Chapter 55A Chronic pancreatitis
Etiology, pathogenesis, and diagnosis
Overview
CP is a well-defined disease on histopathologic grounds, but histology is rarely available for diagnosis; therefore the diagnosis is reached by using a combination of clinical, laboratory, and imaging criteria. The correct diagnosis of CP is easy in late stages but difficult in early stages of the disease. Several imaging methods are available for patients with known or suspected CP. In early stages of the disease, both endoscopic retrograde pancreatography (ERP; see Chapter 18) and endoscopic ultrasonography (EUS; see Chapter 14) are methods with reliable diagnostic accuracy. Initial studies have shown superiority of EUS over ERP for the diagnosis of CP in its early stages. Transabdominal ultrasonography (US) is less sensitive for diagnosis of CP and is limited only to patients with advanced disease. In patients with an early stage of disease, we believe that EUS is the method of choice. In the absence of EUS, the combination of ERP and computed tomography (CT; see Chapter 16) provides the most reliable morphologic information. Among all imaging methods, magnetic resonance imaging (MRI), includingmagnetic resonance cholangiopancreatography (MRCP), has seen the most rapid development over the last years (see Chapter 17). With further improvement of hardware and software, it is likely that these methods will be capable of visualizing even early stages of the disease in the near future. The most common pancreatic function tests do not detect mild to moderate exocrine pancreatic insufficiency with adequate accuracy, therefore functional investigation techniques of the pancreas play only a complementary role in the routine clinical evaluation of CP but are important methods used in clinical research and in specialized pancreatic disease centers.
This chapter is divided into three sections. The first part presents the known etiologies of CP; the second part is a discussion of the most important pathogenic mechanisms of CP. The etiology and pathogenesis of CP are presented based on the TIGAR-O classification (toxic–metabolic, idiopathic, genetic, autoimmune, recurrent severe, obstructive) proposed by Etemad and Whitcomb (2001) and elegantly delineated by Stevens and colleagues (2004). It is inevitable that data must be provided on the pathogenesis of CP while presenting its etiology and vice versa; in this manner, the reader will have a better comprehension of the disease process. The third part of this chapter discusses an approach to the diagnosis of CP, with emphasis on the use of radiologic diagnostic methods, and a brief discussion of the most commonly used pancreatic function tests.
Etiology Of Chronic Pancreatitis
The classification of CP has evolved during the last several decades, and the basis for such classification stems from three consensus conferences: Marseille in 1963, Marseille-Rome in 1986, and Cambridge in 1984 (Axon et al, 1984; Sarles, 1986, 1991; Singer et al, 1985). Subdivisions of CP are based mainly on imaging characteristics, but newer classification systems such as the TIGAR-O categorize CP based on the various known etiologic factors and mechanisms jointly considered to be risk modifiers (Chari & Singer, 1994; Etemad & Whitcomb, 2001). It is likely that these risk modifiers, as well as multiple genetic and environmental cofactors, interact to produce expression of the disease in a given individual (Bourliere et al, 1991; Cavallini et al, 1994; Cavestro et al, 2003; Cohn et al, 1998; Ichimura et al, 2002; Whitcomb, 2001). It is clear that an individual, whose genetic composition is unique, will respond or not respond to an insult of similar etiology in a different morphologic pattern (inflammation or no inflammation) giving rise to disease. Response to an insult results in presence or absence of disease and progression or regression of the disease. Progression is likely to be closely related to the individual’s genetic characteristics and to ongoing environmental toxic or infectious insults. This general concept explains why individuals respond differently to the same amounts of a toxin, such as alcohol, or why less amounts of the same toxin produce disease in a susceptible individual. Furthermore, the low prevalence of CP among alcoholics would seem to suggest other cofactors at play in many with diagnosed “alcoholic” pancreatitis. In fact, the presence of multiple risk factors may be required for progression to fibrosis. The various etiologies of CP based on the TIGAR-O system (Stevens et al, 2004) provide a further advancement in the etiologic and mechanistic classification of CP, although we anticipate it will likely require revision in the future. For example, the category of “idiopathic” will tend to decrease or even disappear as other etiologies are being discovered (Jansen et al, 2002; Mahlke et al, 2005; Teich et al, 2005; Whitcomb, 2001; Whitcomb et al, 1996a; Whitcomb & Schneider, 2002; Witt et al, 2000, 2001).
Toxic and Metabolic
Table 55A.1 shows the multiple toxic and metabolic etiologies involved in CP. The association of alcohol and CP was first described almost 60 years ago by Comfort and colleagues (1946). Alcohol is still the most common cause of CP in Western industrialized countries, but interestingly, only 5% to 10% of alcoholics develop clinically apparent CP; at autopsy, 10% to 20% of alcoholics are found to have evidence of CP (Ammann, 1997; Ammann et al, 1994; Ammann et al, 1999; Angelini et al, 1993; Bernades et al, 1983; Layer & Melle, 2004; Sarles et al, 1989). Therefore, the majority of alcohol abusers never develop CP. Most patients (60% to 90%) with CP have a history of 10 to 15 years of heavy alcohol consumption, but some individuals have drunk less during a shorter period and still develop CP. This is especially true for patients who began to drink alcohol during puberty (Layer & Melle, 2004). The critical threshold of daily alcohol intake has been estimated to be about 40 g daily for women and 80 g daily for men, regardless of the quality or type of the alcoholic beverage (Layer & Melle, 2004; Papachristou & Whitcomb, 2004). Because the tolerance limits for alcohol differ substantially among individuals, smaller amounts of alcohol may be sufficient to induce pancreatic damage in susceptible individuals.
| Etiology/Mechanism of Injury | Pathogenesis |
|---|---|
AIP, autoimmune pancreatitis; PD, pancreas divisum
In the past it was believed that at the time of the initial attack, most patients with alcohol-induced CP already had underlying fibrosis and calcifications of the pancreas, but the Zürich group has demonstrated that acute attacks preceded the development of chronic disease (Ammann & Muellhaupt, 1994). Because only a fraction of alcoholics develop CP, involvements of other factors are actively being investigated. Several lines of evidence have shown that in addition to the direct effects of alcohol, various predisposing factors, such as genetics, smoking, intestinal infection, high-fat diet, compromised immune function, gallstones, gender, hormonal factors, and drinking patterns, may render the pancreas more susceptible to alcohol-induced tissue injury (Angelini et al, 1993; Lankisch et al, 2002; Levy et al, 1995; Lowenfels et al, 1994; Sahel et al, 1986). Many patients thought to have CP as a result of alcohol abuse, may indeed have a higher inherited susceptibility to alcohol-induced pancreatic damage or genetic defects that cause pancreatitis independent of alcohol exposure (Malats et al, 2001; Stevens et al, 2004).
There is convincing evidence that smoking is also independently associated with increased risk for CP, with odds ratios as high as 17.3 (Talamini et al, 1996). CP induced by smoking is particularly associated with pancreatic calcifications. By mechanisms similar to alcohol, tobacco produces alterations in the secretion and composition of pancreatic juice mainly as a result of decreased pancreatic juice and bicarbonate secretion and induction of oxidative stress (Bynum et al, 1972; Cavallini et al, 1994; Crowley-Weber et al, 2003; Stevens et al, 2004). In a large study involving 146 patients with CP, 52 patients with pancreatic cancer, and 235 healthy controls, Ockenga and colleagues (2003) analyzed the genomic DNA for expression of uridine 5′-diphospho (UDP) glucuronosyltransferase (UGT1A7) genes. These proteins are vital biochemical factors for detoxification and cell defense. The incidence of this mutation was much more common in patients with CP and tobacco abuse but not in patients with nonalcoholic CP. This study establishes the possible connection between genetic predisposition and external triggering factors. It is possible that smoking is the main factor of CP in some patients, whereas in others smoking may increase the damage induced by alcohol, and in another group it might potentiate an as yet unidentified factor or pathogen (Crowley-Weber, 2003).
Calcium plays a central role in trypsinogen secretion and trypsin stabilization. Hypercalcemia as a result of primary or secondary hyperparathyroidism results in recurrent acute pancreatitis, which progresses to CP, likely due to trypsinogen activation, which in turn results in necrosis and fibrosis of the parenchyma (Goebell, 1976; Noël-Jorand et al, 1981). Increased serum calcium concentration is believed to induce direct damage to acinar cells, and increased secretion of calcium results in intraductal stone formation. It also appears that hypercalcemia modifies pancreatic secretion and leads to protein plug formation (Goebell, 1976; Noël-Jorand et al, 1981), in turn resulting in varying degrees of pancreatic fibrosis with calcifications. This is likely due to hypercalcemia related to secondary hyperparathyroidism, but other mechanisms msy nr involved, including toxicity of uremic substances to the pancreatic parenchyma (Karanjia et al, 1992; Owyang et al, 1982).
