CHAPTER 37 Blood groups on red cells, platelets and neutrophils
Red cell surface antigens
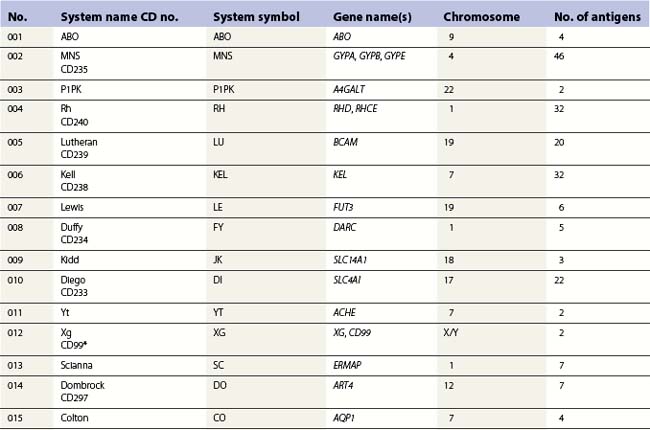
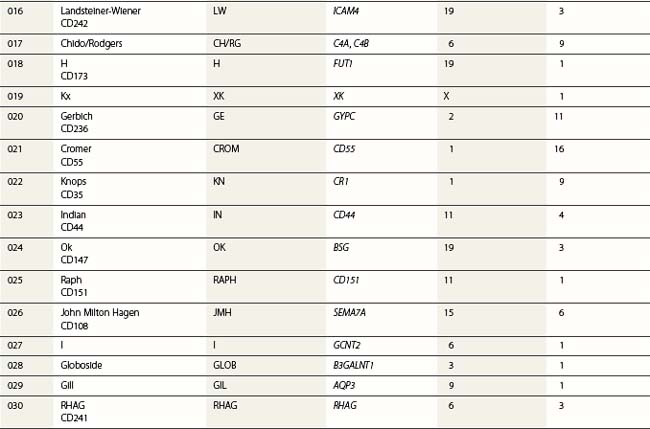
Red cell blood groups have been recognized for over a century, since Landsteiner’s discovery of the ABO system in 1900. Red cell surface antigens are validated and classified by the International Society of Blood Transfusion (ISBT), which currently recognizes 328 antigens, 284 belonging to one of 30 blood-group systems. Each system consists of between one and 52 antigens encoded either by a single gene or by two or three closely linked homologous genes.1,2 The 30 blood group systems are listed in Table 37.1.
Blood-group antigens may be carbohydrate structures on red cell surface glycoproteins or glycolipids, or they may be determined primarily by the amino acid sequence of polypeptides or glycoproteins. At least 23 red cell surface proteins express blood-group polymorphism. The functions of some of these structures are reasonably well understood, such as transport of biologically important molecules in and out of the cell, protection of the cell from autologous complement, and anchoring the membrane to the membrane skeleton. Functions of many, however, can only be speculated on from structural similarity to proteins and glycoprotein of known function.3
Pathology associated with blood-group polymorphism is usually the result of alloimmune destruction of red cells. This might be a hemolytic transfusion reaction following transfusion of red cells to a patient with an alloantibody directed against a determinant present on donor red cells, or hemolytic disease of the fetus and newborn (HDFN), following placental transfer of maternal IgG antibodies into the fetal circulation. Most blood group systems contain a null-phenotype in which the antigens of that system are not expressed. This usually results from an inactivating mutations in the genes encoding the antigen, yet in most cases null-phenotypes are not associated with any pathology, probably due to functional redundancy of cell surface proteins. For reviews on red cell groups see Daniels4 and Reid and Lomas-Francis.5
The ABO and H histo-blood-group systems and other carbohydrate antigens
ABO and H antigens
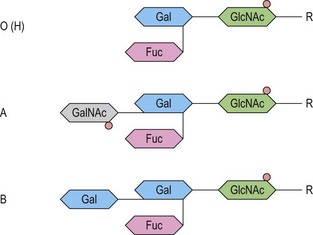
At its most basic level, there are two ABO antigens, A and B, and four phenotypes, A, B, AB and O. O is a null phenotype in which neither A nor B is expressed. The A and B determinants are carbohydrate structures, present on red cell membrane glycoproteins and glycolipids. The major carriers of A and B on red cells are the abundant N-glycosylated glycoproteins, the anion exchanger (band 3) and the glucose transporter (GLUT1). Carbohydrate chains are synthesized by the action of glycosyltransferases, enzymes that catalyse the transfer of specific monosaccharides from a nucleotide donor substrate to an acceptor substrate. The acceptor substrate for A- and B- transferases, products of the A and B alleles, is a terminally fucosylated structure called H antigen (Fig. 37.1). The A gene product is an N-acetylgalactosaminyltransferase that transfers N-acetylgalactosamine from a uridine diphosphate (UDP)-N-acetylgalactosamine donor substrate to the fucosylated galactosyl residue of the H antigen, to produce an A-active structure (Fig. 37.1). B gene product is a galactosyltransferase that transfers galactose from UDP-galactose to the fucosylated galactose of H, to produce B-active structure (Fig. 37.1). The O allele produces no active enzyme, so on group O red cells the H antigen remains unconverted. N-acetylgalactosamine and galactose are the immunodominant sugars of A and B blood groups, respectively.
A- and B-transferases are encoded by a single gene on chromosome 9, cloned by Yamamoto et al6 in 1990. The ABO gene spans about 18–20 kb organized into seven exons of coding sequence. Exons 6 and 7, the two largest, include 77% of the full coding region and encode all of the catalytic domain. The products of the A and B alleles differ by four amino acid substitutions at residues 176, 235, 266 and 268. Leu266Met and Gly268Ala substitutions are most important in determining whether the gene product has predominantly N-acetylgalactosaminyltransferase (A) or galactosyltransferase (B) activity. The sequence of the most common O allele (O1) is identical to the A sequence, apart from a single nucleotide deletion that is responsible for a reading frame shift after codon 86 and the generation of a translation stop signal at codon 117. This allele encodes a truncated protein lacking the catalytic site. There are two other common O alleles: O1v, that, like O1, has the single nucleotide deletion, but differs from O1 by nine nucleotide changes within the coding sequence and O2, that does not have the single nucleotide deletion, but is inactivated by a missense mutation encoding a Gly268Arg substitution (reviewed in 7).
HDFN caused by ABO antibodies
Anti-A and -B are predominantly IgM, but may be IgG. Anti-A,B, which reacts with both A and B antigens, is present in the sera of most group O people and is often partly IgG. ABO HDFN is restricted almost exclusively to group A1 or B fetuses of group O mothers and IgG anti-A,B is generally considered culpable.8
About 15% of pregnancies in women of European origin involve a group O mother with a group A or B fetus. This figure does not vary greatly in most other major ethnic groups, yet ABO HDFN requiring clinical intervention is rare, though minor symptoms involving a small degree of red cell destruction may be relatively common. Hydrops due to ABO HDFN is exceedingly rare, but very occasionally exchange transfusion for the prevention of kernicterus is indicated.8 The main reasons for the low prevalence of clinically significant ABO HDFN is that A and B antigens are present in many tissues. Any antibody crossing the placenta is likely to become bound to placental tissue, reducing the quantity available for destruction of red cells. In addition, immune anti-A,B are mainly IgG2, which does not cause HDFN because there are no Fc receptors for IgG2 on the cells of the mononuclear phagocyte system, and A and B red cell antigens are not fully developed in the fetus or neonate.
Altered expression of ABO antigens in leukemia
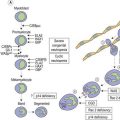
The association of weakened expression of A, B and/or H antigens with myeloid malignancies, usually acute myeloid leukemia (AML), is well documented.4 In some cases all red cells show weakness of the A antigen, whereas in others two populations of red cells are clearly apparent. In a patient with acute monoblastic leukemia, initially only 2% were agglutinated with anti-A, but in remission the proportion of agglutinable cells rose to 65% before falling again shortly before death.9 Depending on the method used for detection, between 17 and 55% of patients with myeloid malignancies have lower ABH antigenic expression compared with healthy controls.4 In all cases the changes represent a loss or diminution of antigen strength and never the expression of a new red cell antigen. In some cases loss of A or B was associated with increase in H, in some A or B loss was secondary to loss of H, and in a third group there appeared to be concommitant loss of A/B and H.10 Modifications of ABH antigens may be detected before diagnosis of malignancy, indicating a preleukemic state. Loss of an ABH antigen in a patient with a hematologic disorder is generally prognostic of AML.
Depression of A or B antigens in AML and in preleukemic states is usually associated with a severe reduction in red cell A- or B-transferase activity, but little or no reduction in red cell H-transferase activity.11 Although there may be multiple mechanisms underlying the loss of ABH antigens, DNA methylation is significantly associated with silencing of the ABO transcript in patients with myeloid malignancies and the ABO transcript can be re-expressed in leukemic cell lines by treating with a demethylating agent.12
Lewis antigens
The Lewis antigens, Lea and Leb, are not synthesized by erythroid cells, but become incorporated into the red cell membrane from the plasma. Their synthesis from H antigen and from its precursor is catalyzed by an α1,3/4-fucosyltransferase, the product of FUT3, a gene on chromosome 19. This fucosyltransferase competes with the H-(FUT2), A-, and B-transferases in endodermal tissue for acceptor substrate. The antigens known as Lex and Ley, which are not detected on red cells, are isomers of Lea and Leb, respectively.13
ABH and Lewis antigens on tumors
ABH antigens are often absent from glycoproteins and glycolipids of malignant tissue of the gastrointestinal tract, oral cavity, uterine cervix, lung, prostate, breast and bladder, despite being present in the surrounding epithelium. In many cases loss of ABH antigens preceded formation of distant metastases and hence a poor prognosis.14,15 Loss of A or B antigens from tumor cells may increase their motility and, consequently, their ability to form metastases. In addition, this absence of A or B antigens generally arises from disappearance of A- or B-transferase activity and results in an accumulation of H, Leb, Ley, sialyl-Lea or sialyl Lex. Sialyl-Lea and sialyl Lex are ligands for selectins, and their presence promotes the metastatic process by facilitating interaction with distant organs.14,15
ABO antigen loss results from downregulated transcription of the ABO gene, as no A or B mRNA can be detected in high-grade tumors. At least two different mechanisms may be involved, possibly occurring in tumors derived from different souces: 1) loss of heterozygosity (allelic loss) involving deletions within chromosome 9q34, which contain the ABO locus in addition to tumor suppressor genes; 2) hypermethylation of the CpG island of the ABO promoter region, which down-regulates transcription.16,17
Up-regulation of glycosyltransferases in tumors may result in increased levels of certain carbohydrate structures in the plasma.18 The quantity of circulating sialylated-Lea, otherwise known as the CA 19-9 antigen, is widely used as a marker to support diagnosis of colorectal, pancreatic and gastric cancer, and as an aid to prognosis after potential curative surgery.19,20
Another phenomenon associated with malignancy is the incompatible A antigen occasionally expressed on tumors of group O or B people. About 10% of colonic tumors from group O patients, shown to be homozygous for the O1 allele, express A antigen and contain A-transferase activity.21,22 The molecular basis of this O to A conversion is not known, but alternative splicing of ABO RNA, resulting in loss of exons 5 and 6, would introduce no frameshift or translation-termination codons, but would eliminate the single nucleotide deletion in exon 6 of O1 and the putative gene product would be a truncated glycosyltransferase with a potential for A-transferase activity.14 The higher incidence of gastric and ovarian adenocarcinomas in group A people could be due to a suppression of development of tumors bearing an A antigen by the anti-A naturally present in group O and B, but not A, patients.23
The Rh blood-group system
Rh is the most complex of the human blood-group systems. It has 52 well-defined antigens, the most immunogenic of which is D (RH1). Between 82% and 88% of Caucasians, about 95% of black Africans, and almost 100% of people from the Far East are D-positive. The other main Rh polymorphisms are C/c and E/e, two pairs of antithetical antigens. C has a frequency of 70% and c a frequency of 80% in Europeans. In black Africans the frequency of c is much higher and the frequency of C much lower, whereas in eastern Asia the opposite is the case, with C approaching 100%. In most populations E has a frequency of about 30% and e about 98%.4,5
The antigens of the Rh system are encoded by two genes, RHD and RHCE (reviewed in 24 and 25). They are highly homologous and have very similar genomic organization, each containing 10 coding exons arranged in opposite orientation on chromosome 1 (Fig. 37.2).26 RHCE encodes the C/c and E/e antigens, plus many others such as Cw, Cx and VS. RHD encodes the many epitopes of the D antigen. In Caucasians the D-negative phenotype almost always results from homozygosity for a deletion of RHD, whereas about 66% of D-negative Africans have RHDψ, an inactive RHD gene.27 The products of the Rh genes are polypeptides that are palmitoylated but, unlike most cell surface proteins, not glycosylated. The D and CcEe proteins differ by only 32–35 amino acids, depending on CcEe phenotype. Hydropathy analysis of the amino acid sequences of the Rh proteins together with immunologic evidence suggests that the Rh proteins span the red cell surface membrane 12 times, with internal termini and six extracellular loops (Fig. 37.2).
