Chapter 85 Cerebrovascular Disease in Children
Overview and Definitions
It is important to distinguish stroke from similar neurological disorders that are discussed elsewhere. Perinatal strokes, including AIS, CSVT, and other varieties, are a common and important variety of cerebrovascular disease discussed in detail in Chapter18. Global hypoxic-ischemic brain injury (HIE) is also common in newborns and children. While the mechanism of cellular injury is similar, these lack the isolated focal distribution of injury that defines vaso-occlusive stroke. Therefore, global HIE diseases in preterm and term neonates (Chapter 17,19), as well as global and watershed HIE (Chapter 17), are covered elsewhere. Specific patterns of intracranial hemorrhage, including germinal matrix bleeding unique to the premature brain (Chapter 19) and traumatic extra-axial intracranial hemorrhage (Chapter 18), also are covered elsewhere. The many conditions that may mimic childhood stroke clinically are reviewed below, with cross-references to corresponding chapters.
The primary objective of this chapter is to provide the reader with a comprehensive, clinically relevant review of the elements of cerebrovascular disease encountered in infants and children. Recent general review articles are available [Pappachan and Kirkham, 2008; Dlamini and Kirkham, 2009; Amlie-Lefond et al., 2008], and section-specific review articles and consensus-based guidelines are referenced below. The information presented is based on the best available evidence to the end of 2009. In order to highlight the limited level of evidence in many areas, relative standards of evidence criteria [Cook et al., 1995] are provided where appropriate. With almost no randomized clinical trails and limited prospective studies, many studies are classified as level IV or V (retrospective case series). As evidence is evolving rapidly, the reader is encouraged to supplement this chapter with searching the current literature regarding specific topics from 2010 on.
Epidemiology of Pediatric Stroke
Stroke is commonly encountered in the practice of pediatric neurology. Hospital administrative data suggest that cerebrovascular disorders are among the top ten causes of death in children, with rates highest in the first year of life [Murphy, 2000]. Stroke-related mortality in young children may have decreased in the past two decades [Fullerton et al., 2003b]. The importance of “stroke in the young” (i.e., <40 years of age) is recognized increasingly and children account for nearly one-quarter of such cases [Kerr et al., 1993]. The burden of pediatric stroke is amplified by the duration of the adverse neurological outcomes suffered by most children, lasting decades. Economic costs of childhood stroke are substantial in the acute [Janjua et al., 2007; Perkins et al., 2009] and medium term [Lo et al., 2008a]. Less studied are the long-term costs, which are likely greater [Perkins et al., 2009]. The life-long stroke-attributable socioeconomic burden per affected individual, therefore, is substantial, and comparable in many regards to adult stroke [Taylor et al., 1996], to which approximately 3 percent of health-care expenditures in developed countries currently is dedicated [Evers et al., 2004]. The burden of pediatric stroke appears global, with recent international research initiatives further defining regional epidemiology [International Pediatric Stroke Study Web-site, 2010].
In the past three decades, incidence rates for pediatric stroke appear to be increasing. Updated summaries of available epidemiological data to date, including comparisons of ischemic versus hemorrhagic stroke, are available [Mallick and O’Callaghan, 2009a; Lynch et al., 2002]. While nearly 20 publications have examined stroke incidence rates, comparability is compromised by differences in methodology, definitions, and inclusion criteria. Factors such as age range (neonates or not), ischemic stroke definitions (CSVT included or not), and hemorrhage (included or not) account for variability in quoted rates. Many studies have relied on health-care administration databases and international classification of disease codes, with very few studies employing true population-based methodology with direct diagnostic classification [Agrawal et al., 2009].
Previously reported combined rates for ischemic and hemorrhagic pediatric stroke in the United States were 2.5–2.7 per 100,000 children per year [Broderick et al., 1993; Schoenberg et al., 1978]. These values are consistent with more recent studies [Fullerton et al., 2003b], including one population-based study using imaging-confirmed diagnoses [Agrawal et al., 2009]. A prospective, population-based Canadian registry estimated total ischemic stroke (AIS and CSVT) incidence at 2.2 per 100,000, including 1.8 for AIS and 0.4 for CSVT (unpublished data). Serial calculation of pediatric stroke rates across the decade ending in 1999 demonstrated a near-doubling from 2.8 to 5.4 per 100,000 [Kleindorfer et al., 2006]. Although less valid rigorous diagnostic criteria were used, even higher rates were reported in the National Hospital Discharge Survey (10.7 per 100,000) [Lynch et al., 2002] and in a truly population-based regional European study (13 per 100,000) [Giroud et al., 1995]. In Victoria, Australia, the incidence of childhood CSVT was estimated at 0.34 per 100,000 children per year [Barnes et al., 2004]. The reported relative proportions of hemorrhagic vs. ischemic stroke in childhood are inconsistent across multiple studies. Regarding which type of stroke predominates, some report hemorrhagic [Broderick et al., 1993; Schoenberg et al., 1978], others ischemic [Lanthier et al., 2000; Lynch et al., 2002; Giroud et al., 1995], and still others, neither [Fullerton et al., 2003b; Earley et al., 1998].
At least two factors have increased the number of diagnosed childhood strokes. First, widespread availability of more sensitive diagnostic tests, particularly magnetic resonance imaging (MRI), has increased [Wiznitzer and Masaryk, 1991; Venkataraman et al., 2004; Warach and Baron, 2004]. Second, more effective treatments have increased survival in previously lethal pediatric diseases predisposing to stroke, including prematurity, congenital heart disease, sickle cell disease, and childhood cancers. Despite improved recognition, ischemic strokes likely remain under-diagnosed, particularly CSVT. A high degree of clinical suspicion and appropriate use of diagnostic tests remains essential.
Arterial Ischemic Stroke
Pathophysiology
Arterial Circulation: Anatomy and Vascular Patterns
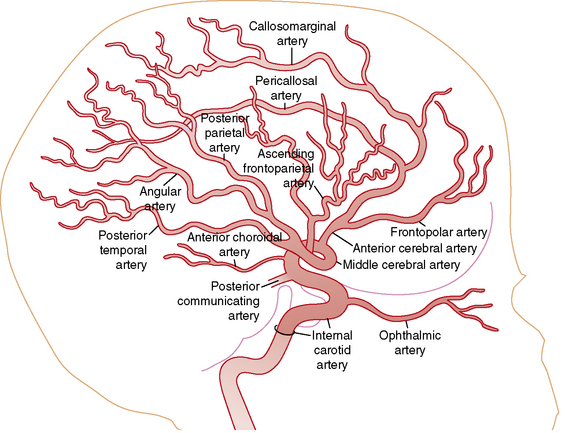
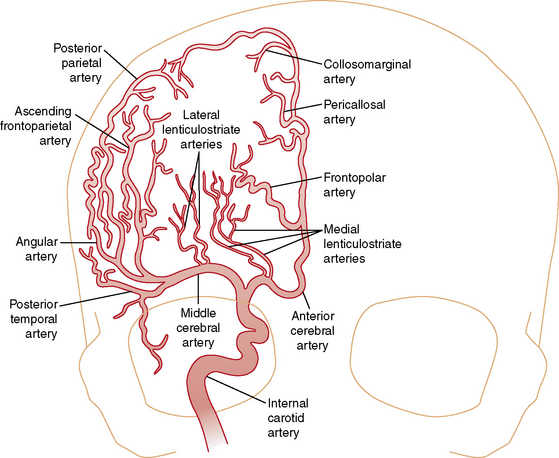
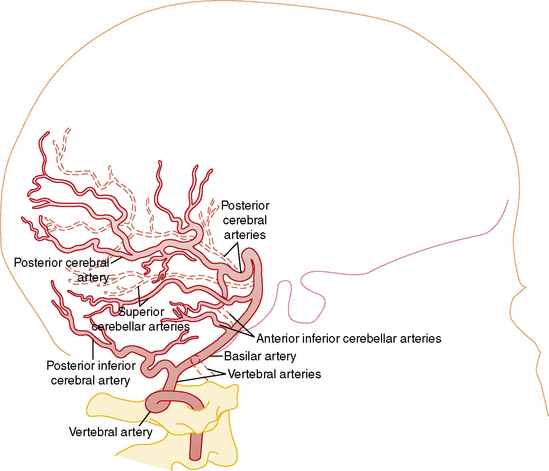
Understanding the pathophysiology, clinical presentation, possible etiologies, and potential outcomes of childhood AIS requires an understanding of craniocervical arterial circulation. The brain receives arterial blood via two principal systems: the “anterior” circulation, consisting of the paired internal carotid arteries, and the “posterior” circulation, consisting of the paired vertebral arteries that join to form the basilar artery (“vertebrobasilar system”) (Figures 85-1 to 85-3). The anterior and posterior communicating arteries link these systems to form the circle of Willis. The major cerebral arteries arising from the circle of Willis are the paired anterior, middle, and posterior cerebral arteries. The anterior and middle cerebral arteries are the major branches of the internal carotid artery, and the posterior cerebral arteries are the major branches of the vertebrobasilar system. Small “perforating arteries” arise from these proximal vessels to supply deep brain regions, such as the basal ganglia and thalamus. These include lenticulostriate branches from the anterior circulation, and thalamostriate vessels from the posterior vessels. Circulation to the posterior fossa includes perforating arteries into the midline of brainstem, and major circumferential arteries supplying the lateral brainstem and cerebellum (the posterior inferior, anterior inferior, and superior cerebellar arteries). Inter-individual variability in the anatomy of the circle of Willis is frequent and can be relevant clinically. Anastomoses, both proximally at the circle of Willis and distally through smaller leptomeningeal vessels, can provide collateral blood flow, the degree of which significantly affects the territory of brain perfused by a major cerebral artery.
Vascular patterns of AIS can be classified as large- or small-vessel infarcts. Infarction due to occlusion of large-caliber cerebral arteries usually results in peripheral wedge-shaped lesions involving the cerebral cortex and subjacent white matter in characteristic distributions (Figure 85-4). Most AIS involves the middle cerebral artery (MCA), which, like the other major vessels, courses to the cerebral surface where pial branches supply the cortex and underlying white matter. AIS often occurs in recognizable patterns within the MCA territory, including:
As end-arteries, lenticulostriate lesions lack collaterals, conferring a higher risk of permanent infarction. Despite being small, such lesions also tend to affect critical structures and functional tracts, such as the internal capsule (Figures 85-5 and 85-6). Such specific recognizable patterns also help predict the underlying etiology, with isolated lenticulostriate AIS commonly associated with parainfectious or inflammatory arteriopathy [Askalan et al., 2001; Chabrier et al., 1998; Braun et al., 2009]. In population-based studies, such small-vessel-distribution infarcts account for 50 percent of childhood AIS. For unknown reasons, AIS more frequently affects the left hemisphere [deVeber, 2000].
Mechanisms of Thromboembolism
Thrombosis is a product of the coagulation and platelet hemostatic systems. In situations of blood stasis or slow flow, such as the venous system, the coagulation system predominates, resulting in fibrin-rich thrombi. However, in arteries, platelet activation with platelet-rich thrombi is a significant contributor to thrombus formation, combining with coagulation activation. The balance of which system predominates varies with flow rate, shear stresses, endothelial integrity, concentrations of circulating anticoagulants (e.g., protein C and S), and other factors. Under physiologic circumstances, arterial endothelium provides an anticoagulant surface, helping to maintain blood in a fluid phase. When the arterial wall is damaged by disease or injury, thrombosis may develop through several mechanisms. Exposure of blood to arterial wall inflammation (e.g., vasculitis) or disruption of the endothelium with exposure to collagen and tissue factor (e.g., dissection) activates both platelets and fibrin formation. In situations of blood stasis or slow blood flow, including arterial occlusion or severe stenosis, the coagulation system may predominate. In contrast, rapid arterial blood flow with high “shear” forces favors platelet activation. Therefore, coagulation- and platelet-mediated thrombotic processes likely coexist in many children with AIS, the relative balance depending on the specific disease. Important developmental differences in both systems through early infancy and childhood are likely also important [Andrew et al., 1990; Carcao et al., 1998; Israels et al., 2001; Roschitz et al., 2001; Monagle, 2008a].
Mechanisms of Infarction and Developmental Brain Injury
Ischemic neuronal dysfunction can be reversible initially, but will shift to irreversible infarction with increasing duration and/or degree of ischemia or increased rates of neuronal metabolic activity. In a transient ischemic attack (TIA), no permanent parenchymal lesion responsible for the symptoms is seen on MRI [Albers et al., 2002]. This is consistent with the transient symptomatology (resolution typically within 60 minutes) and reversible ischemic neuronal dysfunction without permanent damage. In contrast, AIS clinical deficits persist and are accompanied by radiographic confirmation of infarction. Despite this logical clinical-radiographic correlation, pathological processes at the tissue level are certainly more complex and dependent on a number of factors. In AIS, a marked decrease in focal cerebral perfusion results in a central core zone of irreversibly damaged brain. Surrounding this is a less severely compromised penumbral zone where tissue is partially injured but remains potentially viable [Schlaug et al., 1999]. In the penumbra, clinical factors that increase the discrepancy between cerebral metabolic rate and the delivery of oxygen and glucose result in additional tissue injury and cell death. Secondary insults, including seizures and alterations in temperature, blood pressure, and serum glucose, increase this discrepancy between energy supply and demand. The primary objective of acute neuroprotective and fibrinolytic stroke treatment is to rescue this “at-risk” penumbral tissue to salvage functional brain.
Focal brain ischemia results in regional hypoxia and depletion of high-energy compounds (adenosine triphosphate) and carbohydrate stores. Compared to other tissues, the brain has relatively higher baseline metabolic rates and fewer energy storage systems [Trescher, 1992]. The cerebral metabolic rate for oxygen is 3.5 mL/100 g/min and, with virtually no oxygen reserve available, neuronal dysfunction results quickly when supply ceases. Brain glucose reserve is greater, allowing tissue survival up to 90 minutes with adequate oxygenation. The newborn can use lactate as a substrate for the production of energy but this capability is lost quickly. When regional hypoxia develops during ischemia, there is a shift from oxidative metabolism to glycolysis. Glucose is metabolized to lactate, and accumulating acidosis further exacerbates injury. Final infarct volume is determined in part by the balance between the rate of delivery of oxygen and glucose, and regional metabolic brain activity, particularly in the penumbra.
Several elements of pediatric cerebral hemodynamics are relevant in AIS. Cerebral perfusion is a direct function of the mean arterial pressure, intracranial pressure, and vascular resistance, and is age-related [Volpe, 2001a]. Adult cerebral blood flow (CBF) is 50 mL/100 g brain tissue/minute. Neonatal CBF is approximately 40 percent of this, while children under 3 years average 30–50 mL/mg/min. Children of 3–10 years have high CBF (approximately 100 mL/mg/min) that returns toward adult values by the end of adolescence [Altman et al., 1988]. CBF represents 10–20 percent of cardiac output in early infancy, peaks at over 50 percent by 2–4 years, and stabilizes at approximately 15 percent by 8 years [Wintermark et al., 2004]. Cerebral autoregulation refers to homeostatic regional adjustments in cerebral perfusion accomplished through caliber changes in cerebral arteries in response to alterations in perfusion pressure, carbon dioxide levels, and other metabolic factors. Developmental changes in cerebral autoregulation are recognized, though not well studied in AIS. Autoregulation is present across normal blood pressures in term newborns; however, it is impaired in preterm neonates [Volpe, 2001a]. Further maturation of cerebral autoregulatory systems occurs through at least adolescence [Vavilala et al., 2005]. Disturbed cerebrovascular autoregulation surrounding an evolving infarct may further contribute to a mismatch between cerebral metabolic demands and substrate delivery.
Neonatal and juvenile animal stroke models are providing exciting basic science knowledge regarding the cellular pathophysiology of ischemic stroke, including age-related processes distinct from adult stroke [Hou and MacManus, 2002]. Details of the neurotoxic cascade initiated by acute focal hypoxia-ischemia continue to evolve. In AIS, cell death proceeds over time by two primary mechanisms. Acute necrosis occurs over hours, whereas apoptosis (delayed cell death) occurs over days and, in neonates, likely over weeks. More detailed discussion of cell death mechanisms in the brain is found in Chapter 14. Accumulation of extracellular glutamate and activation of glutamate receptors play a major initiating role in this cascade [Aarts et al., 2002]. Accumulation of excess cytosolic calcium results in activation of pathological calcium-mediated intracellular events. The role of nitric oxide and free-radical generation, with resulting damage to critical intracellular components, is also likely important [Wang and Shuaib, 2007]. Complex neuroinflammatory processes have also emerged as potential targets for stroke neuroprotection [Shah et al., 2009].
Pharmacological and other neuroprotective strategies designed to interrupt this multifaceted ischemic cascade in stroke continue to be developed. Unfortunately, none has been successful in human stroke clinical trials, and no such trials have involved children. Hypothermia has shown benefit in the management of neonatal global HIE [Wyatt et al., 2007], but not childhood traumatic brain injury [Hutchison et al., 2008], is unproven in adult stroke [van der Worp et al., 2010] and untested in childhood stroke. Future animal models of stroke in the developing brain will be required to understand the unique aspects of these neuroprotective mechanisms in childhood AIS. Ideally, juvenile animal stroke models would incorporate features relevant to childhood AIS, including inflammatory cerebral arteriopathy, age-dependent coagulation systems and disorders, and inclusion of pediatric age-appropriate outcomes, including achievement of new learning, rather than just relearning previously acquired skills. Since the infant brain relies on programmed cell death for normal development, infant animal models studying anti-apoptotic agents will need to evaluate brain development after treatment. Translation of such results into clinical trials in the pediatric population will be challenging.
Ever since Kennard described better outcomes in younger monkeys following focal brain injury [Kennard, 1936], neuroscientists have sought to understand developmental neuroplasticity and harvest its benefits. Multiple subsequent animal models have confirmed evolving susceptibilities and potentials for recovery across the age spectrum that are dependent on many factors [Yager and Thornhill, 1997; Kolb et al., 2000; Kolb and Cioe, 2003]. AIS represents focal injury in an otherwise healthy brain, providing an ideal model for studies of plasticity. Knowledge regarding plastic changes in the brain following adult stroke has grown exponentially in the past decade [Murphy and Corbett, 2009]. Similar studies have not been performed widely in children. However, the widely held belief that increased plasticity confers an advantage to recovery after brain injury in childhood compared to adults remains unproven, and most children with AIS suffer adverse long-term outcomes. Childhood AIS outcome studies correlating age and recovery show mixed results [Bates et al., 1999; Goodman and Yude, 1996b; Westmacott et al., 2007b, 2009; Max, 2004a; Lansing et al., 2004] Fortunately, modern neurotechnologies, such as functional MRI (fMRI) [Heller et al., 2005], diffusion tensor imaging [Seghier et al., 2004], and transcranial magnetic stimulation [Frye et al., 2008], allow new windows into the study of brain plasticity in childhood AIS. Combining these basic and applied neuroscience methods in children with AIS will be critical to the development of improved interventions and better outcomes.
Clinical Features and Diagnostic Delays
The diagnosis of stroke in children is frequently delayed or missed. Multiple studies have confirmed extended intervals to diagnosis in AIS, often beyond 24 hours from symptom onset [Gabis et al., 2002; Rafay et al., 2009; Hartman et al., 2009; McGlennan and Ganesan, 2008; Srinivasan et al., 2009]. Affected children typically are brought to medical attention quickly with subsequent prolonged delays occurring in the hospital [Rafay et al., 2009]. Diagnostic delays likely reflect a combination of lack of awareness by primary care pediatric physicians, confusing neurological presentations, distracting signs and symptoms leading to erroneous lines of investigation, and a complex differential diagnosis. A false-negative rate for initial computed tomography (CT) scanning of 60–80 percent also contributes to missed and delayed diagnosis of childhood AIS. Yet prompt diagnosis is essential to afford opportunities for emergent neuroprotective management, and interventions to promote recanalization and prevent early recurrence. A high clinical suspicion, coupled with a systematic diagnostic approach, is required for timely diagnosis.
The acute onset of a focal neurological deficit in a child is stroke until proven otherwise. Acute hemiparesis is the most common presenting focal deficit but may be attributed erroneously to other causes that can mimic stroke (migraine, seizure or Todd’s paresis, encephalitis/meningitis, or demyelination) [Shellhaas et al., 2006]. Additional common focal neurological deficits include diplopia, hemianopsia, impaired gaze, dysarthria, vertigo, nystagmus, dysphasia (expressive, receptive, mixed), ataxia, hemisensory symptoms, and neglect. Seizures at the onset of stroke are also frequent in children [Delsing et al., 2001] compared with adults [Trescher, 1992; Yang et al., 1995]. In addition to focal neurological deficits, diffuse or nonfocal neurological signs and symptoms, such as headache, confusion, irritability, and behavioral changes, are frequent, especially in younger children [Trescher, 1992]. Neurological deficits can be subtle, especially in infants who are unable to manifest language deficits or verbalize subjective complaints. Careful consideration of clinical and imaging information is required to distinguish true stroke from the lengthy list of stroke mimics summarized in Table 85-1
Table 85-1 Differential Diagnosis of Strokelike Episodes in Children
| Disease | Clinical Distinction from Stroke | Imaging Distinction from Stroke |
|---|---|---|
| Migraine | Evolving or “marching” symptoms, short duration, complete resolution, headache, personal or family history of migraine | Usually normal. Migrainous infarction is rare |
| Seizure | Positive > negative symptoms. Todd’s paralysis occurs after seizure. Altered level of consciousness | May identify source of focal seizures |
| Infection | Fever, encephalopathy, gradual onset, meningismus | Markers of encephalitis/cerebritis, typically diffuse and bilateral. AIS and CSVT occur in bacterial meningitis |
| Demyelination (ADEM, MS) | Gradual onset, multifocal symptoms, encephalopathy. Past history of DM events (optic neuritis, transverse myelitis) | Multifocal lesions, typical appearance (e.g., patchy in ADEM, ovoid in MS), typical locations (e.g., pericallosal in MS), less likely to show restricted diffusion |
| Hypoglycemia | Risk factor (e.g., insulin therapy), related to meals, additional neuroglycopenic symptoms | Bilateral, symmetrical. May see restricted diffusion. Posterior dominant pattern (neonates) |
| Watershed HIE | Risk factor (e.g., hypotension, sepsis), bilateral deficits | Bilateral, symmetrical, restricted diffusion in arterial border zones |
| Inborn errors of metabolism (MELAS, mito) | Pre-existing delays/regression, multisystem disease, abnormal biochemical profiles | Possible restricted diffusion but often bilateral, not within arterial territories. MR spectroscopy (e.g., high lactate) |
| Hypertensive encephalopathy (PRES) | Documented hypertension, bilateral visual symptoms, encephalopathy | Posterior dominant, bilateral, patchy lesions involving gray and white matter, usually no restricted diffusion |
| Vestibulopathy | Symptoms limited to vertigo. Positional. Nystagmus | Normal |
| Acute cerebellar ataxia | Gradual onset, bilateral, symmetric ataxia, postviral | Normal or diffusely swollen cerebellum |
| Channelopathies (AHC, EA, FHM) | Syndromic cluster of recurring symptoms. Switching sides. Headaches. Family history | Usually normal. Need MR angiography to exclude moyamoya in AHC |
ADEM, acute disseminated encephalomyelitis; AHC, alternating hemiplegia of childhood; AIS, arterial ischemic stroke; CSVT, cerebral sinovenous thrombosis; DM, demyelination; EA, episodic ataxia; FHM, familial hemiplegic migraine; HIE, hypoxic-ischemic encephalopathy; MELAS, mitochondrial encephalopathy with lactic acidosis and strokelike episodes; MR, magnetic resonance; MS, multiple sclerosis; PRES, posterior reversible encephalopathy syndrome.
