[level-membership-for-critical-care-medicine-category]56
Chronic Kidney Disease
Introduction
Chronic kidney disease (CKD) is a common diagnosis in adult medicine.1–6 In order to properly identify patients at risk for adverse outcomes and disease progression, extensive work has focused on early and appropriate classification over the last 10 years. In particular, the National Kidney Foundation (NKF) proposed a classification system for CKD in 2002 using estimated glomerular filtration rate (eGFR) as a method to categorize patients.7,8 Table 56.1 describes current NKF guidelines of CKD. In regard to critical care, CKD patients have higher mortality rate following critical illnesses.9 Understanding this population’s physiology and underlying disease state may aid caregivers in attempting to improve this outcome.
Table 56.1
| Stage | eGFR (mL/min/1.73 m2) |
Urinalysis Findings |
| 1 | ≥90 | Hematuria, proteinuria, or imaging abnormalities at >3 months |
| 2 | 60-89 | Hematuria, proteinuria, or imaging abnormalities at >3 months |
| 3 | 30-59 | ↑ or normal |
| 4 | 15-29 | ↑ or normal |
| 5 | 0-14 | ↑ or normal |
Definition and Etiology
As previously stated, currently CKD is staged in a five-tier system based on eGFR (see Table 56.1). The diagnosis relies on abnormal kidney function or urinalysis findings for 3 months or more. The serum creatinine as well as age, sex, gender, and race is used to generate an eGFR from a quadratic equation derived from the Modification of Diet in Renal Disease Study.7 Most CKD patients do not progress to end-stage renal disease (ESRD), however, because of the very high cardiovascular and noncardiovascular mortality rate associated with this at-risk population.10–13 Proposals exist to more clearly define elderly subjects with CKD stage 3 disease at risk for disease progression, though this practice has not become standard.14–16
Causes for CKD are broad and often multifactorial. Frequently, however, diabetes mellitus and hypertension play a major role. Hypertension may lead to kidney damage in the form of nephrosclerosis or chronic renal ischemia due to atherosclerotic vascular disease. An important recent development is the recognition that the higher prevalence of nondiabetic kidney disease in the African-American population may be due to genetic risk conferred by inherited variants in the apolipoprotein L1 gene (APOL1).17 Additional causes include immune-mediated glomerular diseases such as systemic lupus erythematosus, IgA nephropathy, membranous nephropathy, and other often pre-existing glomerulonephritides. Glomerulonephritis may be active as evidenced by hematuria and proteinuria or may have occurred in the patient’s past, leading to abnormal kidney function but a relatively bland urinalysis. Alternatively, primary tubulointerstitial diseases could exist owing to reflux nephropathy, sarcoidosis, chronic infections, allergic reactions, or side effects from medications such as cyclophosphamide (Cytoxan) and lithium. In general, the diagnosis of CKD should warrant a higher index of suspicion for concurrent, and potentially undiagnosed, cardiovascular disease (CVD).
The CKD population is at risk for deterioration in kidney function during a hospitalization stay, particularly if a critical illness is present. Potential causative factors for this are listed in Box 56.1. Importantly, acute tubular necrosis may occur without obvious hypotension, and patients with reduced kidney function are more at risk for this complication.18
Diagnosis
Intrinsic to the eGFR formula, as well as other estimates of renal function from the serum creatinine, is the presumption that a patient’s serum creatinine is stable. This finding may not be the case in ICU patients; as a result, imprecision should be expected in kidney function estimation in ICU patients.19 Serum creatinine is now recognized to potentially underestimate the level of renal function in a large population of patients, particularly those with low weight, small muscle mass, and liver disease.20 Additionally, both endogenous and exogenous factors can influence the measurement of serum creatinine.21–24 Box 56.2 describes situations in which serum creatinine measurements may not adequately reflect kidney function. Importantly, some vasoactive substances can impart a negative interference on creatinine values if the creatinine sample is obtained from a central line used for vasopressors.25 Nonetheless, creatinine variation has improved since values have been standardized to reference values.26,27
In general, current guidelines recognize the importance of stage 3 through 5 kidney disease for prognostication in outpatient and inpatient outcomes. Proteinuria is now associated with increased cardiovascular risk at all glomerular filtration rates (GFRs), including normal GFRs.28 Even mild kidney dysfunction, defined typically as a serum creatinine level over 1.5 mg/dL or GFR below 60 mL/minute, is associated with worse outcomes after coronary artery bypass graft (CABG), cardiac valvular, and general surgery.29–33
Concern exists about the accuracy of the Modification of Diet in Renal Disease (MDRD) equation at higher GFRs. For example, the newer creatinine-based CKD-EPI formula equation leads to a lower prevalence rate of CKD stage 3 than the MDRD equation, as more patients have an eGFR above 60 mL/minute.34–36 Measurement of serum cystatin C levels, a protein produced by all nucleated cells and freely filtered by the glomerulus, may provide more accurate estimates of kidney function than creatinine-based equations, particularly when kidney function is close to normal.37 Cystatin C may also provide prognostic information about future cardiovascular events.12 In fact, cystatin C has been proposed as a marker of “preclinical” kidney disease based on worse cardiovascular outcomes in elderly subjects with higher cystatin C levels.38 Despite a growing body of literature, use of cystatin C is not routine in clinical practice.
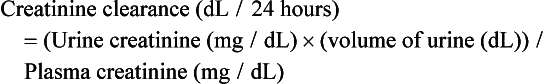
In this equation, units will need to be converted from dL/24 hours to mL/minute by a conversion factor of 0.0694.
If this measure is pursued, clinicians are counseled to pay close attention to units as well as the expectation that men excrete 1500 to 2500 mg/day of creatinine and women excrete 1000 to 1500 mg/day of creatinine.39 If a lower amount of urinary creatinine is obtained, the possibility of an undercollection should be considered. It is also worthwhile to note that historically, drug dosing has been determined by creatinine clearance and not eGFR.
Physiology
Hypertension
Over 80% of CKD patients with eGFR less than 60 mL/minute have hypertension.5 This has several important impacts on the management of ICU patients. First, longstanding hypertension, if uncontrolled, leads to adaptations in autoregulation. Autoregulation refers to the ability of blood vessels to constrict and dilate in the presence of hypertension and hypotension, respectively. Consequently, patients become acclimated to BPs near their “typical” BP and may not tolerate lower BPs despite these apparent lower values appearing in the normal range. A consequence of this may be hypoperfusion to the kidneys as well as the brain, heart, intestine, and other critical organs at BP levels that appear normal. If a history of poorly controlled hypertension is obtained, an astute clinician should aim to keep BP close to the level the patient being treated is used to. This very much requires individualized attention and thorough history taking.
Second, CKD leads to sodium retention.40,41 The most notable ICU impact this will have is the potential for volume retention and the need for diuretics to manage hypertension in the CKD population.42–44 Several mechanisms lead to the development of hypertension in the CKD population. They include upregulation of the renin-angiotensin system, increased sympathetic nervous system activity, and impairment of endothelial-dependent arterial smooth muscle relaxation.45–51 Owing to the preceding mechanisms, CKD patients may develop more edema and volume overload with equivalent amounts of intravenous (IV) fluids. One worthwhile consideration, however, is that sodium retention associated with CKD may be counterbalanced by sodium loss from excessively high BP levels. This phenomenon, known as pressure natriuresis, may explain the rapid drops observed in BP in hypertension emergencies once excess sympathetic activity is controlled.
Electrolyte Disorders
Sodium
Sodium homeostasis in patients with CKD is abnormal. With increased loss of sodium in the urine, but declining numbers of functioning nephrons, sodium balance is maintained.52 Though CKD patients are sodium “avid” and tend toward states of volume overload, they are less able to increase sodium retention in times of need and subsequently are also at risk for volume depletion. In comparison, subjects with normal renal function can quickly increase sodium absorption and avoid volume-depleted states.53 Tubulointerstitial disease and obstructive uropathy may be particularly at risk for episodes of volume depletion due to their recognized inability to increase sodium reabsorption in times of volume depletion.54
When volume overload is present, iatrogenic sources may be playing a role. Potential sources of sodium include maintenance IV fluids (including bicarbonate), antibiotics, and nutritional sources including total parenteral nutrition (TPN). Tracking fluid intake and output (“Is and Os”) does not clearly differentiate between the electrolyte makeup of the various sources. Consequently, volume overload may develop insidiously. When volume overload develops, the standard approach involves minimizing sodium-containing sources and initiating a diuretic regimen. Diuretics range from the more gentle thiazide diuretics, which are ineffective with a GFR below 30 mL/minute, to more potent loop diuretics. In general, if significant volume removal is desired, a diuretic regimen twice a day (every 12 hours) should be prescribed to avoid excess sodium reabsorption in the period after the major diuretic effect.55 Chronic administration of diuretics, prior to ICU admission, may lead to adaptive processes necessitating higher diuretic doses, combination therapy, or alternative approaches to volume overload states.56–59 Torsemide is the loop diuretic that may be closest to lasting 24 hours.60 Doses of loop diuretics need to be increased as renal function declines. Significant variability in loop diuretic kinetics exist; if a dose does not yield an increase in urine output within 2 hours, consideration should be given to increasing the dose. Alternatively, continuous infusion of loop diuretics may provide a better diuresis with less toxicity.61,62 Typically infusion rates of furosemide are 10 to 40 mg/hour. When a loop diuretic drip is ordered, a bolus should be given prior to initiating therapy to avoid a significant delay in efficacy.
