Chapter 17 Cell Biology of the Müller Cell
Introduction
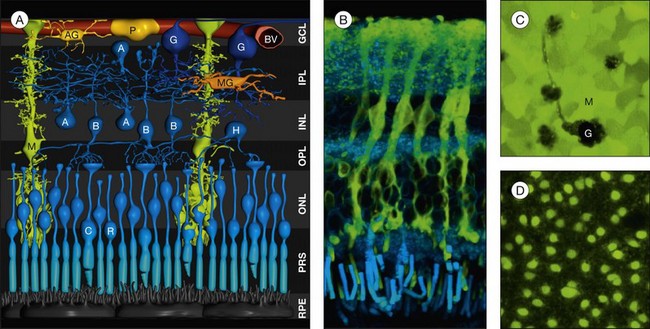
In addition to microglial cells, which are the resident immune cells of the retina, the retina contains two types of macroglial cell: astrocytes and Müller cells (Fig. 17.1A). Macroglial cells support the functioning and metabolism of retinal neurons. For this reason, they constitute an anatomical and functional link between these neurons and the compartments with which they need to exchange molecules (blood vessels, vitreous chamber, and subretinal space). Most nutrients, waste products, ions, water, and other molecules are transported through macroglial cells between the retinal vessels and neurons. As a peculiarity, astrocytes play a crucial role in retinal vascularization; the localization of these cells in the mature retina is restricted to the nerve fiber and ganglion cell layers.
Morphology of müller cells
Heinrich Müller (1820−1864) in 1851 described the “radial fibers” of the retina which later became known as Müller cells. Müller cells are specialized radial glial cells which span the entire thickness of the neural retina, from the subretinal space to the vitreal surface (Fig. 17.1A, B). The somata of the cells are located in the inner nuclear layer, and two stem (trunk) processes radiate from the soma in opposite directions. The outer stem process draws towards the subretinal space; here, microvilli of Müller cells surround the photoreceptor inner segments. Zonulae adherentes between Müller and photoreceptor cells form the outer limiting membrane of the retina. The inner stem process projects towards the vitread surface of the retina; the end of this process is expanded into a funnel-shaped endfoot. Both the basement membrane at the inner retinal surface and the Müller cell endfoot membranes together constitute the inner limiting membrane. Lateral processes extend into the plexiform layers where they form elaborate sheaths around the neuronal synapses, as well as into the nuclear layers where they embed neuronal perikarya. Astrocytes and Müller cells ensheath the vessels of the superficial vascular plexus; the deep capillaries are ensheathed by en passant endfeet of Müller cells. Within the macula, Müller cells display a Z-shaped morphology because the outer processes accompany the centrifugally running cone axons in the Henle fiber layer (Figs 17.2 and 17.3C). The central fovea contains 20–30 atypical Müller cells. These cells are not associated with cone axons; instead, their outer process runs straight towards the inner retinal surface where the soma is located and where their large, flat endfeet contribute to the inner limiting membrane of the fovea proper.1
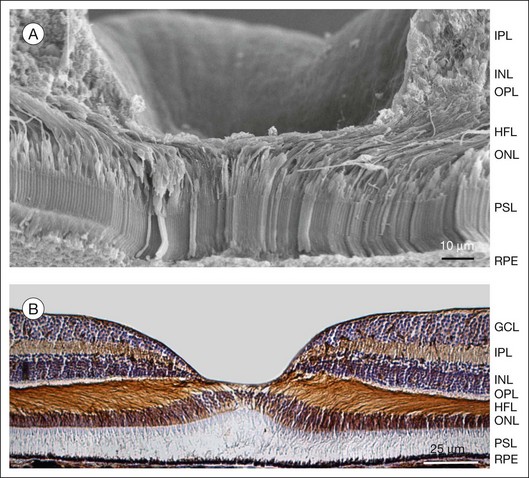
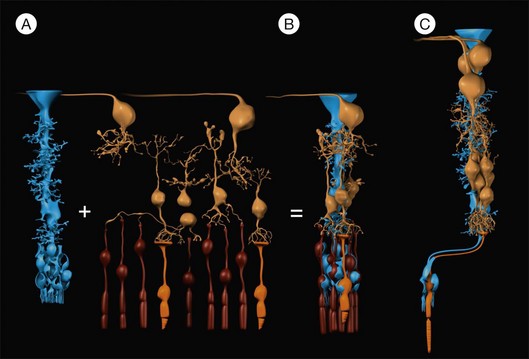
Fig. 17.3 Müller cells form the cores of radial units of retinal neurons. Every Müller cell is surrounded by a distinct group of retinal neurons (A) with which it interacts specifically during development, in the mature functioning retina, and after injury of the retina. These repetitive radial units are almost identical throughout most regions of the retina (B) but differ in the (peri-)fovea (C) by the lack of rods, an increased number of neurons of the inner nuclear and ganglion cell layers, and by an elongation and Z-shaped course of the outer processes of Müller cells (cf. also Fig. 17.2). Blue, Müller cells; yellow, retinal neurons; orange, cones; brown, rods.
Müller cells constitute the cores of functional retinal columns
The human retina contains 8–10 million regularly arranged Müller cells (Figs 17.1D and 17.4C). Each Müller cell constitutes the core of a column of retinal neurons which represents the smallest functional unit for “forward information processing.”2 Such a column contains one cone per Müller cell (Figs 17.3B, C and 17.4B, C) and up to 10 rods, as well as 6 (fovea) and 4 (periphery) inner nuclear layer neurons, and 2.5 (fovea) and 0.3 (periphery) ganglion cell layer neurons, respectively.1
Light guidance
Because the retina is inverted, light has to pass the entire depth of the neuroretina before it arrives at the photoreceptors. In the retina as in any other organ, all cells and their processes and organelles are phase objects which scatter the incoming light. In particular, the synapses in the plexiform layers have dimensions close to 500 nm, i.e., within the wavelength range of visible light; this makes them light-scattering structures.3 The backscattering of light from the retinal cell structures is clinically used by optical coherence tomography. However, light scattering should reduce visual sensitivity and acuity, particularly outside the fovea (where the inner layers are shifted aside, and the light directly hits the cones).
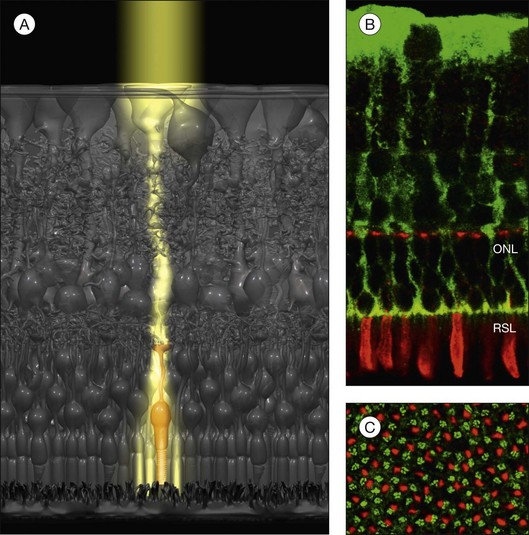
To reduce light scattering, nonfoveal Müller cells act as living optical fibers that guide the light through the inner retinal layers towards the photoreceptors (Fig. 17.4A).3 The funnel-shaped endfeet of Müller cells act as light collectors at the vitread surface of the retina. The refractory index of Müller cells is slightly lower than that of photoreceptor segments (which are also light-guiding fibers) but higher than that of the surrounding retinal tissue.3 On the other hand, the refractory index of Müller cell endfeet is rather low, about halfway between that of Müller cell stem processes and that of the vitreous.3 This allows a “soft coupling” of the light path between the vitreous and the retina, and reduces light reflection at the inner retinal surface. By light guidance, Müller cells transport an image (like a fiberoptic plate) with minimal loss of light intensity from the inner retinal surface to the photoreceptors; this image is resolved in pixels corresponding to individual Müller cells (Fig. 17.4A). Because the local densities of cones and Müller cells are roughly equal (Fig. 17.4C), every cone has its “private” Müller cell which delivers its part of the image, while several (up to 10) rods are illuminated by one Müller cell (Fig. 17.4A).
Recycling of cone photopigments
Müller cells support the function of photoreceptors by other mechanisms, as well. There exist two cycles in the retina that regenerate the photopigments, the rod and the cone visual cycle.4 In the photoreceptor outer segments, all-trans retinal is reduced to all-trans retinol. Rod-derived all-trans retinol is regenerated to 11-cis retinal in the pigment epithelium while cone-derived all-trans retinol is processed by Müller cells. Müller cells convert all-trans retinol to 11-cis retinol which is subsequently oxidized to 11-cis retinal by a retinol dehydrogenase, and released into the extracellular space for uptake by the cone photoreceptors. For the transport of the retinoids, Müller cells express cellular retinol-binding protein and cellular retinal-binding protein (CRALBP).5 Furthermore, Müller cells contribute to the assembly of the photoreceptor outer segments,6 in part by the release of lactose and pigment epithelium-derived factor (PEDF),7,8 and phagocytose outer-segment discs shed from cones.9 Docosahexaenoic acid is required for outer-segment renewal and for the preservation of mitochondria.10 Müller cells incorporate docosahexaenoic acid into phospholipids, and channel it to the photoreceptors.10
Regulation of the synaptic activity by neurotransmitter uptake
Precise shaping (i.e., control in time and space) of synaptic activity depends upon the kinetics of the presynaptic neurotransmitter release and of the (re-)uptake of the transmitter molecules into the cells. In the neuroretina, photoreceptor cells, neurons, and macroglial cells express high-affinity transporters for neurotransmitters. Müller cells possess uptake and exchange systems for various neurotransmitters, particularly for glutamate and γ-aminobutyric acid (GABA) (Fig. 17.5).