Stroke-induced deficits usually begin suddenly and severity is maximal at the onset, aiding differentiation from mimics such as migraine and demyelination. However, symptoms can also have a fluctuating or stuttering onset [Braun et al., 2007]. TIAs are not well studied in children but likely occur prior to AIS in many cases. Previously, the definition of TIA was based on a deficit duration of less than 24 hours. This has now been updated to include negative imaging, since many ischemic episodes of less than 24 hours’ duration do cause acute infarcts seen on MRI. TIA, therefore, is best defined as “a brief episode of neurological dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than one hour, and without imaging (MRI) evidence of acute infarction” [Albers et al., 2002]. Differentiation of TIA from other causes of brief neurological deficits can be challenging. It is essential to recognize TIA symptoms, as prompt treatment may prevent a major stroke.
Risk Factors
The potential causes of AIS in infants and children are myriad, and frequently multiple in individual cases. Causes are also frequently referred to as “risk factors,” reflecting the variable strength of evidence for causality. Case-control and large population-based studies are continuing to provide important information regarding risk factors for childhood stroke. In the following discussion, we have taken an evidence-based approach that highlights well-established risk factors for childhood AIS, followed by a briefer review of additional considerations. Risk factors for perinatal stroke are distinct and discussed elsewhere (see Chapter 18). It is critical in individual children to determine all AIS mechanisms in a timely and thorough manner in order to identify risk of stroke recurrence and determine potentially appropriate therapies [Lanthier et al., 2000; Chabrier et al., 2000; Fullerton et al., 2007b]. This requires a tailored approach to investigations, based on the list of possible etiologies in Table 85-2.
Table 85-2 Potential Risk Factors for Childhood Arterial Ischemic Stroke
| Category | Common/Highly Probable | Uncommon/Possible/Uncertain |
|---|---|---|
| Arteriopathy | Inflammatory/para-infectious cPACNS (nonprogressive or progressive; large-vessel or small-vessel) Transient cerebral arteriopathy Focal cerebral arteriopathy Post-varicella angiopathy Infectious Bacterial meningitis Human immunodeficiency virus Dissection Internal carotid artery Vertebral artery Intracranial arteries Moyamoya Idiopathic Syndrome Neurofibromatosis-1, trisomy 21, other |
Secondary CNS vasculitis SLE, PAN, IBD Takayasu’s arteritis Infectious: mycoplasma, toxoplasmosis, RMSF, Lyme disease, cryptococcosis, chlamydia, Japanese encephalitis, coxsackie B4 and A9, influenza A, enterovirus, parvovirus B19 Postradiation vasculopathy RSCV (Call–Flemming syndrome) Genetic: COL4A1 CT (connective tissue) disease (Marfan’s, Ehlers–Danlos) Pseudoxanthoma elasticum Congenital: PHACES, progeria, Alagille’s syndrome, dwarfism (MOPDII), fibromuscular dysplasias |
| Cardiac | Complex congenital heart disease Cardiac surgery (e.g., Fontan) Cardiac catheterization (e.g., BAS) Bacterial endocarditis Atrial septal aneurysm Atrial septal defect? Patent foramen ovale? Venous thrombosis + right-to-left shunt |
Cardiomyopathy, myocarditis Aortic coarctation Severe ventricular dysfunction Atrial myxoma Valvular disease (e.g., rheumatic fever) Arrhythmia (atrial fibrillation) ECMO Cerebral angiography Embolism (air, fat, amniotic fluid) |
| Prothrombotic | Factor V Leiden Prothrombin gene 20210A Elevated lipoprotein (a) Protein C deficiency Lupus anticoagulant Anticardiolipin antibodies |
MTHFR, hyperhomocysteinemia Protein S deficiency Antithrombin III deficiency Factor VII/IX/XI? Plasminogen deficiency? Dysfibrinogenemia? Sticky platelet syndrome Pregnancy, puerperium |
| Hematological | Sickle cell disease Iron deficiency anemia |
Leukemia Thalassemias? Thrombocytosis? Polycythemia? Hemolytic uremic syndrome Immune thrombocytopenic purpura Thrombotic thrombocytopenic purpura |
| Medications/drugs | Oral contraceptives Chemotherapy (l-asparaginase) |
Cocaine, methamphetamine, ecstasy Ergots, triptans |
| Other | Migraine Inborn errors of metabolism: Fabry’s disease, homocystinuria, mitochondrial |
Metabolic syndrome: hypertension, diabetes, insulin resistance, dyslipidemia, atherosclerosis Cigarette smoking, secondhand smoke |
BAS, balloon atrial septostomy; cPACNS, childhood primary angiitis of the central nervous system; ECMO, extracorporeal membrane oxygenation; IBD, inflammatory bowel disease; MOPDII, Majewski osteodysplastic primordial dwarfism II; MTHFR, methylene tetrahydrofolate reductase; PAN, polyarteritis nodosum; PHACES, posterior fossa abnormalities, hemangiomas, arterial cervicocerebral anomalies, cardiac defects, eye anomalies, sternal defects and/or supraumbilical raphe; RMSF, Rocky Mountain spotted fever; RSCV, reversible segmental cerebral vasoconstriction; SLE, systemic lupus erythematosus.
A substantial proportion of children with AIS remain idiopathic, despite exhaustive investigations. The reported frequency of identifiable risk factors in children with AIS varies widely, depending on the population of children, criteria for risk factors, and extensiveness of etiological investigations. In single-center, tertiary-care studies, one possible risk factor was definable in 80–90 percent of children [Chabrier et al., 2000; Lanthier et al., 2000; Ganesan et al., 2003; deVeber, 2000]. Chronic diseases of childhood frequently underlie childhood AIS; however, approximately half of children are previously healthy [Ganesan et al., 2003]. Multiple risk factors converge in AIS in more than 50 percent of children, emphasizing the need for thorough investigations. Causes for AIS in children differ significantly from those in adults, in whom atrial fibrillation and atherosclerosis (associated with hypertension, dyslipidemia, smoking, and diabetes) predominate [deVeber, 2003]. A comprehensive list of potential risk factors for childhood AIS is included in Table 85-2. However, three main categories of risk factors have emerged as most prominent: arteriopathy, cardiac, and prothrombotic/hematological. These are discussed below, followed by an overview of additional risk factor considerations.
Arteriopathies
Diseases that directly affect the arteries supplying the brain have emerged as the leading cause of childhood AIS and are associated with an increased risk of both recurrence and poor outcome [Goldenberg et al., 2009; Fullerton et al., 2007b]. Recent studies have demonstrated consistently that arteriopathy is present in more than 50 percent of childhood AIS [Chabrier et al., 2000; Ganesan et al., 2003; Fullerton et al., 2007b; Amlie-Lefond et al., 2009a; Sebire et al., 2006; Braun et al., 2009]. Since arteriopathy is likely the single strongest predictor of stroke recurrence, thorough vascular imaging is essential for accurate detection and classification of arteriopathy [Lanthier et al., 2004; Strater et al., 2002; Askalan et al., 2001; Fullerton et al., 2007b; Braun et al., 2009]. Surveillance for a progressive nature in some arteriopathies is important, as it may dictate specific treatment [Danchaivijitr et al., 2006a; Kirkham, 2006; Braun et al., 2009].
Different systems have been proposed to classify childhood cerebral arteriopathies [Sebire et al., 2004], although some disagreement about terminology and proposed underlying pathophysiological processes still exists among experts. The three most important categories are discussed below:
Other, more rare, specific genetic cerebral arteriopathies include PHACES syndrome [Heyer et al., 2008], Alagille’s syndrome [Emerick et al., 2005], progeria [Merideth et al., 2008], primordial dwarfisms [Brancati et al., 2005], childhood forms of fibromuscular dysplasia [Kirton et al., 2006], and dominantly inherited COL4A1 mutations [van der Knaap et al., 2006].
Infectious/inflammatory arteriopathy
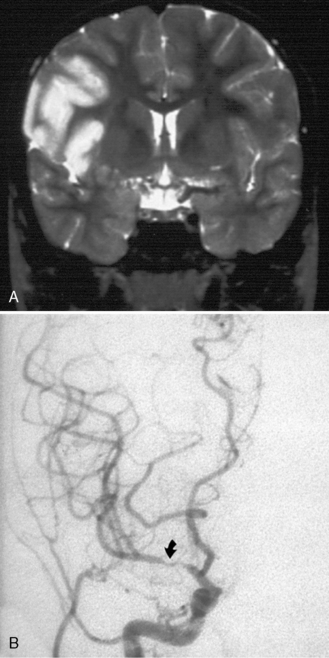
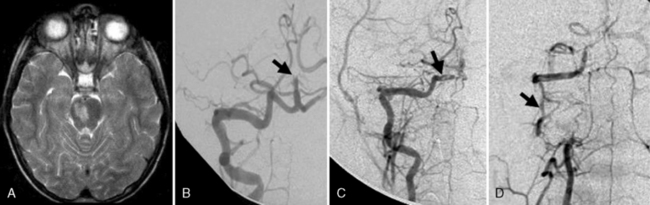
Many descriptions of this condition have been published under the name “transient cerebral arteriopathy” (TCA) [Chabrier et al., 1998; Shirane et al., 1992; Sebire, 2006] An example of the angiographic appearance of TCA is shown in Figure 85-4. Studies have suggested that such intracranial unilateral arteriopathy, even with features suggesting vasculitis on angiography, are non-progressive in more than 90 percent of cases [Braun et al., 2009]. When serial imaging demonstrates increased stenosis or extension to new arterial segments 6 months after onset, a chronic or progressive vasculitis or unilateral moyamoya may be suspected [Sebire, 2004]. Arterial occlusion, moyamoya vessels, and ACA (anterior cerebral artery) involvement may be associated with a progressive course.
Despite being very common, the exact cause or causes of Trasient cerebral Arteriopathy remain undetermined in most cases. This has resulted in the emergence of inconsistent terminology, diagnostic criteria, and treatment considerations; the debate continues. Recent efforts to acknowledge this uncertainty have used the more general term of “focal cerebral arteriopathy” (FCA) for any stenosing arteriopathy without evidence of a well-established arteriopathy, such as moyamoya or dissection [Amlie-Lefond et al., 2009a]. The most consistently suggested etiologies for TCA include infectious, postinfectious, and/or inflammatory mechanisms [Chabrier et al., 1998; Shirane et al., 1992]. TCA is considered by some to be a form of focal and self-limited vasculitis, and the term “nonprogressive childhood primary angiitis of the central nervous system” (cPACNS) has been applied [Elbers and Benseler, 2008]. However, similarities to postvaricella angiopathy (see below) [Askalan et al., 2001] have led to suspicion of parainfectious mechanisms. Still other possibilities, such as intracranial dissection, are often impossible to exclude.
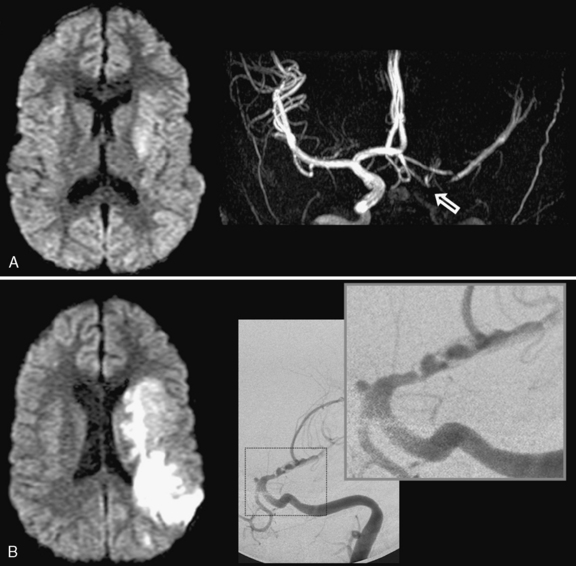
The role of infection in AIS is recognized increasingly [Emsley and Hopkins, 2008; Amlie-Lefond et al., 2009a]. The strongest example in childhood AIS is postvaricella angiopathy (PVA), which demonstrates the imaging features of TCA but occurs within 12 months of clinical varicella infection [Askalan et al., 2001; Lanthier et al., 2005]). PVA also typically affects lenticulostriate arteries, with an irregular, unilateral stenosis centered on the proximal ACA or MCA that resolves over months [Askalan et al., 2001; Lanthier et al., 2005]. An example of this disease is shown in Figure 85-5. PVA is well established as occurring in adults following facial “shingles” [Gilden et al., 2009; Amlie-Lefond et al., 1995]. Proposed mechanisms include viral reactivation from the trigeminal ganglion, with spread along the trigeminovascular bundle that innervates proximal cerebral arteries [Kleinschmidt-DeMasters and Gilden, 2001]. Elevated cerebrospinal fluid antibody titers to varicella zoster virus are found rarely [Ueno et al., 2002], though cerebrospinal fluid polymerase chain reaction and/or antibodies may increase sensitivity [Kronenberg et al., 2002; Gilden et al., 2009]. Histopathological studies can reveal viral particles or antigens in cerebral artery smooth-muscle cells [Eidelberg et al., 1986; Linnemann and Alvira, 1980; Reshef et al., 1985], as was the case in two 4-year-old children who died of PVA-associated stroke [Berger et al., 2000]. With varicella vaccination becoming routine, naturally occurring PVA is expected to decrease. However, AIS has been described in proximity to varicella immunization [Wirrell et al., 2004].
The role of other infections in arteriopathic childhood AIS is undergoing active investigation. Fever and recent “viral infection” symptoms commonly are present in children with AIS. One study of childhood AIS reported fever in 50 percent and leucocytosis in 26 percent [Ganesan et al., 2003]. In another recent international study of 673 children with AIS, an association between viral upper respiratory tract infection and arteriopathy was found, particularly in children with TCA/FCA [Amlie-Lefond et al., 2009a]. Whether a common inflammatory pathophysiology underlies arteriopathy provoked by active viral infection, postviral infection, or “idiopathic” inflammation remains to be established definitively.
More obvious and malignant infections also predispose to ischemic stroke. Arterial or venous stroke associated with bacterial meningitis has been identified in 5–12 percent of children and as many as 75 percent of infants [Chiu et al., 1995; Silverstein and Brunberg, 1995; Chang et al., 2003]. Tuberculous meningitis is an important infectious cause of childhood AIS in certain countries [Springer et al., 2009]. Extension of subarachnoid meningeal infection and inflammation, causing thrombophlebitis of the perforating arteries, is the likely mechanism. Children with human immunodeficiency virus (HIV) often develop a unique arteriopathy that predisposes to both ischemic and hemorrhagic stroke [Patsalides et al., 2002; Visudtibhan et al., 1999; Kleinschmidt-DeMasters and Gilden, 2001]. While limited to small numbers of cases, AIS has been associated with mycoplasma, toxoplasmosis, Rocky Mountain spotted fever, Lyme disease, cryptococcosis, Japanese encephalitis, coxsackie B4 and A9, influenza A, enterovirus, parvovirus B19, and chlamydia [Guidi et al., 2003; Takeoka and Takahashi, 2002]. Regional infectious agents also must be considered, such as malaria, which was commonly associated with stroke in children in Cameroon [Obama et al., 1994]. Only a small portion of these studies have described the possible mechanism, such as TCA with enterovirus or HIV [Ribai et al., 2003; Leeuwis et al., 2007].
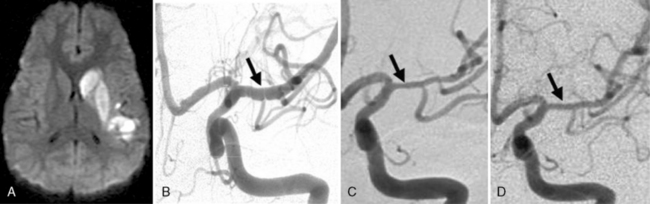
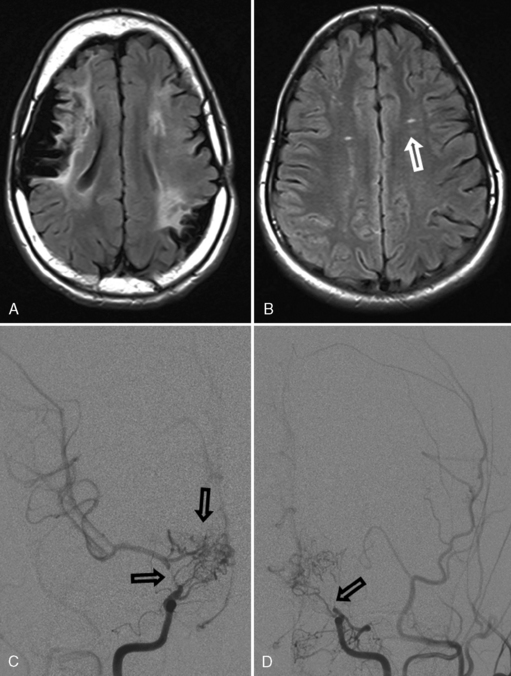
Arterial inflammation without a clear connection to active or previous infection is also a well-established form of cerebral arteriopathy, termed vasculitis. Recent reviews of childhood cerebral vasculitis are available [Elbers and Benseler, 2008]. Primary cerebral vasculitis is increasingly well described [Lanthier et al., 2001], and typically is referred to now as cPACNS [Elbers and Benseler, 2008; Benseler et al., 2006]. In contrast to “nonprogressive” cPACNS, which is very similar to TCA [Benseler et al., 2003; Elbers and Benseler, 2008], “progressive” cPACNS is frequently bilateral, and involves both proximal and distal cerebral arteries. Aggressive treatment may be required to prevent rapid deterioration and multifocal strokes. An example is shown in Figure 85-7. While the anterior circulation is involved more commonly, cPACNS diseases also can be isolated to the posterior circulation (Figure 85-8).
Some forms of cPACNS appear to be limited to distal, small cerebral arteries. Such small-vessel cPACNS lacks the acute stroke presentations described above, typically presenting with insidious symptoms such as headaches, cognitive decline, and personality change, and with MRI changes of multifocal enhancing lesions [Elbers and Benseler, 2008; Rafay et al., 2005; Benseler et al., 2005, 2006]. While conventional angiography is indicated, it may be negative in small-vessel cPACNS and brain biopsy is required for definitive diagnosis [Aviv et al., 2006; Benseler et al., 2005]. Small-vessel vasculitis may also be secondary to a variety of conditions, including infections, collagen vascular diseases, systemic vasculitides, and malignancies [Benseler and Schneider, 2004]. Systemic vasculitides and connective tissue diseases possibly associated with cerebral artery involvement include Kawasaki’s disease, Henoch–Schönlein purpura, polyarteritis nodosa, Wegener’s granulomatosis, systemic lupus erythematosus, Behçet’s syndrome, mixed connective tissue disease, Sjögren’s syndrome, and inflammatory bowel disease [Morfin-Maciel et al., 2002; Belostotsky et al., 2002; Berman et al., 1990; Morales et al., 1991; von Scheven et al., 1998; Graf et al., 1993; Benseler and Schneider, 2004]. Although systemic lupus erythematosus is often associated with AIS, vasculitis as the etiology for stroke is uncommon [Devinsky et al., 1988]. Large arteries can also be affected by secondary vasculitis, as in polyarteritis nodosa, or in Takayasu’s arteritis, where major branches off the aorta, including cervical arteries, are stenosed at their origin. Takayasu’s arteritis presents with asymmetrical and diminished pulses, renovascular hypertension, and stroke [Morales et al., 1991; Ringleb et al., 2005].
The range of infectious/inflammatory arteriopathies must be a primary consideration in all children with AIS, since they have important implications for investigation, treatment, and outcome. Diagnostic investigations should be tailored clinically, but may include screening for specific infections (serology, virology, polymerase chain reaction), in addition to routine hematological blood work, and inflammatory (erythrocyte sedimentation rate, C-reactive protein) and vasculitic (antinuclear antibodies, extractable nuclear antigens, complement, etc.) markers. In primary cPACNS, results of blood and cerebrospinal fluid studies are often negative [Elbers and Benseler, 2008; Riou et al., 2008]. More advanced studies to determine the markers and the relationship between infarction, inflammation, and arteriopathic AIS in children are under way. No specific treatment trials have been performed for the infectious/inflammatory cerebral arteriopathies in children. Whether an antiviral agent like acyclovir could alter the clinical course of PVA has not been determined but should certainly be considered in an immunocompromised child. Once a noninfectious inflammatory vasculitis is diagnosed, immunosuppression, with agents including corticosteroids, cyclophosphamide, azathioprine, or others, should be considered [Benseler and Schneider, 2004; Elbers and Benseler, 2008]. The advice of a pediatric rheumatologist in diagnosing and treating inflammatory cerebral arteriopathy should be sought.
Dissection and other physical injury
Craniocervical dissection represents a breach (usually mechanical) in arterial wall integrity with formation of a subintimal hematoma. AIS complicates arterial dissection by several mechanisms, including arterial occlusion at the dissection site by intraluminal thrombosis or wall hematoma, distal extension of the dissection along the artery, or, most frequently, by artery-to-artery thromboembolism. Dissection in the carotid or posterior vertebrobasilar circulation accounts for up to 20 percent of childhood AIS [Chabrier et al., 2003; Fullerton et al., 2001; Camacho et al., 2001; Rafay et al., 2006]. Extracranial arterial dissection occurs at predictable locations, including the proximal internal carotid artery and in the vertebral arteries at the C1–C2 level. Intracranial dissection can also occur, and carries the added risk of secondary subarachnoid hemorrhage [Fullerton et al., 2001]. Single-center series applying strict diagnostic criteria for dissection report that extracranial dissection is three times more frequent than intracranial dissection in children [Rafay et al., 2006]. A pooled analysis of published cases reported intracranial dissection to be more common [Fullerton et al., 2001], though distinguishing this from TCA is often impossible.
Dissection may result from trauma to the neck, spine, or retropharyngeal area secondary to obvious mechanical injuries or more subtle insults, including external manipulation, physical exertion, or contact sports [Rafay et al., 2006; Chabrier et al., 2003]. However, many appear to occur “spontaneously,” and with minor trauma being so common in children, a definitive causative event is often lacking. Adult studies have suggested that inherent differences in connective tissue structure, detectable on skin biopsy (but not physical examination) may predispose to arterial dissection [Martin et al., 2006]. Nonaccidental trauma should also be considered in small children with dissection [Agner and Weig, 2005]. New-onset head or neck pain is a prominent presenting feature in children, but clinical stroke symptoms may have their onset 7–10 days following pain or the traumatic event. Additional clinical findings may include an ipsilateral Horner’s syndrome, cervical bruit, or musculoskeletal cervical spine abnormalities. Recurrence rates are substantial in dissection, estimated at 20 percent or higher [Rafay et al., 2006; Fullerton et al., 2001, 2007b; Chabrier et al., 2003].
