patients in the ICU, there is no difference in the rate of death from any cause at 28 days’.2 The focused clinical question defines the interventions to be compared, the population of patients to be studied and the primary outcome to be considered. This approach can be formalised using the PICO system. PICO stands for patient, intervention, comparison and outcome. In the example above:
Trials may be designed to answer two quite different questions about the same treatment and the design will be quite different depending on the questions to be answered. An efficacy trial seeks to determine whether a treatment will work under optimal conditions whereas an effectiveness trial seeks to determine the effects of the intervention when applied in normal clinical practice. For a detailed comparison of the features of efficacy and effectiveness trials, please see Hebert et al.3
POPULATION AND SAMPLE SIZE
It is clear that many published trials addressing issues of importance in intensive care medicine are too small to detect clinically important treatment effects;4 fortunately this is now changing.2,5 This has almost certainly given rise to a significant number of false-negative results (type II errors). Type II errors result in potentially beneficial treatments being discarded. In order to avoid these errors, clinical trials have to include a surprisingly large numbers of participants. Examples of sample size calculations based on different baseline incidences, different treatment effects and different power are given in Table 10.1.
RANDOMISATION AND ALLOCATION CONCEALMENT
There are a number of benefits of using a random process to determine treatment allocation. Firstly, it eliminates the possibility of bias in treatment assignment (selection bias). In order for this to be ensured, both a truly random sequence of allocation must be produced and this sequence must not be known to the investigators prior to each participant entering the trial. Secondly, it reduces the chance that the trial results are affected by confounding. It is important that, prior to the intervention in a RCT being delivered, both groups have an equal chance of developing the outcome of interest. A clinical characteristic (such as advanced age, gender or disease severity, as measured by Acute Physiology, Age and Chronic Health Evaluation (APACHE) or Sequential Organ Failure Assessment (SOFA) scores) that is associated with the outcome is known as a confounding factor. Randomisation of a sufficient number of participants ensures that both known and unknown confounding factors (for example, genetic polymorphisms) are evenly distributed between the two treatment groups. The play of chance may result in uneven distribution of known confounding factors between the groups and this is particularly likely in trials with fewer than 200 participants.6 The third benefit of randomisation is that it allows the use of probability theory to quantify the role that chance could have played when differences are found between groups.7 Finally, randomisation with allocation concealment facilitates blinding, another important component in the minimisation of bias in clinical trials.8
Whatever method is used to produce a random allocation sequence, it is important that allocation concealment is maintained. Methods to ensure the concealment of allocation may be as simple as using sealed opaque envelopes,9 or as complex as the centralised automated telephone-based or web-based systems commonly used in large multicentre trials. Appropriate attention to this aspect of a clinical trial is essential as trials with poor allocation concealment produce estimates of treatment effects that may be exaggerated by up to 40%.10
THE INTERVENTIONS
The intervention being evaluated in any clinical trial should be described in sufficient detail that clinicians could implement the therapy if they so desired, or researchers could replicate the study to confirm the results. This may be a simple task if the intervention is a single drug given once at the beginning of an illness, or may be complex if the intervention being tested is the introduction of a process of care, such as the introduction of a medical emergency team.11 There are two additional areas with regard to the interventions delivered in clinical trials that require some thought by those conducting the trial and by clinicians evaluating the results, namely blinding and the control of concomitant interventions.
BLINDING
Blinding, also known as masking, is the practice of keeping trial participants (and, in the case of critically ill patients, their relatives or other legal surrogate decision-makers), care-givers, data collectors, those adjudicating outcomes and sometimes those analysing the data and writing the study reports unaware of which treatment is being given to individual participants. Blinding serves to reduce bias by preventing clinicians from consciously or unconsciously treating patients differently on the basis of their treatment assignment within the trial. It prevents data collectors from introducing bias when recording parameters that require a subjective assessment, for example pain scores and sedation scores or the Glasgow Coma Score. Although many ICU trials cannot be blinded, for example, trials of intensive insulin therapy cannot blind treating staff who are responsible for monitoring blood glucose and adjusting insulin infusion rates, the successful blinding of the Saline versus Albumin Fluid Evaluation (SAFE) trial demonstrated the possibility of blinding even large complex trials if investigators are sufficiently committed and innovative.2 Blinded outcome assessment is also necessary when the chosen outcome measure requires a subjective judgement. In such cases the outcome measure is said to be subject to the potential for ascertainment bias. For example, a blinded outcome assessment committee should adjudicate the diagnosis of ventilator-associated pneumonia (VAP) and blinded assessors should be used when assessing functional neurological recovery using the extended Glasgow Outcome Scale; both the diagnosis of VAP and assessment of the Glasgow Outcome Scale require a degree of subjective assessment and are therefore said to be prone to ascertainment bias.
It has been traditional to describe trials as single-blinded, double-blinded or even triple-blinded. However these terms can be interpreted by clinicians to mean different things, and the terminology may be confusing.12 We recommend that reports of RCTs include a description of who was blinded and how this was achieved, rather than a simple statement that the trial was ‘single-blind’ or ‘double-blind’.13 Blinding is an important safeguard against bias in RCTs, and although not thought to be as essential as maintenance of allocation concealment, empirical studies have shown that unblinded studies may produce results that are biased by as much as 17%.10
CONCOMITANT TREATMENTS
Concomitant treatments are all treatments that are administered to patients during the course of a trial other than the study treatment. With the exception of the study treatment, patients assigned to the different treatment groups should be treated equally. When one group is treated in a way that is dependent on the treatment assignment, but not directly related to the treatment, there is the possibility that this third factor will influence the outcome. An example might be a trial of pulmonary artery catheters (PACs), compared to management without a PAC. If the group assigned to receive management based on the data from a PAC received an additional daily chest X-ray to confirm the position of the PAC, they could conceivably have other important complications noted earlier, such as pneumonia, pulmonary oedema or pneumothoraces, and this may affect outcome in a fashion unrelated to the data available from the PAC. Maintaining balance in concomitant treatments is facilitated by blinding. When trials cannot be blinded, use of concomitant treatments that may alter outcome should be recorded and reported, so that the potential impact of different concomitant treatments can be assessed.
OUTCOME MEASUREMENT
A clinically meaningful outcome is a measure of how patients feel, function or survive.14 Clinically meaningful outcomes are the most credible end-points for clinical trials that seek to change clinical practice. Phase III trials should always use clinically meaningful outcomes as the primary outcome. Examples of clinically meaningful outcomes include mortality and measures of health-related quality of life. In contrast, a surrogate outcome is a substitute for a clinically meaningful outcome; a reasonable surrogate outcome would be expected to predict clinical benefits based upon epidemiologic, therapeutic, pathophysiologic or other scientific evidence.14 Examples of surrogate end-points would include cytokine levels in sepsis trials, changes in oxygenation in ventilation trials or blood pressure and urine output in a fluid resuscitation trial.
[/not-level-membership-for-anesthesiology-category]