Diuretic failure is common in the CKD population. Potential explanations in an ICU population include acute kidney injury, inadequate dosing, and distal tubular hypertrophy due to chronic diuretic use. When high-dose loop diuretics do not achieve adequate diuresis, the addition of metolazone may improve urine output. Historically, metolazone is given 30 minutes prior to the loop diuretics, though it may be efficacious if given at other times as well. Finally, isolated ultrafiltration or potentially dialysis may be required for volume removal. Significant attention has been given to the importance of elevated intra-abdominal pressures leading to a reduction in renal function and, potentially, ability to respond to diuretics.63–68
Water
Water concentration and dilution are abnormal in CKD patients. Normal patients can dilute and concentrate urine within a range of 40 to 1400 mOsm/kg.69 CKD leads to a narrower range of urine osmolality. Specifically, patients with advanced CKD have a urine osmolality much closer to 300 mOsm/kg; the ability to adjust urine osmolality declines as urine function worsens.70 Consequently, patients with CKD are prone both to hypernatremia and hyponatremia. Loop diuretics may further worsen the kidney’s ability to concentrate and dilute urine.71
The approach to hypernatremia and hyponatremia is similar to that in other patients. In hypernatremia cases, concurrent illnesses often lead to patients being unable to obtain water for themselves; a high urine output due to hyperglycemia or a high urea concentration may worsen this. Elevated urea, as with high serum glucose levels, functions as an osmolar agent in the tubular lumen and creates a state of relative antidiuretic hormone (ADH) resistance with the potential for substantial free water loss.72
Hypernatremia can lead to significant agitation and should be treated aggressively with increased water either intravenously, in the form of hypotonic IV fluids, or enterally. When severe, an estimation of free water deficit is appropriate. Unexplained polyuria with a low urine osmolality, hypernatremia, and CKD in an ICU patient with unclear history should warrant an investigation for previous lithium use.73
Hyponatremia reflects irregularities in water excretion in the vast majority of cases. CKD patients are less able to excrete water owing to the inability to lower urine osmolality.74 Sources of water include IV fluids, medications mixed in dextrose, and enteral sources. Patients often are not aware or not forthcoming in the amount of water they drink, though this is less an issue in ICU patients. The approach for most CKD patients is similar to that in other cases of hyponatremia, which involve looking for and reducing causes of increased water intake. Importantly, sodium abnormalities in the CKD patient, both with and without congestive heart failure, are associated with a higher mortality rate.75
Potassium
Potassium (K+) regulation is an essential function of the kidneys in normal states. CKD patients routinely maintain serum K+ in the normal range despite losing up to 90% of renal function.76,77 Mechanisms underlying the maintenance of normal serum K+ in advanced CKD include increased K+ excretion per functioning nephron, increased gastrointestinal (GI) elimination of K+, and increased uptake of K+ by cells.78 Diabetes mellitus, however, may exacerbate the development of hyperkalemia due to the presence of a distal tubular acidosis (type 4) from aldosterone resistance. Hypokalemia may also be present due to current diuretic use or renovascular hypertension.79 Its presence is associated with a higher risk of death and future ESRD needs in the CKD population.80
ICU patients with CKD represent a population prone to irregularities in K+ homeostasis, in particular, hyperkalemia.81,82 Potential causes of hyperkalemia include insulin deficiency, hypoaldosteronism, and a loss of normal intestinal function, which plays a role in increased potassium excretion in CKD patients. Decreased blood flow to muscle and intestines in times of critical illness may impede potassium cellular uptake and excretion, respectively. Tissue ischemia from trauma, hypoxia, or tumor lysis is associated with an elevation in K+ which may occur quickly.83 One additional clinical consideration is that CKD patients who are fasting may develop surprising levels of hyperkalemia without any apparent cause due to low levels of insulin.84 Glucose-containing IV fluids may avoid this.
The evaluation of hyperkalemia in CKD patients begins with assessing the cause. Typically, this includes close scrutiny of medications, IV fluids, and types of nutritional support. Frequently, ICU patients receive nutrition very different from what the body is used to and cannot manage a higher load of K+, especially in states of relative tissue perfusion. Importantly, many CKD patients are on diuretics as part of their routine outpatient medications; simply stopping these medications may lead to states of hyperkalemia. Hyperosmolality leads to a K+ shift out of cells caused by solvent drag; in its most common presentation, hyperglycemia, this can be reversed quickly with the use of insulin. Metabolic acidosis may be associated with hyperkalemia though organic acidosis states appear to cause less hyperkalemia than nonorganic acid–induced states.85
Medications are an important and growing cause of hyperkalemia. Congestive heart failure and proteinuric renal disease remain as two indications for possible dual renin-angiotensin-aldosterone blockade; hyperkalemia has been observed more frequently since the publication of the RALES trial.86 Box 56.3 lists some common causes of hyperkalemia in the CKD population and the associated mechanisms.87 The presence of hyperkalemia should warrant close attention to affected patients as its presence is associated with increased mortality rate.88
Metabolic Acidosis
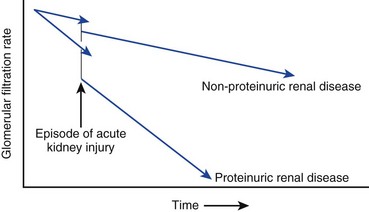
The kidneys and lungs regulate acid excretion by the body. Kidney excretion of acid occurs through two mechanisms: as protons bound to anions such as phosphate and through ammoniagenesis. As kidney function declines below a GFR of 30 mL/minute, the excess excretion of protons by each remaining functional nephron is no longer able to keep serum bicarbonate in the normal range. As a result, bicarbonate levels in CKD patients frequently range in the 18 to 22 mmol/L range due to a modest increase in the serum anion gap.89 A recent trial showed sodium bicarbonate, supplemented orally, could delay kidney disease progression (Fig. 56.1).90
In ICU CKD patients, serum bicarbonate and anion gaps are often further worsened by concurrent conditions including lactic acidosis, ketoacidosis, or dilution due to aggressive volume resuscitation. The latter case, dilutional acidosis, will present with a worsening serum bicarbonate in the days following admission to the ICU and institution of IV fluids; the anion gap should not change significantly. In general, an anion gap over 20 should warrant a workup for other causes of increased acid generation. Medications may also worsen acidosis in the CKD population owing to disturbances in ammonia production as well as excess acid production in the form of lactic acid, keto acids, and pyroglutamic acids.91,92
Mineral Bone Disorder
Substantial information has developed in the last several years about the mechanisms leading to abnormal calcium and bone metabolism in patients with CKD. Historically, this disorder was named “renal osteodystrophy.” The current preferred term is CKD mineral bone disorder.93 The main developments reflects the growing importance of FGF-23 on phosphate and parathyroid regulation and the relaxation of guidelines for strict parathyroid hormone (PTH) control at least in the ESRD population.94
FGF-23 is a phosphaturic protein produced by bone. Phosphaturia occurs as a result of decreased expression of NaPi-2a cotransporters in renal proximal tubular cells.95 In addition, FGF-23 leads to a reduction in 1,25(OH)2D production by the kidney.96 Its concentration increases earlier in the course of CKD than PTH. These adaptations initially keep calcium and phosphorus levels well maintained despite progressively increased serum FGF-23 and PTH levels.