Whether dissection affects the intracranial or extracranial vessels, the medical management of dissections is controversial. Based on theoretical reasons, consideration of an anticoagulation approach is suggested by consensus guidelines [Monagle et al., 2008b; Roach et al., 2008]. Intracranial dissection is considered a relative contraindication due to the risk of subarachnoid hemorrhage. Adult dissection traditionally has been treated with anticoagulation; however, no randomized trials have been conducted and antiplatelet strategies have been suggested recently [Engelter et al., 2007]. Retrospective studies suggest that both are likely safe with generally good outcomes [Engelter et al., 2000]. An adult randomized trial appears logistically unfeasible, suggesting that a pediatric version will never occur. Long-term complications of pediatric dissection, such as recurrent stroke or pseudoaneurysm, require surveillance and further study [Tan et al., 2009a]. Activity restrictions and return-to-play guidelines following dissection are lacking, though pediatric stroke experts appear reluctant to recommend return to high-impact activities [Bernard et al., 2007]. A graded return to activity over months, with avoidance of high-risk activities (contact sports, chiropractic manipulation), is suggested.
Additional acquired vascular injuries may cause childhood AIS. Postradiation vasculopathy manifests with progressive large-vessel stenosis and cerebral ischemia years after irradiation. Younger children with optic chiasm gliomas or other sellar region tumors exposed to higher-dose, focused radiation are at increased risk [Omura et al., 1997]. MRI may demonstrate arterial wall thickening and contrast enhancement [Aoki et al., 2002]. Young adult survivors of childhood cancers recently were shown to harbor a higher risk of stroke [Jordan and Duffner, 2009a]. AIS also may occur in the setting of rapid brain expansion due to diffuse edema, space-occupying lesion, or hydrocephalus, where mechanical compression and occlusion of the anterior (subfalcine) or posterior (tentorial edge) cerebral arteries may occur. Stroke associated with catheterization of cerebral vessels for extracorporeal membrane oxygenation (ECMO) also has been described, with evidence of AIS in 50 percent of 44 autopsied children dying after ECMO [Jarjour and Ahdab-Barmada, 1994].
Moyamoya disease and syndrome
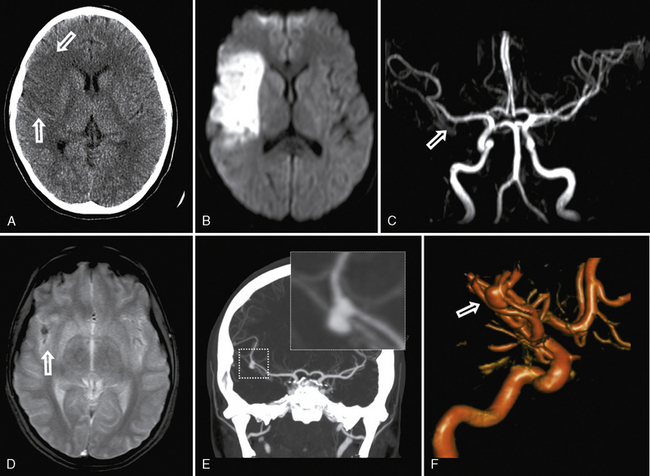
Moyamoya describes the progressive occlusion of the cerebral arteries at the circle of Willis, leading to formation of abnormal networks of small collateral vessels with a “puff of smoke” appearance on angiography (Figure 85-9) [Scott and Smith, 2009; Kuroda and Houkin, 2008]. The classic pattern involves the distal internal carotid arteries bilaterally, but unilateral variants and involvement of additional anterior and posterior circulation vessels are well described. Moyamoya disease is the idiopathic version described primarily in Asian populations, while moyamoya syndrome occurs in association with other conditions, including sickle cell disease (see below), Down syndrome, neurofibromatosis type 1, Williams’ syndrome, postradiation vasculopathy, and congenital arteriopathies (see below) [Jea et al., 2005; Scott and Smith, 2009]. Familial forms of moyamoya increasingly are described, with several potential genetic mechanisms proposed [Ikeda and Yoshimoto, 2005]. Moyamoya is, therefore, a heterogeneous syndrome and not a single disease, but recognizing the shared patterns of presentation is clinically relevant to ensure accurate investigation and treatment.
Moyamoya presents with unique clinical syndromes and patterns of stroke. In contrast to most other forms of AIS, an important mechanism for moyamoya-associated infarction is hypoperfusion. In the setting of internal carotid artery occlusion, perfusion is compromised most severely in the distal perfusion zones of the MCA and ACA in the deep hemispheric white matter. As a result, small (<1 cm) collections of adjacent infarcts accumulate in these “deep watershed” zones. Impaired cerebrovascular autoregulation in chronically underperfused brain territories appears to be an important underlying mechanism for these infarcts [Mikulis et al., 2005]. Therefore, TIA symptoms may be precipitated by alterations in PCO2, such as during hyperventilation (cerebral vasoconstriction) or breath holding (cerebral vasodilatation with “steal”). These infarcts may be subclinical, often with gradual cognitive or behavioral decline, although clear stepwise neurological deteriorations may occur. Moyamoya progresses slowly over time, causing accumulating injury and poor neurological outcomes [Suzuki and Kodama, 1983]. Progression of moyamoya may be faster in preschool-aged children [Kim et al., 2004]. In addition, artery-to-artery embolism of larger cerebral arterial branches can result in more typical AIS in moyamoya. Hemorrhagic stroke also can occur from the compensatory basal collateral vessels (“rete mirabile”), and this risk increases in early adulthood [Yoshida et al., 1993]. Headaches, often with migrainous features, are common in moyamoya and may precede other neurological symptoms.
MRI can suggest moyamoya with small flow voids in the basal ganglia and the infarct patterns described above. MR angiography is highly but incompletely sensitive at detecting moyamoya arteriopathy. Advanced fMRI and other perfusion studies may demonstrate impaired autoregulation [Mikulis et al., 2005; Mori et al., 2009]. However, conventional angiography is required in all children with moyamoya to confirm the presence of collaterals, characterize disease extent and severity, and image the external carotid circulation to determine options for revascularization surgery (see Figure 85-9) [Satoh et al., 1988]. Hypertension is common in moyamoya and may be secondary to associated renal artery stenosis [Kirton et al., 2006]. Doppler ultrasound and/or renal angiography during cerebral studies are indicated in such patients to exclude renovascular involvement.
Treatment of moyamoya is complex. Antiplatelet therapy with acetylsalicylic acid (ASA) often is employed and is likely safe. The chronic, progressive occlusion of distal internal carotid arteries, and consequent low perfusion, result in cerebral autoregulation shifting toward maximal vasodilatation in order to maintain cerebral perfusion. Carbonic anhydrase inhibitors, including acetazolamide, may shift acid–base balance to favor vasodilatation in moyamoya [Ikezaki, 2000]. However, in severe moyamoya, such medications may cause a “steal” or “reverse Robin Hood” phenomenon, whereby dilatation of healthier arteries diverts perfusion away from the most critically affected areas, which subsequently infarct. Such interventions must be considered carefully in severely affected children. Use of antihypertensives in children with moyamoya and hypertension must also be considered carefully, as even small decreases in cerebral perfusion can lead to infarction.
Surgical revascularization procedures are highly successful at improving cerebral perfusion in children with moyamoya [Scott and Smith, 2009]. Direct procedures connect extracranial blood vessels, such as the superficial temporal artery, to distal portions of intracranial vessels, usually the MCA. Direct revascularizations are technically limited in small children and carry some risk of reocclusion. In children, indirect procedures are now employed more frequently where extracranial vessels are approximated to the cortical surface overlying areas of decreased perfusion. Angiogenic signals from ischemic parenchyma subsequently result in neovascularization of the affected areas over a course of months. Encephalodural synangiosis (EDAS) and encephalodural myosynangiosis (EDMS) are common procedures transferring extracranial arteries alone or within scalp muscle, respectively, to the pial surface. A study of 143 children undergoing pial synangiosis found that more than 90 percent enjoyed symptom-free survivals with follow-up of more than 5 years [Scott et al., 2004]. Studies comparing the different surgical approaches are lacking. Indirect procedures are now used widely and sometimes combined with direct methods [Fung et al., 2005]. Perioperative infarctions likely occur in less than 10 percent of cases [Fung et al., 2005; Kim et al., 2004]. Younger age of onset in moyamoya may carry an increased risk of recurrent stroke and poor outcome, suggesting that early surgery is indicated [Kim et al., 2004]. Preliminary evidence suggests revascularization also may be helpful in some cases of sickle-related moyamoya [Hankinson et al., 2008]. Success of revascularization surgery can be evaluated with serial angiography, as well as new cerebrovascular reserve MRI methods [Mikulis et al., 2005].
Cardiac Risk Factors
Heart disease is a major risk factor for AIS in infancy and childhood, present in 12–28 percent of cases [Chabrier et al., 2000; Ganesan et al., 2003; deVeber, 2000]. Abnormalities in cardiac structure, flow, rhythm, endothelium, ventricular function, valves, blood viscosity, and other factors may contribute to thrombus generation and subsequent cerebral embolization. Multiple AIS in multiple, distinct vascular territories (e.g., left and right) suggests a cardiac source. Complete cardiac evaluations should be expedited in acute stroke of unknown etiology, to guide acute medical management and emergency cardiac treatment when indicated.
Complex congenital heart disease, particularly cyanotic lesions, carries the highest stroke risk. Both catheterization procedures and surgical repairs increase risk further. A recent single-center case-control study of more than 5500 children undergoing cardiac surgery documented a symptomatic AIS risk of 5.4 per 1000 (1:185), with reoperation being an independent risk factor [Domi et al., 2008]. This risk is comparable to or higher than the figure in previous studies [Menache et al., 2002]. Certain surgeries carry elevated short- and long-term risk. With the Fontan procedure, risk estimates of immediate or delayed stroke range from 3 to 19 percent [Monagle and Karl, 2002; Barker et al., 2005; Kaulitz et al., 2005]. Risk of symptomatic stroke following cardiac catheterization in children is estimated at 1:600–700 [Liu et al., 2001] with interventional more risky than diagnostic catheterizations [Hamon et al., 2006]. Balloon atrial septostomy prior to transposition switch surgery carries a stroke risk approaching 50 percent, according to one study of 29 patients [McQuillen et al., 2006]. Many ischemic brain lesions related to congenital heart disease are asymptomatic in young children and often are present preoperatively [Miller et al., 2004].
Intracardiac septal defects, such as atrial septal defect and patent foramen ovale, remain controversial risk factors for stroke in the young [Kizer and Devereux, 2005] and have not been well studied in children. Atrial septal lesions create the potential for intermittent right-to-left intracardiac shunting with paradoxical embolization of systemic venous clots to the brain [DiTullio et al., 1992]. When such right-to-left intracardiac shunts are detected, further investigation for systemic venous thrombosis may be indicated [Mas et al., 2001]. Septal lesions such as patent foramen ovale are common in the pediatric population. Associations between patent foramen ovale and otherwise unexplained stroke have been demonstrated repeatedly in young adults [Mas et al., 2001; Handke et al., 2007], although patent foramen ovale may not increase the risk of recurrent stroke [Almekhlafi et al., 2009]. A recent adult analysis suggests that a high proportion of patent foramen ovale in young cryptogenic stroke patients are likely incidental [sheikh-Ali et al., 2009]. Patent foramen ovale associated with atrial septal aneurysm [Mas et al., 2001] and thrombophilias carries increased stroke risk [Pezzini et al., 2003; Rodriguez and Homma, 2003]. The indications for medical and surgical (closure) treatment of uncomplicated patent foramen ovale remain controversial [Tamayo and Harrer, 2009].
Several acquired cardiac etiologies predispose to AIS. Infective endocarditis carries a high risk of multifocal embolic AIS and risk of mycotic aneurysm and intracerebral hemorrhage (Figure 85-10). Detection of signs of infective endocarditis, including fever, physical markers (Janeway lesions, Roth spots), and history of subacute constitutional symptoms or recent dental work, enables early diagnosis and treatment. Anticoagulation is relatively contraindicated in infective endocarditis due to bleed risk from mycotic aneurysms, and recent adult studies have demonstrated frequent subclinical microbleeds with susceptibility-weighted MRI [Klein et al., 2009]. Other acquired cardiac etiologies include poor left ventricular function secondary to cardiomyopathy (infectious, metabolic) [Hays et al., 2006], valvular heart disease (e.g., rheumatic fever, artificial valves), arrhythmia, and atrial myxoma [Omeroglu et al., 2006].
All children with AIS of undetermined cause should have prompt cardiac evaluation, including electrocardiogram and echocardiography. Agitated saline “bubble studies” can help establish the presence of patent foramen ovale and atrial septal defects. In adults, transesophageal echocardiography is superior to transthoracic echocardiography in diagnosing a cardiac stroke source, detecting 31 percent more treatable lesions [de Abreu et al., 2008; de Bruijn et al., 2006]. However in young children with a thin chest wall, transthoracic echocardiography may suffice. The roles of cardiac MRI and ambulatory cardiac rhythm monitoring in childhood AIS have not been investigated.
Consensus across published guidelines suggests that proven or suspected cardiogenic AIS requires specific management [deVeber and Kirkham, 2008]. With a presumed dominant role of the coagulation system, acute anticoagulation with low-molecular-weight heparin (LMWH) usually is indicated. If detected, bacterial endocarditis also requires urgent antibiotic treatment and investigation for mycotic aneurysm [Venkatesan and Wainwright, 2008]. Duration of anticoagulation depends on individual factors that determine recurrence risk, including circumstances of the initial stroke (spontaneous or procedure-related), corrected or uncorrected cardiac defect, prothrombotic testing results, and others.
Prothrombotic and Hematological Disorders
Thrombophilia, or prothrombotic disorders, refers to abnormalities of the coagulation, fibrinolytic, and platelet systems that predispose to pathological thrombus formation. Studies in childhood stroke are complicated by the developmental evolution of the hemostatic system in childhood, a complex variety of disorders, few case-control studies, and inconsistent testing and laboratory methods. With these limitations in mind, a role for prothrombotic disorders likely exists in childhood AIS, though most patients likely have an additional “triggering” risk factor at the time of AIS [Barreirinho et al., 2003; Strater et al., 2002]. Detailed reviews are available [Mackay and Monagle, 2008; Barnes and deVeber, 2006].
Inherited or acquired coagulation disorders have been identified in 20–50 percent of children with AIS [Bonduel et al., 1999; deVeber et al., 1998b; Barnes and deVeber, 2006; Herak et al., 2009]. Factor V Leiden is one of the strongest associations [Strater et al., 2002], with a meta-analysis of five case-control studies suggesting a combined odds ratio of 4.3 for stroke with heterozygote factor V Leiden [Chan et al., 2000]. Elevated levels of lipoprotein (a) and Protein C deficiency are both more common in children with AIS compared to controls [Nowak-Goettl et al; 1999; Barnes and deVeber, 2006]. Although mutations in methylene tetrahydrofolate reductase (MTHFR) are associated with childhood stroke, they are also common in the population and their significance without hyperhomocysteinemia is unclear [Nowak-Goettl, 1999; Ganesan et al., 2003; Mackay and Monagle, 2008]. In contrast to most prothrombotic states that promote venous thrombosis, prothrombin G20210A mutations are associated more frequently with arterial thrombosis in children [Young et al., 2003]. Meta-analysis of studies to date suggests an odds ratio of 3.5 in childhood AIS [Barnes and deVeber, 2006]. Case-control studies also have confirmed an increased occurrence of antiphospholipid antibodies and lupus anticoagulant in childhood AIS [Kenet et al., 2000; Ganesan et al., 2003].
A systematic review of published case-control data has reported increased pooled odds ratios for childhood AIS for protein C deficiency (6.5) and methylene tetrahydrofolate reductase (MTHFR C677T) (1.7), but ratios were not significant for protein S deficiency (1.14), AT (antithrombosis) deficiency (1.02), APCr (activated Protein C resistance) (1.34), prothrombin G20210A (1.10), or elevated plasma homocysteine (1.36) [Haywood et al., 2005]. Presence of certain prothrombotic states increases stroke recurrence risk in children, including factor V Leiden, protein C deficiency, and elevated lipoprotein (a) [Ganesan et al., 2006; Strater et al., 2002], but not elevated ACLA (anticardiolipin antibody) antibodies [Lanthier et al., 2004]. Plasminogen activator inhibitor abnormalities do not appear increased in childhood AIS [Nowak-Gottl et al., 2001]. Additional prothrombotic factors of interest include abnormal levels of plasminogen, fibrinogen, and coagulation factors (VIII, IX, XI), and an increasing number of genetic polymorphisms. Although platelet abnormalities are likely of importance in arterial thrombosis, only limited studies have been conducted in childhood AIS [Herak et al., 2009; Barnes and deVeber, 2006].
Sickle cell disease
Sickle cell disease (SCD) is the most common hematological disorder associated with cerebrovascular disease in children and increases ischemic stroke risk 400-fold. Overall, about 25 percent of SCD children develop cerebrovascular complications, including overt AIS, subclinical small AIS, moyamoya, or hemorrhagic stroke [Pegelow, 2001]. Strokes are ischemic in 75 percent and hemorrhagic in 25 percent, the latter increasing with age [Satoh et al., 1988]. Stroke is responsible for 10 percent of SCD mortality [Manci et al., 2003]. There are two primary mechanisms for AIS in SCD. First, a progressive internal carotid (large-vessel) moyamoya arteriopathy can develop with typical moyamoya-type deep watershed or large-artery territory infarcts (see moyamoya above). The etiology of this arteriopathy is thought to relate to the chronic effects of increased carotid blood-flow rates with sickled red cells damaging endovascular surfaces. Recent studies indicate that proinflammatory genetic polymorphisms also may play a role [Hoppe et al., 2007]. Second, SCD may be associated with occlusion of small cerebral end-arteries by sickled cells and related thrombosis, resulting in randomly distributed multifocal small infarcts. Stroke may occur at any time in SCD, though risk is highest during an acute crisis.
Transcranial Doppler studies are a noninvasive means of following flow rates related to large-vessel vasculopathy, with flows above 200 cm/s predicting increased stroke risk [Adams et al., 1992, 1998]. The STOP (Sickle Cell Transfusion Stroke Prevention) randomized controlled trial demonstrated that regular transfusion therapy (hemoglobin S below 20–30 percent) produced a 90 percent relative risk reduction in initial stroke in children with elevated velocities [Adams et al., 1998]. Recurrent stroke rates were also reduced. A follow-up study showed that stroke risk returns with discontinuation of transfusions [Adams et al., 2005]. This suggests that most families currently need to continue with arduous blood transfusions every 3–6 weeks, including chelation therapy and screening for iron overload [Monagle et al., 2008b; Roach et al., 2008; Aygun et al., 2009]. A similar trial looking at children with small vessel SCD is under way (Silent Infarct Transfusion [SIT] trial; http://sitstudy.wustl.edu/). Increased ultrasound screening and decreased stroke rates have been documented since STOP was published [Fullerton et al., 2004; Armstrong-Wells et al., 2009].
In addition to annual TCD (Trancranial Doppler) screening beginning at age 3, serial MRI should be considered, as this can detect silent infarcts secondary to small-vessel disease. MR angiography should be considered following large-vessel disease and possible moyamoya [Zimmerman, 2005]. Additional stroke prevention strategies in SCD currently under evaluation include hydroxyurea [Heeney and Ware, 2008; Ware et al., 2004] and nocturnal oxygen supplementation [Kirkham et al., 2001b]. Antithrombotic therapy has not been studied but aspirin should be considered, especially in cases with large-vessel vasculopathy, prothrombotic abnormalities, or recurrent stroke [Wolters et al., 1995]. Revascularization surgery may be successful in SCD-related moyamoya [Fryer et al., 2003; Hankinson et al., 2008]. Bone marrow transplantation is feasible in SCD [Walters et al., 1996], but may carry an increased risk of neurological complications in children with cerebrovascular disease [Walters et al., 1995].
Iron deficiency anemia
Another hematological risk factor is iron deficiency anemia. This has emerged as an important and treatable risk factor for AIS, present in up to 40 percent of childhood AIS [Ganesan et al., 2003]. Iron deficiency anemia is a common condition of infancy and early childhood, and previous associations with stroke [Hartfield et al., 1997] now are supported by case-control evidence [Maguire et al., 2007]. The underlying pathophysiology explaining this relation has not been established. Children with low serum hemoglobin and microcytosis should have their iron storage markers checked and be supplemented promptly when these are low.
Additional Considerations
A comprehensive list of probable and possible additional disorders associated with childhood AIS are listed in Table 85-2. Migrainous infarction is uncommon in children [Feucht et al., 1995; Nezu et al., 1997; Santiago et al., 2001] and is a diagnosis of exclusion, requiring strict adherence to international headache society criteria. These include focal neurological symptoms consistent with a migrainous aura that do not resolve in the presence of migraine headache in an at-risk individual (past or family migraine history) with confirmation of acute infarction on MRI. Lesions tend to favor the posterior circulation. Migraine with aura is an important stroke risk factor in young women who are smokers on oral contraceptives [Rothrock, 1993; Bousser and Welch, 2005]. Special treatment considerations in migrainous infarction should address the role of possible vasospasm. Fortunately, good evidence supports the use of flunarizine for migraine prophylaxis in children, as the mechanism of calcium channel blockade appears well suited to preventing migrainous infarction. Such patients also should avoid vasoconstrictor agents, such as triptans, ergots, or sympathomimetics. Secondary stroke prevention in migrainous infarction also includes ASA.
Several inborn errors of metabolism predispose to AIS [Testai and Gorelick, 2010]. Homocystinuria leads to arterial and venous thrombosis, can be recognized by clinical features and laboratory markers, and often is treatable [Bodensteiner et al., 1992]. Fabry’s disease, due to X-linked α-galactosidase deficiency, results in a cerebral arteriopathy and stroke, in addition to ocular, peripheral nerve, and renal complications. Both of these conditions are essential to recognize, as they are highly treatable with vitamin B supplementation and enzyme replacement, respectively. In mitochondrial diseases, such as mitochondrial encephalopathy, lactic acidosis and strokelike episodes (MELAS), clinical presentations mimic AIS. However, evidence supports a metabolic rather than ischemic mechanism [Mudd et al., 1985; Testai and Gorelick, 2010]. Cerebral lesions are bilateral and predominantly posterior and do not respect arterial territories. Elevated serum or cerebrospinal fluid lactates may be present and routine genetic testing is sensitive. Treatment with l-arginine to promote vasodilatation has been suggested but is controversial, given evidence against a primary vascular mechanism [Koga et al., 2005]. The role of other genetic factors in causing stroke is increasingly complex, ranging from single-gene disorders imparting a high risk to complex, multigene polymorphisms influencing additional risk factors. These complexities are beyond the scope of this chapter, and are reviewed elsewhere [Dichgans, 2007]. The interaction of genes with other known risk factors is being explored in childhood stroke [Kirkham, 2004].
