[level-membership-for-neurology-category]
Chapter 13 Brain Plasticity and its Disorders
Introduction
Brain plasticity is an important concept that plays a major role in the expression of many pediatric neurological disorders and strongly influences recovery from brain injuries in neonates, infants, and children. Plasticity refers to the brain’s ability to change in response to experience, whether it is a positive experience such as education or practicing a skill, or an adverse event such as a stroke or other type of brain injury [Johnston, 2004]. The child’s brain exhibits greater plasticity than the adult brain, and common examples of enhanced brain plasticity in children include their ability to learn new motor tasks quickly, such as playing a musical instrument, participating in a sport, or their ability to become fluent in a new language [Meltzoff et al., 2009]. Children and adolescents are also able to form memories more easily than adults and they recover more quickly from brain injuries. The mechanisms responsible for various kinds of brain plasticity are being uncovered at a rapid pace as are the defects in these steps that cause intellectual disability and other pediatric disorders [Johnston, 2009]. This chapter gives a brief overview of normal brain plasticity and its disorders that are relevant to pediatric neurology, and provides insight into the pathogenesis of a variety of disorders described in other sections.
It is useful to consider four aspects or types of plasticity in the developing brain in order to understand how this concept is integrated into many disorders of the child’s nervous system (Box 13-1). The first type is generally referred to as adaptive plasticity, in which the nervous system changes in the process of learning new skills that have an adaptive advantage, such as learning to read or pass a test, learning to play a musical instrument, or learning to play a sport. Adaptive plasticity is also engaged when children recover from a brain injury, such as a stroke or extensive removal of brain tissue to cure epilepsy, and therapists try to harness this form of plasticity when providing speech, occupational, or physical therapy for a variety of disorders. In these cases, it is expected that normal childhood activities, such as attending school, music lessons, athletic practice, or therapies for cerebral palsy or other brain injuries, will activate normal plasticity programs to provide a good outcome that is beneficial for the child [Meltzoff et al., 2009]. Adaptive plasticity is generally enhanced in younger children compared with adults, and the genetically determined programs responsible for this kind of plasticity are heavily influenced by sensory input. Since learning and memory, as well as acquisition of physical skills, are very important for normal childhood development, genetic or acquired defects in the signaling pathways responsible for plasticity are reflected in a variety of developmental disability phenotypes [Johnston, 2004]. These disorders are best understood as examples of impaired plasticity. Many types of intellectual disability are caused by genetic lesions in the signaling cascades that are normally responsible for activity-dependent synaptic plasticity (Table 13-1). For example, in fragile X syndrome (FraX), the most common form of inherited intellectual disability, a trinucleotide repeat in the gene for the fragile X mental retardation protein (FMRP) leads to its absence or reduction in the brain [Penagarikano et al., 2007]. FMRP is responsible for transport of certain messenger RNAs and translation of proteins within dendrites in response to synaptic activity, and its absence causes a reduction in long-term potentiation (LTP), a physiological form of synaptic plasticity involved in learning and memory [Huber et al., 2002]. Accordingly, the intellectual disability and other behavioral features of FraX can be considered to reflect a genetically determined form of impaired plasticity in synapses. Therapies aimed at restoring normal plasticity mechanisms to synapses in FraX show some promise in preclinical and small clinical trials [Wang et al., 2010].




Table 13-1 Pediatric Disorders Caused by Genetic Mutations of Signaling Pathways Involved in Neuronal Plasticity
| Disorder | Mechanism of Disease |
|---|---|
| Neurofibromatosis 1 | Ras too active, enhanced GABA activity |
| Tuberous sclerosis | Upregulated mTOR signaling pathway |
| Rett’s syndrome | Mutations in MeCP2 transcription factor |
| Fragile X syndrome | Upregulated mGluR5 causes LTD |
| Coffin–Lowry syndrome | Mutations in RSK2 in Ras-MAPK pathway |
| Rubinstein–Taybi syndrome | Mutation in CREB binding protein (CBP) |
| X-linked intellectual disabilities | Mutation in PAK3 kinase: links RhoGTPases to dendritic cytoskeleton Mutation in oligophrenin, RhoGTPase Mutation in GluR3 AMPA receptor subunit |
| Costello’s syndrome | Upregulated H-Ras signaling to MAPK (intellectual disability, heart, skeletal disorder) |
| Lead poisoning | Enhanced PKC activity, inhibited NMDA receptors; impairs maturation of dendritic spines |
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CREB, cyclic adenosine monophosphate response element binding protein; GABA, gamma-aminobutyric acid; MAPK, Ras-mitogen activated protein kinase; mTOR, mammalian target of rapamycin; NMDA, N-methyl-d-aspartate; PKC, protein kinase C.
Just as impaired plasticity is responsible for numerous brain disorders in children, maladaptive or excessive plasticity also can cause problems. When this phenomenon occurs, plasticity is misdirected or enhanced in a way that is harmful, or at least not beneficial. For example, children who have had strokes, traumatic brain injuries, or hypoxic-ischemic brain injuries sometimes develop delayed seizures that may result from formation of abnormal neuronal networks in damaged brain tissue [Kadam and Dudek, 2007]. In some cases, neurons in these networks have been shown to have functional changes in voltage-sensitive sodium channels that are similar to those found in certain forms of genetic epilepsy [Graef and Godwin, 2010]. Excessive plasticity may also be responsible for seizures and cognitive impairment in genetic disorders in which intracellular signaling pathways are enhanced, such as tuberous sclerosis and Costello’s syndrome [Crino, 2010; Dileone et al., 2010]. Another example of enhanced, but maladaptive, plasticity that causes neurologic impairment is dystonia in the hand and fingers resulting from over-practice of the piano or another musical instrument requiring intense finger movement [Quartarone et al., 2006]. In patients with this disorder, functional imaging of the contralateral cortex has shown that the somatosensory map of the fingers is blurred in the musicians with dystonia compared to controls [Elbert et al., 1998]. This suggests that the somatosensory cortex is less able to distinguish precisely between individual finger movements, and an attempt to use the fingers leads to dystonic movements instead. Interestingly, selective injection of botulinum toxin into some of the fingers can compensate for the disrupted somatotopic map of the fingers and restore normal movement [Cole et al., 1995]. The immature brain may be especially susceptible to organizational changes in neuronal circuits that lead to acquired disorders associated with maladaptive or excessive plasticity.
A fourth aspect of plasticity that it is important to consider is its potential to create cell-specific selective vulnerabilities, leading to the understanding of plasticity as the brain’s Achilles’ heel. This concept is especially important in the developing brain, where different cells are undergoing dramatic shifts in their composition during growth and development. One example is the subplate neurons that are among the first to reach the cerebral cortex in the second trimester of pregnancy, providing targets for axons projected from neurons in the thalamus [McQuillen et al., 2003]. These neurons are selectively vulnerable to hypoxia-ischemia due to their enhanced expression of excitatory α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors during that time period [Nguyen and McQuillen, 2010]. Immature oligodendroglia are similarly more vulnerable to injury during the second trimester due to the subunit composition of their AMPA receptors that favors permeability to calcium [Volpe, 2009]. N-methyl-d-aspartate (NMDA) receptors also contribute to developmental vulnerability of immature oligodendroglia [Salter and Fern, 2005]. These age-dependent changes in excitatory amino acid receptors on neurons and oligodendroglia that favor excitation have a beneficial role in normal development, but they can also make cells vulnerable to death if they are accidentally exposed to hypoxia or ischemia [McDonald and Johnston, 1990]. In a similar way, developing neurons in the brain closer to term become more vulnerable to excitotoxicity mediated by NMDA receptors due to the fact that the receptors are genetically programmed at that time to be more excitable [Monyer et al., 1993]. NMDA receptors are easier to activate in the developing brain due to their subunit composition, which makes it easier to open their channels. However, this characteristic also creates a vulnerability to injury if the brain is exposed to hypoxia-ischemia, which can result in neuronal depolarization and calcium flooding through opening of the NMDA channels.
In addition, gamma-aminobutyric acid (GABA), which normally has an inhibitory effect on neuronal circuits in older children and adults, mediates excitation in the newborn period because of developmental changes in chloride pumps in GABAergic synapses [Ben-Ari, 2006]. Enhanced activity of the NKCC1 chloride transporter in the neonatal period leads to higher intraneuronal concentrations of chloride than are present later on, so that, when GABA opens chloride channels, the cation leaves the neuron rather than entering [Dzhala et al., 2005]. Therefore, GABA leads to depolarization rather than hyperpolarization, as it does at older ages. Later in development, activity of the NKCC1 pump declines, and expression of the NKCC2 pump, which pushes chloride out of neurons, increases, leading to lower baseline concentrations of chloride inside the neuron compared to outside, and to inhibition. Combined with the expression of NMDA receptors, which are more active and flux more calcium and sodium in the fetal and neonatal brain, developmental changes in the actions of GABA lead to the brain being more excitable. This enhanced excitability during development appears to play an important role in the establishment of normal neuronal circuitry, which is dependent on electrical activity [Penn and Shatz, 1999]. For example, production of growth factors, such as brain-derived neurotropic factor (BDNF), occurs in neurons and is dependent on neuronal activity [Lessmann and Brigadski, 2009]. Prolonged blockade of NMDA receptors, just like too much activity, can cause neuronal death because a minimum baseline amount of channel opening, with entry of calcium and sodium into neurons, is essential for neuronal survival [Hansen et al., 2004]. On the other hand, this bias towards excitement in the developing brain is probably responsible for the higher propensity for seizures in infants and children compared to adults, and the expression of some seizure types such as infantile spasms [Hablitz and Lee, 1992]. Therefore it is quite important for the molecular machinery responsible for balancing excitement and inhibition in the developing brain to operate properly in order to prevent it from becoming an Achilles’ heel during periods of stress, such as hypoxia-ischemia or status epilepticus [McDonald and Johnston, 1990].