From an ICU patient perspective, hypocalcemia will routinely be encountered owing to the preceding developments. Hypocalcemia, in the absence of other causes, suggests renal disease is chronic. Serum phosphorus levels may similarly be elevated as GFR declines. Dietary phosphorus restriction and phosphorus binders are the primary treatments for this. There are limited prospective data evaluating the best management of CKD mineral bone disorder, and therapy is for the most part dictated by nonrandomized studies and consensus guidelines. Elevations of FGF-23 are associated with a higher risk of mortality across multiple levels of kidney function.97 Important issues for anyone caring for CKD patients with abnormalities of calcium bone deposition in the ICU include the following:
• Avoidance of phosphorus-containing GI stimulants because of risk of systemic absorption and acute kidney injury
• Recognition that hypocalcemia may be chronic: typically, patients become adapted to this with a normal ionized calcium due to concurrent metabolic acidosis. Rapidly raising pH may lower serum calcium levels acutely and predispose patients to arrhythmias.98
• Consideration for other causes of hypocalcemia and hyperphosphatemia, in particular, cell death from rhabdomyolysis and tumor lysis syndrome
Anemia and Hematologic Disease
Erythropoietin is produced by the kidney and stimulates hematopoiesis in the bone marrow. CKD is associated with a reduction in erythropoietin levels and consequent anemia. As CKD progresses toward ESRD, anemia becomes more severe.99 As a general rule, most ESRD patients receive supplemental erythropoietin to increase their hemoglobin (Hb) level and avoid transfusions.
Two large trials published in 2006 significantly altered the management of anemia of CKD.100,101 The CHOIR and CREATE trials both showed, in a well-designed format, that randomization to a normal Hb did not improve outcomes and increased some adverse events. As a result, goal Hb levels have dropped closer to 10.0 g/dL in general practice and the use of these medications has fallen substantially. These trials reinforced earlier data that showed that aiming for a normal Hb, achieved through supplemental erythropoietin, in the CKD population was dangerous.102 Similar results were noted with darbapoietin alpha.103 These studies clearly show adverse events from supraphysiologic Hbs in CKD patients. The impact of relative reductions in Hb in critically ill CKD patients is not well studied.
In CKD with critical illnesses the problems of anemia are often magnified. How the anemia of CKD impacts hospital course is not well understood. The current preference of minimizing transfusions and keeping Hbs in a lower range, based on trials for the most part on non-CKD patients, has led to a reduction in Hb in CKD patients as well.104–106 It is important to consider that CKD patients may not be able to recover Hb after acute illness because of reduced erythropoietin production.
Acute drops in Hb in ICU CKD patients should be managed similar to those in non-CKD patients. Chronic anemia should not be presumed to be due to CKD, and routine evaluations should still occur, including stool studies and iron status assessment. CKD patients, when blood urea nitrogen (BUN) levels increase above 100 mg/dL, are more prone to bleeding because of irregularities in platelet functional activity.107–109 Dialysis may reduce the risk of bleeding due to azotemia and “uremic platelets,” though variable data on this point exist.110–112 Additionally, desmopressin will stimulate von Willebrand factor release acutely though tachyphylaxis develops after the first dose.113,114 Finally, conjugated estrogens may provide a sustained reduction in bleeding complications in CKD patients who have recurrent GI bleeding.115 Low-molecular-weight heparin clearance is reduced in patients with low GFRs. Appropriate dose reductions should occur; currently, no guidelines exist for dosing these medications with a GFR below 15 mL/minute, and their use in these settings should be avoided.
End-Stage Renal Disease
Patients with ESRD make up a large and growing percentage of the CKD population. In the United States, 90% of this population receives renal replacement therapy (RRT) in the form of in-center hemodialysis at the time of dialysis initiation.5 Efforts are under way to move more of this care to the patient’s home in the form of home hemodialysis or peritoneal dialysis.
ESRD patients have a higher mortality rate when admitted to ICUs.116,117 Indications for admission may be dialysis specific such as access complications or volume overload or may be due to other comorbid illnesses. Most commonly, indications include cardiovascular causes including acute coronary syndromes and strokes, other infectious causes, and GI hemorrhages. Commonly used ICU prognostic scoring systems such as Apache II and III, Simplified Acute Physiology Score (SAPS) II, and Sequential Organ Failure provide reasonable prognostication of patients with ESRD.118
Attention to a dialysis patient’s access for ESRD is essential in the care of any ICU ESRD patient. Arteriovenous fistulas (AVFs) and arteriovenous grafts (AVGs) may potentially occlude during hypotension.119 Loss of dialysis access due to complications from hypotension or potential iatrogenic causes such as restraints could pose a severe hardship for the patient upon recovery from the current illness.120–122 A relatively recently recognized issue is the need to avoid brachial and subclavian access whenever possible in patient who may currently or imminently need an AVF or AVG. The reason is the frequent development of venous stenosis or thrombosis, which could hamper future access maturation; studies show a high prevalence of thrombosis, over 30%, following peripherally inserted central catheter (PICC) line placement.123 Tunneled intrajugular catheters are an appropriate option in this population.124 Subclavian lines, even temporarily, pose similar complication risks. Currently, the Kidney Disease Outcomes Quality Initiative (KDOQI), sponsored by the National Kidney Foundation, recommends not placing PICC lines in patients with CKD.125
Common causes of infection in the ESRD population are cellulitis, pneumonia, bacteremia, and pycocystitis.126 In the absence of a clear cause of infections, a high index of suspicion for access infection should be maintained for ESRD patients presenting with unexplained sepsis if a tunneled dialysis catheter is being used. Generally, catheters do not show signs of infection. Initially, antibiotic therapy should broadly cover gram-positive and gram-negative organisms as these are the most common organisms associated with infection.127 Vancomycin and gentamicin, prescribed with a loading dose to achieve therapeutic targets based on local sensitivity data, are standard regimens. Considering the prevalence of methicillin-resistant Staphylococcus aureus (MRSA), a cephalosporin-only regimen may significantly delay effective therapy.128 An important consideration is the peritoneal dialysis patient who still makes urine. If feasible, these patients should not receive aminoglycoside therapy in order to maintain their important residual renal function. Instead, a third-generation cephalosporin should be used. Whenever possible, old culture results should be used to guide initial treatment as antibiotic resistance is commonplace. When a tunnel infection is suspected in a hemodialysis patient, as evidenced by an erythematous track around the course of the catheter or purulent drainage from the tunnel itself, catheter removal should urgently occur. When an AVG is infected, urgent removal or resection should also occur. Old, occluded AVGs may represent a source for chronic indolent infections and inflammation.129
Kidney Transplant Patients
Data from the 2008 USRDS survey show that patients with a functioning kidney transplant make up 30% of all ESRD patients.5 Infectious and cardiovascular complications are the most frequent complications requiring admission to an ICU.
The risk of infection in a kidney transplant patient is complex and related to a number of factors including the health of the allograft, specific donor and recipient factors (CMV status, for example), adequate prophylaxis, up-to-date and effective vaccination, and the level of immunosuppression.130 In the early post-transplant period, generally defined as 1 month or less, typical postoperative complications, including Clostridium difficile–associated colitis, are most common. In months 1 to 6 following transplantation, opportunistic infections and viral infections are the predominant source of infection due to the residual effects of induction immunosuppression. After 6 months, typical community-acquired organisms remain the primary source of infection. Infection with cytomegalovirus is an important cause of morbidity in the transplant population and may be present outside the first 6 months. Specific guidelines exist regarding prophylaxis, screening, and treatment for CMV infection in the solid organ transplant patient.131
Drug Dosing
Drug dosing in CKD patients requires close attention to current levels of kidney function and an understanding of the risks and benefits of the medication prescribed. Two points are worthy of specific attention. First, drug dosing has historically been defined by creatinine clearance and not eGFR.132,133 The second is that CKD patients, particularly with advanced disease, are a population that is understudied in terms of medication dosing.134 Antibiotic dosing in the critically ill patient is an area in which the toxicity of overdosage needs to be balanced against the possibility of subtherapeutic dosing. Recognizing that serum creatinine may not reflect true GFR is an important part of this process.
Summary
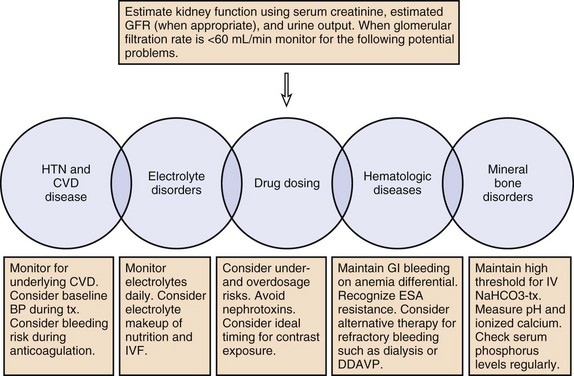
The prevalence of CKD and ESRD in the ICU has increased and this population is prone to poor outcomes. Ideal care of this population includes the recognition that serum creatinine may not reflect GFR, patients are prone to volume overload and volume depletion, and abnormalities in Hb, pH, calcium, and phosphorus may reflect normal and not abnormal physiology. Drug dosing needs to be tailored to individuals, and risks of underdosage as well as overdosage must be considered. See Figure 56-2 for a summary of management principles for CKD.