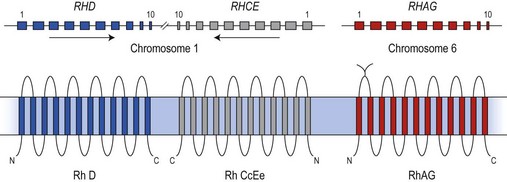
In the D-negative phenotype no D protein is present in the membrane, explaining the high immunogenicity of D compared with other Rh antigens. The C/c and E/e polymorphisms represent amino acid substitutions at different positions in the CcEe protein. Rh epitopes are conformational and may be discontinuous; that is, they are very dependent on the shape of the whole molecule and may involve more than one extracellular loop. Studies with monoclonal antibodies have shown that the D antigen consists of numerous epitopes. There are many variant phenotypes in which some D epitopes are missing, making it possible for a D-like antibody to be produced. Some of these D variants, often referred to as partial D antigens, result from missense mutations in RHD, others because a section of RHD, ranging from part of an exon to several exons, has been replaced by the equivalent region of RHCE. These hybrid RHD-CE-D genes are probably the products of gene misalignment and intergenic recombination during meiosis. RHCE-D-CE hybrid genes are responsible for some CcEe variants.24,25
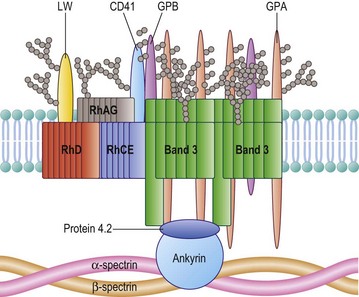
The Rh proteins are closely associated within the membrane with the Rh-associated glycoprotein (RhAG), probably as a heterotrimer.28,29 RhAG has sequence and conformational similarities to the Rh proteins, but has a single N-glycan (Fig. 37.2). This complex is part of a larger protein complex in the red cell membrane, which also includes tetramers of band 3 (the anion transporter, AE1), glycophorins A and B, ICAM-4 (the LW blood group antigen), and CD47, the integrin-associated antigen, in addition to ankyrin and protein 4.2 of the cytoskeleton and some cytosolic proteins, including carbonic anhydrase II and hemoglobin (Fig. 37.3).30 Although the primary function of band 3 is well known – it transfers HCO3− ions across the membrane in exchange for Cl− ions – the function of the Rh protein complex is not known, but there is good evidence that it could act as a gas channel, probably for CO2 and possibly also for O2 and NH3.29–31 The Rh proteins may also be part of another putative membrane complex that includes the glucose transporter (GLUT1), dimers of band 3, glycophorin C, the Duffy (DARC) and Kell glycoproteins, and Xk, which is attached to the cytoskeleton through protein 4.1R, p55, dematin and adducin.32–34
Rh-deficiency syndrome, Rhnull and Rhmod
Red cells with the very rare Rhnull phenotype have no Rh antigens and lack the Rh proteins. The most usual cause of Rhnull is homozygosity for inactivating mutations in RHAG. In the absence of RhAG, no Rh proteins are expressed at the red cell surface. Missense mutations in RHAG may result in reduced Rh antigen expression, a phenotype called Rhmod. Very rarely Rhnull occurs in the presence of RhAG, but with a deletion of RHD and inactivating mutations in RHCE.24,25
Rhnull and Rhmod red cells are morphologically and functionally abnormal. Most Rhnull individuals have some degree of hemolytic anemia associated with stomatocytosis and spherocytosis, which has been referred to as Rh-deficiency syndrome. Rhnull red cells have abnormal organization of their membrane phospholipids, and other anomalies associated with Rhnull include increased cation permeability partially compensated by an increase in the number of K+Na+ pumps, reduced cation and water contents, and significantly reduced CO2 permeability.24,31
HDFN caused by Rh antibodies
The most common form of HDFN results from IgG anti-D crossing the placenta and facilitating the immune destruction of D+ fetal red cells. The severity of anti-D HDFN is highly variable: the most severely affected fetuses die in utero from about the 17th week of gestation onwards; in less severe cases, hydrops fetalis may occur. In those severely affected infants born alive, jaundice may develop rapidly and lead to kernicterus. About 70% of infants who develop kernicterus die within a few days; of those who survive, many have permanent cerebral damage.8
Prior to the late 1960s, HDFN caused by anti-D was a relatively common cause of fetal and neonatal mortality and morbidity. Since then the prevalence of severe anti-D HDFN has been dramatically reduced by anti-D immunoglobulin prophylaxis, in which D− women receive an injection of anti-D IgG within 72 h of delivery of a D+ baby. This prevents immunization of the mother by D+ fetal cells at parturition, protecting D+ fetuses in subsequent pregnancies. In order to reduce the risk of immunization during the pregnancy, it is now policy in some countries to offer one or two doses of anti-D IgG to all D− pregnant women, at around 28–30 weeks’ gestation.35
In Caucasians, only about 60% of fetuses of D− women are D+. When a D− pregnant woman has anti-D, it is beneficial to be able to determine the D phenotype of her fetus in order to determine whether there is any risk from HDFN. Fetal D phenotype can be predicted by polymerase chain reaction-based tests performed on fetal DNA obtained by amniocentesis, chorionic villus sampling or, non-invasively, from cell-free fetal DNA in maternal plasma.36 Basically, the tests determine the presence or absence of RHD in order to predict whether the fetus is D+ or D−, respectively, though modifications are required to prevent errors that would arise from the presence of certain rare variant genes in Caucasians and the relatively common RHDψ in Africans. Where routine antenatal anti-D prophylaxis is offered, it must be offered to all D− pregnant women, as the D type of the fetus is unknown. To prevent unnecessary treatment of pregnant women with blood products, high-throughput technologies suitable for predictng fetal D type from fetal DNA in all D− pregnant women are being developed.37
All antibodies to Rh-system antigens should be considered capable of causing HDFN, but the only Rh antibody other than anti-D that regularly causes severe HDFN is anti-c. Anti-C, -E, -e, and -G have all caused HDFN, but the occurrence is rare and the outcome seldom severe. Methods have been developed for predicting fetal C, c and E from fetal DNA in maternal plasma.36
The Kell system and Xk
The Kell blood group system comprises 32 determinants, almost all of which represent single amino acid substitutions in the Kell glycoprotein.38 The Kell glycoprotein crosses the red cell membrane once, but is unique among blood group proteins as its N-terminal is internal and its large, globular C-terminal domain, which contains six potential N-glycosylation sites and 15 cysteine residues, is external.38 One of the cysteine residues, Cys72, is linked by a disulfide bond to Cys347 of Xk, a multiple membrane spanning protein (see below).39
The Kell glycoprotein is an enzyme; part of the neprilysin family of zinc-dependent endopeptidases. The Kell glycoprotein is able to cleave big endothelin-3, a biologically inactive 40-amino acid peptide, to endothelin-3, a 21-amino acid peptide with vasoconstrictor activity.40 It is not known, however, whether the Kell glycoprotein serves this function in vivo and people who lack the Kell glycoprotein, as a result of inactivating mutations in the KEL gene (Kell-null phenotype), are apparently healthy.
Anti-K and HDFN
Kell-system antibodies can cause severe HDFN. In Caucasian populations anti-K is often the most common immune red cell antibody outside of the ABO and Rh systems.8 Most anti-K appear to be induced by blood transfusion and it is becoming common practice for girls and women of childbearing age to be transfused only with K− red cells.
The pathogenesis of HDFN caused by anti-K differs from that due to anti-D. The severity of the anti-K disease is harder to predict than the anti-D disease. This is because there is very little correlation between anti-K titer and severity of disease and because anti-K HDFN is associated with lower concentrations of amniotic fluid bilirubin than in anti-D HDFN. Postnatal hyperbilirubinemia is not prominent in babies with anemia caused by anti-K. There is also reduced reticulocytosis and erythroblastosis in the anti-K disease, compared with anti-D HDFN. These characteristics suggest that there is less hemolysis in HDFN caused by anti-K, compared with HDFN of comparable severity due to anti-D. This has led to speculation that fetal anemia in anti-K HDFN results predominantly from a suppression of erythropoiesis.41,42 Kell glycoprotein is one of the first erythroid-specific antigens to appear on erythroid progenitors during erythropoiesis, whereas the Rh proteins appear much later.43–45 Vaughan et al46 found that in vitro proliferation of K+ erythroid blast-forming units (BFU-E) and colony-forming units (CFU-E) was specifically inhibited by monoclonal and polyclonal anti-K. They speculated that the Kell glycoprotein might be involved in regulating the growth and differentiation of erythroid progenitors, possibly by enzymatically modulating peptide growth factors on the cell surface. Consequently, binding of anti-K could block the enzymatic activity of the Kell glycoprotein and suppress erythropoiesis. This theory, however, does not take into account the Kell-null phenotype, in which no Kell glycoprotein is present in erythroid cells, yet erythropoiesis is apparently normal. It is likely, therefore, that anti-K suppresses erythropoiesis through the immune destruction of early erythroid progenitors in the fetal liver. Daniels et al.45 have used a functional assay to demonstrate that in the presence of anti-K erythroid progenitors, cultured from cord CD34+ cells derived from a K+ RhD+ baby, elicited a strong response from monocytes; no response was obtained with anti-D because Rh antigens do not appear on erythroid cells before they become hemoglobinized erythroblasts.
The Kell–Xk complex and McLeod syndrome
The 444 amino acid Xk polypeptide is unglycosylated and probably spans the membrane 10 times, with internal N- and C-termini.47 The predicted topographic arrangement is identical to that of members of a family of proteins that cotransport a neurotransmitter together with Na+ and Cl− ions, the amino acid sequence bearing closest resemblance to a Na+-dependent glutamate transporter.47
Xk protein expresses Kx antigen and is covalently linked to the Kell glycoprotein as a disulfide-bonded complex in the red cell membrane.39 The rare absence of Xk gives rise to McLeod syndrome, a multisystem disorder that results from either a gene deletion or from various inactivating mutations within XK, an X-linked gene that encodes the Xk protein.38,47 McLeod syndrome is characterized by weakness of Kell-system antigens and absence of Kx antigen (McLeod phenotype), acanthocytic red cells and elevated serum creatine kinase; late-onset muscular and neurological defects, including muscle wasting, diminished deep tendon reflex, choreiform movements, cardiomyopathy and psychiatric symptoms are common.48 It is likely that absence of Xk from the brain, where it is expressed independently of Kell,49 is responsible for most of the symptoms associated with McLeod syndrome.
