Chapter 69 Cerebral Palsy
Definition and Clinical Characterization
Cerebral palsy is defined as “a group of disorders of the development of movement and posture, causing activity limitation, that are attributed to non-progressive disturbances that occurred in the developing fetal or infant brain” [Bax et al., 2005]. Virtually all such disturbances occur during or before early infancy. Although tone and postural abnormalities may become more pronounced during early childhood, qualitative evolution is uncommon. The full extent of motor disability may not be evident until the age of 3 or 4 years. Intellectual, sensory, and behavioral difficulties may accompany cerebral palsy, and are especially common in patients with spastic quadriplegia and severe motor disability [Shevell et al., 2009]. Children with cerebral palsy often exhibit mental retardation (52 percent), hearing impairment (12 percent), and speech and language disorders (38 percent) [Ashwal et al., 2004], as well as congenital anomalies [Rankin et al., 2009]. Epilepsy occurs in 34–94 percent of children with cerebral palsy, depending on the study population. Although neurologic impairment beyond motor involvement frequently occurs, the diagnosis of cerebral palsy rests upon the presence of motor disability alone.
Cerebral palsy can be classified by the following attributes [adapted from Bax et al., 2005]:
Cerebral palsy is a clinical syndrome that encompasses a wide range of brain disorders associated with impaired motor function. Thus, the definition of cerebral palsy in research studies often varies, depending on the research question being addressed [Badawi et al., 1998c]. The designation of cerebral palsy as an entity, despite the heterogeneity of underlying etiologic conditions, is valuable because afflicted children commonly have similar medical care, rehabilitation, and social services needs.
Epidemiology
Cerebral palsy occurs in 1.2–3.6 children per 1000 live births. Numerous cerebral palsy registries exist throughout the world [Cans et al., 2004], and population prevalence rates from four continents have remained consistent over several decades. In Denmark, there was a declining birth prevalence between the period of 1983–1986 (3.0 per 1000 live births) and the period 1995–1998 (2.1 per 1000 live births) [Ravn et al., 2009]. In the United States, the prevalence of cerebral palsy increased from 1.7 to 2.0 per 1000 live births between the mid-1970s and late 1980s [Winter et al., 2002], and was estimated to be as high as 3.6 per 1000 8-year-old children in 2002 [Yeargin-Allsopp et al., 2008]. Based on these numbers, approximately 12,000 children with cerebral palsy are born annually in the United States.
Prematurity is the single most important risk factor for cerebral palsy. The risk of cerebral palsy in very low birth weight infants is as high as 4–10 percent, whereas the risk in term infants is only 1.0–1.5 per 1000 live births [Grether et al., 1992; Hagberg et al., 2001; Topp et al., 2001; Wu et al., 2003; Ancel et al., 2006; Moster et al., 2008]. Infants born at 24–26 weeks’ gestation may have as high as a 20 percent chance of developing cerebral palsy [Ancel et al., 2006]. In most epidemiologic studies, preterm infants constitute approximately half of all infants with cerebral palsy. An increase in the prevalence rate of cerebral palsy among preterm infants during the mid-1980s was attributed to increased survival of low birth weight infants [Colver et al., 2000; Pharoah et al., 1990; Topp et al., 2001]. Subsequent longitudinal studies, however, suggest that the rate of cerebral palsy among preterm infants has remained constant or even decreased [Clark and Hankins, 2003; Hagberg et al., 2001; O’Shea et al., 1998; Winter et al., 2002; Platt et al., 2007], though this is not a consistent finding in all populations [Vincer et al., 2006].
Term infants represent more than half of all cases of cerebral palsy [Bax et al., 2006]. The prevalence of cerebral palsy among term infants, 1–1.5 per 1000 live births, has not diminished over the past three decades, despite the introduction of electronic fetal monitoring during the 1970s and the subsequent sharp rise in births by cesarean section [Clark and Hankins, 2003; Colver et al., 2000; Topp et al., 2001; Winter et al., 2002]. Although unchanged prevalence is disappointing in view of the fact that perinatal deaths, stillbirths, and birth asphyxia, as measured by low Apgar scores, all have dropped dramatically in recent decades, other data are more encouraging [Ravn et al., 2009]. Longitudinal data in the United States suggest that a slight increase in cerebral palsy prevalence among infants with normal birth weight was responsible for the significant increase in total cerebral palsy prevalence reported over a 15-year period [Winter et al., 2002]. Although the severity of cerebral palsy within an Australian population increased during this period [Blair, 2001], suggesting that perhaps more infants who would have died in previous times are being saved, workers in the United Kingdom have described a decrease in severity of cerebral palsy among term infants over a similar (18-year) period [Colver et al., 2000].
Male infants exhibit a higher risk of cerebral palsy than female infants [Platt et al., 2007; Wu et al., 2009; Johnston and Hagberg, 2007]. Twins, who constitute 2 percent of the population, also carry a higher risk, and contribute 10–12 percent to the overall prevalence of cerebral palsy [Grether et al., 1992; Bax et al., 2006]. The increased risk of cerebral palsy among infants of multiple gestation is partly due to the higher rate of prematurity, though twins born at term may also exhibit an increased risk for cerebral palsy [Petterson et al., 1993; Pharoah, 2001]. Death in utero of a co-twin, such as from twin–twin transfusion, places the surviving twin at particularly high risk for neurologic and other complications [Grether et al., 1993; Szymonowicz et al., 1986].
Blacks have an increased risk of cerebral palsy when compared with whites [Murphy et al., 1993; Yeargin-Allsopp et al., 2008]. Although this finding is due in part to an increased rate of prematurity, the risk of cerebral palsy among black infants born at term also is elevated [Wu et al., 2003]. Furthermore, several studies suggest that infants born to mothers with lower socioeconomic status exhibit an increased risk of cerebral palsy [Sundrum et al., 2005; Yeargin-Allsopp et al., 2008; Dolk et al., 2001]. In one study that adjusted for potential confounding effects of prematurity, the effect of low socioeconomic status on cerebral palsy risk was only partly accounted for by lower birth weight and gestational age [Sundrum et al., 2005].
Life expectancy of patients with cerebral palsy is related to the type of involvement and the severity of motor disability. Severe quadriplegia has been associated with a shortened life expectancy. Other significant variables include associated disabilities and availability of quality medical care. The risk of death is highest in the first 5 years of life [Blair et al., 2001]. As mortality data have become available, it has become clear that, with reasonable medical attention, a majority of affected persons will survive into adult life [Evans et al., 1990; Eyman et al., 1990; Hutton et al., 1994; Plioplys et al., 1998; Strauss et al., 1998; Plioplys, 2003].
Etiology
A wide range of causative disorders and risk factors have been identified for cerebral palsy. Box 69-1 lists some of the more common etiologic categories and risk factors, broadly classified into the following groups: perinatal brain injury, brain injury related to prematurity, developmental abnormalities, prenatal risk factors, and postnatal brain injury. These factors may coexist and interact with each other in contributing to the pathogenesis of brain injury resulting in cerebral palsy.
Box 69-1 Selected Etiologic Disorders and Risk Factors in Cerebral Palsy*















TORCH, toxoplasmosis, other infections, rubella, cytomegalovirus infection, herpes simplex.
Perinatal Brain Injury
Intrapartum hypoxia-ischemia is a well-described cause of cerebral palsy, especially in the setting of an acute intrapartum event such as uterine rupture, placental abruption, or cord prolapse. The extent to which hypoxic-ischemic brain injury is responsible for cerebral palsy, however, has been a major source of controversy. Epidemiologic studies suggest that in a minority (between 6 and 28 percent) of affected children, cerebral palsy is due to perinatal asphyxia [Hagberg et al., 2001; Nelson and Grether 1998]. The form of cerebral palsy most often associated with hypoxic-ischemic brain injury is spastic quadriparesis.