Key Points
• CKD patients frequently die of CVD; a low index of suspicion for occult CVD should guide caregivers.
• Serum creatinine frequently does not accurately reflect current level of kidney function in the ICU patient.
• CKD is associated with sodium retention and a higher prevalence of hypertension. Acute BP control should take into account baseline BP control to avoid tissue underperfusion.
• Electrolyte disorders, in particular hyperkalemia, in the ICU CKD population, are frequent.
• Acute treatment of chronic metabolic acidosis may lead to iatrogenic falls in serum calcium.
• Brachial and subclavian access is relatively contraindicated in the advanced CKD and ESRD population owing to their future impact on maturing vascular access for dialysis.
• Drug dosing in CKD patients is poorly studied. Clinicians should be cautious of under- and overdosage issues.
References
1. Coresh, J, Astor, BC, Greene, T, et al. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis. 2003; 41(1):1–12.
2. Coresh, J, Selvin, E, Stevens, LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007; 298(17):2038–2047.
3. Coresh, J, Byrd-Holt, D, Astor, BC, et al. Chronic kidney disease awareness, prevalence, and trends among U. S. adults, 1999 to 2000. J Am Soc Nephrol. 2005; 16(1):180–188.
4. Castro, AF, Coresh, J. CKD surveillance using laboratory data from the population-based National Health and Nutrition Examination Survey (NHANES). Am J Kidney Dis. 2009; 53(3 Suppl 3):S46–S55.
5. Collins, AJ, Foley, RN, Herzog, C, et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis. 2011; 57(1 Suppl 1):A8.
6. Whaley-Connell, AT, Sowers, JR, Stevens, LA, et al. CKD in the United States: Kidney Early Evaluation Program (KEEP) and National Health and Nutrition Examination Survey (NHANES) 1999-2004. Am J Kidney Dis. 2008; 51(4 Suppl 2):S13–S20.
7. Levey, AS, Coresh, J, Balk, E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Ann Intern Med. 2003; 139(2):137–147.
8. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am J Kidney Dis. 2002; 39(2 Suppl 1):S1–266.
9. Bagshaw, SM, Mortis, G, Doig, CJ, et al. One-year mortality in critically ill patients by severity of kidney dysfunction: A population-based assessment. Am J Kidney Dis. 2006; 48(3):402–409.
10. Go, AS, Chertow, GM, Fan, D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004; 351(13):1296–1305.
11. Henry, RM, Kostense, PJ, Bos, G, et al. Mild renal insufficiency is associated with increased cardiovascular mortality: The Hoorn Study. Kidney Int. 2002; 62(4):1402–1407.
12. Deo, R, Sotoodehnia, N, Katz, R, et al. Cystatin C and sudden cardiac death risk in the elderly. Circ Cardiovasc Qual Outcomes. 2010; 3(2):159–164.
13. Fried, LF, Katz, R, Sarnak, MJ, et al. Kidney function as a predictor of noncardiovascular mortality. J Am Soc Nephrol. 2005; 16(12):3728–3735.
14. Winearls, CG, Glassock, RJ. Dissecting and refining the staging of chronic kidney disease. Kidney Int. 2009; 75(10):1009–1014.
15. Kirsztajn, GM, Suassuna, JH, Bastos, MG. Dividing stage 3 of chronic kidney disease (CKD): 3A and 3B. Kidney Int. 2009; 76(4):462–463.
16. Abutaleb, N. Why we should sub-divide CKD stage 3 into early (3a) and late (3b) components. Nephrol Dial Transplant. 2007; 22(9):2728–2729.
17. Freedman, BI, Kopp, JB, Langefeld, CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010; 21(9):1422–1426.
18. Abuelo, JG. Normotensive ischemic acute renal failure. N Engl J Med. 2007; 357(8):797–805.
19. Hoste, EA, Damen, J, Vanholder, RC, et al. Assessment of renal function in recently admitted critically ill patients with normal serum creatinine. Nephrol Dial Transplant. 2005; 20(4):747–753.
20. Verhave, JC, Fesler, P, Ribstein, J, et al. Estimation of renal function in subjects with normal serum creatinine levels: Influence of age and body mass index. Am J Kidney Dis. 2005; 46(2):233–241.
21. Srisawasdi, P, Chaichanajarernkul, U, Teerakanjana, N, et al. Exogenous interferences with Jaffe creatinine assays: Addition of sodium dodecyl sulfate to reagent eliminates bilirubin and total protein interference with Jaffe methods. J Clin Lab Anal. 2010; 24(3):123–133.
22. Dimeski, G, McWhinney, B, Jones, B, et al. Extent of bilirubin interference with Beckman creatinine methods. Ann Clin Biochem. 2008; 45(Pt 1):91–92.
23. Cameron, SJ, Gerhardt, G, Engelstad, M, et al. Interference in clinical chemistry assays by the hemoglobin-based oxygen carrier, hemospan. Clin Biochem. 2009; 42(3):221–224.
24. Sonntag, O, Scholer, A. Drug interference in clinical chemistry: Recommendation of drugs and their concentrations to be used in drug interference studies. Ann Clin Biochem. 2001; 38(Pt 4):376–385.
25. Saenger, AK, Lockwood, C, Snozek, CL, et al. Catecholamine interference in enzymatic creatinine assays. Clin Chem. 2009; 55(9):1732–1736.
26. Panteghini, M. Enzymatic assays for creatinine: Time for action. Clin Chem Lab Med. 2008; 46(4):567–572.
27. Vickery, S, Stevens, PE, Dalton, RN, et al. Does the ID-MS traceable MDRD equation work and is it suitable for use with compensated Jaffe and enzymatic creatinine assays? Nephrol Dial Transplant. 2006; 21(9):2439–2445.
28. Bello, AK, Hemmelgarn, B, Lloyd, A, et al. Associations among estimated glomerular filtration rate, proteinuria, and adverse cardiovascular outcomes. Clin J Am Soc Nephrol. 2011; 6(6):1418–1426.
29. Anderson, RJ, O’Brien, M, MaWhinney, S, et al. Renal failure predisposes patients to adverse outcome after coronary artery bypass surgery. VA Cooperative Study #5. Kidney Int. 1999; 55(3):1057–1062.
30. Anderson, RJ, O’Brien, M, MaWhinney, S, et al. Mild renal failure is associated with adverse outcome after cardiac valve surgery. Am J Kidney Dis. 2000; 35(6):1127–1134.
31. Ibanez, J, Riera, M, Saez de Ibarra, JI, et al. Effect of preoperative mild renal dysfunction on mortality and morbidity following valve cardiac surgery. Interact Cardiovasc Thorac Surg. 2007; 6(6):748–752.
32. O’Brien, MM, Gonzales, R, Shroyer, AL, et al. Modest serum creatinine elevation affects adverse outcome after general surgery. Kidney Int. 2002; 62(2):585–592.
33. Cooper, WA, O’Brien, SM, Thourani, VH, et al. Impact of renal dysfunction on outcomes of coronary artery bypass surgery: Results from the Society of Thoracic Surgeons National Adult Cardiac Database. Circulation. 2006; 113(8):1063–1070.
34. Korhonen, PE, Kivela, SL, Aarnio, PT, et al. Estimating glomerular filtration rate in hypertensive subjects: Comparison of the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) and Modification of Diet in Renal Disease (MDRD) Study equations. Ann Med. 2012; 44(5):487–493.
35. Earley, A, Miskulin, D, Lamb, EJ, et al. Estimating equations for glomerular filtration rate in the era of creatinine standardization. Annals of Internal Medicine. 2012; 156(11):785–795.
36. White, SL, Polkinghorne, KR, Atkins, RC, Chadban, SJ. Comparison of the prevalence and mortality risk of CKD in Australia using the CKD Epidemiology Collaboration (CKD-EPI) and Modification of Diet in Renal Disease (MDRD) Study GFR estimating equations: The AusDiab (Australian Diabetes, Obesity and Lifestyle) Study. Am J Kidney Dis. 2010; 55(4):660–670.