Idiopathic
Up to 30% of patients with CP have no known risk factors; therefore the CP they have is called idiopathic. It is likely that many of these patients are mislabeled because of underreported alcohol abuse, an underlying genetic abnormality, or other unknown factors (Truninger et al, 2002; Witt, 2000). Pfützer and colleagues (2000) and Witt and colleagues (2000) have described mutations of the serine protease inhibitor Kazal type 1 (SPINK1 gene) in up to 25% of patients with idiopathic CP. Based on the bimodal age of onset of the clinical symptoms, idiopathic pancreatitis is separated into two distinct entities: Early idiopathic CP is seen during the first 2 decades of life, with abdominal pain being the predominant clinical feature, whereas pancreatic calcifications and exocrine and endocrine pancreatic insufficiency are very rare at the time of first diagnosis (Layer et al, 1994). In contrast, the clinical presentation of late-onset idiopathic CP is in patients during their fifth decade of life, usually following a rather painless course but associated with significant exocrine and endocrine pancreatic insufficiency and pancreatic calcifications (Layer et al, 1994; Papachristou & Whitcomb, 2004). Histologically, many cases of idiopathic chronic pancreatitis have T-lymphocyte infiltration, ductal obstruction, acinar atrophy, and fibrosis, raising the possibility of autoimmune etiology.
Tropical or nutritional pancreatitis is considered a form of idiopathic CP. It is the most common form of CP in certain parts of the world, such as India, sub-Saharan Africa, and Brazil, where it is a disease of youth and young adulthood (Schneider et al, 2002). The disease is subdivided into tropical calcific pancreatitis, characterized by severe recurrent and chronic abdominal pain and extensive pancreatic calcifications and fibrocalculous pancreatic diabetes with significant pancreatic endocrine insufficiency. This form of CP is clearly related to mutations in the SPINK1 gene (Chandak et al, 2002). Based on this important fact, this form of CP is likely going to be categorized as genetic. The SPINK1 pancreatic secretory trypsin inhibitory (PSTI) gene, is responsible for the encoding of SPINK1 (Chandak et al, 2002; Schneider et al, 2002). PSTI has the main function of inhibiting activated trypsin. SPINK1 is the major intrapancreatic “deactivator” of activated trypsinogen (Kukor et al, 2002; Teich et al, 2005). Trypsin has a central role in the digestion of dietary proteins and in the activation of other digestive enzymes. If the trypsin inhibitory protein malfunctions or cannot bind itself to trypsin, then trypsin is not properly deactivated or destroyed, and it remains active for a longer period of time. This is called a gain of function of trypsin (Gorry et al, 1997; Kukor et al, 2002; Pfützer et al, 2000; Sahin-Tóth, 2000; Stevens et al, 2004; Whitcomb et al, 1996a). Other genetic alterations have been described in patients with idiopathic CP. Cohn and colleagues (1998) and Durie (1998) independently demonstrated the strong association between cystic fibrosis transmembrane regulator (CFTR) mutations and idiopathic CP. In patients without evidence of cystic fibrosis, the frequency of CFTR mutations was six times that of patients without mutations. Subsequently, Cavestro and colleagues (2003) reported that one third of all patients with idiopathic CP have CFTR mutations. In the future, many patients categorized under idiopathic CP will fall into the categories of the other risk factor groups, specifically in the genetic category. In fact, leading pancreatologists speculate that most CP might be a genetic disease with multifactorial triggering factors (Papachristou & Whitcomb, 2004; Stevens et al, 2004; Whitcomb et al, 1996b).
Genetic
Until a few years ago, data on the genetic basis of CP were scarce. The only well-studied hereditary form of chronic pancreatic insufficiency was cystic fibrosis (Cohn et al, 1998; Durie, 1998). Many cases of CP represent a variable part of the cystic fibrosis syndrome, which is caused by mutations in the gene coding for the cystic fibrosis transmembrane conductance regulator. Several groups have reported an increased prevalence of CFTR mutations in patients with chronic pancreatitis of different etiologies. Later studies demonstrated that the mutations associated with cystic fibrosis (CFTR mutations) were also found with increased frequency in patients with CP (Teich et al, 2005). Interestingly, this mutation was also found to be more frequent in patients with CP thought to be secondary to pancreas divisum (Gelrud et al, 2004).
Other genetic variants predispose for CP. Research has focused on the SPINK1–N34S gene mutation, which is also closely associated with tropical (50%), alcoholic (6%), or idiopathic (20%) CP (Schneider et al, 2002; Sahin-Tóth & Tóth, 2000). One of the major discoveries in CP was the description of the point mutation in patients with autosomal dominant hereditary pancreatitis (HP) (Gorry et al, 1997). Several variants of the mutation of the cationic trypsinogen gene exist, all of which lead to a malfunction of trypsinogen (Chandak et al, 2002; Chen et al, 2000; Kukor et al, 2002; Sahin-Tóth, 2000; Teich et al, 2004). Consequently, premature intracellular activation of trypsinogen within the pancreatic acinar cell leads to activation of other enzymes, which may ultimately result in autodigestion (Kukor et al, 2002). Genetic abnormalities have been described more commonly in HP, which present typically in a bimodal pattern of childhood and adulthood (Teich et al, 2004, 2005). Associated with trypsinogen gene mutations, HP is an autosomal dominant disease that carries an 80% penetrance (Howes et al, 2004; Keim et al, 2001; Sossenheimer et al, 1997). HP is characterized by recurrent episodes of acute pancreatitis or familiar aggregation of CP, but most patients with this genetic mutation are asymptomatic (Teich et al, 2005). The progression of CP is faster in patients with SPINK-N34S mutation than in patients with cationic trypsinogen mutations (Howes et al, 2004; Keim et al, 2003). Patients with HP have a more than 50-fold increased risk of pancreatic ductal cancer compared with the general population (Howes et al, 2004; Lowenfels et al, 1987). Despite great advances in the knowledge of genetics in pancreatitis, currently it is only advised to evaluate for mutations in patients with HP. The genetic-phenotyping correlation of SPINK1 or CFTR mutations has not been studied well enough to allow for guidelines and recommendations to be established regarding its use in general clinical practice (Ellis et al, 2001; Teich et al, 2005).
Autoimmune
Autoimmune pancreatitis (AIP) is a rare but distinct form of CP characterized by specific histopathologic and immunologic features (Klöppel et al, 2005; Külling et al, 2003; Montefusco et al, 1984). In recent years, it has been established as a special type of CP whose morphologic hallmarks are periductal infiltration by lymphocytes and plasma cells and granulocytic epithelial lesions with consequent destruction of the duct epithelium and venulitis (Klöppel et al, 2003).
The pathogenesis of AIP involves both a cellular (CD4+ and CD8+ T cells) and humoral immune-mediated attack to the ductal cells and pancreatic ducts that results in cytokine-mediated inflammation and periductular fibrosis with subsequent obstruction of the pancreatic ducts (see primary duct hypothesis) (Okazaki, 2002). AIP is commonly associated with other autoimmune diseases such as Sjögren syndrome, primary sclerosing cholangitis, and inflammatory bowel disease (Külling et al, 2003; Montefusco et al, 1984). Nevertheless, more than a third of patients with AIP do not have other extrapancreatic autoimmune disorders.
AIP is clinically characterized by minimal abdominal pain and diffuse enlargement of the pancreas without calcifications or pseudocysts, and it most commonly involves the head of the pancreas and the distal bile duct. On occassion, masses have been described as inflammatory myofibroblastic tumors (Klöppel et al, 2005). On laboratory examination patients have hypergammaglobulinemia and autoantibodies, such as antinuclear and anti–smooth muscle antibodies (Bovo et al, 1987; Okazaki et al, 2000). Histopathologic examination of the pancreas reveals inflammatory infiltration of lymphocytes and plasma cells around the pancreatic duct, as well as fibrosis, in a pattern similar to primary sclerosing cholangitis (Montefusco et al, 1984; Okazaki et al, 2000).
In 2002, the Japan Pancreas Society was the first in the world to propose diagnostic criteria for autoimmune pancreatitis. These criteria were revised by an Asian consensus conference a few years later (Otsuki et al, 2008).