The term perinatal asphyxia is confusing and deserves further clarification. Traditionally, this term has been defined by clinical signs and symptoms, including low Apgar scores, meconium-stained amniotic fluid, and low umbilical cord pH. Yet the clinical findings used to define this term are not specific for hypoxic-ischemic brain injury [Blair, 1993]. Although various groups of investigators have defined specific criteria that can be used to establish intrapartum hypoxia-ischemia as the underlying cause of cerebral palsy [MacLennan, 1999], a widely accepted, evidence-based standard for determining when cerebral palsy is due to hypoxia-ischemia is still lacking.
Some investigators prefer the term neonatal encephalopathy to describe the clinical syndrome of perinatal asphyxia because it is an all-embracing term that does not imply a single underlying etiologic disorder [Leviton and Nelson, 1992]. Signs and symptoms of brain dysfunction that constitute neonatal encephalopathy include low Apgar scores, failure to initiate and/or maintain respiration, depressed consciousness, abnormal tone (usually flaccidity), depression of developmental reflexes, and seizures during the first 48 hours of life. The clinical syndrome of neonatal encephalopathy may be preceded by a variety of prenatal and intrapartum risk factors [Badawi et al., 1998b], and usually is associated with acute brain injury occurring around the time of birth [Cowan et al., 2003].
Signs and symptoms of birth asphyxia or neonatal encephalopathy are strongly predictive of cerebral palsy in the child. For instance, term infants whose immediate postpartum course was comprised of a 5-minute Apgar score of 5 or less, continuing neurologic abnormalities, and seizures in the first days of life constitute a group at high risk for chronic motor disability (55 percent) and for death or disability combined (70 percent) [Ellenberg and Nelson, 1988]. The risk of cerebral palsy is increased 30-fold in infants with a 5-minute Apgar score of less than 7, and 80-fold in infants with a 5-minute Apgar score of 3 or less [Moster et al., 2001; Thorngren-Jerneck and Herbst, 2001; Wu et al., 2003].
Neonatal stroke, which encompasses ischemic perinatal infarction and sinovenous thrombosis occuring in the perinatal period (before the age of 7 days) or neonatal period (before 28 days of age), is a particularly important cause of cerebral palsy [Lynch and Nelson, 2001]. Ischemic perinatal stroke may be responsible for 28–50 percent of all cases of hemiplegic cerebral palsy in term infants [Humphreys et al., 2000; Uvebrant, 1988; Wu et al., 2003; Bax et al., 2006]. Among children with cerebral palsy who are referred for neuroimaging, 13–37 percent are diagnosed with a neonatal stroke [Ashwal et al., 2004]. Newborns with ischemic perinatal infarction either may present acutely during the neonatal period, with neurologic symptoms such as seizures, or may be clinically asymptomatic until several months of age, when pathologic handedness or seizures are first noted [Golomb et al., 2001]. The risk of cerebral palsy after ischemic perinatal infarction is especially high among infants with delayed presentation; this finding is not surprising, given the fact that hemiparesis is the most common presenting symptom in this group [Golomb et al., 2001; Wu et al., 2004b]. Involvement of the internal capsule also portends a poorer motor outcome after ischemic perinatal infarction [Mercuri et al., 1999; Wu et al., 2004b].
Neonatal sinovenous thrombosis also can lead to cerebral palsy. In a population-based study of pediatric stroke in Canada, 43 percent of all cases of sinovenous thrombosis occurred during the neonatal period [deVeber and Andrew, 2001]. Risk factors for neonatal sinovenous thrombosis include systemic illness, polycythemia, coagulation abnormalities, and extracorporeal membrane oxygen (ECMO) therapy, and motor sequelae such as cerebral palsy were more common if sinovenous thrombosis led to development of venous infarction [deVeber and Andrew, 2001; Wu et al., 2002]. Periventricular venous infarction [Takanashi et al., 2003], intraparenchymal hemorrhage, and birth trauma also may lead to cerebral palsy. (Please refer to Chapter 18 for further discussion of birth trauma.)
Brain Injury Related to Prematurity
Preterm infants constitute a disproportionate share of the cases of children who develop spastic diplegia but can manifest any cerebral palsy subtype. Both intraventricular hemorrhage and white matter necrosis seen in periventricular leukomalacia may occur before or after birth. The main pathogenetic mechanisms underlying periventricular leukomalacia are hypoxia-ischemia and inflammation [Volpe, 2009; Dammann and Leviton, 1997]. Periventricular leukomalacia and intraventricular hemorrhage are reviewed in Chapters 17–19.
Developmental Abnormalities
Brain malformations originating from intrauterine maldevelopment may underlie the neurologic impairment seen in children with cerebral palsy. As a group, children with cerebral palsy have a higher incidence of congenital malformations both within and outside of the brain [Rankin et al., 2009]. Population-based studies of cerebral palsy suggest that 9–14 percent of affected children have a brain malformation [Croen et al., 2001; Wu et al., 2003]. The most common malformations in term children are cortical dysplasia/polymicrogyria, schizencephaly, and pachygyria/lissencephaly. Complex brain malformations are the most frequent category among preterm infants with cerebral palsy [Ashwal et al., 2004].
A genetic or metabolic disorder may be associated with a specific brain malformation that causes cerebral palsy. For example, children with Zellweger’s syndrome typically have polymicrogyria and other cortical malformations. Miller–Dieker syndrome is a known cause of lissencephaly. Glutaric aciduria type I may masquerade as dyskinetic cerebral palsy [Hauser and Peters, 1998], and arginase deficiency mimics diplegic cerebral palsy [Prasad et al., 1997]. Children with cerebral palsy who demonstrate either progressive decline or atypical features such as dysmorphisms, macrocephaly, or a strong family history should be tested for an underlying genetic or metabolic disorder.
Prenatal Risk Factors
A number of maternal conditions have been associated with an increased risk of cerebral palsy in the offspring. Intrauterine inflammation, or chorioamnionitis, has received increasing attention as a potential risk factor [Grether and Nelson, 1997; Leviton et al., 1999; Wu and Colford, 2000]. Epidemiologic studies suggest that maternal intrapartum fever, a clinical or histologic diagnosis of chorioamnionitis, and serologic markers of inflammation in the fetus all confer an increased risk of cerebral palsy. In term infants, chorioamnionitis, often diagnosed by the presence of intrapartum maternal fever, is a particularly strong risk factor for spastic quadriplegia [Grether and Nelson, 1997; Wu et al., 2003]. Maternal fever during labor also increases the chance that the neonate will have low Apgar scores and characteristics of neonatal encephalopathy [Badawi et al., 1998a; Lieberman et al., 2000]. It is hypothesized that the fetal inflammatory response that occurs in the setting of an inflammatory intrauterine environment is responsible for brain injury leading to cerebral palsy [Dammann et al., 2002; Nelson et al., 1998]. Yet the mechanisms by which intrauterine inflammation might cause cerebral palsy remain controversial. It is postulated that inflammation may interact with a hypoxic-ischemic insult to injure the newborn brain in some children with cerebral palsy. A study has indicated that, in the presence of fetal acidemia, chorioamnionitis has poor predictive value and does not portend additional risk for development of neonatal encephalopathy [Shalak et al., 2005]. It has also been postulated that exposure to neurotropic viruses, such as herpes group B viruses, may also increase the risk of cerebral palsy [Gibson et al., 2006a; Djukic et al., 2009].
Intrauterine growth restriction is associated with an increased risk of cerebral palsy, especially in term infants [Jarvis et al., 2003; Topp et al., 1996]. Infants who are large for gestational age also have been reported to be at higher risk [Uvebrant and Hagberg, 1992]. Prothrombotic abnormalities, including factor V Leiden, methylenetetrahydrofolate reductase (MTHFR) C677T, and presence of anticardiolipin antibodies, have been found with increased frequency among infants with cerebral palsy and perinatal arterial infarction [Gunther et al., 2000; Hagstrom et al., 1998; Gibson et al., 2005], though not all studies have confirmed these associations [Miller et al., 2006]. A history of infertility also has been found to be associated with an increased risk of neonatal encephalopathy, developmental delay, and cerebral palsy [Badawi et al., 1998b; Ericson et al., 2002; Stromberg et al., 2002].