A minority of patients with X-linked chronic granulomatous disease (CGD) also have the McLeod phenotype. CGD, an inherited disorder that may be either autosomal or X-linked, impairs the functioning of phagocytes resulting in severe susceptibility to infection. X-linked CGD results from deletion or inactivity of the gene (CYBB) for the beta subunit of flavocytochrome b558, or from mutations within that gene.50 The locus for X-linked CGD and the XK locus are discrete and the association of McLeod phenotype with CGD results from deletions of part of the X-chromosome that encompass both genes.4
Blood groups on red cell transporters
Membrane transporters facilitate the transfer of biologically important molecules in or out of cells. They are typically polytopic, with an even number of α-helical membrane spanning domains of about 21 amino acids each, and have both termini inside the cytosol. Four red cell membrane transporters have blood group activity: band 3, the anion exchanger is the Diego system antigen; aquaporin 1 (AQP1), a water channel, is the Colton antigen; aquaporin 3 (AQP3), a water and glycerol channel, is the Gill antigen; and HUT11, a urea transporter, is the Kidd antigen.3
Band 3 or anion exchanger 1 (AE1), the Diego blood-group antigen, functions as an anion exchanger. It is an antiporter that permits bicarbonate (HCO3−) ions to cross the membrane in exchange for Cl− ions, rapidly reversing the accumulation of HCO3− in the red cells that would occur when CO2 in the blood is hydrated to HCO3− by carbonic anhydrase located in the red cell cytoplasm. This facilitates transport of HCO3− in the plasma, greatly increasing the quantity of CO2 that the blood can convey to the lungs.51
There is only one report of a Diego-null phenotype, a child homozygous for a band 3 mutation, whose red cells had no band 3, and as a consequence no band 4.2, a glycoprotein of the membrane cytoskeleton associated with band 3. The severely hydropic, anemic baby, was delivered by emergency Cesarean section and resuscitated and kept alive by blood transfusion. A cord blood smear revealed dramatic erythroblastosis and poikilocytosis. After 3 years the child was doing reasonably well on a regimen of regular blood transfusions and daily supplements of sodium bicarbonate.52 So, absence of band 3 is compatible with life, but only with extreme medical intervention.
The Kidd red cell glycoprotein is UT-B, a urea transporter,53 which is also present in endothelial cells of the vasa recta, the vascular supply of the renal medulla.54 A urea transporter in red cells has two main functions: 1) transporting urea rapidly in and out the cells to prevent their shrinkage as they pass through the high urea concentration of the renal medulla and subsequent swelling as they leave; and 2) to prevent the red cells from carrying urea away from the renal medulla, which would decrease the urea concentrating efficacy of the kidney.55 Despite this, a Kidd-null phenotype, resulting from homozygosity for a splice site mutation and exon skipping,56,57 is relatively common in Polynesians, with an incidence of about 1 in 400.58 Individuals with the Kidd-null phenotype have no clinical symptoms, but, like UT-B knockout mice, have urine-concentrating ability reduced by about one-third.59,60 The abundance of UT-A and the water channels AQP2 and AQP3 is increased in the renal medulla of UT-B knockout mice, which could assist in the concentration of urea.61 This apparent compensation may explain why Kidd-null individuals have only a modest reduction in urine-concentrating ability. AQP3, but not AQP2, is present on red cells.
Eleven members of the aquaporin family of water channels are found in mammals; two of these, AQP1 (Colton system) and AQP3 (Gill system), are present in human red cells. Aquaporins can be divided into two groups: those like AQP1 that are permeated mostly by water and those like AQP3, known as the aquaglyceroporins, which are permeated by water, but also by other small solutes, especially glycerol.62 In addition to red cells, the highly water permeable AQP1 is present in kidney, lung, vascular endothelium, brain, and eye. AQP1 deficiency may only become important under stress conditions: individuals homozygous for disrupting mutations in AQP1 have the very rare Colton-null phenotype and about 80% reduction in red cell osmotic permeabilities;63 they were apparently healthy, but were unable to concentrate urine maximally when deprived of water.64 In addition to red cells, AQP3 is present in kidney, skin, lung, eye, and colon.62 The very rare Gill-null phenotype, which results from homozygosity for a splice site mutation in AQP3, is not associated with any obvious clinical syndrome.65
Receptors and adhesion molecules
There are some red cell surface proteins that resemble receptor or adhesion molecules, though their precise functions, at least on red cells, are not clear. Cell surface receptors bind specific ligands, often hormones, and then activate an effector that produces an intracellular signal. In addition to functioning as receptors, adhesion molecules are involved in the adhesion of cells to other cells and to the extracellular matrix.3 It is far from obvious why red cells should display adhesion molecules at their surface and some of these glycoproteins could be vestigial, having served their function during erythropoiesis.
The Duffy antigen receptor for chemokines
The Duffy glycoprotein is DARC, the Duffy antigen receptor for chemokines. It crosses the membrane seven times with a glycosylated extracellular N-terminal domain, which carries Fya or Fyb blood group activity. DARC belongs to the G protein-coupled superfamily of receptors that bind many different ligands, but especially chemokines, pro-inflammatory cytokines that motivate recruitment of leukocytes. DARC, however, lacks the characteristic Asp-Arg-Tyr (DRY) motif on the second cytoplasmic loop required for G-protein coupling and is, therefore, unlikely to be involved in cell signaling. In addition to being on red cells, DARC is abundant on the endothelium of post-capillary venules and veins of many organs.66,67
Chemokines are predominantly secreted chemotactic cytokines, which function primarily to induce the movement of leukocytes along a concentration gradient. DARC is a promiscuous chemokine receptor: it binds with high affinity to 60% of inflammatory chemokines of both CXC and CC classes, but not homeostatic chemokines.68 DARC on red cells has long been considered to function as a sink, binding excess chemokines to prevent inappropriate activation of neutrophils and disrupting chemokine gradients.69 Consequently, red cell DARC may support recruitment of leukocytes by removing ‘desensitizing’ free plasma chemokines and reducing ‘background system noise’, indirectly enhancing the chemokine-encoded signal on endothelial cells at the sites predestined for leukocyte recruitment.67,70
DARC appears to be a negative regulator of growth in prostate, breast, and non-small cell lung carcinoma, probably by the removal of angiogenic CXC chemokines from the tumors and subsequent inhibition of tumor neovascularity.71–73 In addition, DARC on vascular endothelium interacts directly with CD82, a tetraspanin expressed on cancer cells. CD82 is a suppresser of metastasis: interaction between DARC and CD82 inhibits the spread of the cancer to remote sites and also appears to induce cancer cell senescence.74
DARC is exploited by Plasmodium vivax as a receptor and is essential for invasion of the red cells by the parasite. P. vivax is responsible for tertian malaria, a form of malaria prevalent in Africa, but less severe than that caused by P. falciparum.66 A Duffy-null phenotype, Fy(a−b−), in which DARC is absent from red cells, is common in Africans, the frequency reaching 100% in some regions of West Africa.4 Fy(a−b−) red cells are refractory to invasion by P. vivax merozoites and individuals with the Fy(a−b−) phenotype are resistant to vivax malaria. Fy(a−b−) phenotype in Africans results from homozygosity for a single nucleotide substitution in a GATA-1 erythroid-specific transcription factor binding site in the promoter region of DARC.75 This mutation prevents expression of DARC on erythroid cells, although it is still present in other tissues.76 The evolutionary advantages of the Fy(a−b−) phenotype in Africans are obvious. However, the promoter mutation associated with the Fy(a−b−) phenotype is associated with lower leukocyte counts and could be responsible for the so-called ‘benign ethnic neutropenia’ in people of African origin, which could be pertinent to the management of chemotherapy.77
Glycoproteins of the immunoglobulin superfamily
The immunoglobulin superfamily (IgSF) is a large collection of glycoproteins abundant on leukocytes (Fig. 37.4). IgSF glycoproteins contain repeating extracellular domains with sequence homology to immunoglobulin domains. Each IgSF domain consists of approximately 100 amino acids and is structured into two β-sheets stabilized by a conserved disulfide bond. IgSF glycoproteins mostly function as receptors and adhesion molecules, and may be involved in signal transduction.78 IgSF molecules on red cells include the Lutheran (Lu) glycoproteins, ICAM-4 (LW antigen), erythroblast membrane-associated protein (ERMAP, the JMH protein), Scianna antigen, basigin (Ok antigen), and CD47. ICAM-4 and CD47 are part of the band 3/Rh macrocomplex.
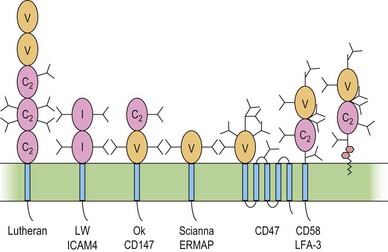
The functions of these proteins on red cells are not known, but the Lu-glycoproteins and ICAM-4 may play roles in erythropoiesis. The Lu-glycoproteins bind specifically and with high affinity to LN-10 and -11, isoforms of laminin, a component of the extracellular matrix (ECM) abundant in basement membranes and also present in vascular endothelia.79 The laminin binding site is formed by Asp312 and a surrounding group of negatively charged residues in the region of the flexible linker between IgSF domains 2 and 3 of the Lu-glycoproteins.80 During ex vivo erythropoiesis the Lu-glycoproteins appear on the erythroid cells at about the orthochromatic erythroblast stage.43,44 The presence of LN-10 and -11 on the bone marrow sinusoidal endothelium has led to speculation that the Lu-glycoproteins are involved in facilitating movement of maturing erythroid cells from the bone marrow, across the sinusoidal endothelium, to the peripheral blood.43,79
Like other ICAMs,78 ICAM-4 is a ligand for integrin adhesion molecules. Although other ICAMs are fairly specific for a single integrin, ICAM-4 interacts with a variety of integrins.81 Also, unlike other ICAMs, ICAM-4 appears to be restricted to erythroid cells and possibly placenta.82 During ex vivo erythropoiesis, LW antigen is detected around the CFU-E to proerythroblast stage.43,44 During the latter stages of erythropoiesis, erythroblasts cluster around bone marrow macrophages to form erythroblastic islands, where the erythroblasts extrude their nuclei, which are ingested by the macrophage. Adhesive interactions between ICAM-4 and α4β1 integrin on adjacent erythroblasts and between ICAM-4 on erythroblasts and αV integrins on macrophages may assist in maintaining the stability of the erythroblastic islands.82,83
Lu-glycoproteins and ICAM-4 are over-expressed on SS red cells in sickle cell disease. Enhanced binding of the Lu-glycoproteins to LN-10/11 and possibly the α4β1 integrin, and ICAM-4 to αVβ3 integrin on the endothelia of damaged blood vessels could contribute to blockage of the vessels and the painful episodes of vaso-occlusion often suffered by sickle cell patients.81,84–86 Peptides representing ICAM-4 reduce adhesion and vessel blockage, and may have therapeutic potential.85
Raph blood group and CD151
The Raph blood group system contains one antigen, MER2, located on the tetraspanin CD151. Tetraspanins associate with integrins within basement membranes to generate complexes that bind laminin, and are important in maintaining the integrity of basement membranes. The extremely rare MER2-null blood group phenotype arises from homozygosity for a single nucleotide insertion in exon 5 of the CD151 gene.87 The resulting disruption of basement membranes causes hereditary nephritis (all MER2-null patients required dialysis and kidney transplants), epidermolysis bullosa, and neurosensory deafness.87 The function of CD151 in the red cell membrane, which does not contain integrins, remains unknown.