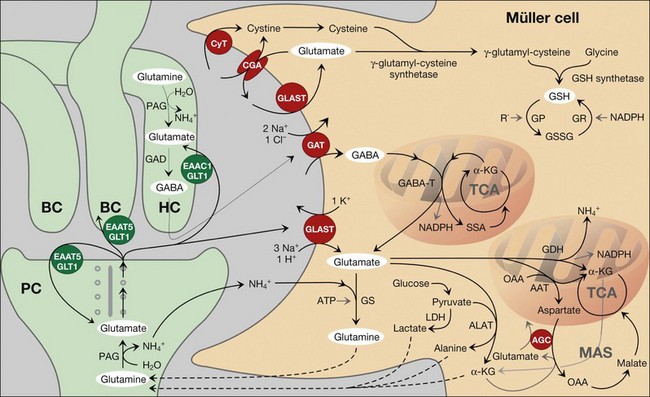
The major glutamate uptake carrier of Müller cells is the glutamate aspartate transporter (GLAST, or excitatory amino acid transporter-1, EAAT1).11–13 In addition, EAAT2 (GLT1) and EAAT3 were found in human Müller cells.14,15 EAATs mediate the cotransport of three sodium ions and one proton, and the countertransport of one potassium ion, with each glutamate anion.16 The transport of an excess of sodium into the cell generates an inward current (the transporter is “electrogenic”; Figs 17.6A and 17.7A, B) and cellular depolarization. The amplitude of the glutamate transporter currents is voltage-dependent (Fig. 17.6A); a very negative membrane potential is essential for an efficient uptake of glutamate.17 Sodium-dependent carriers allow uphill transport of substrates into the cells against a concentration gradient. The driving force for the transport is the electrochemical sodium gradient across the plasma membrane, generated by the Na+,K+-ATPase. Other transporters, e.g., for GABA, are also electrogenic (Figs 17.6B and 17.7F). The subcellular distribution of the GABA transporter currents (Fig. 17.6B) corresponds with the observation that GABA is taken up by amacrine and Müller cells in the inner retina but exclusively by Müller cells in the outer retina.18
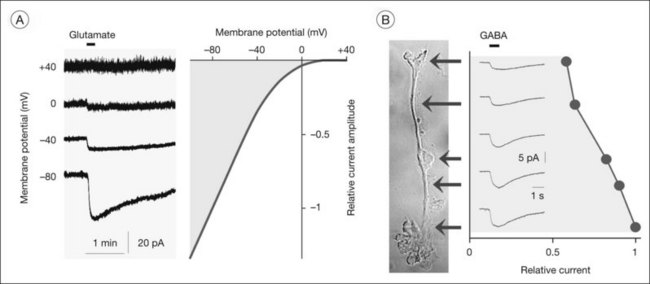
In the outer plexiform layer, the photoreceptor-derived glutamate is mainly removed by photoreceptor, horizontal, and bipolar cells.12,19 whereas Müller cells prevent the lateral spread of neurotransmitters beyond the synapse(s) where they are released, thus ensuring visual resolution (Fig. 17.5). In the inner plexiform layer, Müller cell-provided glutamate uptake takes a more active part in shaping the synaptic responses. The rapid termination of synaptic glutamate action in nonspiking inner retinal neurons and in retinal ganglion cells is predominantly mediated by the uptake of glutamate into Müller cells.20,21 Generally, Müller cells remove the bulk of extracellular glutamate in the inner retina.13,22,23
Malfunction of glial glutamate uptake contributes to glutamate toxicity
Glutamate toxicity is a major cause of neuronal loss in many retinal disorders, including glaucoma, ischemia, diabetes, and inherited photoreceptor degeneration. A malfunction and/or downregulation of GLAST may cause, or contribute to, a rise in extracellular glutamate towards excitotoxic levels.23–25 Hypoxia facilitates the formation of free radicals in mitochondria; the resulting lipid peroxidation disrupts glutamate transport into Müller cells.26 This mechanism is probably implicated in the neuronal dysfunction in diabetic retinopathy27 and in ganglion cell death in Leber hereditary optic neuropathy.28 Inflammatory lipids such as arachidonic acid, a reduction in extracellular pH as occurs in ischemia, and zinc ions released from photoreceptors29 all inhibit glial glutamate uptake.30,31
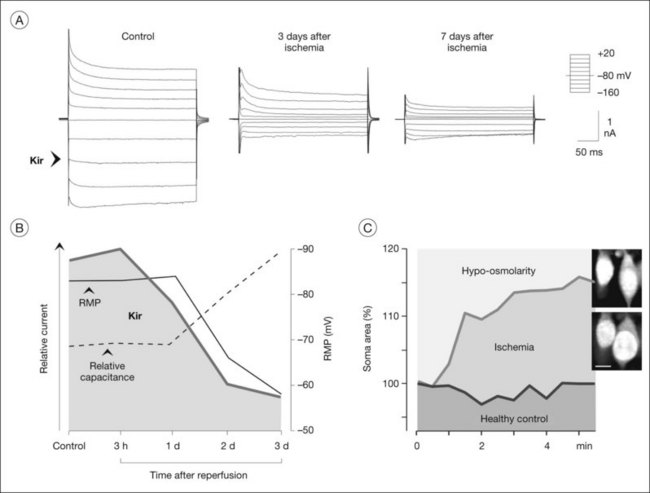
Because glutamate transport is voltage-dependent (Fig. 17.6A), depolarization decreases the efficiency of glial glutamate uptake.17,32 Depolarization of Müller cells occurs in response to pathological rises in extracellular potassium (as occurs in ischemia and glaucoma), to the opening of cation channels in Müller cells (Fig. 17.7C), and to a decrease in the potassium permeability of glial membranes due to inactivation and/or downregulation of inwardly rectifying potassium (Kir) channels (Fig. 17.8A, B). An inactivation of Kir channels was observed in animal models of various retinopathies (see below). Depolarization of Müller cells can also be evoked by inflammatory lipids such as arachidonic acid and prostaglandins which potently inhibit Na+,K+-ATPase. Depolarization of Müller cells may even cause a reversal of the glutamate transport.33 A nonvesicular release of glutamate and aspartate via reversed operation of glial glutamate transporters was implicated in the excitotoxic damage of retinal ganglion cells.34,35 A release of glial glutamate might also be mediated by the cystine glutamate antiporter (which is activated under oxidative stress conditions when an elevated production of glutathione requires an increased uptake of cystine; Fig. 17.5) and by exocytosis of glutamate-containing secretory vesicles.1,36 Under various pathological conditions including ischemia, glaucoma, retinal detachment, and proliferative retinopathies, an enhanced expression or activity of GLAST was observed.11,37 This might counterbalance the depolarization-induced malfunction of glial glutamate uptake.
Production of neurotransmitter precursors
To sustain the mechanism of transmitter recycling at the synapses, the Müller cells are endowed with specific enzymes. After GABA has been taken up by Müller cells, it is readily converted to glutamate by the GABA transaminase, and glutamate is converted to glutamine by the glutamine synthetase (Fig. 17.5). Glutamine synthetase is exclusively localized to glial cells.38 Glutamine is released from Müller cells, and taken up by neurons as a precursor for the resynthesis of glutamate and GABA (glutamate–glutamine cycle; Fig. 17.5).39 In the photoreceptor cells, this cycle accounts only for a part of glutamate supply; they directly take up glutamate from the synaptic cleft, and, in addition, synthesize glutamate from α-ketoglutarate as well as from Müller cell-derived glutamine.19,40 By contrast, the production of glutamate in bipolar and ganglion cells almost entirely depends on Müller cell-derived glutamine.40 Pharmacological blockade of the glutamine synthetase in Müller cells causes a rapid loss of the glutamate content of bipolar and ganglion cells, and (within 2 minutes) the animals become functionally blind.40 Likewise, a significant amount of GABA in amacrine cells is synthesized from Müller cell-derived glutamine.40
Downregulation of the glial glutamine synthetase contributes to neuronal dysfunction and glutamate toxicity in inherited photoreceptor degeneration and retinal detachment.41,42 The downregulation is mediated (at least in part) by the basic fibroblast growth factor (bFGF).43 bFGF is rapidly released within the retina after detachment, and its expression level increases under ischemic conditions, after light injury, and in response to inherited photoreceptor degeneration and mechanical injury.1 Though bFGF is a neurotrophic factor which supports the survival of photoreceptors and neurons,44–46 the bFGF-induced downregulation of glutamine synthetase might rather aggravate neuronal degeneration. Under ischemic conditions, the GABA transaminase activity is decreased, and GABA accumulates in Müller cells.47,48 This impairs the efficiency of glial GABA uptake due to the decrease in the extra- to intracellular gradient as a driving force.
Trophic support of photoreceptors and neurons
The uptake of glutamate and GABA by Müller cells links neuronal excitation with the release of metabolic substrates, as well as with the defense against oxidative stress. Müller cells produce various substrates of the oxidative neuronal metabolism such as glutamine, lactate, alanine, and α-ketoglutarate (Fig. 17.5)49; these substrates are used by photoreceptors and neurons in periods of metabolic stress in the dark. The metabolic support is regulated by neuron-derived glutamate (which stimulates the uptake of glucose and the production of lactate) and potassium (which induces hydrolyzation of glycogen in Müller cells).1,50
Antioxidative support of photoreceptors and neurons
Glutamate in Müller cells is also utilized for the production of the antioxidant, glutathione (Fig. 17.5).39 The retina has a high need of antioxidant protection; this results from light exposure together with high oxygen consumption and the presence of high polyunsaturated fatty acid levels in photoreceptors. In addition, the outer retina underlies circadian periods of hypoxia (dark) and hyperoxia (light)51 which both increase oxidative stress. Usually, retinal glutathione is confined to glial and horizontal cells.39,52 In Müller cells, glutathione constitutes about 2% of the total protein.1 In response to oxidative stress such as during ischemia, glutathione is rapidly released from Müller cells and provided to the neurons,52 where it acts as cofactor of the enzyme glutathione peroxidase. The glial release of antioxidants like glutathione, and of neuroprotective factors like bFGF and adenosine, is implicated in the protection of photoreceptors from the harmful effects of circadian light exposure.