Obstructive
Obstruction of the main pancreatic duct is well known to result in CP. The most common etiologies causing CP as a result of obstruction of the pancreatic duct include scars of the pancreatic duct, tumors of the ampulla of Vater and head of the pancreas, and trauma (see Table 55A.1). Other disorders, such as sphincter of Oddi dysfunction and pancreas divisum, have a more tenuous connection with CP. Obstruction of the main pancreatic duct produces changes of CP within weeks in several animal models (Boerma et al, 2003; Reber et al, 1999). Main pancreatic duct obstruction may lead to stagnation and stone formation of pancreatic juice (stone and duct obstruction theory) or acute recurrent pancreatitis and periductular fibrosis (necrosis-fibrosis theory). Histopathologic characteristics of human CP as a result of obstruction include uniform distribution of interlobular and intralobular fibrosis and marked destruction of the exocrine parenchyma in the territory of obstruction, without significant protein plugs and calcifications (Suda et al, 1990). Experiments in cats with obstructive chronic pancreatitis have demonstrated impaired pancreatic blood flow in addition to elevated tissue pressure (Karanjia et al, 1992; Reber et al, 1992). In contrast to the normal hyperemic response, these cats show a decrease in blood flow after secretin injection that constitutes a form of compartment syndrome, as the normal postprandial augmentation of blood flow and oxygen supply is prevented because of decreased tissue compliance and increased interstitial pressures.
Pathogenesis Of Chronic Pancreatitis
Necrosis-Fibrosis Hypothesis
The hypothesis originally proposed by Comfort and colleagues almost 60 years ago (Comfort et al, 1946) fits most with the understanding of the pathogenesis of chronic pancreatitis. The histologic changes and pathogenesis of CP have been extensively studied by Klöppel and Maillet (1991, 1992). The hypothesis of necrosis-fibrosis views the development and course of CP as a consequence of acute pancreatitis, emphasizing that fibrosis is a late development resulting from repeated attacks of acute (alcoholic) pancreatitis, which initially lead to inflammation and necrosis (Demols, 2002; Klöppel & Maillet, 1991). Ongoing inflammation is replaced by fibrosis in the parenchyma and in the area around the pancreatic ducts, resulting in scarring and sacculations of the ducts, which in turn obstruct the flow of juice and facilitate the precipitation of proteins and subsequent calcification (Kennedy et al, 1987). This process in turn leads to further stasis, plugging, and stone formation, further obstruction, additional fibrosis, and, finally, atrophy of the gland (Klöppel & Maillet, 1992). The necrosis-fibrosis hypothesis has significant supporting evidence from epidemiologic and large follow-up studies that have shown CP to result from recurrent attacks of acute pancreatitis. Several histopathologic studies have demonstrated that mild perilobular fibrosis is common in resolving acute pancreatitis; marked fibrosis with ductal distortion is seen more frequently in patients with advanced CP (Ammann et al, 1984, 1994, 1996). In one study by Ammann and colleagues (1984), 245 patients were prospectively followed after their first episode of alcoholic pancreatitis, and investigators found that the higher the severity and frequency of attacks, the more rapid the progression to CP. Studies of ERBB2 oncogene expression also support the acute pancreatitis–CP sequence (Standop et al, 2002). The recurrent attacks of acute pancreatitis in HP are also supportive of the necrosis-fibrosis hypothesis. One important aspect that partially negates this hypothesis is the fact that the type of fibrosis that follows acute attacks of pancreatitis involves short-lived collagen type III and procollagen type IV, not the long-lasting collagen types I and IV (Casini et al, 2000).
Protein-Plug (Stone/Ductal Obstruction) Hypothesis
Sarles of France has proposed and extensively studied the hypothesis that states that CP results from plugging of the pancreatic duct (Multigner et al, 1985; Sarles, 1986). These investigators proposed that the origin of CP was within the lumen of the pancreatic ductules, in contrast to the origins of acute pancreatitis, which tends to be inside the acinar cell. The investigators proposed that increased lithogenicity of pancreatic fluid leads to the formation of eosinophilic proteinaceous aggregates that precipitate and obstruct the pancreatic ductules (Guy et al, 1983). These plugs then become rich in calcium and precipitate in the ductules because of deficiency of lithostatin or pancreatic stone protein (PSP); this protein is synthesized in the acinar cell and is an important factor in avoiding calcification within the ductules (Bimmler et al, 2001; Cavallini et al, 1998; Guy et al, 1983; Sarles, 1986; Sarles et al, 1990). Indeed, several studies have proven that alcohol decreases the formation and secretion of pancreatic juice, making it more viscous; lower in bicarbonate; richer in protein, enzymes, and calcium crystals; and deficient in lithostatin (Sarles et al, 1989; Suda et al, 1990).
The mechanism by which heavy alcohol use leads to a decrease in PSP synthesis has not been fully elucidated. Alcohol has also been shown to mediate the release of gastrointestinal hormones by increasing cholecystokinin (CCK)-releasing factor, which in turn affects pancreatic juice formation and flow. The pancreatic stones and plugs are believed to produce ulceration of the ductal epithelial cells that results in inflammation, fibrosis, obstruction, stasis, and further stone formation. Parenchymal damage in the form of inflammation and fibrosis then follows and is usually worst proximal to the obstruction (Boerma et al, 2003).
Another protein believed to induce pancreatic plug formation is glycoprotein 2 (GP-2), a major component of the zymogen granule cell membranes (Fukuoka et al, 1991). The chain of events of decreased flow, decreased lithostatin production, plug formation, calcification, stone precipitation, ductal ulceration, parenchymal and periductular inflammation, stenosis, and stasis repeats itself in a constant vicious cycle (Guy et al, 1983; Reber et al, 1992). Although pancreatic stones and plugs are found in late stages of pancreatitis, it has not been proven whether their formation represents a primary event or is only part of the sequence of CP.
Oxidative Stress Theory
Braganza (1983) from England postulated that the original pathogenic mechanism of CP was a dysregulation and overactivity of the hepatic mixed-function oxidases, mechanisms leading to oxidative stress. This theory places the major area of injury by oxidative stress at the acinar cell, usually as a result of steady exposure of xenobiotics that induce the cytochrome P-450 enzymatic system and deplete glutathione (Braganza, 1998). The hepatic mixed-function oxidases are part of the hepatic detoxification system, and as a consequence of the metabolization process, several “waste products”—such as toxic epoxides, free radicals, and lipid peroxidation products—are produced and are then released into the systemic circulation and reach the pancreatic parenchyma; or they are secreted into the bile and end up refluxing into the pancreatic duct, where they induce inflammatory damage of the acinar and ductular cell. Each burst of oxidative stress affects exocytosis, resulting in fragility of the lysozomes. Pancreatitis is triggered through interference of the methionine–glutathione transulfuration pathway, resulting in diversion of free radicals into the pancreatic tissue with subsequent activation of inflammation and fibrosis of the ductules and resultant low flow of pancreatic juice, inhibition of lithostatin, and precipitation of proteins and calcium (Braganza, 1998; Wilson et al, 1990). Alcohol may also contribute to oxidative stress as a result of the depletion of scavengers such as selenium, vitamins E and C, and riboflavin; such depletion helps to induce or propagate the damage (Atten et al, 2003; De las Heras Castano et al, 2000; Wilson & Apte, 2003).
Toxic-Metabolic Theory
Bordalo and colleagues from Portugal (1977) have proposed the toxic-metabolic hypothesis of CP. They described how alcohol and its toxic metabolites cause accumulation of intracellular lipids and fatty acid ethyl esters, which damage the acinar cell. The alterations of intracellular lipid metabolism lead to fatty degeneration, apoptosis, and scarring of the pancreatic parenchyma with impairment of the pancreatic microcirculation. In a landmark study, biopsies from 42 chronic alcoholics with and without established CP were evaluated by histology and electron microscopy (Bordalo et al, 1977). Even though many of these patients did not have CP, many changes were found to be due to cellular damage, such as cytoplasmic fat droplets of the acinar cells, decreased zymogen granules, and increased mitochondrial size.
Several studies in animals, as well as in human pancreatic tissue, have demonstrated that the toxic or metabolic insult to the fat cells (Kupffer cells) plays an important role in the pathogenesis of pancreas fibrosis in a similar fashion as Ito cells do in the liver (Jaskiewicz et al, 2003; Luttenberger et al, 2000; McCarroll et al, 2003; Phillips et al, 2003). It was demonstrated that these fat cells exist in the human pancreas; they can migrate into the periacinar spaces, and they are activated by alcohol and acetylaldehyde, transforming into scar-producing cells (Vogelmann et al, 2001; Xie et al, 2001; Yokota et al, 2002). As demonstrated in immunohistochemical analysis of pancreatic tissue, a clear correlation exists between the expression of activated Kupffer cells and the degree of fibrosis. These cells have been shown to deposit collagen very early in the process of CP (Haber et al, 1999); therefore an analogy with cirrhosis of the liver can be made when talking about macronodular and micronodular fibrosis in CP.