Complement regulatory glycoproteins
DAF helps to protect red cells from lysis by autologous complement, a role it shares with CD59, another GPI-linked glycoprotein, which is not polymorphic and does not have blood group activity.88 DAF functions by inhibiting the action of C3-convertases, whereas CD59 prevents assembly of the membrane-attack complex. Patients with paroxysmal nocturnal hemoglobinuria (PNH)89 have red cells that lack both DAF and CD59 as a result of defective somatic mutations in PIGA, an X-linked gene that encodes a subunit of an enzyme essential for the biosynthesis of the GPI anchor.89 These patients often have severe hemolysis, whereas DAF-deficient, Cromer-null red cells show no signs of hemolysis and individuals with a very rare CD59-deficient phenotype had only mild hemolytic anemia.4
The major function of red cell CR1 is to bind and process C3b/C4b-coated immune complexes and transport them to the liver and spleen for removal from the circulation.90,91 No true Knops-null phenotype has been identified, though red cells of some individuals have very low CR1 copy numbers.4
Red cell glycoproteins that anchor the membrane to its skeleton
Band 3, the Diego blood-group antigen, is the red cell anion exchanger, but also has an important structural function. Band 3 has an extended N-terminal domain, which interacts with the red cell membrane cytoskeleton, a submembranous matrix of glycoproteins that is responsible for maintaining the shape and integrity of the red cell. Band 3 interacts with the membrane skeleton through ankyrin and proteins band 4.2 and band 4.1R.32–34 Heterozygosity for a 27 basepair deletion in the band 3 gene, encoding a nine amino acid deletion, is responsible for Southeast Asian ovalocytosis (SAO), a condition common in the southern Pacific region and among Melanesians. About 20% of cases of hereditary spherocytosis, a common, familial hemolytic anemia characterized by small spheroid red cells, result from mutations within the band 3 gene and an absence or decrease of the mutant protein from the red cell membrane.51
The Gerbich blood-group antigens are located on two red cell glycoproteins, glycophorins C and D (GPC and GPD), encoded by a single gene (GYPC) by initiation of translation at two sites.4,5 The C-terminal cytoplasmic domains of GPC and GPD form a complex with the cytoskeletal protein 4.1R. GPC and GPD may be part of a proposed membrane complex that includes the glucose transporter (GLUT1), dimers of band 3, Rh proteins, the Duffy (DARC) and Kell glycoproteins, and Xk, attached to the cytoskeleton through protein 4.1R, p55, dematin and adducin.32–34
In addition there is evidence that RhAG interacts with ankyrin, the Lutheran and Xk proteins with spectrin, and CD44 with protein 4.1R. Red cells lacking RhAG or Xk, or with reduced levels of Lutheran glycoproteins and CD44, have some degree of abnormal morphology.34
Associations with infectious disease
Cell surface antigens may be exploited by pathogenic microorganisms for attachment to the cells and subsequent invasion. Many such associations have been reported involving antigens present on red cells, although the targets for the pathogens are often cells other than erythroid cells. Examples of these are associations are Leb and Helicobacter pylori, P antigen and the Dra antigen of CD55 and Escherichia coli, CD35 (Knops antigen) and Mycobacterium leprae, and AnWj (an antigen associated with CD44) and Hemophilus influenzae (reviewed in 92 and 93). Mononuclear cells of individuals with the Pk red cell phenotype, which arises from deficiency of the glycosyltransferase that converts the glycolipid antigen Pk (Gb3) to P (Gb4), were resistant to HIV-1 infection.94 P antigen (globoside, Gb4) is a cellular receptor for parvovirus B19, a human pathogen that replicates in erythroid progenitor cells. B19 is the cause of fifth disease, a common childhood illness, and occasionally more severe disorders of erythropoiesis, particularly in immunocompromised patients.95 Individuals with P deficiency (p phenotype) appear to be naturally resistant to parvovirus B19 infection. Fumagalli et al.96 concluded that ‘blood group antigens have been playing a central role in the host-pathogen arms race during human evolutionary history and no other gene category shows similar levels of widespread selection, with the only exception of loci involved in antigen recognition’.
Red cells of the rare phenotypes in which the sialic acid-rich red cell glycoproteins glycophorins A, B, or C/D are absent are relatively resistant to invasion by Plasmodium falciparum.97 These structures are all exploited as ligands for P. falciparum receptors.98–100 Absence of the Sla antigen of the Knops blood group, which is expressed on CR1 (CD35), and red cells with low expression of CD35, demonstrate reduced rosetting when parasitized with P. falciparum, a characteristic associated with severe disease.101 Sl(a−) phenotype and CD35 deficiency are relatively rare in Caucasians, but are present in about 70% of West Africans and 80% of Papua New Guineans, respectively.4 The association between the malarial parasite Plasmodium vivax and the Duffy antigen is described in a previous section of this chapter.
A major selection pressure responsible for the abundance of O alleles of ABO, which almost certainly evolved from A by a single nucleotide deletion, could be Plasmodium falciparum malaria. Despite some apparent discrepancies and contradictions, the overall impression from the literature is that individuals of blood group O are relatively resistant to severe malaria caused by infection with Plasmodium falciparum.102,103 The geographic distribution of group O is consistent with selection by P. falciparum in favor of group O individuals in malaria endemic regions.104
Platelet antigens
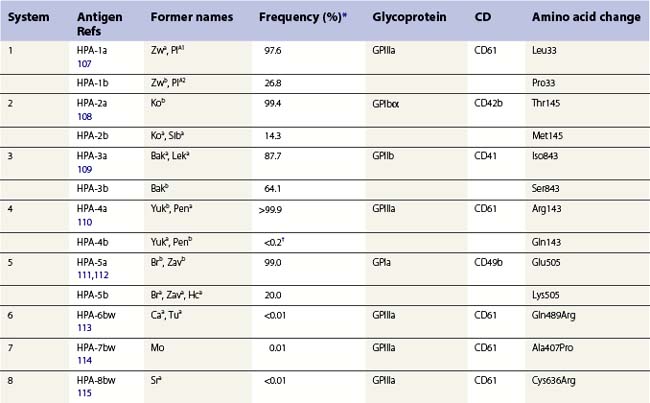
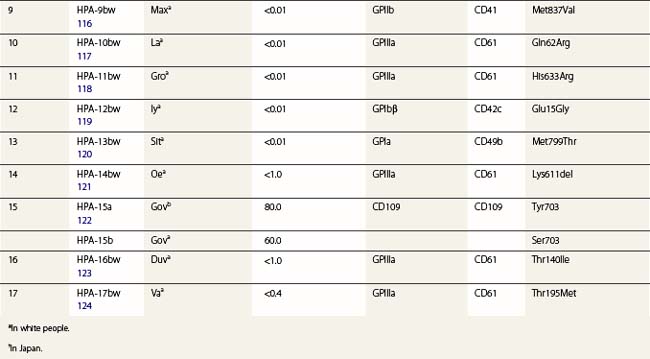
The normal hemostatic function of platelets involves membrane glycoproteins, which function as receptors during platelet adhesion and aggregation. Several of the key platelet glycoproteins are polymorphic. Many of these polymorphisms are alloantigenic and were first recognized because of feto-maternal incompatibility characterized by the production of platelet-specific alloantibodies in the mother and fetal and neonatal alloimmune thrombocytopenia (NAIT). In addition to the well-established role of platelet antigens in the pathogenesis of NAIT, some alloantigenic polymorphisms may affect platelet function and the predisposition to thrombotic disease. Although platelet alloantigens have been identified by many groups around the world, resulting in a variety of different terminologies, the human platelet antigen (HPA) nomenclature has been almost universally adopted.105,106 Currently, 17 systems are recognized, six containing a pair of alloantigens (1 to 5 and 15) and the remaining 11 containing a single antigen of low frequency (Table 37.2).125 Unlike the red cell blood group systems, HPA systems represent antithetical antigens, so several different systems can be encoded by the same gene and their antigens present on the same protein. The letters a and b designate the higher and lower frequency antigens, respectively. The letter w (workshop) is added when the antithetical alloantigen has not been identified. Alleles of genes encoding six platelet membrane glycoproteins result in the expression of alloantigens on the cell surface (Table 37.2).
Serological methods for detecting platelet antigens generally involve the use of fluorescent-labeled antibodies. A 10-year review on external quality assessment of these methods was published in 2008.126 Molecular methods, often involving the application of DNA microarray technology, are also available to predict platelet phenotypes.127
Glycoproteins expressing platelet antigens
Antigens on the GPIIb/IIIa (CD41/CD61 or αIIbβ3) complex
GPIIb/IIIa is the most abundant platelet integrin, present at approximately 80 000 copies per platelet. Integrins are a family of hererodimeric proteins, composed of non-covalently associated α- and β-subunits.128 Upon platelet activation, GPIIb/IIIa is involved in platelet aggregation by binding adhesive proteins such as fibrinogen, fibrin, fibronectin, vitronectin, thrombospondin and von Willebrand factor (vWF).128,129 Glanzmann’s thrombasthenia, a bleeding disorder, results when GPIIb/IIIa is absent or dysfunctional.
Three alloantigenic forms of GPIIb have been identified (HPA-3a, -3b, -9bw). Carbohydrate residues on GPIIb appear to contribute to the binding site of HPA-3 antibodies.130
GPIIIa is the most polymorphic molecule on the platelet surface apart from HLA class I. The molecular bases of 12 antigens on GPIIIa have been determined (Table 37.2). Ten of the antigens (HPA-1b, -4b, -6bw, -7bw, -8bw, -10bw, -11bw, 14bw, 16bw and 17bw) are encoded by relatively rare single nucleotide substitutions in the common form of GPIIIa. The exception is the HPA-14bw, which results from the deletion of three nucleotides, encoding a single amino acid deletion.
HPA-1a is the most frequent cause of maternal alloimmunization to fetal platelets and is responsible for most cases of NAIT in Caucasians (discussed below). Although only 5–10% of HPA-1a-negative women with HPA-1a-positive fetuses produce anti-HPA-1a, the presence of the HLA-DRB3*0101 allele increases the risk of alloimmunization by a factor of 140.131 HPA-1b results from a Leu33Pro substitution. The immune response to HPA-1b does not appear to be HLA restricted.132 An explanation for this observation was provided when Wu et al.133 used an in vitro peptide-binding assay to show that leucine, but not proline, at position 33 anchors the peptide to the HLA-DRB3*0101-encoded molecule.
Antigens on the GPIa/IIa (CD49/CD29 or α2β1)complex
GPIa/IIa, also known as VLA-2 (very late antigen-2), is an integrin expressed at relatively low density on platelets as well as on activated T-lymphocytes. The GPIa/IIa heterodimer comprises a 167 kDa α2 polypeptide and a 130 kDa β1 polypeptide. GPIa/IIa integrin is a receptor for collagen and functions in collagen-mediated platelet activation.128 A mild bleeding disorder may result when the complex is absent.134
Three alloantigens (HPA-5a, -5b and -13w) reside on GPIa; none on GPIIa (Table 37.2). Alloimmunization to the HPA-5b antigen could be responsible for approximately 15% of cases of NAIT in Caucasians (described below). Silent SNPs within codons 224 and 246 of the GPIa gene are associated with up to tenfold variation in expression of the GPIa/IIa complex.135 Collagen-induced aggregation responses of HPA-13bw-positive platelets were diminished relative to HPA-13bw-negative platelets, indicating that the Met799Thr substitution affects the function of the GPIa/IIa complex.120 None of the HPA-13bw-positive individuals studied, however, had any signs of altered hemostasis.