Müller cells from aged animals contain reduced levels of glutathione; this is associated with mitochondrial damage, membrane depolarization, and reduced cell viability.53 An age-dependent decrease in retinal glutathione may accelerate the pathogenesis of retinopathies in the elderly. In diabetic retinopathy, the decreased glutamate uptake of Müller cells results in reduced glutathione synthesis54,55 and in an upregulation of glutaredoxin which catalyzes the deglutathionylation of proteins; the latter mechanism induces nuclear translocation of nuclear factor-κB and upregulation of proinflammatory factors.56 Other antioxidants provided by Müller cells include pyruvate, α-ketoglutarate, metallothionein, lysozyme, ceruloplasmin, heme oxygenase, and reduced ascorbate.1
Removal of carbon dioxide
Photoreceptor cells display the highest rate of oxidative metabolism of all cells in the body. This high metabolic activity is associated with high oxygen and glucose demands. The active glucose metabolism, in turn, generates much carbon dioxide and water. Via transcellular transport, Müller cells redistribute metabolic waste into the blood and vitreous. Carbon dioxide is cleared by a process called carbon dioxide siphoning57; it is rapidly hydrated to bicarbonate and protons by carbonic anhydrases.58 Carbonic anhydrase II is localized within Müller and amacrine cells, whereas the membrane-bound carbonic anhydrase XIV is present in glial cells and vascular endothelia.57,59,60 Bicarbonate is transported to the blood and vitreous by sodium bicarbonate cotransporters and anion exchangers, both enriched in glial endfoot membranes.57
Regulation of the extracellular pH
By the removal of carbon dioxide, Müller cells also regulate the extracellular pH. Light-evoked neuronal activity is associated with extracellular alkalinization.61 The pH shift is balanced by an efflux of protons from photoreceptors62 and by the consecutive action of carbonic anhydrases and glial sodium bicarbonate cotransporters.63 Because sodium bicarbonate transporters are electrogenic, depolarization of Müller cells by potassium released from active neurons causes an influx of bicarbonate into the cells.63 The carbonic anhydrase-generated protons remain outside the cells and cause an extracellular acidification.63 Because extracellular acidification inhibits synaptic transmission,64 Müller cell-mediated pH regulation may protect neurons against overexcitation.
Spatial potassium buffering
Neuronal activity is associated with rapid ion shifts between the intra- and extracellular spaces. Sodium, chloride, and calcium ions flow into active neurons, and potassium ions are released from neurons. Light onset causes increases in extracellular potassium in the plexiform layers, and a decrease in the subretinal space.65,66 If not corrected, increased potassium will cause neuronal depolarization and hyperexcitation. Müller cells buffer imbalances in the extracellular potassium concentration via permission of transcellular potassium currents, a process termed “spatial potassium buffering” or “potassium siphoning.”67,68 They take up excess potassium from the extracellular fluid, and release equal amounts of potassium into fluid-filled spaces outside the neuroretina where the potassium concentration is constant or decreased, i.e., into blood vessels, the vitreous, and the subretinal space (Fig. 17.9C).
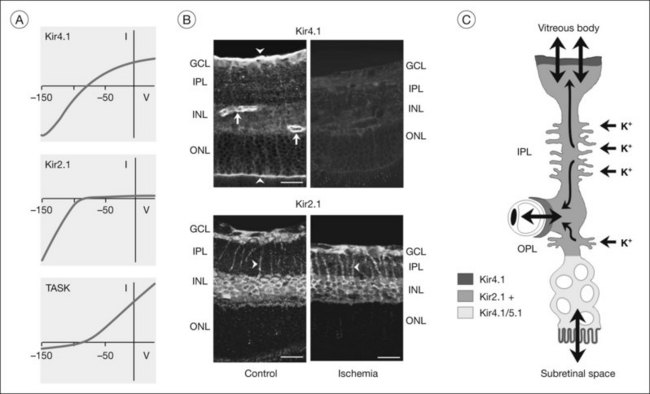
Because Müller cell membranes are highly permeable to potassium, Müller cells have a very negative resting membrane potential, close to the potassium equilibrium potential, around –80 mV.69,70 The potassium permeability is mainly mediated by special potassium channels belonging to the Kir subfamily of K+ channels.71 In particular, Kir4.1 channels (which mediate bidirectional potassium currents; Fig. 17.9A) and Kir2.1 channels (which mediate inward potassium currents) are implicated in potassium siphoning.72,73 The channel proteins are localized in a polarized fashion in the Müller cell membrane (Fig. 17.9B). Homomeric Kir4.1 channels are mainly localized in membrane domains across which the cells extrude potassium into spaces outside the neuroretina, i.e., in perivascular and vitread endfoot membranes, and in the microvilli that extend into the subretinal space.72,74 Membranes which abut neuronal cell structures contain Kir channels which mediate inward potassium currents, i.e., Kir2.1 (Fig. 17.9B) and heterotetrameric Kir4.1/Kir5.1 channels.73,75 Thus, the direction of the transcellular potassium currents is determined by the subcellular localization of different Kir channel subtypes. Local increases in extracellular potassium cause the driving force for passive currents through Kir channels. Müller cells also express other types of potassium channels which may contribute to potassium buffering, including voltage-gated potassium (KA, KDR), calcium-activated big-conductance potassium (BK) channels (Figs 17.7E and 17.10A), and tandem-pore domain potassium channels.76 In addition, the Na+,K+-ATPase and transporter molecules, e.g., the K/Na/2Cl cotransporter, contribute to Müller cell-mediated potassium homeostasis.1
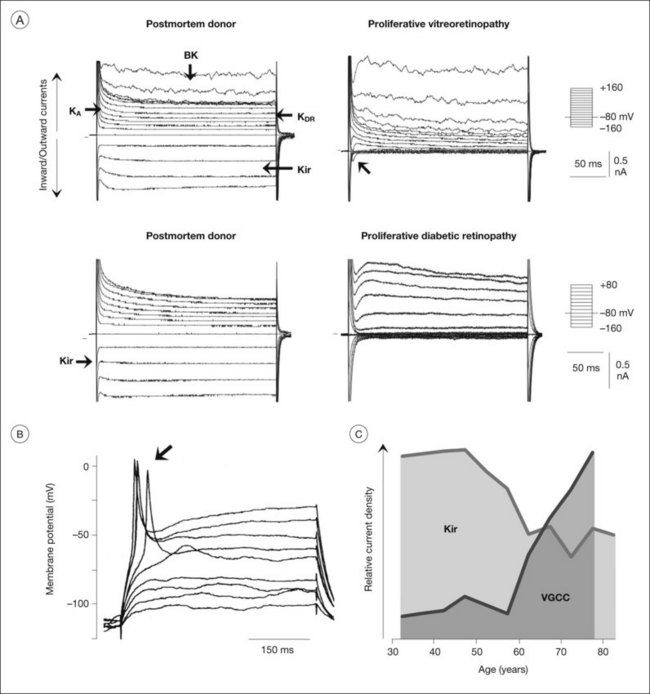
Many of the homeostatic functions of Müller cells, including potassium siphoning and neurotransmitter uptake, depend on the very negative membrane potential constituted by the Kir4.1 channels.72 Kir4.1 channels are also involved in cell volume regulation which compensates for changes in the osmotic conditions.1,77 Under various pathological conditions, Kir4.1 channels of Müller cells are downregulated and/or dislocated (Fig. 17.9B); this is associated with functional inactivation and results in a decrease of the potassium currents through Müller cells (Fig. 17.8A, B). Such alterations have been observed in animal models of various ischemic-hypoxic and inflammatory diseases, including ischemia-reperfusion, ocular inflammation, diabetic retinopathy, blue-light injury, retinal detachment, retinal vein occlusion, and proliferative vitreoretinopathy, as well as in Müller cells from patients with proliferative retinopathies (Fig. 17.10A).1,77 The impaired transcellular potassium transport and the depolarization of the cells (which impairs the glutamate uptake) contribute to neuronal hyperexcitation and glutamate toxicity.32 Human Müller cells display an age-dependent decrease in Kir currents, on average by 50% between the ages of 40 and 80 years (Fig. 17.10C). This may enhance the susceptibility for age-dependent retinopathies because dysfunctional Müller cells cannot efficiently buffer pathological complications such as neuronal hyperexcitation due to hypoxia caused by diabetic alterations of the vessels.