37. Roos, JF, Doust, J, Tett, SE, Kirkpatrick, CM. Diagnostic accuracy of cystatin C compared to serum creatinine for the estimation of renal dysfunction in adults and children—A meta-analysis. Clin Biochem. 2007; 40(5-6):383–391.
38. Shlipak, MG, Katz, R, Sarnak, MJ, et al. Cystatin C and prognosis for cardiovascular and kidney outcomes in elderly persons without chronic kidney disease. Ann Intern Med. 2006; 145(4):237–246.
39. Bingham, SA, Williams, R, Cole, TJ, et al. Reference values for analytes of 24-h urine collections known to be complete. Ann Clin Biochem. 1988; 25(Pt 6):610–619.
40. Schrier, RW, Howard, RL. Unifying hypothesis of sodium and water regulation in health and disease. Hypertension. 1991; 18(5 Suppl):III164–168.
41. Rodriguez-Iturbe, B, Johnson, RJ. The role of renal microvascular disease and interstitial inflammation in salt-sensitive hypertension. Hypertens Res. 2010; 33(10):975–980.
42. Sander, GE, Giles, TD. Resistant hypertension: Concepts and approach to management. Curr Hypertens Rep. 2011; 13(5):347–355.
43. Sica, DA. Pharmacologic issues in treating hypertension in CKD. Adv Chronic Kidney Dis. 2011; 18(1):42–47.
44. Sica, DA. Hypertension, renal disease, and drug considerations. J Clin Hypertens (Greenwich). 2004; 6(10 Suppl 2):24–30.
45. Grassi, G, Bertoli, S, Seravalle, G. Sympathetic nervous system: Role in hypertension and in chronic kidney disease. Curr Opin Nephrol Hypertens. 2012; 21(1):46–51.
46. Baylis, C. Nitric oxide synthase derangements and hypertension in kidney disease. Curr Opin Nephrol Hypertens. 2012; 21(1):1–6.
47. Crowley, SD, Zhang, J, Herrera, M, et al. Role of AT receptor-mediated salt retention in angiotensin II-dependent hypertension. Am J Physiol Renal Physiol. 2011; 301(5):F1124–F1130.
48. Augustyniak, RA, Tuncel, M, Zhang, W, et al. Sympathetic overactivity as a cause of hypertension in chronic renal failure. J Hypertens. 2002; 20(1):3–9.
49. Bidani, AK, Griffin, KA. Pathophysiology of hypertensive renal damage: Implications for therapy. Hypertension. 2004; 44(5):595–601.
50. Campese, VM. Neurogenic factors and hypertension in renal disease. Kidney Int Suppl. 2000; 75:S2–S6.
51. Neumann, J, Ligtenberg, G, Koomans, HA, et al. Sympathetic hyperactivity in chronic kidney disease: Pathogenesis, clinical relevance, and treatment. Kidney Int. 2004; 65(5):1568–1576.
52. Yee, J, Parasuraman, R, Narins, RG. Selective review of key perioperative renal-electrolyte disturbances in chronic renal failure patients. Chest. 1999; 115(5 Suppl):149S–157S.
53. Earley, LE, Daugharty, TM. Sodium metabolism. N Engl J Med. 1969; 281(2):72–86.
54. Nzerue, C, Schlanger, L, Jena, M, et al. Granulomatous interstitial nephritis and uveitis presenting as salt-losing nephropathy. Am J Nephrol. 1997; 17(5):462–465.
55. De Bruyne, LK. Mechanisms and management of diuretic resistance in congestive heart failure. Postgrad Med J. 2003; 79(931):268–271.
56. Ellison, DH. Diuretic resistance: Physiology and therapeutics. Semin Nephrol. 1999; 19(6):581–597.
57. Stanton, BA, Kaissling, B. Adaptation of distal tubule and collecting duct to increased Na delivery. II. Na+ and K+ transport. Am J Physiol. 1988; 255(6 Pt 2):F1269–F1275.
58. Loon, NR, Wilcox, CS, Unwin, RJ. Mechanism of impaired natriuretic response to furosemide during prolonged therapy. Kidney Int. 1989; 36(4):682–689.
59. Kaissling, B, Stanton, BA. Adaptation of distal tubule and collecting duct to increased sodium delivery. I. Ultrastructure. Am J Physiol. 1988; 255(6 Pt 2):F1256–F1268.
60. Vasavada, N, Saha, C, Agarwal, R. A double-blind randomized crossover trial of two loop diuretics in chronic kidney disease. Kidney Int. 2003; 64(2):632–640.
61. Rudy, DW, Voelker, JR, Greene, PK, et al. Loop diuretics for chronic renal insufficiency: A continuous infusion is more efficacious than bolus therapy. Ann Intern Med. 1991; 115(5):360–366.
62. Sanjay, S, Annigeri, RA, Seshadri, R, et al. The comparison of the diuretic and natriuretic efficacy of continuous and bolus intravenous furosemide in patients with chronic kidney disease. Nephrology (Carlton). 2008; 13(3):247–250.
63. Mohmand, H, Goldfarb, S. Renal dysfunction associated with intra-abdominal hypertension and the abdominal compartment syndrome. J Am Soc Nephrol. 2011; 22(4):615–621.
64. Mufarrej, FA, Abell, LM, Chawla, LS. Understanding intra-abdominal hypertension: From the bench to the bedside. J Intensive Care Med. 2012; 27(3):145–160.
65. Malbrain, ML, Cheatham, ML. Definitions and pathophysiological implications of intra-abdominal hypertension and abdominal compartment syndrome. Am Surg. 2011; 77(Suppl 1):S6–11.
66. Chalkias, A, Xanthos, T. Intra-abdominal hypertension: A potent silent killer of cardiac arrest survivors. Am J Emerg Med. 2012; 30(3):502–504.
67. Kim, IB, Prowle, J, Baldwin, I, Bellomo, R. Incidence, risk factors and outcome associations of intra-abdominal hypertension in critically ill patients. Anaesth Intensive Care. 2012; 40(1):79–89.
68. Sandhu, G, Mankal, P, Gupta, I, et al. Pathophysiology and management of acute kidney injury in the setting of abdominal compartment syndrome. Am J Ther. 2012.
69. Rose, BD. Regulation of plasma osmolality. In: Rose BD, ed. Clinical Physiology of Acid-Base and Electrolyte Disorders. 4th ed. New York: McGraw-Hill; 1994:261–273.
70. Alcazar Arroyo, R. [Electrolyte and acid-base balance disorders in advanced chronic kidney disease]. Nefrologia. 2008; 28(Suppl 3):87–93.
71. Brater, DC. Use of diuretics in chronic renal insufficiency and nephrotic syndrome. Semin Nephrol. 1988; 8(4):333–341.
72. Ishikawa, S, Sakuma, N, Fujisawa, G, et al. Opposite changes in serum sodium and potassium in patients in diabetic coma. Endocr J. 1994; 41(1):37–43.
73. Garofeanu, CG, Weir, M, Rosas-Arellano, MP, et al. Causes of reversible nephrogenic diabetes insipidus: A systematic review. Am J Kidney Dis. 2005; 45(4):626–637.
74. Prough, DS. Physiologic acid-base and electrolyte changes in acute and chronic renal failure patients. Anesthesiol Clin North Am. 2000; 18(4):809–833.
75. Kovesdy, CP, Lott, EH, Lu, JL, et al. Hyponatremia, hypernatremia, and mortality in patients with chronic kidney disease with and without congestive heart failure. Circulation. 2012; 125(5):677–684.
76. Drion, I, Joosten, H, Dikkeschei, LD, et al. eGFR and creatinine clearance in relation to metabolic changes in an unselected patient population. Eur J Intern Med. 2009; 20(7):722–727.
77. Hsieh, MF, Wu, IW, Lee, CC, et al. Higher serum potassium level associated with late stage chronic kidney disease. Chang Gung Med J. 2011; 34(4):418–425.
78. Panese, S, Martin, RS, Virginillo, M, et al. Mechanism of enhanced transcellular potassium-secretion in man with chronic renal failure. Kidney Int. 1987; 31(6):1377–1382.
79. Bunchman, TE, Sinaiko, AR. Renovascular hypertension presenting with hypokalemic metabolic alkalosis. Pediatr Nephrol. 1990; 4(2):169–170.
80. Korgaonkar, S, Tilea, A, Gillespie, BW, et al. Serum potassium and outcomes in CKD: Insights from the RRI-CKD cohort study. Clin J Am Soc Nephrol. 2010; 5(5):762–769.