The pathogenesis of cerebral palsy is complex and multifactorial. For instance, among infants with perinatal ischemic infarction in the newborn period, multiple risk factors often are observed [Gunther et al., 2000; Wu et al., 2002]. When signs of hypoxia-ischemia and markers of infection both are present in a newborn, the risk for cerebral palsy is markedly heightened [Nelson and Grether, 1998]. Studies of placental abnormalities in cerebral palsy also suggest that a combination of thrombotic and inflammatory findings confers the highest risk of adverse neurologic outcome [Redline and O’Riordan, 2000].
A number of toxins have been described as causing cerebral palsy, such as benzyl alcohol preservative and in utero alcohol exposure [Benda et al., 1986; Burd et al., 2003]. Ingestion of methyl mercury by pregnant women in the Minamata Bay disaster resulted in the birth of children who were spastic, microcephalic, and cognitively impaired. Congenital infections, often referred to as TORCH infections (toxoplasmosis, rubella, cytomegalovirus, and herpes simplex infection), also can infect the fetus and produce serious encephalitides with motor sequelae [Sever, 1985].
Recent literature suggests that the risk of cerebral palsy may be modified by relatively common genetic polymorphisms. Inherited cytokine polymorphisms, including several found in the tumor necrosis factor-alpha and mannose-binding lectin genes, have been associated with increased risk of cerebral palsy in term infants [Gibson et al., 2006b, 2008]. Furthermore, a functional promotor region polymorphism in the interleukin (IL)-6 gene has been associated with cerebral palsy in term infants, as well as with the extent of brain injury and gray-matter volume in premature infants [Wu et al., 2009; Harding et al., 2004; Reiman et al., 2008; Djukic et al., 2009].
Postnatal Brain Injury
Bilirubin toxicity is a well-known cause of dyskinetic cerebral palsy, and continues to be a significant problem, in spite of recent progress in management of hyperbilirubinemic neonates [Shapiro, 2003]. Although bilirubin levels below 25 mg/dL rarely may be associated with kernicterus [Soorani-Lunsing et al., 2001], more commonly, bilirubin levels greater than 30 mg/dL are responsible for this problem. Symptoms of kernicterus in a jaundiced newborn usually are present by the second or third day of life. The child becomes listless and sucks poorly. Fever may be present, and crying becomes weak. The Moro and deep tendon reflexes become difficult to elicit, and general muscle tone becomes decreased. After several weeks, tone increases, and the infant manifests extension of the back with opisthotonos and marked extension of the extremities [Van Praagh, 1961].
The full clinical pattern of kernicterus infrequently is present in a single affected patient. Signs and symptoms consist of choreoathetosis, dystonia, tremors, and rigidity. Upward gaze and, on rare occasions, horizontal gaze may be impaired. Sensorineural hearing loss is common. Mental retardation, microcephaly, and spasticity may be present. A clinical score for assessing bilirubin-induced dysfunction in newborns has been recommended [Johnson et al., 1999]. Preterm infants with athetotic cerebral palsy have relatively uniform features, similar to term infants with kernicterus [Okumura et al., 2009]. Unconjugated bilirubin damages mitochondria and is toxic to neurons and to astrocytes [Ostrow et al., 2004]. Microscopic examination reveals yellow granules that are found not only in neurons, but also in the interstitial tissue. These changes occur in the first few days after injury. Neurons are grossly pyknotic, and in children who live for longer periods, large areas of cell loss, gliosis, and demyelination are evident.
The primary management problem for children affected with kernicterus is the lack of provision of the multiple services needed by affected infants and, more importantly, the failure to recognize early at-risk infants and manage severe hyperbilirubinemia in an efficient and timely fashion [Johnson et al., 2009; Maisels, 2009].
Clinical Features and Diagnosis
The clinical findings often change with maturation; both severity and distribution may be altered. A child with cerebral palsy who has been hypotonic may become hypertonic. A number of infants with mild abnormalities subsequently demonstrate a decrease or, in some instances, a disappearance of motor dysfunction [Nelson and Ellenberg, 1982]. The clinical picture should not include evidence of progressive disease or loss of skills previously acquired. Any suggestion of progression of disability should result in expansion of the differential diagnosis to explain the progression. The history should focus on an attempt to identify a specific cause and should especially investigate familial or metabolic disease that might have implications for treatment or family counseling [Barabas and Taft, 1986].
Further Diagnostic Evaluation
A useful algorithm for the evaluation of the child with cerebral palsy has been published (Figure 69-1) [Ashwal et al., 2004]. An abnormality is documented on cranial magnetic resonance imaging (MRI; in 89 percent of cases) or computed tomography (CT; in 77 percent) in a large percentage of patients; therefore, all children with cerebral palsy should undergo a neuroimaging study, preferably MRI [Accardo et al., 2004]. Because of the reported low incidence of metabolic and genetic disorders among children with cerebral palsy, testing for these disorders is indicated only in children in whom the history or clinical examination includes atypical features, or if a specific diagnosis is not established with neuroimaging. Children with a brain malformation also warrant consideration for further testing to determine if an underlying genetic or metabolic disorder is present. The presence of microcephaly at birth, dysmorphic features, and congenital anomalies outside the nervous system suggests early developmental defects. Microcephaly is one of the most common brain malformations and is caused by mutations of a number of different genes [Ashwal et al., 2009; Mochida, 2009]. The high incidence rates for mental retardation (52 percent), epilepsy (45 percent), ophthalmologic defects (28 percent), speech and language disorders (38 percent), and hearing impairment (12 percent) [Ashwal et al., 2004] make it imperative that all children with cerebral palsy be screened for mental retardation, ophthalmologic and hearing impairments, and speech and language disorders; nutrition, growth, and swallowing also should be closely monitored. Unfortunately, although neuronal plasticity likely aids improvement in brain dysfunction, significant injury persists [Johnston, 2009].
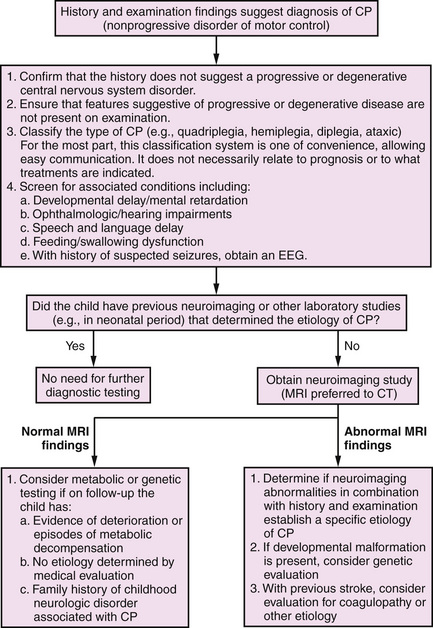
Fig. 69-1 Algorithm for the evaluation of the child with cerebral palsy (CP).
(From Ashwal S et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2004;62:851–863.)
Functional Classification
The validated Gross Motor Function Classification System (GMFCS) provides an easy and straightforward way to classify severity of motor impairment into one of the five levels [Palisano et al., 1997]. The GMFCS scale has been found to predict Gross Motor Function Measure (GMFM) scores [Palisano et al., 2000] and is simpler to use.
Some evidence suggests that family reporting may be utilized to enhance the use of the GMFCS [Morris et al., 2004]. In recent years, the GMFCS has been applied to develop five distinct motor development curves for use in prognostication [Rosenbaum et al., 2002]. Use of the GMFCS has indicated that there is no functional decline in levels I and II. However, in levels III, IV, and V, function peaked from 6 years 11 months to 7 years 11 months, and declined thereafter [Hanna et al., 2009]. Many other scales and inventories are in use, however [Shapiro, 2004]. These include the modified Ashworth scale, passive Range of Motion measurements, Selective Motor Control scale, and the Pediatric Evaluation of Disability Inventory [Ostensjo et al., 2004]. Adequacy of performance of everyday activities is best predicted by the child’s ability to perform gross motor tasks [Ostensjo et al., 2004].