Water clearance
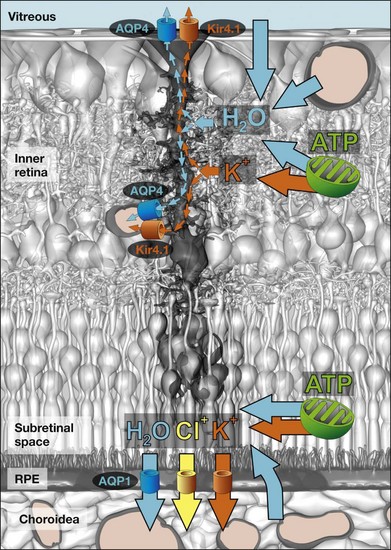
Retinal water clearance is mediated by an osmotically driven transcellular water transport coupled to transport of osmolytes, and in particular to potassium transport.1,74 The Müller cell-mediated clearance of the retinal tissue from excess potassium will simultaneously redistribute metabolic water out of the neuroretina. The transmembrane water transport is facilitated by water-selective channels, the aquaporins. Aquaporin-4 is mainly localized in glial membranes within the inner retina and the outer plexiform layer,74 while aquaporin-1 is present in photoreceptor and pigment epithelial cells (Fig. 17.11).1,78 In Müller cell membranes that contact fluid-filled spaces outside the neuroretina, aquaporin-4 (Fig. 17.11) and Kir4.1 channels (Fig. 17.9B) are colocalized.74 This suggests that osmotic gradients between the neuroretina on the one hand and the blood, vitreal fluid, and subretinal fluid on the other hand are compensated by bidirectional potassium and water flux across Müller cell membranes. In addition, Müller cells express aquaporin-4 in their perisynaptic membranes within the plexiform layers. Because the plexiform layers are high-resistance barriers for paracellular fluid movement,79 any water flowing into the neurons (associated with ion flux through ionotropic receptors) will be delivered from Müller cells through aquaporin-4.1 Aquaporin-4 knockout mice display reduced electroretinogram b-waves,80 suggesting that Müller cell-mediated water transport is implicated in the maintenance of regular neuronal activity. Ischemia-reperfusion results in a decreased thickness of the inner retina (Fig. 17.9B) due to neuronal degeneration caused by reactive oxygen radicals and glutamate toxicity.81 Deletion of aquaporin-4 in mice protects against ischemic injury of the retina,82 suggesting that Müller cell-mediated water transport is implicated in the pathomechanisms of neuronal cell death after ischemia.
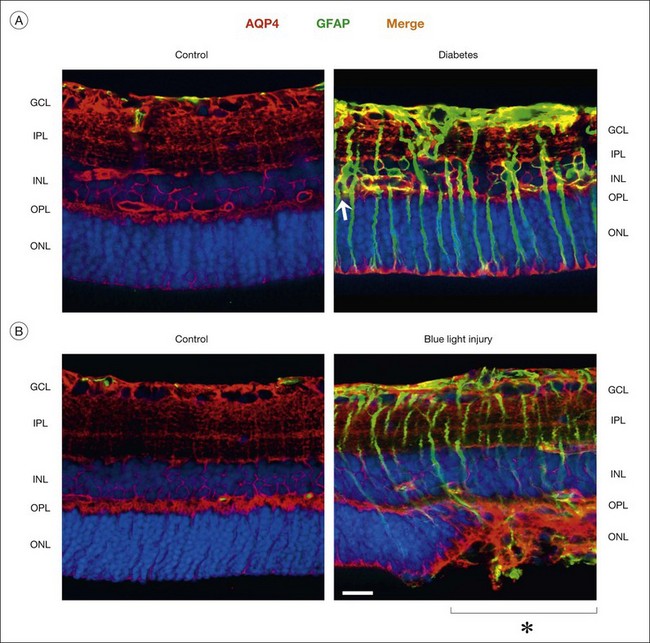
Contribution to edema development and resolution
Müller cells in animal models of retinopathies, which are known to be associated with the development of edema in the human retina, undergo an altered expression of aquaporins.1 In response to outer retinal and/or subretinal edema caused by blue-light illumination, Müller cells strongly increase their expression of aquaporin-4 in the outer nuclear layer (Fig. 17.12B). In ischemia and diabetes, Müller cells and astrocytes express aquaporin-1 in membranes which contact the vitreous and the innermost blood vessels.1 These alterations might be helpful for edema resolution. On the other hand, dysfunctional Müller cells might also contribute to edema development. Edema is characterized by a thickening of the retinal tissue, caused by water accumulation due to vascular leakage and/or by a swelling of neuronal and glial cells. Whereas neuronal swelling is observed early after ischemia-hypoxia, and is mainly caused by an overexcitation of ionotropic glutamate receptors,81 Müller cell swelling occurs within days of hypoxia.83 Because the water transport through Müller cells is coupled to the potassium currents,74 downregulation or functional inactivation of Kir4.1 channels observed under pathological conditions (Fig. 17.8A, B) should also disturb the transcellular water transport. Indeed, Müller cell bodies in slices of diseased retinas swell upon hypo-osmotic challenge which is not observed in control tissues (Fig. 17.8C). The swelling of Müller cells reflects an alteration in the osmotically driven water transport across Müller cell membranes. In the absence of functional Kir4.1 channels, the cells are not capable of rapidly releasing potassium and, thus, to compensate the osmotic gradient between the Müller cell interior and the hypo-osmotic environment. This results in water influx and cellular swelling. Because Müller cells are still capable of taking up excess potassium from the extracellular space through Kir2.1 channels which are not altered in their expression after ischemia (Fig. 17.9B), the impairment in the release of potassium from Müller cells will result in a potassium accumulation within the cells and, thus, in increased intracellular osmotic pressure.77 The increased osmotic pressure draws water from fluid-filled spaces outside the neuroretina (blood, vitreous) into the perivascular and endfeet regions of Müller cells, resulting in Müller cell swelling. The water influx from blood and vitreous is supported by aquaporin-4 water channels which are not altered, or even increased, in their expression (Fig. 17.12).83 Thus, dysfunctional water clearance, and possibly Müller cell swelling, contribute to the development of edema.
Further factors which induce osmotic Müller cell swelling are reactive oxygen radicals and inflammatory lipids (arachidonic acid, prostaglandins).1 Increased vascular permeability is associated with the extravasation of albumin; in the presence of a hypo-osmotic environment, albumin induces Müller cell swelling.84 The effect of albumin is mediated by activation of transforming growth factor-ß receptor type II, oxidative stress, production of inflammatory lipids, and intracellular sodium overload.84 Thus, extravasated albumin may represent one factor which links vascular leakage, Müller cell swelling, and dysregulation of retinal fluid clearance. Other blood-derived factors such as thrombin and glutamate (as a constituent of the plasma), which close Kir channels and depolarize Müller cells, might be involved.85,86 A reduced efficiency of glial glutamate uptake, due to cellular depolarization, may explain in part the clinical observation that the presence of hemorrhages is associated with an aggravated reduction in retinal function.87
Regulation of the blood–retinal barrier
Vascular leakage caused by opening of the tight junctions between vascular endothelial cells and/or by increased vesicular transport of serum proteins across vascular endothelia is an important pathogenic mechanism of retinal edema.88 Retinal capillaries are closely ensheathed by glial processes. Usually, Müller cells enhance the barrier function of vascular endothelia89 by the secretion of factors such as PEDF, thrombospondin-1, and glial cell line-derived neurotrophic factor (GDNF).90,91 However, in response to hypoxia, inflammation, or glucose deprivation, Müller cells produce factors such as vascular endothelial growth factor (VEGF) and tumor necrosis factor that increase vascular permeability.90,92,93 Müller cells are also a source of matrix metalloproteinases93,94 which degrade the tight junction protein, occludin.95
Mediation of neurovascular coupling
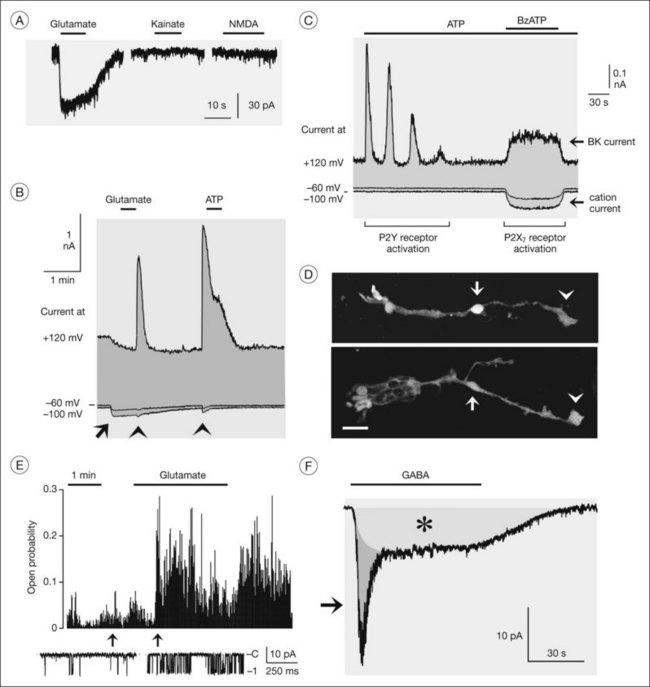
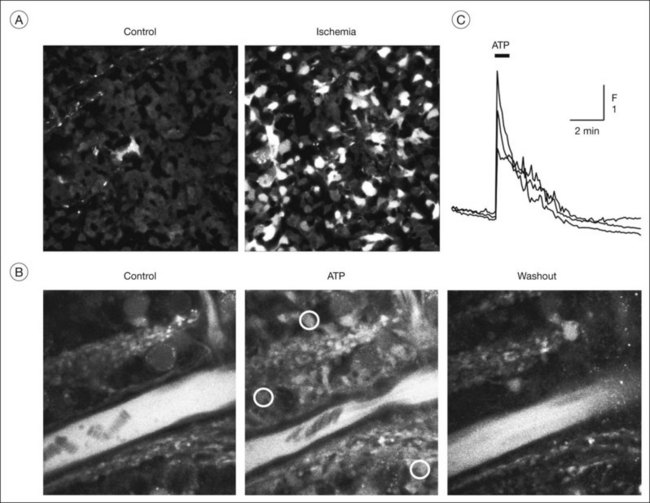
Changes in neuronal activity induce different responses in Müller cells, including alterations in their membrane potential and intracellular calcium responses triggered by activation of transmitter receptors.1 Müller cells in the rat retina generate spontaneous calcium transients that start within the inner plexiform layer and propagate to the endfeet of the cells.96 Flickering light increases the frequency of the calcium transients; this is likely mediated by adenosine 5’-triphosphate (ATP) released from neurons (amacrine and ganglion cells) and subsequent activation of metabotropic purinergic (P2Y) receptors on Müller cells.96 Such purinergic neuron-to-glia signaling is involved in the activity-dependent regulation of the local retinal blood flow.97 Calcium responses in Müller cells evoked by neuron-derived ATP (Fig. 17.13B, C) activate the calcium-dependent phospholipase A2; arachidonic acid metabolites are then released from perivascular glial membranes which cause dilation (epoxyeicosatrienoic acids) or constriction (20-hydroxyeicosatetraenoic acid) of arterioles.97 Whether vasodilating or vasoconstricting responses are produced depends on nitric oxide.97 ATP and adenosine, which are released from both neurons and glial cells,98 evoke constriction and relaxation, respectively, of pericytes.99,100
Regulation of the extracellular space volume
Neuronal activity is also associated with alterations in the volume of neuronal cell compartments (in particular, of synapses).1 The ion flux through ionotropic receptors is (for osmotic reasons) coupled to a water flux. The ion flux into neurons (and of excess potassium into Müller cells) decreases the osmolarity of the extracellular fluid,101 and the influx of water into neurons results in a swelling of neuronal cell processes and, thus, in a decrease in the extracellular space volume.1 To avoid a decrease in the extracellular space volume (which would cause neuronal hyperexcitation), Müller cells inhibit their swelling even if the extracellular fluid is hypo-osmotic, which rather favors a water influx. In addition, Müller cells elongate and reduce the thickness of their inner stem processes, at least in vitro.1 The rapid, activity-dependent alterations in cell shape are supported by the viscoelastic properties of Müller cells (see below).