Use of medications and illicit drugs should be determined. The risk of ischemic stroke is increased in young women using oral contraceptives, even with newer low-estrogen preparations [Gillum et al., 2000]. The role of typical metabolic and lifestyle factors that are important in adult stroke – diabetes, hypertension, dyslipidemia, smoking, and obesity – is undetermined in children. Hypercholesterolemia has been found in up to 9 percent of children with stroke [Ganesan et al., 2003]. Fasting serum cholesterol, triglycerides, and high-density and low-density lipoprotein fractions should be obtained and compared to aged norms [Muhonen and Lauer, 1996]. Although unstudied, long-term preventative care after childhood AIS should include attention to modifiable risk factors to promote arterial health, particularly in children with arteriopathy. This would include healthy lifestyle measures, such as regular exercise, a balanced diet with minimization of saturated and trans fats, and the avoidance of smoking and street drugs. Such simple interventions might mitigate decades of added insult to an already injured cerebral artery [Roach et al., 2008].
Diagnosis: Neuroimaging
Computed Tomography
Head CT usually is the initial type of neuroimaging performed in childhood AIS but is insufficient to confirm or exclude the diagnosis. Initial CT scan may miss childhood AIS diagnosis in an overwhelming proportion (84 percent) of children with AIS [Srinivasan et al., 2009], consistent with the 63 percent false-negative rate for initial CT in a Canadian pediatric AIS study [Rafay et al., 2009]. CT findings of AIS in children are otherwise similar to adults, with infarction represented by focal, increasing hypodensity conforming to an arterial territory (see Figure 85-10). CT can also demonstrate intra-arterial thrombus (“hyperdense artery sign”), hemorrhagic transformation, and malignant cerebral edema (see below). Contrast-enhanced CT angiography, available at most centers, can provide detailed images of cerebral arteries down to third-order arteries or better, and may confirm the characteristics of an arteriopathy or embolic occlusion (see Figure 85-10). Small but significant age-related risks of radiation exposure need to be considered with CT use in children [Brenner and Hall, 2007].
Magnetic Resonance Imaging
MRI has revolutionized the diagnosis, management, and study of cerebrovascular disease and avoids radiation exposure [Muir et al., 2006; Hunter, 2002]. Diffusion-weighted MRI (DWI) is exquisitely sensitive at detecting early brain ischemia (see Figures 85-5, 85-7, and 85-10). DWI is the gold standard for acute AIS imaging for diagnosis, and is the investigation of choice when time and access permit [Groenendaal et al., 1995; Warach and Baron, 2004]. Acute ischemic lesions demonstrate restricted diffusion, evidenced by combined DWI hyperintensity and apparent diffusion coefficient map hypointensity. This pattern typically normalizes over 1–2 weeks, helping to determine infarct age. Traditional MR sequences, including T2-weighted images and fluid-attenuated inversion recovery (FLAIR), can demonstrate AIS within several hours, and abnormalities persist into the long term. MRI is unquestionably superior to CT in detecting strokes in children, especially early and small lesions and those located in the posterior fossa [Ameri and Bousser, 1992; Fiebach et al., 2002; Kidwell et al., 2004; Paonessa et al., 2009]. Limitations of MRI include longer completion times, technical complexities, contraindications such as implanted metallic devices, and the need for sedation in younger children.
Advanced MR procedures are improving our understanding and assessment of pediatric stroke. Modalities such as gradient ECHO and susceptibility-weighted methods can detect small amounts of blood, possibly predicting the risk of hemorrhagic transformation (see Figure 85-10). Perfusion-weighted imaging demonstrates regional brain perfusion abnormalities but pediatric studies have been limited [Huisman and Sorensen, 2004]. Arterial spin labeling MRI also may assess perfusion, with abnormalities correlating with final infarct volume [Chen et al., 2009]. MR perfusion using dynamic susceptibility contrast reveals focal perfusion deficits in children with SCD [Kirkham et al., 2001a]. Single-photon emission computed tomography (SPECT) and CT perfusion also can evaluate perfusion. The ultimate goal of such modalities is to detect “at-risk” brain tissue, represented by diffusion–perfusion “mismatch,” though this method remains unproven, even for adult stroke. Other advanced MRI techniques include fMRI of cerebrovascular reserve to detect impaired autoregulation in the setting of chronic hypoperfusion [Mikulis et al., 2005; Mori et al., 2009], diffusion tensor imaging (DTI) and tractography, and localization of brain activity with fMRI. MRI “wall imaging” may demonstrate concentric vessel-wall enhancement, suggesting inflammation [Swartz et al., 2009], a potentially valuable marker in pediatric arteriopathies.
MR angiography of the intracranial and neck vessels should be performed simultaneously with brain MRI in children with AIS. MR angiography can provide reasonable images of the major first- and second-order cerebral arteries (see Figures 85-7 and 85-10) [Husson and Lasjaunias, 2004; Koelfen et al., 1995; Wiznitzer and Masaryk, 1991], but remains limited in smaller-vessel diseases [Ganesan et al., 1999; Husson et al., 2002]. MR angiography can suggest the nature and extent of underlying arteriopathy. An abrupt arterial “cutoff” suggests embolic stroke, while irregular stenosis favors arteriopathy. However, time-of-flight MR angiography relies on flow signal and, in areas of turbulent flow or stenosis, signal dropout can mimic occlusion. Gadolinium RA (MR angiography) has better resolution and specificity than “time-of-flight” imaging.
Conventional Angiography
Despite advances in noninvasive methods, conventional angiography (CA) remains the gold standard for cerebrovascular imaging [Hill et al., 2007] (see Figures 85-4, 85-5,and 85-7 to 85-9). Specific arteriopathies may be diagnosed only by seeing the detail that CA provides; these arteriopathies include “string,” “double lumen,” or intimal flap signs in dissection, “string of beads” in vasculitis, and arteriopathies limited to small-caliber arteries. CA should be performed in children with AIS when a specific etiology remains undetermined after initial MR angiography or CT angiography, and considered in others to characterize the nature and extent of a probable arteriopathy better. Ideally, CA should be performed where pediatric expertise is available. Several large experiences recently published suggest that the rate of combined intra- and postprocedural complications for diagnostic CA is below 0.5 percent [Burger et al., 2006; Wolfe et al., 2009], a lower rate than in adults [Johnston et al., 2001]. An additional concern with CA is radiation exposure, with recent estimates suggesting that a child undergoing diagnostic neuroangiography may incur a lifetime attributable risk of cancer approaching 1 percent [Raelson et al., 2009].
Certain causes of childhood AIS merit special imaging considerations. Inflammatory or postinfectious arteriopathies can be screened for and followed on MR angiography [Aviv et al., 2006]. However, CA will reveal the details and extent of disease, particularly when smaller vessels are affected. Small-vessel cerebral vasculitis presenting without AIS is typically negative on CA, requiring diagnosis with brain biopsy [Benseler et al., 2005]. Diagnosing arterial dissection can be challenging and may be missed on CT angiography and MR angiography [Tan et al., 2009b]. Axial fat saturation T1 can reveal wall hematoma (“crescent sign”) but, otherwise, CA may be required. Moyamoya patterns generally are detectable on MR angiography, but CA is required to visualize collaterals and the full stage and extent of disease, as well as for surgical planning [Scott and Smith, 2009].
Treatment
These guidelines are predominantly concordant for major treatment recommendations, though they differ in certain areas. Comparison of these guidelines [deVeber and Kirkham, 2008] and additional reviews of childhood stroke treatment [Bernard et al., 2008] are available. With these limitations in mind, the following provides a chronological approach to the comprehensive treatment of the child with stroke. This evolves from emergent, hyperacute treatments in the first hours, including neuroprotective care and thrombolysis, to issues of antithrombotic therapy. Disease-specific treatments are discussed under their respective headings above. Chronic issues of secondary stroke prevention, rehabilitation, and support are discussed in the context of outcomes. Many of the same principles apply to later sections on CSVT and hemorrhagic stroke, where specific additional differences will be highlighted.
Emergent Treatment
Neuroprotection
Immediate measures to prevent secondary brain injury are an essential, easily overlooked aspect of emergency care in children with stroke [Hutchison et al., 2004]. The mantra of “time is brain” certainly also applies to children. and is supported by evidence that nearly 2 million neurons may die per minute of arterial occlusion [Saver, 2006]. Neuroprotective therapy is aimed at delivering an adequate supply of oxygen and glucose to the ischemic tissue in the penumbra (optimizing blood oxygenation and glucose, blood pressure, and cerebral perfusion) while reducing the neuronal metabolic demand (aggressive control of fever and seizures). An improved understanding of the cellular and molecular processes that mediate ischemic brain injury has generated numerous neuroprotective trials in adult stroke [Sugawara et al., 2004]. Results have been disappointing and, even with successful medication development in the future, unique aspects of the developing brain require additional consideration [Ferriero, 2004]. Despite the failure of neuroprotective trials to discover a “magic bullet,” simple attention to physiological variables and application of conservative, supportive measures may help accomplish the same objective. Existing evidence on such strategies comes from adult stroke [Becker, 2000], but is likely applicable to children in most regards.
Both hyperglycemia and hypoglycemia are associated with poorer neurological outcomes in adult stroke [Baird et al., 2003; Yager and Thornhill, 1997] and pediatric critical illness [Faustino and Apkon, 2005; Srinivasan et al., 2004]. Therefore, maintenance of normoglycemia, potentially including insulin for marked hyperglycemia, appears reasonable in children with acute stroke. Ideal blood pressure parameters in acute childhood stroke are unknown, and extrapolation from adult guidelines is problematic because adults frequently have chronic pre-existing hypertension. Malignant hypertension is a relative contraindication to anticoagulation therapy [Monagle et al., 2008b], but modest increases in blood pressure in the days following stroke may be a normal physiological response. Maintenance of blood pressure between the 50th and 90th percentiles seems reasonable in most cases. Elevated pressures can be lowered gradually with titration of short-acting agents (e.g., labetalol), while fluids and pressor agents may correct relative hypotension to avoid penumbral hypoperfusion. Maintenance of normal blood oxygen and carbon dioxide levels is prudent in the face of tenuous cerebral perfusion and acute alterations in autoregulation [Singhal, 2007a; Paediatric Stroke Working Group, 2004]. The role of supplemental oxygen in normoxic patients is controversial, with mixed evidence from adult stroke studies [Singhal et al., 2007b; Ronning and Guldvog, 1999].
Therapeutic hypothermia has not been studied in childhood AIS and mixed effects have been reported in neonatal HIE [Thoresen and Whitelaw, 2005; Gluckman et al., 2005], childhood traumatic brain injury [Hutchison et al., 2008], and adult stroke [Olsen et al., 2003]. Ischemic brain injury clearly is exacerbated by hyperthermia in animal models and adult stroke [Saini et al., 2009; Hajat et al., 2000; Jorgensen et al., 1996; Volpe, 2001b]. Both adult and pediatric stroke care guidelines [Adams et al., 2007; Paediatric Stroke Working Group, 2004] recommend temperature control to avoid hyperthermia, including aggressive treatment of infection and regular dosing of antipyretics. The addition of simple external cooling measures to attain normothermia also may be indicated. Acute seizures are common in children with AIS and likely worsen ischemic brain injury [Wirrell et al., 2001]. A low threshold for clinical detection and electroencephalographic (EEG) investigation should lead to quick treatment with anticonvulsants. Additional specific treatments influencing outcomes in adult stroke that are unstudied in children include aspiration prevention, optimal nutrition, skin care, and deep venous thrombosis prophylaxis. Specialized acute care markedly reduces the risk of death or dependency in adult stroke, and should be offered to children with stroke whenever possible [Teasell et al., 2006; Paediatric Stroke Working Group, 2004; Stroke Unit Trialists’ Collaboration, 2000]. Children with suspected acute stroke should be stabilized and transferred promptly to an institution that offers specialized pediatric neurovascular care [Hutchison et al., 2004; Paediatric Stroke Working Group, 2004].
Thrombolysis
Thrombolytic therapy has revolutionized acute management and improved outcomes in adult stroke. Thrombolytic agents, such as tissue plasminogen activator (tPA), promote the conversion of plasminogen into plasmin to dissolve occlusive arterial thrombi. The benefits of the original National Institute of Neurological Disorders and Stroke (NINDS) thrombolysis trial remain generally representative in adults with an absolute risk reduction in poor outcome of approximately 15 percent (number needed to treat, 7–8) [NINDS TPA, 1995]. The risk of bleeding related to thrombolysis outweighs the benefits after several hours post onset (4.5 hours for intravenous). The safety and efficacy of thrombolysis in children remains unstudied. Despite benefits in adults and similar fundamental pathophysiology in childhood AIS, other unique components of pediatric stroke must be considered, including disease types, thrombosis and intrinsic fibrinolysis systems, cerebrovascular autoregulation, and others. These differences may alter both the benefits and anticipated risks of thrombolysis substantially in children with AIS.
Thrombolysis for childhood AIS has been reported in limited administrative database studies [Janjua et al., 2007; Shi et al., 2008] and isolated case reports [Gruber et al., 2000; Carlson et al., 2001; Noser et al., 2001; Kirton et al., 2003; Golomb et al., 2003c; Benedict et al., 2007; Cannon et al., 2001; Thirumalai and Shubin, 2000; Arnold et al., 2009]. In the only published observational study of thrombolysis in childhood stroke, among 680 children with AIS from the International Pediatric Stroke Study, 15 (2 percent) received intravenous or intra-arterial tPA by individual center treatment methods [Amlie-Lefond et al., 2009c]. Violations of adult tPA protocols were common. Outcomes included death (2), neurological deficit (12), and normal recovery (1), while 4 out of 14 patients (29 percent) had post-tPA bleeds. When these 15 IPSS cases were compared to individual pediatric AIS thrombolysis case reports, a publication bias toward normal outcomes was observed in the latter. Among all published childhood AIS cases receiving tPA, all those with normal outcomes were teenagers, with both deaths occurring in children below 10 years of age.
Many challenges need to be overcome before thrombolysis can be used in childhood AIS. Foremost among these is the generation of age-appropriate safety data. Delays to diagnosis represent an important limitation, though studies suggest that many children with AIS arrive to hospital within thrombolysis-eligible timeframes (i.e., 3–6 hours) [Ichord et al., 2007]. Tools to assist in identifying children with the worst prognosis are becoming available, including a pediatric modification of the National Institutes of Health Stroke Scale (PedNIHSS) [Ichord et al., 2005] and acute imaging markers [Boardman et al., 2005; Domi et al., 2009]. In adults, tPa-associated bleeding risk is lower in younger patients [Hill and Buchan, 2005]. However, tPA contraindications specific for children will require completion of a safety study and cannot be extrapolated directly from adult evidence. Intra-arterial administration of thrombolytics and mechanical therapies such as clot-retrieval devices [Lutsep, 2008] have not yet proven superior in adult stroke. Risk profiles of such interventions still are being established, and the availability of neurointerventionalists experienced with children and pediatric angiography is another limiting factor. How the unique differences in children’s vascular and coagulation systems and stroke mechanisms affect thrombolysis safety and efficacy also is unknown. Extending adult thrombolysis protocols to older adolescents seems possible but must be considered risky in younger children. Prospective, multicenter safety trials of thrombolysis in pediatric AIS are now in development [Amlie-Lefond et al., 2009b; Whelan et al., 2008].
Urgent Treatment
Antithrombotic therapies
Managing childhood AIS without antithrombotic agents is no longer considered a reasonable option in most cases, with evidence supporting safety and high rates of early recurrence when no antithrombotic treatment is provided. However, choosing between antiplatelet and anticoagulation treatments in the acute setting is not well informed by evidence, with no randomized clinical trials to date. This lack of evidence is reflected in discordant practices among pediatric stroke experts and differing recommendations across the three consensus guidelines [deVeber, 2000; Fullerton et al., 2007; Goldenberg et al., 2009]. A recent international observational cohort study of 661 children with AIS found that anticoagulation was provided fairly consistently to children with cardiac and dissection etiologies [Goldenberg et al., 2009]. Across six international regions, however, anticoagulation rates ranged from 27 to 69 percent. With good safety evidence for all agents [deVeber, 2000; Strater et al., 2001; Ng et al., 2002; Burak et al., 2003], the early initiation of anticoagulation or antiplatelet agents should be considered strongly in all children with AIS.
The decision to employ acute anticoagulation in childhood AIS rests on balancing the potential benefit of reducing thrombus extension or recurrence with risk of hemorrhage. Decisions should be based on some suspicion of pathological fibrin clot formation (coagulation system) playing a dominant role. Preliminary evidence suggests that clinically significant hemorrhagic transformation of childhood AIS is rare and not significantly increased with anticoagulation [Kirton et al., 2007]. Relative contraindications to anticoagulation therapy include intracranial hemorrhage, uncontrollable hypertension, or bleeding diathesis [Monagle et al., 2008b]. Acute anticoagulation has an established role in certain adult stroke situations, including those with a high risk of thrombus extension or embolic recurrence, such as nonvalvular atrial fibrillation [Sandercock et al., 1993; Albers et al., 2001]. Multiple large adult clinical trials across different stroke populations have shown only modestly improved efficacy of heparin over ASA, which generally is negated by an increased risk of hemorrhagic complications [Chaves and Caplan, 2000; International Stroke Trial Collaborative Group, 1997]. However, subanalysis of the Warfarin Aspirin Recurrent Stroke Study (WARSS) among adults with cryptogenic strokes found a 30 percent risk reduction for recurrent stroke with warfarin over aspirin [Mohr, 2004]. These findings may be relevant to children with strokes not related to hypertension and atherosclerosis.
While there are no large-scale clinical trials of anticoagulation therapies in children with AIS, extensive pediatric experience has established safety [deVeber, 2000; Monagle et al., 2008b]. New evidence suggests that anticoagulation approaches are safe in children with arteriopathy [Bernard et al., 2009]. Both the Chest and American Heart Association guidelines support the initial use of anticoagulation in childhood AIS [Monagle et al., 2008b; Roach et al., 2008], while the UK guidelines favor initial use only in selected circumstances, such as cardiogenic stroke [Paediatric Stroke Working Group, 2004]. Duration of therapy varies by clinical indication. Age-dependent differences in thrombin and antithrombin levels complicate heparin therapy in neonates and young children [Andrew et al., 1994]. The advice of an expert colleague in pediatric hematology or thrombosis can prove invaluable. Once a decision for anticoagulation therapy is reached, there are two acute alternatives: unfractionated heparin and LMWH.
Unfractionated Heparin
Unfractionated heparin (UFH) is a large polysaccharide complex that opposes thrombosis by inhibiting specific coagulation pathway enzymes. The primary action is the potentiation of antithrombin’s inactivation of thrombin and other enzymes [Hirsh et al., 1998]. Children require lower doses than adults [Andrew et al., 1994]. Dosing guidelines for heparin in children with AIS are available [Monagle et al., 2008b]. Typically, an initial heparin bolus is not given, theoretically to limit risk of hemorrhagic transformation of a bland infarct, though this is controversial. Maintenance doses are 28 U/kg/hr in infants, 20 U/kg/hr in children over 12 months, and the adult dose of 18 U/kg/hr in older children. Dosing can be monitored according to activated partial thromboplastin times (APTT) and heparin levels. Duration of therapy depends on multiple factors, including the suspected or confirmed etiology and severity of impending stroke. Acute risks of heparin include both intracranial and systemic hemorrhage, as well as heparin-induced thrombocytopenia, which occurs in less than 4 percent of children [Newall et al., 2003]. Alternatives such as danaparoid can be considered. Rapid and complete reversal of the anticoagulation effects of heparin with protamine confers a potential advantage in situations of higher bleeding risk or impending surgery.
Low Molecular Weight Heparins
LMWH are molecular subcomponents of unfractionated heparin. Advantages of LMWH include more reproducible pharmacokinetics [Massicotte et al., 1996], subcutaneous injection, less frequent monitoring, and modestly increased safety profiles [Massicotte et al., 1996]. Efficacy profiles in acute adult stroke appear comparable to unfractionated heparin [Kay et al., 1995] and ASA [Bath et al., 2001]. Safety data specific to childhood AIS are also available [Burak et al., 2003; Ng W et al., 2002; Strater et al., 2001; deVeber, 2000]. As a result of this favorable balance, LMWH has become the treatment of choice for acute anticoagulant therapy in childhood AIS. Updated dosing and administration guidelines are available [Monagle et al., 2008b]. Subcutaneous injections once or twice daily are generally well tolerated. Monitoring of efficacy with anti-factor Xa levels (target range 0.5–1.0) also is less frequent, though the clinical utility of this is controversial. LMWH is not monitored reliably by the APTT. The most common LMWH used in children is enoxaparin (1 mg/kg subcutaneously every 12 hours) [Massicotte et al., 1996]. Other options, including dalteparin and tinzaparin, can be given once daily, as reviewed in the Chest guidelines [Monagle et al., 2008]. LMWH carries a low risk of thrombocytopenia but must be monitored carefully in patients with renal failure. It can be administered by parents, enabling early discharge. Subcutaneous injection ports can reduce discomfort from skin injections and simplify administration.
Warfarin
Warfarin generally is reserved for children requiring long-term anticoagulation, such as those with complex congenital heart diseases or severe prothrombotic disorders. Warfarin antagonizes the activity of vitamin K-dependent coagulation factors in a dose-dependant manner. A favorable risk:benefit ratio is established for secondary stroke prevention in adults with atrial fibrillation. Pediatric dosing and administration guidelines for warfarin are available [Monagle et al., 2008b]. Target INR of 2.0–3.0 is appropriate for most children (2.5–3.5 for mechanical heart valves). Potential contraindications include children with unpredictable vitamin K levels, such as breastfed infants, children with gastrointestinal disorders, and older children unwilling to forgo contact sports.
Antiplatelet therapy
Platelet-mediated thrombosis may be a primary mechanism in many cases of childhood AIS, particularly those in whom arteriopathy has emerged as the predominant cause [Fullerton et al., 2007b; Ganesan et al., 2006; Danchaivijitr et al., 2006a; Amlie-Lefond et al., 2009a]. Initial antiplatelet treatment has been suggested by only one guideline [Paediatric Stroke Working Group, 2004], representing the single largest discrepancy between published consensus guidelines [deVeber and Kirkham, 2008]. While the safety profiles for ASA are even more favorable, suggested relative contraindications include evidence of intracranial hemorrhage and, possibly, SCD [Paediatric Stroke Working Group, 2004]. While the acute use of antiplatelet agents is controversial, they are the mainstay of long-term secondary stroke prevention in most circumstances.