Primary Duct Hypothesis
Cavallini (1993) from Italy proposed that CP represents a primary autoimmune or inflammatory condition beginning in the pancreatic duct and suggests that the primary pathogenic factor leading to CP is an outflow obstruction, likely the result of duct inflammation, destruction, and fibrosis; these are likely the result of an immunologic attack to a specific genetic, structural, or acquired antigen of the periductular epithelium. The target of this attack may be some specific genetic or acquired antigen on the duct epithelium. Cavallini proposes that the immune-type mechanism occurs via two channels; one is due to aberrant expression of major histocompatibility antigens by the ductal epithelium, the other to infiltration of activated lymphocytes that produce a periductular cytotoxic response. Several reports have shown a defect of ductal epithelial aberrant expression, which leads to periductular lymphocyte infiltration (Bedossa et al, 1990; Bovo, 1987; Jaskiewicz et al, 2003). Therefore, it appears that CP is an autoimmune or “duct-destroying” disease, analogous to primary sclerosing cholangitis (PSC). This is supported by several observations: the radiologic and histologic similarity of chronic PC and PSC, the activation of cytotoxic T lymphocytes in the periductular areas of the pancreas in patients with alcoholic CP, and the occasional association of CP and PSC (Külling et al, 2003; Montefusco et al, 1984; Okazaki, 2001).
Diagnosis Of Chronic Pancreatitis
CP is a dynamic disease characterized by a progressive loss of pancreatic parenchyma caused by inflammation and tissue destruction and subsequent synthesis of fibrotic tissue (Braganza et al, 1982; DiMagno et al, 1973; Lankisch, 1993; Malfertheiner & Büchler, 1989). According to Amman, the course of the disease can be classified in three distinct stages (Table 55A.2). Stage A is the early stage, characterized by recurrent acute attacks, without any or with only mild impairment of pancreatic function. Stage B comes later in the course of the disease, when complications are seen—pseudocysts, cholestasis, segmental portal hypertension—along with increasing intensity of pain, more frequent acute painful attacks, and significantly impaired pancreatic function. Stage C represents end-stage disease, characterized by less frequent episodes and less intense pain but with marked impairment of endocrine and exocrine pancreatic function (“burnout”).
Table 55A.2 Stages of Chronic Pancreatitis: Typical Clinical and Morphologic Pictures, Pancreatic Function, and Recommended Diagnostic Procedures
In all stages of the disease, clinical symptoms, such as pain, weight loss, steatorrhea, diabetes mellitus, and local complications, can be observed in various combinations and degrees. Abdominal pain is usually located in the epigastrium and radiates to the back. Weight loss is due to two factors: at first, patients fear eating because of the accompanying pain, whereas later in the disease process, weight loss occurs as a result malabsorption related to pancreatic insufficiency (Mahlke et al, 2005). Close correlation between morphologic changes and impairment of pancreatic function is demonstrated in late stages of the disease, but in the early course of CP, no correlation is seen between morphologic changes and pancreatic function (Antman et al, 1994). In various stages of the disease, different morphologic examinations and function tests are necessary to establish the diagnosis of CP (Glasbrenner et al, 1997). In all cases, a complete staging of the disease is only possible by the assessment of the clinical setting and evaluation of morphologic changes and functional impairment. For patients presenting with advanced CP after lifelong alcohol abuse, staging is not difficult; most patients are initially seen in earlier stages of the disease, and the diagnosis of CP in these cases is challenging. Idiopathic CP is diagnosed by exclusion of known causes of CP, including nutritional or hereditary causes, hypercalcemia, trauma with residual duct injury, hyperlipidemia, autoimmunity, pancreas divisum, ampullary and duodenal diseases that cause obstructive CP, and primary pancreatic tumors that cause obstructive pancreatitis.
Two scoring systems are widely used for aiding in the diagnosis of CP: the Luneburg and Mayo Clinic scores. The Luneburg score appears to provide a more complete evaluation than the Mayo score, because it includes more aspects, such as US, EUS, CT, and indirect pancreatic function tests (Mahlke et al, 2005).
Imaging Methods
Noninvasive imaging methods are preferred for diagnosing CP in most cases (Malfertheiner & Büchler, 1989). Sensitivity and specificity of different imaging methods vary significantly, depending on the imaging modality used, the stage of the disease, and the experience of the investigator. ERP is still the gold standard among all imaging methods, but in the future, it may be replaced by other methods, such as EUS (see Chapter 14), or after a further significant refinement by magnetic resonance cholangiography (MRC; see Chapter 17).
Transabdominal Ultrasonography (See Chapter 13)
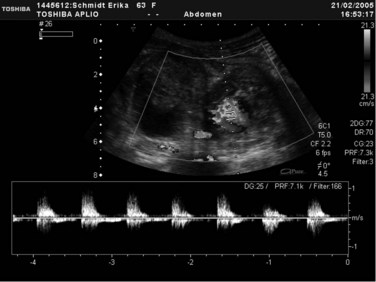
Transabdominal ultrasonography (US) is an essential tool to visualize the entire pancreas. It is useful for the detection of calcifications and pseudocysts (Figs. 55A.1 to 55A.4). It is inexpensive, simple, noninvasive, widely distributed, well tolerated, and therefore often the first imaging method in patients with abdominal complaints. The sensitivity of US to detect CP is low: it varies from 48% to 96% and increases in advanced stages of CP; the specificity is described between 75% and 90% (Bolondi et al, 1987, 1989b; Freeny & Lawson, 1982; Hessel et al, 1982; Table 55A.3). If US detects changes of CP, diagnosis is certain (high specificity), but if the pancreas is not completely visualized or appears normal, further examinations are necessary (low sensitivity). US is the easiest method to detect complications of CP and to follow up patients with CP. The major disadvantage of US is the difficult examination based on poor visualization of the pancreas, mainly because of overlying gas-filled bowel loops and technical and operator-dependent factors, such as experience of the operator and the stage of disease. Reliance on US for the diagnosis of CP is therefore limited to advanced stages (see Fig. 55A.2). Diagnostic criteria for the diagnosis of CP by transabdominal US are 1) irregular contour (lobulation), 2) pancreatic duct dilation and irregularity of the main pancreatic duct, 3) loss or reduction of pancreatic parenchyma echogenicity (echo-poor or echo-rich areas), 4) cysts or cavities, and 5) calcifications (Alpern et al, 1985; Niederau & Grendell, 1985).
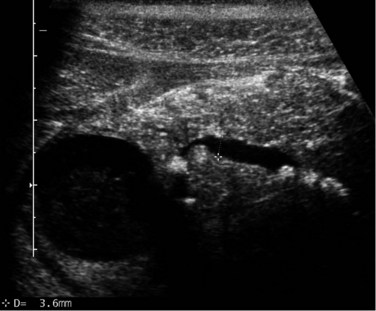
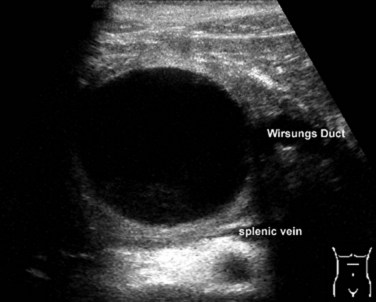
FIGURE 55A.2 Ultrasound showing a large solitary pancreatic pseudocyst complicating chronic pancreatitis.
| Sensitivity (%) | Specificity (%) | |
|---|---|---|
| Transabdominal ultrasound | 48-96 | 75-90 |
| Computed tomography | 56-95 | 85-100 |
| Endoscopic retrograde pancreatography | 68-100 | 89-100 |
| Endoscopic ultrasound | 85-100 | 85-100 |
Computed Tomography (See Chapter 16)
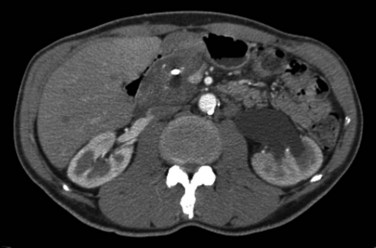
CT findings of CP include pancreatic duct dilatation, calcifications, and cystic lesions (Fig. 55A.5). Other significant findings include heterogeneous density of the pancreatic gland with atrophy or enlargement. CT is as specific as US but is more sensitive (80%). Early parenchymal changes and effects on small pancreatic ducts cannot be detected by CT, but advanced stages and complications of the disease can be evaluated with high reliability. After oral and intravenous administration of contrast medium, the pancreas can be completely visualized with spiral CT and an optimal scanning technique using a slice thickness of 5 mm (Lieb & Dragonov, 2008). In this technical setting, CT is the most sensitive method to detect calculi. Dilation of the main pancreatic duct is visualized with high sensitivity, whereas side branches are only detectable in advanced stages of the disease. CT is an excellent method to detect advanced stages, but discrete changes of the early stages of CP can be easily missed. In summary, CT shows a sensitivity of 56% to 95% with a specificity of 85% to 100% (Robinson & Sheridan, 2000; Lieb & Dragonov, 2008) (see Table 55A.3).
