[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 6
Biosynthesis, Processing, and Secretion of the Islet Hormones
Insulin, Islet Amyloid Polypeptide (Amylin), Glucagon, Somatostatin, and Pancreatic Polypeptide
Morphologic Organization of the Insulin Biosynthetic Machinery
Structure and Properties of Proinsulin
Enzymatic Basis of the Conversion of Proinsulin to Insulin
Regulation of Insulin Production
Insulin Secretion and Its Regulation
Insulin Gene Mutations as a Cause of Neonatal Diabetes
Although diabetes had long been recognized as a serious metabolic disorder, it was the classic experiments of Von Mering and Minkowski in 1890 that demonstrated the important role of the pancreas in its prevention.1 A number of investigators attempted to prepare antidiabetic pancreatic extracts in the early 20th century, but it was not until the work of the Canadians—Frederick Banting, Charles Best, and John Collip, working in the laboratory of Professor J.J.R. Macleod at the University of Toronto in 1921-1922—that potent preparations of insulin were routinely made.2 The name insulin was based on the belief that the hormone was derived from the islets of Langerhans and actually was suggested as early as 1909 by deMayer and by Sir Edward Sharpey-Schafer in 1917. Preparative methods based on the early work of Scott,3 as well as refinements by Banting et al.,4 were rapidly developed for the commercial preparation of the hormone, and within about 1 year, insulin began to be administered to patients with diabetes, often with dramatic effects.2 The chemical nature of insulin was a more elusive problem, although the fact that it was destroyed by proteolytic enzymes suggested that it was a protein.5 When J.J. Abel first crystallized insulin in 1926, there was great controversy as to whether the crystals of protein he obtained were actually the active biological principle or merely the vehicle for a smaller active moiety.6 Today we know that insulin occurs throughout the vertebrate kingdom, and insulin-like substances are present in the brains and/or digestive systems of many invertebrates.7 Many of the modern techniques of protein chemistry were developed in part through the clinical need for insulin, which made it abundantly available to biochemists as a model protein for study.8,9
This chapter reviews the isolation, structure, and properties of insulin and other islet hormones, including islet amyloid polypeptide (IAPP or amylin), glucagon, somatostatin, pancreatic polypeptide (PP) and their precursor forms, and the mechanisms of their biosynthesis and secretion. Studies on the mechanism of biosynthesis of insulin via preproinsulin and proinsulin in the pancreatic β cells have provided a useful paradigm for understanding the production of many other neuroendocrine peptides and a basis for advances in the diagnosis and treatment of diabetes and related endocrine disorders. The recent identification of the enzymes that catalyze the proteolytic cleavage of prohormones and proneuropeptides—prohormone convertases—has revealed a new family of processing enzymes that act on a larger subset of precursor proteins, including growth factors, their receptors, and many other proteins that traverse the secretory pathway.10
The Biosynthesis of Insulin
The two-chain structure of insulin was the first complete protein structure to be elucidated through the pioneering work of Fred Sanger in 1955.9 In 1967, the discovery of the single-chain insulin precursor molecule, proinsulin,11 then solved the problem of assembly of the insulin chains (Fig. 6-1). The human proinsulin polypeptide begins with the 30-amino-acid B chain domain, extends through a 35-amino-acid connecting segment, and ends with the 21-amino-acid A chain domain, comprising a single 9000-D polypeptide chain.11–14 It contains paired basic residue cleavage sites (Arg-Arg or Lys-Arg) at each end of the connecting segment which are removed during the proteolytic conversion of proinsulin to insulin within maturing secretory granules. However, cell-free translation of the initial polypeptide products encoded in insulin messenger ribonucleic acid (mRNA) extracted from islets or islet cell tumors led in 1976 to the discovery of preproinsulin (Fig. 6-2). This extended form of the prohormone has a hydrophobic N-terminal 24-residue signal peptide.15 Such prepeptide extensions interact with the signal recognition particle (SRP), a cytosolic ribonucleoprotein particle that assists in transfer of the nascent secretory protein from the cytosolic compartment, where its synthesis is initiated, into the secretory pathway via a complex series of molecular interactions that result in the translocation of the nascent peptide across the membrane of the rough endoplasmic reticulum (RER) into its internal compartments, or cisternae.16 During or shortly after translocation, the signal sequence is cleaved by the signal peptidase, which is located on the inner surface of the RER membrane, and the signal peptide is rapidly degraded.17 The proinsulin molecule then folds and undergoes rapid formation of disulfide bonds to achieve its native structure. Evidence suggests that this process is catalyzed by protein disulfide isomerase,18 a resident protein of the RER having a C-terminal Lys-Glu-Asp-Leu (KDEL) localization sequence.19 The folded proinsulin is then transferred in small coated vesicles from the endoplasmic reticulum (ER) to the cis region of the Golgi apparatus.20,21 It then passes from the cis to the trans Golgi, where it is sorted into immature secretory vesicles, or progranules, where it is proteolytically cleaved to release insulin and the connecting segment, minus the four basic residues, from the cleavage sites. The connecting peptide fragment released by cleavage of proinsulin is designated the C peptide. Both insulin and C peptide are stored in the secretory granules along with small amounts of residual proinsulin and partially cleaved intermediate forms22 as well as a variety of other minor β cell secretory products.23 Stimulation of secretion results in the release of the granule contents into the hepatic portal circulation.
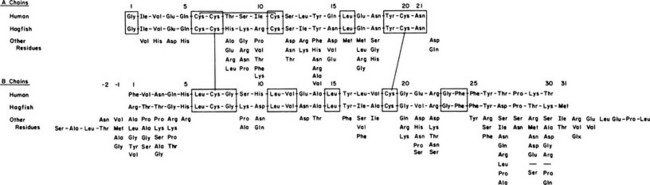
FIGURE 6-1 Amino acid sequence of human and hagfish insulins, with substitutions occurring at each position in 70 other known vertebrate insulins shown below for comparison. Invariant residues are enclosed in boxes.
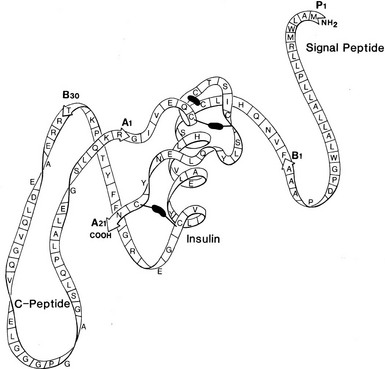
FIGURE 6-2 Schematic structure of human preproinsulin. Rapid removal of the first 24 amino acids (signal peptide) gives rise to proinsulin, which then folds to form the native insulin domain stabilized by three disulfide bonds (black bars). The connecting segment is not structurally organized. Cleavage after B30 (T) and before A1 (G) then gives rise to insulin and the free C peptide. (See text for details.)
Morphologic Organization of the Insulin Biosynthetic Machinery
The β cells of the islets of Langerhans share many features with other endocrine and neurosecretory cells. Preproinsulin is the initial translational product of insulin mRNA, as discussed earlier, and is rapidly cleaved to proinsulin in the RER. After folding and disulfide oxidation, proinsulin is transported from the ER to the Golgi apparatus in small coated vesicles20 (Fig. 6-3) in an energy-dependent process that requires about 20 minutes.22,24,25 The biochemical basis of the energy requirement for the intracellular transport of secretory proteins is associated with the budding and/or fusion of small vesicles that transfer secretory products from the ER to and through the cis to trans Golgi cisternae.26,27
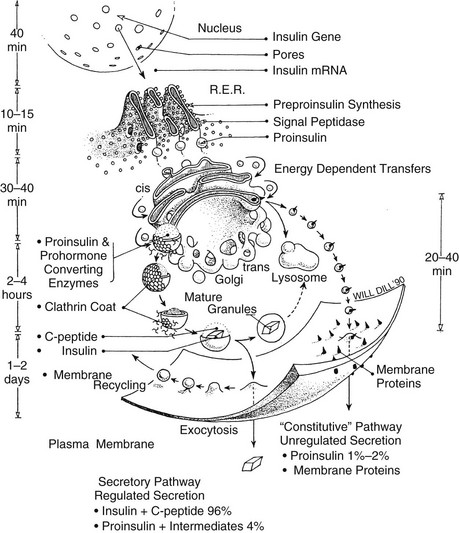
FIGURE 6-3 Schematic model of the subcellular transport of preproinsulin after its synthesis on membrane-bound ribosomes of the rough endoplasmic reticulum (RER) and rapid cleavage to proinsulin (within 1 to 2 minutes). Proinsulin is then released into the intracisternal spaces of the RER, where it folds and forms the native disulfide bonds of insulin. It is then transported to the Golgi apparatus by an energy-dependent process. The clathrin-coated early granules budding from the trans-Golgi cisternae are rich in proinsulin and contain the converting proteases, PC2 and PC1/3. Processing occurs mainly, if not exclusively, in the early secretory granules24,30 giving rise to the more condensed mature granules. Fractionation studies have confirmed that the mature granule-dense cores consist almost entirely of insulin, often in crystalline arrays (see Fig. 6-9), whereas the granule-soluble phase that surrounds the inclusion consists mainly of C peptide and small amounts of proinsulin.84 The release of newly synthesized proinsulin and insulin begins only about 1 hour after synthesis in the RER; hence, granules must undergo a maturation process that renders them competent for secretion. There is no evidence for significant nongranular routes of secretion of either proinsulin or insulin in normal islets. Exocytosis of granules is regulated by glucose and many other factors (see section on insulin secretion), and in humans and dogs, it results in the release of insulin and C peptide in approximately equimolar proportions under both basal and stimulated conditions.95 The mechanism of recycling of the granule membrane and its components is not well understood.
The role of the Golgi apparatus in forming insulin-containing secretory vesicles, or β-granules, was first recognized in the 1940s and subsequently confirmed and extended by Munger,28 who used electron microscopy to identify pale, homogeneous-appearing, immature “progranules” near the Golgi body. The structural organization and various functions of this complex organelle continue to engage the attention of cell biologists and biochemists.29 Orci and coworkers demonstrated that these newly formed clathrin-clad vesicles are derived from the trans-Golgi network (TGN) cisternae and contain a high proportion of proinsulin, definitively confirming their proposed role as the major sites of proinsulin processing to insulin.30 Conversion of proinsulin to insulin in vivo behaves kinetically as a pseudo–first-order reaction having a half-time ranging from about 20 minutes to 1 hour.22,31,32 Peak labeling of proteins in the Golgi apparatus occurs 30 to 40 minutes after pulse labeling islets with 3H-amino acids, and relatively little radioactivity remains in this region after 1 hour.33 A similar pattern for proinsulin is observed by means of electron microscopic immunocytochemistry. Thus, although small amounts of proinsulin conversion may be initiated in the TGN, most of it occurs in newly formed secretory vesicles, or progranules, which mature biochemically in the cytosol30 (see Fig. 6-3). Numerous subsequent studies have confirmed that newly synthesized neuroendocrine precursor peptides pass via the Golgi apparatus into secretory granules of the regulated secretory pathway, where they are cleaved and processed to their mature products and then stored until secretion in response to signals such as glucose.10
Structure and Properties of Proinsulin
Mammalian proinsulins range in size from 81 (cow) to 86 (human, horse, rat) amino acid residues, owing to variations in the length of the C peptide. Various C-peptide sequences are compared in Fig. 6-4. Proinsulin is closely similar to insulin in many properties, including solubility, isoelectric point,22 self-associative properties,34 and reactivity with insulin antisera.11,35 Indeed, the conformation of the insulin moiety in proinsulin is essentially identical to that of insulin itself.36 It is noteworthy that the highly flexible C peptide is much longer than necessary simply to bridge the short 8-Å gap between the ends of the B and A chains as they exist in the folded insulin molecule (see Fig. 6-2). The C-peptide segment overlays a portion of the surface of the insulin monomer, reducing its potency to about 3% to 5% of that of insulin in vitro,22,37 and it undergoes little if any proteolysis or activation in the circulation or tissues.38 The presence of the C peptide in proinsulin also does not prevent the dimerization or hexamerization of its insulin moiety39 (Fig. 6-5). It lies outside the zinc-stabilized globular insulin hexamer in a disordered loop, as assessed by nuclear magnetic resonance (NMR) spectroscopy. Since low levels of proinsulin can form mixed hexamers with insulin, it can be efficiently incorporated into insulin crystals.40 This property accounts for the presence of 1% to 2% of proinsulin in most crystalline preparations of insulin prepared from animal pancreata.41 However, x-ray analysis of crystals of proinsulin have failed to reveal any details regarding the structure of the C-peptide segment.42,43

FIGURE 6-4 Compilation of amino acid sequences of proinsulin C peptides in vertebrates, amphioxus, and in a mollusk, Lymnae stagnalis (for sources see Ref. 96).
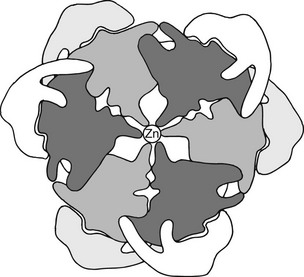
FIGURE 6-5 Hypothetical 2-Zn proinsulin hexamer as viewed along the threefold axis. The connecting peptide is shown in light gray and white around the periphery of the darker outline of an insulin hexamer arranged according to the structural data of Blundell et al.88 The central density represents two zinc atoms in coordination linkage to the six (three above and three below hexamer plane) histidine side chains at position 10 in the B chain.
The major intermediate cleavage products of proinsulin consist mainly of forms cleaved at only one site and lacking the exposed basic residues (i.e., des-31,32 or des-64,65 intermediates).12,22,41,44,45 Their relative ratios in the circulation differ among species, depending on differences in the amino acid sequences surrounding the cleavage sites.46 Proinsulin-like proteins having comparably sized C peptides have been found throughout the vertebrates and also in a variety of invertebrates. Although it seems likely that the single-chain insulin-like growth factors, IGF-1 and IGF-2, arose from a single proinsulin-like gene in protochordates,47 some invertebrates, such as Caenorhabditis elegans,48 have numerous insulin-like peptides that lack defined C peptides and typical paired basic residue sites for cleavage by the evolutionarily well-conserved prohormone convertases.
Enzymatic Basis of the Conversion of Proinsulin to Insulin
The major proteolytic cleavages required for converting proinsulin to insulin are summarized in Fig. 6-6. In early studies, it was shown that the joint action of pancreatic trypsin and carboxypeptidase B gives rise to the naturally occurring conversion products—C peptide and native insulin in high yields—quantitatively converting proinsulin to insulin in vitro.49 This model also explains the major intermediate forms found in pancreatic extracts,44,50 and it led to a search for a cellular trypsin-like convertase, assumed to be a serine protease related to trypsin,22 and for carboxypeptidases related to carboxypeptidase B, an exocrine pancreatic exopeptidase with specificity for C-terminal basic residues.49 Early studies with isolated islet secretory granules revealed that these were major sites of proinsulin conversion, and that conversion resulted in the release of insulin along with free arginine, rather than basic dipeptides,51 confirming the likely participation of both trypsin-like and carboxypeptidase B–like proteases in maturing secretory granules.52 Carboxypeptidase E (or H), a homolog of carboxypeptidase B but with a more acidic pH optimum, was first identified in islets52 and subsequently by Fricker in brain.53 Molecular cloning confirmed its structural and evolutionary relationship to the pancreatic carboxypeptidases. Additional processing carboxypeptidases have been uncovered in brain and other tissues.54
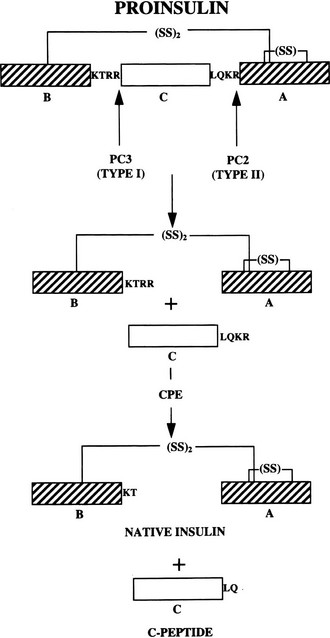
FIGURE 6-6 The cleavage of proinsulin to insulin by the combined action of the subtilisin-like prohormone convertases, PC2 and PC1/PC3, and carboxypeptidase E. (See text for details.)
The first authentic processing endoprotease, kexin, was identified through studies on the processing of the yeast alpha mating–factor precursor and proved to be a calcium-dependent serine protease related to bacterial subtilisin and the subtilases rather than to trypsin.55–57 This information enabled the discovery of mammalian homologs, which form a family of seven closely related endoproteases with differing points of action within the secretory pathway.10,58,59 These have been designated as subtilisin-like proprotein convertases (SPCs), or more simply as PCs. Two members of this family, PC1/3 and PC2, have proven to be the principal effectors of neuroendocrine peptide precursor processing and are largely confined in their expression to brain and endocrine tissues in most animal species.10 For further information on the extended SPC/PC family and its multiple functions, please see the reviews in Refs. 60 to 62.