Malignant cerebral edema
Post-stroke cerebral edema is a life-threatening early complication of childhood AIS. Higher brain tissue:intracranial volume ratios in children increase risk compared to adults, in whom reduced extra-axial space is available to accommodate brain swelling. High-risk lesions must be recognized at the time of diagnosis to initiate anticipatory management and surveillance. Complete MCA syndromes carry the highest risk, but MCA trunk infarcts, as well as large hemorrhagic transformations, can generate acute transtentorial herniation. Large cerebellar AIS may swell within the posterior fossa to create brainstem compression, cerebellar herniation (up or down), or obliteration of the fourth ventricle and obstructive hydrocephalus. While most common at 48–72 hours, malignant edema can develop in less than 24 hours. Any neurological deterioration within this timeframe should raise suspicion, particularly if accompanied by signs of herniation, such as decreased level of consciousness or unilateral pupil dilatation. Despite its importance, studies of malignant cerebral edema in pediatric AIS have been limited [Ramaswamy et al., 2008].
Management consists of patient stabilization followed by emergent imaging (CT) to confirm malignant edema and exclude hemorrhage. Measures to reduce increased intracranial pressure should be considered, including adequate sedation, paralysis, manipulation of PCO2, and osmotic therapy, such as mannitol or hypertonic saline. The role of intracranial pressure monitoring is unstudied. A study combining three randomized trials of decompressive hemicraniectomy in younger adults with stroke suggests the procedure is life-saving and generally beneficial [Vahedi et al., 2007]. These findings are consistent with previous smaller studies [Cho et al., 2003; Gupta et al., 2004], although procedural complications and concerns that some patients simply are shifted from outcomes of death to severe disability merit consideration. While unstudied in children with stroke, malignant cerebral edema is a neurological emergency and early decompression should be considered strongly [Paediatric Stroke Working Group, 2004].
Acetylsalicylic Acid
ASA is the primary antiplatelet agent for children and has a well-established safety profile [Monagle, 2006; Strater et al., 2001]. ASA inhibits platelet aggregation by decreasing thromboxane A2 production. In adults, acute ASA confers a modest benefit [International Stroke Trial Collaborative Group, 1997; Bousser and Russell, 1997]. There have been no clinical trials of ASA in childhood AIS. Recommended dosing varies from 2 to 5 mg/kg/day, usually administered once daily. The loading doses of ASA used in adults have not been studied in children with stroke. ASA is generally well tolerated, with easy bruising, nosebleeds, and stomach upset generally being mild. Reye’s syndrome is a fair concern consistently raised by parents but current evidence supports an extremely low risk. Reye’s syndrome is likely related to rare inborn errors in metabolism (not associated with pediatric stroke) and has never been reported at the low doses used in pediatric stroke [Hall, 1990; Schror, 2007]. To minimize Reye’s syndrome concerns, some physicians recommend reduction of therapy during a febrile illness and annual influenza vaccination. The role of ASA resistance is controversial and does not appear routinely measurable by modern methods [Seok et al., 2008].
Other Agents
Alternative antiplatelet agents studied in stroke include clopidogrel and extended-release dipyridamole. Current adult stroke guidelines for non-cardioembolic stroke advise the use of either combined ASA and dipyridamole (25/200 mg b.i.d.), or clopidogrel over aspirin (grade 1A), and avoiding long-term combined ASA and clopidogrel [Albers et al., 2008]. Preliminary evidence suggests that clopidogrel is safe and well tolerated in children with stroke [Soman et al., 2006a]. Previous studies extrapolated adult doses of 1 mg/kg/day, but new evidence suggests children may achieve similar platelet inhibition at doses 20–50 percent lower [Li et al., 2008]. Clopidogrel between 0.5 and 1 mg/kg/day appears to be a reasonably safe alternative in children with AIS who require antiplatelet treatment but cannot take ASA. Other antiplatelet agents employed in adult stroke, such as dipyridamole, ticlopidine, or the glycoprotein IIb–IIIa antagonists, have not been studied in children.
Outcomes and Chronic Management
Most survivors of childhood stroke suffer neurological morbidity. Moderate to severe neurological deficits or epilepsy occur in over 50 percent of children after AIS [deVeber, 2000; Delsing et al., 2001; Ganesan et al., 2000]. Mortality rates are estimated at 5–13 percent [Delsing et al., 2001; deVeber, 2000]. Risk of adverse outcome may be associated with age below 12 months, presentation with altered level of consciousness or seizures, and infarct location and size [Delsing et al., 2001; Ganesan et al., 2000; Max, 2004a; Lansing et al., 2004; Mercuri et al., 2004; Golomb et al., 2003a]. Aggressive multimodal treatment and rehabilitation must target the physical, occupational, language, cognitive/behavioral, and psychosocial consequences of stroke for both the child and family [Paediatric Stroke Working Group, 2004]. Accumulating evidence suggests that earlier and more intense rehabilitation usually is beneficial [Teasell et al., 2006]. Though not yet available for children, recent evidence-based practice guidelines for adult stroke rehabilitation are helpful [Khadilkar et al., 2006]. Many approaches remain based on adult stroke rationale and data, long-standing historical practices, and simple theory, underscoring the need for additional age-appropriate evidence.
Enhanced plasticity often is assumed to afford better outcomes from brain injury in the young [Lynch et al., 2001]. However, evidence supporting the accuracy of such assumptions, and the mechanisms that might underlie it, remains limited. The continuous acquisition of new abilities during child development complicates the objective evaluation of rehabilitation interventions. Rehabilitation treatments, therefore, must incorporate knowledge of normal age-related developmental skills, trying to distinguish differences between relearning a brain function and acquiring it for the first time. Modern understanding of brain reorganization mechanisms after adult stroke is providing new avenues and strategies to promote recovery [Ward, 2004; Teasell et al., 2006]. Such knowledge needs to be acquired in children with stroke and translated into new rehabilitative approaches to promote better functional outcomes across the full range of pediatric stroke morbidities. The following summarizes the issues and management approaches relating to the physical and neuropsychological consequences of stroke, and is followed by review of associated medical morbidities and psychosocial outcomes.
Motor Disabilities
Motor disabilities after childhood AIS are common, functionally important, and readily amenable to evaluation [deVeber, 2000; Ganesan et al., 2000]. Common involvement of the MCA territory damages both cortical and subcortical motor systems, resulting in hemiparesis affecting the upper more than the lower extremity (see Figure 85-6). Problems of gait and lower-extremity morbidity, as well as effects of cerebellar and basal ganglia lesions, also occur and should not be overlooked. Neuroimaging may facilitate early prediction of poor motor outcomes in childhood AIS [Boardman et al., 2005; Domi et al., 2009], possibly identifying those in need of aggressive rehabilitation. Historical, commonsense physical and occupational therapy approaches typically are applied to enhance motor recovery after stroke [Aoyagi and Tsubahara, 2004; Dobkin, 2004]. Programs that combine strength and aerobic conditioning, task-specific training, and assistive devices form the basis of post-stroke motor rehabilitation in adults [Teasell et al., 2006], and generally are recommended in children [Paediatric Stroke Working Group, 2004]. Fortunately, evidence is now emerging to establish and expand the benefits of both traditional and novel strategies to improve motor function.
Constraint-induced movement therapy
Constraint-induced movement therapy (CIMT) is a therapeutic approach to promote functional use of an impaired limb after stroke. Originating from studies of motor learning in nonhuman primates, CIMT has developed into a promising stroke neurorehabilitation technique [Taub et al., 1998, 2002; Wolf et al., 2006]. Constraint of the normal hand is combined with repetitive motor practice and “shaping” of functional activities in the impaired hand. Evidence supports CIMT effectiveness in adult stroke, in whom weeks of therapy can generate motor gains lasting months [Wolf et al., 2006, 2008; Hakkennes and Keating, 2005]. Pediatric CIMT studies, limited to children with perinatal stroke and hemiplegic cerebral palsy [Charles and Gordon, 2005], include positive randomized controlled trials [Taub et al., 2004, 2007; Willis et al., 2002; Charles and Gordon, 2006a; Deluca et al., 2006; Sung et al., 2005]. While results are encouraging, proper clinical trials in childhood stroke are required [Sakzewski et al., 2009; Brady and Garcia, 2009]. Two positive randomized controlled trials in children with hemiplegia have demonstrated that delivering daily therapy for 2 weeks is well tolerated [Taub et al., 2007; Charles et al., 2006b]. Studies of developmental plastic organization and the neurophysiological effects of motor rehabilitational interventions with fMRI and transcranial magnetic stimulation are clarifying, and even predicting, how such interventions work [Hoare, 2008; Liepert et al., 2000; Liepert, 2006].
Transcranial magnetic stimulation
Transcranial magnetic stimulation (TMS) mapping of brain physiology has advanced our understanding of post-stroke reorganization and function [Rossini and Pauri, 2005]. TMS is safe and well tolerated in children [Garvey and Gilbert, 2004; Gilbert et al., 2004]. In addition, repetitive TMS (rTMS) carries therapeutic potential, possibly producing lasting modulations in cortical function [Pascual-Leone et al., 1998; Khedr et al., 2005; Naeser et al., 2005]. Clinical trials of rTMS may improve motor functions in chronic adult stroke [Mansur et al., 2005; Fregni et al., 2006; Khedr et al., 2005; Boggio et al., 2006]. A randomized clinical trial in childhood AIS found that daily application of inhibitory rTMS to the non-lesioned motor cortex could improve motor hand function in children with chronic subcortical stroke [Kirton et al., 2008a]. The ability of TMS to measure and modulate functional areas of human cortex noninvasively suggests that it will play an increasing role in the study and treatment of childhood stroke.
Other physical rehabilitation possibilities
Complementing CIMT are new “bimanual” approaches, focusing on function of the unaffected extremity [Charles and Gordon, 2006a]. Both peripheral electrical stimulation and transcutaneous electrical nerve stimulation are recommended in adult stroke rehabilitation clinical practice guidelines [Khadilkar et al., 2006], but remain essentially untested in children. Robot-assisted rehabilitation [Lum et al., 2002; Khadilkar et al., 2006] and virtual reality training strategies [Lam et al., 2006] continue to be explored in adult stroke. Other physical rehabilitation possibilities unstudied to date in childhood stroke include biofeedback, specific gait and balance training, therapeutic ultrasound, and acupuncture [Khadilkar et al., 2006].
Abnormal Tone
Abnormal tone and the need for assistive devices should be considered in children with stroke. Both rate-dependent (spasticity) and rate-independent (e.g., dystonia) hypertonia complicate the lives of and limit function in children with AIS. Such morbidities can be complex and require a systematic approach, often with the collaboration of experts in physical medicine and orthopedics [Sommerfeld et al., 2004]. Spasticity interventions must include comprehensive, objective, and blinded pre- and post-interventional outcome measures of efficacy. Botulinum toxin may alleviate increased tone, though effects on motor function and quality of life in children have been questioned [Brashear et al., 2002; Redman et al., 2008], and issues of cost, frequency, and tolerability often are significant. Pharmacological spasticity therapies are not well evaluated in childhood stroke and may be complicated by systemic toxicity. Careful selection criteria and better evidence of efficacy also are required for surgical interventions, such as tendon-lengthening procedures. Assistive devices, including ankle-foot orthoses and Lycra hand splints, may facilitate joint positioning and function, and are supported by evidence in adult stroke [Lannin and Herbert, 2003; Aoyagi and Tsubahara, 2004]. Consensus pediatric stroke guidelines suggest splinting for carefully selected children [Paediatric Stroke Working Group, 2004]. An added challenge in young pediatric stroke patients is resulting asymmetries in physical growth, with leg-length discrepancy and scoliosis that may require orthopedic intervention.
Neuropsychological Impairments
Neuropsychological impairments are common in children with stroke, limiting academic, social, and independent functional success [Friefeld et al., 2004; Nass and Trauner, 2004]. Regular, age-appropriate neuropsychological evaluations should be considered in all at-risk children to determine educational and support needs. Serial assessments across development are required, as defects in higher brain function often emerge over time in young children [Westmacott et al., 2007a, 2009]. On average, childhood stroke survivors exhibit moderately compromised global intellectual function [Hetherington et al., 2005; McLinden et al., 2007]. Lesion side, size, and age at injury may influence outcome, though accurate prediction is challenging [Lansing et al., 2004; Max, 2004a; Ricci et al., 2008]. Younger age at stroke does not appear to predict better cognitive outcomes [Stiles, 2000; Lansing et al., 2004; Westmacott et al., 2007b]. Language function often is impaired also, usually related to dominant (left) hemisphere injury [Chilosi et al., 2001; Nass and Trauner, 2004]. While children may recover quickly from acute aphasias, residual deficits in complex language are common [Hertz-Pannier et al., 2002; Van Dongen et al., 1994]. Verbal learning and memory often are impaired in children following stroke [Max, 2004a; Lansing et al., 2004]. Comprehensive, serial testing of cognitive, language, and other higher brain functions by pediatric neuropsychologists using standardized measures is an important component of a childhood stroke rehabilitation program.
Emotional and behavioral disorders after childhood stroke are poorly understood but potentially affect all aspects of quality of life. Limited studies suggest that social function commonly is impaired following stroke in childhood [Mosch et al., 2005]. In children with perinatal stroke, rates of emotional and behavioral disturbances are increased, the effects often not emerging until preschool or later [Goodman and Graham, 1996a]. Lesion location has not been correlated well with neuropsychological outcomes [Max et al., 2002b; Trauner et al., 2001], but bilateral lesions may elevate the risk of emotional or behavioral problems [Nass and Trauner, 2004]. Rates of attention-deficit hyperactivity disorder also are increased [Max et al., 2002a, 2003, 2004b], particularly with lesions involving the putamen [Teicher et al., 2000]. Such potentially treatable psychiatric comorbidities should be screened for in children with stroke.
Stroke Recurrence
Stroke recurrence is significant, mandating a thorough approach to secondary stroke prevention over the long term. Previous studies suggested that stroke and TIA recurrence risks range from 7 to 35 percent [Isler, 1984; Chabrier et al., 2000; Lanthier et al., 2000, 2004; De Schryver et al., 2000; deVeber, 2000]. The rate of recurrence approaches 50 percent when antithrombotic agents are not given [Lanthier et al., 2004]. A more recent population-based study confirmed an overall 5-year recurrence risk of 20 percent and, more importantly, established that the majority of risk is carried by children with arteriopathy, in whom recurrence was 66 percent [Fullerton et al., 2007b]. That arteriopathy predicts recurrence is consistent with previous studies [Chabrier et al., 2000; Lanthier et al., 2004]. With no randomized trials, the relative efficacy of various secondary stroke prevention interventions is unknown. Consistent with clinical practice [Goldenberg et al., 2009] and published guidelines [deVeber and Kirkham, 2008], antiplatelet therapy with ASA (1–5 mg/kg/day) is the mainstay of long-term secondary stroke prevention in childhood AIS. Exceptions exist, however, and therapy must be tailored and adjusted over time to the individual patient. Insensitivity or intolerance of ASA may require an alternative antiplatelet agent, like clopidogrel. Certain cardiac and prothrombotic conditions may require long-term anticoagulation with warfarin. Additional treatments may include immunosuppression for inflammatory vasculitis, transfusions for SCD, and revascularization surgery for moyamoya. Attention to arterial health, with balanced diet, exercise, avoidance of smoking and drugs, and surveillance for hypertension, dyslipidemia, or insulin resistance, may also help prevent recurrence over the long term.
Medical Comorbidities
Epilepsy
Epilepsy will complicate childhood AIS in approximately 15–20 percent of cases [deVeber, 2000; deVeber et al., 2001; Yang et al., 1995; Fitzgerald et al., 2007]. Seizures likely arise from partially injured, perilesional brain parenchyma, and partial or secondary generalized events are expected. However, bilateral EEG abnormalities and generalized epileptic encephalopathies, including infantile spasms, can occur after unilateral, focal stroke in infants and young children [Kirton et al., 2008b; Soman et al., 2006b; Fitzgerald et al., 2007]. Concerns that on-going clinical seizures, or even subclinical pathological EEG activity, can affect plastic neurodevelopment and recovery adversely in children after stroke are theoretical but important. While some predictors of childhood epilepsy are appreciated in perinatal stroke [Fitzgerald et al., 2007; Kirton et al., 2008b], studies in older children are limited. Children with large cortical lesions may harbor the highest risk [Yang et al., 1995; deVeber, 2000]. Screening for seizures on history and use of supportive investigations, such as EEG, are required. Treatment requires anticonvulsants and attention to the extensive additional psychosocial impact of epilepsy on the entire family. As stroke typically represents a focal lesion in an otherwise healthy brain, children with AIS and epilepsy may be particularly good candidates for epilepsy surgery [Wyllie et al., 2007; Guzzetta et al., 2006].
Hyperkinetic Movement Disorders
Hyperkinetic movement disorders can follow childhood AIS, often not presenting for weeks or months after stroke [Kwak and Jankovic, 2002; Boardman et al., 2005; Nardocci et al., 1996]. Basal ganglia strokes carry the greatest risk. Prevalence may be underestimated because dystonia can be difficult to distinguish from spasticity. History, examination, and physical therapy consultation should be integrated to determine the relative balance of weakness, spasticity, and dystonia in an individual patient with motor dysfunction after AIS. Treatment options differ, based on this, and, as there has been little study specific to stroke, should employ the general principles of managing pediatric movement disorders (see Chapter 68) [Edgar, 2003]. Options include anticholinergic (e.g., trihexyphenidyl), antidopamine (e.g., pimozide), or dopamine-depleting (e.g., tetrabenazine) agents, as well as other medications and botulinum toxin injections. Such interventions should begin at low doses, with gradual titration to minimize side effects, combined with objective assessments of efficacy by blinded observers. The role of newer movement disorder medications and therapies, such as deep brain stimulation, have not been determined in children with stroke.
Headache Disorders
Headache disorders also appear to be increased following pediatric stroke, occurring in 33 percent of cases [deVeber, 2000]. Risk factors include pre-existing conditions (e.g., migraine), the nature of the underlying disease (e.g., moyamoya), and psychosocial stressors. Screening is important, as new or worsening headaches may be a marker of disease progression, including progression of small-vessel cPACNS, moyamoya, or migrainous infarction. Therefore, diagnoses of primary headache disorders, such as migraine or tension-type headache, must be made cautiously in accordance with International Headache Society criteria. Most established pharmacological and nonpharmacological interventions for headache (see Chapter 63) are safe in children with AIS, with a few exceptions. Regular use of nonsteroidal anti-inflammatory medications can decrease the efficacy of ASA modestly, but occasional use for abortive headache therapies is likely safe if alternatives such as acetaminophen are ineffective. In children with migrainous infarction, the calcium channel blocker flunarizine theoretically provides both headache and stroke prevention. Vasoconstricting agents, including triptans, probably should be avoided in those with migrainous infarction or arteriopathy. Attention to lifestyle, triggers, and psychological factors remains essential.
Psychosocial Family Support
Psychosocial family support also must be considered. Complete care of childhood stroke centers on the child and family. Complex psychosocial challenges are likely common and under-estimated. While resources are slowly improving, there is an on-going need for the development of educational and supportive resources to improve pediatric stroke care [Paediatric Stroke Working Group, 2004]. Adverse effects of stroke on quality of life are measurable in most childhood stroke survivors and their family [Gordon et al., 2002], and relate to both neurological deficits and psychosocial factors [Friefeld et al., 2004]. Social factors and environment have been shown to affect adult stroke recovery and may be even more important in children [Teasell et al., 2006]. Similarly, coping strategies may influence adult stroke outcome [Donnellan et al., 2006] but have not been studied in childhood, when such challenges are likely even greater. Each element of treatment and rehabilitation needs to consider the individual child’s lifestyle, goals, and level of functioning, and be well integrated into the school and home environments [Paediatric Stroke Working Group, 2004]. Parents require education and may need encouragement to avoid “overprotection” and to treat their children as normal. Alleviation of parental guilt also may provide substantial benefit. As the assignment of an exact etiology for many childhood strokes often is lacking, parents may assume unjustly that some modifiable factor within their control is somehow responsible. It is important, therefore, to screen parents for such misplaced interpretations and feelings of guilt, and to offer supportive education, reassurance, and discussion.
Family support also includes guidance in the interpretation of a growing number of unproven therapeutic options. Stem-cell therapies are an increasingly controversial topic [Patoine, 2009], and are many years away from consideration in the treatment of childhood stroke [Kurtzberg, 2009]. Giving more cause for concern is the preponderance of individuals and operations that prey on the vulnerable families of children with neurological disabilities [Rosenbaum, 2003]. Many “pseudotherapies” are marketed unfairly to families of children with stroke, offering incredible but unsubstantiated claims of efficacy. Hyperbaric oxygen lacks theoretical basis, risks multiple adverse effects [Nuthall et al., 2000; Muller-Bolla et al., 2006], and repeatedly has been proven ineffective [Collet et al., 2001; Hardy et al., 2002]. Despite this, it continues to be promoted at high cost by flagrantly conflicted organizations. The use of complementary and alternative medicines is common in pediatric stroke, despite no proof of efficacy and potential interference with medical therapies [Golomb et al., 2003b]. Few such pseudotherapies will ever undergo proper testing for safety and efficacy. The treating physician must, therefore, guide families to make informed decisions, respecting their motivation to help their children, while advocating on their behalf for responsible and equal regulation of all therapies offered to children with disabilities.
Cerebral Sinovenous Thrombosis
CSVT includes thrombosis within the cerebral venous system, whether symptomatic or not. Approximately half of patients will have no visible parenchymal lesions at diagnosis, while the remainder have venous infarction that often is hemorrhagic. Though CSVT incidence is likely underestimated, epidemiology studies suggest CSVT affects approximately 1 in 100,000 children per year, including neonates, and accounts for 1 in 4 cases of childhood ischemic stroke [Mallick and O’Callaghan, 2009a]. Similar to AIS, CSVT risk is highest in the neonatal period (see Chapter 18) and in males [Golomb et al., 2009; Wasay et al., 2008). The seminal epidemiological paper, a population-based Canadian study of 160 consecutive cases of pediatric CSVT [deVeber et al., 2001] has been supplemented by recent large national and international pooled case series [Heller et al., 2003; Kenet et al., 2004; Sebire et al., 2005; Wasay et al., 2008; Vieira et al., 2009; Jordan et al., 2010].
Pathophysiology
Sinovenous Circulation: Anatomy and Vascular Patterns
Cerebral venous drainage of the brain occurs through a network of cerebral veins and sinuses (Figure 85-11). A general pattern of distal venous drainage toward midline channels that flow in a posterior direction is recognized. In the superficial venous system, cortical veins drain cortical and subcortical areas medially toward the superior sagittal sinus. The deep venous network includes internal cerebral, medullary, thalamic, and choroidal veins, which converge centrally at the vein of Galen; this, in turn, drains into the straight sinus. These superficial and deep systems converge posteriorly at the torcula. Bilateral, often asymmetrical, transverse sinuses then drain laterally to the sigmoid sinuses, which connect to the jugular veins. Though variable, the superficial system frequently drains predominantly into a larger right transverse sinus, while the deep system empties via a smaller left transverse sinus. Additional venous drainage pathways connecting with this system at recognizable locations include the anterior cavernous and petrosal sinuses, as well several large superficial veins (e.g., Trolard, Labbé). Pathological thrombosis can occur anywhere within this system; however, the superficial venous system is involved more frequently [deVeber et al., 2001; Sebire et al., 2005].