81. Acker, CG, Johnson, JP, Palevsky, PM, Greenberg, A. Hyperkalemia in hospitalized patients: Causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Arch Intern Med. 1998; 158(8):917–924.
82. Paice, B, Gray, JM, McBride, D, et al. Hyperkalaemia in patients in hospital. Br Med J (Clin Res Ed). 1983; 286(6372):1189–1192.
83. Lameire, N, Van Biesen, W, Vanholder, R. Electrolyte disturbances and acute kidney injury in patients with cancer. Semin Nephrol. 2010; 30(6):534–547.
84. Allon, M, Takeshian, A, Shanklin, N. Effect of insulin-plus-glucose infusion with or without epinephrine on fasting hyperkalemia. Kidney Int. 1993; 43(1):212–217.
85. Oster, JR, Perez, GO, Vaamonde, CA. Relationship between blood pH and potassium and phosphorus during acute metabolic acidosis. Am J Physiol. 1978; 235(4):F345–F351.
86. Juurlink, DN, Mamdani, MM, Lee, DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351(6):543–551.
87. Preston, RA, Hirsh, MM, Oster, MD, Jr., Oster, HM. University of Miami Division of Clinical Pharmacology therapeutic rounds: Drug-induced hyperkalemia. Am J Ther. 1998; 5(2):125–132.
88. Einhorn, LM, Zhan, M, Hsu, VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009; 169(12):1156–1162.
89. Schwartz, WB, Relman, AS. Acidosis in renal disease. N Engl J Med. 1957; 256(25):1184–1186.
90. de Brito-Ashurst, I, Varagunam, M, Raftery, MJ, Yaqoob, MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol. 2009; 20(9):2075–2084.
91. Tsai, IC, Huang, JW, Chu, TS, et al. Factors associated with metabolic acidosis in patients receiving parenteral nutrition. Nephrology (Carlton). 2007; 12(1):3–7.
92. Liamis, G, Milionis, HJ, Elisaf, M. Pharmacologically-induced metabolic acidosis: A review. Drug Safety. 2010; 33(5):371–391.
93. Moe, S, Drueke, T, Cunningham, J, et al. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006; 69(11):1945–1953.
94. Moe, SM, Drüeke, TB, Block, GA, et al, KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease—Mineral and Bone Disorder (CKD-MBD) (Kidney Disease: Improving Global Outcomes CKD-MBD Work Group). Kidney Int Suppl. 2009(113):S1–130.
95. Miyamoto, K, Ito, M, Tatsumi, S, et al. New aspect of renal phosphate reabsorption: The type IIc sodium-dependent phosphate transporter. Am J Nephrol. 2007; 27(5):503–515.
96. Liu, S, Gupta, A, Quarles, LD. Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Curr Opin Nephrol Hypertens. 2007; 16(4):329–335.
97. Isakova, T, Xie, H, Yang, W, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011; 305(23):2432–2439.
98. Knockel, JP. Biochemical alterations in advanced uremic failure. In: Jacobson HR, Striker GE, Klahr S, eds. The Principles and Practice of Nephrology. Philadelphia: B. C. Decker; 1991:682–684.
99. McGonigle, RJ, Wallin, JD, Shadduck, RK, Fisher, JW. Erythropoietin deficiency and inhibition of erythropoiesis in renal insufficiency. Kidney Int. 1984; 25(2):437–444.
100. Singh, AK, Szczech, L, Tang, KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006; 355(20):2085–2098.
101. Drueke, TB, Locatelli, F, Clyne, N, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006; 355(20):2071–2084.
102. Besarab, A, Bolton, WK, Browne, JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998; 339(9):584–590.
103. Pfeffer, MA, Burdmann, EA, Chen, CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009; 361(21):2019–2032.
104. Shander, A, Javidroozi, M, Ozawa, S, Hare, GM. What is really dangerous: Anaemia or transfusion? Br J Anaesth. 2011; 107(Suppl 1):41–59.
105. Palazzuoli, A, Antonelli, G, Nuti, R. Anemia in cardio-renal syndrome: Clinical impact and pathophysiologic mechanisms. Heart Fail Rev. 2011; 16(6):603–607.
106. Hebert, PC, Wells, G, Blajchman, MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999; 340(6):409–417.
107. Castaldi, PA, Rozenberg, MC, Stewart, JH. The bleeding disorder of uraemia. A qualitative platelet defect. Lancet. 1966; 2(7454):66–69.
108. Noris, M, Remuzzi, G. Uremic bleeding: Closing the circle after 30 years of controversies? Blood. 1999; 94(8):2569–2574.
109. Deykin, D. Uremic bleeding. Kidney Int. 1983; 24(5):698–705.
110. Hedges, SJ, Dehoney, SB, Hooper, JS, et al. Evidence-based treatment recommendations for uremic bleeding. Nat Clin Pract Nephrol. 2007; 3(3):138–153.
111. Lindsay, RM, Moorthy, AV, Koens, F, Linton, AL. Platelet function in dialyzed and non-dialyzed patients with chronic renal failure. Clin Nephrol. 1975; 4(2):52–57.
112. Remuzzi, G, Livio, M, Marchiaro, G, et al. Bleeding in renal failure: Altered platelet function in chronic uraemia only partially corrected by haemodialysis. Nephron. 1978; 22(4-6):347–353.
113. Mannucci, PM, Remuzzi, G, Pusineri, F, et al. Deamino-8-D-arginine vasopressin shortens the bleeding time in uremia. N Engl J Med. 1983; 308(1):8–12.
114. Chen, KS, Huang, CC, Leu, ML, et al. Hemostatic and fibrinolytic response to desmopressin in uremic patients. Blood Purif. 1997; 15(2):84–91.
115. Livio, M, Mannucci, PM, Vigano, G, et al. Conjugated estrogens for the management of bleeding associated with renal failure. N Engl J Med. 1986; 315(12):731–735.
116. Clermont, G, Acker, CG, Angus, DC, et al. Renal failure in the ICU: Comparison of the impact of acute renal failure and end-stage renal disease on ICU outcomes. Kidney Int. 2002; 62(3):986–996.
117. Sood, MM, Roberts, D, Komenda, P, et al. End-stage renal disease status and critical illness in the elderly. Clin J Am Soc Nephrol. 2011; 6(3):613–619.
118. Uchino, S, Morimatsu, H, Bellomo, R, et al. End-stage renal failure patients requiring renal replacement therapy in the intensive care unit: Incidence, clinical features, and outcome. Blood Purif. 2003; 21(2):170–175.
119. Smith, GE, Gohil, R, Chetter, I. Factors affecting the patency of arteriovenous fistulas for dialysis access. J Vasc Surg. 2012; 55(3):849–855.
120. Develter, W, De Cubber, A, Van Biesen, W, et al. Survival and complications of indwelling venous catheters for permanent use in hemodialysis patients. Artif Organs. 2005; 29(5):399–405.
121. Allon, M, Daugirdas, J, Depner, TA, et al. Effect of change in vascular access on patient mortality in hemodialysis patients. Am J Kidney Dis. 2006; 47(3):469–477.
122. Ng, LJ, Chen, F, Pisoni, RL, et al. Hospitalization risks related to vascular access type among incident US hemodialysis patients. Nephrol Dial Transplant. 2011; 26(11):3659–3666.
123. Abdullah, BJ, Mohammad, N, Sangkar, JV, et al. Incidence of upper limb venous thrombosis associated with peripherally inserted central catheters (PICC). Br J Radiol. 2005; 78(931):596–600.
124. Sasadeusz, KJ, Trerotola, SO, Shah, H, et al. Tunneled jugular small-bore central catheters as an alternative to peripherally inserted central catheters for intermediate-term venous access in patients with hemodialysis and chronic renal insufficiency. Radiology. 1999; 213(1):303–306.
125. Clinical practice guidelines for vascular access. Am J Kidney Dis. 2006; 48(Suppl 1):S176–S247.
126. Arulkumaran, N, Montero, RM, Singer, M. Management of the dialysis patient in general intensive care. Br J Anaesth. 2012; 108(2):183–192.
127. Al-Solaiman, Y, Estrada, E, Allon, M. The spectrum of infections in catheter-dependent hemodialysis patients. Clin J Am Soc Nephrol. 2011; 6(9):2247–2252.
128. Parker, MG, Doebbeling, BN. The challenge of methicillin-resistant Staphylococcus aureus prevention in hemodialysis therapy. Semin Dial. 2012; 25(1):42–49.