Basic Mechanisms for Plasticity in the Developing Brain
In addition to these four broad types of plasticity in the developing brain, there are at least six basic cellular mechanisms for plasticity (Box 13-2). The earliest mechanism is the overproduction of neurons and glia from stem cells, and then reduction of this population by apoptosis in the fetus [Haydar et al., 1999]. Most neurogenesis ceases after birth, but it continues in selected niches of stem-cell production in the subventricular zone of the lateral ventricles and the subgranular zone of the dentate gyrus of the hippocampus [Kernie and Parent, 2010]. These restricted zones of neurogenesis may contribute to recovery from brain injuries throughout life. Additional mechanisms for plasticity are:







Mechanisms of Synaptic Plasticity
Synaptic plasticity is the most important mechanism for everyday activities such as learning and memory, and acquiring new skills (Figure 13-1). The strength of the signals between axons and dendrites across synapses can be increased or decreased through processes called long-term potentiation (LTP) or long-term depression (LTD), and these processes are strongly influenced by previous synaptic activity [Malenka and Nicoll, 1993]. The most prominent examples of LTP and LTD occur in excitatory glutamate synapses, which account for about 75 percent of all synapses in the brain. However, LTP and LTD are also present in many different types of neurons, including inhibitory GABAergic neurons and dopaminergic neurons [Nugent and Kauer, 2008]. One of the best examples of LTP is mediated by the NMDA-type glutamate receptors, which are voltage-dependent, meaning that NMDA channel opening is dependent in part on membrane depolarization [Gilland et al., 1998]. Channel opening is also dependent on occupancy of receptors for glutamate and glycine by these amino acids. When multiple excitatory axons depolarize the neuronal membrane, and glycine and glutamate occupy their receptors, the NMDA-activated calcium channel opens, allowing a pulse of calcium to enter the neurons and, in turn, activating a cascade of biochemical events that encode a memory of the event [Johnston, 2004]. The NMDA receptor has been referred to as a coincidence detector because it requires both membrane depolarization, stimulated by multiple presynaptic inputs, and activation of glutamate and glycine receptors to open its channel [Brown and Milner, 2003]. NMDA receptor-mediated LTP results in a “step up” in the signal mediated by the synapse during subsequent synapse activations, and it becomes easier to activate. Neuronal stimulation can also activate another type of a glutamate receptor, called a metabotropic gluR5 (mGluR5) receptor because it activates the second messenger phosphoinositol turnover, rather than opening an ion channel, as NMDA receptors do [Antion et al., 2008].
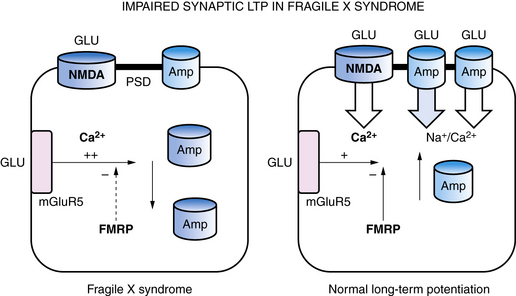
Within synapses, a variety of mechanisms contribute to LTP and LTD, including an increase or decrease in neurotransmitter release from presynaptic nerve terminals, a change in reuptake of neurotransmitter by astroglia that surround each synapse, a change in the number of neurotransmitter receptors in the postsynaptic membrane, changes in the flux of calcium and other ions through membrane channels, and an activation of intracellular signaling cascades [Citri and Malenka, 2008]. One of the common mechanisms that synapses use to increase or decrease synaptic strength is to alter the number of receptors in the postsynaptic membrane through a process called “receptor trafficking” (Figure 13-2) [Keifer and Zheng, 2010; Conboy and Sandi, 2010]. In this process, receptors shuttle back and forth between the cytoplasm, where they do not have access to neurotransmitter in the synaptic cleft, postsynaptic density, and postsynaptic membrane, where they are positioned to interact with neurotransmitter. One excitatory receptor that is heavily regulated by receptor trafficking in synapses is the AMPA receptor, which is the ionotropic glutamate receptor that carries most of the fast excitatory current in the brain. AMPA receptors shuttle between the dendritic cytoplasm, where they are inaccessible to glutamate, to the postsynaptic membrane, where they can be stimulated by glutamate [Keifer and Zheng, 2010]. The number of receptors within the synaptic membrane determines the strength of the synapse when it is activated. Accordingly, LTP is associated with an increase in AMPA receptors inserted into the postsynaptic membrane, while a decrease in AMPA receptors in the postsynaptic membrane is associated with LTD. LTP can be induced by rapid stimulation of the postsynaptic membrane, leading to voltage-dependent opening of NMDA receptors and trafficking of AMPA receptors from the cytoplasm into the postsynaptic membrane. This makes the synapse better able to conduct a signal, in effect creating a fragment of a memory about that synapse’s past experience. The mGluR5 metabotropic glutamate receptor also controls AMPA receptor trafficking, and an increase in mGluR5 activity leads to internalization of AMPA receptors, LTD, and weaker synaptic strength. In experimental models of fragile X syndrome, the activity of mGluR5 receptors has been reported to be increased, along with enhanced synaptic LTD, and antagonists of the mGluR5 receptor are able to restore LTP and improve abnormal behaviors and cognition (see Figure 13-2) [Muddashetty et al., 2007]. Fragile X syndrome is one example of a developmental brain disorder that can be understood at the level of the synapse. Synapses are thought to be the primary site of memory storage across large networks in which the strength of individual synapses is increased or decreased by the processes of LTP or LDP.
Production of nerve growth factors like BDNF is another mechanism by which previous synaptic activity can lead to plasticity in neuronal connections. BDNF is produced in neurons in response to neuronal activity, and it has been shown to increase and stabilize synaptic connections, as well as contribute to their maturation and integration into complex neuronal circuits [Yoshii and Constantine-Paton, 2010]. Learning has also been shown to increase neurotrophin signaling through the TrκB receptor, which mediates the actions of BDNF in the hippocampus in mice. BDNF has also been associated with cortical plasticity in humans. Kleim et al. reported that individuals with a val66met polymorphism (i.e., methionine codon substitution for valine codon at position 66 in the BDNF gene) showed reduced ability to reorganize their cortical motor map in response to a training exercise involving the fingers of one hand [Kleim et al., 2006]. In vitro studies showed that this mutation impairs the secretion of BDNF from neurons, but its expression is normal. McHughen et al. used functional magnetic resonance imaging (fMRI) to show that subjects with this BDNF mutation made more errors and had poorer retention on a driver-based learning task, and had altered short-term plasticity [McHughen et al., 2010]. BDNF genotype has also been shown to be associated with anxiety and memory problems, as well as with impaired functional connectivity from the hippocampus to the amygdala, insula, and striatal regions in children studied with functional connectivity (FC) analysis on data obtained through whole-brain fMRI [Thomason et al., 2009].
Plasticity of Dendrites and Dendritic Spines
Activity-dependent plasticity in excitatory synapses is associated with physical changes in dendritic spines that are thought to reflect information storage (Figure 13-3) [Hotulainen and Hoogenraad, 2010]. Immature dendritic spines are long with a small head, but repeated activity during development, including LTP, leads to shortening of spine shafts and enlargement of spine heads. New learning in animals also has been shown to be associated with localized enlargement of some dendritic spines and loss of others. Remodeling of dendrite morphology occurs continuously in association with synaptic plasticity, and is mediated by the actin-rich cytoskeleton that fills dendrites [Hotulainen and Hoogenraad, 2010]. The actin cytoskeleton within spines containing excitatory synapses provides support for the postsynaptic density (PSD), which anchors glutamate receptors and cell adhesion molecules that span the synaptic cleft (see Figure 13-3). NMDA receptors within the PSD regulate actin signaling pathways within an endocytic zone just below the PSD. The purpose of this zone is to recycle the synaptic pool of AMPA-type glutamate receptors involved in trafficking between the cytoplasm and the PSD. Therefore, the actin cytoskeleton plays an important role in LTP, and it is not surprising that mutations in proteins involved in regulation of this cytoskeleton, such as small Rho and Ras GTPases, alter spine morphology and cause cognitive disorders and autistic spectrum disorders [Pinto et al., 2010]. Similarly, key proteins that make up the PSD and the scaffolding proteins beneath the PSD, such as SHANK and the cell adhesion proteins – neurexins, neuroligins, and cadherins – are associated with clinical disorders that impair learning (Table 13-2) [Durand et al., 2007; Johnston et al., 2001].
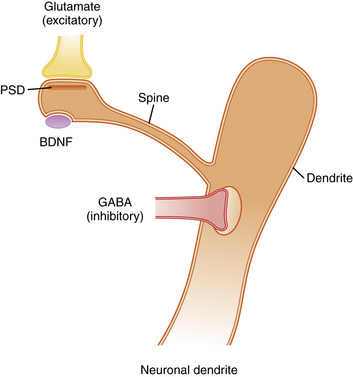
Excitatory axons that contain glutamate synapse on the tips of dendritic spines, while inhibitory axons that contain gamma-aminobutyric acid (GABA) synapse on the shafts of dendrites. Excitatory synapses are asymmetric and contain a postsynaptic density (PSD), which anchors neurotransmitter receptors and a variety of other proteins that provide scaffolding for receptors (see Table 13-2). Typically, each dendritic spine contains only one excitatory synapse, while the shaft of the dendrite contains multiple inhibitory GABAergic synapses. This arrangement allows a single dendrite to receive and process information from many excitatory synapses on its dendritic spines. However, the summed output for each dendrite is controlled by inhibitory receptors on the shaft. The morphology of dendritic spines is controlled in part by age and activity, so that young spines are long and thin with narrow spine heads, while older spines that have experienced considerable activity become shorter with a broader spine head. These shape changes are mediated by an actin cytoskeleton that fills the spines, allowing them to change shape in response to synaptic activity. Abnormal morphology of dendrites, spines, and PSD proteins is commonly associated with syndromes that include intellectual disability, autism, and behavior disorders. BDNF, brain-derived neurotropic factor.