To regulate cellular volume, Müller cells possess a purinergic signaling mechanism which is triggered by glutamate released from neurons or Müller cells.1 The final step of this autocrine cascade is the release of adenosine from Müller cells, which activates their adenosine A1 receptors, resulting in the opening of potassium and chloride channels. The ion efflux compensates the osmotic gradient across the Müller cell membrane and thus prevents cellular swelling. This mechanism might also accelerate the clearance of excess fluid from the retina through Müller cells.1 The anti-inflammatory corticosteroid triamcinolone acetonide, which is clinically used to induce a rapid clearance of retinal edema, was shown to inhibit the osmotic swelling of Müller cells, via inducing a release of endogenous adenosine.102 The adenosine-induced opening of ion channels may re-establish the ion and water transport through Müller cells under conditions when the Kir4.1 channels are inactivated.
Responses to mechanical stress
Neuronal activity is associated with rapid alterations in the size of neuronal synapses and cell bodies, in particular, in the plexiform layers which contain many ionotropic receptors. Müller cells respond by reshaping their processes; this is supported by the viscoelastic properties of the cells. Müller cells are softer than neurons; in particular, the processes within the plexiform layers are very soft.103 Because growing neurites prefer soft substrates,104 soft glial processes may support the growth of neuronal processes implicated in synaptic plasticity.103 Gliotic Müller cells display an increase in their stiffness due to upregulation of intermediate filaments, particularly, of glial fibrillary acidic protein (GFAP) (Fig. 17.12); this will inhibit neurite growth and might be one reason for the aberrant tissue repair after retinal injury.105 The increased stiffness of gliotic Müller cells might be also responsible for the fact that, in retinal neovascularization, new blood vessels grow towards the vitreous rather than within the retinal tissue.106
Müller cells sense mechanical deformations of the retina by calcium-dependent mechanisms. Stretching of the retinal tissue induces calcium responses, activation of extracellular signal-regulated kinases (ERK1/2), and upregulation of the transcription factor c-Fos and of bFGF in Müller cells.107 This may have implications also for postnatal eye growth. Because bFGF counteracts the development of form deprivation-induced axial myopia,108 Müller cell-derived bFGF might prevent retinal overstretching and the development of myopia during ocular growth.107
Müller cells have stretch-activated calcium-permeable cation channels.109 Activation of the channels results in an increased activity of calcium-activated BK channels.109 The potassium efflux through BK channels is associated with an efflux of cell water and results in decreased Müller cell volume.109 Stretch-induced Müller cell responses may be implicated in epiretinal membrane formation. Vitreous fibers, which adhere to Müller cell endfeet at sites of vitreoretinal attachment after partial detachment of the vitreous from the retina, exert tractional forces on to the cells; this activates Müller cells and results in cell hypertrophy and proliferation, as well as in vascular leakage.110 Mechanically stressed Müller cells release growth factors (e.g., bFGF) and ATP.107,111,112 Calcium influx (through stretch-activated and/or voltage-gated channels) and activation of BK channels are required for the growth factor- and ATP-induced proliferation of Müller cells.1,113,114
Regulation of neuronal activity by release of gliotransmitters
In addition to the uptake of neurotransmitters, Müller cells regulate the synaptic transmission by the release of “gliotransmitters,” in particular of glutamate and of the purines, ATP and adenosine. Gliotransmitters provide excitatory (glutamate, ATP) and inhibitory effects (adenosine) on neighboring neurons.98,115
Retinal glial cells release glutamate in response to electrical and mechanical stimuli, and after activation of receptors which are coupled to intracellular calcium responses, including VEGF receptor-2 and P2Y receptors.1,116 Glia-derived glutamate activates inhibitory amacrine cells which results in a depression of the light-evoked activity in a population of ganglion cell layer neurons.116,117 Glutamate released from Müller cells by calcium-dependent exocytosis directly activates ganglion cell layer neurons.1 Glia-derived glutamate (acting at ionotropic glutamate receptors) and ATP (acting at ionotropic purinergic P2X7 receptors) might contribute to the swelling and degeneration of retinal ganglion cells, e.g., in glaucoma.1,118,119 In addition, Müller cell-derived glutathione and polyamines, which increase the currents of ionotropic glutamate receptors, might contribute to glutamate-induced neuronal degeneration.120,121
Receptor agonists such as glutamate as well as electrical and mechanical stimuli trigger calcium waves in the glial network of the rat retina.111,116 The propagation of the waves among Müller cells depends upon the release of ATP and autocrine/paracrine activation of P2Y receptors.111,116 The glial calcium waves are associated with both enhancement and depression of light-evoked activity of distinct ganglion cells.117 Glial ATP released into the inner plexiform layer is converted extracellularly to adenosine that activates A1 receptors in a population of ganglion cells, resulting in reduced spontaneous activity.112 Müller cell-derived adenosine, which is also released via nucleoside transporters,1 may act as a negative-feedback regulator of neurotransmission; this adapts the level of neuronal activity to the capacity of Müller cells to maintain retinal homeostasis.
Müller cells are involved in setting the sensitivity of ganglion and amacrine cells by the release of d-serine, which potentiates N-methyl-d-aspartate receptor currents.122 They are implicated in providing the direction selectivity of direction-selective ganglion cells by the secretion of acyl coenzyme A-binding protein.123 Müller cells also produce nitric oxide; the production of nitric oxide is strongly enhanced during dark adaptation.124 Nitric oxide diffuses out of the cells and has multiple effects on neighboring neurons, including a potentiation of phototransduction, closure of gap junctions and N-methyl-d-aspartate receptor channels, and an increase of calcium channel currents.1 Under ischemic-hypoxic conditions, glial cells are a major source of retinal nitric oxide. Nitric oxide dilates blood vessels, prevents platelet aggregation, and protects neurons from apoptosis by closure of N-methyl-d-aspartate receptor channels.125 However, production of excess nitric oxide and subsequnt formation of free nitrogen radicals and protein nitrosylation have toxic effects on surrounding neurons; these effects are implicated in the pathogenesis of diabetic retinopathy, for example.126–128
Ionotropic receptors of Müller cells
Müller cells can sense neuronal activity,129 primarily by membrane depolarization upon activation of receptor channels. Human Müller cells express GABAA and P2X7 receptors (Fig. 17.7D),37,130 which are ligand-gated chloride and cation channels, respectively. GABA evokes two types of inward currents in Müller cells, a fast transient GABAA receptor current and a sustained current mediated by electrogenic GABA transporters (Fig. 17.7F).37 Both currents depolarize the Müller cells. Zinc ions released from photoreceptors29 increase the GABAA receptor currents.37 GABAA receptors may have different functional roles; because the receptor channels are also permeable for bicarbonate, they may be implicated in the regulation of the extracellular pH. The chloride efflux through the receptor channels may also stimulate GABA uptake by the cells (which is driven by a cotransport of sodium and chloride) and may compensate the decrease in the extracellular chloride level caused by chloride influx into activated neurons.
P2X7 receptor channels are calcium-permeable cation channels. Opening of the channels by extracellular ATP results in a calcium influx into the cells, calcium release from intracellular stores, and activation of BK channels (Fig. 17.7B, C).1,130 Moreover, the opening of P2X7 channels causes cellular depolarization which decreases the efficiency of the electrogenic neurotransmitter uptake.130 However, the depolarization is counterregulated by hyperpolarization due to a potassium flux through BK channels. The BK channel-mediated hyperpolarization increases the driving force for the calcium influx. P2X7 receptor currents are increased when the extracellular fluid contains low concentrations of divalent cations,130 suggesting that the light-induced decrease in extracellular calcium will facilitate the gating of P2X7 receptors in Müller cells.