Role of PC2 and PC1/3 in Proinsulin Processing
Both PC1/3 and PC2 are required for proinsulin processing and have been shown to be identical with the calcium-dependent type I and type II insulinoma granule-processing activities, respectively, originally described by Davidson et al.63 The importance of calcium for the transport and proteolytic maturation of proinsulin has been well documented.64 The role and order of action of PC2 and PC1/3 in proinsulin conversion has also been carefully studied.65 Rhodes et al.66 demonstrated that the type II convertase (PC2) prefers the proinsulin intermediate that has already been processed at the B chain–C peptide junction (des-31,32 intermediate) as a substrate. This observation has led to the scheme for conversion outlined in (Fig. 6-7), in which PC1/3 acts first to generate the des-31,32 intermediate, which is then cleaved at the C peptide–A chain junction by PC2. This order of cleavage is consistent with observations that PC1/3 achieves an enzymatically active form more rapidly than PC2 and has a somewhat higher pH optimum. Thus, PC1/3 may begin cleaving proinsulin as it is concentrated into immature secretory vesicles in the TGN, whereas PC2 acts only in the maturing granules as the pH decreases to around 5.5. According to this scheme, PC1/3 plays a more important role in proinsulin processing, as born out by observations on islets from mice lacking PC2 or PC1/3.46,67–69
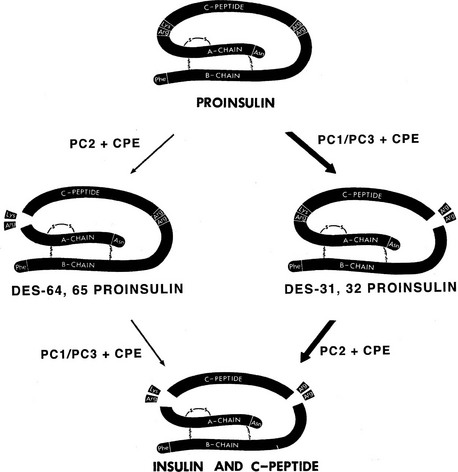
FIGURE 6-7 Routes of processing of proinsulin in the pancreatic β cell. The pathway on the right is probably more dominant under normal conditions because des-31,32 proinsulin is a preferred substrate for PC2,66 and the more acidic pH optimum and slower maturation of this enzyme may delay its action during the initial phases of secretory granule maturation. The C-terminal basic residues are removed by CPE (CPH) after endoproteolytic cleavage by the PCs. (Modified from Rouillé Y, Duguay SJ, Lund K et al: Proteolytic processing mechanisms in the biosynthesis of neuroendocrine peptides: the subtilisin-like proprotein convertases, Front Neuroendocrinol 16:322–361, 1995.)
PC2-null mice are not diabetic, but they exhibit significant hyperproinsulinemia, with plasma proinsulin levels in the range of 60%.67 Pancreatic extracts also show increased proinsulin levels, but only in the homozygous nulls, as indicated in Table 6-1. Pulse-chase studies of insulin biosynthesis in isolated islets, comparing PC2(−/−) mice with wild-type (WT) controls, also confirm significantly slower processing of proinsulin to insulin, with accumulation of significant amounts of des-31,32 intermediate proinsulin.46,67 Approximately a third of the labeled proinsulin remains after a 3- or 4-hour chase, consistent with the levels found by radioimmunoassay in pancreatic extracts. Thus in normal mice, PC2 converts at most about a third of the available proinsulin, while PC1/3 is responsible for processing the remaining two-thirds.69
Table 6-1
Percent Proinsulin-Like Immunoreactivity in Pancreatic Extracts of Wild-Type, PC2-Null, and PC1/3-Null Mice

A human subject with inactivating mutations in both copies of the PC1/3 gene has been reported.70 This 43-year-old woman is obese and had gestational diabetes. Examination of the subject’s serum revealed no detectable circulating insulin, in association with greatly elevated intact proinsulin and significant amounts of des-64,65 intermediate proinsulin but little or no des-31,32 intermediate proinsulin. Similar results with respect to proinsulin processing have been reported for the PC1/3-null mouse, which has very high levels of proinsulin and elevated des-64,65 intermediate proinsulin68,69 (see Table 6-1). This picture is consistent with the conversion scheme outlined in Fig. 6-7 and indicates that the pathway shown on the right side of the diagram is the predominant one. It also confirms the likelihood that PC1/3 is the more important convertase in the processing of proinsulin. The PC1/3-null mice, however, are not obese like the human subject; instead, they exhibit a severe growth defect, most likely due to lack of normally processed growth hormone–releasing hormone.69 These mice are also unable to produce GLP-1 but did not have altered intraperitoneal glucose tolerance. Altered proopiomelanocortin processing and other endocrine abnormalities, as well as intestinal malfunction, also are present in these mice.68
Significance of Circulating Proinsulin and Des-31,32 Intermediate in Man
An important clinical issue concerns observations that in man, des-31,32 intermediate proinsulin is a major intermediate, making up a very significant proportion of the circulating proinsulin-like material.71 It has been suggested that the elevated accumulation of proinsulin and des-31,32 proinsulin in diabetics might be due to defective action of PC2.72 Isolated normal human islets of Langerhans also have been reported to convert proinsulin to insulin with significant accumulation of des-31,32 intermediate, despite the presence of normal levels of PC2.73 However, des-31,32 intermediate only reaches levels of 15% to 20% of total immunoreactive insulin-like material during biosynthetic pulse-chase studies in the islets of PC2-null mice.46 Thus, even the complete absence of PC2 should not in itself give rise to such high levels of this intermediate as are seen in human serum samples. It has recently been clarified that this phenomenon arises due to the faster rate of cleavage by the PCs of substrates having a basic residue in the fourth position upstream (P4 position) of the usual dibasic cleavage sites. Such an upstream basic residue is present at the B chain–C peptide junction in human proinsulin (B29 is Lys) but is lacking at the A chain–C peptide junction (C62 of proinsulin is Leu) (see Fig. 6-6). This causes an imbalance in the relative susceptibility of these two sites to either of the two convertases and tends to favor the accumulation of the des-31,32 intermediate.46 On the other hand, in the dual proinsulins of rats and mice,22 other P4 sequence changes in these cleavage sites produce different ratios of intermediates than those seen in man.46
Studies on the regulation of the biosynthesis of PC2 and PC1/3 in β cells suggest that the rates of translation of both of these enzymes are up-regulated by glucose, similar to that of proinsulin on glucose stimulation.74 Also both PC2 and PC1/3 mRNA levels are equally elevated along with insulin mRNA during more prolonged stimulation of islets with glucose.75 However, under conditions of chronic stimulation, a relative deficiency of PC2 could develop that could exaggerate the abnormalities in circulating proinsulin intermediates seen in diabetics. Accordingly, the normal accumulation of des-31,32 intermediate proinsulin in human subjects merely reflects sequence differences in the two cleavage sites of human proinsulin, whereas increased levels of both proinsulin and des-31,32 proinsulin in prediabetics and diabetics may result from a deficiency of convertase action when islets are stressed by hyperglycemia. Genetic studies have not yet indicated a major role for mutations in the PC1/3,76 PC2,77 or CPE78 genes in susceptibility to any form of diabetes, although common amino acid polymorphisms in PC1/3 may increase risk of obesity.79 However, loss-of-function mutations in the transcription factor PAX6 have recently been described which result in reduced expression of PC1/3 in the islets and are associated with hyperproinsulinemia and mild age-related diabetes.80
Secretory Granule Formation and Maturation
One of the unsolved puzzles of neuroendocrine and other secretory cells is the nature of the mechanism underlying the efficient sorting of proteins destined for regulated secretion into immature secretory vesicles in the TGN. In the β cell, this process is remarkably efficient, resulting in very low levels of unregulated or “constitutive” release of proinsulin (<1% to 2%). However, the newly formed secretory granules have a clathrin coat (Fig. 6-8) which appears to be involved in some reorganization of the granule contents after and/or during their formation in the TGN.81 This passive sorting presumably occurs via small clathrin-clad vesicles that transport some proteins that are excluded from the condensing granule cores into endosomal pathways that either recycle to the TGN or to the cell surface. As a consequence of this “constitutive-like” pathway, proteins such as furin, procathepsin B, and possibly others briefly pass through the immature granule compartment, where they may play an active albeit transient role (e.g., furin may participate in processing of some prohormones).81,82 Also, small amounts of abundant soluble granule components such as proinsulin and/or C peptide may exit the granules within these vesicles.83 Passive sorting may also play a role in maintaining synchrony between granule membrane area and granule volume as maturation proceeds.81
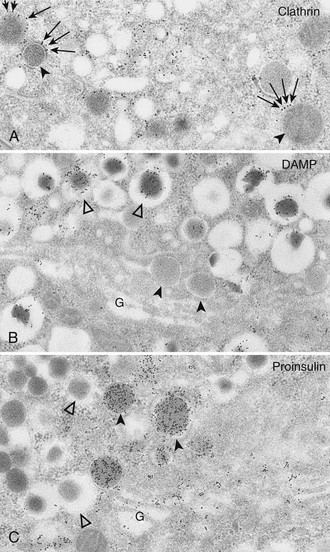
FIGURE 6-8 A, Clathrin immunolabeling; B, 3-(2,4-dinitroanilino)-3′-amino-N-methyldipropylamine (DAMP) immunolabeling; and C, proinsulin immunolabeling of Golgi areas (G) of B cells (protein A–gold techniques). These electron micrographs show that the population of secretory granules with tightly fitting cores (black arrowheads) is clathrin coated (arrows in A), DAMP poor (B), and proinsulin rich (C). These granules correspond to the maturing coated secretory granules freshly released from the Golgi complex. Conversely, secretory granules with wide, clear halos (white arrowheads) are deprived of clathrin (not shown here), are DAMP rich (B). and are proinsulin poor (C). These correspond to the noncoated mature (storage) insulin-containing secretory granules. Because DAMP immunoreactivity is assumed to represent an indirect measure of intraorganelle acidity, this may indicate a decreasing pH gradient between proinsulin-rich and insulin-rich granules (i.e., between the converting and the storage compartments) (A, ×28,000; B and C, ×27,000). (From Orci L: The insulin cell: its cellular environment and how it processes proinsulin, Diabetes Metab Rev 2:71, 1986.)
Newly formed secretory vesicles in neuroendocrine cells undergo biochemical and morphologic maturation to typical dense-core granules. In β cells, the progranules are larger, less dense, and more uniform in appearance.29,30 The most important biochemical change taking place as these progranules mature is the proteolytic conversion of proinsulin to insulin, accompanied by reorganization of the products.30 Electron microscopic studies indicate that they progressively acquire a crystalline dense central core (Fig. 6-9). High magnification reveals repeat-unit spacings in the cores that are closely similar to those observed in ordinary zinc insulin crystals.84 Thus, as insulin is liberated from proinsulin, it tends to crystallize with zinc that is concentrated by the β cells. Recent studies have demonstrated that zinc is taken up into maturing granules via the ZnT8 zinc transporter, and they do not assume the normal dense-core appearance in its absence, even though proinsulin conversion is not impaired.85 Biochemical fractionation of mature islet secretory granules confirms that the cores contain only insulin, whereas the C peptide liberated in the conversion process remains in solution in the clear fluid space surrounding the dense crystalline core.84 There is no evidence for co-crystallization of the C peptide with insulin under these conditions or in vitro.
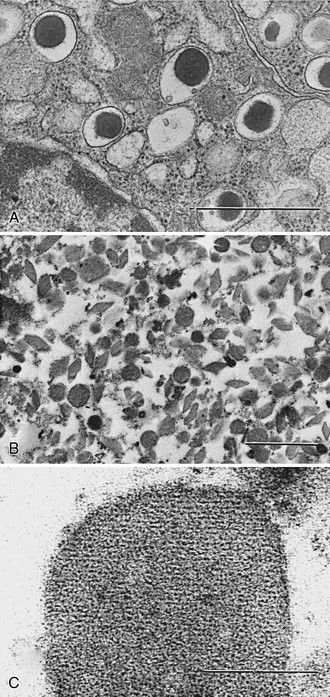
FIGURE 6-9 A, Photomicrograph of normal rat β cells (×28,000) showing morphology of mature granules (bar = 1 µm). B, Isolated rat β-granule cores (×17,000)236 (bar = 1 µm). C, High-magnification view (×250,000) showing repeat unit structure of a crystalline core (bar = 0.1 µm). The cores are made up of both rat insulin I and II in approximately equal proportions (Michael J and Steiner DF, unpublished data). Samples were fixed with Karnovsky’s solution and stained with osmium tetroxide. (Electron micrographs courtesy Hewson H. Swift.)
Most of the zinc in islets is present in the β granules and is liberated proportionately to insulin during secretion, in keeping with its role in crystallization of the hormone.86 The insulins of some species, including the guinea pig, coypu, and other hystricomorph rodents42 and the primitive hagfish,87 lack the histidine residue at position 10 of the B chain required for zinc binding during the association of insulin dimers into hexamers.88 A human subject heterozygous for a mutation at B10 (His B10 Asp) had elevated levels of this proinsulin in the blood because it tended to be released constitutively at much higher levels than normal proinsulin.89 However, in a transgenic mouse model of this mutation, there was no defect in its conversion to insulin in the granules, despite its altered self-association and sorting properties.90
The pH of the interior of the mature secretory granule appears to be between pH 5.0 and 6.0,51,91 an optimal pH range for insulin crystallization in vitro. The neutral or slightly alkaline pH in the cisternal spaces of the RER favors proinsulin folding and sulfhydryl oxidation. The pH remains near neutral throughout the Golgi apparatus but becomes more acidic (pH 6.1) in the TGN as the secretory products are sorted into granules and proteolytic processing begins. Vesicular proton pumps may begin to increase the uptake of protons, which then displace the cationic arginine and lysine residues liberated during conversion. As these move out of the granules and are replaced by hydrogen ions, a downward shift in intragranular pH may occur. Thus the initially mildly acidic progranules91 undergo gradual acidification as they mature in the cytosol (see Fig. 6-8), creating appropriate conditions for the conversion and crystallization of the newly formed insulin. One recently discovered role of the constitutive pathway convertase, furin, in the β cell is to activate the proton pump, which acidifies the secretory granules. As a result, proinsulin processing is impaired in β cells lacking furin.92
The cellular processes related to the biosynthesis of insulin via preproinsulin and proinsulin and their intracellular transport, sorting, proteolysis, and ultimate storage in secretory granules are remarkably well integrated, both topologically and biochemically. This delicately poised integration of processes leading to the formation and storage of insulin is disturbed in islet cell tumors, which often show unregulated release of insulin together with large amounts of proinsulin; measurements of the latter can provide a useful diagnostic indicator93 (see Chapter 21).