Table 13-2 Proteins Associated with the Postsynaptic Density of Excitatory Synapses and Possible Role in Neurologic Disease
| Protein | Function | Connection with Neurological Disease |
|---|---|---|
| α-Actinin | Spine morphogenesis | Associated with faster-sprinting athletes |
| CaMKIIα/β | Activates gene expression | Major pathway to gene expression |
| Synapsins | Regulate neurotransmitter release | Seizures, cognitive disorders |
| N-cadherin | Adhesion molecule | Deafness, Usher’s syndrome, epilepsy |
| Protocadherin | Adhesion molecule | Seizures, intellectual disability in females |
| GluR1,2,3 | AMPA receptors | Cognitive impairment |
| Homer | Bind to mGluR5 receptors | Interacts with mGluR5 in fragile X syndrome |
| Neuroligin | Binds to presynaptic neurexin | Associated with autism |
| NR1-2A,B | NMDA glutamate subunits | Involved in learning and memory, autoimmune encephalopathy |
| PSD-95 | Regulates synaptic plasticity | Major regulator of synapse development |
| Shank1/2/3 | Scaffolding protein, promotes maturation of synapse | Associated with autism |
| SynGAP Ras GTPase activating protein | Controls signaling between synapse and nucleus to activate transcription | Regulates MAP kinases |
In addition to the microscopic changes in synapses that occur during development, Huttenlocher has shown that there are large expansions of the number of synapses that occur during childhood, so that, by 2 years of age, there are approximately twice the number of synapses that will be present by age 16, when the number approximates adult levels [Huttenlocher, 1997]. This wave of expansion, overshoot, and then pruning begins earlier in the occipital lobe than in the frontal lobe, and pruning of synapses continues longest in the frontal lobes. Chugani and colleagues have shown, using positron emission tomography (PET), that glucose consumption of the cortex during childhood follows a curve that is similar to synapse number, consistent with the fact that synapses are the major site of energy consumption in the brain [Chugani et al., 1987]. These dynamic changes in synapse number probably support brain plasticity and growth of cognitive power in children because they allow synapses to be chosen from a surplus fairly late in childhood, based on synaptic activity provided by experience, and provide for redundancy of synapses if some injury does occur. Shaw et al. provided some evidence that these dynamic changes in synapse number may be functionally relevant in a study that used sequential MRI to measure cortical thickness in children of different intelligence levels [Shaw et al., 2006]. They found that children with superior intelligence had, on average, a higher peak and greater duration of peak cortical thickness than children of lower intelligence. The major differences in cortical thickness between groups of children with different levels of intelligence were found in the frontal cortex, which is the area that normally matures last. These results, as well as the earlier studies of glucose consumption and postmortem synaptic counting, suggest that cortical maturation in children moves like a wave of synapse proliferation and then pruning from back to front, and a higher, more prolonged wave is correlated with higher intelligence [Johnston, 2004]. These studies suggest that neuronal plasticity at multiple levels contributes to learning, memory, and higher intellectual function.
Intracellular Signaling Cascades and Gene Transcription
The changes in synapses, dendrites, and dendritic spines that accompany information storage in the developing brain require gene transcription and translation of proteins (Figure 13-4) [Johnston, 2004]. Several signaling cascades from the synaptic membrane to the nucleus are especially important in learning and memory; these include the calcium/calmodulin kinase II (CaMKII) pathway, the Ras-mitogen activated protein kinase II (MAP kinase II) pathway, and the protein kinase A (PKA) pathway. These pathways can all stimulate phosphorylation of the master transcription factor, cyclic adenosine monophosphate response element binding protein (CREB), which activates transcription of many genes that are important for learning, memory, and synaptic plasticity [Shalin et al., 2006]. CREB works closely with CREB binding protein (CBP), which has intrinsic histone acetylase activity that is required for DNA to unwind from histones in order to allow gene transcription [Roth et al., 2010]. The action of CREB is antagonized by transcriptional repressor proteins, including thyroid hormone receptor without thyroid hormone and MeCP2, the transcriptional factor that is mutated in Rett’s syndrome. MeCP2 recruits transcriptional repressor Sin3 to block transcription of BDNF, although it has been shown also to activate many genes in the hypothalamus (see Figure 13-4) [Kaufmann et al., 2005]. Many other signaling cascades probably participate in activity-dependent transcription of genes that are involved in learning and memory and other aspects of brain plasticity.
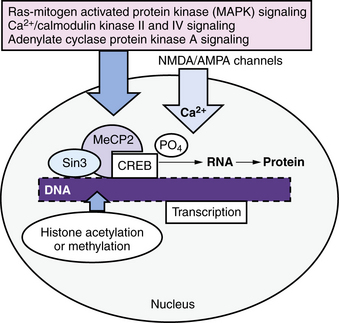
This cellular machinery allows information to be stored for the long term within a distributed network of synapses, and it is essential for long-term memory. The pink box shows several important signaling cascades that activate transcription by phosphorylation of the master memory transcription factor, cyclic adenosine monophosphate response element binding protein (CREB). CREB enables the transcription of multiple genes and is assisted by CREB binding protein (CBP, not shown), which has histone acetylase activity that opens up DNA to allow transcription. Another important transcription factor is MeCP2, a transcriptional repressor that recruits other repressors such as Sin3, and controls activity-dependent transcription of brain-derived neurotropic factor (BDNF). Mutations in MeCP2 are the main cause of Rett’s syndrome. Mutations in proteins involved in activity-dependent signaling within neurons are important causes of intellectual disability, autism, behavior disorders, and epilepsy (see Figure 13-2). AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-d-aspartate.
Adaptive Plasticity
Adaptive plasticity includes the brain’s ability to learn and remember information, and its capacity to change in response to practicing physical skills, such as playing the piano or a sport, as well as the ability to recover from a brain injury. The developing human brain has a greater capacity for adaptive plasticity than the adult brain, and several interesting examples have been reported. One of the earliest studies of morphologic changes in the developing brain in response to environment involved the ocular dominance columns in the visual cortex of monkeys in which one eye was occluded. These studies showed that reducing the visual input from one eye led to a reduction in thalamocortical axons from that eye, while the territory assigned to axons from the opposite eye expanded [Tropea et al., 2008]. This process corresponds to the clinical scenario in which young children with strabismus develop amblyopia in one eye. Amblyopia can be corrected before about 10 years of age if the opposite eye is patched to restore balance in the visual inputs from the two eyes. Studies of plasticity in the occipital cortex of persons who were blind from birth have also provided insights into adaptive plasticity in the developing brain. Functional imaging using PET demonstrated that adults who were blind from birth activated the visual cortex in the occipital lobe, as well as somatosensory cortex representing their Braille-reading fingers [Sadato et al., 1996]. Other studies have shown that the occipital cortex of congenitally blind subjects is activated by nonvisual tasks, such as auditory processing, semantic processing, and verbal memory. Occipital lobe activity in these subjects seems to be functionally important for these nonvisual tasks because occipital strokes and temporary inactivation with repetitive transcranial magnetic stimulation (rTMS) have been shown to impair nonvisual skills such as Braille reading [Kupers et al., 2007]. It has also been shown that auditory association cortex is appropriated for visual processing in deaf children whose disability began at an age of less than 14 years [Weeks et al., 2000]. These examples demonstrate plasticity within one modality (vision) in response to differences between the eyes, and transmodal plasticity in which cortex assigned to another modality can be re-assigned if its intended sensory input is lost at an early age.
Although visual plasticity was one of the first types to be examined in humans, plasticity of the somatosensory cortex in response to the use of hands in musicians has also been explored. Taub and colleagues compared the differences in the contralateral somatosensory cortical maps for the fingers and thumb of the left hand between musicians who had played stringed instruments for many years versus nonplayers [Elbert et al., 1995]. Using a technique called magnetic source imaging to monitor effects of finger stimulation, they found that the expanse of cortex assigned to the fingers and thumb of the left hand used to finger the strings, as well as the strength of the signal, was greater than for nonplayers in direct proportion to the number of years they had been practicing. These data suggest that longer practice shapes and expands the cortical map in proportion to the amount of activity in the left hand. In another study of musicians, Schlaug et al. also reported that intense musical training for 29 months in children of 5–7 years of age, who played either piano or stringed instruments, was associated with an increase in volume of the anterior mid-body of the corpus callosum [Schlaug et al., 2009], compared to the same area of the corpus callosum in non-musician controls or musicians who practiced infrequently. The mid-body connects premotor and supplementary motor areas. Like the study of changes in the somatotopic map for string players, these results support the hypothesis that engagement of both hemispheres is important for musicians to process and play music, and that practice has a structural impact on the brain. Bengtsson et al. also found that extended piano practicing from childhood onward was strongly correlated with improved white-matter organization in the pyramidal and other white-matter tracts, as measured by increased fractional anisotropy on diffusion tensor imaging (DTI) [Bengtsson et al., 2005]. These results are consistent with classic experiments by Buonomano and Merzenich in primates, showing that the somatosensory map can be expanded by increased somatosensory stimulation of the fingers [Buonomano and Merzenich, 1998]. Nudo et al. showed that sensory stimulation and retraining of the weak hand in a primate model of focal stroke prevented the shrinkage of the somatosensory area assigned to that hand that normally occurred [Nudo and Milliken, 1996]. As mentioned above, experiments in which subjects with a val66met polymorphism in the BDNF gene showed reduced ability to reorganize their cortical motor map in response to a training exercise involving the fingers of one hand show that adaptive plasticity is partially under genetic control [Kleim et al., 2006].
These results also agree with studies of the “barrel” field cortex in mice and rats, which represents a map of the animal’s whiskers across the tangential expanse of somatosensory cortex [Nishimura et al., 2002]. Rodents depend on whiskers to navigate in the dark places where they live, as their visual system is less developed. Many experiments have shown that clipping a row of whiskers or enhancing whisker activity in neonatal rodents results in reassignment of the cortical map assigned to these whiskers. Specific neurotransmitters, such as glutamate, the inhibitory neurotransmitter GABA, and acetylcholine within the nucleus basalis projection from the basal forebrain to the cerebral cortex, have been shown to be important in this form of activity-dependent cortical plasticity [Inan and Crair, 2007]. Similar experiments, focused on the area of cortex that receives auditory inputs in developing rodents, have shown that a tonotopic map corresponding to sounds of different pitches is distributed across the surface of cortex, and can be altered markedly by exposing the animals to repeated monotonal sounds, together with a behavioral program and stimulation of the nucleus basalis [Barak et al., 2003]. These data are in agreement with those from children described above. The experiments in immature rodents show that structural brain plasticity is highly conserved across species and probably contributes to the enhanced functional plasticity seen in children.