Endoscopic Retrograde Pancreatography (See Chapter 18)
Endoscopic retrograde pancreatography (ERP) is the gold standard among all imaging methods for diagnosis and staging of CP, because it has 90% sensitivity and 100% specificity in diagnosis (Axon et al, 1984; Forsmark & Toskes, 1984). An ERP staging system based on pancreatic ductal changes has been developed for the diagnosis of CP, and international definitions are based on ERP findings published in 1984 as the Cambridge criteria (Table 55A.4; Axon et al, 1984). Nevertheless, it is important to mention that changes of early CP may not be seen on ERP, which may assess ductal changes that occur in advanced CP, such as irregularity, dilation, tortuosity, stenosis, cysts, and calculi (see Fig. 55A.5). These findings may culminate in a “chain of lakes” appearance in the main pancreatic duct with intermittent points of obstruction in a dilated pancreatic duct (Figs. 55A.6 and 55A.7). In large multicenter trials, the sensitivity of ERP was described to be 68% to 100%, with a specificity of 89% to 100% (Braganza et al, 1982; Malfertheiner & Büchler, 1989; Sica et al, 1999).
| Stage | Typical Changes |
|---|---|
| Normal | Normal appearance of side branches and main pancreatic duct |
| Equivocal | Dilation or obstruction of less than three side branches; normal main pancreatic duct |
| Mild | Dilation or obstruction of more than three side branches; normal main pancreatic duct |
| Moderate | Additional stenosis and dilatation of main pancreatic duct |
| Severe | Additional obstructions, cysts, and stenosis of main pancreatic duct; calculi |
From Axon AT, et al, 1984: Pancreatography in chronic pancreatitis: international definitions. Gut 25:1107-1112.
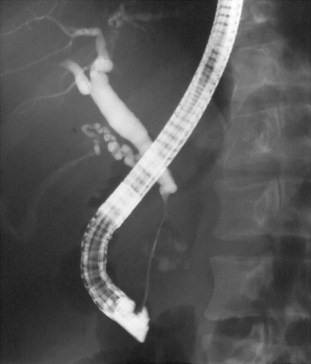
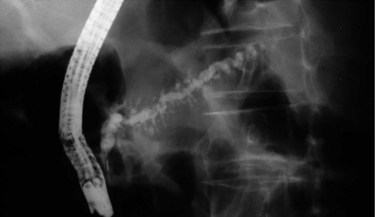
FIGURE 55A.7 Pancreatogram shows a dilated and tortuous pancreatic duct with multiple side branches.
It must be kept in mind, however, that ERP is an invasive method with a low but important rate of post-ERP pancreatitis in 3% to 7% (Sherman & Lehman, 1993). For successful ERP, specially trained personnel are necessary to perform the procedure and to interpret the pancreatograms. In special cases, typical changes of CP detected by ERP are not pathognomonic for CP: in patients examined early after AP, ductal changes might be reversible; in patients older than 65 years, these changes can be obvious without having CP (Lieb & Dragonov, 2008). In general, correlation is good between the changes observed on ERP and results of a secretin stimulation test. ERP may also be useful in distinguishing CP from pancreatic cancer. A dominant stricture as opposed to ductular ectasia—with multiple stenosis, irregular branching ducts, and intraductal calculi—is highly suggestive of pancreatic cancer rahter than CP.
Abnormal pancreatograms in patients with upper abdominal pain should not always lead to a diagnosis of CP. Occasionally, patients have suffered a previous episode of acute pancreatitis that resulted in fibrotic damage to the pancreatic duct. In these patients, it is likely that the pancreatic changes are from the initial acute inflammatory episode, and they do not have CP (Mahlke et al, 2005).
Endoscopic Ultrasonography
Endoscopic ultrasonography (EUS) visualizes both the pancreatic ducts and the parenchyma (see Chapter 14; Nattermann et al, 1993, 1995; Tenner et al, 1997; Vazquez & Wiersema, 2000; Wiersema et al, 1993). Wiersema and colleagues (1993)described diagnostic criteria of CP detectable by EUS (Stevens & Parsi, 2011; Table 55A.5). These Milwaukee criteria were further refined with the Rosemont classification (Catalano et al, 2009). Parenchymal and ductal changes were divided into major and minor criteria, based on their importance. To diagnose CP based on the Rosemont classification criteria, a sum score consisting of different major and minor changes must be calculated. The advantage of this classification system, compared with further descriptions, is to support the endosonographer with weighted criteria.
Table 55A.5 Endoscopic Ultrasound Criteria of Chronic Pancreatitis
| Parenchymal Features |
From Wiersema MJ, et al, 1993: Prospective evaluation of endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in patients with chronic abdominal pain of suspected pancreatic origin. Endoscopy 25(9):555-564.
In contrast to other imaging methods, which detect CP only in its advanced stages, EUS has the ability to detect CP in patients with early stages of the disease as well as in those with advanced CP (Wiersema et al, 1993; Mönkemüller et al, 2004). EUS features of CP include ductal and parenchymal changes, such as echotexture of the gland, calcification, lobulation, and bands of fibrosis (Figs. 55A.8 and 55A.9). A prospective evaluation comparing EUS with ERP and a secretin stimulation test in the diagnosis of CP showed good correlation in normal subjects and in patients with moderate disease, showing three or four features, or severe disease, in which more than five features are evident. The agreement was poor for mild EUS changes (one to two features) compared with ERP and a secretin stimulation test; moderate EUS criteria for CP also correlated poorly with secretin test results but had a 92% concordance with ERP in moderate disease. In summary, compared with ERP, EUS is accurate and at least as sensitive for detection of moderate to severe CP.
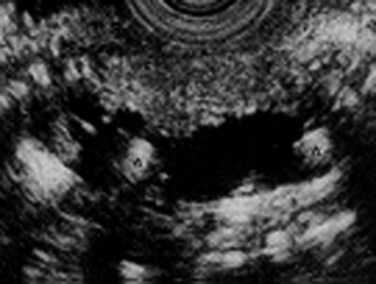
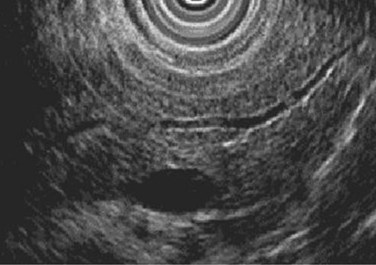
In early stages of CP, EUS has a number of advantages compared with other imaging modalities. In a series of patients with a history of chronic alcohol abuse and recurrent abdominal pain with normal ERP, we detected typical changes of CP by EUS. These patients were included in a follow-up program. After a median follow-up of 18 months, we detected typical changes of CP by repeat ERP in 68.8% of patients initially presenting with normal ERP but who showed typical changes of CP on EUS. Our conclusion is that EUS has a higher sensitivity to detect CP compared with ERP in a typical clinical and patient history setting (Kahl et al, 2002). The disadvantage of this method is the need for expert EUS endoscopists and a dedicated EUS unit. The advantages of EUS are the reliable visualization of the pancreatic parenchyma and ductal system without any risk for pancreatitis.
Tests of Exocrine Pancreatic Function
Today, function tests play only a minor, complementary role in the diagnosis of CP. The two main reasons are that noninvasive tests of exocrine pancreatic function show high sensitivity only in advanced stages of CP (Glasbrenner et al, 1997; Lankisch, 2000; Leodolter et al, 2000) and any clinical manifestation of an exocrine pancreatic insufficiency occurs late in the course of the disease, after approximately 90% of the exocrine parenchyma has been destroyed (DiMagno et al, 1973). The two types of function tests are direct (invasive) and indirect (noninvasive) (Table 55A.6; Mahlke et al, 2005). Nevertheless, one of the most sensitive tests to detect exocrine insufficiency is an invasive test, which requires duodenal intubation and aspiration of duodenal juice after pancreatic stimulation. Only this test is able to detect functional impairment in early stages of CP (Malfertheiner & Büchler, 1989), but invasive tests are time consuming, expensive, and require additional manpower and technical equipment to provide little information that is of use in the clinical routine. Therefore such tests are only used in specialized centers or to answer scientific questions. For clinical decision making, such as that regarding enzyme supplementation, pancreatic function tests can provide important information and can allow follow-up of patients with CP (Malfertheiner & Büchler, 1989). In clinical routine today, noninvasive tests, such as the pancreolauryl test and determination of fecal elastase-1 (FE-1) play a more relevant role. With the exception of secretin cerulein test function, tests of exocrine pancreatic function do not play any role in the initial diagnosis of CP because of their decreased sensitivity to detect early functional impairment (see Table 55A.6).