Müller cell gliosis
In response to virtually every pathological stimulus, Müller cells become activated.1 Reactive gliosis is thought to represent a cellular attempt to protect the tissue from further damage, to promote tissue repair, and to limit tissue remodeling. It includes morphological, biochemical, and physiological changes; these responses vary with the type and severity of the insult. An upregulation of the intermediate filaments, vimentin and GFAP in glial cells (Fig. 17.12) is a very sensitive and early retinal stress indicator.1,131
In fact, Müller cell gliosis has both cytoprotective and cytotoxic effects on retinal neurons.1 Particularly under conditions of massive (proliferative) gliosis, the regular glial–neuronal interactions are disrupted and the retina degenerates (Fig. 17.14). An inverse relation between Müller cell proliferation and the expression of neuron glia symbiosis-mediating proteins has been observed. However, also in the case of “conservative” gliosis (associated with short-term, or low-level, Müller cell proliferation), a dysfunction of Müller cells may contribute to neurodegeneration. After retinal detachment, for example, the downregulation of CRALBP and of glutamine synthetase132,133 disrupts glia–neuronal interactions involved in photopigment and neurotransmitter recycling. The downregulation of carbonic anhydrase132,133 and Kir channels (Fig. 17.8A, B)1 results in disturbances of the retinal acid–base, ion, and water homeostasis. Because the electrogenic uptake carriers are voltage-dependent (Fig. 17.6A), membrane depolarization reduces the efficiency of glial neurotransmitter recycling which, together with an increase in extracellular potassium, will aggravate neurotoxicity. Because the potassium currents of Müller cells are also a major driving force for the water transport through the cells,1 downregulation of functional Kir channels also impairs retinal water homeostasis, contributing to the development of edema.134
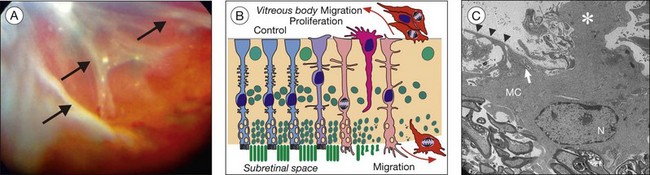
The involvement of reactive Müller cells in immune and inflammatory responses may also exert detrimental effects. After retinal injuries such as retinal detachment, Müller cells upregulate inflammatory factors, including monocyte chemoattractant protein (MCP)-1, which recruit phagocytotic monocytes/macrophages and microglial cells to the injured area.135,136 Blood-derived monocytes/macrophages and neutrophils release oxygen free radicals and cytotoxic cytokines which play critical roles in the photoreceptor apoptosis after retinal detachment137 and in neuronal degeneration after ischemia-reperfusion.138 Mice deficient in vimentin and GFAP display an attenuation of the detachment-induced glial cell responses (activation of ERK1/2 and c-Fos, induction of MCP-1) and, as a consequence, a decrease in monocyte infiltration and photoreceptor apoptosis was observed.139
In contrast, reactive Müller cells might protect photoreceptors and neurons from cell death by various mechanisms, including the secretion of neurotrophic and growth factors, in particular bFGF.1,46,140 Various cytokines might reduce (e.g., nerve growth factor) or increase (e.g., GDNF) the production of bFGF by Müller cells.46,141,142 GDNF induces phosphorylation of ERK1/2 in Müller cells, resulting in a transcriptional upregulation of bFGF that in turn supports photoreceptor survival.141,142 GDNF, which is produced by photoreceptor and glial cells,141,143 also protects photoreceptors and ganglion cells from apoptosis through upregulation of the glial glutamate transporter GLAST.144,145 Retinal preconditioning with bright light or mechanical stress protects photoreceptors from apoptosis because these stimuli induce an upregulation of bFGF and ciliary neurotrophic factor in Müller cells.45,140 Likewise, argon laser photocoagulation slows photoreceptor degeneration by induction of bFGF in Müller cells.146
Müller stem cells
As a precondition for proliferation and migration, Müller cells reactivate distinct cellular programs normally used in ontogenetic development. In the injured retina, a population of Müller cells dedifferentiates to cells with properties similar to pluripotent retinal progenitor/stem cells, and may even express distinct neuronal and photoreceptor markers.147,148 The decrease in Kir currents (Fig. 17.10A) and the increase in voltage-gated sodium currents (Fig. 17.10B) observed in Müller cells from patients with proliferative retinopathies37 may reflect such a transdifferentiation of the cells. However, the neuron-regenerating capability of mammalian Müller cells in situ is very restricted. Attempts to facilitate the neurogenic program of Müller cells, e.g., by transdifferentiation of cultured Müller cells, are ongoing.148,149 The tedious progress of this approach underlines the general issue that a better understanding of gliotic mechanisms is essential for the development of efficient therapeutic strategies which increase the protective and regenerative, and decrease the destructive, roles of reactive Müller cells.
1 Reichenbach A, Bringmann A. Müller cells in the healthy and diseased retina. New York: Springer; 2010.
2 Reichenbach A, Robinson SR. Phylogenetic constraints on retinal organization and development: an Haeckelian perspective. Prog Retin Eye Res. 1995;15:139–171.
3 Franze K, Grosche J, Skatchkov SN, et al. Müller cells are living optical fibers in the vertebrate retina. Proc Natl Acad Sci U S A. 2007;104:8287–8292.
4 Muniz A, Villazana-Espinoza ET, Hatch AL, et al. A novel cone visual cycle in the cone-dominated retina. Exp Eye Res. 2007;85:175–184.
5 Bunt-Milam AH, Saari JC. Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J Cell Biol. 1983;97:703–712.
6 Jablonski MM, Iannaccone A. Targeted disruption of Müller cell metabolism induces photoreceptor dysmorphogenesis. Glia. 2000;32:192–204.
7 Jablonski MM, Wohabrebbi A, Ervin CS. Lactose promotes organized photoreceptor outer segment assembly and preserves expression of photoreceptor proteins in retinal degeneration. Mol Vis. 1999;5:16.
8 Wang X, Iannaccone A, Jablonski MM. Contribution of Müller cells toward the regulation of photoreceptor outer segment assembly. Neuron Glia Biol. 2005;1:1–6.
9 Long KO, Fisher SK, Fariss RN, et al. Disc shedding and autophagy in the cone-dominant ground squirrel retina. Exp Eye Res. 1986;43:193–205.
10 Polit L, Rotstein N, Carri N. Effects of docosahexaenoic acid on retinal development: cellular and molecular aspects. Lipids. 2001;36:927–935.
11 Otori Y, Shimada S, Tanaka K, et al. Marked increase in glutamate-aspartate transporter (GLAST/GluT-1) mRNA following transient retinal ischemia. Mol. Brain Res. 1994;27:310–314.
12 Rauen T, Rothstein JD, Wässle H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996;286:325–336.
13 Rauen T, Taylor WR, Kuhlbrodt K, et al. High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998;291:19–31.
14 Kugler P, Beyer A. Expression of glutamate transporters in human and rat retina and rat optic nerve. Histochem Cell Biol. 2003;120:199–212.
15 Ward MM, Jobling AI, Kalloniatis M, et al. Glutamate uptake in retinal glial cells during diabetes. Diabetologia. 2005;48:351–360.
16 Barbour B, Brew H, Attwell D. Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature. 1988;335:433–435.
17 Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 1987;327:707–709.
18 Marc RE. Structural organization of GABAergic circuitry in ectotherm retinas. Prog Brain Res. 1992;90:61–92.
19 Hasegawa J, Obara T, Tanaka K, et al. High-density presynaptic transporters are required for glutamate removal from the first visual synapse. Neuron. 2006;50:63–74.
20 Matsui K, Hosoi N, Tachibana M. Active role of glutamate uptake in the synaptic transmission from retinal nonspiking neurons. J Neurosci. 1999;19:6755–6766.
21 Higgs MH, Lukasiewicz PD. Glutamate uptake limits synaptic excitation of retinal ganglion cells. J Neurosci. 2002;19:3691–3700.
22 Pow DV, Barnett NL, Penfold P. Are neuronal transporters relevant in retinal glutamate homeostasis? Neurochem Int. 2000;37:191–198.
23 Holcombe DJ, Lengefeld N, Gole GA, et al. The effects of acute intraocular pressure elevation on rat retinal glutamate transport. Acta Ophthalmol Scand. 2008;86:408–414.
24 Naskar R, Vorwerk CK, Dreyer EB. Concurrent downregulation of a glutamate transporter and receptor in glaucoma. Invest Ophthalmol Vis Sci. 2000;41:1940–1944.
25 Barnett NL, Pow DV, Bull ND. Differential perturbation of neuronal and glial glutamate transport systems in retinal ischaemia. Neurochem Int. 2001;39:291–299.
26 Müller A, Maurin L, Bonne C. Free radicals and glutamate uptake in the retina. Gen Pharmacol. 1998;30:315–318.
27 Li Q, Puro DG. Diabetes-induced dysfunction of the glutamate transporter in retinal Müller cells. Invest Ophthalmol Vis Sci. 2002;43:3109–3116.
28 Beretta S, Wood JP, Derham B, et al. Partial mitochondrial complex I inhibition induces oxidative damage and perturbs glutamate transport in primary retinal cultures. Relevance to Leber hereditary optic neuropathy (LHON). Neurobiol Dis. 2006;24:308–317.
29 Wu SM, Qiao X, Noebels JL, et al. Localization and modulatory actions of zinc in vertebrate retina. Vision Res. 1993;33:2611–2616.
30 Barbour B, Szatkowski M, Ingledew N, et al. Arachidonic acid induces a prolonged inhibition of glutamate uptake into glial cells. Nature. 1989;342:918–920.
31 Spiridon M, Kamm D, Billups B, et al. Modulation by zinc of the glutamate transporters in glial cells and cones isolated from the tiger salamander retina. J Physiol. 1998;506:363–376.