Common Cerebral Palsy Syndromes
Spastic Hemiplegia
Although children may manifest obvious hemiplegia in the second year of life, specific difficulties may not be observed during the first 3–5 months of life. After a perinatal stroke, an infant may be neurologically normal until the development of pathologic handedness at approximately 4–6 months of age. For unexplained reasons, the left hemisphere (right side of the body) is affected in two-thirds of patients, and perinatal stroke is more common on the left than on the right [Crothers and Paine, 1959; Nelson and Lynch, 2004].
During the examination, the child exhibits impaired gross and fine motor coordination, has difficulty moving the hand quickly, and frequently is unable to grasp small items with a pincer grasp. The obligate palmar grasp reflex, which usually is absent by age 6 months and frequently rudimentary after age 4 months, may remain obligate. Weakness of the wrist and forearm often is associated with limitation of range of motion of supination. The range of elbow extension may be restricted. Attempts at reaching for objects may be accompanied by athetotic posturing with flexion of the wrist and hyperextension of the fingers (avoidance reaction). Facial involvement is unusual. Only 10 percent of affected patients, including those with extensive hemiplegia, have homonymous hemianopia [Black, 1988]. Children with hemiparesis may have a circumductive gait with a variable degree of abnormality. Most commonly, the child walks on the toes and swings the affected leg over a nearly semicircular arc during the course of each step. In contrast with the leg, the affected arm usually moves less than normal and does not participate in normal reciprocal motion during ambulation. An equinovarus positioning of the foot is seen; weakness and lack of full range of motion of dorsiflexion often are present. Further evidence of upper motor neuron involvement on the hemiplegic side includes hyperreflexia of the deep tendon reflexes, ankle clonus, and extensor toe signs.
Although frequently overlooked, corticosensory impairment and hemineglect of the affected side are common. Examination for the integrity of stereognosis and graphesthesia usually reveals varying degrees of compromise [Brown et al., 1987; Skatvedt, 1960].
Spastic Quadriplegia
Little [1861] first described cerebral palsy. He used the term spastic rigidity in place of the modern term spasticity. As part of his original treatise, he wrote: “Spastic cerebral palsy has the characteristics of upper motor unit involvement, such as hyperreflexia and increased tone, often with ankle clonus, crossed adductor reflexes, and extensor toe signs. Both lower extremities are more or less generally involved. Sometimes the affection of one limb only is observed by the parent, but examination usually shows a smaller degree of affection in the limb supposed to be sound. The contraction in the hips, knees, and ankles is often considerable. The flexors and adductors of thighs, the flexors of knees, and the gastrocnemii, preponderate. In most cases, after a time, owing to structural shortening of the muscles and of the articular ligaments, and perhaps to some changes of form of articular surface, the thighs cannot be completely abducted or extended, the knees cannot be straightened, nor can the heels be properly applied to the ground. The upper extremities are sometimes held down by preponderating action of pectorals, teres major and teres minor, and latissimus dorsi; the elbows are semiflexed, the wrists partially flexed, pronated, and the fingers incapable of perfect voluntary direction. Sometimes the upper extremities appear unaffected with spasm or want of volition, sometimes a mere awkwardness in using them exists.”
Ophthalmologic evaluation of children with spastic quadriplegia more commonly reveals visual impairment in these children than in children with athetoid cerebral palsy [Preakey et al., 1974]. The incidence of auditory, visual, motor, and learning disability is much higher in children with spastic quadriplegia than in children with spastic hemiplegia, spastic diplegia, and ataxic cerebral palsy [Robinson, 1973; Shevell et al., 2009].
Spastic Diplegia
Spastic diplegia is characterized by bilateral leg involvement and, commonly, some degree of upper extremity impairment. Preterm infants are particularly prone to spastic diplegia. Approximately 80 percent of preterm infants who manifest motor abnormalities have spastic diplegia [McDonald, 1963]. In recent years, the survival of very small preterm infants has resulted in a larger group of more severely neurologically impaired survivors [Hagberg et al., 1989a, b]. Diffusion tensor imaging in preterm infants has demonstrated disruption of thalamocortical connections, as well as descending corticospinal pathways [Hoon et al., 2009].
Extrapyramidal Cerebral Palsy
Extrapyramidal (dyskinetic) cerebral palsy can be divided arbitrarily into two primary clinical subtypes – choreoathetotic and dystonic. Patients are unable to perform meaningful movements smoothly because of interfering movements and involvement of inappropriate agonist and antagonist muscles (see Chapter 68). Extrapyramidal cerebral palsy involves defects of posture and involuntary movement (e.g., athetosis, ballismus, chorea, dystonia); increased tone usually is associated with these conditions and is of the “lead pipe” or rigid variety. Children with this form of cerebral palsy have more severe cognitive and motor impairments than children with bilateral spastic quadriplegia [Himmelmann et al., 2009].
Dystonic cerebral palsy
In one report, patients with severe athetoid cerebral palsy originating perinatally were divided into two groups neuropathologically: the “globo-Luysian group” and the “thalamoputaminal group.” The major abnormal sites in the globo-Luysian group were the pallidum and subthalamic nucleus, and in the thalamoputaminal group, the thalamus and putamen. The causative pathologic condition in the globo-Luysian group was primarily severe perinatal jaundice, and the cause in the thalamoputaminal group was predominantly neonatal asphyxia. The patients in the thalamoputaminal group demonstrated lower mental ability and suffered from more intractable convulsions than those in the globo-Luysian group. In the globo-Luysian group, rigidity and spasticity were frequently demonstrated, with fluctuation of athetoid movements, whereas in the thalamoputaminal group, various abnormalities of muscle tone and rather restricted athetosis were observed [Hayashi et al., 1991].
MRI studies in 22 children with athetotic cerebral palsy frequently revealed high-intensity areas in the thalamus and putamen in T2-weighted images. Of 16 children with known perinatal asphyxia, 14 had lesions in the basal ganglia, thalamus, and/or cerebral white matter. MRI findings were normal in 7 of the 22 children [Yokochi et al., 1991].
Hypotonic (Atonic) Cerebral Palsy
Diagnosis is difficult because of the plethora of diagnostic possibilities. Most children with generalized hypotonia have so-called central hypotonia (see Chapter 5), resulting from inadequate control of the motor pathways and subsequent disruption of gamma loop function. Others, with absent or hypoactive deep tendon reflexes, may have involvement of the lower motor neuron unit (i.e., anterior horn cell, peripheral nerve, neuromuscular junction, muscle). Extrapyramidal (choreoathetotic and dystonic) cerebral palsy may be preceded by a hypotonic phase.
Although, in the past, it has been thought that muscle tone almost always increases with maturation in this form of cerebral palsy [Ingram, 1964], experience has taught that in a sizable number of cases, spasticity does not develop, but the child remains hypotonic.
Ataxic Cerebral Palsy
The least common form of cerebral palsy is the ataxic form. It sometimes coexists with spastic diplegia [Hagberg et al., 1975]. This form usually is associated with other motor abnormalities; however, the diagnosis is applied only when the predominant manifestation is cerebellar dysfunction. Patients with ataxic cerebral palsy may have impairment of intellectual ability, but they are rarely grossly delayed [Clement et al., 1984]. Motor difficulties often are not apparent until late in the first year of life. Early manifestations include hypotonia, truncal ataxia with sitting, dysmetria, and gross incoordination. The motor involvement results in delayed attainment of motor skills; independent walking may not occur until age 3 or 4 years, and then may be performed only with great difficulty and frequent falling. Compromise of writing skills and other skills that demand good fine motor coordination often adversely affects educational endeavors.
Because of the large number of conditions associated with ataxia, the clinician must exclude conditions in which ataxia predominates in early childhood (see Chapter 67). Ataxia, especially if accompanied by mental retardation, may not be properly included among cerebral palsy conditions but may be the result of one of many inherited conditions [Hagberg et al., 1984].