Plasticity and Epilepsy in Children
A number of studies suggest that chronic epilepsy in children can disrupt learning and memory, as well as motor function [Holmes et al., 2002]. On the other hand, the child’s brain can reorganize in response to epilepsy or extensive surgery for epilepsy to a greater extent than that of adults. Several studies indicate that motor and sensory functions in areas affected by focal epilepsy can be transferred from one hemisphere to the other. Boatman et al. reported a series of children who gained abilities in receptive language when a left hemispherectomy was performed for Rasmussen’s syndrome in children aged 7–14 years [Boatman et al., 1999]. Hertz-Pannier et al. also reported a case of 9-year-old boy who had considerable improvement in speech after receiving a cortical resection in the left hemisphere for Rasmussen’s encephalopathy [Hertz-Pannier et al., 2002]. In that case, speech improvement was associated with increased activity on fMRI scanning in the right hemisphere and both frontal lobes, suggesting that the epileptic activity in the left hemisphere led to migration of speech programs to the opposite side but was also impairing their expression. In this case, we can hypothesize that chaotic electrical activity impairs the expression of plasticity associated with movement of speech to the right hemisphere, and removal of the seizure focus is required to regain speech. Lippe et al. also recently reported that young children who underwent parieto-occipital lobe resection for intractable epilepsy due to cortical dysplasia had improved verbal and neuropsychological outcome when evaluated 3–7 years later [Lippe et al., 2010]. Roulet-Perez et al. also reported on cognitive functioning in a group of young children who had early surgery for severe intractable epilepsy, and found that cessation of epileptic activity can be associated with substantial cognitive gains, although not in all children [Roulet-Perez et al., 2010].
Plasticity in Older Children and Adults
Although children appear to have a greater degree of brain plasticity than adults, there is also evidence that the adult brain undergoes structural changes in response to experience and motor practice. For example, an MRI study of licensed taxi drivers in London found that their posterior hippocampi were larger than those in control subjects who did not drive taxis, and enlargement in the right, but not the left, hippocampus was directly related to their time as a taxi driver [Maguire et al., 2000]. The posterior hippocampus is an area that is associated with spatial representation of the environment, and this study suggests that a “mental map” of the city is stored there. Another study of young adults used voxel-based morphometry to examine their brains before and after they received instruction in learning to juggle for the first time [Draganski et al., 2004]. This study found an increase in parietal-occipital cortical gray matter assigned to visual motor processing after 3 months of practice compared with before practice. These changes persisted for several weeks after practice ended. Similar studies have revealed increases in gray matter in the auditory and visual-spatial cortex in professional musicians compared to non-musicians, increases in parietal lobe cortex in professional mathematicians, and thickening in the cortex of medical students studying for examinations [Draganski et al., 2006; Aydin et al., 2007].
Facilitating Adaptive Plasticity with Therapy Programs
Anecdotal and scientific evidence for plasticity in the nervous system has led to attempts to harness it to improve recovery from a variety of brain and spinal cord injuries and other disorders. One approach is generally referred to as constraint-based therapy or constraint-induced movement therapy (CIMT), used for patients with hemiparesis [Gauthier et al., 2008]. Another approach used for patients with spinal cord injury uses programmed, multi-electrode, direct stimulation of muscles above and below the level of injury in paralyzed individuals with spinal cord injury [Sadowsky and McDonald, 2009]. This type of stimulation simulates walking in order to improve muscle tone and cardiovascular function, as well as to enhance remyelination and neural stem-cell production at the site of partial spinal cord injury.
CIMT was developed for patients with hemiparesis for unilateral lesions such as stroke, and is based on the theory that using the good arm impairs movement of the hemiparetic side. Accordingly, the theory suggests that the good side should be constrained while movement of the hemiparetic arm is encouraged. In CIMT sessions, the less impaired hand is restrained using a mitt over the hand or a cast covering the arm and hand, and a therapist directs repetitive movement of the hemiparetic side, along with behavioral shaping to encourage real-world use. EXCITE (Effect of Constraint-Induced Movement Therapy on Upper Extremity Function) was one of the first randomized controlled trials of this type of therapy, and it evaluated adults who had strokes more than 6–9 months prior to intervention [Park et al., 2008]. This trial demonstrated clinically relevant improvement in arm movement that lasted for more than a year. Subsequent trials in adults with stroke have shown that a behavioral program to promote use of the arm in real-world activities is important for a beneficial effect. Similar studies in rodents suggested that a behavioral program was important for changing the tonotopic map for sound, and this could be substituted by stimulation of the nucleus basalis that projects to the cortex [Buonomano and Merzenich, 1998].
MRI studies using voxel-based morphometry have shown that CIMT with a behavioral intervention produced bilateral gray matter increases in sensory and motor areas, as well as in the hippocampus [Gauthier et al., 2008]. In contrast to this study of CIMT, rehabilitation trials using bilateral arm movement while walking on a treadmill with auditory cueing did not improve arm use in adults with stroke, although it benefited their walking and cardiovascular fitness [Luft et al., 2004, 2008]. CIMT is also being applied to children with cerebral palsy with hemiparesis and has had some success [Cope et al., 2010; Eliasson et al., 2005]. One of the determinants of efficacy of CIMT in children with hemiplegia appears to be whether or not there is persistence of an ipsilateral corticospinal tract. Ipsilateral corticospinal tracts are normally present in the fetus and newborn, along with crossed corticospinal tracts, but then regress in infancy. Eyre et al. used TMS to study the development of the corticospinal tracts in infants who had unilateral or bilateral cerebral injuries in the neonatal period, and found that those with strong persistence of an ipsilateral CST had worse motor outcomes than those in which the ipsilateral tract regressed and the contralateral tract persisted [Eyre et al., 2007; Eyre, 2007]. Kuhnke et al. studied children with congenital hemiparesis and found that those with a single functional contralateral corticospinal tract responded better to CIMT than those with a persistent ipsilateral tract. Walther et al. recently reported a group of children with hemiparesis due to perinatal stroke, whose hand movements improved after CIMT [Walther et al., 2009]. TMS stimulation of the hemisphere contralateral to the hemiparetic limb in this group produced a stronger motor-evoked amplitude in the hemiparetic hand after CIMT treatment, suggesting increased synaptic excitability in the corticospinal tract pathway [Kuhnke et al., 2008]. These data suggest that CIMT is able to enhance plasticity and functional improvement in children with hemiparetic cerebral palsy, especially those who have an intact contralateral CST without input from a persistent ipsilateral CST.
Brain Stimulation and Adaptive Plasticity
As already discussed, brain plasticity is an activity-dependent process in which neurons that receive greater stimulation from the primary senses or other parts of the brain build larger, stronger network connections, but neurons deprived of activity lose their connections [Fregni and Pascual-Leone, 2007]. A relatively new form of therapy based on neural plasticity theory is referred to as electrical brain stimulation, and uses the techniques of TMS and transcranial direct current stimulation (tDCS) to control neuronal excitability in the brain [Fregni and Pascual-Leone, 2007]. TMS refers to the pulsatile magnetic stimulation delivered through the scalp and skull by an electromagnetic coil specially constructed for this purpose. The TMS coil is held over the scalp and generates an electrical current within the cortex that flows in the opposite direction to the current in the coil. The TMS pulse frequency can be controlled and pulse sequences have been discovered empirically that can be either inhibitory or excitatory for the cortex. This technique has been found generally to be safe in children and adults. One theory for the mechanism of impairment in patients with a hemiparesis after a stroke is that reduced function of the damaged hemisphere results in part from excessive neuronal inhibition from the opposite undamaged hemisphere. This is supported by reports that delivering inhibitory TMS sequences to the good hemisphere can improve function controlled by the opposite injured hemisphere. Excitatory TMS sequences delivered to the injured hemisphere, combined with paired retrograde stimulation of the same hemisphere via the contralateral median nerve, have also been found to produce functional improvement in patients with hemiparesis [Celnik et al., 2007]. The technique of tDCS uses a device that provides a constant level of direct current through the skull and brain, and has been shown to enhance neuronal plasticity in a variety of paradigms, including motor, language, and memory performance, as well as in the ability to form and retain motor memories [Galea and Celnik, 2009; Fregni and Pascual-Leone, 2007]. Experiments in mice show that tDCS depends on activity-dependent secretion of BDNF and augmentation of synaptic plasticity associated with TrκB receptor activation [Fritsch et al., 2010]. These electrical techniques appear to hold promise for improving outcome in children and adults with brain injury by enhancing brain plasticity.
Impaired Plasticity due to Genetic or Acquired Disorders
Many genetic and acquired disorders disrupt plasticity processes in the brain, leading to impaired learning, memory, behavior, and motor function, as well as brain and somatic growth abnormalities. Table 13-1 lists some prominent examples of disorders in which plasticity mechanisms are targeted. Neurofibromatosis 1 (NF-1) is a dominantly inherited disorder that is one of the most common single-gene abnormalities and is known for its café au lait skin lesions, plexiform neuromas, optic gliomas, skeletal manifestations, and learning problems. It is caused by mutations in neurofibromin, an oncogene and GTPase activating protein (GAP) that regulates the activity of Ras, a G-protein that mediates membrane receptor-mediated activation of MAPK signaling (see Figure 13-4). The Ras-MAPK signaling cascade normally leads to learning and memory formation and cellular growth, but excessive upregulation can lead to tumor formation and impaired cognition. At the synaptic level, mice with neurofibromin mutations show memory loss on water maze testing, and impaired LTP that is related to hyperactivity of inhibitory GABAergic interneurons [Staley and Anderson, 2009]. Cui et al. showed that GABAergic neurons in these animals released excessive GABA from inhibitory synapses due to phosphorylation of synapsin 1, a protein that regulates neurotransmitter release [Cui et al., 2008]. Administration of low concentrations of a GABA antagonist has been shown to improve their memory problems and impaired LTP [Costa et al., 2002]. Lovastatin, an approved drug used to treat high cholesterol in adults, has also been shown to improve cognitive function by reducing elevated Ras-MAPK activity, and it is being studied as a treatment for cognitive and other manifestations of NF1 [Li et al., 2005].