Antigens on the GPIb/IX/V (CD42) complex
GPIb/IX/V binds VWF and mediates the adhesion of platelets to exposed vascular subendothelium surfaces under conditions of high shear stress, and interaction essential for slowing circulating platelets, allowing formation of arterial thrombi. The GPIb-VWF interaction can also activate platelets through intracellular signals.128 GPIb comprises two disulfide-bonded subunits, GPIbα (CD42b) and GPIbβ (CD42c). GPIb is non-covalently associated with GPIX (CD42a) and GPV (CD42d). There are approximately 25 000 GPb/IX molecules and 12 000 GPV molecules per platelet. Absence of the complex results in the inherited bleeding disorder Bernard–Soulier syndrome.
Three antigens (HPA-2a, -2b and -12bw) on GPIb have been described (Table 37.2), but none on GPIX or GPV. In addition to these antigenic variations, there are four size variants of GPIbα: A, B, C and D (from largest to smallest). The differences arise from the number of 13-amino acid tandem repeats within the GPIbα (A = 4, B = 3, C = 2, D = 1).136 Only the relatively rare A and B forms of glycoprotein Ibα contain Met145, which is required for the expression of the HPA-2b epitope.137
Antigens on CD109
CD109 is a member of the α2-macroglobulin/complement (AMCOM) superfamily, which contains α2-macroglobulin and the complement components C3, C4 and C5. CD109 is a 175 kDa GPI-anchored glycoprotein expressed on platelets, activated T-lymphocytes, cultured endothelial cells and several tumor cell lines.138–140 CD109 expresses the antithetical alloantigens HPA-15a and -15b (Table 37.2).141 Antibodies to both antigens may cause NAIT, but they are encountered rarely.
Fetal and neonatal alloimmune thrombocytopenia
A variety of names and acronyms have been used for the condition characterized by thrombocytopenia in a fetus or neonate caused by maternal antibodies crossing the placenta and facilitating the immune destruction of fetal platelets. Because of the analogy with HDFN, the term fetal and neonatal alloimmune thrombocytopenia, but with the more usual acronym NAIT, will be used here. For reviews see.142–144
Pathophysiology
In Caucasians, NAIT occurs in about 1 in 1000–2000 births. All the HPAs listed in Table 37.2 have been associated with NAIT. Anti-HPA-1a is the dominant cause of NAIT in Caucasians, responsible for about 80% of cases.145 After anti-HPA-1a, anti-HPA-5b and -15a are most frequently responsible, followed by anti-HPA-1b, -3a, and -2a/2b.143,145,146 In people from Africa and eastern Asia HPA-1b is very rare, so anti-HPA-1a antibodies are also rare.147 In Japanese people, for example, anti-HPA-4a and -4b cause NAIT more frequently than other platelet antibodies.148
The natural history of NAIT differs from HDFN in that maternal sensitization to fetal platelet antigens often occurs in the first pregnancy. Approximately 40% of cases of NAIT occur in primiparae. Although the mechanisms involved in immunization remain unclear,142 GPIIIa, which expresses HPA-1 antigens, is present on placental syncytiotrophoblast microvilli from the first trimester.149 Syncytiotrophoblasts are naturally shed into the maternal circulation and could be the source of HPA-1a antigen responsible for early immunization of HPA-1a-negative primigravidae.
Maternal HPA antibodies are predominantly IgG1150 and are transported across the placenta to the fetus. Studies of autoimmune thrombocytopenia in adults suggest that antibody-mediated platelet destruction involves the sequestration of platelets by splenic macrophages in a manner analogous to the extravascular destruction of IgG-sensitized red cells.151 Fcγ receptor III (FcγRIII) may be involved in the recognition of sensitized platelets: administration of monoclonal anti-FcγRIII to a patient with autoimmune thrombocytopenia was associated with a rise in platelet count.152
Clinical features
Fetal thrombocytopenia may start early in pregnancy: almost 50% of affected fetuses have a platelet count of less than 20 × 109/l.153 There is no spontaneous remission of the thrombocytopenia in utero and, in the absence of therapy, platelet counts usually fall as gestation progresses. In the absence of screening programs, alloimmune thrombocytopenia is usually recognized at birth when the majority of affected cases have petechiae, purpura or overt bleeding. Intracerebral hemorrhage (ICH) occurs in about 20% of affected infants and about one-third of these events are fatal. If non-fatal, mental retardation, cerebral palsy, cortical blindness and seizures often result. Up to 80% of ICH occur prenatally, as early as 16 weeks’ gestation, and may lead to structural and metabolic central nervous system abnormalities.8,143
Clinically, NAIT is a diagnosis of exclusion. Infants have no signs of disseminated intravascular coagulation, infection, or congenital anomalies that may be associated with thrombocytopenia. The mother has no history of autoimmune disease, thrombocytopenia, or ingestion of drugs that may cause thrombocytopenia. At delivery, standard laboratory tests show that neonatal platelet counts are low. Fetal hemoglobin concentration may be low if bleeding has occurred. Bone marrow biopsy usually reveals normal levels of megakaryocytes. Cranial ultrasound and computerized tomographic scans may reveal dilated cerebral ventricles and evidence of intraventricular hemorrhage.154 Once suspected on clinical grounds, a provisional diagnosis of NAIT should be confirmed by establishing the presence of platelet-specific alloantibodies in the maternal serum that react with fetal platelets.
Neutrophil antigens
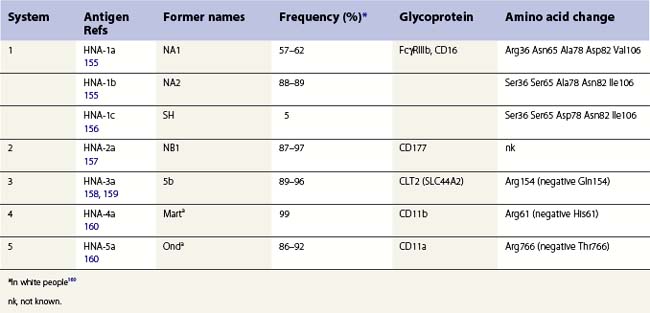
Granulocyte-specific polymorphisms are less well characterized than those on red cells and platelets. Nevertheless, it is well established that some polymorphisms are antigenic and capable of eliciting alloimmune responses resulting in, for example, neonatal alloimmune neutropenia (NAIN). Most neutrophil antigens are expressed on membrane receptors involved in inflammation and the clearance of immune complexes and there is some evidence that these polymorphisms might affect cell function and the risk of some infectious or autoimmune diseases. An HNA terminology, analogous to the HPA terminology for platelets, has been established by the International Society for Blood Transfusion for granulocyte-specific antigens and those shared with a limited number of other cell types (Table 37.3).161,162
Glycoproteins expressing neutrophil antigens
Antigens classified in the HNA terminology are located on FcγRIIIb (CD16), CD177, CD11a, CD11b, and CLT2 (SLC44A2) (Table 37.3). Neutrophils also express some antigens with a wide tissue distribution. These include the I and P1 blood group antigens, Lex and sialyl-Lex (CD15), and HLA class I, and are not discussed here.
Antigens on Fcγ receptor III (FcγRIIIb, CD16)
HNA-1a, -1b and -1c are located on Fcγ RIIIb,155,156 a low-affinity receptor for complexed IgG, which is selectively expressed on neutrophils.163 FcγRIIIb has an extracellular region consisting of two disulfide-bonded immunoglobulin-like domains and is linked to the plasma membrane by a GPI-anchor. When subjected to polyacrylamide gel electrophoresis, Fcγ RIIIb migrates as a broad band with a molecular weight between 50 kDa and 80 kDa. The differences in electrophoretic migration arise from amino acid differences between HNA-1a and HNA-1b at positions 65 and 82, which result in two additional glycosylation sites in HNA-1b (Ser65, Asn82).155 FCGR3B*01 and FCGR3B*02, the alleles encoding HNA-1a and HNA-1b, differ at five SNPs, but only four of these give rise to amino acid changes (Table 37.3). HNA-1c has an identical amino acid sequence to HNA-1b, except for a Ala78Asp substitution.156 HNA-1c expression is often associated with the presence of an additional Fcγ RIIIb gene, probably as the result of gene duplication and giving rise to an increased quantity of Fcγ RIIIb.164 Frequencies for HNA-1 alloantigens in Caucasians are shown in Table 37.3. In eastern Asia HNA-1a is more common (88–91%), HNA-1b less common (51–54%) and HNA-1c is relatively rare; in Africans HNA-1c is more common (23–31%) and HNA-1b is less common (46–66%) than in Caucasian, with HNA-1a about the same.161
LAN and SAR are high frequency antigens on Fcγ RIIIb, but their molecular genetic bases are unknown.165,166
Approximately 0.1% of Caucasians and 4% of black Africans do not have FcγRIIIb as a result of gene deletion. Women with this HNA-1 null phenotype can form isoantibodies causing severe neonatal neutropenia.167
Antigens on CD11/CD18
The HNA-4a and -5a alloantigens are located on α chains of β2 integrins: HNA-4a on αMβ2 (CD11b/CD18, Mac-1); HNA-5a on αLβ2 (CD11a/CD18, LFA-1). CD11a is expressed on all leukocytes while CD11b is expressed on granulocytes, monocytes and natural killer cells. Both alloantigens differ from their relatively uncommon, antigen-negative alternatives by a single amino acid (Table 37.3).
Neutrophil antigens on other glycoproteins
HNA-2a is present on granulocytes from between 87% and 99% of Caucasians, Asians, and Africans.161 Antibodies to HNA-2a recognize CD177, a 56–64 kDa GPI-linked glycoprotein present on the neutrophil surface and secondary granules.168 HNA-2a-negative phenotype may result from splicing defects in CD177.169 Expression of HNA-2a is reduced on neutrophils from patients with PNH or chronic myelocytic leukemia, but this appears to be of no clinical relevance.8 The antibody named NB2 does not appear to be antithetical to anti-HNA-2a (initially named anti-NB1), and may detect human monocyte antigen 1.125
HNA-3a is carried on choline transporter-like protein-1 (SLC44A2), a glycoprotein of unknown function with homology to choline transporters. HNA-3-negative phenotype results from Arg154Gln.158,159
Clinical syndromes associated with neutrophil antibodies
There are five main clinical syndromes that may be caused by antibodies to HNAs: neonatal alloimmune and isoimmune neutropenia; autoimmune neutropenia; transfusion-related alloimmune neutropenia; transfusion-related acute lung injury; and febrile reactions following transfusion.8,125
Neonatal alloimmune neutropenia (NAIN)
NAIN occurs in approximately 1 in 1500 births. It is characterized by neutropenia in a fetus or infant caused by placental transfer of neutrophil IgG antibodies. HNAs are well expressed from before birth and the condition may occur in the first-born infant.170 Anti-HNA-1a and 2a are the most common causes of NAIN.
NAIN usually has a relatively benign course with infants developing mild infections, predominantly of the skin and mucous membranes. Occasionally, more serious infections resulting in pneumonia and meningitis develop. Hematologic investigation reveals a severe neutropenia, which may persist for up to 6 months.170 NAIN is probably under-diagnosed as infants are usually asymptomatic at birth and white cell counts might not be performed routinely. Diagnosis is largely one of exclusion. In the absence of disease processes, drugs and environmental factors that can cause neutropenia or impairment of neutrophil function, a provisional diagnosis of NAIN should be confirmed by demonstrating the presence of antibodies in the maternal plasma that react with the infant’s neutrophils.