Biosynthetic Role and Biological Actions of the C Peptide
Because of its cosecretion with insulin in essentially equimolar amounts,22,94,95 the C peptide has been of great value as a marker of insulin secretion in humans under a variety of conditions. Representative vertebrate C-peptide amino acid sequences are compared in Fig. 6-4.96 These peptides exhibit a 15-fold higher rate of mutation acceptance than do the corresponding insulins, a finding that has often been interpreted as indicating that this region in the proinsulin molecule is unlikely to have any specific hormonal function. Nonetheless, several acidic residues are consistently present at certain positions in mammalian C peptides. These offset the added cationic charges due to the pairs of basic residues at the proinsulin cleavage sites, such that the isoelectric pH of proinsulin is nearly the same as that of insulin (i.e., in the range of pH 5.1 to 5.5).22
A large body of evidence supports a biosynthetic role for the C-peptide segment in proinsulin. Primarily, it converts the insulin A and B chain interaction from an inefficient bimolecular reaction to a highly efficient and concentration-independent unimolecular reaction.97 Certain regions of the connecting peptide may also facilitate the folding of the proinsulin polypeptide chain and the formation of the correct disulfide bonds, or guide the enzymatic cleavage of proinsulin to insulin by helping to orient the basic residue pairs for efficient binding and cleavage by the convertases.98,99 Recent molecular modeling studies indicate that the C peptide may assist in achieving important conformational changes in the N-terminal helical region of the A-chain segment, aiding its interaction with the convertases during processing.100
The conservation of a length of 30 to 35 amino acids in almost all proinsulin C peptides, including those of insects, mollusks, and nematodes, despite the short 8- to 10-angstrom separation between A1 and B30 in the native insulin molecule88 (see Fig. 6-2), supports the existence of an important length-related function. Since much shorter connecting segments do not impair sulfhydryl oxidation and formation of the native structure,99,101 it is clear that this is not a critical constraint. A highly plausible reason for the evolutionary retention of a relatively long C peptide in proinsulin may be to facilitate the translocation of proinsulin across the RER membrane during its synthesis. The length of polypeptide chain required to span the large ribosomal subunit and the RER membrane has been estimated to be about 65 residues in extended configuration.102 Moreover, the arrest of translation by the signal recognition particle (SRP) occurs only after synthesis of a nascent chain of about this length and thus may play a role in translational control of insulin biosynthesis.102 Efficient intracellular transport and correct targeting to secretory granules may impose additional demands on the primary (and tertiary) structure of proinsulin and other precursor proteins. However, miniproinsulins with deleted or greatly truncated C peptides do not fold efficiently but are correctly targeted to the regulated secretory pathway.98,101,103
Another very important function of the C-peptide is to facilitate its removal from proinsulin after it has served its biosynthetic functions, as mentioned earlier. The kinetics of insulin metabolism in vivo, as well as its biological activity, are both dependent upon its high affinity for the insulin receptor. Proinsulin’s much lower receptor-binding affinity leads to a much slower rate of clearance, which explains its relatively higher apparent biological activity in vivo. The rapid clearance of insulin is adaptively as important a characteristic as its bioactivity. To assure its interaction with the convertases, proinsulin must present more than just a pair of basic residues to the convertases. Their active sites require an extended β strand that is at least eight residues in length for recognition at each cleavage site. Molecular modeling studies have suggested how the full length of the connecting segment contributes to achieving the necessary conformations for effective proinsulin binding and cleavage by the convertases.100
Since the early 1990s, a number of reports have appeared describing a variety of biological actions of the C peptide and/or smaller peptides derived from it.104 These putative effects include enhancement of glucose transport and utilization; improvements in microcirculation in muscle, skin, retina, and nerve in diabetics; and stimulation of renal tubular Na+,K+-adenosine triphosphatase (ATPase) activity and other parameters of renal function. It has been suggested that tissue receptors for C peptide might exist, and that the circulating C peptide may contribute to improved glycemic control and help to slow the development of the vascular and neural complications of diabetes.105 Further study is needed in this area in view of reports that some C-peptide effects do not follow the usual rules of ligand-receptor chemistry (i.e., chirality does not matter). Thus, a C peptide synthesized entirely of d amino acids is equally as active as a peptide made with l amino acids in reversed order,106 whereas a random sequence of the same amino acids leads to loss of activity. These tantalizing findings suggest the need for a controlled clinical trial of combined insulin and C-peptide therapy in diabetics to fully evaluate its therapeutic potential.
The availability of biosynthetic human proinsulin and insulin, as well as of human C peptide, has opened many new possibilities for studies of the role, metabolism, and antigenicity of these peptides.107–109 In recent years, active insulin analogs with shortened linking peptides have also been developed. Such miniproinsulins and/or “single-chain insulins” appear to readily oxidize to form correct disulfide bridges and either can be used intact or after cleavage to insulin.98,99,101 Other efforts have been directed towards synthesis of modified insulins with improved absorption, aggregation, and/or pharmacokinetic properties for therapeutic use.
Regulation of Insulin Production
Under normal circumstances the pancreatic β cell retains a remarkable state where insulin secreted in response to a stimulus is rapidly replenished at the biosynthetic level in parallel, thus keeping intracellular insulin stores optimal. The vast majority (98% to 99%) of insulin is secreted via the “regulated” secretory pathway110 and only occurs in response to increases in specific nutrients, certain hormones, neuropeptides, and some pharmacologic reagents.111,112 In general, most nutrients that regulate insulin secretion also control proinsulin biosynthesis,111 and the most physiologically relevant of these is glucose.112 Glucose metabolism is required for both glucose-induced insulin secretion and production,111 but the secondary stimulus-coupling mechanisms are quite distinct. For example, stimulated insulin secretion is Ca2+ dependent, but glucose-induced proinsulin biosynthesis is Ca2+ independent.112,113 There are effective inhibitors of glucose-induced insulin secretion, such as diazoxide, epinephrine, and somatostatin, but these have no effect on glucose-induced proinsulin biosynthesis.111 In contrast, effective stimulators of insulin secretion, such as sulfonylureas and fatty acids, have no effect on proinsulin biosynthesis.111,114 This differential regulation of insulin secretion and proinsulin biosynthesis indicates that glucose-induced proinsulin biosynthesis is not regulated by a positive feedback of locally secreted insulin.113 For stimulated insulin secretion, the intracellular secondary signaling mechanisms are relatively well defined,115 but those for glucose-induced proinsulin biosynthesis, other than a need for mitochondrial metabolism and subsequent succinate production,116 are not currently well understood. However, it is known that both glucose-induced insulin secretion and production can be potentiated by an elevation in [cAMP]i secondary to the action of incretins such as GLP-1.117 The glucose-dependent effect of GLP-1 to increase proinsulin biosynthesis is predominately at the translational level, not at the transcriptional level.118
Glucose regulation of proinsulin biosynthesis is relatively specific and occurs independently of the control of general protein synthesis in the β cell.119 There is a minor subset of β-cell proteins (~50) that are also specifically regulated by glucose at the biosynthetic level in parallel to proinsulin, with these mostly being insulin secretory-granule proteins, including the proinsulin-processing endopeptidases, PC1/3 and PC2.74,75,120,121 This provides a mechanism whereby the enzymes that process proinsulin and the intracellular storage compartment in which insulin is stored are coordinately increased in response to the up-regulation of proinsulin biosynthesis.
The mechanism for specific regulation of glucose-induced proinsulin biosynthesis is complex and can occur at multiple levels depending on the temporal exposure to elevated glucose levels. Under normal fluctuations in glucose concentrations that occur over periods of ≤3 hours, glucose-induced proinsulin biosynthesis is entirely mediated at the translational level.111,113,122,123 For periods of ≥12 hours, this translational regulation is supplemented by glucose promoting preproinsulin mRNA stability124 to provide more preproinsulin mRNA template for translational regulation.113 Longer-term increased proinsulin biosynthesis can also be contributed to by increased insulin gene transcription.125,126
However, even under these longer-term circumstances, the primary control of glucose-induced proinsulin biosynthesis is mediated at the translational level. Yet despite glucose-induced translational regulation of proinsulin biosynthesis being known for nearly 40 years,22,122,127 the molecular mechanism remains unresolved. There are adaptive responses of the general translational machinery to up-regulate preproinsulin mRNA translation that have been shown to occur at the initiation and elongation phases of translation,22,123,127 as well as reduction in the duration of SRP signal peptide–mediated arrest of nascent preproinsulin-chain elongation prior to ribosome docking in the early phases of RER translocation.123 However, the specific translational regulation of proinsulin biosynthesis lies in the preproinsulin mRNA itself, particularly in the untranslated regions (UTRs).128 The 3′ UTR of preproinsulin mRNA contains elements such as a polypyrimidine-rich tract (PRT)129 and a highly conserved UUGAA element111,128 that likely control its stability. The control of preproinsulin mRNA stability via the 3′ UTR PRT also involves a glucose-responsive association of the polypyrimidine tract binding protein (PTB).128 The 5′ UTR of preproinsulin mRNA contains a conserved translational control cis element, ppIGE (for preproinsulin glucose element), that is required for glucose-induced translational control of proinsulin biosynthesis.128,130 The ppIGE has a highly conserved ppIGE “mirrored” core of GUCxnCUG or GUUxnUUG (where n ≤ 4 bases). There is a protein trans-acting factor (ppIE-BP) found in islet cytosolic extracts that binds to this translational control ppIGE cis element of preproinsulin mRNA in a glucose-dependent manner that parallels the characteristics of glucose-regulated proinsulin biosynthesis.130 The identity of the ppIGE-BP is currently unknown. Nonetheless, it is intriguing that the ppIGE is also conserved in the 5′ UTR of the mRNA templates of other secretory granule proteins whose biosynthesis is specifically controlled at the translational level in β cells, including PC1/3 and PC2.98,99 As such, the glucose-induced specific translational control of proinsulin biosynthesis and that for other insulin secretory-granule protein is likely via the same mechanism. For the longer-term transcriptional regulation of insulin gene expression by glucose, this is known to be a Ca2+-dependent mechanism131 involving the putative phosphorylation of key β-cell transcription factors, Pdx-1 and MafA.125 Prolonged glucose stimulation can also lead to increased β-cell hyperplasia and hypertrophy,132 which might also be additionally influenced by a wide variety of growth factors and other nutrients.133 Increased numbers of β cells will also increase insulin production, as occurs in nondiabetic obesity and pregnancy.133
Insulin secretory-granule turnover should also be considered in the long-term regulation of insulin production and availability. An insulin secretory granule has an estimated half-life of 3 to 5 days,94 and if it does not undergo exocytosis, it is degraded intracellularly. Degradation of intracellular organelles like insulin secretory granules is mediated via autophagic mechanisms.134,135 Under normal circumstances, autophagy plays a janitorial role, removing aged secretory granules.136 However, under some circumstances where insulin secretion is compromised and proinsulin biosynthesis is not, then autophagy is up-regulated to prevent overaccumulation of intracellular insulin stores.94,136–138 It is thought that glucose also influences the autophagic degradation of intracellular insulin, but the mechanism is largely unknown94; recent studies suggest that LAMP2, ATG-7, and VMP1 may be involved.136,139,140
Insulin Secretion and Its Regulation
Insulin regulates glucose disposal in humans, maintaining blood glucose levels between approximately 3.8 and 6.1 mM in the fasting state; one definition of diabetes mellitus is a fasting blood sugar greater than 7 mM (see Chapter 13 in this volume). In normal, healthy individuals, the glucose level may rise to about 10 mM after a meal and decline to 3.3 mM during a prolonged fast. A sophisticated glucose-sensing and control mechanism controls insulin secretion, primarily linked to blood sugar concentrations but also dependent on numerous other factors, such as the incretins, GLP-1 and GIP, that all act to maintain blood sugar levels within these limits.
Similar to other neuroendocrine cells, insulin-secreting cells are electrically excitable.141 This means that insulin secretion in response to glucose is dependent on initiation of electrical activity from a basal electro-negative resting state. A summary of the many factors that regulate the secretory activity of the β cell is illustrated in Fig. 6-10. The importance of electrical control is most dramatically illustrated in the cases of neonatal diabetes caused by mutations in the two subunits of the ATP-dependent potassium channel (KATP), Kir6.2 and SUR1.142,143 In the normal course of glucose excitation coupling to insulin secretion, glucose enters the cell via GLUT2 (and GLUT1) glucose transporters and is trapped in the cell by phosphorylation via glucokinase, a specialized high-Km hexokinase. The high Km of glucokinase for glucose underlies its role as the “glucose sensor” of the β cell, in that its enzyme activity reflects physiologic circulating glucose concentrations. There is good evidence from studies of patients with loss-of-function mutations in glucokinase that this enzyme determines the insulin secretory response to changes in glucose concentration across the physiologic range.144 Following glucose phosphorylation, further metabolism via glycolysis, the Krebs (tricarboxylic acid [TCA]) cycle, and oxidative phosphorylation in the mitochondria leads to an increase in the ATP/ADP ratio in the cytoplasm.
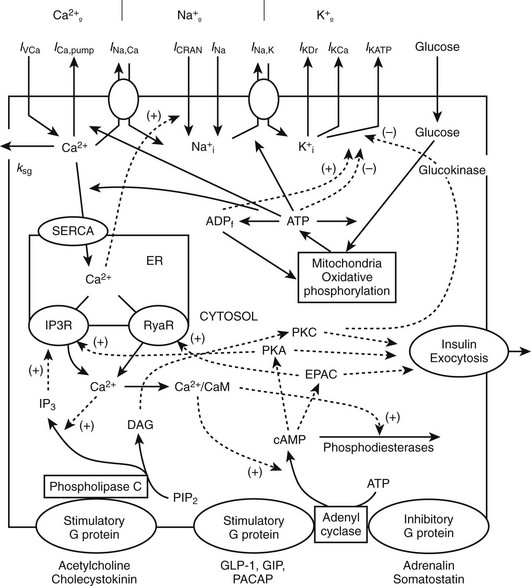
FIGURE 6-10 Interaction between membrane channels, ions, receptors, and selected mechanisms of cAMP and IP3 pathway regulation in insulin secretion. Top, plasma membrane currents: voltage-dependent Ca2+ current (IVCa), a calcium pump current (ICa,pump), Na+/Ca2+ exchange current (INa,Ca), Ca2+ release-activated nonselective cation current (ICRAN); inward Na+ currents (INa); a sodium-potassium pump current (INa,K), a delayed rectifying K+ current (IKDr), the small conductance Ca2+-activated K+ current (IKCa), ATP-sensitive K+ current (IKATP). Note that ksg is a coefficient of the sequestration rate of Ca2+ by the secretory granules, SERCA is the endoplasmic reticulum calcium pump, Ca2+ leaks out of the ER through IP3 receptor (IP3R) and ryanodine receptor (RyaR). Signals originating from fuel metabolism increase cytosolic calcium. Bottom, CaM, calmodulin, Ca2+/CaM is Ca2+-bound calmodulin. Synthesis and degradation of cAMP are catalyzed by adenylyl cyclase (AC) and phosphodiesterase (PDE), respectively. Epac is a guanine exchange protein activated by cAMP; PKA, protein kinase A; GLP-1 is glucagon-like peptide-1; PACAP is pituitary adenylate cyclase–activating polypeptide; GIP is glucose-dependent insulinotropic peptide. Synthesis of IP3 and diacylglycerol (DAG) from phosphatidylinositol-4,5-bisphosphate (PIP2) is catalyzed by phospholipase C (PLC). A variety of kinases activates exocytosis of insulin granules. Solid lines indicate fluxes, and dashed lines indicate inhibitory or stimulatory influences on currents or fluxes. (Courtesy L. Fridlyand.)
While there are alternative hypotheses, it is the ATP/ADP ratio increase driven by metabolism that leads to KATP channel inhibition, in turn leading to accumulation of positive charge (K+ and Na+) inside the cell, causing the cell membrane to depolarize. As the membrane potential reaches about −20 mV from the resting level of about −70 mV, voltage-dependent calcium channels open, allowing entry of Ca2+. The rise in Ca2+ is soon opposed by opening of voltage-dependent and Ca2+-dependent K+ channels, contributing to the appearance of tonic or periodic transients of both membrane potential and Ca2+.145 The action potentials following glucose administration occur in grouped bursts of spikes on a depolarized plateau, with a variable quiet interval between bursts (Fig. 6-11). Some studies have shown that Ca2+ entry can lead to release of Ca2+ from intracellular (presumably endoplasmic reticulum) pools where it has been sequestered by the action of the sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCA). Release of intracellular calcium through the action of inositol trisphosphate (IP3) on the IP3 receptors, as well as via ryanodine receptors and additional messengers, may all play complex roles in shaping intracellular Ca2+ transients. Mitochondria are also important stores of calcium and take up Ca2+ during metabolic activity, in part to regulate the key dehydrogenases in the TCA cycle. The elevation in intracellular free Ca2+ activates multiple proteins (e.g., small G proteins such as Rabs and soluble N-ethylmaleimide attachment protein receptor [SNARE] pathways) regulating the Ca2+-triggered fusion of the (predocked) insulin-containing granules with the cell membrane, resulting in the first phase of insulin secretion. As in neurotransmitter release, β-cell granule fusion depends on interactions of synaptosome-associated proteins, v-SNAREs (VAMP2 and synaptotagmin), with plasma membrane receptors such as SNAP-25, a t-SNARE (target localized SNAP receptor), and syntaxins. The importance of the depolarization step in insulin secretion is indicated by activating mutations in Kir6.2 and SUR1 that keep the KATP channel from closing in response to glucose metabolism, completely preventing physiologic insulin secretion and resulting in permanent neonatal diabetes.142,143 In contrast, loss-of-function mutations in these proteins that result in a persistently closed channel cause continued insulin release, leading to the condition known as hyperinsulinemia of infancy.146 Second-phase insulin secretion refers to the continued release of insulin following the initial peak. The kinetics of second-phase secretion at the β cell or islet level are variable and species specific. Granules are recruited via microtubules and associated kinases, chaperones, and small GTP-binding proteins (syntaxin4, Munc18) through the actin network until they too can dock with the plasma membrane at t-SNARE sites.147
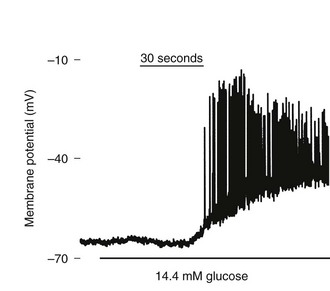
FIGURE 6-11 Effect of glucose on membrane potential in a mouse islet. The graph shows the onset of electrical bursting activity in a mouse islet, where the patch electrode was placed on one cell still connected to other islet cells. (Courtesy D. Jacobson.)