Tuberous sclerosis (TSC) shares some features of NF-1, in that it is also an autosomal-dominant neurocutaneous disorder that results from overactivity in a signaling pathway that links extracellular signals, such as glucose, glutamate, other amino acids, and insulin, with pathways that regulate intracellular protein synthesis, cell growth, proliferation, and survival [Crino, 2010; Costa et al., 2002]. Two-thirds of individuals with TSC harbor sporadic mutations in one of two tumor suppressor genes, hamartin (TSC1) or tuberin (TSC2). These mutations result in upregulation of the small GTPase Rheb and its downstream binding partner, mammalian target of rapamycin (mTOR), an enzyme that regulates cell growth and transcription. Upstream signals that act on TSC proteins come from the Ras-MAPK and the phosphoinositide-3-kinase pathways. TSC is associated with skin and eye manifestations, as well as epilepsy, intellectual disability, and autism in a high percentage of cases. Subependymal nodules and tubers are typically present in the brain, and tumors or hamartomas are often found in the lung, kidney, and heart. mTOR has been implicated in synaptic plasticity through experiments showing that its inhibitor rapamycin can inhibit LTP and LTD in snails, crayfish, and mice [Ehninger et al., 2009]. Mice with mutations in Tsc1 and Tsc2 have abnormalities in synaptic plasticity, as well as in dendritic aborization, axonal outgrowth, neuronal migration, and behavior [Ehninger et al., 2009; Shilyansky et al., 2010]. It is interesting that a model of TSC has also been produced with homozygous mutations in the Tsc1 gene in glia only, and these mice have low expression of the glutamate-1 transporter [Zeng et al., 2007]. The hypothesis that mTOR controls synaptic plasticity in brain is also supported by recent data showing that upregulation of mTOR by the anesthetic ketamine increases spine density and synaptic activity associated with an antidepressant effect in rats [Li et al., 2010]. Rapamycin is a clinically approved drug for immunosuppression for organ transplantation, and has been used in clinical trials to reduce tumor size in patients with TSC [Wong, 2010].
Impaired Plasticity in Fragile X and Rett’s Syndromes
Fragile X syndrome and Rett’s syndrome are X-linked disorders that cause intellectual disability, autistic-like behaviors, and other neuropsychiatric abnormalities, as well as abnormalities in synapse structure and plasticity in the brain. As described earlier, FraX is caused by a reduction in the FMRP protein, which binds to RNAs in the synapse, leading to abnormalities in translation of proteins regulated by synaptic activity (see Figure 13-2) [Penagarikano et al., 2007]. In contrast, Rett’s syndrome is due to mutations in the MeCP2 transcription factor, which regulates gene transcription in the nucleus [Amir et al., 1999]. The primary role of MeCP2 is to silence expression of certain genes, including the growth factor BDNF; UBE3A, the gene involved in Angelman’s syndrome; and the homeobox gene DLX5, which stimulates development of GABAergic neurons in the brain (see Figure 13-4) [Zhou et al., 2006]. Upregulation of glucocorticoid-regulated genes has also been reported in a mouse model of Rett’s syndrome. In resting neurons, MeCP2 acts like a brake for BDNF transcription, but when neurons are activated, calcium-mediated phosphorylation of MeCP2 leads to de-repression of BDNF transcription and increased protein [Nuber et al., 2005; Stornetta and Zhu, 2010]. Both FraX and Rett’s syndrome are associated with abnormalities in dendritic spines and synapses and with synaptic plasticity [Cruz-Martin et al., 2010; Fukuda et al., 2005]. In FraX, there is an excess of dendritic spines that are abnormally long and slender, probably reflecting their relative immaturity secondary to diminished excitatory neurotransmission (see Figure 13-2) [Pfeiffer et al., 2010]. Huber et al. also reported that LTD is selectively enhanced in the FraX mouse [Huber et al., 2002]. Several experimental drugs that inhibit mGluR5 receptors, such as fenobam, or their downstream signaling pathways, such as lithium or minocycline, have been reported to promote spine maturation and improve behavior in animal models [Berry-Kravis et al., 2008, 2009; Bilousova et al., 2009]. Some functional benefits have also been reported for these drugs in early clinical studies in adults with FraX [Wang et al., 2010]. In Rett’s syndrome, the immature brain contains an excess of glutamate receptors that decrease later in life, and synaptic contacts in mice with Rett’s syndrome have been reported to be immature, with reduced cross-sectional length of PSDs [Fukuda et al., 2005; Johnston et al., 2005; Blue et al., 1999]. Levels of glutamate have also been reported to be elevated in girls with Rett’s syndrome, consistent with the seizures and hyperkinetic behavior they show [Horska et al., 2009]. Studies in Rett mice also indicate that neurons develop to the point of beginning to make synaptic connections but then cannot stabilize them [Belichenko et al., 2008; Smrt et al., 2007; Palmer et al., 2008]. Learning and memory, as well as synaptic plasticity as measured by LTP and LTD, are diminished in the hippocampus in a mouse model of Rett’s syndrome [Moretti et al., 2006; Asaka et al., 2006]. Both insulin like growth factor (IGF-1) and BDNF have been reported partially to rescue the phenotype of Rett’s syndrome [Kline et al., 2010; Tropea et al., 2009] Therefore, these two disorders represent variations on the theme of X-linked plasticity disorders.
Similar disorders that involve cell signaling pathways involved in neuronal plasticity are listed in Table 13-1, and others are likely to be described in the future. In addition to lesions in intracellular signaling cascades or defects in transcription or translation, recent genome microarray studies of children with intellectual disability or autistic spectrum disorders have found deletions and other copy number variations that include genes for glutamate receptors and other synaptic proteins [Miller et al., 2010]. These include presynaptic proteins or those localized to the PSD and associated scaffolding proteins, including SHANK2, SHANK3, SHANK4, SYNGAP1, NLGN3, NLGN4X, NRXN1, or DLGAP2. The Autism Genome Project Consortium (AGP) found rare gene deletions in individuals with autism that are enriched in functional networks, including those for GTPase/Ras signaling, cell proliferation and motility, central nervous system development, and cytoskeleton organization [Pinto et al., 2010] These genes can often be linked to both autism and intellectual disability.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Amir H., van der W.L., Wan R., et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein. 1999. 2:185–186
Antion M.D., Hou L., Wong H., et al. mGluR-dependent long-term depression is associated with increased phosphorylation of S6 and synthesis of elongation factor 1A but remains expressed in S6K-deficient mice. Mol Cell Biol. 2008;28:2996-3007.
Asaka Y., Jugloff D.G., Zhang L., et al. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217-227.
Aydin K., Ucar A., Oguz K.K., et al. Increased gray matter density in the parietal cortex of mathematicians: a voxel-based morphometry study. Am J Neuroradiol. 2007;28:1859-1864.
Barak B., Chang M., Davisson M.T., et al. Progressive degradation and subsequent refinement of acoustic representations in the adult auditory cortex. 2003. 10765–10775
Belichenko N.P., Belichenko P.V., Li H.H., et al. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. J Comp Neurol. 2008;508:184-195.
Ben-Ari Y. Basic developmental rules and their implications for epilepsy in the immature brain. Epileptic Disord. 2006;8:91-102.
Bengtsson S.L., Nagy Z., Skare S., et al. Extensive piano practicing has regionally specific effects on white matter development. Nat Neurosci. 2005;8:1148-1150.
Berry-Kravis E., Hessl D., Coffey S., et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266-271.
Berry-Kravis E., Sumis A., Hervey C., et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29:293-302.
Bilousova T.V., Dansie L., Ngo M., et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94-102.
Blue M.E., Naidu S., Johnston M.V. Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol. 1999;45:541-545.
Boatman D., Freeman J., Vining E., et al. Language recovery after left hemispherectomy in children with late-onset seizures. Ann Neurol. 1999;46:579-586.
Brown R.E., Milner P.M. The legacy of Donald O. Hebb: more than the Hebb synapse. Nat Rev Neurosci. 2003;4:1013-1019.
Buonomano D.V., Merzenich M.M. Cortical plasticity: from synapses to maps. Annu Rev Neurosci. 1998;21:149-186.
Celnik P., Hummel F., Harris-Love M., et al. Somatosensory stimulation enhances the effects of training functional hand tasks in patients with chronic stroke. Arch Phys Med Rehabil. 2007;88:1369-1376.
Chugani H.T., Philip A.G., Maziotta J.C.. Positron emission tomography study of human brain functional development. 1987. 487–497
Citri A., Malenka R.C. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18-41.
Cole R., Hallett M., Cohen L.G. Double-blind trial of botulinum toxin for treatment of focal hand dystonia. Mov Disord. 1995;10:466-471.
Conboy L., Sandi C. Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology. 2010;35:674-685.
Cope S.M., Liu X.C., Verber M.D., et al. Upper limb function and brain reorganization after constraint-induced movement therapy in children with hemiplegia. Dev Neurorehabil. 2010;13:19-30.
Costa R.M., Federov N.B., Kogan J.H., et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526-530.
Crino P.B. The pathophysiology of tuberous sclerosis complex. Epilepsia. 2010;51(Suppl 1):27-29.
Cruz-Martin A., Crespo M., Portera-Cailliau C. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci. 2010;30:7793-7803.
Cui Y., Costa R.M., Murphy G.G., et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. 2008. pp 549–560
Dileone M., Profice P., Pilato F., et al. Enhanced Human Brain Associative Plasticity in Costello Syndrome. J Physiol. 2010.
Draganski B., Gaser C., Busch V., et al. Neuroplasticity: changes in grey matter induced by training. Nature. 2004;427:311-312.