Autoimmune neutropenia of infancy
In addition to their role in the pathogenesis of NAIN, some alloantigenic polymorphisms on neutrophils are also the target of autoantibodies implicated in autoimmune neutropenia of infancy. Disease onset usually occurs between 4 months and 2 years with spontaneous remission in 95% of patients within 7–24 months.171,172 It is a relatively benign condition associated with infections of the skin, middle ear, throat, and respiratory and digestive tracts. The incidence of autoimmune neutropenia of infancy is approximately 1 in 100 000.173 Pathogenesis may involve a deficiency of suppresser T-lymphocytes, which results in the production of granulocyte-specific IgG or IgM autoantibodies, usually specific for HNA-1a.171
Transfusion-related acute lung injury (TRALI)
Characterized by acute respiratory distress and non-cardiogenic lung edema developing during or within 6 hours of blood transfusion, TRALI is one of the most common causes of transfusion-associated morbidity and death.174,175 Antibody-mediated TRALI is triggered by leukocyte antibodies, either HLA class I or II antibodies or HNA antibodies.176,177 In most cases TRALI results from antibodies in transfused plasma, but occasionally the reaction occurs between the recipient’s neutrophil antibodies and transfused neutrophils. Neutrophils stimulated by leukocyte antibodies aggregate and release cytotoxic enzymes that damage the endothelial cells of the lung capillaries. Antibodies to HNA-1a, 1b, 2a and 3a have all been incriminated in TRALI.8 HNA-3a antibodies are rare and difficult to detect, but have been associated with several fatal cases.178,179
1 Daniels GL, Fletcher A, Garratty G, et al. Blood group terminology 2004. From the ISBT Committee on Terminology for Red Cell Surface Antigens. Vox Sanguinis. 2004;87:304-316.
2 Daniels G, Castilho L, Flegel WA, et al. International Society of Blood Transfusion Committee on Terminology for Red Cell Surface Antigens: Macao Report. 3. Vox Sanguinis. 2009;96:153-156. Also see online http://ibgrl.blood.co.uk/
3 Daniels G. Functions of red cell surface proteins. Vox Sanguinis. 2007;93:331-340.
4 Daniels G. Human blood groups, 2nd ed. Oxford: Blackwell Science; 2002.
5 Reid ME, Lomas-Francis C. The blood group antigen facts book, 2nd ed. London: Academic Press; 2004.
6 Yamamoto F, Clausen H, White T, et al. Molecular genetic basis of the histo-blood group ABO system. Nature. 345, 1990. 229–223
7 Chester MA, Olsson ML. The ABO blood group gene: a locus of considerable genetic diversity. Transfusion Medicine Reviews. 2001;15:177-200.
8 Klein HG, Anstee DJ. Mollison’s Blood Transfusion in Clinical Medicine, 11th ed. Oxford: Blackwell Publishing; 2005.
9 Gold ER, Tovey GH, Benney WE, et al. Changes in the group A antigen in a case of leukaemia. Nature. 1959;183:892-893.
10 Bianco T, Farmer BJ, Sage RE, Dobrovic A. Loss of red cell A, B, and H antigens is frequent in myeloid malignancies. Blood. 2001;97:3633-3639.
11 Salmon C, Cartron JP, Lopez M, et al. Level of the A, B and H blood group glycosyltransferases in red cell membranes from patients with malignant hemopathies. Revue Française de Transfusion et Immuno-Hématologie. 1984;27:625-637.
12 Bianco-Miotto T, Hussey DJ, Day TK, et al. DNA methylation of the ABO promoter underlies loss of ABO allelic expression in a significant proportion of leukemic patients. PloS ONE. 2009;4:e4788.
13 Henry S, Oriol R, Samuelsson B. Lewis histo-blood group system and associated secretory phenotypes. Vox Sanguinis. 1995;69:166-182.
14 Hakomori S. Antigen structure and genetic basis of histo blood groups A, B and O: their changes associated with human cancer. Biochimica et Biophysica Acta. 1999;1473:246-266.
15 Le Pendu J, Marionneau S, Cailleau-Thomas A, et al. ABH and Lewis histo-blood group antigens in cancer. APMIS. 2001;109:9-31.
16 Dabelsteen E, Gao S. ABO blood-group antigens of oral cancer. Journal of Dental Research. 2004;84:21-28.
17 Chihara Y, Sugano K, Kobayashi A, et al. Loss of blood group A antigen expression in bladder cancer caused by allelic loss and/or methylation of the ABO gene. Laboratory Investigation. 2005;85:895-907.
18 Ørntoft TF, Bech E. Circulating blood group related carbohydrate antigens as tumour markers. Glygoconjugate Journal. 1995;12:200-205.
19 Steinberg W. The clinical utility of the CA 19-9 tumor-associated antigen. American Journal of Gastroenterology. 1990;85:350-355.
20 Grote T, Logsdon CD. Progress on molecular markers of pancreatic cancer. Current Opinion in Gastroenterology. 2007;23:508-514.
21 Clausen H, Hakomori S, Graem N, et al. Incompatible A antigen expressed in tumors of blood group O individuals: immunochemical, immunohistologic, and enzymatic characterization. Journal of Immunology. 1986;136:326-330.
22 David L, Leitao D, Sobrinho-Simoes M, et al. Biosynthetic basis of incompatible histo-blood group A antigen expression: anti-A transferase antibodies reactive with gastric cancer tissue of type O individuals. Cancer Research. 1993;53:5494-5500.
23 Hakomori S. Immunochemical and molecular genetic basis of the histo-blood group ABO(H) and related antigen system. Baillière’s Clinical Haematology. 1991;4:957-997.
24 Avent ND, Reid ME. The Rh blood group system: a review. Blood. 2000;95:375-387.
25 Huang C-H, Liu PZ, Cheng JG. Molecular biology and genetics of the Rh blood group system. Seminars in Hematology. 2000;37:150-165.
26 Wagner FF, Flegel WA. The RHD gene deletion occurred in the Rhesus box. Blood. 2000;95:3662-3668.
27 Singleton BK, Green CA, Avent ND, et al. The presence of an RHD pseudogene containing a 37 base pair duplication and a nonsense mutation in most Africans with the Rh D-negative blood group phenotype. Blood. 2000;95:12-18.
28 Conroy MJ, Bullough PA, Merrick M, Avent ND. Modelling the human rhesus proteins: implications for structure and function. British Journal of Haematology. 2005;131:543-551.
29 Burton NM, Anstee DJ. Nature, function and significance of Rh proteins in red cells. Current Opinion in Hematology. 2008;15:625-630.
30 Bruce LJ, Beckmann R, Ribeiro ML, et al. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101:4180-4188.
31 Endeward V, Cartron J-P, Ripoche P, Gros G. RhAG protein of the Rhesus complex is a CO2 channel in the human red cell membrane. FASEB Journal. 2008;22:64-73.
32 Salomao M, Zhang X, Yang Y, et al. Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proceedings of the National Academy of Sciences. 2008;105:8026-8031.
33 Khan AA, Toshihiko H, Mohseni M, et al. Dematin and adducin provide a novel link between the spectrin cytoskeleton and human erythrocyte membrane by directly interacting with glucose transporter-1. Journal of Biological Chemistry. 2008;238:14600-14609.
34 Mohandas N, Gallagher PG. Red cell membrane: past, present, and future. Blood. 2008;112:3939-3948.
35 Pilgrim H, Lloyd-Jones M, Rees A. Routine antenatal anti-D prophylaxis for RhD-negative women: a systematic review and economic evaluation. Health Technology Assessment. 2009;13:1-103.
36 Daniels G, Finning K, Martin P, Massey E. Non-invasive prenatal diagnosis of fetal blood group phenotypes: current practice and future prospects. Prenatal Diagnosis. 2009;29:101-107.
37 Finning K, Martin P, Summers J, et al. Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti-RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. British Medical Journal. 2008;336:816-818.
38 Lee S, Russo D, Redman C. Functional and structural aspects of the Kell blood group system. Transfusion Medicine Reviews. 2000;14:93-103.
39 Russo D, Redman C, Lee S. Association of XK and Kell blood group proteins. Journal of Biological Chemistry. 1998;273:13950-13956.
40 Lee S, Lin M, Mele A, et al. Proteolytic processing of big endothelin-3 by the Kell blood group protein. Blood. 1999;94:1440-1450.
41 Vaughan JI, Warwick R, Letsky E, et al. Erythropoietic suppression in fetal anemia because of Kell alloimmunization. American Journal of Obstetrics and Gynecology. 1994;171:247-252.
42 Weiner CP, Widness JA. Decreased fetal erythropoiesis and hemolysis in Kell hemolytic anemia. American Journal of Obstetrics and Gynecology. 1996;174:547-551.
43 Southcott MJG, Tanner MJA, Anstee DJ. The expression of human blood group antigens during erythropoiesis in a cell culture system. Blood. 1999;93:4425-4435.
44 Bony V, Gane P, Bailly P, et al. Time-course expression of polypeptides carrying blood group antigens during human erythroid differentiation. British Journal of Haematology. 1999;107:263-274.
45 Daniels G, Hadley A, Green CA. Causes of fetal anemia in hemolytic disease due to anti-K. Transfusion. 2003;43:115-116.
46 Vaughan JI, Manning M, Warwick RM, et al. Inhibition of erythroid progenitor cells by anti-Kell antibodies in fetal alloimmune anemia. New England Journal of Medicine. 1998;338:798-803.
47 Ho M, Chelly J, Carter N, et al. Isolation of the gene for McLeod syndrome that encodes a novel membrane transport protein. Cell. 1994;77:869-880.
48 Danek A, Rubio JP, Rampoldi L, et al. McLeod neuroacanthocytosis: genotype and phenotype. Annals of Neurology. 2001;50:755-764.
49 Clapéron A, Hattab C, Armand V, et al. The Kell and Xk proteins of the Kell blood group are not co-expressed in the central nervous system. Brain Research. 2007;1147:12-24.
50 Roos D, de Boer M, Kuribayashi F, et al. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood. 1996;87:1663-1681.
51 Bruce L. Mutations in band 3 and cation leaky red cells. Blood Cells and Molecular Disease. 2006;36:331-336.
52 Ribeiro ML, Alloisio N, Almeida H, et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with total absence of band 3. Blood. 2000;96:1602-1604.
53 Olivès B, Mattei M-G, Huet M, et al. Kidd blood group and urea transport function of human erythrocytes are carried by the same protein. Journal of Biological Chemistry. 1995;270:15607-15610.
54 Sands JM. Renal urea transporters. Current Opinions in Nephrological Hypertension. 2004;13:525-532.
55 Macey RI, Yousef LW. Osmotic stability of red cells in renal circulation required rapid urea transport. American Journal of Physiology. 1988;254:C669-C674.
56 Lucien N, Sidoux-Walter F, Olivès B, et al. Characterization of the gene encoding the human Kidd blood group/urea transporter protein. Evidence for splice site mutations in Jknull individuals. Journal of Biological Chemistry. 1998;273:12973-12980.
57 Irshaid NM, Henry SM, Olsson ML. Genomic characterization of the Kidd blood group gene: different molecular basis of the Jk(a–b–) phenotype in Polynesians and Finns. Transfusion. 2000;40:69-74.
58 Henry S, Woodfield G. Frequencies of the Jk(a−b−) phenotype in Polynesian ethnic groups. Transfusion. 1995;35:277.
59 Sands JM, Gargus JJ, Fröhlich O, et al. Urinary concentrating ability in patients with Jk(a−b−) blood type who lack carrier-mediated urea transport. Journal of the American Society of Nephrology. 1992;2:1689-1696.
60 Yang B, Bankir L, Gillespie A, et al. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. Journal of Biological Chemistry. 2002;277:10633-10637.