Mutations in the insulin gene or the genes encoding several transcription factors, ion-channel subunits, and glucokinase have all been linked to impaired insulin secretion in monogenic diabetes. These include the KATP subunits KCNJ11 (Kir6.2), ABCC8 (SUR1), HNF1A, HNF4A, HNF1B, PDX1, and mitochondrial genomic mutations, among others.148–150 Activating mutations of glucokinase,144,149 ABCC8, KCNJ11, glutamate dehydrogenase, or the mitochondrial enzyme short-chain 3-hydroxyacyl-CoA dehydrogenase can all cause hyperinsulinemia and hypoglycemia.151 In diabetes due to transcription-factor mutations, β cells gradually lose the ability to secrete insulin, yet in some forms (mutations in the transcription factor genes HNF1A and HNF4A) retain responsiveness to sulfonylurea drugs, at least for some time. It is likely that multiple pathways are affected by heterozygous mutations in these genes. Mutations in glucokinase (GCK) are interesting in that the activating mutations can also cause hypoglycemia, whereas those that decrease enzyme activity cause a (usually) stable fasting hyperglycemia. Not all GCK mutations causing hyperglycemia have been demonstrated to cause changes to Km or Vmax.144,152 The discovery of glucokinase activators153 to potentially treat type 2 diabetes may provide a specific treatment in those cases where the glucokinase mutation–related hyperglycemia is progressive, but usually these mutations require no treatment at all.
The ZnT8 transporter, Slc30A8, is a Zn2+ transporter that is relatively highly expressed in β-cell granules154 and is an antigen in type 1 diabetes.155 Mutations in the coding region of this gene have been linked to type 2 diabetes in genomewide association studies.156 Zn2+ coordinates the formation of the insulin crystal hexamer, facilitating the storage and stability of insulin crystals, and may also act as a messenger in secretion, affecting ion channels on nearby α or β cells, for example. The role of Zn2+ in insulin granules and the mechanism of the linkage of ZnT8 to type 2 diabetes may be clarified by further study of ZnT8-null mice.85,155,157
While glucose is the key physiologic stimulator of the insulin-secreting system, it can be replaced or enhanced by other energy-providing metabolites (for example, leucine and succinic acid monomethyl ester) that either feed into glycolysis, the TCA cycle, or related oxidative phosphorylation pathways.158 The mitochondrial production of ATP, along with decrements in ADP, are key steps in regulating KATP, but other anaplerotic molecules coming from the mitochondria, glutamate in particular, have been implicated in regulation of secretion.159,160 Naturally occurring or experimentally induced mutations in the mitochondrial genome or in the proteins that regulate mitochondrial energetics have dramatic effects on insulin secretion.161
Once KATP closes and Ca2+ entry begins via the closure of voltage-dependent calcium channels, a variety of mechanisms control intracellular Ca2+.162 These include other K+ channels, voltage- and Ca2+-dependent, that serve to help repolarize the plasma membrane and voltage-dependent Na+ channels that accelerate plasma membrane depolarization, especially in human, canine, and porcine islets.163 Calcium-dependent Ca2+ release couples extracellular Ca2+ entry to intracellular Ca2+ release from the endoplasmic reticulum or other stores such as the insulin granules. Key intracellular regulators include the IP3 receptor and NAADP receptors that activate ryanodine pathways from either specialized areas of the ER, the insulin granules, mitochondria, or all of these. Spatial and temporal control of Ca2+ signals, as well as other second messengers regulating secretion such as cAMP, may be highly regulated in plasma membrane microdomains where insulin granules fuse.164,165 Although cytoplasmic calcium signals, often oscillatory, are highly correlated with insulin secretion, they are separable from insulin secretion in experimental situations, showing that changes in intracellular Ca2+ can be both bypassed and/or augmented.166
G protein–coupled receptors (GPCR) function in β cells in both stimulatory and inhibitory mechanisms involving multiple protein kinase pathways.167 The incretin agonists, principally GLP-1, GIP, and glucagon, activate receptors that primarily couple to adenylate cyclase.168 The subsequent increase in cAMP has multiple effects, from activating gene expression and inhibiting cell death pathways169 to increasing insulin secretion in a glucose-dependent manner, both by positive effects on ion channels but primarily through augmentation of downstream exocytotic pathways, including granule fusion. Both protein kinase A and the guanine exchange protein, EPAC, play a role in this pathway.170 Beta- and alpha-adrenergic GPCRs also regulate insulin secretion; for example, β2-agonists result in increased insulin secretion, also via a PKA mechanism.171 Other agents that affect the half-life of cAMP, including pertussis toxin,172 isobutylmethylxanthine (IBMX), a nonspecific inhibitor of phosphodiesterase (PDE),173 and specific PDE inhibitors, have been shown to augment glucose-dependent insulin secretion.174 Levels of cAMP in the β cell may be oscillatory in response to oscillatory changes in adenylyl cyclase, PDE activity, and intracellular Ca2+, highlighting a role for microdomains within the cytoplasmic compartment.164,175,176
The muscarinic M3 receptor is activated by acetyl choline (ACh or its analog, carbachol) through the vagal autonomic pathways.177 Muscarinic activation has an adjunctive role in glucose-stimulated insulin secretion, but the ability of transplanted islets and whole pancreas to maintain normoglycemia in the absence of vagal innervation suggests that this pathway is dispensable. The putative fatty acid receptor termed GPR40178 has a positive effect on glucose-dependent insulin secretion, but whether activation of this pathway leads to useful increases in insulin secretion as a compensation for insulin resistance is not yet clear.179,180
Insulin Gene Mutations as a Cause of Permanent Neonatal Diabetes
The gene for insulin was among the first to be isolated. Its structure in man and several other species181 is summarized in Fig. 6-12. The single-copy human gene is located on the short arm of chromosome 11 in band p15. It is flanked on the 5′ side by a unique polymorphic region composed of tandem repeats that lies beyond the upstream regulatory region; this polymorphic region does not seem to influence the gene’s expression in the pancreas, but it may influence its transient expression in the thymus during development.182 It also provides a useful marker for genetic studies.183 Previous studies of mutations in the human insulin gene identified several missense mutations of proinsulin and insulin (all in the heterozygous state) that influence either the biological activity of the hormone or the conversion of proinsulin to insulin. These mutations may cause mild diabetes in the presence of insulin resistance (for a review, see Ref. 184).
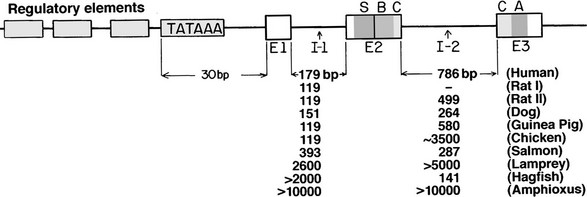
FIGURE 6-12 Diagrammatic representation of the insulin gene in vertebrates. Exons (E) appearing in mature preproinsulin mRNA are shown as bars, and the sizes of the two introns or intervening sequences (I) in various species are tabulated below. A, A-chain coding region; B, B-chain coding region; C, C-peptide coding region; S, signal peptide. A typical TATA box signaling transcription initiation is shown approximately 30 base pairs upstream from the messenger start site, preceded by a promoter region (unfilled boxes). The human insulin gene is located on the short arm of chromosome 11 in the region p15.183
Permanent neonatal diabetes (PNDM) is a rare form of diabetes that usually develops in the first few months of life, with an incidence of 1 in 100,000 to 300,000 live births. Activating mutations in Kir6.2 or SUR1 are the most common cause,142,143 and patients with mutations in these two proteins often respond to treatment with sulfonylureas, which reverse the inhibition of insulin secretion, freeing patients from the need for insulin injections.185 Recently, mutations in insulin and its precursors, proinsulin and preproinsulin, have been found to be another important cause of neonatal diabetes. The first such mutation found was a missense mutation that changed Gly B8 to Ser (G32S) in four affected members in a family.148 Since substitution of Gly B8 by l amino acids is known to compromise formation of the A7-B7 disulfide bond and diminish in vitro insulin folding yields, it likely acts in a dominant-negative fashion in vivo to disrupt insulin production and precipitate neonatal diabetes. A variety of other mutations in the preproinsulin molecule have since been identified in children with unexplained neonatal diabetes and/or early-onset antibody-negative type 1b diabetes, as well as in patients with a diagnosis of maturity-onset diabetes of the young (MODY).148,186–188 Among the mutations identified was one identical to that found to produce early-onset diabetes in the Akita mutant mouse (C96Y), thus connecting the human INS mutations associated with neonatal diabetes with dominant negative–acting Ins2 mutations in mice that impair the folding of proinsulin and induce severe ER stress.148 Another similar study from Italy identified an additional seven mutations in 10 probands.189 Expression of these mutant proinsulins in HEK293 cells demonstrated intracellular retention and greatly reduced secretion in six of the seven mutations studied. Other workers also confirmed that substitution of B5 His by Asp (H29D)186 impairs insulin folding.190 A variety of functional studies have shown that many of these mutant preproinsulins are retained in the ER, resulting in little or no secretion.190a,190b Their findings also confirm the suppressive effect of the mutant forms on the production of normal insulin, consistent with their dominant-negative character.
All currently known preproinsulin mutations are shown in Fig. 6-13, including earlier ones that do not affect folding but have reduced biological activity or cannot be processed to insulin, resulting in hyperproinsulinemia.184 It is noteworthy that three of the mutations that cause neonatal diabetes occur in the signal peptide.148 The first, R6C, replaces a basic residue that is frequently present just before the hydrophobic central core in signal peptides, and this results in a milder, later onset (ages 15 and 65) MODY-like form of diabetes. This basic residue may be required to properly align the signal peptide for its interactions with signal peptide recognition particle (SRP) and/or other translocon components. The next, A23S, was identified in a child diagnosed with type 1b diabetes at 6 years, 8 months of age.191 This mutation probably disrupts cleavage of the signal peptide. A third mutation, A24D, is far more common, having been found so far in seven patients. It prevents cleavage of the signal peptide at the normal A·X·A↓ cleavage site resulting in marked ER retention.190b
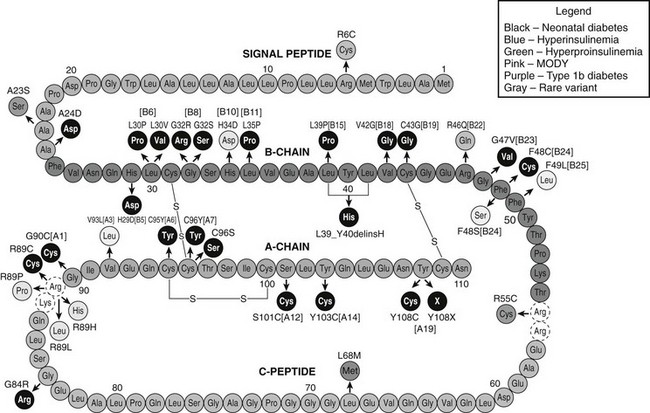
FIGURE 6-13 Diagrammatic representation of the amino acid sequence of human preproinsulin (green, signal peptide; red, B chain; orange, C peptide; dark blue, A chain) indicating sites of mutations identified in patients with diabetes as well as hyperinsulinemia and hyperproinsulinemia. Mutations shown in black disrupt proinsulin folding and/or disulfide-bond formation, leading to permanent neonatal diabetes (PNDM). Mutations in light blue do not impair folding but are associated with reduced insulin-receptor binding potency (hyperinsulinemia). Mutations in light green are associated with hyperproinsulinemia and either impair proteolytic processing to insulin or, in the case of H34D, aggregation and sorting into dense-core granules of the regulated secretory pathway. Mutations in pink and purple were found in patients with a diagnosis of MODY and type 1b diabetes, respectively. The mutation shown in gray, L68M, is a rare variant without functional effects on proinsulin/insulin biosynthesis (see text for details).
Many of the A- and B-chain mutations are substitutions of Cys residues involved in interchain disulfide bonds by non–sulfur containing amino acids or additions of Cys at other sites, resulting in unpaired cysteines that may tend to clog ER pathways involved in disulfide-bond formation and leading to ER stress. One of the most severe mutations is C43G, which prevents formation of the disulfide bond at B19-A20, the first to form as proinsulin folds. The absence of this critical disulfide bond prevents the formation of an important folding intermediate that normally assists the later folding steps.192 Other mutations replace conserved Gly residues in the B chain at positions 8 and 24. Glycine residues provide greater flexibility to peptide chains because they lack side chains that sterically interfere with unusual peptide-chain conformations forbidden for l amino acids. These two Gly residues participate in β turns that are necessary to properly align the cysteines at B7 or B19 for disulfide-bond formation with the A7 or A20 cysteines, respectively.148,193 The highly flexible C peptide contains a high proportion of Gly residues (7 of 31), and one of these is the site of another mutation, G84R, that could interfere in proinsulin folding, most likely due to steric hindrance from the bulky Arg side chain. Other B-chain mutations that replace conserved Leu residues with Pro or Val likely disrupt folding by preventing correct packing of the hydrophobic core of the insulin molecule.189
Mutations that impair proinsulin biosynthesis and folding can cause diabetes in both humans and mice.194,195 However, the pathogenesis of the ER stress and the mechanism of the dominant-negative effect are not well understood. An important factor is the unmet but rising demand on the β cell for increased insulin output as hyperglycemia develops. The failure to fold proinsulin leads to increased traffic over the ER sulfhydryl oxidation pathway, owing to repeated redox cycles involving protein disulfide isomerase (PDI) and its various isoforms.196 The electrons removed during oxidative folding by PDI are ultimately transferred to molecular oxygen by two mammalian ER flavoproteins, Ero1α and β,197 thereby generating increased amounts of reactive oxygen species (ROS). The buildup of misfolded mutant proinsulin likely blocks the folding of normal proinsulin and other nascent proteins in the ER. The accumulation of misfolded protein in the ER then triggers the unfolded protein response (UPR), leading to increased levels of ER chaperonins such as BIP, GRP94, and PDI196 and enhanced phosphorylation of eukaryotic initiation factor 2α by PERK (dsRNA-activated protein kinase-like ER kinase) to adaptively attenuate protein translation.198 It is possible that β-cell-specific isoforms of PDI or other specialized chaperonins are required for efficient proinsulin folding in vivo.199 Increased ER-associated degradation (ERAD) of misfolded protein also plays an important role in alleviating ER stress but may not be adequate to the task with some mutant proinsulins.200 Whether the development of diabetes is due to apoptotic β-cell death or this is a later complication due to chronic hyperglycemia is unclear. However, early therapy with insulin and/or with GLP-1 receptor agonists or related agents may help prevent β cell loss in affected individuals.
Biosynthesis and Processing of Other Islet Hormones
β Cells normally secrete small amounts of a number of other peptides and proteins in addition to insulin.33,201 Some of these are unique to the islet β cells, whereas others are expressed in other neuroendocrine cells and neoplasms. Chromogranins A, B, and C are members of a granin family of closely related acidic peptides that are expressed widely in the neuroendocrine system.202 Chromogranin A is found in the β, α, and γ cells in the islets, whereas chromogranins B and C are only present in the α cell.202–204 These proteins are thought to play a role in the formation and organization of secretory granules and/or give rise to bioactive peptides. Chromogranin A is processed to release at least two peptides of interest: (1) pancreastatin, a 49-amino-acid amidated peptide from the central region of chromogranin A, which was originally isolated from porcine pancreas and which inhibits insulin secretion205; and (2) β-granin, a 24-kD peptide derived from the N-terminal region of chromogranin A.201,203 Both peptides are stored in insulin granules and are released along with insulin. Contrary to earlier proposals, disruption of the chromogranin A gene does not prevent the formation of neuroendocrine secretory granules but is accompanied by compensatory up-regulation of other granin proteins.206
Islet Amyloid Polypeptide
Another intriguing islet protein is the 37-amino-acid neuropeptide-like molecule, islet amyloid polypeptide (IAPP), or amylin, a major protein constituent of the amyloid deposits that occur in the islets of patients with type 2 diabetes and in many benign insulinomas of the pancreas, as well as in the normal pancreases of the aged23,207,208 (Fig. 6-14). Although first noted in specimens of human pancreas as early as 1901,209 it was not until 1986 that efforts to solubilize islet amyloid were successful, revealing a single peptide, IAPP. Its amino acid sequence was related to the 37-amino-acid calcitonin gene–related peptide types 1 and 2 (CGRP-1, CGRP-2).207,208 Cloning of its mRNA and gene further support its evolutionary relationship to CGRP/calcitonin.23,210,211 Its precursor has a signal peptide, followed by a short propeptide ending in Lys-Arg at the N terminus of IAPP, and on its C-terminal side is followed by Gly-Lys-Arg and a second short propeptide (Fig. 6-15). The presence of the glycine residue in the latter cleavage site indicates that IAPP is normally carboxyamidated,212 as is CGRP.23 The human IAPP gene is related to the genes encoding CGRP-1 and CGRP-2 and is located on the short arm of chromosome 12.210
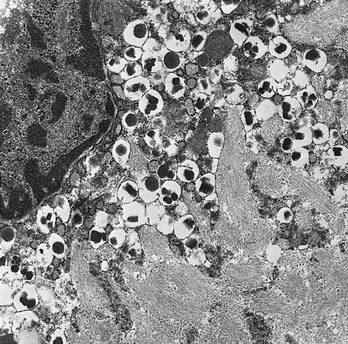
FIGURE 6-14 Photomicrograph showing extensive islet amyloid deposits with adjacent β cells in a human diabetic pancreas (×28,000). (Electron photomicrograph courtesy Dr. Per Westermark, Linköping, Sweden.)