The cerebral sinuses are external to the brain, enclosed between two layers of fibrous dura mater. Attachment of the outer layer to the calvarium maintains a fixed open lumen with venous sinus caliber unresponsive to changes in systemic blood pressure, facilitating passive venous drainage [Capra and Anderson, 1984]. However, the fixed lumen and absence of valves or surrounding muscles that characterize systemic veins results in slow, reversible flow within the sinuses. Gravity compensates in part for these differences; however, cerebral venous circulation remains sensitive to impaired venous return. In supine neonates and young infants with open posterior fontanels, mechanical compression of the superior sagittal sinus by the occipital bone may compromise venous drainage, resulting in venous stasis [Newton and Gooding, 1975; Tan et al., 2007]. As the superior sagittal sinus also is the primary site for cerebrospinal fluid reabsorption via the arachnoid granulations, thrombosis or venous hypertension here can result in communicating hydrocephalus [Bousser, 1997].
With a working knowledge of the above venous anatomy, clinical patterns of CSVT and venous infarction can be recognized. Bilateral, parasagittal infarcts, especially with hemorrhagic transformation, are likely secondary to sagittal sinus thrombosis. Deep venous thrombosis involving the internal cerebral veins, straight sinus, and/or vein of Galen will produce venous edema and infarction of the deep white matter, basal ganglia, and thalamus. Such hemorrhagic infarction in deep locations immediately adjacent to the ventricular system (e.g., thalamic lesions by third ventricle) can produce intraventricular hemorrhage (IVH). In term neonates with IVH, nearly one-third have an underlying deep CSVT [Wu et al., 2003]. As both venous drainage pathways and infarction mechanisms are heterogeneous, the imaging and clinical presentations of venous infarcts and hemorrhage lack the more predictable patterns of arterial infarcts, necessitating a higher index of clinical suspicion.
Mechanisms of Thromboembolism
Factors mediating thrombosis in cerebral venous channels differ in some respects to those underlying thrombosis in cerebral arteries. In contrast to AIS, the coagulation cascade and thrombin-rich thrombosis predominate in CSVT, with platelets playing a less significant role [Furie and Furie, 2008]. Inherited or acquired coagulation abnormalities (see below), including prothrombotic medications, are important in CSVT pathogenesis. There is a relative absence of thrombomodulin in the endothelial lining of cerebral sinuses compared with cerebral arteries, theoretically increasing the tendency for thrombosis [Lin et al., 1994]. Consideration of Virchow’s triad is highly relevant in the cerebral venous system, with thrombosis resulting from abnormalities of blood components, blood flow, or vessel wall. In many children with CSVT, the exact mechanism of thrombosis is not evident. Dehydration is a frequent and potent risk factor for CSVT, where decreased blood volume and hemoconcentration decrease venous flow rates, favoring thrombus formation and propagation. In septic CSVT, head or neck infection in tissues immediately adjacent to venous channels provokes cerebral venous thrombophlebitis. With mechanical damage or compression of venous structures, venous stasis and vessel wall damage may combine to cause thrombosis. Examples include trauma, neurosurgical procedures, insertion of venous catheters, compressive mass lesions, and occipital bone compression in supine young infants [Deitcher et al., 2004; Newton and Gooding, 1975; Tan et al., 2007].
Mechanisms of Infarction
Cerebral venous sinus occlusion causes a rise in venous pressure, with consequences developing over time in cerebral tissue. This may occur focally in a single venous territory, or more globally as a syndrome of diffuse increased intracranial pressure. In focal injury, the failure of venous emptying results in local tissue pressure elevation with transudation of fluid across venous or capillary channels. Initially, this may produce parenchymal vasogenic edema without true infarction, accompanied by focal clinical symptoms. However, further increases in regional tissue pressure eventually can exceed the incoming arterial perfusion pressure, resulting in permanent infarction. On MRI, DWI may differentiate venous edema from infarction [Forbes et al., 2001; Carvalho et al., 2001], although, in CSVT, some restricted diffusion may be reversible [Ducreux et al., 2001]. Venous drainage can occur via alternate routes, including redundant pathways and pial collaterals, but these may require time to develop. Therefore, very rapid or complete thrombotic venous occlusion carries an increased risk of tissue infarction [Bousser, 1997]. The increased “back pressure” of CSVT accounts for the high rate of transformation of infarcts from initially bland to hemorrhagic. The presence of hydrocephalus, either from IVH or communicating hydrocephalus, creates a further increase in compression of already compromised brain tissue, possibly accelerating injury.
Clinical Features
Prompt diagnosis of childhood CSVT is clinically challenging but essential to effecting the best possible outcome. Neonates (see Chapter 18) and infants less than 1 year comprise approximately 50 percent of pediatric CSVT patients [deVeber et al., 2001; Carvalho et al., 2001]. As in other varieties of pediatric stroke, clinical features are age-dependent and more difficult to recognize in infants and young children. In contrast to AIS, diffuse neurological signs and seizures are more common than focal deficits. Signs and symptoms also are more likely to develop gradually over many hours, days, or even weeks, although symptoms also may begin or worsen abruptly. Common clinical presentations include headache, lethargy, nausea and vomiting, and signs of increased intracranial pressure, including papilledema and sixth nerve palsy. This syndrome is clinically indistinguishable from idiopathic intracranial hypertension (IIH) and the latter diagnosis should not be made without excluding CSVT [Reul et al., 1997; Ameri and Bousser, 1992]. Narrowing of the lateral sinuses has been documented in a high proportion of adults with IIH, but whether this is a cause or results from increased pressure is undetermined [Farb et al., 2003b]. Seizures are more common in CSVT compared to AIS, with estimates ranging from 40 to 90 percent [Wasay et al., 2008]. Young infants tend to present with seizures or diffuse encephalopathy, and focal signs are uncommon [Barron et al., 1992; Rivkin et al., 1992; deVeber et al., 2001; Carvalho et al., 2001]. Children in the Canadian Pediatric Ischemic Stroke Registry had at least one of the following at presentation: seizure (48 percent), headache (54 percent), papilledema (22 percent), altered consciousness (49 percent), and focal signs (53 percent) [deVeber et al., 2001].
Risk Factors
Risk factors are definable in most children and frequently are multiple. CSVT risk factors are age-related. In neonates (see Chapter 18) and young infants, dehydration and perinatal events are common, and the proportion without evident risk factors is higher. Head and neck infections are present in nearly one-third of preschool children, while other associations, including systemic disease and trauma, are clustered in the older age groups. The largest prospective, population-based study of 160 children and neonates with CSVT described the following proportions: prothrombotic disorders (41 percent), dehydration (25 percent), systemic infection (9 percent), head and neck infection (18 percent), other head and neck disorders (12 percent), hematological disorders (12 percent), malignancy (8 percent), and cardiac disease (5 percent). Sixty-five percent of patients had multiple risk factors [deVeber et al., 2001]. Therefore, the approach to investigation must be broad and systematic, beginning with consideration of major categories: infection, prothrombotic states, and both acute and chronic systemic diseases.
Infection is a major risk factor for childhood CSVT. Septic CSVT is defined as thrombosis of venous sinuses associated with head and neck infections, including otitis media, mastoiditis, sinusitis, meningitis, and others [Byers and Hass, 1933]. Septic CSVT may account for 18–50 percent of cases [deVeber et al., 2001; Wasay et al., 2008], with recent studies suggesting even higher rates [Barnes et al., 2004; Sebire et al., 2005]. The usual mechanism involves spread of infection from adjacent tissues into the venous sinuses, causing thrombophlebitis. However, acute systemic bacterial infections and sepsis impart multiple risks, including dehydration, systemic inflammation, and coagulopathy, in addition to local thrombophlebitis. Lemierre’s syndrome occurs secondary to human necrobacillosis, and recognizing the unique syndrome of pharyngitis and neck pain progressing to jugular venous thrombosis is required for proper treatment [Jaremko et al., 2003]. Thorough history of infections, and examination for middle ear infection, mastoid or air sinus tenderness, and meningismus are important, and diagnostic investigations, including cerebrospinal examination, may be required.
Prothrombotic disorders are reported in 20–80 percent of pediatric patients with CSVT [Ganesan et al., 1996; Riikonen et al., 1996; Vielhaber et al., 1998; deVeber et al., 1998a, 2001]. These figures greatly exceed estimates of 15–20 percent in adult CSVT [Deschiens et al., 1996; Zuber et al., 1996]. A large, prospective case-control study of 149 children with CSVT undergoing standardized coagulation testing found abnormalities in 56 percent, compared to 21 percent of controls [Heller et al., 2003]. A smaller case-control study found that thrombophilic markers were increased only in children with otherwise idiopathic CSVT [Kenet et al., 2004]. A number of specific prothrombotic disorders have been reported in pediatric CSVT, including factor V Leiden, anticardiolipin antibodies, elevated lipoprotein (a), and deficiencies of protein C, protein S, and antithrombin [deVeber et al., 2001; Ganesan et al., 1996; Gouault-Heilman et al., 1994; Martinelli et al., 1996; Riikonen et al., 1996; Vielhaber et al., 1998; Heller et al., 2003]. Both hyperhomocysteinemia and MTHFR homozygosity have been associated with childhood and adult CSVT [Bonduel et al., 1999; Hillier et al., 1998; Vielhaber et al., 1998; Carhuapoma et al., 1997; Buoni et al., 2001]. Recurrent CSVT is associated with heterozygosity for the factor II G20210A mutation [Kenet et al., 2007]. A rapidly expanding list of novel prothrombotic disorders, such as the factor XII C46T polymorphism associated with adult CSVT [Reuner et al., 2008], remain untested in children. A formal prothrombotic evaluation is recommended for most children with CSVT. Elevated d-dimers may be present in CSVT, and in adults are considered an option for screening for venous thrombosis, but their clinical utility in children has not been determined [Cucchiara et al., 2005].
Other acute illnesses should be considered, including dehydration, which is a common risk factor for pediatric CSVT. Head trauma or cranial surgery may damage the dural sinuses, leading to CSVT [Muthukumar, 2004; Sousa et al., 2004; Huisman et al., 2001]. A study of 131 children with head injury requiring CT found CSVT in 6 percent of patients [Stiefel et al., 2000]. Mechanical compression of the superior sagittal sinus in supine infants also may predispose to CSVT [Tan et al., 2007]. Chronic systemic diseases also increase the risk of childhood CSVT, especially in older school-age children. Childhood cancers and their treatments carry multiple potential risks, including chemotherapy-related hypercoagulable states (l-asparaginase), infections secondary to compromised immunity, direct CNS involvement of cancer and neurosurgery, and placement of jugular venous catheters [Deitcher et al., 2004]. Acute lymphoblastic leukemia is a common association, and a retrospective study of over 2300 children found a CSVT prevalence of 1:200 [Santoro et al., 2005]. In inflammatory bowel disease, systemic inflammation may combine with gastrointestinal disease factors, such as dehydration, medications, and iron deficiency [Standridge and de los, 2008]. Nephrotic syndrome may lead to relative deficiencies in coagulation factors like antithrombin III and predispose to CSVT [Fluss et al., 2006]. Other systemic diseases may share this mechanism of an acquired prothrombotic state, such as lupus anticoagulant and antiphospholipid antibodies in systemic lupus erythematosus. An association between iron deficiency anemia and childhood CSVT is now well established in infants and toddlers, and should be excluded [Meena et al., 2000; Belman et al., 1990; Sébire et al., 2005; Hartfield et al., 1997; Maguire et al., 2007]. Cardiac disease is present in 4–9 percent of children with CSVT, and thrombosis may be due to impaired venous return (cerebral venous hypertension) triggered by cardiac surgery or other treatments (jugular lines, ECMO, etc.) [deVeber et al., 2001; Barnes et al., 2004; Sebire et al., 2005].
Diagnosis: Neuroimaging
The imaging diagnosis of childhood CSVT is challenging and, unless specific venous imaging is performed, the disease frequently is missed. Radiographic features and diagnostic pitfalls are reviewed elsewhere [Shroff and deVeber, 2003; Leach et al., 2006]. Although parenchymal imaging can be diagnostic (Figure 85-12), the primary objective is to image flow and/or luminal filling defect(s) within the venous system as evidence of pathological thrombosis. CSVT location can be diffuse, multifocal, or focal within the venous channels. Approximately half of children have multiple sinuses or veins affected [deVeber et al., 2001]. The extent (i.e., partial or complete occlusion) and location of thrombosis determine the parenchymal consequences and relative clinical severity. The superficial system, including superior sagittal sinus, is affected most commonly. Demonstrating thrombosis here is relatively easy; however, lesions isolated to cortical veins can be missed easily (Figure 85-13). A small thrombosis in a critical location can have catastrophic consequences. For example, complete occlusion of the straight sinus or vein of Galen can lead to bilateral thalamic infarction with IVH.
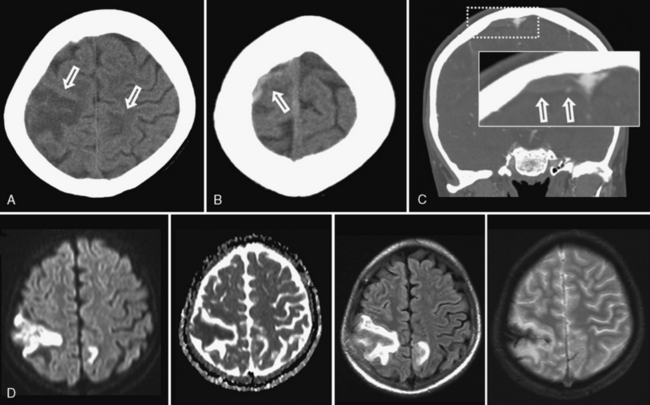
Consistent with the pathophysiology described earlier, CSVT brain lesions can be of variable severity. Approximately half of children have CSVT without parenchymal changes. In others, focal, potentially reversible edema is difficult to distinguish from early venous infarction, though restricted diffusion within the lesion likely increases risk of the latter greatly. A high proportion of venous infarcts will undergo hemorrhagic transformation, sometimes mimicking a primary intracranial hemorrhage. While venous infarcts occur in less predictable territories than AIS, recognizable patterns of supply, according to known venous anatomy, are established [Teksam et al., 2008]. Modern neuroimaging with CT and MRI has advanced significantly the ability to diagnose CSVT and to make these clinically important distinctions. In neonates and young infants, trans-fontanel Doppler ultrasound can detect and possibly monitor CSVT, but has not been well studied [Bezinque et al., 1995; Dean and Taylor, 1995]. CA is employed rarely in CSVT [deVeber et al., 2001]. Similar to CT venography (CTV), CA demonstrates either reduced or absent contrast opacification of venous channels [Ameri and Bousser, 1992].
Computed Tomography
Plain (non-contrast) CT is inadequate to exclude CSVT, and also may underestimate early edema or infarction [Ameri and Bousser, 1992; Barron et al., 1992; Bousser, 1997; Jacewicz and Plum, 1990]. Plain CT also can produce false-positive results [Hamburger et al., 1990], particularly in neonates and young infants, in whom higher hematocrit, slower venous flow, and unmyelinated brain hypodensity may create an illusion of sinus hyperdensity [Ludwig et al., 1980; Kriss, 1998; Davies and Slavotinek, 1994]. Tentorial subdural hemorrhage is common in the newborn and may mimic transverse sinus thrombosis. Despite these limitations, CSVT can appear as hyperdense sinuses or individual veins on noncontrast CT (see Figure 85-13). The combination of hyperdense straight sinus, thalamic hypodensity/bleeding, and IVH is a recognizable, life-threatening syndrome due to deep system CSVT [Linn et al., 2009].
Contrast-enhanced CTV is highly sensitive and specific for childhood CSVT diagnosis. Thrombus appears as a filling defect in the vein or sinus, where contrast may be either absent (complete occlusion) or focally reduced (incomplete occlusion) (Figures 85-13 and 85-14). The “empty triangle” or “empty delta” sign represents such a filling defect at the torcula. Multislice contrast-enhanced CT, with submillimeter slices and multiplanar reformations for axial, coronal, and sagittal views, has improved the ability to diagnose CSVT significantly. As CTV requires additional imaging and most children will require serial scanning, age-adjusted radiation dosages must be considered.
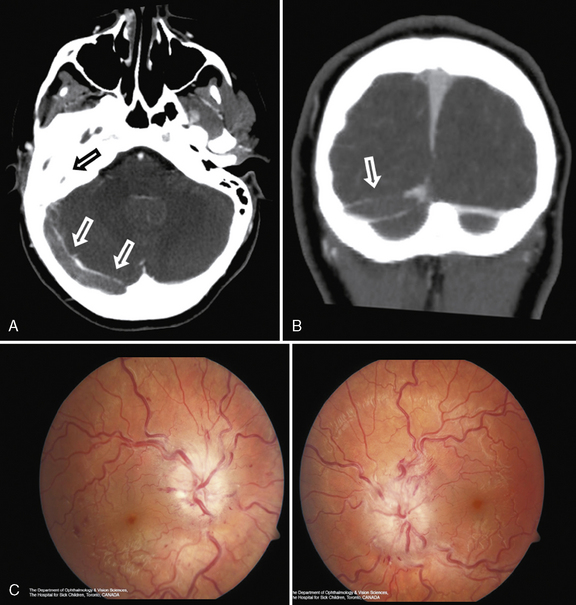
Magnetic Resonance Imaging
In most circumstances, MRI is the diagnostic modality of choice in pediatric CSVT. MRI lacks radiation and may provide additional details of both brain parenchyma and the venous system. Combining MRI modalities sensitive to vasogenic edema (e.g., FLAIR), cytotoxic edema and infarction (e.g., diffusion), and blood product (e.g., gradient echo) can both confirm and characterize CSVT-related parenchymal changes (Figures 85-12, 85-13, and 85-15). MRI may also image thrombosis itself, possibly providing more information about the age and nature of the lesion. Subacute thrombus often appears hyperintense on T1- and T2-weighted images (see Figure 85-12). This hyperdense appearance can be particularly helpful in diagnosing cortical vein thrombosis, where a filling defect may be hard to demonstrate, however, numerous artifactual mimics also must be excluded [Leach et al., 2006]. Restricted diffusion within a thrombus appears to predict a longer time to recanalization [Favrole et al., 2004]. Susceptibility-weighted MRI, which is exquisitely sensitive to blood, also may help detect small, early, and/or isolated cortical vein thrombosis [Idbaih et al., 2006].
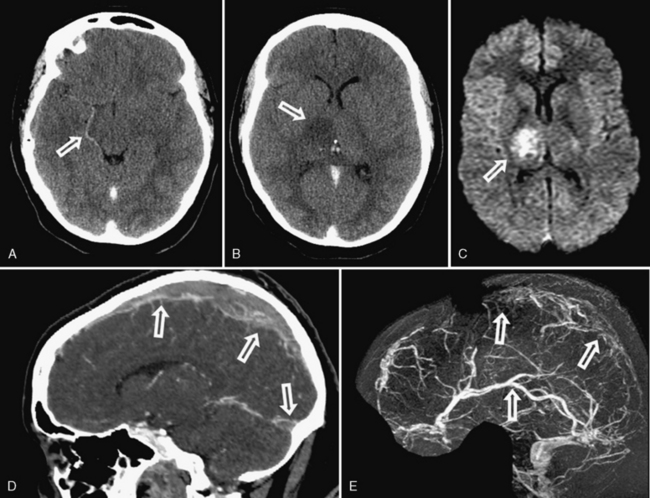
MR venography can employ time-of-flight or contrast-enhanced methods to image the cerebral venous system in a comparable fashion to CTV (see Figure 85-15). Time-of-flight venography depends on flow-signal characteristics and is therefore subject to significant artifacts, especially slow or turbulent flow [Ayanzen et al., 2000]. Thus, apparent “filling defects” must be interpreted with caution. Expert interpretation is essential and confirmatory CTV may be required. Gadolinium-enhanced MR venography is superior to time-of-flight venography and, possibly, to CTV in adults, and shows promise in children [Farb et al., 2003a; Rollins et al., 2005].
Treatment
Many of the overall approaches to the treatment of the child with stroke described above, including urgent supportive and neuroprotective care, apply in CSVT. Antithrombotic interventions to prevent thrombus propagation and allow intrinsic fibrinolytic systems to work are the mainstay of treatment. Subacute treatment includes treatment of intracranial hypertension. In the longer term, attention to secondary prevention is important. Several treatment guidelines for pediatric CSVT are available [Paediatric Stroke Working Group, 2004; Monagle et al., 2008b; Roach et al., 2008]. Additional CSVT treatment guidelines for children and adults have been published by the American Heart Association.
Emergent Treatment
Thrombolysis
Emergent recanalization therapies in CSVT are poorly studied, particularly in children, and are considered experimental. Due to the potential risks, endovascular thrombolysis is reserved for children with progressive deterioration, despite an adequate trial of anticoagulation. When it is being considered, a consensus of experts in childhood stroke, acute stroke care, neurointerventionalists, and the family is required. A regional, small burden of clot theoretically is more easily amenable to thrombolysis, compared to the large volumes of thrombus often seen in childhood CSVT. In adults, limited evidence supports possible efficacy of endovascular thrombolysis [Ferro and Canhao, 2008; Ciccone et al., 2004; Sujith et al., 2008]. However, in one study, endovascular thrombolysis was associated with increased hemorrhage in one-quarter of treated adults, and death or handicap in 40 percent [Stam et al., 2008]. Published experience in children has been limited to case reports or small case series [Griesemer et al., 1994; Wong et al., 1987; Prasad et al., 2006]. One large series of childhood CSVT reported three treated with thrombolysis but no details were provided [Wasay et al., 2008]. A recent single-center case series of 21 children with CSVT reported thrombolysis treatment in 4 (19 percent) children, all with severe progression of thrombosis despite anticoagulation [Mallick et al., 2009b]. In each case, local sinus application of thrombolytics was combined with guidewire manipulation, and no major adverse events were reported.
Urgent Treatment
Anticoagulation
Anticoagulant treatment of childhood CSVT is supported by adult CSVT clinical trials and large cohort studies, smaller pediatric CSVT cohort studies, and research and clinical experience in extracranial venous thrombosis. Medical therapy of neonatal CSVT also is discussed elsewhere (see Chapter 18). Three properly powered randomized controlled trials in adult CSVT have reported significant increases in survival and improved neurological outcome in patients treated with anticoagulation [Einhaupl et al., 1991; Maiti and Chakrabarti, 1997; Nagaraja et al., 1995]. One large study was under-powered to find a difference [de Bruijn and Stam, 1999]. Meta-analysis of the adult data suggests, at worst, good safety and a trend for efficacy, and, at best, a significant reduction in death and disability [Stam et al., 2003]. The safety of anticoagulation in adults with CSVT and intracerebral hemorrhage also is supported, with only 1 of 52 patients receiving heparin, despite initial intracerebral hemorrhage, having extension of the hemorrhage [Nagaraja et al., 1995]. Additional large cohort studies report safety and improved outcomes in adults with CSVT treated with anticoagulation [Milandre et al., 1989; Ameri and Bousser, 1992; Preter et al., 1996].