Table 55A.6 Sensitivity and Specificity of Imaging Methods and Tests of Exocrine Pancreatic Function
| Sensitivity (%) | Specificity (%) | |
|---|---|---|
| Invasive Tests | ||
| Spiral computed tomography | ≥90-100 | >90 |
| Noninvasive Tests | ||
| Pancreolauryl | 70-85 | 75 |
| Fecal elastase-1 | 35-85 | 83 |
Invasive Function Tests
Duodenal intubation to measure pancreatic juice production after humoral stimulation of the pancreas (secretin-cerulein test, Lundh test) is the only test able to detect functional impairment in all stages of disease (Mahlke et al, 2005). Even though this test has some limitations, it is considered the gold standard in the evaluation of exocrine pancreatic function, because of its acceptable sensitivity and specificity (75% to 95%; see Table 55A.5). The secretin stimulation test, with or without concomitant cholecystokinin (CCK 8) or cerulein administration, measures the volume of secretion and the concentration of output of bicarbonate and pancreatic enzymes via aspiration of duodenal contents in response to injection of secretin. Bicarbonate levels less than 50 mEq/L are consistent with CP; levels greater than 50 mEq/L but less than 75 mEq/L are normal. The disadvantages of this test are its invasiveness, as it requires duodenal tube insertion for collection of secretions. False-positive results may be associated with primary diabetes mellitus, Billroth II gastrectomy, celiac sprue, and cirrhosis.
Until recently, standardization of the secretin-cerulein test has not been done. Most of the investigators use a maximal stimulation of the pancreas with CCK 8 or cerulein, which leads to maximal pancreatic stimulation and output over 60 to 90 minutes (Lankisch, 1993). Direct function tests are not widely employed in clinical routine despite the fact that the costs and time involved in their performance are similar or less than imaging tests such as ERCP, MRCP, or CT (Mahlke et al, 2005).
Noninvasive Function Tests
Measurement of stool quantity and weight over a period of 72 hours has been considered essential for the diagnosis of CP with malabsorption, but this complicated and malaodorous process has not gained popularity in clinical routine (Mahlke et al, 2005). Stool tests for the quantification of fat (with subsequent correction after pancreatic enzyme replacement), chymotrypsin, and FE-1 have been well studied. Today, evaluation of stool fat plays no further role in the diagnosis of CP, because fat malabsorption occurs only in later stages, after more than 90% of the exocrine pancreatic parenchyma has been destroyed (DiMagno et al, 1973). Therefore only a minority of patients will have significant steatorrhea even with marked exocrine insufficiency (DiMagno et al, 1973). Two stool tests are commercially available for use in clinical routine: chymotrypsin (Immunodiagnostik AG, Bensheim, Germany), and Elastase-1 (ScheBo Biotech, Wettenberg, Germany, and BioServ Diagnostics, Rostock, Germany). Measurement of fecal chymotrypsin is still popular today, but the diagnostic accuracy of this test is not acceptable. Before performing this test, it is important to discontinue pancreatic enzyme supplement therapy. The best accuracy among all stool tests is obtained by measurement of FE-1. Besides high stability of pancreatic elastase during passage throughout the intestine, the commercial test measures only human elastase. This is of advantage in cases of therapeutic oral enzyme supplementation, because this lipase is of porcine origin. Even if FE-1 measurement is superior to other stool tests, its diagnostic value should not be overrated; sensitivity and specificity are low in early stages of the disease, with only mild to moderate exocrine insufficiency (Lankisch et al, 1998; Leodolter et al, 2000).
Endocrine Pancreatic Function
For clinical decision making, the differentiation between normal endocrine function, impaired oral glucose tolerance, and overt diabetes mellitus is sufficient. This differentiation succeeds with determination of fasting blood glucose level and, if normal, with an oral glucose tolerance test (Glasbrenner et al, 1997).
Alpern MB, et al. Chronic pancreatitis: ultrasonic features. Radiology. 1985;155(1):215-219.
Ammann RW. A clinically based classification system for alcoholic chronic pancreatitis: summary of an international workshop on chronic pancreatitis. Pancreas. 1997;14(3):215-221.
Ammann RW, et al. Course and outcome of chronic pancreatitis: longitudinal study of a mixed medical-surgical series of 245 patients. Gastroenterology. 1984;86(5 Pt 1):820-828.
Ammann RW, et al. Alcoholic nonprogressive chronic pancreatitis: prospective long-term study of a large cohort with alcoholic acute pancreatitis (1976-1992). Pancreas. 1994;9(3):365-373.
Ammann RW, et al. Course of alcoholic chronic pancreatitis: a prospective clinico-morphological long-term study. Gastroenterology. 1996;111(1):224-231.
Ammann RW, et al. The “two-hit” pathogenetic concept of chronic pancreatitis. Int J Pancreatol. 1999;25(3):251.
Ammann RW, Muellhaupt B. Progression of alcoholic acute to chronic pancreatitis. Gut. 1994;35(4):552-556.
Ammann RW, Muellhaupt B. The natural history of pain in alcoholic chronic pancreatitis. Gastroenterology. 1999;116(5):1132-1140.
Angelini G, et al. Long-term outcome of acute pancreatitis: a prospective study with 118 patients. Digestion. 1993;54(3):143-147.
Atten MJ, et al. Antioxidants up-regulate PPAR and decrease fibrosis in chronic pancreatitis. Am J Gastroenterol. 2003;98:A149.
Axon AT, et al. Pancreatography in chronic pancreatitis: international definitions. Gut. 1984;25(10):1107-1112.
Bedossa P, et al. Lymphocyte subset and HLA-DR expression in normal pancreas and in chronic pancreatitis. Pancreas. 1990;5(4):415-420.
Bernades P, et al. Natural history of chronic pancreatitis: a study of 120 cases. Gastroenterol Clin Biol. 1983;7(1):8-13.
Bimmler D, et al. Pancreatic stone protein (lithostathine), a physiologically relevant pancreatic calcium carbonate crystal inhibitor? J Biol Chem. 2001;272(5):3073-3082.
Boerma D, Straatsburg IH, Offerhaus GJ. Experimental model of obstructive chronic pancreatitis in pigs. Dig Surg. 2003;20(6):520-526.
Bolondi L, et al. Relationship between morphological changes detected by ultrasonography and pancreatic exocrine function in chronic pancreatitis. Pancreas. 1987;2(2):222-229.
Bolondi L, et al. Impaired response of main pancreatic duct to secretin stimulation in early chronic pancreatitis. Dig Dis Sci. 1989;34(6):834-840.
Bolondi L, et al. Sonography of chronic pancreatitis. Radiol Clin North Am. 1989;27(4):815-833.
Bordalo O, et al. Newer concept for the pathogenesis of chronic alcoholic pancreatitis. Am J Gastroenterol. 1977;68(3):278-285.
Bourliere M, et al. Is tobacco a risk factor for chronic pancreatitis and alcoholic cirrhosis? Gut. 1991;32(11):1392-1395.
Bovo P, et al. HLA molecule expression on chronic pancreatitis specimens: is there any role for autoimmunity? A preliminary study. Pancreas. 1987;2(3):350-356.
Braganza JM. A framework for the aetiogenesis of chronic pancreatitis. Digestion. 1998;59(Suppl4):1-12.
Braganza JM. Pancreatic disease: a casualty of hepatic “detoxification”? Lancet. 1983;2(8357):1000-1003.
Braganza JM, Hunt LP, Warwick F. Relationship between pancreatic exocrine function and ductal morphology in chronic pancreatitis. Gastroenterology. 1982;82(6):1341-1347.
Bynum TE, et al. Inhibition of pancreatic secretion in man by cigarette smoking. Gut. 1972;13(5):361-365.
Casini A, et al. Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J Pathol. 2000;192(1):81-89.
Catalano MF, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc. 2009;69(7):1251-1261.
Cavallini G. Is chronic pancreatitis a primary disease of the pancreatic ducts? A new pathogenetic hypothesis. Ital J Gastroenterol. 1993;25(7):391-396.
Cavallini G, et al. Effect of alcohol and smoking on pancreatic lithogenesis in the course of chronic pancreatitis. Pancreas. 1994;9(1):42-46.
Cavallini G, et al. Lithostathine messenger RNA expression in different types of chronic pancreatitis. Mol Cell Biochem. 1998;185(1-2):147-152.
Cavestro GM, et al. Association of HLA-DRB1*0401 allele with chronic pancreatitis. Pancreas. 2003;26(4):388-391.
Chandak GR, et al. Mutations in the pancreatic secretory trypsin inhibitor gene (PSTI/SPINK1) rather than the cationic trypsinogen gene (PRSS1) are significantly associated with tropical calcific pancreatitis. J Med Genet. 2002;39(5):347-351.
Chari ST, Singer MV. The problem of classification and staging of chronic pancreatitis: proposals based on current knowledge of its natural history. Scand J Gastroenterol. 1994;29(10):949-960.
Chen JM, et al. A CGC > CAT gene conversion-like event resulting in the R122H mutation in the cationic trypsinogen gene and its implication in the genotyping of pancreatitis. J Med Genet. 2000;37(11):E36.
Cohn JA, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339(10):653-658.