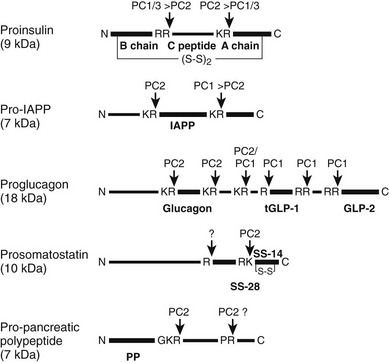
FIGURE 6-15 Schematic structure and processing by prohormone convertases of the major prohormones produced in the islets of Langerhans. Vertical arrows indicate sites of processing by PC1/3 or PC2. Heavier lines indicate active products, thinner lines denote propeptide regions, and tGLP-1 denotes the active truncated form of GLP-1 produced by cleavage at the single R site in proglucagon (see text for further details).
Biosynthesis and Levels of IAPP in Islets
Immunocytochemical studies indicate that IAPP normally is localized to the secretory granules of the β cells and some delta (δ) cells.23,213 In β cells, proIAPP is synthesized at levels of a few percent that of proinsulin and transferred with it into newly forming secretory vesicles in the TGN, where it is then processed into the mature 37-residue carboxyamidated peptide, stored, and subsequently cosecreted with insulin.214 Like proinsulin, the efficient processing of proIAPP requires the actions of both β-cell convertases, PC2215 and PC1/3.216 However, PC2 is more critical, since it alone is able to process the N-terminal cleavage site of proIAPP, whereas both PC2 and PC1/3 can process the C-terminal site, although PC2 does so more efficiently.217 The dominant role of PC2 explains how IAPP can be produced in the δ cells of the islet, which express only PC2.10,67 It has been proposed that the greater dependence of IAPP upon PC2 for its processing in the β cell, unlike proinsulin where PC1/3 plays the major role, may tend to allow greater accumulation of incompletely processed proIAPP in the diabetic β cell, where granule maturation processes may be perturbed by hyperglycemia.218
The expression of IAPP is stimulated by glucose in parallel with insulin under normal conditions, but relative expression levels may be altered in pathologic states.219 Very low levels of IAPP mRNA have also been detected in the stomach and other regions of the gastrointestinal tract, lung, and in dorsal root ganglia of the spinal cord.220 The relative levels of IAPP to insulin in the β cell are very low. HPLC analysis of freshly isolated rat islets shows amounts of IAPP in the range of 1% to 2% of insulin.23 Most studies221–223 agree that the levels of IAPP are in the range of 0.2% to 3% of the level of insulin in normal adult rat islets or normal human pancreas. Studies with isolated rat islets have shown that IAPP secretion is stimulated by glucose, and that IAPP amounts to about 5% of the amount of insulin released in 1 hour at 16.7 mM glucose.223
Biological Actions of IAPP
Cooper and others have shown that IAPP inhibits glucose uptake and glycogen synthesis in muscle exposed to IAPP in vitro, an effect it shares with CGRP.224 In whole animals, efforts to modify glucose tolerance with IAPP infusion have met with mixed success. However, euglycemic glucose clamp studies with dogs have demonstrated that the amidated form of IAPP at high levels inhibits insulin-stimulated glucose disposal over short infusion periods of 1 to 2 hours.225 Other studies indicate that the actions of IAPP may be complementary to those of insulin and may include delayed nutrient delivery and suppression of postprandial glucagon secretion.226 However, mice lacking IAPP due to a knockout of the gene show increased insulin secretion and more rapid glucose disappearance, suggesting that its normal role is inhibitory with respect to insulin secretion and action.227,228
Both nonamidated and amidated forms of IAPP have serum calcium–lowering effects in animals in vivo as well as in cell culture systems.229 MacIntyre230 has proposed that IAPP may be secreted along with insulin to promote the utilization of ingested calcium. A direct effect on uptake of calcium by bone tissue also has been demonstrated, but it is not clear whether this effect is mediated via calcitonin or IAPP receptors.229 However, in 1999, studies of cells with high IAPP binding activity led to the discovery that calcitonin receptors were involved but required the coexpression of receptor activity–modifying proteins (RAMPs) to enhance the binding of IAPP and attenuate calcitonin binding.231 In the past few years, a great deal of new information has accumulated on the pharmacologic properties of at least six IAPP receptors existing at high levels in the hypothalamus, as well as elsewhere in the brain and possibly in some peripheral tissues. These result from the combination of RAMPs 1 to 3 with two spliced isoforms (a and b) of the calcitonin receptor. The RAMPs also interact with several other related G protein–coupled receptors.232 The high levels of IAPP binding in the hypothalamus are consistent with the anoretic properties of the peptide, which may account for a tendency to obesity of IAPP-null mice.227 This interesting knockout confirmed that IAPP is not necessary for normal islet development and function in this species; both null males and females exhibited improved glucose tolerance in vivo. Other effects of IAPP include reduced gastrointestinal motility, inhibition of glucagon secretion, and delayed gastric emptying, favorable actions for improving glucose levels in diabetes and an area that is beginning to be exploited therapeutically in man.233
Mechanism of Amyloid Formation
Numerous studies have shown that islet amyloid formation (see Fig. 6-14) occurs more prominently in spontaneously diabetic animals and in certain species more so than others. Diabetes-prone animals include several species of nonhuman primates as well as cats and raccoons.234 It is interesting to note that the IAPP sequences in these species differ most significantly in the region that has been defined as amyloidogenic (residues 20-29) in studies by Glenner et al.235 and O’Brien et al.234 Synthetic peptides from this region have the greatest tendency to form fibrillar, stacked β-pleated sheet structures similar to those occurring in amyloid. However, in the Octodon degus, or degu, a New World rodent related to the guinea pig, islet amyloid deposits consist mainly of degu insulin, which differs significantly from most other mammalian insulins.237
Antibodies raised to various regions of IAPP have verified its presence in islet amyloid by both light and electron microscopic immunocytochemical analysis.238 Although in normal β cells it is localized within the insulin secretory granules,214,238 fibrillar immunoreactive amyloid deposits have also been noted within the cytoplasm of β cells of some patients with type 2 diabetes by Clark and co-workers.239 Others have also noted the proximity of amyloid deposits to the β cells, suggesting that it has arisen from these cells either by secretion or by some other means of deposition.234 Clark et al.240 have found IAPP immunoreactivity in lysosomes and lipofuscin bodies within the β cells of the islets of both normal and diabetic individuals and have suggested that amyloid may begin to form during the intracellular degradation of secretory granules, as occurs in the normal turnover of unused secretory products, a process known as crinophagy.
The factors leading to amyloid deposition in diabetes remains unclear,241 but recent work with transgenic mice hypersecreting human IAPP have demonstrated amyloid deposition under some circumstances, especially with a high-fat diet.242 Genetic studies of patients with type 2 diabetes suggest that mutations do not contribute to the formation of islet amyloid deposits.243 However, recent reports of increased amyloid deposits in transplanted human islets in both human subjects and animal models suggest that amyloid formation may be an important contributing factor to islet transplant failure.244 Thus there is growing interest in inhibitors of amyloid fiber propagation, which might help delay transplant failure in type 1 diabetics and slow the progression of β-cell deficiency in type 2 diabetes.245,246
Biosynthesis and Processing of Glucagon, GLP1, GLP2, Somatostatin, Pancreatic Polypeptide, and Ghrelin
The hormones of the alpha (α), delta (δ), gamma (γ), and epsilon (ε) cells of the islets are glucagon, somatostatin, pancreatic polypeptide (PP), and ghrelin, respectively. All these peptides are derived from larger protein precursors (preproproteins) that traverse the regulated secretory pathway and are processed and stored in dense-core granules analogous to the insulin/IAPP storage granules in the β cells.247 But unlike the β cells, the other islet endocrine cells normally express only PC2 and not PC1/3.248 Not surprisingly then, as illustrated in Fig. 6-15, all of these hormones with the exception of ghrelin are cleaved from their precursors by PC2 acting alone. Thus in the PC2-null mouse, there is a total absence of mature glucagon in the islets and circulation and the presence only of large amounts of unprocessed inactive precursor forms.67 As a consequence of the lack of active glucagon, there is chronic hypoglycemia and extensive hyperplasia of both the α and δ cells, which form a greatly enlarged mantle surrounding the β cells in the islets.67,249 Similar changes are seen in mice lacking the glucagon receptor.250 In the case of the PC2-null mice, this hyperplastic state can be reversed by the intraabdominal administration of glucagon via minipumps over a period of several weeks.251
Proglucagon is a multifunctional precursor that contains two additional peptides related to glucagon10 (see Fig. 6-15). The two glucagon-like peptides, GLP-1 and GLP-2, are located within the inactive C-terminal half of the precursor molecule (major proglucagon fragment [MPGF], residues 72 to 160), which follows the 69-residue inactive N-terminal half, known as glicentin. Glicentin contains the 29-amino-acid glucagon molecule, preceded by an N-terminal propeptide of 30 amino acids and followed by another short propeptide segment which links it via a Lys-Arg doublet (residues 70 and 71) to MPGF. This “interdomain cleavage site,” as it is known, is the most rapidly cleaved site in proglucagon and can be cleaved by either PC1/3 or PC2. The other cleavage sites are then cleaved by either PC2 or PC1/3 but usually not both, as illustrated in Fig. 6-15, which allows the α cell to produce only glucagon by virtue of expressing only PC2, and the intestinal proglucagon-expressing l cells to produce only GLP-1 and GLP-2 via the exclusive expression of PC1/3.10,247 Thus a single precursor embodies a glucose-elevating hormone (glucagon), a β cell–stimulating hormone or “incretin” (GLP-1), and a third hormone which augments intestinal function and growth (GLP2). Selectivity of secretory product(s) is then determined by the convertase available to process the precursor at various sites. However, in the embryonic pancreas, the early-developing α cells express both PC1/3 and PC2,252 and this also has been reported to occur in the α cells in some poorly controlled diabetics with elevated numbers of islet α cells that express some PC1/3 and thus can secrete some active GLP-1.253 Perhaps this local production of GLP-1 may protect or augment β-cell function via its actions as an incretin. (For further discussion of the important actions of the glucagon peptide family, see Chapter 9 in this volume.)
Somatostatin, the product of the islet δ cells, is a widely distributed inhibitory peptide that influences endocrine secretion, intestinal smooth muscle contractility, and islet, brain, and gastrointestinal function generally.247 The islets and the brain in PC2-null mice contain only somatostatin 28 (SS-28) instead of the normally predominant somatostatin 14 (SS-14).67,247 This quite dramatic change is not likely to have much functional impact, however, inasmuch as somatostatin receptors do not appear to discriminate greatly between these two forms.254 As shown in Fig. 6-15, prosomatostatin can be processed only by PC2 at the unusual but highly conserved RK↓G cleavage site within SS-28 at the N-terminus of SS-14. The presence of SS-28 in the PC2-null mice indicates that cleavage at the single arginine required for its production still occurs in the absence of any other known convertase in the δ-cell secretory granules.247,248 This cleavage also occurs in yeast that lack kexin and is believed to be due to a different type of convertase, possibly an aspartyl protease.255
Pancreatic polypeptide (PP), the product of the γ cells of the islets, mainly regulates gastrointestinal functions, including gall-bladder emptying and pancreatic secretion, rather than influencing carbohydrate metabolism.247 PC2-null mice fail to process propancreatic polypeptide (A. Zhou and D.F. Steiner, unpublished results), which indicates that both of the cleavages required for its maturation are accomplished by PC2 (see Fig. 6-15). Cleavage of proPP at the Pro-Arg↓ site by PC2 is rather surprising, since in proghrelin, a similar Pro-Arg↓ site is cleaved only by PC1/3.256
The ghrelin prohormone256 (not shown in the figure) is similar to propancreatic polypeptide in having the ghrelin sequence at the N-terminus, but it is followed by a considerably larger C-terminal segment that contains another putative hormonal sequence called obestatin. However, in contrast to proPP, it requires PC1/3 for its processing, as demonstrated in extracts of stomach in PC1/3-null mice.256 An unusual feature of the mature ghrelin peptide that is required for most of its known biological functions is the modification of serine 3 by O-acylation with octanoic acid, a modification carried out by a specialized acyltransferase.257 Ghrelin is produced mainly in the stomach and functions as an orexigenic and metabolic hormone. Only small numbers of ghrelin-positive ε cells occur in normal adult human and rodent pancreatic islets.258 Although ghrelin cells are more prominent during developmental phases, ghrelin has not been shown to play a significant role in normal pancreatic islet development.258
Conclusions
The more complex NE polyprotein precursors often contain multiple cleavage sites that may differ considerably in their susceptibility to the major NE prohormone convertases, PC2 and PC1/3. Thus, multifunctional precursors such as proglucagon or proopiomelanocortin10 may be processed differentially into functionally divergent product mixtures in different tissue sites in the organism; for example, proglucagon gives rise to glucagon in the islet α cells and GLP-1 and GLP-2 in the intestinal l cells. All the peptide products of proteolytic cleavage are usually retained in the secretory vesicles and cosecreted by exocytosis in response to stimuli (e.g., insulin and C peptide are retained in the storage granules and secreted together in equimolar amounts along with small amounts of proinsulin from the pancreatic islets). Peptide fragments such as the proinsulin C peptide can be utilized as useful markers of secretory activity, although differences in their half-lives in the circulation due to divergent pathways of metabolism/degradation/excretion must be taken into consideration. Circulating immunoreactive peptides are often heterogeneous, consisting of larger or smaller sequence-related or overlapping peptides arising from the incomplete processing of a common precursor. These may have varying potencies, metabolic properties, and immunologic reactivities that can cause discrepancies in the assessment of circulating hormonal bioactivity by radioimmunoassay.
The biosynthesis of insulin and other islet hormones is regulated at multiple levels, including transcription, mRNA stabilization, and translation, in response to glucose and other signals. Recent studies with β cells indicate that glucose also coordinately up-regulates many other cellular genes, among them those encoding many components of the secretory pathway.259 Controlled degradation of newly synthesized260 or stored hormone may also play an important regulatory role in some systems.
References
1. Von Mering, J, Minkowski, O. Diabetes mellitus nach pankreas extirpation. Arch Exp Pathol Pharmacol Leipzig. 1890;26:371.
2. Bliss, M. The discovery of insulin. Chicago: University of Chicago Press; 1982.
3. Scott, EL. On the influence of intravenous injections of an extract of the pancreas on experimental pancreatic diabetes. Am J Physiol. 1912;29:306.
4. Banting, FG, Best, CH, Collip, JB. Insulin patent. Chem Abstracts. 1923;17:3571.
5. Macleod, JJR, Campbell, WR. Insulin: Its use in the treatment of diabetes. In: Medicine monographs, vol VI, parts I and II. Baltimore: Williams & Wilkins; 1925.
6. Murnaghan, JH, Talalay, P. John Jacob Abel and the crystallization of insulin. Perspect Biol Med. 1967;10:334–380.
7. Falkmer, S, El-Salhy, M, Titlbach, M. Evolution of the neuroendocrine system in vertebrates: a review with particular reference to the phylogeny and postnatal maturation of the islet parenchyma. In: Falkmer S, Håkanson R, Sundler F, eds. Evolution and tumour pathology of the neuroendocrine system. Amsterdam: Elsevier; 1984:59.