32 Napper GA, Pianta MJ, Kalloniatis M. Reduced glutamate uptake by retinal glial cells under ischemic/hypoxic conditions. Vis Neurosci. 1999;16:149–158.
33 Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–446.
34 Maguire G, Simko H, Weinreb RN, et al. Transport-mediated release of endogenous glutamate in the vertebrate retina. Pflügers Arch. 1998;436:481–484.
35 Marcaggi P, Hirji N, Attwell D. Release of L-aspartate by reversal of glutamate transporters. Neuropharmacology. 2005;49:843–849.
36 Kato S, Ishita S, Sugawara K, et al. Cystine/glutamate antiporter expression in retinal Müller glial cells: implications for DL-α-aminoadipate toxicity. Neuroscience. 1993;57:473–482.
37 Reichelt W, Pannicke T, Biedermann B, et al. Comparison between functional characteristics of healthy and pathological human retinal Müller glial cells. Surv Ophthalmol. 1997;42:S105–S117.
38 Riepe RE, Norenburg MD. Müller cell localisation of glutamine synthetase in rat retina. Nature. 1977;268:654–655.
39 Pow DV, Crook DK. Direct immunocytochemical evidence for the transfer of glutamine from glial cells to neurons: use of specific antibodies directed against the D-stereoisomers of glutamate and glutamine. Neuroscience. 1996;70:295–302.
40 Pow DV, Robinson SR. Glutamate in some retinal neurons is derived solely from glia. Neuroscience. 1994;60:355–366.
41 Fletcher EL, Kalloniatis M. Neurochemical architecture of the normal and degenerating rat retina. J Comp Neurol. 1996;376:343–360.
42 Marc RE, Murry RF, Fisher SK, et al. Amino acid signatures in the detached cat retina. Invest Ophthalmol Vis Sci. 1998;39:1694–1702.
43 Kruchkova Y, Ben-Dror I, Herschkovitz A, et al. Basic fibroblast growth factor: a potential inhibitor of glutamine synthetase expression in injured neural tissue. J Neurochem. 2001;77:1641–1649.
44 Faktorovich EG, Steinberg RH, Yasumura D, et al. Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factor. Nature. 1990;347:83–86.
45 Liu C, Peng M, Laties AM, et al. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci. 1998;18:1337–1344.
46 Harada T, Harada C, Nakayama N, et al. Modification of glial–neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron. 2000;26:533–541.
47 Barnett NL, Osborne NN. Redistribution of GABA immunoreactivity following central retinal artery occlusion. Brain Res. 1995;677:337–340.
48 Ishikawa A, Ishiguro S, Tamai M. Changes in GABA metabolism in streptozotocin-induced diabetic rat retinas. Curr Eye Res. 1996;15:63–71.
49 Poitry S, Poitry-Yamate C, Ueberfeld J, et al. Mechanisms of glutamate metabolic signaling in retinal glial (Müller) cells. J Neurosci. 2000;20:1809–1821.
50 Poitry-Yamate CL, Poitry S, Tsacopoulos M. Lactate released by Müller glial cells is metabolized by photoreceptors from mammalian retina. J Neurosci. 1995;15:5179–5191.
51 Linsenmeier RA. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J Gen Physiol. 1986;88:521–542.
52 Schütte M, Werner P. Redistribution of glutathione in the ischemic rat retina. Neurosci Lett. 1998;246:53–56.
53 Paasche G, Gärtner U, Germer A, et al. Mitochondria of retinal Müller (glial) cells: the effects of aging and of application of free radical scavengers. Ophthalmic Res. 2000;32:229–236.
54 Kern TS, Kowluru RA, Engerman RL. Abnormalities of retinal metabolism in diabetes or galactosemia: ATPases and glutathione. Invest Ophthalmol Vis Sci. 1994;35:2962–2967.
55 Reichelt W, Stabel-Burow J, Pannicke T, et al. The glutathione level of retinal Müller glial cells is dependent on the high-affinity sodium-dependent uptake of glutamate. Neuroscience. 1997;77:1213–1224.
56 Shelton MD, Kern TS, Mieyal JJ. Glutaredoxin regulates nuclear factor κ-B and intercellular adhesion molecule in Müller cells: model of diabetic retinopathy. J Biol Chem. 2007;282:12467–12474.
57 Newman EA. A physiological measure of carbonic anhydrase in Müller cells. Glia. 1994;11:291–299.
58 Oakley B, Wen R. Extracellular pH in the isolated retina of the toad in darkness and during illumination. J Physiol. 1989;419:353–378.
59 Linser PJ, Moscona AA. Carbonic anhydrase-C in the neural retina: transition from generalized to glia-specific cell localization during embryonic development. Proc Natl Acad Sci U S A. 1981;78:7190–7194.
60 Nagelhus EA, Mathiisen TM, Bateman AC, et al. Carbonic anhydrase XIV is enriched in specific membrane domains of retinal pigment epithelium, Müller cells, and astrocytes. Proc Natl Acad Sci U S A. 2005;102:8030–8035.
61 Borgula GA, Karwoski CJ, Steinberg RH. Light-evoked changes in extracellular pH in frog retina. Vision Res. 1989;29:1069–1077.
62 DeVries SH. Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron. 2001;32:1107–1117.
63 Newman EA. Acid efflux from retinal glial cells generated by sodium bicarbonate cotransport. J Neurosci. 1996;16:159–168.
64 Barnes S, Merchant V, Mahmud F. Modulation of transmission gain by protons at the photoreceptor output synapse. Proc Natl Acad Sci U S A. 1993;90:10081–10085.
65 Steinberg RH, Oakley B, II., Niemeyer G. Light-evoked changes in [K+]0 in retina of intact cat eye. J Neurophysiol. 1980;44:897–921.
66 Karwoski CJ, Newman EA, Shimazaki H, et al. Light-evoked increases in extracellular K+ in the plexiform layers of amphibian retinas. J Gen Physiol. 1985;86:189–213.
67 Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175.
68 Karwoski CJ, Lu HK, Newman EA. 1989. Spatial buffering of light-evoked potassium increases by retinal Müller (glial) cells. Science. 1989;244:578–580.
69 Newman EA. Regional specialization of retinal glial cell membrane. Nature. 1984;309:155–157.
70 Brew H, Gray PT, Mobbs P, et al. Endfeet of retinal glial cells have higher densities of ion channels that mediate K+ buffering. Nature. 1986;324:466–468.
71 Newman EA. Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci. 1993;13:3333–3345.
72 Kofuji P, Ceelen P, Zahs KR, et al. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740.
73 Kofuji P, Biedermann B, Siddharthan V, et al. Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia. 2002;39:292–303.
74 Nagelhus EA, Horio Y, Inanobe A, et al. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54.
75 Ishii M, Fujita A, Iwai K, et al. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol. 2003;285:C260–C267.
76 Skatchkov SN, Eaton MJ, Shuba YM, et al. Tandem-pore domain potassium channels are functionally expressed in retinal (Müller) glial cells. Glia. 2006;53:266–276.
77 Pannicke T, Iandiev I, Uckermann O, et al. A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci. 2004;26:493–502.
78 Stamer WD, Bok D, Hu J, et al. Aquaporin-1 channels in human retinal pigment epithelium: role in transepithelial water movement. Invest Ophthalmol Vis Sci. 2003;44:2803–2808.
79 Antcliff RJ, Hussain AA, Marshall J. Hydraulic conductivity of fixed retinal tissue after sequential excimer laser ablation: barriers limiting fluid distribution and implications for cystoid macular edema. Arch Ophthalmol. 2001;119:539–544.
80 Li J, Patil RV, Verkman AS. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Invest Ophthalmol Vis Sci. 2002;43:573–579.
81 Osborne NN, Casson RJ, Wood JP, et al. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147.
82 Da T, Verkman AS. Aquaporin-4 gene disruption in mice protects against impaired retinal function and cell death after ischemia. Invest Ophthalmol Vis Sci. 2004;45:4477–4483.
83 Kaur C, Sivakumar V, Yong Z, et al. Blood–retinal barrier disruption and ultrastructural changes in the hypoxic retina in adult rats: the beneficial effect of melatonin administration. J Pathol. 2007;212:429–439.
84 Löffler S, Wurm A, Kutzera F, et al. Serum albumin induces osmotic swelling of rat retinal glial cells. Brain Res. 2010;1317:268–276.
85 Puro DG, Stuenkel EL. Thrombin-induced inhibition of potassium currents in human retinal glial (Müller) cells. J Physiol. 1995;485:337–348.
86 Kusaka S, Kapousta-Bruneau NV, Puro DG. Plasma-induced changes in the physiology of mammalian retinal glial cells: role of glutamate. Glia. 1999;25:205–215.
87 Gass JD. Stereoscopic atlas of macular diseases: diagnosis and treatment. St Louis, MO: Mosby; 1997. p.1061
88 Cunha-Vaz JG, Travassos A. Breakdown of the blood–retinal barriers and cystoid macular edema. Surv Ophthalmol. 1984;28:S485–S492.
89 Tout S, Chan-Ling T, Hollander H, et al. The role of Müller cells in the formation of the blood–retinal barrier. Neuroscience. 1993;55:291–301.
90 Eichler W, Yafai Y, Wiedemann P, et al. Angiogenesis-related factors derived from retinal glial (Müller) cells in hypoxia. Neuroreport. 2004;15:1633–1637.
91 Nishikiori N, Osanai M, Chiba H, et al. Glial cell-derived cytokines attenuate the breakdown of vascular integrity in diabetic retinopathy. Diabetes. 2007;56:1333–1340.