Comfort MW, Gambill EE, Baggenstos AM. Chronic pancreatitis: a study of 29 cases without associated disease of the biliary or gastro-intestinal tract. Gastroenterology. 1946;6:239. passim
Crowley-Weber CL, et al. Nicotine increases oxidative stress, activates NF-kappaB and GRP78, induces apoptosis and sensitizes cells to genotoxic/xenobiotic stresses by a multiple stress inducer, deoxycholate: relevance to colon carcinogenesis. Chem Biol Interact. 2003;145(1):53-66.
De las Heras Castano G, et al. Use of antioxidants to treat pain in chronic pancreatitis. Rev Esp Enferm Dig. 2000;92(6):375-385.
Demols A, et al. Endogenous interleukin-10 modulates fibrosis and regeneration in experimental chronic pancreatitis. Am J Phys. 2002;282(6):G1105-G1112.
DiMagno EP, Go VL, Summerskill WH. Relations between pancreatic enzyme ouputs and malabsorption in severe pancreatic insufficiency. N Engl J Med. 1973;288(16):813-815.
Dominguez-Muñoz JE, Malfertheiner P. Optimized serum pancreolauryl test for differentiating patients with and without chronic pancreatitis. Clin Chem. 1998;44(4):869-875.
Durie PR. Pancreatitis and mutations in the cystic fibrosis gene. N Engl J Med. 1998;339(10):687-688.
Ellis I, et al. Genetic testing for hereditary pancreatitis: guidelines for indications, counselling, consent and privacy issues. Pancreatology. 2001;1(5):405-415.
Etemad B, Whitcomb DC. Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology. 2001;120(3):682-707.
Forsmark CE, Toskes PP. What does an abnormal pancreatogram mean? Gastrointest Endosc Clin North Am. 1995;5(1):105-123.
Freeny PC, Lawson T. Radiology of the Pancreas. New York: Springer-Verlag; 1982.
Fukuoka S, Freedman S, Scheete G. A single gene encodes membrane-bound and free forms of GP2, the major glycoprotein in pancreatic secretory (zymogen) granule membranes. Proc Natl Acad Sci U S A. 1991;88(7):2898-2902.
Gelrud A, et al. Analysis of cystic fibrosis gene product (CFTR) function in patients with pancreas divisum and recurrent acute pancreatitis. Am J Gastroenterol. 2004;99(8):1557-1562.
Glasbrenner B, et al. Endocrine pancreatic function in the diagnosis and staging of chronic pancreatitis. In: Malfertheiner, P, et al. Diagnostic Procedures in Chronic Pancreatitis. Berlin/Heidelberg/New York: Springer Verlag; 1997:303-309.
Goebell H. The role of calcium in pancreatic secretion and disease. Acta Hepatogastroenterol (Stuttg). 1976;23(2):151-161.
Gorry MC, et al. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology. 1997;113(4):1063-1068.
Guy O, et al. Protein content of precipitates present in pancreatic juice of alcoholic subjects and patients with chronic calcifying pancreatitis. Gastroenterology. 1983;84(1):102-107.
Haber P, et al. Activation of pancretic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol. 1999;155(4):1087-1095.
Hessel SJ, et al. A prospective evaluation of computed tomography and ultrasound of the pancreas. Radiology. 1982;143(1):129-133.
Howes N, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2(3):252-261.
Ichimura T, et al. Primary sclerosing cholangitis associated with autoimmune pancreatitis. Hepatogastroenterol. 2002;49(47):1221-1224.
Jansen JB, et al. Genetic basis of chronic pancreatitis. Scand J Gastroenterol Suppl. 2002;236:91-94.
Jaskiewicz K, et al. Immunocytes and activated stellate cells in pancreatic fibrogenesis. Pancreas. 2003;26(3):239-242.
Kahl S, et al. Endoscopic ultrasonography in the diagnosis of early chronic pancreatitis: a prospective follow-up study. Gastrointest Endosc. 2002;55(4):507-511.
Karanjia ND, et al. Pancreatic ductal and interstitial pressures in cats with chronic pancreatitis. Dig Dis Sci. 1992;37(2):268-273.
Keim V, et al. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am J Med. 2001;111(8):622-626.
Keim V, et al. The course of genetically determined chronic pancreatitis. JOP. 2003;4(4):146-154.
Kennedy RH, et al. Pancreatic extracellular matrix alterations in chronic pancreatitis. Pancreas. 1987;2(1):61-72.
Klöppel G, Maillet B. Chronic pancreatitis: evolution of the disease. Hepatogastroenterology. 1991;38(5):408-412.
Klöppel G, Maillet B. The morphological basis for the evolution of acute pancreatitis into chronic pancreatitis. Virchows Arch A Pathol Anat Histopathol. 1992;420(1):1-4.
Klöppel G, et al. Autoimmune pancreatitis: pathological, clinical, and immunological features. Pancreas. 2003;27(1):14-19.
Klöppel G, et al. Autoimmune pancreatitis: pathological findings. JOP. 2005;6(1 Suppl):97-101.
Kukor Z, et al. Human cationic trypsinogen. Arg(117) is the reactive site of an inhibitory surface loop that controls spontaneous zymogen activation. J Biol Chem. 2002;277(8):6111-6117.
Külling D, Tresch S, Renner E. Triad of sclerosing cholangitis, chronic pancreatitis, and Sjögren’s syndrome: case report and review. Gastrointest Endosc. 2003;57(1):118-120.
1993 Lankisch PG. Function tests in the diagnosis of chronic pancreatitis: critical evaluation. Int J Pancreatol. 1993;14(1):9-20.
Lankisch PG, et al. Faecal elastase 1: not helpful in diagnosing chronic pancreatitis associated with mild to moderate exocrine pancreatic insufficiency. Gut. 1998;42(4):551-554.
Lankisch PG, et al. Fecal elastase 1 is not the indirect pancreatic function test we have been waiting for. Dig Dis Sci. 2000;45(1):166-167.
Lankisch PG, et al. Epidemiology of pancreatic diseases in Lüneburg County: a study in a defined german population. Pancreatology. 2002;2(5):469-477.
Layer P, Melle U. Chronic pancreatitis: definition and classification for clinical practice. In: Dominguez-Muñoz, JE, editor. Clinical Pancreatology for Practicing Gastroenterologists and Surgeons. Oxford, UK: Blackwell; 2004:180-191.
Layer P, et al. The different courses of early- and late-onset idiopathic and alcoholic chronic pancreatitis. Gastroenterology. 1994;107(5):1481-1487.
Leodolter A, et al. Comparison of two tubeless function tests in the assessment of mild-to- moderate exocrine pancreatic insufficiency. Eur J Gastroenterol Hepatol. 2000;12(12):1335-1338.
Levy P, et al. A multidimensional case-control study of dietary, alcohol, and tobacco habits in alcoholic men with chronic pancreatitis. Pancreas. 1995;10(3):231-238.
Lieb JG, Dragonov. Pancreatic function testing: here to stay for the 21st century. World J Gastroenterol. 2008;14(20):3149-3158.
Lowenfels AB, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1987;89(6):442-446.
Lowenfels AB, et al. Prognosis of chronic pancreatitis: an international multicenter study. International Pancreatitis Study Group. Am J Gastroenterol. 1994;89(9):1467-1471.
Luttenberger T, et al. Platelet-derived growth factor stimulates proliferation and extracellular matrix synthesis of pancreatic stellate cells: Implications in pathogenesis of pancreas fibrosis. Lab Invest. 2000;80(1):47-55.
Mahlke R, Lübbers H, Lankisch PG. Diagnosis and therapy of chronic pancreatitis. Internist. 2005;46(2):145-156.
Malats N, et al. Cystic fibrosis transmembrane regulator (CFTR) DeltaF508 mutation and 5T allele in patients with chronic pancreatitis and exocrine pancreatic cancer. PANKRAS II Study Group. Gut. 2001;48(1):70-74.
Malfertheiner P, Büchler MW. Correlation of imaging and function in chronic pancreatitis. Radiol Clin North Am. 1989;27(1):51-64.
Malfertheiner P, et al. Fluorescein dilaurate serum test: a rapid tubeless pancreatic function test. Pancreas. 1987;2(1):53-60.
Malfertheiner P, et al. Pancreatic morphology and function in relationship to pain in chronic pancreatitis. Int J Pancreatol. 1987;2(1):59-66.
McCarroll JA, et al. Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas. 2003;27(2):150-160.
Mönkemüller K, Kahl S, Malfertheiner P. Endoscopic therapy of chronic pancreatitis. Dig Dis Sci. 2004;22:280-291.
Montefusco PP, et al. Sclerosing cholangitis, chronic pancreatitis, and Sjogren’s syndrome: A syndrome complex. Am J Surg. 1984;147(6):822-826.
Multigner L, et al. Pancreatic stone protein II: implication in stone formation during the course of chronic calcifying pancreatitis. Gastroenterology. 1985;89(2):387-391.