The pathologic features of ataxic cerebral palsy are poorly defined and inconstant. Discussion of these features is confounded by the fact that total absence of the vermis may not give rise to cerebellar symptoms in certain congenital conditions, whereas aplasia of the vermis may be associated with nonprogressive ataxia [Bordarier and Aicardi, 1990]. Cerebellar hemispheric lesions may or may not be present in patients with ataxic cerebral palsy. The lack of correlation of evident structural changes with functional impairment is emphasized by CT studies. In one report, CT evaluation of patients with ataxic cerebral palsy revealed that the posterior fossa was normal in 38 percent and abnormal in 28 percent. By contrast, the cerebral hemispheres were abnormal in 55 percent of the patients [Miller and Cala, 1989].
General Prognosis for Motor Function
A number of factors affect the prognosis of the child with cerebral palsy: the clinical type of cerebral palsy; the degree of delay in meeting milestones noted at evaluation; the pathologic reflexes present; and the degree of associated deficits in intelligence, sensation, and emotional adjustment [Sala and Grant, 1995]. Cognitive level is difficult to assess in the young child with motor impairments but can be gauged even in the severely affected child [McCarty et al., 1986]. It is necessary to consider the cognitive level, despite the challenges posed in assessment, because the level of mental function may be the factor that really determines the quality of life the child will enjoy.
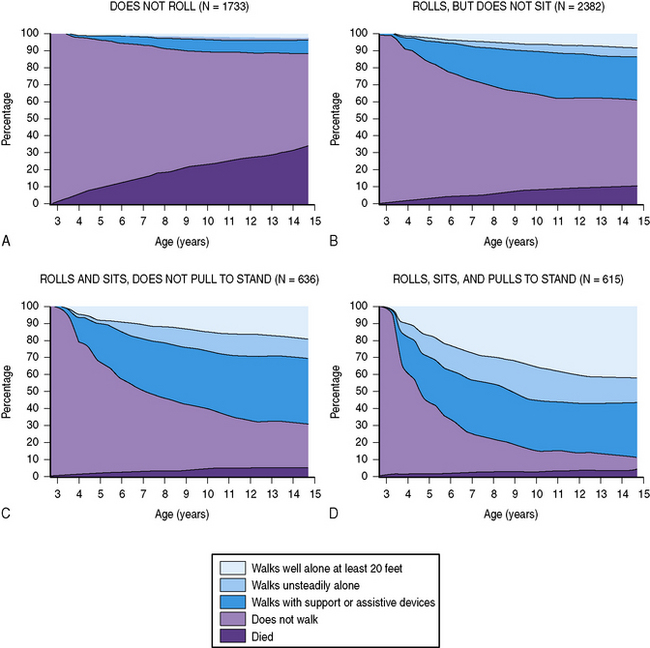
The prognosis for walking has been evaluated recently in large populations of children with cerebral palsy. The validated GMFCS can help predict whether the child will eventually achieve the ability to walk 10 steps unsupported [Rosenbaum et al., 2002]. Furthermore, a child’s motor function at age 2 years can be used to evaluate the prognosis for future ambulation at three levels of competency, with the help of ambulation charts reflecting the combined experience with more than 5000 children with cerebral palsy (Figure 69-2) [Wu et al., 2004a].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Accardo J., Kammann H., Hoon A.H.Jr. Neuroimaging in cerebral palsy. J Pediatr. 2004;145(Suppl 2):S19.
Ancel P.Y., Livinec F., Larroque B., et al. Cerebral palsy among very preterm children in relation to gestational age and neonatal ultrasound abnormalities: the EPIPAGE cohort study. Pediatrics. 2006;117:828.
Ashwal S., Michelson D., Plawner L., et alQuality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2009;73(11):887-897.
Ashwal S., Russman B.S., Blasco P.A., et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62:851.
Badawi N., Kurinczuk J.J., Keogh J.M., et al. Intrapartum risk factors for newborn encephalopathy: The Western Australian case-control study. BMJ. 1998;317:1554.
Badawi N., Kurinczuk J.J., Keogh J.M., et al. Antepartum risk factors for newborn encephalopathy: The Western Australian case-control study. BMJ. 1998;317:1549.
Badawi N., Watson L., Petterson B., et al. What constitutes cerebral palsy? Dev Med Child Neurol. 1998;40:520.
Barabas G., Taft L.T. The early signs and differential diagnosis of cerebral palsy. Pediatr Ann. 1986;15:203.
Bax M., Goldstein M., Rosenbaum P., et al. Proposed definition and classification of cerebral palsy. Dev Med Child Neurol. 2005;47:571.
Bax M., Tydeman C., Flodmark O. Clinical and MRI correlates of cerebral palsy: the European Cerebral Palsy Study. JAMA. 2006;296:1602.
Benda G.I., Hiller J.L., Reynolds J.W. Benzyl alcohol toxicity: Impact on neurologic handicaps among surviving very low birthweight infants. Pediatrics. 1986;77:507.
Black P.D. Ocular defects in children with cerebral palsy. BMJ. 1988;281:487.
Blair E. A research definition for “birth asphyxia”? Dev Med Child Neurol. 1993;35:449.
Blair E., Watson L., Badawi N., et al. Life expectancy among people with cerebral palsy in Western Australia. Dev Med Child Neurol. 2001;43(8):508-515.
Blair E. Trends in cerebral palsy. Indian J Pediatr. 2001;68:433.
Bordarier C., Aicardi J. Dandy-Walker syndrome and agenesis of the cerebellar vermis: Diagnostic problems and genetic counseling. A review. Dev Med Child Neurol. 1990;32:285.
Brown J.K., Van Rensburg F., Walsh G., et al. A neurological study of hand function of hemiplegic children. Dev Med Child Neurol. 1987;29:287.
Burd L., Cotsonas-Hassler T.M., Martsolf J.T., et al. Recognition and managment of fetal alcohol syndrome. Neurotoxicol Teratol. 2003;25:681-688.
Cans C., Surman G., McManus V., et al. Cerebral palsy registries. Semin Pediatr Neurol. 2004;11:18-22.
Clark S.L., Hankins G.D. Temporal and demographic trends in cerebral palsy—fact and fiction. Am J Obstet Gynecol. 2003;188:628.
Clement M.C., Briard J.L., Ponsot G., et al. Ataxies cerebelleuses congenitales nonprogressives. Arch Fr Pediatr. 1984;41:685.
Colver A.F., Gibson M., Hey E.N., et al. Increasing rates of cerebral palsy across the severity spectrum in north-east England 1964–1993. The North of England Collaborative Cerebral Palsy Survey. Arch Dis Child Fetal Neonatal Ed. 2000;83:F7.
Cowan F., Rutherford M., Groenendaal F., et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736.
Croen L.A., Grether J.K., Curry C.J., et al. Congenital abnormalities among children with cerebral palsy: More evidence for prenatal antecedents. J Pediatr. 2001;138:804.
Crothers B., Paine R.S. The natural history of cerebral palsy. Cambridge: Harvard University Press; 1959.
Dammann O., Kuban K.C., Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev. 2002;8:46.
Dammann O., Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1.
deVeber G., Andrew M. Cerebral sinovenous thrombosis in children. Canadian Pediatric Ischemic Stroke Study Group. N Engl J Med. 2001;345:417.
Djukic M., Gibson C.S., Maclennan A.H., et al. Genetic susceptibility to viral exposure may increase the risk of cerebral palsy. Aust N Z J Obstet Gynaecol. 2009;49:247.
Dolk H., Pattenden S., Johnson A. Cerebral palsy, low birthweight and socio-economic deprivation: inequalities in a major cause of childhood disability. Paediatr Perinat Epidemiol. 2001;15:359.
Ellenberg J.H., Nelson K.B. Cluster of perinatal events identifying infants at high risk for death or disability. J Pediatr. 1988;113:546.
Ericson A., Nygren K.G., Olausson P.O., et al. Hospital care utilization of infants born after IVF. Hum Reprod. 2002;17:929.
Evans P.M., Evans S.J.W., Alberman E. Cerebral palsy: Why we must plan for survival. Arch Dis Child. 1990;65:1329.
Eyman R.K., Grossman H.J., Chaney R.H., et al. The life expectancy of profoundly handicapped people with mental retardation. N Engl J Med. 1990;323:584.