The safety of anticoagulation therapy in children with CSVT previously was supported by numerous studies [deVeber et al., 1998a, 2001; Dix et al., 2000; Streif et al., 1999; Sebire et al., 2005; Mallick et al., 2009b; Vieira et al., 2009]. The safety of standardized anticoagulant treatment has been established further now in a recent single-center study of 162 infants and children [Moharir et al., 2006]. The 85 patients who received anticoagulation therapy at diagnosis included 29 of 83(35 percent) neonates and 56 of 79 (71 percent) children. Major hemorrhage occurred in 6 percent (6 of 99) of treated patients, all nonfatal, and the proportion with favorable clinical outcome was similar to the remaining patients. The frequency with which children with CSVT are anticoagulated is unknown, although current practice patterns, even among pediatric stroke experts, likely vary widely [Fullerton et al., 2007]. The three consensus-based pediatric stroke guidelines are in general agreement regarding the indications, contraindications, and duration of anticoagulation therapy [Monagle et al., 2008b; Roach et al., 2008; Paediatric Stroke Working Group, 2004; deVeber and Kirkham, 2008]. Only “major” intracerebral or systemic bleeding is considered a relative contraindication. If acute anticoagulation therapy is not initiated for this or another reason, early repeat venous imaging (5–7 days) is indicated to exclude thrombus propagation. Recent evidence suggests that thrombus propagation occurs in 20–30 percent of children with CSVT who are not treated with anticoagulation in the first week, and that propagation is associated with new parenchymal infarction and worse outcome [Moharir et al., 2006].
Anticoagulation therapy options include unfractionated heparin, LMWH, and, possibly, coumadin. Dosing and administration guidelines for all are available [Monagle et al., 2008b]. The duration of therapy follows common adult practice and is planned for 3–6 months [deVeber and Kirkham, 2008]. Repeat cerebral venous imaging is performed at 3 months. If complete recanalization has occurred, and no severe risk factors remain, therapy often is discontinued. In the face of on-going risk or incomplete recanalization, continuation of therapy to 6 months is suggested. If risks have resolved, therapy often is discontinued at this point, regardless of the degree of recanalization, though the evidence for this approach is minimal.
Urgent Treatment
Non-antithrombotic therapies
In addition to antithrombotic and neuroprotective treatments, both specific underlying etiologies and early consequences of CSVT must be addressed. Screening, recognition, surveillance, and management of increased intracranial pressure are a top priority in children with CSVT (see Figure 85-14). Malignant intracranial pressure and resulting ischemic optic neuropathy can occur acutely or over the long term, and can be secondary to thrombosis, narrowed venous sinuses, or communicating hydrocephalus. Children with globally impaired venous drainage (e.g., bilateral transverse sinus disease) are at increased risk; however, all patients should be monitored. Regular funduscopy is required, including retinal photography by an ophthalmologist if possible. At-risk patients may require serial visual field testing and imaging. Treatment options include the temporary use of carbonic anhydrase inhibitors to lower cerebrospinal fluid production, serial lumbar punctures, and, in resistant cases, lumboperitoneal shunting or optic nerve sheath fenestration.
Septic CSVT requires aggressive, pathogen-specific antibiotics, usually at meningitic doses. Surgical mastoidectomy sometimes is required. Unique infection-related CSVT, like Lemierre’s syndrome, requires specific antimicrobial agents. Adult evidence suggests that corticosteroids are generally not beneficial, and possibly even harmful, in CSVT, so routine use in children is not supported. However, this may need to be balanced for children with systemic inflammatory diseases responsible for their CSVT, such as inflammatory bowel disease or nephrotic syndrome. Other modifiable risk factors include removing jugular venous lines, altering chemotherapy protocols, or possibly replacing depleted anticoagulation factors. Simple measures to minimize risk factors should not be forgotten, such as ensuring full hydration and replacement of iron stores. Recently, urgent decompressive hemicraniectomy has been employed as a life-saving measure and was associated with excellent outcome in several cases [Coutinho et al., 2009].
Outcome
Outcome after pediatric CSVT has been described now in numerous cohort studies. While in general agreement, few have employed standardized outcome measures and many lack the long-term duration of follow-up required to appreciate the full spectrum of morbidity, including late-appearing deficits, from early childhood brain injury. Consistent with this, 25 percent of children with CSVT followed in the Canadian Pediatric Ischemic Stroke Registry showed increased severity of their neurological deficits over time [deVeber et al., 2001].
The nine available pediatric CSVT outcome studies report favorable outcomes ranging from 26 to 75 percent, suggesting not only inconsistency in methods, but also a generally worse outcome as compared to adults [Wasay et al., 2008]. Several large studies suggest that only 10–15 percent of adult CSVT patients suffer adverse neurological outcomes [Preter et al., 1996; Ferro et al., 2004]. Pediatric cohort studies report mortality rates of 4–25 percent, though the primary causes of death often are not described. Adult predictors of poor outcome include presentation with stupor or coma, intracranial hemorrhage, and deep system involvement [Mehraein et al., 2003; Ferro et al., 2004]. Pediatric studies suggest similar clinical outcome predictors [Wasay et al., 2008; deVeber et al., 2001]. The largest population-based, prospective cohort study of childhood CSVT (mean follow-up 1.6 years) reported unfavorable outcomes in 48 percent, including 35 percent with neurological deficit, 20 percent with epilepsy, and a 9 percent mortality [deVeber et al., 2001]. The recent single-center prospective study of 162 infants and children reported unfavorable outcome in 47 percent. In the 68 children beyond 1 month of age at diagnosis, there were no CSVT-related deaths and 63 percent were normal, with 16 percent having mild neurological deficit and the remainder having moderate or severe deficits [Moharir et al., 2010]. Children with deep CSVT and bilateral infarctions of central structures can be impaired more severely. Persistent intracranial hypertension, communicating hydrocephalus, and visual sequelae may occur in up to 30 percent of individuals [Sebire et al., 2005; Mallick et al., 2009b]. The comprehensive studies required to characterize the long-term neuropsychological outcomes from childhood CSVT have not been completed.
In one large study, 13 percent of children with CSVT developed recurrent cerebral or systemic thrombosis [deVeber et al., 2001]. This rate appears comparable to that in adults, in whom symptomatic recurrence usually occurs within the first year, including CSVT (12 percent) and pulmonary embolism (11 percent) [Bousser, 1997; Preter et al., 1996]. Due to the risk of recurrence, screening for chronic risk factors (e.g., prothrombotic state) and long-term follow-up are advisable. As many studies have included a high proportion of anticoagulated patients, the natural history of recurrence cannot be extrapolated. The relative influence of sinus recanalization on CSVT outcome is not well known. In adults, recanalization rates appear maximal after 4 months of anticoagulation therapy [Baumgartner et al., 2003]. An association between recanalization and favorable outcome has been suggested in adults [Strupp et al., 2002]. In childhood CSVT, incomplete recanalization is seen frequently [Vieira et al., 2009; Sebire et al., 2005], and the rate of recanalization may correlate with outcome [Moharir et al., 2010]. All children with CSVT require the same comprehensive, family-centered care approach to rehabilitation, education, and support as do patients with AIS.
Hemorrhagic Stroke
Hemorrhagic stroke (HS) occurs when cerebral blood vessels rupture. These are often abnormal blood vessels (e.g., vascular malformation), although normal vessels can also hemorrhage (e.g., bleeding diatheses). The result is the extravasation of blood into intracranial compartments and these locations are employed to classify HS. HS is a neurological emergency with a high mortality rate, acutely and secondary to a high recurrence risk. Two major types of HS are discussed here: intracerebral (i.e., intraparenchymal) hemorrhage (ICH) and subarachnoid hemorrhage (SAH). Relative rates of each location in pediatric HS are compared infrequently [Giroud et al., 1995; Lanthier et al., 2000], but ICH likely accounts for 50–75 percent, while SAH is 25 percent or less [Blom et al., 2003]. Intraventricular hemorrhage (IVH) is more common in the neonate (see Chapter 18) but can occur, often via extension of ICH or SAH [Kumar et al., 2009]. The close approximation of these three spaces often means blood will be found in multiple locations, though certain lesions are more likely to occur in each location. Extra-axial intracranial bleeding is subdivided into subdural and epidural hematomas. As these are usually associated with trauma, they are addressed elsewhere (see Chapters 18 and 74).
Important differences exist between hemorrhagic and ischemic stroke in children. Although studies have varied, incidence rates of the two are probably comparable overall, estimated at 1–5/100,000 children/year [Mallick and O’Callaghan, 2009a]. A specific cause is more likely to be found for hemorrhagic compared to ischemic stroke, though many still remain idiopathic. Etiologies are entirely different, in general, though overlap certainly exists and conditions like moyamoya, SCD, and postinfectious and congenital vasculopathies can predispose to both [Schoenberg et al., 1978; Danchaivijitr et al., 2006b; Heyer et al., 2008; Scott and Smith, 2009]. In contrast to ischemic stroke, definitive treatment for HS often requires neurosurgical intervention. Compared with AIS, HS is associated with an increased mortality but survivors tend to have better neurological outcomes.[Lo et al., 2008b] Long-term recurrence risk is often high, necessitating close follow-up and curative therapy when possible [Fullerton et al., 2007a].
Clinical research in childhood HS has lagged behind ischemic stroke. Information provided here is summarized from a growing collection of single-center, retrospective cohort studies and case series [Livingston and Brown, 1986; Lin et al., 1999; Blom et al., 2003; Lanthier et al., 2000; Meyer-Heim and Boltshauser, 2003; Lo et al., 2008b; de Ribaupierre et al., 2008; Kumar et al., 2009]. While not a formal systematic review and recognizing significant methodological differences, an overview of the nine largest non-neonatal HS studies published in the CT era is provided in Table 85-3. Larger, multicenter, prospective cohort and case-control studies are required. Given the more generalized available evidence regarding clinical presentations, diagnostic imaging, treatment, and outcomes, these elements are discussed here collectively, with reference to ICH and SAH specifics as indicated. This is followed by a review of specific diseases predisposing to HS in childhood.
Table 85-3 Summary of the Main Findings from the Largest Published Case Series of Childhood Hemorrhagic Stroke

Pathophysiology
Hemorrhage into intracerebral spaces occurs when arteries rupture. Lesion type will influence location, such as medium arteries in arteriovenous malformations (AVMs), larger arteries in congenital aneurysms, and distal vessels in mycotic aneurysms. In contrast to adult HS, predicting etiology by location is less precise in children. The majority of events are supratentorial, with approximately equal involvement of left and right hemispheres, while cortical versus subcortical patterns are not well described [Meyer-Heim and Boltshauser, 2003; Livingston and Brown, 1986; Blom et al., 2003; de Ribaupierre et al., 2008]. An intraparenchymal hematoma can disrupt neuronal structures directly by mechanical means, but may also compromise local perfusion and metabolism. However, in contrast to ischemic stroke, “lesional” brain tissue in ICH may also be displaced, rather than damaged. This may explain, in part, the generally better outcomes seen in survivors of HS compared to AIS and CSVT. The presence of blood products and damage to the blood–brain barrier promotes cerebral edema, resulting in acute neurological deficits, seizures, and malignant increases in intracranial pressure. Resulting herniation will depend on lesion location and size, but includes transtentorial, cerebellar, central, and subfalcine syndromes. Resulting pressure on critical structures can be quickly fatal, particularly in the posterior fossa, where limited space can cause immediate brainstem compression, as well as obstructive hydrocephalus.
Clinical Features
An instantaneous, severe headache is the hallmark of HS. A thunderclap headache, particularly if accompanied by nausea/vomiting or neurological symptoms, should be considered HS until proven otherwise. Headache with nausea/vomiting is the most consistently reported presentation of HS in children, described in more than 50 percent. In a large series of childhood HS where 78 percent of children presented with headache, only 30 percent were described as “sudden” in onset [de Ribaupierre et al., 2008]. Children with aneurysms and SAH are more likely to present acutely than those with AVM [Meyer-Heim and Boltshauser, 2003]. The significance of warning or “sentinel” bleed symptoms is well established in adult SAH, and should be extrapolated to children, in whom prompt evaluation still is required, even when symptoms are transient. Although less studied than in AIS, diagnostic delays appear common in childhood HS, and may relate to decreased awareness of primary care physicians, nonspecific clinical presentations, and delayed investigations [Meyer-Heim and Boltshauser, 2003; de Ribaupierre et al., 2008].
Alterations in level of consciousness and focal neurological deficits also are common, while seizures are initially present in 15–37 percent [Livingston and Brown, 1986; Meyer-Heim and Boltshauser, 2003; Lo et al., 2008b; de Ribaupierre et al., 2008; Kumar et al., 2009]. However, HS presentations also can be subtle and slowly progressive, described in up to 40–50 percent of cases [Meyer-Heim and Boltshauser, 2003; Livingston and Brown, 1986]. In posterior fossa hemorrhage, bulbar signs, ataxia, and rapid deterioration to coma are typical. An elevated blood pressure at presentation may be found in up to 45 percent [Lo et al., 2008b] but simply may represent a physiological response. Careful systemic examination can reveal the etiology, including cranial bruits, skin lesions (e.g., purpura, telangiectasias, hemangiomas), markers of infective endocarditis, or recognizable genetic syndromes. Meningeal signs and fever may occur with subarachnoid bleeding and need to be distinguished from infection. In SAH, retinal examination may reveal subhyaloid retinal hemorrhages. As in ischemic stroke, specific signs and symptoms are more difficult to appreciate in young infants, in whom unexplained irritability, behavior changes, or a bulging fontanel may be evident [Lo et al., 2008b]. Unique presentations of cerebrovascular malformations include infants with vein of Galen malformations, who may develop congestive heart failure and macrocephaly [Lasjaunias et al., 1996]. All children presenting with symptoms suggestive of ICH require rapid assessment, stabilization, and imaging.
Diagnosis
Emergent investigation of suspected HS in a child begins with CT. Adult SAH studies suggest sensitivities of 92–100 percent in the first 24–48 hours, falling to 85 percent at 3 days, and only 50 percent at 1 week [Sames et al., 1996; Edlow et al., 2008]. These observations are extrapolated reasonably to children and other locations of hemorrhage with blood appearing hyperdense, although very acute or active bleeding can be hypodense or isodense to brain. Addition of acute CTA may add useful information in adult stroke, in which both the “spot sign” and contrast extravasation may predict early hematoma expansion [Thompson et al., 2009; Ederies et al., 2009]. Mechanisms of childhood HS are different, but responsible lesions, such as an AVM or aneurysm, can be detected by CTA (Figures 85-16 to 85-19).
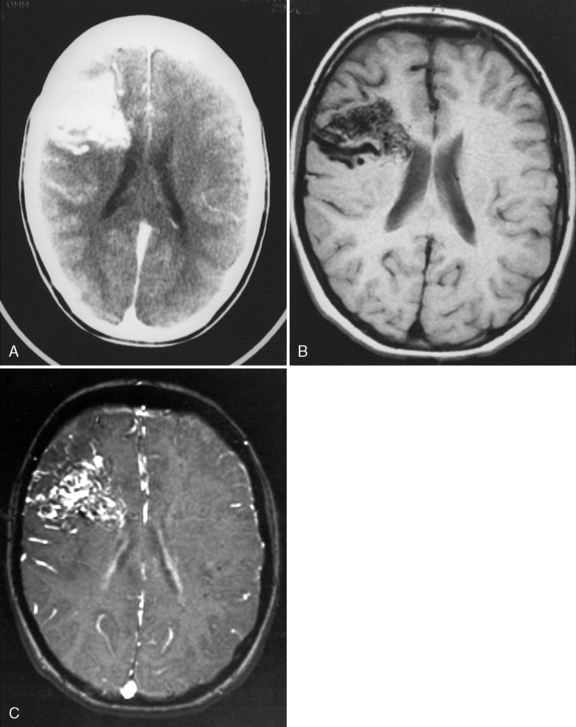
Fig. 85-16 Large hemispheric arteriovenous malformation in an 8-year-old child presenting with seizures.
(Courtesy of Karel Ter Brugge and Division of Neuroradiology, Hospital for Sick Children, Toronto, Ontario, Canada.)
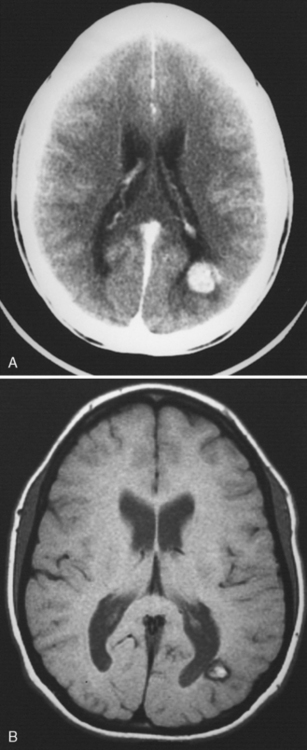
Fig. 85-17 Cavernous malformation in an 18-year-old male presenting with seizures.
(Courtesy of Karel Ter Brugge and Division of Neuroradiology, Hospital for Sick Children, Toronto, Canada.)
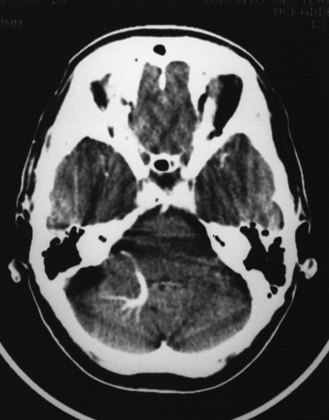
Fig. 85-18 Venous angioma of the cerebellum of a 6-year-old male presenting with migraine headache.
(Courtesy of Karel Ter Brugge and Division of Neuroradiology, Hospital for Sick Children, Toronto, Ontario, Canada.)
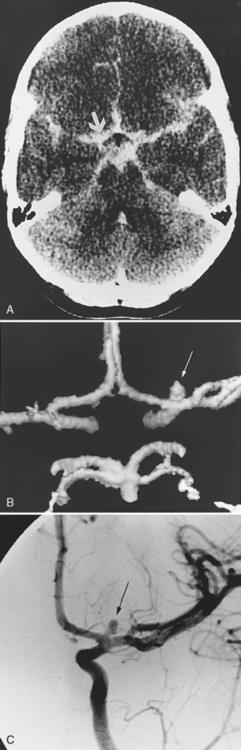
Fig. 85-19 Saccular aneurysm of the left anterior cerebral artery manifesting with subarachnoid hemorrhage.
(Courtesy of Karel Ter Brugge and Division of Neuroradiology, Hospital for Sick Children, Toronto, Ontario, Canada.)
A child with unexplained thunderclap headache or other suspicion of HS and a normal CT requires lumbar puncture to exclude xanthochromia [Sidman et al., 1996]. The precise temporal evolution in hemoglobin degradation required to produce visible xanthochromia in children is unknown, but adult studies suggest it occurs within hours and is nearly always detectable by 6–12 hours [Edlow et al., 2008]. Dilution of cerebrospinal fluid red blood cell counts from first to last tube does not distinguish a bloody tap from intracranial hemorrhage reliably [Shah and Edlow, 2002]. Additional abnormalities in cerebrospinal fluid opening pressure, glucose (low), or protein (high) also may be present in HS patients, while elevated white cells could suggest uncommon etiologies, such as infection.
As most published pediatric HS studies have spanned long periods of time, the availability of MR applications was often limited and their role remains to be defined (see Figures 85-16, 85-17, and 85-20). Advantages of MRI are many, including the ability to image the parenchyma and define areas of injury, as well as the arterial (MR angiography) and venous (MR venography) systems to detect vascular lesions. An imaging study of 50 children with nontraumatic HS found that MRI was able to define the etiology in 65 percent of cases, with a false-negative rate of only 6 percent [Liu et al., 2006]. Newer MRI methods with exquisite sensitivity to both acute and chronic blood products, such as gradient echocardiography and susceptibility-weighted imaging, may be particularly advantageous in childhood HS [Santhosh et al., 2009]. Such modalities can detect extremely small “microbleeds,” allowing the study of previously unrecognized HS syndromes in multiple diseases, including nonaccidental trauma, cavernomas, and endocarditis [Klein et al., 2009].
Fig. 85-20 Vein of Galen malformation in a 3-month-old infant presenting with cardiac failure.
(Courtesy of Karel Ter Brugge and Division of Neuroradiology, Hospital for Sick Children, Toronto, Ontario, Canada.).
CA remains the gold standard in childhood HS (see Figure 85-19). One large case series found a definable etiology in 97 percent of children undergoing CA, compared to 80 percent who did not [Al Jarallah et al., 2000]. Despite this, studies suggest that only 53–67 percent of children with HS undergo CA [Livingston and Brown, 1986; Meyer-Heim and Boltshauser, 2003; Lin et al., 1999; Al Jarallah et al., 2000]. Acutely negative CA may need to be repeated weeks later, as vasospasm, thrombosis, or hemorrhage may alter the lesion, making it temporarily undetectable. A small study of children with SAH suggests that CA and MR angiography are well correlated and of comparable clinical utility [Fasulakis and Andronikou, 2003]. Unless a clear alternative etiology is evident, such as a severe hematological disturbance, children with HS should undergo CA, either to define fully the characteristics of vascular pathology, or to exclude definitively it. As most children will require general anesthesia for CA, a combined diagnostic and interventional procedure may be planned beforehand.
Risk Factors
A probable cause for HS can be found in 85–90 percent of children (Box 85-1). Vascular diseases, both congenital and acquired, as well as intravascular factors, must be considered. Congenital vascular causes predominate, and are collectively responsible for more than half of all cases of childhood HS [Livingston and Brown, 1986; Lin et al., 1999; Blom et al., 2003; Lanthier et al., 2000; Meyer-Heim and Boltshauser, 2003; Lo et al., 2008b; de Ribaupierre et al., 2008; Kumar et al., 2009]. The most common and unique varieties – AVM, cavernoma, aneurysm, and vein of Galen malformation – and their specific management are discussed in detail below. Prior to this, a brief summary of the causes, management, and outcomes of the remaining 40–50 percent of cases is provided.
Box 85-1 Disorders Associated with Hemorrhagic Stroke in Children







































Congenital Arteriopathies
In addition to AVM, cavernoma, and some aneurysms, numerous less common congenital arteriopathies have been associated with childhood HS. Hereditary hemorrhagic telangiectasia (HHT) or Osler–Weber–Rendu syndrome features systemic vascular malformations affecting the lung and liver, as well as the nervous system [Krings et al., 2005]. Dominantly inherited mutations in several genes within the transforming growth factor (TGF)-β signaling superfamily are responsible [Govani and Shovlin, 2009]. Connective tissue disorders, such as dominantly inherited COL4A1 mutations, can predispose to recurrent HS [van der Knaap et al., 2006]. Pediatric versions of fibromuscular dysplasia include intracranial aneurysms and HS [Kirton et al., 2006]. An increasing number of congenital conditions associated with cerebral vasculopathy and HS includes Majewski osteodysplastic primordial dwarfism (MOPD) II [Brancati et al., 2005], PHACES [Heyer et al., 2008], and Alagille’s [Emerick et al., 2005] syndromes.