61 Klein JD, Sands JM, Qian L, et al. Upregulation of urea transporter UT-A2 and water channels AQP2 and AQP3 in mice lacking urea transporter UT-B. Journal of the American Society of Nephrology. 2004;15:1161-1167.
62 King LS, Kozono D, Agre P. From structure to disease: the evolving tale of aquaporin biology. Nature Reviews Molecular Cell Biology. 2004;5:687-698.
63 Preston GM, Smith BL, Zeidel ML, et al. Mutations in aquaporin-1 in phenotypically normal humans without functional CHIP water channels. Science. 1994;265:1585-1587.
64 King LS, Choi M, Fernandez PC, et al. Defective urinary concentrating ability due to a complete deficiency of aquaporin-1. New England Journal of Medicine. 2001;345:175-179.
65 Roudier N, Ripoche P, Gane P. AQP3 deficiency in humans and the molecular basis of a novel blood group system, GIL. Journal of Biological Chemistry. 2002;48:45854-45859.
66 Hadley TJ, Peiper SC. From malaria to chemokine receptor: the emerging physiologic role of the Duffy blood group antigen. Blood. 1997;89:3077-3091.
67 Rot A. Contribution of Duffy antigen to chemokine function. Cytokine and Growth Factor Reviews. 2005;16:687-694.
68 Gardner L, Patterson AM, Ashton BA, et al. The human Duffy antigen binds selected inflammatory but not homeostatic chemokines. Biochemical Biophysical Research Communications. 2004;32:306-312.
69 Darbonne WC, Rice GC, Mohler MA, et al. Red blood cells are a sink for interleukin 8, a leukocyte chemotaxin. Journal of Clinical Investigation. 1991;88:1362-1369.
70 Pruenster M, Rot A. Throwing light on DARC. Biochemical Society Transactions. 2006;34:1005-1008.
71 Shen H, Schuster R, Stringer KF, et al. The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB Journal. 2006;20:59-64.
72 Wang J, Ou Z-L, Hou Y-F. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene. 2006;25:7201-7211.
73 Addison CL, Belperio JA, Burdick MD, Strieter RM. Overexpression of the duffy antigen receptor for chemokines (DARC) by NSCLC tumor cells results in increased tumor necrosis. BMC Cancer. 2004;4:28.
74 Bandyopadhyay S, Zhan R, Chaudhuri A. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nature Medicine. 2006;12:933-938.
75 Tournamille C, Colin Y, Cartron JP, et al. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nature Genetics. 1995;10:224-228.
76 Peiper SC, Wang Z, Neote K, et al. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy-negative individuals who lack the erythrocyte receptor. Journal of Experimental Medicine. 1995;181:1311-1317.
77 Afenyi-Annan A, Ashley-Koch A, Telen MJ. Duffy (Fy), DARC, and neutropenia among women from the United States, Europe and the Caribbean. British Journal of Haematology. 2009;145:266-267.
78 Isacke CM, Horton MA. The Adhesion Molecule Facts Book, 2nd ed. London: Academic Press; 2000.
79 Parsons SF, Lee G, Spring FA. Lutheran blood group glycoprotein and its newly characterized mouse homologue specifically bind α5 chain-containing human laminin with high affinity. Blood. 2001;97:312-320.
80 Mankelow TJ, Burton N, Stefansdottir FO, et al. The laminin 511/521 binding site on the Lutheran blood group glycoprotein is located at the flexible junction of Ig domains 2 and 3. Blood. 2007;110:3398-3406.
81 Delahunty M, Zennadi R, Telen MJ. LW protein: a promiscuous integrin receptor activated by adrenergic signaling. Transfusion Clinical Biology. 2006;13:44-49.
82 Parsons SF, Spring FA, Chasis JA, et al. Erythroid cell adhesion molecules Lutheran and LW in health and disease. Baillière’s Clinical Haematology. 1999;12:729-745.
83 Lee G, Lo A, Short SA. Targeted gene deletion demonstrates that cell adhesion molecule ICAM-4 is critical for erythroblastic island formation. Blood. 2006;108:2064-2071.
84 Eyler CE, Telen MJ. The Lutheran glycoprotein: a multifunctional adhesion receptor. Transfusion. 2006;46:668-677.
85 Kaul DK, Liu X, Zhanmg X. Peptides based on αV-binding domains of erythrocyte ICAM-4 inhibit sickle red cell-endithelial interactions and vaso-occlusion in the microcirculation. American Journal of Physiology and Cell Physiology. 2006;291:C922-C930.
86 El Nemer W, Wautier M-P, Rahuel C. Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell α4β1 integrin: role in adhesion of sickle red blood cells to endothelial cells. Blood. 2007;109:3544-3551.
87 Karamatic Crew V, Burton N, Kagan A. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood. 2004;104:2217-2223.
88 Lublin DM. Review: Cromer and DAF: role in health and disease. Immunohematology. 2005;21:39-47.
89 Johnson RJ, Hillmen P. Paroxysmal nocturnal hemoglobinuria: nature’s gene therapy? Molecular Pathology. 2002;55:145-152.
90 Rao N, Ferguson DJ, Lee S-F, et al. Identification of human erythrocyte blood group antigens on the C3b/C4b receptor. Journal of Immunology. 1991;146:3502-3507.
91 Moulds JM, Nickells MW, Moulds JJ, et al. The C3b/C4b receptor is recognized by the Knops, McCoy, Swain-Langley, and York blood group antisera. Journal of Experimental Medicine. 1991;173:1159-1163.
92 Eder AF, Spitalnik SL. Blood group antigens as receptors for pathogens. In: Blancher A, Klein J, Socha WW, editors. Molecular biology and evolution of blood group and MHC antigens in primates. Berlin: Springer; 1997:268-304.
93 Rios M, Bianco C. The role of blood group antigens in infectious diseases. Seminars in Hematology. 2000;37:177-185.
94 Lund N, Olsson ML, Ramkumar S. The human Pk histo-blood group antigen provides protection against HIV-1 infection. Blood. 2009;113:4980-4991.
95 Brown KE, Young NS. Parvovirus B19 infection and hematopoiesis. Blood Reviews. 1995;9:176-182.
96 Fumagalli M, Cagliani R, Pozzoli U. Widespread balancing selection and pathogen-driven selection at blood group antigen genes. Genome Research. 2009;19:199-212.
97 Pasvol G, Wainscoat JS, Weatherall DJ. Erythrocytes deficient in glycophorin resist invasion by the malarial parasite Plasmodium falciparum. Nature. 1982;297:64-66.
98 Sim BKL, Chitnis CE, Wasniowska K, et al. Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science. 1994;264:1941-1944.
99 Mayer DCG, Jiang L, Achur RN, et al. The glycophorin C N-linked glycan is a critical component of the ligand for the Plasmodium falciparum erythrocyte receptor BAEBL. Proceeding of the National Academy of Science of the United States of America. 2006;103:2358-2362.
100 Mayer DCG, Jiang L, Achur RN, et al. The glycophorin C N-linked glycan is a critical component of the ligand for the Plasmodium falciparum erythrocyte receptor BAEBL. Proceeding of the National Academy of Science of the United States of America. 2006;103:2358-2362.
101 Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388:292-295.
102 Uneke CJ. Plasmodium falciparum malaria and ABO blood group: is there any relationship? Parasitology Research. 2007;100:759-765.
103 Loscertales M-P, Owens S, O’Donnell J, et al. ABO blood group phenotypes and Plasmodium falciparum malaria: unlocking a pivotal mechanism. Advances in Parasitology. 2007;65:1-50.
104 Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007;110:2250-2258.
105 von dem Borne AEGKr, Decary F. Nomenclature of platelet specific antigens. British Journal of Haematology. 1990;74:239-240.
106 Metcalfe P, Watkins NA, Ouwehand WH, et al. Nomenclature of human platelet antigens. Vox Sanguinis. 2003;85:240-245.
107 Newman PJ, Derbes RS, Aster RH. The human platelet alloantigens, P1A1 and P1A2, are associated with a leucine33/proline33 amino acid polymorphism in membrane glycoprotein IIIa, and are distinguishable by DNA typing. Journal of Clinical Investigation. 1989;83:1778-1781.
108 Kuijpers RW, Faber NM, Cuypers HT, et al. NH2-terminal globular domain of human platelet glycoprotein Ibα has a methionine 145/threonine 145 amino acid polymorphism, which is associated with the HPA-2 (Ko) alloantigens. Journal of Clinical Investigation. 1992;89:381-384.
109 Lyman S, Aster RH, Visentin GP, et al. Polymorphism of human platelet membrane glycoprotein IIb associated with the Baka/Bakb alloantigen system. Blood. 1990;75:2343-2348.
110 Wang R, Furihata K, McFarland JG, et al. An amino acid polymorphism within the RGD binding domain of platelet membrane glycoprotein IIIa is responsible for the formation of the Pena/Penb alloantigen system. Journal of Clinical Investigation. 1992;90:2038-2043.
111 Santoso S, Kalb R, Walka M, et al. The human platelet alloantigens Bra and Brb are associated with a single amino acid polymorphism on glycoprotein Ia (integrin subunit α2). Journal of Clinical Investigation. 1993;92:2427-2432.
112 Simsek S, Gallardo D, Ribera A, et al. The human platelet alloantigens, HPA-5(a+, b−) and HPA-5(a−, b+), are associated with a Glu505/Lys505 polymorphism of glycoprotein Ia (the α2 subunit of VLA-2). British Journal of Haematology. 1994;86:671-674.
113 Wang R, McFarland JG, Kekomaki R, et al. Amino acid 489 is encoded by a mutational ‘hot spot’ on the β3 integrin chain: the CA/TU human platelet alloantigen system. Blood. 1993;82:3386-3391.
114 Kuijpers RW, Simsek S, Faber NM, et al. Single point mutation in human glycoprotein IIIa is associated with a new platelet-specific alloantigen (Mo) involved in neonatal alloimmune thrombocytopenia. Blood. 1993;81:70-76.
115 Santoso S, Kalb R, Kroll H, et al. A point mutation leads to an unpaired cysteine residue and a molecular weight polymorphism of a functional platelet beta 3 integrin subunit. The Sra alloantigen system of GPIIIa. Journal of Biological Chemistry. 1994;18:8439-8444.
116 Noris P, Simsek S, de Bruijne-Admiraal LG, et al. Maxa, a new low-frequency platelet-specific antigen localized on glycoprotein IIb, is associated with neonatal alloimmune thrombocytopenia. Blood. 1995;86:1019-1026.
117 Peyruchaud O, Bourre F, Morel-Kopp MC, et al. HPA-10w(b) (Laa): genetic determination of a new platelet-specific alloantigen on glycoprotein IIIa and its expression in COS-7 cells. Blood. 1997;89:2422-2428.
118 Simsek S, Folman C, van der Schoot CE, et al. The Arg633His substitution responsible for the private platelet antigen Groa unravelled by SSCP analysis and direct sequencing. British Journal of Haematology. 1997;97:330-335.
119 Sachs UJ, Kiefel V, Bohringer M, et al. Single amino acid substitution in human platelet glycoprotein Ibbeta is responsible for the formation of the platelet-specific alloantigen Iya. Blood. 2000;95:1849-1855.
120 Santoso S, Amrhein J, Hofmann HA, et al. A point mutation Thr(799)Met on the α2 integrin leads to the formation of new human platelet alloantigen Sita and affects collagen-induced aggregation. Blood. 1999;94:4103-4111.
121 Santoso S, Kiefel V, Richter IG, et al. A functional platelet fibrinogen receptor with a deletion in the cysteine-rich repeat region of the β3 integrin:the Oea alloantigen in neonatal alloimmune thrombocytopenia. Blood. 2002;99:1205-1214.