Gibson C.S., Maclennan A.H., Dekker G.A., et al. Candidate genes and cerebral palsy: a population-based study. Pediatrics. 2008;122:1079.
Gibson C.S., MacLennan A.H., Goldwater P.N., et al. Neurotropic viruses and cerebral palsy: population based case-control study. BMJ. 2006;332:76.
Gibson C.S., MacLennan A.H., Goldwater P.N., et al. The association between inherited cytokine polymorphisms and cerebral palsy. Am J Obstet Gynecol. 2006;194:674.
Gibson C.S., MacLennan A.H., Hague W.M., et al. Associations between inherited thrombophilias, gestational age, and cerebral palsy. Am J Obstet Gynecol. 2005;193:1437.
Golomb M.R., MacGregor D.L., Domi T., et al. Presumed pre- or perinatal arterial ischemic stroke: Risk factors and outcomes. Ann Neurol. 2001;50:163.
Grether J.K., Cummins S.K., Nelson K.B. The California Cerebral Palsy Project. Paediatr Perinat Epidemiol. 1992;6:339.
Grether J.K., Nelson K.B.. Maternal infection and cerebral palsy in infants of normal birth weight. [published erratum appears in JAMA 1998;279:118] JAMA. 1997;278:207.
Grether J.K., Nelson K.B., Cummins S.K. Twinning and cerebral palsy: Experience in four northern California counties, births 1983 through 1985. Pediatrics. 1993;92:854.
Gunther G., Junker R., Strater R., et al. Symptomatic ischemic stroke in full-term neonates: Role of acquired and genetic prothrombotic risk factors. Stroke. 2000;31:2437.
Hagberg B., Hagberg G., Beckung E., et al. Changing panorama of cerebral palsy in Sweden. VIII. Prevalence and origin in the birth year period 1991–94. Acta Paediatr. 2001;90:271.
Hagberg B., Hagberg G., Olow I. The changing panorama of cerebral palsy in Sweden 1954–1970. II. Analysis of the various syndromes. Acta Paediatr Scand. 1975;64:193.
Hagberg B., Hagberg G., Olow I. The changing panorama of cerebral palsy in Sweden: IV. Epidemiological trends 1959–78. Acta Paediatr Scand. 1984;73:433.
Hagberg B., Hagberg G., Olow I., et al. The changing panorama of cerebral palsy in Sweden: V. The birth year period 1979–52. Acta Paediatr Scand. 1989;78:283.
Hagberg B., Hagberg G., Zetterstrom R. Decreasing perinatal mortality – increase in cerebral palsy morbidity? Acta Paediatr Scand. 1989;78:664.
Hagstrom J.N., Walter J., Bluebond-Langner R., et al. Prevalence of the factor V Leiden mutation in children and neonates with thromboembolic disease. J Pediatr. 1998;133:777.
Hanna S.E., Rosenbaum P.L., Bartlett D.J., et al. Stability and decline in gross motor function among children and youth with cerebral palsy aged 2 to 21 years. Dev Med Child Neurol. 2009;51:295.
Harding D.R., Dhamrait S., Whitelaw A., et al. Does interleukin-6 genotype influence cerebral injury or developmental progress after preterm birth? Pediatrics. 2004;114:941.
Hauser S.E., Peters H. Glutaric aciduria type 1: An underdiagnosed cause of encephalopathy and dystonia-dyskinesia syndrome in children. J Paediatr Child Health. 1998;34:302.
Hayashi M., Satoh J., Sakamoto K., et al. Clinical and neuropathological findings in severe athetoid cerebral palsy: A comparative study of globo-Luysian and thalamo-putaminal groups. Brain Dev. 1991;13:47.
Himmelmann K., McManus V., Hagberg G., et al. Dyskinetic cerebral palsy in Europe: trends in prevalence and severity. Arch Dis Child. 2009;94:921.
Hoon A.H.Jr, Stashinko E.E., Nagae L.M., et al. Sensory and motor deficits in children with cerebral palsy born preterm correlate with diffusion tensor imaging abnormalities in thalamocortical pathways. Dev Med Child Neurol. 2009;51:697.
Humphreys P., Whiting S., Pham B. Hemiparetic cerebral palsy: Clinical pattern and imaging in prediction of outcome. Can J Neurol Sci. 2000;27:210.
Hutton J.L., Cooke T., Pharoah P.O.D. Life expectancy in children with cerebral palsy. BMJ. 1994;309:431.
Ingram T.T.S. Paediatric aspects of cerebral palsy. Edinburgh: Churchill-Livingstone; 1964.
Jarvis S., Glinianaia S.V., Torrioli M.G., et al. Cerebral palsy and intrauterine growth in single births: European collaborative study. Lancet. 2003;362:1106.
Johnson L., Brown A.K., Bhutania V.K. BIND – a clinical score for bilirubin induced neurologic dysfunction in newborns. Pediatrics. 1999;104:746.
Johnson L., Bhutani V.K., Karp K., et al. Clinical report from the pilot USA Kernicterus Registry (1992 to 2004). J Perinatol. 2009;29(Suppl 1):S25.
Johnston M.V. Plasticity in the developing brain: implications for rehabilitation. Dev Disabil Res Rev. 2009;15:94.
Johnston M.V., Hagberg H. Sex and the pathogenesis of cerebral palsy. Dev Med Child Neurol. 2007;49:74.
Leviton A., Nelson K.B. Problems with definitions and classifications of newborn encephalopathy. Pediatr Neurol. 1992;8:85.
Leviton A., Paneth N., Reuss M.L., et al. Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Developmental Epidemiology Network Investigators. Pediatr Res. 1999;46:566.
Lieberman E., Lang J., Richardson D.K., et al. Intrapartum maternal fever and neonatal outcome. Pediatrics. 2000;105:8.
Little W.J. On the influence of abnormal parturition, difficult labours, premature birth, and asphyxia neonatorum on the mental and physical condition of the child, especially in relation to deformities. Trans Obstet Soc London. 1861;3:293.
Lynch J.K., Nelson K.B. Epidemiology of perinatal stroke. Curr Opin Pediatr. 2001;13:499.
MacLennan A. A template for defining a causal relation between acute intrapartum events and cerebral palsy: International consensus statement. BMJ. 1999;319:1054.
Maisels M.J. Neonatal hyperbilirubinemia and kernicterus – not gone but sometimes forgotten. Early Hum Dev. 2009;85:72732.
McCarty S.M., St. James P., Berninger V.W., et al. Assessment of intellectual functioning span in severe cerebral palsy. Dev Med Child Neurol. 1986;28:364.
McDonald A.D. Cerebral palsy in children of very low birth weight. Arch Dis Child. 1963;38:579.
Mercuri E., Rutherford M., Cowan F., et al. Early prognostic indicators of outcome in infants with neonatal cerebral infarction: A clinical, electroencephalogram, and magnetic resonance imaging study. Pediatrics. 1999;103:39.
Miller G., Cala L.A. Ataxic cerebral palsy—clinico-radiologic correlations. Neuropediatrics. 1989;20:84.
Miller S.P., Wu Y.W., Lee J., et al. Candidate gene polymorphisms do not differ between newborns with stroke and normal controls. Stroke. 2006;37:2678.
Moster D., Lie R.T., Irgens L.M., et al. The association of Apgar score with subsequent death and cerebral palsy: A population-based study in term infants. J Pediatr. 2001;138:798.
Moster D., Lie R.T., Markestad T. Long-term medical and social consequences of preterm birth. N Engl J Med. 2008;359:262.
Murphy C.C., Yeargin-Allsopp M., Decoufle P., et al. Prevalence of cerebral palsy among ten-year-old children in metropolitan Atlanta, 1985 through 1987. J Pediatr. 1993;123:S13.
Nelson K.B., Dambrosia J.M., Grether J.K., et al. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998;44:665.
Nelson K.B., Ellenberg J.H. Children who “outgrew” cerebral palsy. Pediatrics. 1982;69:529.
Nelson K.B., Grether J.K. Potentially asphyxiating conditions and spastic cerebral palsy in infants of normal birth weight. Am J Obstet Gynecol. 1998;179:507.