8. Humbel, RE, Bosshard, HR, Zahn, H. Chemistry of insulin. In: Steiner DF, Freinkel N, eds. Handbook of physiology, endocrinology I. Baltimore: Williams & Wilkins; 1972:111.
9. Sanger, F. Chemistry of insulin. Science. 1959;129:1340.
10. Rouillé, Y, Duguay, SJ, Lund, K, et al. Proteolytic processing mechanisms in the biosynthesis of neuroendocrine peptides: the subtilisin-like proprotein convertases. Front Neuroendocrinol. 1995;16:322–361.
11. Steiner, DF, Oyer, PE. The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc Natl Acad Sci U S A. 1967;57:473.
12. Chance, RE, Ellis, RM, Bromer, WW. Porcine proinsulin: characterization and amino acid sequence. Science. 1968;161:165.
13. Steiner, DF, Clark, JL, Nolan, D, et al. Proinsulin and the biosynthesis of insulin. Recent Prog Horm Res. 1969;25:207.
14. Oyer, PE, Cho, E, Peterson, JD, et al. Studies on human proinsulin: isolation and amino acid sequence of the human pancreatic C-peptide. J Biol Chem. 1971;246:1375.
15. Chan, SJ, Keim, P, Steiner, DF. Cell-free synthesis of rat preproinsulins: characterization and partial amino acid sequence determination. Proc Natl Acad Sci U S A. 1976;73:1964.
16. Sanders, SL, Schekman, R. Polypeptide translocation across the endoplasmic reticulum membrane. J Biol Chem. 1992;267:13791.
17. Patzelt, C, Labrecque, AD, Duguid, JR, et al. Detection and kinetic behavior of preproinsulin in pancreatic islets. Proc Natl Acad Sci U S A. 1978;75:1260.
18. Frand, AR, Cuozzo, JW, Kaiser, CA. Pathways for protein disulphide bond formation. Trends Cell Biol. 2000;10:203.
19. Munro, S, Pelham, HRB. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899.
20. Orci, L, Perrelet, A, Ravazzola, et al. Coatomer-rich endoplasmic reticulum. Proc Natl Acad Sci U S A. 1994;91:11924.
21. Pagano, A, Letourneur, F, Garcia-Estefania, D, et al. Sec24 proteins and sorting at the endoplasmic reticulum. J Biol Chem. 1999;274:7833.
22. Steiner, DF, Kemmler, W, Clark, JL, et al. The biosynthesis of insulin. In: Steiner DF, Freinkel N, eds. Handbook of physiology, Endocrinology I. Baltimore: Williams & Wilkins; 1972:175.
23. Nishi, M, Sanke, T, Nagamatsu, S, et al. Islet amyloid polypeptide: a new β cell secretory product related to islet amyloid deposits. J Biol Chem. 1990;265:4173.
24. Steiner, DF, Clark, JL, Nolan, C, et al. The biosynthesis of insulin and some speculation regarding the pathogenesis of human diabetes. In: Cerasi E, Luft R, eds. The pathogenesis of diabetes mellitus. New York: John Wiley & Sons; 1970:57.
25. Howell, SL. Role of ATP in the intracellular translocation of proinsulin and insulin in the rat pancreatic B cell. Nat New Biol. 1972;235:85.
26. Wattenberg, BW, Rothman, JE. Multiple cytosolic components promote intra-Golgi protein transport: resolution of a protein acting at a late state, prior to membrane fusion. J Biol Chem. 1986;61:2208.
27. Chappell, TG, Welch, WF, Schlossman, DM, et al. Uncoating ATPase is a member of the 70 kDa family of stress proteins. Cell. 1986;45:3.
28. Munger, BL. A light and electron microscopic study of cellular differentiation in the pancreatic islets of the mouse. Am J Anat. 1958;103:275.
29. Mogelsvang, S, Howell, KE. Global approaches to study Golgi function. Curr Opin Cell Biol. 2006;18:438–443.
30. Orci, L, Ravazzola, M, Amherdt, M, et al. Direct identification of prohormone conversion site in insulin-secreting cells. Cell. 1985;42:671.
31. Steiner, DF. Evidence for a precursor in the biosynthesis of insulin. Trans N Y Acad Sci. 1967;30:60.
32. Nagamatsu, S, Bolaffi, JL, Grodsky, GM. Direct effects of glucose on proinsulin synthesis and processing during desensitization. Endocrinology. 1987;120:1225.
33. Orci, L, Lambert, AE, Kanazawa, Y, et al. Morphological and biochemical studies of B cells in fetal rat endocrine pancreas in organ culture: evidence for proinsulin biosynthesis. J Cell Biol. 1971;50:565.
34. Frank, BH, Veros, AJ. Physical studies on proinsulin: association behavior and conformation in solution. Biochem Biophys Res Commun. 1968;32:155.
35. Rubenstein, AH, Mako, M, Welbourne, WP, et al. Comparative immunology of bovine, porcine, and human proinsulin and C-peptides. Diabetes. 1970;19:546.
36. Weiss, MA, Frank, BH, Khait, I, et al. NMR and photo-CIDNP studies of human proinsulin and prohormone processing intermediates with application to endopeptidase recognition. Biochemistry. 1990;29:8389.
37. Gliemann, J, Sorenson, HH. Assay of insulin-li activity by the isolated fa cell method: IV. The biological activity of proinsulin. Diabetologia. 1970;6:499.
38. Given, BD, Cohen, RM, Shoelson, SE, et al. Biochemical and clinical implications of proinsulin conversion intermediates. J Clin Invest. 1985;76:1398.
39. Frank, BH, Veros, AJ. Interaction of zinc with proinsulin. Biochem Biophys Res Commun. 1970;38:284.
40. Steiner, DF. Cocrystallization of proinsulin and insulin. Nature. 1973;243:528.
41. Steiner, DF, Hallund, O, Rubenstein, AH, et al. Isolation and properties of proinsulin, intermediate forms and other minor components from crystalline bovine insulin. Diabetes. 1968;17:725.
42. Blundell, T, Wood, S. The conformation, flexibility, and dynamics of polypeptide hormones. Annu Rev Biochem. 1982;51:123–154.
43. Low, BW, Fullerton, WW, Rosen, LS. Insulin/proinsulin, a new crystalline complex. Nature. 1974;248:339.
44. Nolan, C, Margoliash, E, Peterson, JD, et al. The structure of bovine proinsulin. J Biol Chem. 1971;246:2780.
45. Kuzuya, J, Chance, RE, Steiner, DF, et al. On the preparation and characterization of standard materials for natural human proinsulin and C-peptide. Diabetes. 1978;27:161.
46. Furuta, M, Carroll, R, Martin, S, et al. Incomplete processing of proinsulin to insulin accompanied by elevation of des-31,32 proinsulin intermediates in islets of mice lacking active PC2. J Biol Chem. 1998;273:3431.
47. Chan, SJ, Cao, Q-P, Steiner, DF. Evolution of the insulin superfamily: cloning of a hybrid insulin/insulin-like growth factor cDNA from amphioxus. Proc Natl Acad Sci U S A. 1990;87:9319.
48. Li, C, Kim, K. Neuropeptides. WormBook. 2008;25:1–36.
49. Kemmler, W, Peterson, JD, Steiner, DF. Studies on the conversion of proinsulin to insulin: I. Conversion in vitro with trypsin and carboxypeptidase B. J Biol Chem. 1971;246:6786.
50. Steiner, DF, Cho, S, Oyer, PE, et al. Isolation and characterization of proinsulin C-peptide from bovine pancreas. J Biol Chem. 1971;246:1365.
51. Kemmler, W, Steiner, DF, Borg, J. Studies on the conversion of proinsulin to insulin: III. Studies in vitro with a crude secretion granule fraction isolated from islets of Langerhans. J Biol Chem. 1973;248:4544.
52. Zühlke, H, Steiner, DF, Lernmark, NA, et al. Carboxypeptidase B-like and trypsin-like activities in isolated rat pancreatic islets. In: CIBA Foundation, ed. Polypeptide hormones: molecular and cellular aspects. Amsterdam: Excerpta Medica North-Holland; 1976:183.
53. Fricker, LD, Evans, CJ, Esch, FS, et al. Cloning and sequence analysis of cDNA for bovine carboxypeptidase E. Nature. 1986;323:461.
54. Song, W, Fricker, LD, Day, R. Carboxypeptidase D is a potential candidate to carry out redundant processing functions of carboxypeptidase E based on comparative distribution studies in the rat central nervous system. Neuroscience. 1999;89:1301.
55. Julius, D, Brake, A, Blair, L, et al. Isolation of the putative structural gene for the lysine-arginine-cleavage endopeptidase required for processing of yeast prepro-α-factor. Cell. 1984;37:1075.
56. Fuller, RS, Sterne, RE, Thorner, J. Enzymes required for yeast prohormone processing. Annu Rev Physiol. 1988;50:345.
57. Mizuno, K, Nakamura, T, Ohshima, T, et al. Yeast KEX2 gene encodes an endopeptidase homologous to subtilisin-like serine proteases. Biochem Biophys Res Commun. 1988;156:246.
58. Smeekens, SP, Steiner, DF. Identification of a human insulinoma cDNA encoding a novel mammalian protein structurally related to the yeast diabasic processing protease Kex2. J Biol Chem. 1990;265:2997.
59. Smeekens, SP, Avruch, AS, LaMendola, J, et al. Identification of a cDNA encoding a second putative prohormone convertase related to PC2 in AtT20 cells and islets of Langerhans. Proc Natl Acad Sci U S A. 1991;88:340.
60. Seidah, NG, Chrétien, M, Day, R. The family of subtilisin/kexin like pro-protein and pro-hormone convertases: divergent or shared functions. Biochimie. 1994;76:197.
61. Zhou, A, Webb, G, Zhu, X, et al. Proteolytic processing in the secretory pathway. J Biol Chem. 1999;274:20745.
62. Seidah, NG, Prat, A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J Mol Med. 2007;85(7):685–696. [Jul. Epub Mar 10].
63. Davidson, HW, Rhodes, CJ, Hutton, JC. Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic β cell via two distinct site-specific endopeptidases. Nature. 1988;333:93.
64. Guest, PC, Bailyes, EM, Hutton, JC. Endoplasmic reticulum Ca2+ is important for the proteolytic processing and intracellular transport of proinsulin in the pancreatic β-cell. Biochem J. 1997;323:445.
65. Smeekens, SP, Albiges-Rizo, C, Carroll, R, et al. Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2 and PC3. Proc Natl Acad Sci U S A. 1992;89:8822.
66. Rhodes, CJ, Lincoln, B, Shoelson, SE. Preferential cleavage of des-31,32-proinsulin over intact proinsulin by the insulin secretory granule type II endopeptidase: implications of a favored route for prohormone processing. J Biol Chem. 1992;267:22719.
67. Furuta, M, Yano, H, Zhou, A, et al. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci U S A. 1997;94:6646.
68. Zhu, X, Zhou, A, Dey, A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A. 2002;99:10293.
69. Zhu, X, Orci, L, Carroll, R, et al. Severe block in processing of proinsulin to insulin accompanied by elevation of des-64,65 proinsulin intermediates in islets of mice lacking prohormone convertase 1/3. Proc Natl Acad Sci U S A. 2002;99:10299.
70. Jackson, RS, Creemers, JWM, Ohagi, S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303.
71. Zethelius, B, Byberg, L, Hales, CN, et al. Proinsulin and acute insulin response independently predict type 2 diabetes mellitus in men—report from 27 years of follow-up study. Diabetologia. 2003;46:20.
72. Rhodes, CJ, Alarcon, C. What β-cell defect could lead to hyperproinsulinemia in NIDDM? Some clues from recent advances made in understanding the proinsulin-processing mechanism. Diabetes. 1994;43:511.
73. Sizonenko, S, Irminger, J-C, Buhler, L, et al. Kinetics of proinsulin conversion in human islets. Diabetes. 1993;42:933.
74. Skelly, R, Schuppin, G, Ishihara, H, et al. Glucose-regulated translational control of proinsulin biosynthesis with that of the proinsulin endopeptidases PC2 and PC3 in the insulin-producing MIN6 cell line. Diabetes. 1996;45:37.
75. Schuppin, GT, Rhodes, CJ. Specific co-ordinated regulation of PC3 and PC2 gene expression with that of preproinsulin in insulin-producing beta TC3 cells. Biochem J. 1996;313(Pt 1):259–268.
76. Kalidas, K, Dow, E, Saker, P, et al. Prohormone convertase 1 in obesity, gestational diabetes mellitus, and NIDDM: no evidence for a major susceptibility role. Diabetes. 1998;47:287.
77. Yoshida, H, Ohagi, S, Sanke, T, et al. Association of the prohormone convertase 2 gene (PCSK2) on chromosome 20 with NIDDM in Japanese subjects. Diabetes. 1995;44:389.
78. Utsunomiya, N, Ohagi, S, Sanke, T, et al. Organization of the human carboxypeptidase E gene and molecular scanning for mutations in Japanese subjects with NIDDM or obesity. Diabetologia. 1998;41:701.
79. Benzinou, M, Creemers, JW, Choquet, H, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat. Genet. 2008;40:943–945.
80. Wen, JH, Chen, YY, Song, SJ, et al. Paired box 6 (PAX6) regulates glucose metabolism via proinsulin processing mediated by prohormone convertase 1/3 (PC1/3). Diabetologia. 2009;52:504–513.
81. Arvan, P, Castle, D. Protein sorting and secretion granule formation in regulated secretory cells. Trends Cell Biol. 1992;2:327.
82. Kuliawat, R, Klumperman, J, Ludwig, T, et al. Differential sorting of lysosomal enzymes out of the regulated secretory pathway in pancreatic beta-cells. J Cell Biol. 1997;137:595.
83. Arvan, P, Kuliawat, R, Prabakaran, D, et al. Protein discharge from immature secretory granules displays both regulated and constitutive characteristics. J Biol Chem. 1991;266:14171.
84. Michael, J, Carroll, R, Swift, H, et al. Studies on the molecular organization of rat insulin secretory granules. J Biol Chem. 1987;262:16531.
85. Nicolson, TJ, Bellomo, EA, Wijesekara, N, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes.. 2009. [[Epub ahead of print]].
86. Logothetopoulos, J, Maneko, M, Wrenshall, GA, et al, Zinc, granulation, and extractable insulin of islet cells following hyperglycemia or prolonged treatment with insulin. The structure and metabolism of the pancreatic islets Wenner-Gren Center International Symposium Series. Brolin, SE, Hellman, B, Knutson, H, eds. The structure and metabolism of the pancreatic islets Wenner-Gren Center International Symposium Series, vol 3. Oxford: Pergamon Press, 1964:333.
87. Peterson, JD, Steiner, DF, Emdin, SO, et al. The amino acid sequence of the insulin from a primitive vertebrate, the Atlantic hagfish (Myxine glutinosa). J Biol Chem. 1975;250:5183.
88. Blundell, TL, Dodson, GG, Hodgkin, DC, et al. Insulin: the structure in the crystals and its reflection in chemistry and biology. Adv Protein Chem. 1972;26:279.
89. Chan, SJ, Seino, S, Gruppuso, PA, et al. A mutation in the B chain coding region of the human insulin gene is associated with impaired proinsulin conversion in a family with hyperproinsulinemia. Proc Natl Acad Sci U S A. 1987;84:2194.
90. Carroll, RJ, Hammer, RE, Chan, SJ, et al. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc Natl Acad Sci U S A. 1988;85:8943.
91. Orci, L. The insulin factory: a tour of the plant surroundings and a visit to the assembly line. Diabetolgia. 1985;28:528.
92. Louagie, E, Taylor, NA, Flamez, D, et al. Role of furin in granular acidification in the endocrine pancreas: identification of the V-ATPase subunit Ac45 as a candidate substrate. Proc Natl Acad Sci U S A. 2008;105(34):112319–112324.
93. Cohen, RM, Given, BD, Licinio-Paixao, J, et al. Proinsulin radioimmunoassay in the evaluation of insulinomas and familial hyperproinsulinemia. Metabolism. 1986;35:1137.
94. Halban, PA. Structural domains and molecular lifestyles of insulin and its precursors in the pancreatic beta cell. Diabetologia. 1991;34:767.
95. Polonsky, K, Rubenstein, AH. Current approaches to measurement of insulin secretion. Diabetes Metab Rev. 1986;2:315.
96. Steiner, DF:, The biosynthesis of insulin. Insulin—handbook of experimental pharmacology. Cuatrecasas, P, Jacobs, S, eds. Insulin—handbook of experimental pharmacology, Vol. 92. Berlin: Springer-Verlag, 1990:67–92.
97. Steiner, DF, Clark, JL. The spontaneous reoxidation of reduced beef and rat proinsulins. Proc Natl Acad Sci U S A. 1968;60:622.