122 Schuh AC, Watkins NA, Nguyen Q, et al. A tyrosine703serine polymorphism of CD109 defines the Gov platelet alloantigens. Blood. 2002;99:1692-1698.
123 Jallu V, Meunier M, Brément M, Kaplan C. A new platelet polymorphism Duva+, localized within the RGD blinding domain of glycoprotein IIIa, is associated with neonatal thrombocytopenia. Blood. 2002;99:4449-4456.
124 Stafford P, Garner SF, Rankin A, et al. A single-nucleotide polymorphism in the human ITBG3 gene is associated with the platelet-specific alloantigen Vaa (HPA-17bw) involved in fetal maternal alloimmune thrombocytopenia. Transfusion. 2008;48:1432-1438.
125 Allen DL, Lucas GF, Murphy MF, Ouwehand WH. Platelet and neutrophil antigens. Murphy MF, Pamphilon DH, editors. Practical Transfusion Medicine. 3rd ed. 2009:44-59.
126 Porcelijn L, van Beers W, Gratama JW, et al. External quality assessment of platelet serology and human platelet antigen genotyping: a 10-year review. Transfusion. 2008;48:1699-1706.
127 Veldhuisen B, van der Schoot CE, de Haas M. Blood group genotyping: from patient to high-throughput donor screening. Vox Sanguinis. 2009;97:198-206.
128 Kasirer-Friede A, Kahn ML, Shattil SJ. Platelet integrins and immunoreceptors. Immunological Reviews. 2007;218:247-264.
129 Calvete JJ. Platelet integrin GPIIb/IIIa: structure-function correlations. An update and lessons from other integrins. Proceedings of the Society of Experimental Biology and Medicine. 1999;222:29-38.
130 Calvete JJ, Muniz-Diaz E. Localization of an O-glycosylation site in the alpha-subunit of the human platelet integrin GPIIb/IIIa involved in Baka (HPA-3a) alloantigen expression. FEBS Letters. 1993;328:30-34.
131 Williamson LM, Hackett G, Rennie J, et al. The natural history of fetomaternal alloimmunization to the platelet-specific antigen HPA-1a (PLA1, Zwa) as determined by antenatal screening. Blood. 1998;92:2280-2287.
132 Kuijpers RWAM, von dem Borne AEG, Kiefel V, et al. Leucine33-proline33 substitution in human platelet glycoprotein IIIa determines HLA-DR52a (Dw24) association of the immune response against HPA-1a (Zwa/P1A1) and HPA-1b (Zwb/P1A2). Human Immunology. 1992;34:253-356.
133 Wu S, Maslanka K, Gorski J. An integrin polymorphism that defines reactivity with alloantibodies generates an anchor for MHC class II peptide binding: a model for unidirectional alloimmune responses. Journal of Immunology. 1997;158:3221-3226.
134 Nieuwenhuis HK, Akkerman JW, Houdijk WP, et al. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature. 1985;318:470-472.
135 Kunicki TJ, Kritzik M, Annis DS, et al. Hereditary variation in platelet integrin α2β1 density is associated with two silent polymorphisms in the α2 gene coding sequence. Blood. 1997;89:1939-1943.
136 Lopez JA, Ludwig EH, McCarthy BJ. Polymorphism of human glycoprotein Ibα results from a variable number of tandem repeats of a 13-amino acid sequence in the mucin-like macroglycopeptide region. Structure/function implications. Journal of Biological Chemistry. 1992;267:10,055-61.
137 Simsek S, Bleeker PM, van der Schoot CE, et al. Association of a variable number of tandem repeats (VNTR) in glycoprotein Ibα and HPA-2 alloantigens. Thrombosis and Haemostasis. 1994;72:757-761.
138 Smith JW, Hayward CP, Horsewood P, et al. Characterization and localization of the Gova/b alloantigens to the glycosylphosphatidylinositol-anchored protein CDw109 on human platelets. Blood. 1995;86:2807-2814.
139 Lin M, Sutherland R, Horsfall W, et al. Cell surface antigen CD109 is a novel member of the α2 macroglobulin/C3, C4, C5 family of thioester-containing proteins. Blood. 2002;99:1683-1691.
140 Solomon KR, Sharma P, Chan M, et al. CD109 represents a novel branch of the α2-macroglobulin/complement gene family. Gene. 2004;327:171-183.
141 Schuh AC, Watkins NA, Nguyen Q, et al. A tyrosine703serine polymorphism of CD109 define the Gov platelet alloantigens. Blood. 2002;99:1692-1698.
142 Kaplan C. Les thrombopénies foetales et néonatales allo-immunes: problèmes en suspens. Tranfusion Clinique et Biologique. 2005;12:131-134.
143 Arnold DM, Smith JW, Kelton JG. Diagnosis and management of neonatal alloimmune thrombocytopenia. Transfusion Medicine Reviews. 2008;22:255-267.
144 van den Akker ESA, Oepkes D. Fetal and neonatal alloimuune thrombocytopenia. Best Practice and Research Clinical Obstetrics and Gynaecology. 2008;22:3-14.
145 Davoren A, Curtis BR, Aster RH, McFarland JG. Human platelet antigen-specific alloantibodies implicated in 1162 cases of neonatal alloimmune thrombocytopenia. Trnafsusion. 2004;44:1220-1225.
146 Berry JE, Murphy CM, Smith GA, et al. Detection of Gov system antibodies by MAIPA reveals an immunogenicity similar to the HPA-5 alloantigens. British Journal of Haematology. 2000;110:735-742.
147 Ramsey G, Salamon DJ. Frequency of P1A1 in blacks. Transfusion. 1986;26:531-532.
148 Shibata Y, Matsuda I, Miyaji T, et al. Yuka, a new platelet antigen involved in two cases of neonatal alloimmune thrombocytopenia. Vox Sanguinis. 1986;50:177-180.
149 Kumpel BM, Sibley K, Jackson DJ, et al. Ultrastructural localization of glycoprotein IIIa (GIIIa, β3 integrin) on placental syncytiotrophoblast microvilli: implications for platelet alloimmunization during pregnancy. Transfusion. 2008;48:2077-2086.
150 Proulx C, Filion M, Goldman M, et al. Analysis of immunoglobulin class, IgG subclass and titre of HPA-1a antibodies in alloimmunized mothers giving birth to babies with or without neonatal alloimmune thrombocytopenia. British Journal of Haematology. 1994;87:813-817.
151 McMillan R, Longmire RL, Tavassoli M, et al. In vitro platelet phagocytosis by splenic leukocytes in idiopathic thrombocytopenic purpura. New England Journal of Medicine. 1974;290:249-251.
152 Clarkson SB, Bussel JB, Kimberley RP, et al. Treatment of refractory immune thrombocytopenic purpura with an anti-Fcγ-receptor antibody. New England Journal of Medicine. 1986;314:1236-1239.
153 Bussel JB, Zabusky MR, Berkowitz RL, et al. Fetal alloimmune thrombocytopenia. New England Journal of Medicine. 1997;337:22-26.
154 Burrows RF, Caco CC, Kelton JG. Neonatal alloimmune thrombocytopenia: spontaneous in utero intracranial hemorrhage. American Journal of Hematology. 1988;28:98-102.
155 Ory PA, Clark MR, Kwoh EE, et al. Sequences of complementary DNAs that encode the NA1 and NA2 forms of Fc receptor III on human neutrophils. Journal of Clinical Investigation. 1989;84:1688-1691.
156 Bux J, Stein E-L, Bierling P, et al. Characterization of a new alloantigen (SH) on the human neutrophil Fcγ receptor IIIb. Blood. 1997;89:1027-1034.
157 Stroncek DF, Shankar RA, Plachta LB, et al. Polyclonal antibodies against the NB1-bearing 58- to 64-kDa glycoprotein of human neutrophils do not identify an NB2-bearing molecule. Transfusion. 1993;33:399-404.
158 Greinacher A, Wesche J, Hammer E, et al. Characterization of the human neutrophil alloantigen-3a. Nature Medicine. 2010;16:45-48.
159 Curtis BR, Cox NJ, Sullivan MJ, et al. The neutrophil alloantigen HNA-3a (5b) is located on choline transporter-like protein 2 and appears to be encoded by an R>Q154 amino acid substitution. Blood. 2010;115:2073-2076.
160 Simsek S, van der Schoot CE, Daams M, et al. Molecular characterization of antigenic polymorphisms (Onda and Marta) of the beta 2 family recognized by human leukocyte alloantisera. Blood. 1996;88:1350-1358.
161 Bux J. Human neutrophil alloantigens. Vox Sanguinis. 2008;94:277-285.
162 Bux J. Nomenclature of granulocyte alloantigens. Transfusion. 1999;39:662-663.
163 van de Winkel JGJ, Capel PJA. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunology Today. 1993;14:215-221.
164 Koene HR, Kleijer M, Roos D, et al. FcγRIIIB genes in NA(1+,2+)SH(+)individuals. Blood. 1998;91:673-679.
165 Metcalfe P, Waters AH. Location of the granulocyte-specific antigen LAN on the Fc-receptor III. Transfusion Medicine. 1993;2:283-287.
166 Bux J, Hartmann C, Mueller-Eckhardt C. Alloimmune neonatal neutropenia resulting from immunization to a high frequency antigen on the granulocyte Fcγ receptor III. Transfusion. 1994;34:608-611.
167 de Haas M, Kleijer M, van Zwieten R, et al. Neutrophil FcγRIII deficiency, nature and clinical consequences: a study of 21 individuals from 14 families. Blood. 1995;86:2403-2413.
168 Stroncek DF, Skubitz KM, McCullough JJ. Biochemical characterization of the neutrophil-specific antigen NB1. Blood. 1990;75:744-755.
169 Kissel K, Scheffler S, Kerowgan M, Bux J. Molecular basis of NB1 (HNA-2a, CD177) deficiency. Blood. 2002;99:4231-4233.
170 Bux J, Jung D, Kauth T, et al. Serological and clinical aspects of granulocyte antibodies leading to alloimmune neonatal alloimmune neutropenia. Transfusion Medicine. 1992;2:143-149.
171 Lalezari P, Jiang AF, Yegen L, et al. Chronic autoimmune neutropenia due to anti-NA2 antibody. New England Journal of Medicine. 1975;293:744-747.
172 Bux J, Behrens G, Jaeger G, et al. Diagnosis and clinical course of autoimmune neutropenia in infancy: analysis of 240 cases. Blood. 1998;91:181-186.
173 Lyall EGH, Lucas GF, Eden OB. Autoimmune neutropenia of infancy. Journal of Clinical Pathology. 1992;45:431-434.
174 Popovsky MA, Moore SB. Diagnostic and pathogenic considerations in transfusion-related lung injury. Transfusion. 1985;25:573-577.
175 Kleinman S, Caulfield T, Chan P, et al. Toward an understanding of transfusion-related lung injury: statement of a consensus panel. Transfusion. 2004;44:1774-1789.
176 Bux J. Transfusion-related lung injury (TRALI): a serious adverse event of blood transfusion. Vox Sanguinis. 2005;89:1-10.
177 Bux J, Sachs UJH. The pathogenesis of transfusion-related lung injury (TRALI). British Journal of Haematology. 2007;136:788-799.
178 Kopko PM, Marshall CS, MacKenzie MR, et al. Transfusion-related lung injury. Report of a clinical look-back investigation. Journal of the American Medical Association. 2002;15:1968-1971.
179 Muniz M, Sheldon S, Schuller RM, et al. Patient-specific transfusion-related lung injury. Vox Sanguinis. 2008;94:70-73.