Draganski B., Gaser C., Kempermann G., et al. Temporal and spatial dynamics of brain structure changes during extensive learning. J Neurosci. 2006;26:6314-6317.
Durand C.M., Betancur C., Boeckers T.M., et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25-27.
Dzhala V.I., Talos D.M., Sdrulla D.A., et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205-1213.
Ehninger D., de Vries P.J., Silva A.J. From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J Intellect Disabil Res. 2009;53:838-851.
Elbert T., Candia V., Altenmuller E., et al. Alteration of digital representations in somatosensory cortex in focal hand dystonia. Neuroreport. 1998;9:3571-3575.
Elbert T., Pantev C., Wienbruch C., et al. Increased cortical representation of the fingers of the left hand in string players. Science. 1995;270:305-307.
Eliasson A.C., Krumlinde-sundholm L., Shaw K., et al. Effects of constraint-induced movement therapy in young children with hemiplegic cerebral palsy: an adapted model. Dev Med Child Neurol. 2005;47:266-275.
Eyre J.A. Corticospinal tract development and its plasticity after perinatal injury. Neurosci Biobehav Rev. 2007;31:1136-1149.
Eyre J.A., Smith M., Dabydeen L., et al. Is hemiplegic cerebral palsy equivalent to amblyopia of the corticospinal system? Ann Neurol. 2007;62:493-503.
Fregni F., Pascual-Leone A. Technology insight: noninvasive brain stimulation in neurology-perspectives on the therapeutic potential of rTMS and tDCS. Nat Clin Pract Neurol. 2007;3:383-393.
Fritsch B., Reis J., Martinowich K., et al. Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron. 2010;66:198-204.
Fukuda T., Itoh M., Ichikawa T., et al. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J Neuropathol Exp Neurol. 2005;64:537-544.
Galea J.M., Celnik P. Brain polarization enhances the formation and retention of motor memories. J Neurophysiol. 2009;102:294-301.
Gauthier L.V., Taub E., Perkins C., et al. Remodeling the brain: plastic structural brain changes produced by different motor therapies after stroke. Stroke. 2008;39:1520-1525.
Gilland E., Puka-Sundvall M., Hillered L., et al. Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature rat brain: involvement of NMDA-receptors. J Cereb Blood Flow Metab. 1998;18:297-304.
Graef J.D., Godwin D.W. Intrinsic Plasticity in Acquired Epilepsy: Too Much of a Good Thing? Neuroscientist. 2010.
Hablitz J.J., Lee W.L. NMDA receptor involvement in epileptogenesis in the immature neocortex. Epilepsy Res Suppl. 1992;8:139-145.
Hansen H.H., Briem T., Dzietko M., et al. Mechanisms leading to disseminated apoptosis following NMDA receptor blockade in the developing rat brain. Neurobiol Dis. 2004;16:440-453.
Haydar T.F., Kuan C.Y., Flavell R.A., et al. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex. 1999;9:621-626.
Hertz-Pannier L., Chiron C., Jambaque I., et al. Late plasticity for language in a child’s non-dominant hemisphere: a pre- and post-surgery fMRI study. Brain. 2002;125:361-372.
Holmes G.L., Khazipov R., Ben-Ari Y. Seizure-induced damage in the developing human: relevance of experimental models. Prog Brain Res. 2002;135:321-334.
Horska A., Farage L., Bibat G., et al. Brain metabolism in Rett syndrome: age, clinical, and genotype correlations. 2009. pp 90–97
Hotulainen P., Hoogenraad C.C. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619-629.
Huber K.M., Gallagher S.M., Warren S.T., et al. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746-7750.
Huttenlocher P.R.D.A.S.. Regional differences in synaptogenesis in human cerebral cortex. 1997. pp 167–178
Inan M., Crair M.C. Development of cortical maps: perspectives from the barrel cortex. Neuroscientist. 2007;13:49-61.
Johnston M.V. Clinical disorders of brain plasticity. Brain Dev. 2004;26:73-80.
Johnston M.V. Plasticity in the developing brain: implications for rehabilitation. Dev Disabil Res Rev. 2009;15:94-101.
Johnston M.V., Blue M.E., Naidu S. Rett syndrome and neuronal development. J Child Neurol. 2005;20:759-763.
Johnston M.V., Nishimura A., Harum K., et al. Sculpting the developing brain. Adv Pediatr. 2001;48:1-38.
Kadam S.D., Dudek F.E. Neuropathogical features of a rat model for perinatal hypoxic-ischemic encephalopathy with associated epilepsy. J Comp Neurol. 2007;505:716-737.
Kaufmann W.E., Johnston M.V., Blue M.E. MeCP2 expression and function during brain development: implications for Rett syndrome’s pathogenesis and clinical evolution. Brain Dev. 2005;27(Suppl 1):S77-S87.
Keifer J., Zheng Z. AMPA receptor trafficking and learning. Eur J Neurosci. 2010.
Kernie S.G., Parent J.M. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis. 2010;37:267-274.
Kleim J.A., Chan S., Pringle E., et al. BDNF val66met polymorphism is associated with modified experience-dependent plasticity in human motor cortex. Nat Neurosci. 2006;9:735-737.
Kline D.D., Ogier M., Kunze D.L., et al. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303-5310.
Kuhnke N., Juenger H., Walther M., et al. Do patients with congenital hemiparesis and ipsilateral corticospinal projections respond differently to constraint-induced movement therapy?. 2008. pp 898–903
Kupers R., Pappens M., de Noordhout A.M., et al. rTMS of the occipital cortex abolishes Braille reading and repetition priming in blind subjects. Neurology. 2007;68:691-693.
Lessmann V., Brigadski T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res. 2009;65:11-22.
Li N., Lee B., Liu R.J., et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959-964.
Li W., Cui Y., Kushner S.A., et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15:1961-1967.
Lippe S., Bulteau C., Dorfmuller G., et al. Cognitive outcome of parietooccipital resection in children with epilepsy. Epilepsia. 2010.
Luft A.R., Combe-Waller S., Whitall J., et al. Repetitive bilateral arm training and motor cortex activation in chronic stroke: a randomized controlled trial. JAMA. 2004;292:1853-1861.
Luft A.R., Macko R.F., Forrester L.W., et al. Treadmill exercise activates subcortical neural networks and improves walking after stroke: a randomized controlled trial. Stroke. 2008;39:3341-3350.
Maguire E.A., Gadian D.G., Johnsrude I.S., et al. Navigation-related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci USA. 2000;97:4398-4403.
Malenka R.C., Nicoll R.A. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521-527.
McDonald J.W., Johnston M.V. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev. 1990;15:41-70.
McHughen S.A., Rodriguez P.F., Kleim J.A., et al. BDNF val66met polymorphism influences motor system function in the human brain. Cereb Cortex. 2010;20:1254-1262.
McQuillen P.S., Sheldon R.A., Shatz C.J., et al. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci. 2003;23:3308-3315.
Meltzoff A.N., Kuhl P.K., Movellan J., et al. Foundations for a new science of learning. Science. 2009;325:284-288.
Miller D.T., Adam M.P., Aradhya S., et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749-764.
Monyer H., Brunashev N., Laurie D.J.. Development and regional expression in the rat brain and functional properties of four NMDA receptors. 1993. pp 529–540
Moretti P., Levenson J.M., Battaglia F., et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319-327.
Muddashetty R.S., Kelic S., Gross C., et al. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338-5348.
Nguyen V., McQuillen P.S. AMPA and metabotropic excitoxicity explain subplate neuron vulnerability. Neurobiol Dis. 2010;37:195-207.
Nishimura A., Hohmann C.F., Johnston M.V., et al. Neonatal electrolytic lesions of the basal forebrain stunt plasticity in mouse barrel field cortex. Int J Dev Neurosci. 2002;20:481-489.
Nuber U.A., Kriaucionis S., Roloff T.C., et al. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:2247-2256.
Nudo R.J., Milliken G.W. Reorganization of movement representations in primary motor cortex following focal ischemic infarcts in adult squirrel monkeys. J Neurophysiol. 1996;75:2144-2149.
Nugent F.S., Kauer J.A. LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol. 2008;586:1487-1493.
Palmer A., Qayumi J., Ronnett G. MeCP2 mutation causes distinguishable phases of acute and chronic defects in synaptogenesis and maintenance, respectively. Mol Cell Neurosci. 2008.
Park S.W., Wolf S.L., Blanton S., et al. The EXCITE Trial: Predicting a Clinically Meaningful Motor Activity Log Outcome. Neurorehabil Neural Repair. 2008;22:486-493.
Penagarikano O., Mulle J.G., Warren S.T. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet. 2007;8:109-129.
Penn A.A., Shatz C.J. Brain waves and brain wiring: the role of endogenous and sensory-driven neural activity in development. Pediatr Res. 1999;45:447-458.
Pfeiffer B.E., Zang T., Wilkerson J.R., et al. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 2010;66:191-197.
Pinto D., Pagnamenta A.T., Klei L., et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368-372.
Quartarone A., Siebner H.R., Rothwell J.C. Task-specific hand dystonia: can too much plasticity be bad for you? Trends Neurosci. 2006;29:192-199.
Roth T.L., Roth E.D., Sweatt J.D. Epigenetic regulation of genes in learning and memory. Essays Biochem. 2010;48:263-274.
Roulet-Perez E., Davidoff V., Mayor-Dubois C., et al. Impact of severe epilepsy on development: recovery potential after successful early epilepsy surgery. Epilepsia. 2010;51:1266-1276.
Sadato N., Pascual-Leone A., Grafman J., et al. Activation of the primary visual cortex by Braille reading in blind subjects. Nature. 1996;380:526-528.
Sadowsky C.L., McDonald J.W. Activity-based restorative therapies: concepts and applications in spinal cord injury-related neurorehabilitation. Dev Disabil Res Rev. 2009;15:112-116.
Salter M.G., Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167-1171.
Schlaug G., Forgeard M., Zhu L., et al. Training-induced neuroplasticity in young children. Ann N Y Acad Sci. 2009;1169:205-208.