Nattermann C, Goldschmidt AJ, Dancygier H. Endosonography in chronic pancreatitis: a comparison between endoscopic retrograde pancreatography and endoscopic ultrasonography. Endoscopy. 1993;25(9):565-570.
Nattermann C, Goldschmidt AJ, Dancygier H. [Endosonography in the assessment of pancreatic tumors. A comparison of the endosonographic findings of carcinomas and segmental inflammatory changes.]. Dtsch Med Wochenschr (in German). 1995;120(46):1571-1576.
Niederau C, Grendell JH. Diagnosis of chronic pancreatitis. Gastroenterology. 1985;88(6):1973-1995.
Noël-Jorand MC, Verine HJ, Sarles H. Dose-dependent and long-lasting effects of repeated intravenous injections of calcium on canine secretin-stimulated pancreatic juice secretion. Eur J Clin Invest. 1981;11(1):25-31.
Ockenga J, et al. UDP glucuronosyltransferase (UGT1A7) gene polymorphisms increase the risk of chronic pancreatitis and pancreatic cancer. Gastroenterology. 2003;124(7):1802-1808.
Okazaki K. Autoimmune-related pancreatitis. Curr Treat Options Gastroenterol. 2001;4(5):369-375.
Okazaki K. Clinical relevance of autoimmune-related pancreatitis. Best Pract Res Clin Gastroenterol. 2002;16(3):365-378.
Okazaki K, et al. Autoimmune-related pancreatitis is associated with autoantibodies and Th1/Th2-type cellular immune response. Gastroenterology. 2000;118(3):573-581.
Otsuki M, et al. Asian diagnostic criteria for autoimmune pancreatitis: consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis. J Gastroenterol. 2008;43(6):403-408.
Owyang C, et al. Pancreatic exocrine function in severe human chronic renal failure. Gut. 1982;23(5):357-361.
Papachristou GI, Whitcomb DC. Etiopathogenesis of chronic pancreatitis: a genetic disease with some precipitating factors? In: Dominguez-Muñoz, JE, editor. Clinical Pancreatology for Practicing Gastroenterologists and Surgeons. Oxford, UK: Blackwell Publishing; 2004:192-200.
Pfützer RH, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology. 2000;119(3):615-623.
Phillips PA, et al. Cell migration: a novel aspect of pancreatic stellate cell biology. Gut. 2003;52(5):677-682.
Reber HA, et al. Pancreatic blood flow in cats with chronic pancreatitis. Gastroenterology. 1992;103(2):652-659.
Reber PU, et al. Feline model of chronic obstructive pancreatitis: effects of acute pancreatic duct decompression on blood flow and interstitial pH. Scand J Gastroenterol. 1999;34(4):439-444.
Robinson PJ, Sheridan MB. Pancreatitis: computed tomography and magnetic resonance imaging. Eur Radiol. 2000;10(3):401-408.
Sahai AV, et al. The decision-making value of magnetic resonance cholangiopancreatography in patients seen in a referral center for suspected biliary and pancreatic disease. Am J Gastroenterol. 2001;96(7):2074-2080.
Sahel J, et al. Multicenter pathological study of chronic pancreatitis. Morphological regional variations and differences between chronic calcifying pancreatitis and obstructive pancreatitis. Pancreas. 1986;1(6):471-477.
Sahin-Tóth M. Human cationic trypsinogen: role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem. 2000;275:22750-22755.
Sahin-Tóth M, Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun. 2000;278(2):286-289.
Sarles H. Etiopathogenesis and definition of chronic pancreatitis. Dig Dis Sci. 1986;31(9 Suppl):91S-107S.
Sarles H. Definitions and classifications of pancreatitis. Pancreas. 1991;6(4):470-474.
Sarles H, Bernard JP, Johnson C. Pathogenesis and epidemiology of chronic pancreatitis. Ann Rev Med. 1989;40:453-468.
Sarles H, et al. Renaming pancreatic stone protein as “lithostathine.”. Gastroenterology. 1990;99(3):900-901.
Schneider A, et al. SPINK1/PSTI mutations are associated with tropical pancreatitis and type II diabetes mellitus in Bangladesh. Gastroenterology. 2002;123(4):1026-1030.
Sica GT, et al. Comparison of endoscopic retrograde cholangiopancreatography with MR cholangiopancreatography in patients with pancreatitis. Radiology. 1999;210(3):605-610.
Sherman S, Lehman GA. Endoscopic therapy of pancreatic disease. Gastroenterologist. 1993;1(1):5-17.
Singer MV, Gyr K, Sarles H. Revised classification of pancreatitis: report of the Second International Symposium on the Classification of Pancreatitis in Marseille, France, March 28-30, 1984. Gastroenterology. 1985;89(3):683-685.
Sossenheimer MJ, et al. Clinical characteristics of hereditary pancreatitis in a large family, based on high-risk haplotype: the Midwest Multicenter Pancreatic Study Group (MMPSG). Am J Gastroenterol. 1997;92(7):1113-1116.
Soto JA, et al. MR cholangiopancreatography: findings on 3D fast spin-echo imaging. Am J Roentgenol. 1995;165(6):1397-1401.
Soto JA, et al. Pancreatic duct: MR cholangiopancreatography with a three-dimensional fast spin-echo technique. Radiology. 1995;196(2):459-464.
Standop J, et al. ErbB2 oncogene expression supports the acute pancreatitis-chronic pancreatitis sequence. Virchows Arch. 2002;441(4):385-391.
Stevens T, Conwell DL, Zuccaro G. Pathogenesis of chronic pancreatitis: an evidence-based review of past theories and recent developments. Am J Gastroenterol. 2004;99(11):2256-2270.
Suda K, et al. Histopathologic and immunohistochemical studies on alcoholic pancreatitis and chronic obstructive pancreatitis: special emphasis on ductal obstruction and genesis of pancreatitis. Am J Gastroenterol. 1990;85(3):271-276.
Takehara Y, et al. Breath-hold MR cholangiopancreatography with a long-echo-train fast spin-echo sequence and a surface coil in chronic pancreatitis. Radiology. 1994;192(1):73-78.
Talamini G, et al. Cigarette smoking: an independent risk factor in alcoholic pancreatitis. Pancreas. 1996;12(2):131-137.
Teich N, Keim V, Mossner J. Clinical implications of genetic risk factors of chronic pancreatitis. Internist. 2005;46(2):123-130.
Teich N, et al. Interaction between trypsinogen isoforms in genetically determined pancreatitis: mutation E79K in cationic trypsin (PRSS1) causes increased transactivation of anionic trypsinogen (PRSS2). Hum Mutat. 2004;23(1):22-31.
Tenner SM, et al. Evaluation of pancreatic disease by endoscopic ultrasonography. Am J Gastroenterol. 1997;92(1):18-26.
Truninger K, et al. Mutations of the serine protease inhibitor, Kazal type 1 gene, in patients with idiopathic chronic pancreatitis. Am J Gastroenterol. 2002;97(5):1133-1137.
Vazquez SE, Wiersema MJ. The role of endoscopic ultrasonography in diagnosis, staging, and management of pancreatic disease states. Curr Gastroenterol Rep. 2000;2(2):125-132.
Vogelmann R, et al. Effects of fibrogenic mediators of the development of pancreatic fibrosis in a TGF-1 transgenic mouse-model. Am J Physiol Gastrointest Liver Physiol. 2001;280(1):G164-G172.
Whithcomb DC. Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut. 1999;45(3):317-322.
Whitcomb DC. Hereditary pancreatitis: a model for understanding the genetic basis of acute and chronic pancreatitis. Pancreatology. 2001;1(6):565-570.
Whitcomb DC, Schneider A. Hereditary pancreatitis: a model for inflammatory disease of the pancreas. Best Pract Res Clin Gastroenterol. 2002;16(3):347-363.
Whitcomb DC, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14(2):141-145.
Whitcomb DC, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology. 1996;110(6):1975-1980.
Witt H, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25(2):213-216.
Witt H, et al. Mutation in the SPINK1 trypsin inhibitor gene, alcohol use, and chronic pancreatitis. JAMA. 2001;285(21):2716-2717.
Wilson JS, Apte MV. Role of alcohol metabolism in alcoholic pancreatitis. Pancreas. 2003;27(4):311-315.
Wilson JS, et al. Both ethanol consumption and protein deficiency increase the fragility of pancreatic lysosomes. J Lab Clin Med. 1990;115(6):749-755.
Xie MJ, et al. Expression of tumor necrosis factor-alpha, interleukin-6, and interferon-gamma in spontaneous chronic pancreatitis in the WBN/Kob rat. Pancreas. 2001;22(4):400-408.
Yokota T, et al. Pancreatic stellate cell activation and MMP production in experimental pancreatic fibrosis. J Sur Res. 2002;104(2):106-111.