Nelson K.B., Lynch J.K. Stroke in newborn infants. Lancet Neurol. 2004;3:150-158.
Okumura A., Kidokoro H., Shoji H., et al. Kernicterus in preterm infants. Pediatrics. 2009;123:e1052.
O’Shea T.M., Preisser J.S., Klinepeter K.L., et al. Trends in mortality and cerebral palsy in a geographically based cohort of very low birth weight neonates born between 1982 to 1994. Pediatrics. 1998;101:642.
Ostensjo S., Carlberg E.B., Vollestad N.K. Motor impairments in young children with cerebral palsy: Relationship to gross motor function and everyday activities. Dev Med Child Neurol. 2004;46:580.
Ostrow J.D., Pascolo L., Brites D., et al. Molecular basis of bilirubin-induced neurotoxicity. Trends Mol Med. 2004;10:65-70.
Palisano R., Rosenbaum P., Walter S., et al. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol. 1997;39:214-223.
Palisano R.J., Hanna S.E., Rosenbaum P.L., et al. Validation of a model of gross motor function for children with cerebral palsy. Phys Ther. 2000;80:974-985.
Petterson B., Nelson K.B., Watson L., et al. Twins, triplets, and cerebral palsy in births in Western Australia in the 1980s. BMJ. 1993;307:1239.
Pharoah P.O. Twins and cerebral palsy. Acta Paediatr Suppl. 2001;90:6.
Pharoah P.O., Cooke T., Cooke R.W., et al. Birthweight specific trends in cerebral palsy. Arch Dis Child. 1990;65:602.
Platt M.J., Cans C., Johnson A., et al. Trends in cerebral palsy among infants of very low birthweight (<1500 g) or born prematurely (<32 weeks) in 16 European centres: a database study. Lancet. 2007;369:43.
Plioplys A.V. Survival rates of children with severe neurologic disabilities. A review. Semin Pediatr Neurol. 2003;10:120.
Plioplys A.V., Kasnicka I., Lewis S., et al. Survival rates among children with severe neurologic disabilities. South Med J. 1998;91:161.
Prasad A.N., Breen J.C., Ampola M.G., et al. Argininemia: A treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: Case reports and literature review. J Child Neurol. 1997;12:301.
Preakey A., Wilson J., Wilson B. Sensory and perceptual function in the cerebral palsied; perceptual relationships. J Nerv Ment Dis. 1974;158:70.
Rankin J., Cans C., Garne E., et al. Congenital anomalies in children with cerebral palsy: a population-based record linkage study. Dev Med Child Neurol. 2009. Epub ahead of print
Ravn S.H., Flachs E.M., Uldall P. Cerebral palsy in eastern Denmark: Declining birth prevalence but increasing numbers of unilateral cerebral palsy in birth year period 1986–1998. Eur J Paediatr Neurol. 2009. Epub ahead of print
Redline R.W., O’Riordan M.A. Placental lesions associated with cerebral palsy and neurologic impairment following term birth. Arch Pathol Lab Med. 2000;124:1785.
Reiman M., Kujari H., Ekholm E., et al. Interleukin-6 polymorphism is associated with chorioamnionitis and neonatal infections in preterm infants. J Pediatr. 2008;153:19.
Robinson R. The frequency of other handicaps in children with cerebral palsy. Dev Med Child Neurol. 1973;15:305.
Rosenbaum P.L., Walter S.D., Hanna S.E., et al. Prognosis for gross motor function in cerebral palsy: Creation of motor development curves. JAMA. 2002;288:1357.
Sala D.A., Grant A.D. Prognosis for ambulation in cerebral palsy. Dev Med Child Neurol. 1995;37:1020.
Sever J.L. TORCH tests and what they mean. Am J Obstet Gynecol. 1985;152:495.
Shalak L., Johnson-Welch S., Perlman J.M. Chorioamnionitis and neonatal encephalopathy in term infants with fetal acidemia: Histopathologic correlations. Pediatr Neurol. 2005;33:162.
Shapiro B.K. Cerebral palsy: A reconceptualization of the spectrum. J Pediatr. 2004;145(2 Suppl):S3.
Shapiro S.M. Bilirubin toxicity in the developing nervous system. Pediatr Neurol. 2003;29:410.
Shevell M.I., Dagenais L., Hall N. Comorbidities in cerebral palsy and their relationship to neurologic subtype and GMFCS level. Neurology. 2009;72:2090.
Skatvedt M. Sensory, perceptual and other non-motor defects in cerebral palsy. Little Club Clin Dev Med. 1960;1:115.
Soorani-Lunsing I., Woltil H.A., Hadders-Algra M. Are moderate degrees of hyperbilirubinemia in healthy term neonates really safe for the brain? Pediatr Res. 2001;50:701.
Strauss D.J., Shavelle R.M., Anderson T.W. Life expectancy of children with cerebral palsy. Pediatr Neurol. 1998;18:143.
Stromberg B., Dahlquist G., Ericson A., et al. Neurological sequelae in children born after in-vitro fertilisation: A population-based study. Lancet. 2002;359:461.
Sundrum R., Logan S., Wallace A., et al. Cerebral palsy and socioeconomic status: a retrospective cohort study. Arch Dis Child. 2005;90:15.
Szymonowicz W., Preston H., Yu V.Y. The surviving monozygotic twin. Arch Dis Child. 1986;61:454.
Takanashi J., Barkovich A.J., Ferriero D.M., et al. Widening spectrum of congenital hemiplegia: Periventricular venous infarction in term neonates. Neurology. 2003;61:531.
Thorngren-Jerneck K., Herbst A. Low 5-minute Apgar score: A population-based register study of 1 million term births. Obstet Gynecol. 2001;98:65.
Topp M., Langhoff-Roos J., Uldall P., et al. Intrauterine growth and gestational age in preterm infants with cerebral palsy. Early Hum Dev. 1996;44:27.
Topp M., Uldall P., Greisen G. Cerebral palsy births in eastern Denmark, 1987–90: Implications for neonatal care. Paediatr Perinat Epidemiol. 2001;15:271.
Uvebrant P. Hemiplegic cerebral palsy. Aetiology and outcome. Acta Paediatr Scand Suppl. 1988;345(Suppl):1.
Uvebrant P., Hagberg G. Intrauterine growth in children with cerebral palsy. Acta Paediatr. 1992;81:407.
Van Praagh R. Diagnosis of kernicterus in the neonatal period. Pediatrics. 1961;28:870.
Vincer M.J., Allen A.C., Joseph K.S., et al. Increasing prevalence of cerebral palsy among very preterm infants: a population-based study. Pediatrics. 2006;118:e1621.
Volpe J.J. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110.
Winter S., Autry A., Boyle C., et al. Trends in the prevalence of cerebral palsy in a population-based study. Pediatrics. 2002;110:1220.
Wu Y.W., Colford J.M. Chorioamnionitis as a risk factor for cerebral palsy: A meta-analysis. JAMA. 2000;284:1417.
Wu Y.W., Day S.M., Strauss D.J., et al. Prognosis for ambulation in cerebral palsy: A population-based study. Pediatrics. 2004;114:1264.
Wu Y.W., March W.M., Croen L.A., et al. Perinatal stroke in children with motor impairment: A population-based study. Pediatrics. 2004;114:612.
Wu Y.W., Escobar G.J., Grether J.K., et al. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA. 2003;290:2677.
Wu Y.W., Miller S.P., Chin K., et al. Multiple risk factors in neonatal sinovenous thrombosis. Neurology. 2002;59:438.
Wu Y.W., Croen L.A., Torres A.R., et al. Interleukin-6 genotype and risk of cerebral palsy in term and near-term infants. Ann Neurol. 2009;66(5):663-670.
Yeargin-Allsopp M., Van Naarden Braun K., Doernberg N.S., et al. Prevalence of cerebral palsy in 8-year-old children in three areas of the United States in 2002: a multisite collaboration. Pediatrics. 2008;121:547.
Yokochi K., Aiba K., Kodama M., et al. Magnetic resonance imaging in athetotic cerebral palsied children. Acta Paediatr Scand. 1991;80:818.