98. Guo, ZY, Qiao, ZS, Feng, YM. The in vitro oxidative folding of the insulin superfamily. Antioxid Redox Signal. 2008;10(1):127–139.
99. Kjeldsen, T, Ludvigsen, S, Diers, I, et al. Engineering-enhanced protein secretory expression in yeast with application to insulin. J Biol Chem. 2002;24:277. [(21):18245–18248, Epub Mar 28].
100. Lipkind, G, Steiner, DF. Predicted structural alterations in proinsulin during its interactions with prohormone convertases. Biochemistry. 1999;38:890.
101. Liu, M, Ramos-Cstaneda, J, Arvan, P. Role of the connecting peptide in insulin biosynthesis. J Biol Chem. 2003;278:14798.
102. Okun, MM, Shields, D. Translocation of preproinsulin across the endoplasmic reticulum membrane. J Biol Chem. 1992;267:11476.
103. Powell, S, Orci, L, Craik, CS, et al. Efficient targeting to storage granules of human proinsulins with altered propeptide domain. J Cell Biol. 1988;106:1843.
104. Wahren, J, Johansson, B-L, Wallberg-Henriksson, H. Does C-peptide have a physiological role? Diabetologia. 1994;37:99.
105. Wahren, J, Ekberg, K, Jörnvall, H. C-peptide is a bioactive peptide. Diabetologia. 2007;50:503–509.
106. Ido, Y, Vindigni, A, Chang, K, et al. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science. 1997;277:563.
107. Galloway, JA, Hooper, SA, Spradlin, CT, et al. Biosynthetic human proinsulin—review of chemistry, in vitro and in vivo receptor binding, animal and human pharmacology studies, and clinical experience. Diabetes Care. 1992;15:666.
108. Glauber, HS, Henry, RR, Wallace, P. The effects of biosynthetic human proinsulin on carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N Engl J Med. 1987;316:443.
109. Madsen, OD, Frank, BH, Steiner, DF. Human proinsulin specific antigenic determinants identified by monoclonal antibodies. Diabetes. 1984;33:1012.
110. Rhodes, CJ, Halban, PA. Newly-synthesized proinsulin/insulin and stored insulin are released form pancreatic B-cells via a regulated, rather than a constitutive pathway. J Cell Biol. 1987;105:145–153.
111. Uchizono, Y, Alarcon, C, Wicksteed, BL, et al. The balance between proinsulin biosynthesis and insulin secretion: where can imbalance lead? Diabetes Obes Metab. 2007;9(Suppl 2):56–66.
112. Rhodes, CJ. Processing of the insulin molecule. In: LeRoith D, Taylor SI, Olefsky JM, eds. Diabetes mellitus: a fundamental and clinical text. Philadelphia: Lippincott-Raven Publishers; 2004:27–50.
113. Wicksteed, BL, Alarcón, C, Briaud, I, et al. Glucose-induced translational control of proinsulin biosynthesis is proportional to preproinsulin mRNA levels, but not regulated via a positive feedback of secreted insulin on in islet β-cells. J Biol Chem. 2003;278:42080–42090.
114. Bollheimer, LC, Skelly, RH, Chester, M, et al. Chronic exposure to free fatty acid reduces pancreatic β-cell insulin content by increasing basal insulin secretion that is not compensated for a corresponding increase in proinsulin biosynthesis translation. J Clin Invest. 1998;101:1094–1101.
115. Prentki, M, Matchinsky, FM. Ca++, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev. 1987;67:1185–1248.
116. Alarcón, C, Wicksteed, BL, Prentki, M, et al. Elevation in pancreatic β-cell cytosolic succinyl-CoA is a candidate for specific metabolic stimulus coupling of proinsulin biosynthesis translation, but not insulin secretion. Diabetes. 2002;51:2496–2504.
117. Drucker, DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–165.
118. Alarcon, C, Wicksteed, B, Rhodes, CJ. Exendin 4 controls insulin production in rat islet beta cells predominantly by potentiation of glucose-stimulated proinsulin biosynthesis at the translational level. Diabetologia. 2006;49:2920–2929.
119. Herbert, TP, Alarcón, C, Skelly, RH, et al. Regulation of prohormone conversion by coordinated control of processing endopeptidase biosynthesis with that of the prohormone substrate. In: Hook VYH, ed. proteolytic and cellular mechanisms in prohormone and proprotein processing. Austin, TX.: R.G. Landes Company; 1998:105–120.
120. Guest, PC, Bailyes, EM, Rutherford, NG, et al. Insulin secretory granule biogenesis: co-ordinate regulation of the biosynthesis of the majority of constituent proteins. Biochem J. 1991;274:73–78.
121. Alarcón, C, Lincoln, B, Rhodes, CJ. The biosynthesis of the subtilisin-related proprotein convertase PC3, but not that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J Biol Chem. 1993;268:4276–4280.
122. Itoh, N, Okamoto, H. Translational control of proinsulin synthesis by glucose. Nature. 1980;283:100–102.
123. Welsh, M, Scherberg, N, Gilmore, R, et al. Translational control of insulin biosynthesis. Evidence for regulation of elongation, initiation and signal-recognition-particle-mediated translational arrest by glucose. Biochem J. 1986;235:459–467.
124. Welsh, M, Nielsen, DA, MacKrell, AJ, et al. Control of insulin gene expression in pancreatic β-cells and in an insulin producing cell line, RIN-5F cells. 2. Regulation of insulin mRNA stability. J Biol Chem. 1985;260:13590–13594.
125. German, M. Insulin gene regulation. In: LeRoith D, Taylor SI, Olefsky JM, eds. Diabetes mellitus—a fundamental and clinical text. Philadelphia: Lippincott Williams & Wilkins; 2004:15–26.
126. Van Lommel, L, Janssens, K, Quintens, R, et al. Probe-independent and direct quantification of insulin mRNA and growth hormone mRNA in enriched cell preparations. Diabetes. 2006;55:3214–3220.
127. Permutt, MA, Kipnis, DM. Insulin biosynthesis and secretion. Fed Proc. 1975;34:1549–1555.
128. Wicksteed, B, Herbert, TP, Alarcón, C, et al. Cooperation between 5′- and 3′-untranslated regions of preproinsulin mRNA control proinsulin biosynthesis. J Biol Chem. 2001;276:22553–22558.
129. Fred, RG, Tillmar, L, Welsh, N. The role of PTB in insulin mRNA stability control. Curr Diabetes Rev. 2006;2:363–366.
130. Wicksteed, BL, Uchizono, Y, Alarcon, C, et al. A cis-element in the 5′-untranslated region of the preproinsulin mRNA (ppIGE) is required for glucose regulation of proinsulin translation. Cell Metab. 2007;5:221–227.
131. German, MS, Moss, LG, Rutter, WJ. Regulation of gene expression by glucose and calcium in transfected primary islet cultures. J Biol Chem. 1990;265:22063–22066.
132. Swenne, I. The role of glucose in the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic β-cells. Diabetes. 1982;31:754–760.
133. Lingohr, MK, Buettner, R, Rhodes, CJ. Pancreatic beta-cell growth and survival—a role in obesity-linked type 2 diabetes? Trends Mol Med. 2002;8:375–384.
134. Orci, L, Ravazzola, M, Amherdt, M, et al. Insulin, not C-peptide (proinsulin), is present in crinophagic bodies of the pancreatic B-cell. J Cell Biol. 1984;98:222–228.
135. Schnell, AH, Swenne, I, Borg, LA. Lysosomes and pancreatic islet function. A quantitative estimation of crinophagy in the mouse pancreatic B-cell. Cell Tissue Res. 1988;252:9–15.
136. Marsh, BJ, Soden, C, Alarcón, C, et al. Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine β-cells. Mol Endocrinol. 2007;21:2255–2269.
137. Halban, PA, Wollheim, CB. Intracellular degradation of insulin stores by rat pancreatic islets in vitro. An alternative pathway for homeostasis of pancreatic insulin content. J Biol Chem. 1980;255:6003–6006.
138. Skoglund, G, Ahren, B, Lundquist, I. Biochemical determination of islet lysosomal enzyme activities following crinophagy-stimulating treatment with diazoxide in mice. Diabetes Res. 1987;6:81–84.
139. Ebato, C, Uchida, T, Arakawa, M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–332.
140. Grasso, D, Sacchetti, ML, Bruno, L, et al. Autophagy and VMP1 expression are early cellular events in experimental diabetes. Pancreatology. 2008;9:81–88.
141. Ashcroft, FM, Rorsman, P. Electrophysiology of the pancreatic beta cell. Prog Biophys Mol Biol. 1989;54:87–143.
142. Gloyn, AL, Pearson, ER, Antcliff, JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849.
143. Babenko, AP, Polak, M, Cave, H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–466.
144. Byrne, MM, Sturis, J, Clément, K, et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest. 1994;93:1120–1130.
145. Jacobson, DA, Kuznetsov, A, Lopez, JP, et al. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6:229–235.
146. Aguilar-Bryan, L, Bryan, J. Neonatal diabetes mellitus. Endocr Rev. 2008;29:265–291.
147. Hou, JC, Min, L, Pessin, JE. Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm. 2009;80:473–506.
148. Stoy, J, Edghill, EL, Flanagan, SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040–15044.
149. Murphy, R, Ellard, S, Hattersley, AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:200–213.
150. Sayed, S, Langdon, DR, Odili, S, et al. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58:1419–1427.
151. De Leon, DD, Stanley, CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57–68.
152. Miller, SP, Anand, GR, Karschnia, EJ, et al. Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes. 1999;48:1645–1651.
153. Matschinsky, FM, Magnuson, MA, Zelent, D, et al. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes. 2006;55:1–12.
154. Chimienti, F, Devergnas, S, Favier, A, et al. Identification and cloning of a beta-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;53:2330–2337.
155. Wenzlau, JM, Juhl, K, Yu, L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A. 2007;104:17040–17045.
156. Sladek, R, Rocheleau, G, Rung, J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885.
157. Fu, Y, Tian, W, Pratt, EB, et al. Down-regulation of ZnT8 expression in INS-1 rat pancreatic beta cells reduces insulin content and glucose-inducible insulin secretion. PLoS One. 2009;4:e5679.
158. Conget, I, Zhang, TM, Eizirik, DL, et al. SAM prevents impairment of glucose-stimulated insulin secretion caused by hexose deprivation or starvation. Am J Physiol. 1995;268:E580–E587.
159. Maechler, P, Kennedy, ED, Pozzan, T, et al. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta-cells. EMBO J. 1997;16:3833–3841.
160. Maechler, P, Wollheim, CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402:685–689.
161. Wiederkehr, A, Wollheim, CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology. 2006;147:2643–2649.
162. Ashcroft, FM, Proks, P, Smith, PA, et al. Stimulus-secretion coupling in pancreatic β-cells. J Cell Biochem. 1994;55S:54–65.
163. Misler, S, Barnett, DW, Gillis, KD, et al. Electrophysiology of stimulus-secretion coupling in human B-cells. Diabetes. 1992;41:1221–1228.
164. Fridlyand, LE, Harbeck, MC, Roe, MW, et al. Regulation of cAMP dynamics by Ca2+ and G protein–coupled receptors in the pancreatic beta-cell: a computational approach. Am J Physiol Cell Physiol. 2007;293:C1924–C1933.
165. Rutter, GA, Tsuboi, T, Ravier, MA. Ca2+ microdomains and the control of insulin secretion. Cell Calcium. 2006;40:539–551.
166. Henquin, JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751.
167. Nesher, R, Anteby, E, Yedovizky, M, et al. Beta-cell protein kinases and the dynamics of the insulin response to glucose. Diabetes. 2002;51(Suppl 1):S68–S73.
168. Drucker, DJ. The biology of incretin hormones. Cell Metab. 2006;3:153–165.
169. Wek, RC, Anthony, TG. Extending beta cell survival by upregulating ATF4 translation. Cell Metab. 2006;4:333–334.
170. Holz, GG, Kang, G, Harbeck, M, et al. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5–15.
171. Philipson, LH. Beta-agonists and metabolism. J Allergy Clin Immunol. 2002;110:S313–S317.
172. Toyota, T, Kakizaki, M, Kimura, K, et al. Islet activating protein (IAP) derived from the culture supernatant fluid of Bordetella pertussis: effect on spontaneous diabetic rats. Diabetologia. 1978;14:319–323.
173. Draznin, B, Steinberg, JP, Goodman, M, et al. Control of secretion, vesicle margination, and lysis by glucose, IBMX, and glyburide. Am J Physiol. 1985;248:E375–E380.
174. Dov, A, Abramovitch, E, Warwar, N, et al. Diminished phosphodiesterase-8B potentiates biphasic insulin response to glucose. Endocrinology. 2008;149:741–748.
175. Landa, LR, Jr., Harbeck, M, Kaihara, K, et al. Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J Biol Chem. 2005;280:31294–31302.
176. Dyachok, O, Isakov, Y, Sagetorp, J, et al. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting beta-cells. Nature. 2006;439:349–352.
177. Gilon, P, Henquin, JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22:565–604.
178. Covington, DK, Briscoe, CA, Brown, AJ, et al. The G-protein-coupled receptor 40 family (GPR40-GPR43) and its role in nutrient sensing. Biochem Soc Trans. 2006;34:770–773.
179. Brownlie, R, Mayers, RM, Pierce, JA, et al. The long-chain fatty acid receptor, GPR40, and glucolipotoxicity: investigations using GPR40-knockout mice. Biochem Soc Trans. 2008;36:950–954.
180. Tan, CP, Feng, Y, Zhou, YP, et al. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes. 2008;57:2211–2219.
181. Steiner, DF, Chan, SJ, Welsh, JM, et al. Structure and evolution of the insulin gene. Annu Rev Genet. 1985;19:463.
182. Pugliese, A, Zeller, M, Fernandez, A, Jr., et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet.. 1997;15:293–297.
183. Bell, GI, Selby, MJ, Rutter, WJ. The highly polymorphic region near the human insulin gene is composed of simple tandemly repeating sequences. Nature. 1982;295:31.
184. Steiner, DF, Tager, HS, Chan, SJ, et al. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care. 1990;13:600–609.
185. Rafiq, M, Flanagan, SE, Patch, AM, et al. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31:204–209.
186. Edghill, E, Flanagan, S, Patch, AM, et al. Insulin mutation screening in 1044 patients with diabetes mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042.
187. Molven, A, Ringdal, M, Nordbø, AM, et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type I diabetes. Diabetes. 2008;57:1131–1135.
188. Polak, M, Dechaume, A, Cave, H, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy. Diabetes. 2008;57:1115–1119.
189. Colombo, C, Porzio, O, Liu, M, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J Clin Invest. 2008;118:2148–2156.
190. Hua, QX, Liu, M, Hu, SQ, et al. A conserved histidine in insulin is required for the foldability of human proinsulin: structure and function of an ALAB5 analog. J Biol Chem. 2006;281:24889–24899.
190a. Rajan, S, Eames, SC, Park, SY, et al. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab. 2009 Dec 1. [[Epub ahead of print]].
190b. Park, SY, Ye, H, Steiner, DF, et al. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2009 Dec 23. [[Epub ahead of print]].
191. Bonfanti, R, Colombo, C, Nocerino, V, et al. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies. Diabetes Care. 2009;32:123–125.
192. Qiao, ZS, Min, CY, Hua, QX, et al. In vitro refolding of human proinsulin. Kinetic intermediates, putative disulfide-forming pathway, folding initiation site and potential role of C-peptide in folding process. J Biol Chem. 2003;278:17800–17809.
193. Nakagawa, SH, Hua, QX, Hu, SQ, et al. Chiral mutagenesis of insulin. Contribution of the B20-B23 beta-turn to activity and stability. J Biol Chem. 2006;281:22386–22396.
194. Wang, J, Takeuchi, T, Tanaka, S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the MODY mouse. J Clin Invest. 1999;103:27–37.
195. Herbach, N, Rathkolb, B, Kemter, E, et al. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe β-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56:1268–1276.
196. Shimizu, Y, Hendershot, LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal. 2009. [Feb 25. [Epub ahead of print]].
197. Sevier, CS, Kaiser, CA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta. 2008;1783:549–556.
198. Back, SH, Scheuner, D, Han, J, et al. Translation attenuation through elF2α phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26.
199. Zhu, YL, Abdo, A, Gesmonde, JF, et al. Aggregation and lack of secretion of most newly synthesized proinsulin in non-β-cell lines. Endocrinology. 2004;145:3840–3849.