Acquired Arteriopathies
The list of acquired arteriopathies predisposing children to HS overlaps with those causing AIS. Ischemic infarcts may undergo significant hemorrhagic transformation, some extensive enough to mimic primary HS. This is more common in CSVT ischemic stroke, of which at least 50 percent of cases are hemorrhagic [deVeber et al., 2001], but may also occur in 10–20 percent of childhood AIS [Kirton et al., 2007]. Intracranial arterial dissection may be more common in children as compared to adults in whom failure of arterial wall integrity leads to SAH [Fullerton et al., 2001]. In moyamoya disease, HS risk is lower than that of ischemic stroke, but still is substantial and may relate to unstable collateral vessels, diffuse arteriopathy, hypertension, and other factors. Both morbidity and long-term recurrence risk of moyamoya HS are high [Yoshida et al., 1999]. Additional arteriopathies that typically lead to AIS but in which HS has been reported include SCD, postradiation vasculopathy, and primary and secondary CNS vasculitides. Several infectious arteriopathies carry a much higher risk of HS, including mycotic aneurysms secondary to bacterial endocarditis (see Figure 85-8) [Venkatesan and Wainwright, 2008] and HIV vasculopathy [Shah et al., 1996]. The generalized cerebral arteriopathies and risk factors so prominent in adult HS, such as hypertension, amyloid, atherosclerosis, and smoking, are not seen in children. Hypertension is listed as a potential factor in several studies, with frequencies ranging from 5 to10 percent [Livingston and Brown, 1986; Meyer-Heim and Boltshauser, 2003]. However, most do not report the timing of measurement and a large study reporting acute hypertension in 45 percent of children with ICH did not identify it as a causative risk factor in any of 85 cases [Lo et al., 2008b].
Traumatic Hemorrhage
Traumatic hemorrhage can occupy epidural, subdural, subarachnoid, intracerebral, or intraventricular intracranial compartments (see Chapter 74). Traumatic ICH can appear as one or multiple hemorrhagic contusions or multifocal small hemorrhagic foci throughout the white matter. Traumatic SAH can result from the direct rupture of small subarachnoid vessels, or can occur as a complication of post-traumatic dissection of cerebral arteries. Any unexplained ICH in infants must raise the consideration of nonaccidental trauma. A large cohort study of shaken baby syndrome described the following hemorrhage patterns: subdural (94 percent), SAH (16 percent), ICH (8 percent), and epidural (1 percent) [Morad et al., 2002]. Retinal hemorrhages were present in 75 percent and were correlated with intracranial bleed severity. Dural arteriovenous fistulas (DAVF) are abnormal arteriovenous shunts within the dura mater of the sinuses that can predispose to HS and be either spontaneous or post-traumatic [Neumaier-Probst, 2009].
Hematological Factors
Hematological factors are a major consideration in any child with intracerebral bleeding. Compared to adult ICH, children are more likely to harbor a bleeding diathesis that may be related to numerous underlying conditions [Eyster et al., 1978; Roach and Riela, 1995]. A hematological factor is identified as the primary cause in 5–20 percent of childhood HS [Livingston and Brown, 1986; Blom et al., 2003; Lanthier et al., 2000; Meyer-Heim and Boltshauser, 2003; Kumar et al., 2009]. One large study identified hematological factors in 32 percent of cases, with examples including thrombocytopenias, multiple coagulation factor deficiencies, and SCD [Al Jarallah et al., 2000]. Additional intravascular considerations include coagulopathies secondary to hematological malignancies and their treatment, hepatic dysfunction, and factor deficiencies such as hemophilia. Thrombocytopenia may be secondary to infectious, immune, or neoplastic diseases, as well as their treatments. All children with otherwise unexplained HS require an immediate evaluation of their hemostatic system, including complete blood count, prothrombin and partial thromboplastin times, and specific hematological studies as indicated.
Additional etiologies for childhood HS include bleeding into brain neoplasms, described as the cause in 2–15 percent of cases [Al Jarallah et al., 2000; Lo et al., 2008b]. Iatrogenic causes include neurosurgical procedures and intravascular interventions, such as angioplasty or stenting and, occasionally, CA. Finally, exposure to medications and illicit substances should be considered. Potential substances include thrombolytics, anticoagulants, antiplatelet agents, sympathomimetic drugs, cocaine, and amphetamine [Forman et al., 1989; Levine et al., 1990]. Thorough history, examination, and appropriate investigations usually will uncover the cause of childhood HS, with less than 10 percent remaining idiopathic in most studies.
Treatment
Management of a child with HS rests on the same principles described above for ischemic stroke, with a few important distinctions. Evidence to date is minimal and the three pediatric stroke consensus guidelines do not comment on pediatric HS [deVeber and Kirkham, 2008]. Neurosurgical intervention often is required in childhood HS [Punt, 2004; Smith et al., 2002], reported in 60–80 percent of cases [Blom et al., 2003; Meyer-Heim and Boltshauser, 2003; Kumar et al., 2009; Livingston and Brown, 1986; de Ribaupierre et al., 2008]. Emergency procedures often include evacuation of a hematoma, although the indications for this in adult HS are controversial, and no consensus has been established for children. Early placement of intraventricular drains and intracranial pressure monitoring devices also may be indicated. Depending on the underlying cause, additional surgical procedures include clipping or coiling of aneurysms, removal or embolization of AVMs (see below), removal of neoplasms, placement of ventriculoperitoneal shunts for post-hemorrhagic hydrocephalus, and revascularization surgery for moyamoya.
Medical therapies for the treatment of HS must not be overlooked and are evolving. A randomized clinical trial of 399 adults with HS treated with recombinant factor VIIa (rfVIIa) within 4 hours found decreased rates of hematoma growth, poor neurological outcome (approximately 70 percent vs. approximately 50 percent), and death. However, this was offset by a more than 3-fold increased risk of serious thrombotic events [Mayer et al., 2005]. The role of rfVIIa in adult HS remains controversial and results are minimally applicable to children, in whom mechanisms of HS and the coagulation systems are very different. While such therapies likely will evolve further, their use in children with HS must be considered entirely experimental. In contrast, medical management of underlying diseases predisposing to HS is essential. Examples include antibiotics for infections (e.g., endocarditis), replacement of deficient factors (e.g., factor VIII), reversal of offending drugs (e.g., vitamin K), and immunomodulatory therapy for inflammatory conditions (e.g., immune thrombocytopenic purpura). All of the above must be accompanied by the neuroprotective strategies outlined above.
Outcomes
Compared to ischemic stroke, mortality rates are higher but long-term morbidities are less common in HS. The most comprehensive, systematic outcome study in childhood HS followed 56 cases for a mean of 10 years [Blom et al., 2003]. In the 65 percent of children surviving, approximately 50 percent had a favorable outcome. However, this study was unique in that it documented abnormalities on neuropsychological testing and these accounted for most of the morbidity, present to some degree in 52 percent. Hemiparesis was present in 31 percent, though usually was not severe, and 17 percent had epilepsy. These results are generally consistent with the adverse outcomes reported in other studies (see Table 85-3), including mortality rates ranging from 6 to 54 percent, epilepsy in 0–17 percent, and overall “good” outcome in 50–75 percent. Additional adverse outcomes include visual deficits, language disorders, ataxia after posterior fossa HS, and hydrocephalus requiring shunting after IVH. One study found that 40 percent of children required a formal rehabilitation program [Meyer-Heim and Boltshauser, 2003].
Studies to date have not been adequately rigorous or powered to determine predictors of outcome from childhood HS accurately. However, several factors potentially have been associated with poor outcome, including decreased level of consciousness at presentation, delay in diagnosis, and posterior fossa location [Kumar et al., 2009; Higgins et al., 1991]. Mortality is likely higher in acute posterior fossa hemorrhages, where critical brainstem compression often develops before neurosurgical intervention can be carried out [Livingston and Brown, 1986]. Recurrence rates are substantial in childhood HS, mandating timely management of amenable lesions and long-term imaging surveillance in many cases. A population-based study of 116 children with nontraumatic HS years found a 5-year cumulative recurrence rate of 10 percent. Most recurrences (64 percent) occurred in the first 6 months and none was in children with idiopathic HS [Fullerton et al., 2007a]. The largest long-term follow-up study of childhood HS documented recurrent hemorrhage in 21 percent over a 10-year period [Blom et al., 2003]. A specific AVM study found that recurrence rates in children are comparable to adults: approximately 2 percent per year [Fullerton et al., 2005]. All children require imaging surveillance over time, according to the presumed cause of their HS. Secondary stroke prevention may include medical or surgical treatment of the primary condition, while additional measures, such as the avoidance of contact sports, have not been well studied.
Specific Vascular Diseases Underlying Hemorrhagic Stroke
Vascular Malformations and Aneurysms
Vascular malformations traditionally have been defined as vascular developmental anomalies with preservation of abnormal connections between normally structured vessels. Such lesions are classically divided into four types, based on the caliber and histological structure of component vessels: AVMs, cavernous malformations, venous angiomas and capillary telangiectasias [Russell and Rubenstein, 1989]. Capillary telangiectasias are an infrequent cause of HS and usually are associated with neurocutaneous disorders (see Chapter 40). However, additional malformations of the vascular system do not meet these classical criteria entirely. They include vein of Galen malformations (below) and several diseases alluded to above, including DAVF [Neumaier-Probst, 2009] and many congenital arteriopathies (see Box 85-1). Many aneurysms in children are considered congenital and, as an important cause of HS, they also are included here.
Arteriovenous malformations
A true AVM is an admixture of arteries and veins and can range in size from a few millimeters to several centimeters. Development of an AVM is thought to result from failure of the normal capillary bed between primitive cerebral arteries and veins formed during the first trimester. Most AVMs are isolated and sporadic, although familial cases are reported [Yokoyama et al., 1991]. AVM is the leading cause of HS in children, accounting for 13–55 percent of cases (see Table 85-3). The incidence of childhood AVM diagnosis is approximately 1 in 100,000/year, though only 10–20 percent of all AVMs are thought to become symptomatic during childhood [Menovsky and van Overbeeke, 1997].
AVMs tend to cause supratentorial ICH, though SAH and infratentorial or intraventricular involvement is possible. Consistent with this, bleeding AVMs tend to present with sudden onset of headaches, nausea/vomiting, and focal deficits. However, about 20 percent of AVMs will present prior to rupture [Humphreys et al., 1996], typically with less acute headaches, recurrent focal seizures, or pulsatile tinnitus. CT is sensitive to acute blood and CT angiography can demonstrate characteristic features, such as feeding arteries or draining veins (see Figure 85-16). Advantages of MRI may include the ability to detect parenchymal injury and smaller, older bleeds with blood-sensitive sequences [Liu et al., 2006]. However, all children with suspected AVM require CA to define the anatomy, location, arterial supply, and venous drainage of the lesion accurately so that treatment options can be defined.
Definitive AVM treatment usually is required due to a high rate of recurrence [Fullerton et al., 2005]. Treatment options are complex and include surgical resection, interventional neuroradiological techniques, and radiosurgery [Burrows et al., 1987; Brown et al., 2005]. Successful surgical resection confers immediate protection against recurrent hemorrhage. One study of 36 children with AVMs treated with surgical resection found an HS recurrence rate of 5.5 percent (mean follow-up of 9 years), attributed to undetected residual nidus [Andaluz et al., 2004]. Endovascular treatment of AVM typically involves the injection of occlusive materials into feeding arteries to diminish or potentially cure the lesion. In addition to possibly curing an AVM with a less invasive procedure, endovascular interventions can facilitate subsequent surgical resection or radiosurgery by reducing nidus volume [Gailloud, 2005]. Stereotactic radiosurgery is another option, in which focused irradiation (e.g., gamma knife, proton beam) potentially can obliterate small lesions or decrease the size of larger ones. A study of 31 children treated with radiosurgery found a dose-dependent effect, with an overall obliteration rate of 63 percent [Smyth et al., 2002]. Disadvantages of radiosurgery include long effect times (1–2 years), with an intercurrent risk of bleeding, incomplete effectiveness in some, and limited availability of technology [Loeffler et al., 1990; Levy et al., 2000; Smyth et al., 2002; Shin et al., 2002]. A team of neurosurgeons, neurointerventionalists, radiation specialists, and others expert in pediatric AVM is required to determine the best treatment plan.
Cavernous malformations
A cavernous malformation is a dense collection of thin, dilated, sinusoidal vascular channels without intervening brain parenchyma. These are “dynamic” lesions with recurrent microbleeds, thrombosis, and calcification occurring over time. Only a handful of cavernous malformation studies specific to children have been published [Acciarri et al., 2009; Consales et al., 2009; Mottolese et al., 2001; Lee et al., 2008]. Etiologies are poorly understood but many genetic familial cavernous malformation syndromes are now described [Sirvente et al., 2009]. With dominant inheritance, familial cavernous malformation may include multiple cerebral and retinal lesions, a higher predilection for bleeding, and skin lesions [Malik et al., 1992; Sirvente et al., 2009]. An increased risk of cavernous malformation in children treated with cranial irradiation also has been reported [Burn et al., 2007]. The risk of cavernous malformation hemorrhage is lower than AVM and aneurysm, estimated at less than 0.25 percent annually [Del Curling et al., 1991]. The size of bleeds also is much smaller, and life-threatening hemorrhage is rare, unless it occurs in a vital structure such as the brainstem. Symptomatic presentations in children are approximately one-third seizures, one-third focal neurological deficits, and one-third intracerebral hemorrhage [Simard et al., 1986; Lee et al., 2008]. Compared to cavernous malformation in adults, pediatric cavernous malformation may be larger, more prone to bleeding, and more often associated with additional vascular anomalies [Acciarri et al., 2009].
The advent of MRI, particularly microbleed-sensitive modalities, has increased the number of cavernous malformations diagnosed greatly, leading to a higher proportion of asymptomatic individuals. Typical appearance is a sharply demarcated, spherical, heterogeneous “popcorn” lesion, with evidence of blood (see Figure 85-17) [Mottolese et al., 2001]. Angiography typically is normal, although associated vascular malformations can be found [Rogner, 1976]. With a low bleeding risk, unruptured cavernous malformations in noneloquent areas usually are managed expectantly. For those with epilepsy, focal deficits, or critically located lesions, surgical resection often is preferred [Roach and Riela, 1995; Punt, 2004]. In 42 children with cavernous malformation undergoing surgical resection, 69 percent had a favorable outcome, infrequent morbidities (7 percent) being associated with deep or critically located lesions [Acciarri et al., 2009]. Children operated on for cavernous malformation-associated epilepsy may be cured [Consales et al., 2009]. The role of radiosurgery is limited [Mottolese et al., 2001].
Developmental venous anomalies
Developmental venous anomalies or venous angiomas probably are the most common and most benign cerebrovascular anomalies. They rarely become symptomatic, unless associated with other vascular malformations [Sarwar and McCormick, 1978]. Seizures are the most common presentation of isolated venous angiomas, although a causal relation needs to be established by clinical semiology and investigation [Roach and Riela, 1995]. Both CT/CT angiography and MR/MR angiography can demonstrate venous angiomas and associated vascular lesions [Awad et al., 1993]. Vascular imaging typically shows a “caput medusae” pattern, consisting of a main vein draining to the ventricle or cortical surface with a cluster of tributary veins in a radial configuration (see Figure 85-18) [Agnoli and Hildebrandt, 1985]. Conservative management of isolated venous angiomas usually is recommended, as removal can increase neurological deficit. A recent review of developmental venous anomalies is available elsewhere [Ruiz et al., 2009].
Aneurysms
Aneurysms are focal arterial enlargements secondary to disruption of wall integrity. All three layers of the artery are involved (intima, media, adventitia); if not, the term pseudoaneurysm is used. The size, shape, and underlying causes of aneurysms vary greatly, particularly in children, in whom pathophysiology often is not understood. Many aneurysms in childhood are of the saccular variety and are believed to arise from a focal congenital weakness within the elastic and muscular layers of cerebral arteries [Ahlsten et al., 1985; Rogner, 1976; Khoo and Levy, 1999].
Some pediatric aneurysms are associated with definable congenital or acquired vascular diseases (see Box 85-1). Not surprisingly, these include disorders of connective tissue, such as COL4A1, Ehlers–Danlos syndrome, fibromuscular dysplasia, and polycystic kidney disease. Inherited aneurysms tend to bleed earlier and more often, and multiple syndromes are being defined through the Familial Intracranial Aneurysms Study [Broderick et al., 2009]. Children with dissection may develop pseudoaneurysms with an intracranial location, risking SAH [Tan et al., 2009a]. One large case series suggests dissection may account for over 50 percent of childhood (pseudo)aneurysms [Lasjaunias et al., 2005]. Post-traumatic aneurysms typically occur at vulnerable locations and may be under-recognized in children [Ventureyra and Higgins, 1994]. Acquired aneurysms also may be the result of bacterial endocarditis (see Figure 85-8), where cerebral embolization of infected material leads to localized arteritis and compromised arterial wall integrity. Aneurysms may develop over days, weeks, or months, and surveillance is required [Brust et al., 1990], though whether the increased evidence of microbleeds on MRI seen in adults with endocarditis [Klein et al., 2009] predicts mycotic aneurysm formation is not known. Other infectious arteriopathies may lead to aneurysms, particularly HIV vasculopathy in children [Shah et al., 1996].
Approximately 1–5 percent of cerebral aneurysms present in childhood. In large case series of childhood HS, aneurysms are reported to account for 2–34 percent of individuals (see Table 85-3). A recent population-based study confirmed that aneurysms cause most childhood SAH and are responsible for over 10 percent of all pediatric HS [Jordan et al., 2009b]. Aneurysms in childhood are more common in males, whereas the reverse is true in adults [Ostergaard, 1991]. Giant aneurysms (greater than 10 cm) are found in 20–50 percent of pediatric patients, particularly infants [Kasahara et al., 1996]. Pediatric aneurysms involve the posterior circulation in 20–27 percent, nearly three times more often than in adults [Khoo and Levy, 1999]. A bimodal distribution of aneurysmal HS in childhood has been described [Adner et al., 1969; Jordan et al., 2009b]. In children under 2 years, large aneurysms (over 1 cm) affecting large vessels (anterior or middle cerebral artery, internal carotid artery) tend to predominate [Crisostomo et al., 1986; Kasahara et al., 1996]. Children over 10 years and adolescents tend to have aneurysms at the ICA bifurcation [Khoo and Levy, 1999].
Most childhood aneurysms present with acute SAH, although extension into the parenchyma and intraventricular and extra-axial spaces is common [Lasjaunias et al., 2005]. Headache and encephalopathy were most common in a recent population-based study [Jordan et al., 2009b]. Early warning signs and sentinel bleed symptoms often may be missed in children, as they are in adults. Giant aneurysms may present with slowly progressive neurological deficits secondary to parenchymal or cranial nerve compression. CT/CT angiography and lumbar puncture are required to confirm SAH, as described above. MRI can help identify small, older bleeds and parenchymal injury, but MR angiography can detect only aneurysms of a significant size. CA therefore remains the gold standard for the diagnosis and characterization of cerebral aneurysms (see Figure 85-19). In addition to defining size, CA will detect additional smaller aneurysms and underlying arteriopathies [Ross et al., 1990].
Conservative medical measures to minimize risk of early recurrent hemorrhage include sedation, adequate analgesia, and seizure prophylaxis. Vasospasm is a common cause of neurological morbidity in adults with SAH. Supportive “triple H” therapy includes maintenance of hypervolemia, hypertension, and hemodilution [Komotar et al., 2007]. A Cochrane review concluded that calcium channel blockers (namely, nimodipine) reduce the risk of poor outcome and secondary ischemia in adult SAH [Dorhout Mees et al., 2007a]. Additional measures that potentially reduce the risk of post-SAH vasospasm and infarction include magnesium, vasodilator and antiplatelet therapies, balloon angioplasty, and shunting for hydrocephalus [Bakker et al., 2007; Dorhout Mees et al., 2007a, b; Komotar et al., 2007]. Post-SAH hydrocephalus is a serious but potentially treatable complication associated with adverse outcomes in adults [Bakker et al., 2007]. These interventions are unstudied in childhood SAH. Early aneurysmal rebleeding rates are substantial and may be increased in children compared to adults [Wojtacha et al., 2001]. Prompt curative intervention therefore is preferred but may need to be delayed in unstable patients [Kassell et al., 1990]. Options include direct surgical clipping and interventional procedures such as “coiling,” in which small metal pieces are inserted into the aneurysm via a catheter, resulting in thrombosis and resolution of the aneurysm. The relative risks and benefits are as above, with no pediatric studies comparing them.
Vein of galen malformations
The vein of Galen aneurysmal malformation (VGAM) is the most important of several malformations in this location and is a serious condition of infancy. VGAMs form early in embryogenesis, are fed by choroidal arteries, and do not connect to the deep venous system. VGAM typically presents in early infancy with progressive high-output congestive heart failure secondary to the large intracranial circulatory shunt. Progressive macrocephaly secondary to hydrocephalus occurs due to increased venous pressure. ICH is uncommon. However, children with severe lesions may incur progressive, bilateral cerebral infarction (“melting brain syndrome”) due to a complex, poorly understood pathophysiology. MRI demonstrates a dilated venous structure in the location of the vein of Galen (Figure 85-20) and is sensitive to evolving parenchymal injury. Vascular imaging with MR angiography/MR venography can add information, but CA is required to confirm the origin and number of feeding arteries and venous drainage patterns. Antenatal ultrasound diagnosis is made in about 30 percent of cases and may predict worse outcome, but usually is not an indication for early delivery or cesarean section [Rodesch et al., 1994; Alvarez et al., 2007].
Management of VGAM, particularly the decision to treat surgically or not, often is difficult. A scoring system, considering neurological, cardiac, and systemic elements combined with lesion characteristics, appears to distinguish reliably those children most likely to benefit from intervention from those who require palliative care [Alvarez et al., 2007; Geibprasert et al., 2010]. Neurologically and hemodynamically stable children generally are followed to an older age before definitive treatment – ideally, between 4 and 5 months. Infants with refractory heart failure, hydrocephalus, or neurological deterioration typically are treated with an earlier embolization procedure. Injection of various materials into feeding arteries need only remove effects on acute stability or long-term brain development, and complete obliteration is not always the goal. In the largest published series of 317 children with VGAM, 74 percent underwent embolization, 21 percent were managed conservatively, and only 5 percent were lost to follow-up [Alvarez et al., 2007]. In those requiring neonatal intervention, mortality was 52 percent (many of whom would now be managed conservatively) but less than 10 percent of survivors had severe disability. In contrast, stable children treated later had normal neurological outcomes in 76 percent of cases. Neurological outcome in those with complete obliteration is particularly good, with less than 10 percent having severe sequelae [Alvarez et al., 2007]. These results are consistent with previous studies [Fullerton et al., 2003a; Lasjaunias et al., 1996, 2006]. Staged microsurgical procedures for VGAM generally have fallen out of use [Moodie et al., 1983]. Due to the complex hydrodynamics underlying hydrocephalus in VGAM, cerebrospinal fluid shunting procedures generally are not successful and are even potentially harmful [Zerah et al., 1992]. Children with VGAM will often require a comprehensive chronic care program like that outlined above.