129. Nassar, GM, Fishbane, S, Ayus, JC. Occult infection of old nonfunctioning arteriovenous grafts: A novel cause of erythropoietin resistance and chronic inflammation in hemodialysis patients. Kidney Int Suppl. 2002; 80:49–54.
130. Fishman, JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007; 357(25):2601–2614.
131. Kotton, CN, Kumar, D, Caliendo, AM, et al. International consensus guidelines on the management of cytomegalovirus in solid organ transplantation. Transplantation. 2010; 89(7):779–795.
132. Spruill, WJ, Wade, WE, Cobb, HH, 3rd. Comparison of estimated glomerular filtration rate with estimated creatinine clearance in the dosing of drugs requiring adjustments in elderly patients with declining renal function. Am J Geriatr Pharmacother. 2008; 6(3):153–160.
133. Doogue, MP, Polasek, TM. Drug dosing in renal disease. Clin Biochem Rev. 2011; 32(2):69–73.
134. Cantu, TG, Ellerbeck, EF, Yun, SW, et al. Drug prescribing for patients with changing renal function. Am J Hosp Pharm. 1992; 49(12):2944–2948.
135. Antoniou, T, Gomes, T, Mamdani, MM, et al. Trimethoprim-sulfamethoxazole induced hyperkalaemia in elderly patients receiving spironolactone: Nested case-control study. BMJ. 2011; 343:d5228.
136. Alappan, R, Perazella, MA, Buller, GK. Hyperkalemia in hospitalized patients treated with trimethoprim-sulfamethoxazole. Ann Intern Med. 1996; 124(3):316–320.
137. Oster, JR, Singer, I, Fishman, LM. Heparin-induced aldosterone suppression and hyperkalemia. Am J Med. 1995; 98(6):575–586.
138. Arthur, S, Greenberg, A. Hyperkalemia associated with intravenous labetalol therapy for acute hypertension in renal transplant recipients. Clin Nephrol. 1990; 33(6):269–271.
139. Papadakis, MA, Wexman, MP, Fraser, C, Sedlacek, SM. Hyperkalemia complicating digoxin toxicity in a patient with renal failure. Am J Kidney Dis. 1985; 5(1):64–66.
140. Martyn, JA, Richtsfeld, M. Succinylcholine-induced hyperkalemia in acquired pathologic states: Etiologic factors and molecular mechanisms. Anesthesiology. 2006; 104(1):158–169.
141. Thiele, A, Rehman, HU. Hyperkalemia caused by penicillin. Am J Med. 2008; 121(8):e1-2.
142. Mercer, CW, Logic, JR. Cardiac arrest due to hyperkalemia following intravenous penicillin administration. Chest. 1973; 64(3):358–359.
143. Graber, M, Subramani, K, Corish, D, Schwab, A. Thrombocytosis elevates serum potassium. Am J Kidney Dis. 1988; 12(2):116–120.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]56
Chronic Kidney Disease
Introduction
Chronic kidney disease (CKD) is a common diagnosis in adult medicine.1–6 In order to properly identify patients at risk for adverse outcomes and disease progression, extensive work has focused on early and appropriate classification over the last 10 years. In particular, the National Kidney Foundation (NKF) proposed a classification system for CKD in 2002 using estimated glomerular filtration rate (eGFR) as a method to categorize patients.7,8 Table 56.1 describes current NKF guidelines of CKD. In regard to critical care, CKD patients have higher mortality rate following critical illnesses.9 Understanding this population’s physiology and underlying disease state may aid caregivers in attempting to improve this outcome.
Table 56.1
| Stage | eGFR (mL/min/1.73 m2) |
Urinalysis Findings |
| 1 | ≥90 | Hematuria, proteinuria, or imaging abnormalities at >3 months |
| 2 | 60-89 | Hematuria, proteinuria, or imaging abnormalities at >3 months |
| 3 | 30-59 | ↑ or normal |
| 4 | 15-29 | ↑ or normal |
| 5 | 0-14 | ↑ or normal |
Definition and Etiology
As previously stated, currently CKD is staged in a five-tier system based on eGFR (see Table 56.1). The diagnosis relies on abnormal kidney function or urinalysis findings for 3 months or more. The serum creatinine as well as age, sex, gender, and race is used to generate an eGFR from a quadratic equation derived from the Modification of Diet in Renal Disease Study.7 Most CKD patients do not progress to end-stage renal disease (ESRD), however, because of the very high cardiovascular and noncardiovascular mortality rate associated with this at-risk population.10–13 Proposals exist to more clearly define elderly subjects with CKD stage 3 disease at risk for disease progression, though this practice has not become standard.14–16
Causes for CKD are broad and often multifactorial. Frequently, however, diabetes mellitus and hypertension play a major role. Hypertension may lead to kidney damage in the form of nephrosclerosis or chronic renal ischemia due to atherosclerotic vascular disease. An important recent development is the recognition that the higher prevalence of nondiabetic kidney disease in the African-American population may be due to genetic risk conferred by inherited variants in the apolipoprotein L1 gene (APOL1).17 Additional causes include immune-mediated glomerular diseases such as systemic lupus erythematosus, IgA nephropathy, membranous nephropathy, and other often pre-existing glomerulonephritides. Glomerulonephritis may be active as evidenced by hematuria and proteinuria or may have occurred in the patient’s past, leading to abnormal kidney function but a relatively bland urinalysis. Alternatively, primary tubulointerstitial diseases could exist owing to reflux nephropathy, sarcoidosis, chronic infections, allergic reactions, or side effects from medications such as cyclophosphamide (Cytoxan) and lithium. In general, the diagnosis of CKD should warrant a higher index of suspicion for concurrent, and potentially undiagnosed, cardiovascular disease (CVD).
The CKD population is at risk for deterioration in kidney function during a hospitalization stay, particularly if a critical illness is present. Potential causative factors for this are listed in Box 56.1. Importantly, acute tubular necrosis may occur without obvious hypotension, and patients with reduced kidney function are more at risk for this complication.18
Diagnosis
Intrinsic to the eGFR formula, as well as other estimates of renal function from the serum creatinine, is the presumption that a patient’s serum creatinine is stable. This finding may not be the case in ICU patients; as a result, imprecision should be expected in kidney function estimation in ICU patients.19 Serum creatinine is now recognized to potentially underestimate the level of renal function in a large population of patients, particularly those with low weight, small muscle mass, and liver disease.20 Additionally, both endogenous and exogenous factors can influence the measurement of serum creatinine.21–24 Box 56.2 describes situations in which serum creatinine measurements may not adequately reflect kidney function. Importantly, some vasoactive substances can impart a negative interference on creatinine values if the creatinine sample is obtained from a central line used for vasopressors.25 Nonetheless, creatinine variation has improved since values have been standardized to reference values.26,27
In general, current guidelines recognize the importance of stage 3 through 5 kidney disease for prognostication in outpatient and inpatient outcomes. Proteinuria is now associated with increased cardiovascular risk at all glomerular filtration rates (GFRs), including normal GFRs.28 Even mild kidney dysfunction, defined typically as a serum creatinine level over 1.5 mg/dL or GFR below 60 mL/minute, is associated with worse outcomes after coronary artery bypass graft (CABG), cardiac valvular, and general surgery.29–33
Concern exists about the accuracy of the Modification of Diet in Renal Disease (MDRD) equation at higher GFRs. For example, the newer creatinine-based CKD-EPI formula equation leads to a lower prevalence rate of CKD stage 3 than the MDRD equation, as more patients have an eGFR above 60 mL/minute.34–36 Measurement of serum cystatin C levels, a protein produced by all nucleated cells and freely filtered by the glomerulus, may provide more accurate estimates of kidney function than creatinine-based equations, particularly when kidney function is close to normal.37 Cystatin C may also provide prognostic information about future cardiovascular events.12 In fact, cystatin C has been proposed as a marker of “preclinical” kidney disease based on worse cardiovascular outcomes in elderly subjects with higher cystatin C levels.38 Despite a growing body of literature, use of cystatin C is not routine in clinical practice.
In this equation, units will need to be converted from dL/24 hours to mL/minute by a conversion factor of 0.0694.
If this measure is pursued, clinicians are counseled to pay close attention to units as well as the expectation that men excrete 1500 to 2500 mg/day of creatinine and women excrete 1000 to 1500 mg/day of creatinine.39 If a lower amount of urinary creatinine is obtained, the possibility of an undercollection should be considered. It is also worthwhile to note that historically, drug dosing has been determined by creatinine clearance and not eGFR.