Bleck E.E. Orthopaedic management in cerebral palsy. In Clinics in Developmental Medicine. Philadelphia: Lippincott-Raven; 1987. No. 99
Bobath K. Neurophysiological basis for the treatment of cerebral palsy. In Clinics in Developmental Medicine. Philadelphia: Lippincott-Raven; 1991. No. 75
Broggi G., Angelini L., Bono R., et al. Long-term results of stereotactic thalmotomy for cerebral palsy. Neurosurgery. 1983;12:195.
Burke J.P., O’Keefe M., Bowell R. Optic nerve hypoplasia, encephalopathy, and neurodevelopmental handicap. Br J Ophthalmol. 1991;75:236.
Colditz P.B., Henderson-Smart D.J. Electronic fetal heart rate monitoring during labour: Does it prevent perinatal asphyxia and cerebral palsy? Med J Aust. 1990;153:88.
Cooper I.S., Riklan M., Amin I., et al. Chronic cerebellar stimulation in cerebral palsy. Neurology. 1976;26:744.
Crofts B.J., King R., Johnson A. The contribution of low birth weight to severe vision loss in a geographically defined population. Br J Ophthalmol. 1998;82:9.
Davidoff R.A. Antispasticity drugs: Mechanism of action. Ann Neurol. 1985;17:107.
Dennis J., Johnson M.A., Mutch L.M.M., et al. Acid-base status at birth in term infants and outcome at 4.5 years. Am J Obstet Gynecol. 1989;161:213.
Dinnage R. The child with cerebral palsy. In National Children’s Bureau Bibliographies. Albany, NY: Delmar; 1986. 2
Faust D., Ziskin J. The expert witness in psychology. Science. 1988;239:31.
Fenichel G.M., Lane D.A., Livengood J.R., et al. Adverse events following immunization: Assessing probability of causation. Pediatr Neurol. 1989;5:287.
Gahm N.H., Russman B.S., Cerciello R.L., et al. Chronic cerebellar stimulation for cerebral palsy: A double-blind study. Neurology. 1981;31:87.
Galjaard H., Prechtl H.F.R., Velickovic M. Early detection and management of cerebral palsy. In Topics in the Neurosciences. Norwell, Mass: Kluwer Academic Press; 1988.
Geralis E. Children with cerebral palsy: A parents’ guide. New York: Cambridge University Press; 1992.
Gorter J.W., Rosenbaum P.L., Hanna S.E., et al. Limb distribution, motor impairment, and functional classification of cerebral palsy. Dev Med Child Neurol. 2004;46:461.
Gross H., Jellinger K., Kaltenback E., et al. Infantile cerebral disorders: Clinical neuropathological correlations to elucidate the aetiological factors. J Neurol Sci. 1968;7:551.
Hoyme H.E., Higginbottom M.C., Jones K.L. Vascular etiology of disruptive structure defects in monozygotic twins. Pediatrics. 1981;67:288.
Ingram T. A study of cerebral palsy in the childhood population of Edinburgh. Arch Dis Child. 1955;117:395.
Koeda T., Suganuma I., Kohno Y., et al. MR imaging of spastic diplegia: Comparative study between preterm and term infants. Neuroradiology. 1990;32:187.
Krageloh-Mann I., Hagberg B., Petersen D., et al. Bilateral spastic cerebral palsy – pathogenetic aspects from MRI. Neuropediatrics. 1992;23:46.
Landau W.M., Hunt C.C. Dorsal rhizotomy, a treatment of unproven efficacy. J Child Neurol. 1990;5:174.
Leviton A., Paneth N. White matter damage in preterm newborns – an epidemiologic perspective. Early Hum Dev. 1990;24:1.
Malamud N., Itabashi H.H., Castor J., et al. An etiologic and diagnostic study of cerebral palsy. J Pediatr. 1964;65:270.
Melone P.J., Ernest J.M., O’Shea M.D., et al. Appropriateness of intrapartum fetal heart rate management and risk of cerebral palsy. Am J Obstet Gynecol. 1991;165:272.
Mochida G.H. Genetics and biology of microcephaly and lissencephaly. Semin Pediatr Neurol. 2009;16:120.
Meyer B.A., Dickinson J.E., Chambers C., et al. The effect of fetal sepsis on umbilical cord blood gases. Am J Obstet Gynecol. 1992;166:612.
Miller F., Bachrach S.J. Cerebral palsy: A complete guide for caregiving. In: Johns Hopkins Press Health Book. Baltimore: Johns Hopkins University Press; 1998.
Miller G., Clark C.D. The cerebral palsies: Causes, consequences, and management. Boston: Butterworth-Heinemann; 1998.
Morris C., Bartlett D. Gross Motor Function Classification System: Impact and utility. Dev Med Child Neurol. 2004;46:60.
Morris C., Galuppi B.E., Rosenbaum P.L. Reliability of family report for the Gross Motor Function Classification System. Dev Med Child Neurol. 2004;46:455.
Narayan R.K., Loubser P.G., Jankovic J., et al. Intrathecal Baclofen for intractable axial dystonia. Neurology. 1991;41:1141.
Nelson K.B. Prenatal origin of hemiparetic cerebral palsy: How often and why? Pediatrics. 1991;88:1059.
Nelson K.B., Ellenberg J.H. Apgar scores as predictors of chronic neurologic disability. Pediatrics. 1981;68:36.
Nelson K.B., Ellenberg J.H. Cerebral palsy. 1. Univariate analysis of risks. Am J Dis Child. 1985;139:1031.
Nelson K.B., Ellenberg J.H. Predictors of low and very low birth weight and the relation of these to cerebral palsy. JAMA. 1985;254:1473.
Nelson K.B., Leviton A. How much of neonatal encephalopathy is due to birth asphyxia? Am J Dis Child. 1991;145:1325.
Paludetto R. Neonatal complications specific to twin (multiple) births (twins transfusion syndrome, intrauterine death of cotwin). J Perinat Med. 1991;19(Suppl 1):246.
Peacock W., Staudt L.A. Spasticity in cerebral palsy and the selective posterior rhizotomy procedure. J Child Neurol. 1990;5:179.
Perlman J.M., Tack E.D. Renal injury in the asphyxiated newborn infant: Relationship to neurologic outcome. J Pediatr. 1988;113:875.
Phelan J.P., Ock Ahn M., Korst L., et al. Intrapartum fetal asphyxial brain injury with absent multiorgan system dysfunction. J Matern Fetal Med. 1998;7:19.
Scrutton D. Management of the motor disorders of children with cerebral palsy. In Clinics in Developmental Medicine. New York: Cambridge University Press; 1991. No. 90
Seidman D.S., Paz I., Laor A., et al. Apgar scores and cognitive performance at 17 years of age. Obstet Gynecol. 1991;77:875.
Stanley F. Epidemiology of the cerebral palsies. In Clinics in Developmental Medicine. Philadelphia: Lippincott-Raven; 1991. No. 87
Sussman M.D. The use of casts as an adjunct to physical therapy management of cerebral palsy patients. In: Proceedings: Orthopedic Aspects of Developmental Disabilities. Chapel Hill: University of North Carolina School of Medicine; 1978:47.
Vogt C., Vogt O. Zur psychiatrischen Wurdingung der anatonschen Entdekung und Wertung des Status marmoratus striat. J Psychol Neurol. 1928;37:387.
Volpe J.J. Value of MR in definition of the neuropathology of cerebral palsy in vivo. Am J Neuroradiol. 1992;13:79.
Volpe J.J. Neurology of the newborn, ed 3. Philadelphia: WB Saunders; 1995.
Watt J., Sims D., Harchom F. A controlled study of inhibition casting as an adjunct to physiotherapy for cerebral-palsied children. Dev Med Child Neurol. 1986;28:480.
Wiley M.E., Damiano D.L. Lower-extremity strength profiles in spastic cerebral palsy. Dev Med Child Neurol. 1998;40:100.
Young R.R. Physiologic and pharmacologic approaches to spasticity. Neurol Clin. 1987;5:529.