Shalin S.C., Egli R., Birnbaum S.G., et al. Signal transduction mechanisms in memory disorders. Prog Brain Res. 2006;157:25-41.
Shaw P., Greenstein D., Lerch J., et al. Intellectual ability and cortical development in children and adolescents. Nature. 2006;440:676-679.
Shilyansky C., Karlsgodt K.H., Cummings D.M., et al. Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc Natl Acad Sci USA. 2010;107:13141-13146.
Smrt R.D., Eaves-Egenes J., Barkho B.Z., et al. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis. 2007;27:77-89.
Staley K.J., Anderson A.E. Hyperactive interneurons impair learning in a neurofibromatosis model. Nat Neurosci. 2009;12:8-10.
Stornetta R.L., Zhu J.J. Ras and Rap Signaling in Synaptic Plasticity and Mental Disorders. Neuroscientist. 2010.
Thomason M.E., Yoo D.J., Glover G.H., et al. BDNF genotype modulates resting functional connectivity in children. Front Hum Neurosci. 2009;3:55.
Tropea D., Giacometti E., Wilson N.R., et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA. 2009;106:2029-2034.
Tropea D., Van W.A., Sur M. Review. Molecular mechanisms of experience-dependent plasticity in visual cortex. Philos Trans R Soc Lond B Biol Sci. 2008.
Volpe J.J. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110-124.
Walther M., Juenger H., Kuhnke N., et al. Motor cortex plasticity in ischemic perinatal stroke: a transcranial magnetic stimulation and functional MRI study. Pediatr Neurol. 2009;41:171-178.
Wang L.W., Berry-Kravis E., Hagerman R.J. Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics. 2010;7:264-274.
Weeks R., Horwitz B., ziz-Sultan A., et al. A positron emission tomographic study of auditory localization in the congenitally blind. J Neurosci. 2000;20:2664-2672.
Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27-36.
Yoshii A., Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70:304-322.
Zeng L.H., Ouyang Y., Gazit V.. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis. 2007. pp 184–196
Zhou Z., Hong E.J., Cohen S., et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255-269.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 13 Brain Plasticity and its Disorders
Introduction
Brain plasticity is an important concept that plays a major role in the expression of many pediatric neurological disorders and strongly influences recovery from brain injuries in neonates, infants, and children. Plasticity refers to the brain’s ability to change in response to experience, whether it is a positive experience such as education or practicing a skill, or an adverse event such as a stroke or other type of brain injury [Johnston, 2004]. The child’s brain exhibits greater plasticity than the adult brain, and common examples of enhanced brain plasticity in children include their ability to learn new motor tasks quickly, such as playing a musical instrument, participating in a sport, or their ability to become fluent in a new language [Meltzoff et al., 2009]. Children and adolescents are also able to form memories more easily than adults and they recover more quickly from brain injuries. The mechanisms responsible for various kinds of brain plasticity are being uncovered at a rapid pace as are the defects in these steps that cause intellectual disability and other pediatric disorders [Johnston, 2009]. This chapter gives a brief overview of normal brain plasticity and its disorders that are relevant to pediatric neurology, and provides insight into the pathogenesis of a variety of disorders described in other sections.
It is useful to consider four aspects or types of plasticity in the developing brain in order to understand how this concept is integrated into many disorders of the child’s nervous system (Box 13-1). The first type is generally referred to as adaptive plasticity, in which the nervous system changes in the process of learning new skills that have an adaptive advantage, such as learning to read or pass a test, learning to play a musical instrument, or learning to play a sport. Adaptive plasticity is also engaged when children recover from a brain injury, such as a stroke or extensive removal of brain tissue to cure epilepsy, and therapists try to harness this form of plasticity when providing speech, occupational, or physical therapy for a variety of disorders. In these cases, it is expected that normal childhood activities, such as attending school, music lessons, athletic practice, or therapies for cerebral palsy or other brain injuries, will activate normal plasticity programs to provide a good outcome that is beneficial for the child [Meltzoff et al., 2009]. Adaptive plasticity is generally enhanced in younger children compared with adults, and the genetically determined programs responsible for this kind of plasticity are heavily influenced by sensory input. Since learning and memory, as well as acquisition of physical skills, are very important for normal childhood development, genetic or acquired defects in the signaling pathways responsible for plasticity are reflected in a variety of developmental disability phenotypes [Johnston, 2004]. These disorders are best understood as examples of impaired plasticity. Many types of intellectual disability are caused by genetic lesions in the signaling cascades that are normally responsible for activity-dependent synaptic plasticity (Table 13-1). For example, in fragile X syndrome (FraX), the most common form of inherited intellectual disability, a trinucleotide repeat in the gene for the fragile X mental retardation protein (FMRP) leads to its absence or reduction in the brain [Penagarikano et al., 2007]. FMRP is responsible for transport of certain messenger RNAs and translation of proteins within dendrites in response to synaptic activity, and its absence causes a reduction in long-term potentiation (LTP), a physiological form of synaptic plasticity involved in learning and memory [Huber et al., 2002]. Accordingly, the intellectual disability and other behavioral features of FraX can be considered to reflect a genetically determined form of impaired plasticity in synapses. Therapies aimed at restoring normal plasticity mechanisms to synapses in FraX show some promise in preclinical and small clinical trials [Wang et al., 2010].
Table 13-1 Pediatric Disorders Caused by Genetic Mutations of Signaling Pathways Involved in Neuronal Plasticity
| Disorder | Mechanism of Disease |
|---|---|
| Neurofibromatosis 1 | Ras too active, enhanced GABA activity |
| Tuberous sclerosis | Upregulated mTOR signaling pathway |
| Rett’s syndrome | Mutations in MeCP2 transcription factor |
| Fragile X syndrome | Upregulated mGluR5 causes LTD |
| Coffin–Lowry syndrome | Mutations in RSK2 in Ras-MAPK pathway |
| Rubinstein–Taybi syndrome | Mutation in CREB binding protein (CBP) |
| X-linked intellectual disabilities | Mutation in PAK3 kinase: links RhoGTPases to dendritic cytoskeleton Mutation in oligophrenin, RhoGTPase Mutation in GluR3 AMPA receptor subunit |
| Costello’s syndrome | Upregulated H-Ras signaling to MAPK (intellectual disability, heart, skeletal disorder) |
| Lead poisoning | Enhanced PKC activity, inhibited NMDA receptors; impairs maturation of dendritic spines |
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CREB, cyclic adenosine monophosphate response element binding protein; GABA, gamma-aminobutyric acid; MAPK, Ras-mitogen activated protein kinase; mTOR, mammalian target of rapamycin; NMDA, N-methyl-d-aspartate; PKC, protein kinase C.
Just as impaired plasticity is responsible for numerous brain disorders in children, maladaptive or excessive plasticity also can cause problems. When this phenomenon occurs, plasticity is misdirected or enhanced in a way that is harmful, or at least not beneficial. For example, children who have had strokes, traumatic brain injuries, or hypoxic-ischemic brain injuries sometimes develop delayed seizures that may result from formation of abnormal neuronal networks in damaged brain tissue [Kadam and Dudek, 2007]. In some cases, neurons in these networks have been shown to have functional changes in voltage-sensitive sodium channels that are similar to those found in certain forms of genetic epilepsy [Graef and Godwin, 2010]. Excessive plasticity may also be responsible for seizures and cognitive impairment in genetic disorders in which intracellular signaling pathways are enhanced, such as tuberous sclerosis and Costello’s syndrome [Crino, 2010; Dileone et al., 2010]. Another example of enhanced, but maladaptive, plasticity that causes neurologic impairment is dystonia in the hand and fingers resulting from over-practice of the piano or another musical instrument requiring intense finger movement [Quartarone et al., 2006]. In patients with this disorder, functional imaging of the contralateral cortex has shown that the somatosensory map of the fingers is blurred in the musicians with dystonia compared to controls [Elbert et al., 1998]. This suggests that the somatosensory cortex is less able to distinguish precisely between individual finger movements, and an attempt to use the fingers leads to dystonic movements instead. Interestingly, selective injection of botulinum toxin into some of the fingers can compensate for the disrupted somatotopic map of the fingers and restore normal movement [Cole et al., 1995]. The immature brain may be especially susceptible to organizational changes in neuronal circuits that lead to acquired disorders associated with maladaptive or excessive plasticity.
A fourth aspect of plasticity that it is important to consider is its potential to create cell-specific selective vulnerabilities, leading to the understanding of plasticity as the brain’s Achilles’ heel. This concept is especially important in the developing brain, where different cells are undergoing dramatic shifts in their composition during growth and development. One example is the subplate neurons that are among the first to reach the cerebral cortex in the second trimester of pregnancy, providing targets for axons projected from neurons in the thalamus [McQuillen et al., 2003]. These neurons are selectively vulnerable to hypoxia-ischemia due to their enhanced expression of excitatory α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors during that time period [Nguyen and McQuillen, 2010]. Immature oligodendroglia are similarly more vulnerable to injury during the second trimester due to the subunit composition of their AMPA receptors that favors permeability to calcium [Volpe, 2009]. N-methyl-d-aspartate (NMDA) receptors also contribute to developmental vulnerability of immature oligodendroglia [Salter and Fern, 2005]. These age-dependent changes in excitatory amino acid receptors on neurons and oligodendroglia that favor excitation have a beneficial role in normal development, but they can also make cells vulnerable to death if they are accidentally exposed to hypoxia or ischemia [McDonald and Johnston, 1990]. In a similar way, developing neurons in the brain closer to term become more vulnerable to excitotoxicity mediated by NMDA receptors due to the fact that the receptors are genetically programmed at that time to be more excitable [Monyer et al., 1993]. NMDA receptors are easier to activate in the developing brain due to their subunit composition, which makes it easier to open their channels. However, this characteristic also creates a vulnerability to injury if the brain is exposed to hypoxia-ischemia, which can result in neuronal depolarization and calcium flooding through opening of the NMDA channels.