Physiology
Hypertension
Over 80% of CKD patients with eGFR less than 60 mL/minute have hypertension.5 This has several important impacts on the management of ICU patients. First, longstanding hypertension, if uncontrolled, leads to adaptations in autoregulation. Autoregulation refers to the ability of blood vessels to constrict and dilate in the presence of hypertension and hypotension, respectively. Consequently, patients become acclimated to BPs near their “typical” BP and may not tolerate lower BPs despite these apparent lower values appearing in the normal range. A consequence of this may be hypoperfusion to the kidneys as well as the brain, heart, intestine, and other critical organs at BP levels that appear normal. If a history of poorly controlled hypertension is obtained, an astute clinician should aim to keep BP close to the level the patient being treated is used to. This very much requires individualized attention and thorough history taking.
Second, CKD leads to sodium retention.40,41 The most notable ICU impact this will have is the potential for volume retention and the need for diuretics to manage hypertension in the CKD population.42–44 Several mechanisms lead to the development of hypertension in the CKD population. They include upregulation of the renin-angiotensin system, increased sympathetic nervous system activity, and impairment of endothelial-dependent arterial smooth muscle relaxation.45–51 Owing to the preceding mechanisms, CKD patients may develop more edema and volume overload with equivalent amounts of intravenous (IV) fluids. One worthwhile consideration, however, is that sodium retention associated with CKD may be counterbalanced by sodium loss from excessively high BP levels. This phenomenon, known as pressure natriuresis, may explain the rapid drops observed in BP in hypertension emergencies once excess sympathetic activity is controlled.
Electrolyte Disorders
Sodium
Sodium homeostasis in patients with CKD is abnormal. With increased loss of sodium in the urine, but declining numbers of functioning nephrons, sodium balance is maintained.52 Though CKD patients are sodium “avid” and tend toward states of volume overload, they are less able to increase sodium retention in times of need and subsequently are also at risk for volume depletion. In comparison, subjects with normal renal function can quickly increase sodium absorption and avoid volume-depleted states.53 Tubulointerstitial disease and obstructive uropathy may be particularly at risk for episodes of volume depletion due to their recognized inability to increase sodium reabsorption in times of volume depletion.54
When volume overload is present, iatrogenic sources may be playing a role. Potential sources of sodium include maintenance IV fluids (including bicarbonate), antibiotics, and nutritional sources including total parenteral nutrition (TPN). Tracking fluid intake and output (“Is and Os”) does not clearly differentiate between the electrolyte makeup of the various sources. Consequently, volume overload may develop insidiously. When volume overload develops, the standard approach involves minimizing sodium-containing sources and initiating a diuretic regimen. Diuretics range from the more gentle thiazide diuretics, which are ineffective with a GFR below 30 mL/minute, to more potent loop diuretics. In general, if significant volume removal is desired, a diuretic regimen twice a day (every 12 hours) should be prescribed to avoid excess sodium reabsorption in the period after the major diuretic effect.55 Chronic administration of diuretics, prior to ICU admission, may lead to adaptive processes necessitating higher diuretic doses, combination therapy, or alternative approaches to volume overload states.56–59 Torsemide is the loop diuretic that may be closest to lasting 24 hours.60 Doses of loop diuretics need to be increased as renal function declines. Significant variability in loop diuretic kinetics exist; if a dose does not yield an increase in urine output within 2 hours, consideration should be given to increasing the dose. Alternatively, continuous infusion of loop diuretics may provide a better diuresis with less toxicity.61,62 Typically infusion rates of furosemide are 10 to 40 mg/hour. When a loop diuretic drip is ordered, a bolus should be given prior to initiating therapy to avoid a significant delay in efficacy.
Diuretic failure is common in the CKD population. Potential explanations in an ICU population include acute kidney injury, inadequate dosing, and distal tubular hypertrophy due to chronic diuretic use. When high-dose loop diuretics do not achieve adequate diuresis, the addition of metolazone may improve urine output. Historically, metolazone is given 30 minutes prior to the loop diuretics, though it may be efficacious if given at other times as well. Finally, isolated ultrafiltration or potentially dialysis may be required for volume removal. Significant attention has been given to the importance of elevated intra-abdominal pressures leading to a reduction in renal function and, potentially, ability to respond to diuretics.63–68
Water
Water concentration and dilution are abnormal in CKD patients. Normal patients can dilute and concentrate urine within a range of 40 to 1400 mOsm/kg.69 CKD leads to a narrower range of urine osmolality. Specifically, patients with advanced CKD have a urine osmolality much closer to 300 mOsm/kg; the ability to adjust urine osmolality declines as urine function worsens.70 Consequently, patients with CKD are prone both to hypernatremia and hyponatremia. Loop diuretics may further worsen the kidney’s ability to concentrate and dilute urine.71
The approach to hypernatremia and hyponatremia is similar to that in other patients. In hypernatremia cases, concurrent illnesses often lead to patients being unable to obtain water for themselves; a high urine output due to hyperglycemia or a high urea concentration may worsen this. Elevated urea, as with high serum glucose levels, functions as an osmolar agent in the tubular lumen and creates a state of relative antidiuretic hormone (ADH) resistance with the potential for substantial free water loss.72
Hypernatremia can lead to significant agitation and should be treated aggressively with increased water either intravenously, in the form of hypotonic IV fluids, or enterally. When severe, an estimation of free water deficit is appropriate. Unexplained polyuria with a low urine osmolality, hypernatremia, and CKD in an ICU patient with unclear history should warrant an investigation for previous lithium use.73
Hyponatremia reflects irregularities in water excretion in the vast majority of cases. CKD patients are less able to excrete water owing to the inability to lower urine osmolality.74 Sources of water include IV fluids, medications mixed in dextrose, and enteral sources. Patients often are not aware or not forthcoming in the amount of water they drink, though this is less an issue in ICU patients. The approach for most CKD patients is similar to that in other cases of hyponatremia, which involve looking for and reducing causes of increased water intake. Importantly, sodium abnormalities in the CKD patient, both with and without congestive heart failure, are associated with a higher mortality rate.75
Potassium
Potassium (K+) regulation is an essential function of the kidneys in normal states. CKD patients routinely maintain serum K+ in the normal range despite losing up to 90% of renal function.76,77 Mechanisms underlying the maintenance of normal serum K+ in advanced CKD include increased K+ excretion per functioning nephron, increased gastrointestinal (GI) elimination of K+, and increased uptake of K+ by cells.78 Diabetes mellitus, however, may exacerbate the development of hyperkalemia due to the presence of a distal tubular acidosis (type 4) from aldosterone resistance. Hypokalemia may also be present due to current diuretic use or renovascular hypertension.79 Its presence is associated with a higher risk of death and future ESRD needs in the CKD population.80
ICU patients with CKD represent a population prone to irregularities in K+ homeostasis, in particular, hyperkalemia.81,82 Potential causes of hyperkalemia include insulin deficiency, hypoaldosteronism, and a loss of normal intestinal function, which plays a role in increased potassium excretion in CKD patients. Decreased blood flow to muscle and intestines in times of critical illness may impede potassium cellular uptake and excretion, respectively. Tissue ischemia from trauma, hypoxia, or tumor lysis is associated with an elevation in K+ which may occur quickly.83 One additional clinical consideration is that CKD patients who are fasting may develop surprising levels of hyperkalemia without any apparent cause due to low levels of insulin.84 Glucose-containing IV fluids may avoid this.
The evaluation of hyperkalemia in CKD patients begins with assessing the cause. Typically, this includes close scrutiny of medications, IV fluids, and types of nutritional support. Frequently, ICU patients receive nutrition very different from what the body is used to and cannot manage a higher load of K+, especially in states of relative tissue perfusion. Importantly, many CKD patients are on diuretics as part of their routine outpatient medications; simply stopping these medications may lead to states of hyperkalemia. Hyperosmolality leads to a K+ shift out of cells caused by solvent drag; in its most common presentation, hyperglycemia, this can be reversed quickly with the use of insulin. Metabolic acidosis may be associated with hyperkalemia though organic acidosis states appear to cause less hyperkalemia than nonorganic acid–induced states.85
Medications are an important and growing cause of hyperkalemia. Congestive heart failure and proteinuric renal disease remain as two indications for possible dual renin-angiotensin-aldosterone blockade; hyperkalemia has been observed more frequently since the publication of the RALES trial.86 Box 56.3 lists some common causes of hyperkalemia in the CKD population and the associated mechanisms.87 The presence of hyperkalemia should warrant close attention to affected patients as its presence is associated with increased mortality rate.88