92 Aiello LP, Northrup JM, Keyt BA, et al. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol. 1995;113:1538–1544.
93 Noda K, Ishida S, Shinoda H, et al. Hypoxia induces the expression of membrane-type 1 matrix metalloproteinase in retinal glial cells. Invest Ophthalmol Vis Sci. 2005;46:3817–3824.
94 Behzadian MA, Wang XL, Windsor LJ, et al. TGF-ß increases retinal endothelial cell permeability by increasing MMP-9: possible role of glial cells in endothelial barrier function. Invest Ophthalmol Vis Sci. 2001;42:853–859.
95 Giebel SJ, Menicucci G, McGuire PG, et al. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood–retinal barrier. Lab Invest. 2005;85:597–607.
96 Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005;25:5502–5510.
97 Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26:2862–2870.
98 Housley GD, Bringmann A, Reichenbach A. Purinergic signaling in special senses. Trends Neurosci. 2009;32:128–141.
99 Li Q, Puro DG. Adenosine activates ATP-sensitive K+ currents in pericytes of rat retinal microvessels: role of A1 and A2A receptors. Brain Res. 2001;907:93–99.
100 Peppiatt CM, Howarth C, Mobbs P, et al. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704.
101 Dmitriev AV, Govardovskii VI, Schwahn HN, et al. Light-induced changes of extracellular ions and volume in the isolated chick retina-pigment epithelium preparation. Vis Neurosci. 1999;16:1157–1167.
102 Uckermann O, Kutzera F, Wolf A, et al. The glucocorticoid triamcinolone acetonide inhibits osmotic swelling of retinal glial cells via stimulation of endogenous adenosine signaling. J Pharmacol Exp Ther. 2005;315:1036–1045.
103 Lu Y-B, Franze K, Seifert G, et al. Viscoelastic properties of individual glial cells and neurons in the CNS. Proc Natl Acad Sci U S A. 2006;103:17759–17764.
104 Flanagan LA, Ju YE, Marg B, et al. Neurite branching on deformable substrates. Neuroreport. 2002;13:2411–2415.
105 Lu YB, Iandiev I, Hollborn M, et al. Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. FASEB J. 2011;25:624–631.
106 Lundkvist A, Reichenbach A, Betsholtz C, et al. Under stress, the absence of intermediate filaments from Müller cells in the retina has structural and functional consequences. J Cell Sci. 2004;117:3481–3488.
107 Lindqvist N, Liu Q, Zajadacz J, et al. Retinal glial (Müller) cells: sensing and responding to tissue stretch. Invest Ophthalmol Vis Sci. 2010;51:1683–1690.
108 Rohrer B, Stell WK. Basic fibroblast growth factor (bFGF) and transforming growth factor ß (TGF-ß) act as stop and go signals to modulate postnatal ocular growth in the chick. Exp Eye Res. 1994;58:553–561.
109 Puro DG. Stretch-activated channels in human retinal Müller cells. Glia. 1991;4:456–460.
110 Schubert HD. Cystoid macular edema: the apparent role of mechanical factors. Prog Clin Biol Res. 1989;312:277–291.
111 Newman EA. Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. J Neurosci. 2001;21:2215–2223.
112 Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666.
113 Puro DG, Mano T. Modulation of calcium channels in human retinal glial cells by basic fibroblast growth factor: a possible role in retinal pathobiology. J Neurosci. 1991;11:1873–1880.
114 Puro DG, Roberge F, Chan C-C. Retinal glial cell proliferation and ion channels: a possible link. Invest Ophthalmol Vis Sci. 1989;30:521–529.
115 Newman EA. Glial modulation of synaptic transmission in the retina. Glia. 2004;47:268–274.
116 Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847.
117 Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028.
118 Zhang X, Zhang M, Laties AM, et al. Stimulation of P2X7 receptors elevates Ca2+ and kills retinal ganglion cells. Invest Ophthalmol Vis Sci. 2005;46:2183–2191.
119 Resta V, Novelli E, Vozzi G, et al. Acute retinal ganglion cell injury caused by intraocular pressure spikes is mediated by endogenous extracellular ATP. Eur J Neurosci. 2007;25:2741–2754.
120 Janaky R, Ogita K, Pasqualotto BA, et al. Glutathione and signal transduction in the mammalian CNS. J Neurochem. 1999;73:889–902.
121 Pernet V, Bourgeois P, Di Polo A. A role for polyamines in retinal ganglion cell excitotoxic death. J Neurochem. 2007;103:1481–1490.
122 Miller RF. D-Serine as a glial modulator of nerve cells. Glia. 2004;47:275–283.
123 Barmack NH, Bilderback TR, Liu H, et al. Activity-dependent expression of acyl-coenzyme A-binding protein in retinal Müller glial cells evoked by optokinetic stimulation. J Neurosci. 2004;24:1023–1033.
124 Zemel E, Eyal O, Lei B, et al. NADPH diaphorase activity in mammalian retinas is modulated by the state of visual adaptation. Vis Neurosci. 1996;13:863–871.
125 Goldstein IM, Ostwald P, Roth S. Nitric oxide: a review of its role in retinal function and disease. Vision Res. 1996;36:2979–2994.
126 Roth S. Role of nitric oxide in retinal cell death. Clin Neurosci. 1997;4:216–223.
127 Goureau O, Regnier-Ricard F, Courtois Y. Requirement for nitric oxide in retinal neuronal cell death induced by activated Müller glial cells. J Neurochem. 1999;72:2506–2515.
128 Koeberle PD, Ball AK. Nitric oxide synthase inhibition delays axonal degeneration and promotes the survival of axotomized retinal ganglion cells. Exp Neurol. 1999;158:366–381.
129 Rillich K, Gentsch J, Reichenbach A, et al. Light stimulation evokes two different calcium responses in Müller glial cells of the guinea pig retina. Eur J Neurosci. 2009;29:1165–1176.
130 Pannicke T, Fischer W, Biedermann B, et al. P2X7 receptors in Müller glial cells from the human retina. J Neurosci. 2000;20:5965–5972.
131 Bignami A, Dahl D. The radial glia of Müller in the rat retina and their response to injury: an immunofluorescence study with antibodies to the glial fibrillary acidic (GFA) protein. Exp Eye Res. 1979;28:63–69.
132 Lewis GP, Erickson PA, Guerin CJ, et al. Changes in the expression of specific Müller cell proteins during long-term retinal detachment. Exp Eye Res. 1989;49:93–111.
133 Lewis GP, Mervin K, Valter K, et al. Limiting the proliferation and reactivity of retinal Müller cells during experimental retinal detachment: the value of oxygen supplementation. Am J Ophthalmol. 1999;128:165–172.
134 Jackson TL, Hillenkamp J, Williamson TH, et al. An experimental model of rhegmatogenous retinal detachment: surgical results and glial cell response. Invest Ophthalmol Vis Sci. 2003;44:4026–4034.
135 Nakazawa T, Matsubara A, Noda K, et al. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. 2006;12:867–878.
136 Hollborn M, Francke M, Iandiev I, et al. Early activation of inflammation- and immune response-related genes after experimental detachment of the porcine retina. Invest Ophthalmol Vis Sci. 2008;49:1262–1273.
137 Nakazawa T, Hisatomi T, Nakazawa C, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A. 2007;104:2425–2430.
138 Neufeld AH, Kawai S, Das S, et al. Loss of retinal ganglion cells following retinal ischemia: the role of inducible nitric oxide synthase. Exp Eye Res. 2002;75:521–528.
139 Nakazawa T, Takeda M, Lewis GP, et al. Attenuated glial reactions and photoreceptor degeneration after retinal detachment in mice deficient in glial fibrillary acidic protein and vimentin. Invest Ophthalmol Vis Sci. 2007;48:2760–2768.
140 Wen R, Song Y, Cheng T, et al. Injury-induced upregulation of bFGF and CNTF mRNAs in the rat retina. J Neurosci. 1995;15:7377–7385.
141 Harada C, Harada T, Quah HMA, et al. Potential role of glial cell line-derived neurotrophic factor receptors in Müller glial cells during light-induced retinal degeneration. Neuroscience. 2003;122:229–235.
142 Hauck SM, Kinkl N, Deeg CA, et al. GDNF family ligands trigger indirect neuroprotective signaling in retinal glial cells. Mol Cell Biol. 2006;26:2746–2757.
143 Igarashi Y, Chiba H, Utsumi H, et al. Expression of receptors for glial cell line-derived neurotrophic factor (GDNF) and neurturin in the inner blood–retinal barrier of rats. Cell Struct Funct. 2000;25:237–241.
144 Delyfer MN, Simonutti M, Neveux N, et al. Does GDNF exert its neuroprotective effects on photoreceptors in the rd1 retina through the glial glutamate transporter GLAST? Mol Vis. 2005;11:677–687.
145 Koeberle PD, Bähr M. The upregulation of GLAST-1 is an indirect antiapoptotic mechanism of GDNF and neurturin in the adult CNS. Cell Death Differ. 2008;15:471–483.
146 Chu Y, Humphrey MF, Alder VV, et al. Immunocytochemical localization of basic fibroblast growth factor and glial fibrillary acidic protein after laser photocoagulation in the Royal College of Surgeons rat. Aust N Z J Ophthalmol. 1998;26:87–96.
147 Fischer AJ, Reh TA. Müller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci. 2001;4:247–252.
148 Ooto S, Akagi T, Kageyama R, et al. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc Natl Acad Sci U S A. 2004;101:13654–13659.
149 Lawrence JM, Singhal S, Bhatia B, et al. MOI-M1 cells and similar Müller glial cell lines derived from adult human retina exhibit neural stem cell characteristics. Stem Cells. 2007;25:2033–2043.