In addition, gamma-aminobutyric acid (GABA), which normally has an inhibitory effect on neuronal circuits in older children and adults, mediates excitation in the newborn period because of developmental changes in chloride pumps in GABAergic synapses [Ben-Ari, 2006]. Enhanced activity of the NKCC1 chloride transporter in the neonatal period leads to higher intraneuronal concentrations of chloride than are present later on, so that, when GABA opens chloride channels, the cation leaves the neuron rather than entering [Dzhala et al., 2005]. Therefore, GABA leads to depolarization rather than hyperpolarization, as it does at older ages. Later in development, activity of the NKCC1 pump declines, and expression of the NKCC2 pump, which pushes chloride out of neurons, increases, leading to lower baseline concentrations of chloride inside the neuron compared to outside, and to inhibition. Combined with the expression of NMDA receptors, which are more active and flux more calcium and sodium in the fetal and neonatal brain, developmental changes in the actions of GABA lead to the brain being more excitable. This enhanced excitability during development appears to play an important role in the establishment of normal neuronal circuitry, which is dependent on electrical activity [Penn and Shatz, 1999]. For example, production of growth factors, such as brain-derived neurotropic factor (BDNF), occurs in neurons and is dependent on neuronal activity [Lessmann and Brigadski, 2009]. Prolonged blockade of NMDA receptors, just like too much activity, can cause neuronal death because a minimum baseline amount of channel opening, with entry of calcium and sodium into neurons, is essential for neuronal survival [Hansen et al., 2004]. On the other hand, this bias towards excitement in the developing brain is probably responsible for the higher propensity for seizures in infants and children compared to adults, and the expression of some seizure types such as infantile spasms [Hablitz and Lee, 1992]. Therefore it is quite important for the molecular machinery responsible for balancing excitement and inhibition in the developing brain to operate properly in order to prevent it from becoming an Achilles’ heel during periods of stress, such as hypoxia-ischemia or status epilepticus [McDonald and Johnston, 1990].
Basic Mechanisms for Plasticity in the Developing Brain
In addition to these four broad types of plasticity in the developing brain, there are at least six basic cellular mechanisms for plasticity (Box 13-2). The earliest mechanism is the overproduction of neurons and glia from stem cells, and then reduction of this population by apoptosis in the fetus [Haydar et al., 1999]. Most neurogenesis ceases after birth, but it continues in selected niches of stem-cell production in the subventricular zone of the lateral ventricles and the subgranular zone of the dentate gyrus of the hippocampus [Kernie and Parent, 2010]. These restricted zones of neurogenesis may contribute to recovery from brain injuries throughout life. Additional mechanisms for plasticity are:
Mechanisms of Synaptic Plasticity
Synaptic plasticity is the most important mechanism for everyday activities such as learning and memory, and acquiring new skills (Figure 13-1). The strength of the signals between axons and dendrites across synapses can be increased or decreased through processes called long-term potentiation (LTP) or long-term depression (LTD), and these processes are strongly influenced by previous synaptic activity [Malenka and Nicoll, 1993]. The most prominent examples of LTP and LTD occur in excitatory glutamate synapses, which account for about 75 percent of all synapses in the brain. However, LTP and LTD are also present in many different types of neurons, including inhibitory GABAergic neurons and dopaminergic neurons [Nugent and Kauer, 2008]. One of the best examples of LTP is mediated by the NMDA-type glutamate receptors, which are voltage-dependent, meaning that NMDA channel opening is dependent in part on membrane depolarization [Gilland et al., 1998]. Channel opening is also dependent on occupancy of receptors for glutamate and glycine by these amino acids. When multiple excitatory axons depolarize the neuronal membrane, and glycine and glutamate occupy their receptors, the NMDA-activated calcium channel opens, allowing a pulse of calcium to enter the neurons and, in turn, activating a cascade of biochemical events that encode a memory of the event [Johnston, 2004]. The NMDA receptor has been referred to as a coincidence detector because it requires both membrane depolarization, stimulated by multiple presynaptic inputs, and activation of glutamate and glycine receptors to open its channel [Brown and Milner, 2003]. NMDA receptor-mediated LTP results in a “step up” in the signal mediated by the synapse during subsequent synapse activations, and it becomes easier to activate. Neuronal stimulation can also activate another type of a glutamate receptor, called a metabotropic gluR5 (mGluR5) receptor because it activates the second messenger phosphoinositol turnover, rather than opening an ion channel, as NMDA receptors do [Antion et al., 2008].
Within synapses, a variety of mechanisms contribute to LTP and LTD, including an increase or decrease in neurotransmitter release from presynaptic nerve terminals, a change in reuptake of neurotransmitter by astroglia that surround each synapse, a change in the number of neurotransmitter receptors in the postsynaptic membrane, changes in the flux of calcium and other ions through membrane channels, and an activation of intracellular signaling cascades [Citri and Malenka, 2008]. One of the common mechanisms that synapses use to increase or decrease synaptic strength is to alter the number of receptors in the postsynaptic membrane through a process called “receptor trafficking” (Figure 13-2) [Keifer and Zheng, 2010; Conboy and Sandi, 2010]. In this process, receptors shuttle back and forth between the cytoplasm, where they do not have access to neurotransmitter in the synaptic cleft, postsynaptic density, and postsynaptic membrane, where they are positioned to interact with neurotransmitter. One excitatory receptor that is heavily regulated by receptor trafficking in synapses is the AMPA receptor, which is the ionotropic glutamate receptor that carries most of the fast excitatory current in the brain. AMPA receptors shuttle between the dendritic cytoplasm, where they are inaccessible to glutamate, to the postsynaptic membrane, where they can be stimulated by glutamate [Keifer and Zheng, 2010]. The number of receptors within the synaptic membrane determines the strength of the synapse when it is activated. Accordingly, LTP is associated with an increase in AMPA receptors inserted into the postsynaptic membrane, while a decrease in AMPA receptors in the postsynaptic membrane is associated with LTD. LTP can be induced by rapid stimulation of the postsynaptic membrane, leading to voltage-dependent opening of NMDA receptors and trafficking of AMPA receptors from the cytoplasm into the postsynaptic membrane. This makes the synapse better able to conduct a signal, in effect creating a fragment of a memory about that synapse’s past experience. The mGluR5 metabotropic glutamate receptor also controls AMPA receptor trafficking, and an increase in mGluR5 activity leads to internalization of AMPA receptors, LTD, and weaker synaptic strength. In experimental models of fragile X syndrome, the activity of mGluR5 receptors has been reported to be increased, along with enhanced synaptic LTD, and antagonists of the mGluR5 receptor are able to restore LTP and improve abnormal behaviors and cognition (see Figure 13-2) [Muddashetty et al., 2007]. Fragile X syndrome is one example of a developmental brain disorder that can be understood at the level of the synapse. Synapses are thought to be the primary site of memory storage across large networks in which the strength of individual synapses is increased or decreased by the processes of LTP or LDP.
Production of nerve growth factors like BDNF is another mechanism by which previous synaptic activity can lead to plasticity in neuronal connections. BDNF is produced in neurons in response to neuronal activity, and it has been shown to increase and stabilize synaptic connections, as well as contribute to their maturation and integration into complex neuronal circuits [Yoshii and Constantine-Paton, 2010]. Learning has also been shown to increase neurotrophin signaling through the TrκB receptor, which mediates the actions of BDNF in the hippocampus in mice. BDNF has also been associated with cortical plasticity in humans. Kleim et al. reported that individuals with a val66met polymorphism (i.e., methionine codon substitution for valine codon at position 66 in the BDNF gene) showed reduced ability to reorganize their cortical motor map in response to a training exercise involving the fingers of one hand [Kleim et al., 2006]. In vitro studies showed that this mutation impairs the secretion of BDNF from neurons, but its expression is normal. McHughen et al. used functional magnetic resonance imaging (fMRI) to show that subjects with this BDNF mutation made more errors and had poorer retention on a driver-based learning task, and had altered short-term plasticity [McHughen et al., 2010]. BDNF genotype has also been shown to be associated with anxiety and memory problems, as well as with impaired functional connectivity from the hippocampus to the amygdala, insula, and striatal regions in children studied with functional connectivity (FC) analysis on data obtained through whole-brain fMRI [Thomason et al., 2009].
Plasticity of Dendrites and Dendritic Spines
Activity-dependent plasticity in excitatory synapses is associated with physical changes in dendritic spines that are thought to reflect information storage (Figure 13-3) [Hotulainen and Hoogenraad, 2010]. Immature dendritic spines are long with a small head, but repeated activity during development, including LTP, leads to shortening of spine shafts and enlargement of spine heads. New learning in animals also has been shown to be associated with localized enlargement of some dendritic spines and loss of others. Remodeling of dendrite morphology occurs continuously in association with synaptic plasticity, and is mediated by the actin-rich cytoskeleton that fills dendrites [Hotulainen and Hoogenraad, 2010]. The actin cytoskeleton within spines containing excitatory synapses provides support for the postsynaptic density (PSD), which anchors glutamate receptors and cell adhesion molecules that span the synaptic cleft (see Figure 13-3). NMDA receptors within the PSD regulate actin signaling pathways within an endocytic zone just below the PSD. The purpose of this zone is to recycle the synaptic pool of AMPA-type glutamate receptors involved in trafficking between the cytoplasm and the PSD. Therefore, the actin cytoskeleton plays an important role in LTP, and it is not surprising that mutations in proteins involved in regulation of this cytoskeleton, such as small Rho and Ras GTPases, alter spine morphology and cause cognitive disorders and autistic spectrum disorders [Pinto et al., 2010]. Similarly, key proteins that make up the PSD and the scaffolding proteins beneath the PSD, such as SHANK and the cell adhesion proteins – neurexins, neuroligins, and cadherins – are associated with clinical disorders that impair learning (Table 13-2) [Durand et al., 2007; Johnston et al., 2001].
Excitatory axons that contain glutamate synapse on the tips of dendritic spines, while inhibitory axons that contain gamma-aminobutyric acid (GABA) synapse on the shafts of dendrites. Excitatory synapses are asymmetric and contain a postsynaptic density (PSD), which anchors neurotransmitter receptors and a variety of other proteins that provide scaffolding for receptors (see Table 13-2
[/not-level-membership-for-neurology-category]