200. Zuber, C, Fan, JY, Guhl, B, et al. Misfolded proinsulin accumulates in expanded pre-Golgi intermediates and endoplasmic reticulum subdomains in pancreatic beta cells of Akita mice. FASEB J. 2004;18:917–999.
201. Hutton, JC. The insulin secretory granule. Diabetologia. 1989;32:271.
202. Taupenot, L, harper, KL, O’Connor, DT. The chromogranin-secretogranin family. N Engl J Med. 2003;348:1134.
203. Hutton, JC, Nielsen, E, Kastern, W. The molecular cloning of the chromogranin A-like precursor of β-granin and pancreastatin from the endocrine pancreas. FEBS Lett. 1988;236:269.
204. Schmidt, WE, Siegel, EG, Kratzin, H, et al. Isolation and primary structure of tumor-derived peptides related to human pancreastatin and chromogranin A. Proc Natl Acad Sci U S A. 1988;85:8231.
205. Tatemoto, K, Efendic, S, Mutt, V, et al. Pancreastatin, a novel pancreatic peptide that inhibits insulin secretion. Nature. 1986;324:476.
206. Hendy, GN, Li, T, Girard, M, et al. Targeted ablation of the chromogranin a (CHGA) gene: normal neuroendocrine dense-core secretory granules and increased expression of other granins. Mol Endocrinol. 2006;8:1935–1947.
207. Westermark, P, Wernstedt, C, Wilander, E, et al. A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem Biophys Res Commun. 1986;140:827–831.
208. Cooper, GJS, Willis, AC, Clark, A, et al. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A. 1987;84:8628.
209. Opie, E. The relation of diabetes mellitus to lesions of the pancreas: hyaline degeneration of the islands of Langerhans. J Exp Med. 1901;5:527.
210. Nishi, M, Sanke, T, Seino, S, et al. Human islet amyloid polypeptide gene: sequence, chromosomal localization and evolutionary history. Mol Endocrinol. 1989;3:1775.
211. Mosselman, S, Höppener, JWM, Lips, CJM, et al. The complete islet amyloid polypeptide precursor is encoded by two exons. FEBS Lett. 1989;247:154.
212. Prigge, ST, Eipper, BA, Mains, RE, et al. New insignts into copper monoxygenases and peptide amidation: structure, mechanism and function. Cell Mol Life Sci. 2000;57:1236–1259.
213. Iki, K, Pour, PM. Distribution of pancreatic endocrine cells including IAPP-expressing cells in non-diabetic and type 2 diabetic cases. J Histochem Cytochem. 2007;55:111–118.
214. Nagamatsu, S, Nishi, M, Steiner, DF. Biosynthesis of islet amyloid polypeptide. J Biol Chem. 1991;266:13737.
215. Wang, J, Xu, J, Finnerty, J, et al. The prohormone convertase enzyme 2 (PC2) is essential for processing pro-amyloid polypeptide at the NH2-terminal cleavage site. Diabetes. 2001;50:534.
216. Marzban, L, Trigo-Gonzalez, G, Zhu, X, et al. Role of beta-cell prohormone convertase (PC) 1/3 in processing of pro-islet amyloid polypeptide. Diabetes. 2004;53:141.
217. Marzban, L, Park, K, Verchere, CB. Islet amyloid polypeptide and type 2 diabetes. Exp Gerontol. 2003;38:347–351.
218. Marzban, L, Rhodes, CJ, Steiner, DF, et al. Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes. 2006;55:2192–2201.
219. Mulder, H, Ahren, B, Stridsberg, M, et al. Non-parallelism of islet amyloid polypeptide (amylin) and insulin gene expression in rat islets following dexamethasone treatment. Diabetologia. 1995;38:395.
220. Ferrier, GJLM, Pierson, AM, Jones, PM, et al. Expression of the rat amylin (IAPP/DAP) gene. J Mol Endocrinol. 1989;3:R1.
221. Nakazato, M, Asai, J, Kangawa, K, et al. Establishment of radioimmunoassay for human islet amyloid polypeptide and its tissue content and plasma concentration. Biochem Biophys Res Commun. 1989;164:394.
222. Asai, J, Nakazato, M, Kangawa, K, et al. Regional distribution and molecular forms of rat islet amyloid polypeptide. Biochem Biophys Res Commun. 1990;169:788.
223. Kanatsuka, A, Makino, H, Ohsawa, H, et al. Secretion of islet amyloid polypeptide in response to glucose. FEBS Lett. 1989;259:199.
224. Cooper, GJS, Amylin and related proteins: physiology and pathology. Handbook of physiology, The Endocrine System. Jefferson, LS, Cunningham, AD, eds. Handbook of physiology, The Endocrine System, Vol. II. Oxford University Press, 2001:303–396. [Chapter 10].
225. Sowa, R, Sanke, T, Hirayama, J, et al. Islet amyloid polypeptide amide causes peripheral insulin resistance in vivo. Diabetologia. 1990;33:118.
226. Scherbaum, WA. The role of amylin in the physiology of glycemic control. Exp Clin Endocrinol Diabetes. 1998;106:97.
227. Gebre-Medhin, S, Mulder, H, Pekny, M, et al. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem Biophys Res Commun. 1998;250:271.
228. Ahrén, B, Oosterwijk, C, Lips, CJM, et al. Transgenic overexpression of human islet amyloid polypeptide inhibits insulin secretion and glucose elimination after gastric glucose gavage in mice. Diabetologia. 1998;41:1374.
229. Datta, HK, Saidi, M, Wimalawansa, SJ, et al. In vivo and in vitro effects of amylin and amylin-amide on calcium metabolism in the rat and rabbit. Biochem Biophys Res Commun. 1989;162:876.
230. MacIntyre, I. Amylinamide, bone conservation, and pancreatic β-cells. Lancet. 1989;2:1026.
231. Christopoulos, G, Perry, KJ, Morfis, M, et al. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol. 1999;56:235.
232. Morfis, M, Tilakaratne, N, Furness, SG, et al. Receptor activity-modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology. 2008;149:5423–5431.
233. Schmitz, O, Brock, B, Rungby, J. Amylin agonists: a novel approach in the treatment of diabetes. Diabetes. 2004;53:S233–S238.
234. O’Brien, TD, Butler, PC, Westermark, P, et al. Islet amyloid polypeptide: a review of its biology and potential roles in the pathogenesis of diabetes mellitus. Vet Pathol. 1993;30:317–332.
235. Glenner, GG, Eanes, ED, Wiley, CA. Amyloid fibrils formed from a segment of the pancreatic islet amyloid protein. Biochem Biophys Res Commun. 1988;155:608.
236. Hallman, U, Wernstedt, C, Westermark, P, et al. Amino acid sequence from degu islet amyloid-derived insulin shows unique sequence characteristics. Biochem Biophys Res Commun. 1980;169:571.
237. Nishi, M, Steiner, DF. Cloning of complementary DNAs encoding islet amyloid polypeptide, insulin, and glucagon precursors from a New World rodent, the degu, Octodon degus. Mol Endocrinol. 1990;4:1192.
238. Lukinius, A, Wilander, E, Westermark, GT, et al. Co-localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic islets. Diabetologia. 1989;32:240.
239. Clark, A, Wells, CA, Buley, ID, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas. Diabetes Res. 1988;9:1519.
240. Clark, A, Edwards, CA, Ostle, LR, et al. Localisation of islet amyloid peptide in lipofuscin bodies and secretory granules of human β-cells and in islets of type-2 diabetic subjects. Cell Tissue Res. 1989;259:179.
241. Westermark, G, Westermark, P, Eizirik, DL, et al. Differences in amyloid deposition in islets of transgenic mice expressing human islet amyloid polypeptide versus human islets implanted into nude mice. Metabolism. 1999;48:448.
242. Kahn, SE, Andrikopoulos, S, Verchere, CB. Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48:241.
243. Nishi, M, Bell, GI, Steiner, DF. Islet amyloid polypeptide (amylin): no evidence of an abnormal precursor sequence in 25 type 2 (non–insulin-dependent) diabetic patients. Diabetologia. 1990;33:628.
244. Anderson, A, Bohman, S, Borg, LA, et al. Amyloid deposition in transplanted human pancreatic islets: a conceivable cause of their long-term failure. Exp Diabetes Res. Epub 2009.
245. Porat, Y, Mazor, Y, Efrat, S, et al. Inhibition of islet amyloid polypeptide fibril formation: a potential role for heteroaromatic interactions. Biochemistry. 2004;43:14454–14462.
246. Tatarek-Nossol, M, Yan, L-M, Schmauder, A, et al. Inhibition of hIAPP amyloid-fibril formation and apoptotic cell death by a designed hIAPP amyloid-core-containing hexapeptide. Chem and Biol. 2005;12:797–809.
247. Steiner, DF, Bell, GI, Rubenstein, AH, et al, Chemistry and biosynthesis of the islet hormones: insulin, islet amyloid polypeptide (amylin), glucagon, somatostatin, and pancreatic polypeptide, ed 5. DeGroot L, Jameson JL, eds. Endocrinology. W.B. Saunders: Philadelphia, 2006:925–960. [Chapter 48].
248. Tanaka, S, Kurabuchi, S, Mochida, H, et al. Immunocytochemical localization of prohormone convertases PC1/PC3 and PC2 in rat pancreatic islets. Arch Histol Cytol. 1996;59:261–271.
249. Furuta, M, Zhou, A, Webb, G, et al. Severe defect in proglucagon processing in islet A-cells of prohormone convertase 2 null mice. J Biol Chem. 2001;276:27197–27202.
250. Gelling, RW, Du, XQ, Dichmann, DS. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A. 2003;100:1438–1443.
251. Webb, GC, Akbar, MS, Zhao, C, et al. Glucagon replacement via micro-osmotic pump corrects hypoglycemia and alpha-cell hyperplasia in PC2 knockout mice. Diabetes. 2002;51:398–405.
252. Wilson, ME, Kalamaras, JA, German, MS. Expression pattern of IAPP and prohormone convertase 1/3 reveals a distinctive set of endocrine cells in the embryonic pancreas. Mech Dev. 2002;115:171–176.
253. Nie, Y, Nakashima, M, Brubaker, PL, et al. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest. 2000;105:955–965.
254. Csaba, Z, Dournaud, P. Cellular biology of somatostatin receptors. Neuropeptides. 2001;35:1.
255. Egel-Mitani, M, Flygenring, HP, Hansen, MT. A novel aspartyl protease allowing KEX2-independent MFα propheromone processing in yeast. Yeast. 1990;6:127.
256. Zhu, X, Cao, Y, Voogd, K, et al. On the processing of proghrelin to ghrelin. J Biol Chem. 2006;281:38867–38870.
257. Yang, J, Brown, MS, Liang, G, et al. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell. 2008;132:387–396.
258. Andralojc, KM, Mercalli, A, Nowak, KW, et al. Ghrelin-producing epsilon cells in the developing and adult human pancreas. Diabetologia. 2009;52:486–493.
259. Webb, GB, Akbar, MS, Zhao, C, et al. Expression profiling of pancreatic beta cells: glucose regulation of secretory and metabolic pathway genes. Proc Natl Acad Sci U S A. 2000;97:5773.
260. Canaff, L, Bennett, HPJ, Hou, Y, et al. Proparathyroid hormone processing by the proprotein convertase-7: comparison with furin and assessment of modulation of parathyroid convertase messenger ribonucleic acid levels by calcium and 1,25-dihydroxyvitamin D3. Endocrinology. 1999;140:3633–3642.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 6
Biosynthesis, Processing, and Secretion of the Islet Hormones
Insulin, Islet Amyloid Polypeptide (Amylin), Glucagon, Somatostatin, and Pancreatic Polypeptide
Morphologic Organization of the Insulin Biosynthetic Machinery
Structure and Properties of Proinsulin
Enzymatic Basis of the Conversion of Proinsulin to Insulin
Regulation of Insulin Production
Insulin Secretion and Its Regulation
Insulin Gene Mutations as a Cause of Neonatal Diabetes
Although diabetes had long been recognized as a serious metabolic disorder, it was the classic experiments of Von Mering and Minkowski in 1890 that demonstrated the important role of the pancreas in its prevention.1 A number of investigators attempted to prepare antidiabetic pancreatic extracts in the early 20th century, but it was not until the work of the Canadians—Frederick Banting, Charles Best, and John Collip, working in the laboratory of Professor J.J.R. Macleod at the University of Toronto in 1921-1922—that potent preparations of insulin were routinely made.2 The name insulin was based on the belief that the hormone was derived from the islets of Langerhans and actually was suggested as early as 1909 by deMayer and by Sir Edward Sharpey-Schafer in 1917. Preparative methods based on the early work of Scott,3 as well as refinements by Banting et al.,4 were rapidly developed for the commercial preparation of the hormone, and within about 1 year, insulin began to be administered to patients with diabetes, often with dramatic effects.2 The chemical nature of insulin was a more elusive problem, although the fact that it was destroyed by proteolytic enzymes suggested that it was a protein.5 When J.J. Abel first crystallized insulin in 1926, there was great controversy as to whether the crystals of protein he obtained were actually the active biological principle or merely the vehicle for a smaller active moiety.6 Today we know that insulin occurs throughout the vertebrate kingdom, and insulin-like substances are present in the brains and/or digestive systems of many invertebrates.7 Many of the modern techniques of protein chemistry were developed in part through the clinical need for insulin, which made it abundantly available to biochemists as a model protein for study.8,9
This chapter reviews the isolation, structure, and properties of insulin and other islet hormones, including islet amyloid polypeptide (IAPP or amylin), glucagon, somatostatin, pancreatic polypeptide (PP) and their precursor forms, and the mechanisms of their biosynthesis and secretion. Studies on the mechanism of biosynthesis of insulin via preproinsulin and proinsulin in the pancreatic β cells have provided a useful paradigm for understanding the production of many other neuroendocrine peptides and a basis for advances in the diagnosis and treatment of diabetes and related endocrine disorders. The recent identification of the enzymes that catalyze the proteolytic cleavage of prohormones and proneuropeptides—prohormone convertases—has revealed a new family of processing enzymes that act on a larger subset of precursor proteins, including growth factors, their receptors, and many other proteins that traverse the secretory pathway.10
The Biosynthesis of Insulin
The two-chain structure of insulin was the first complete protein structure to be elucidated through the pioneering work of Fred Sanger in 1955.9 In 1967, the discovery of the single-chain insulin precursor molecule, proinsulin,11 then solved the problem of assembly of the insulin chains (Fig. 6-1). The human proinsulin polypeptide begins with the 30-amino-acid B chain domain, extends through a 35-amino-acid connecting segment, and ends with the 21-amino-acid A chain domain, comprising a single 9000-D polypeptide chain.11–14 It contains paired basic residue cleavage sites (Arg-Arg or Lys-Arg) at each end of the connecting segment which are removed during the proteolytic conversion of proinsulin to insulin within maturing secretory granules. However, cell-free translation of the initial polypeptide products encoded in insulin messenger ribonucleic acid (mRNA) extracted from islets or islet cell tumors led in 1976 to the discovery of preproinsulin (Fig. 6-2). This extended form of the prohormone has a hydrophobic N-terminal 24-residue signal peptide.15 Such prepeptide extensions interact with the signal recognition particle (SRP), a cytosolic ribonucleoprotein particle that assists in transfer of the nascent secretory protein from the cytosolic compartment, where its synthesis is initiated, into the secretory pathway via a complex series of molecular interactions that result in the translocation of the nascent peptide across the membrane of the rough endoplasmic reticulum (RER) into its internal compartments, or cisternae.16 During or shortly after translocation, the signal sequence is cleaved by the signal peptidase, which is located on the inner surface of the RER membrane, and the signal peptide is rapidly degraded.17 The proinsulin molecule then folds and undergoes rapid formation of disulfide bonds to achieve its native structure. Evidence suggests that this process is catalyzed by protein disulfide isomerase,18 a resident protein of the RER having a C-terminal Lys-Glu-Asp-Leu (KDEL) localization sequence.19 The folded proinsulin is then transferred in small coated vesicles from the endoplasmic reticulum (ER) to the cis region of the Golgi apparatus.20,21 It then passes from the cis to the trans Golgi, where it is sorted into immature secretory vesicles, or progranules, where it is proteolytically cleaved to release insulin and the connecting segment, minus the four basic residues, from the cleavage sites. The connecting peptide fragment released by cleavage of proinsulin is designated the C peptide. Both insulin and C peptide are stored in the secretory granules along with small amounts of residual proinsulin and partially cleaved intermediate forms22 as well as a variety of other minor β cell secretory products.23 Stimulation of secretion results in the release of the granule contents into the hepatic portal circulation.
FIGURE 6-1 Amino acid sequence of human and hagfish insulins, with substitutions occurring at each position in 70 other known vertebrate insulins shown below for comparison. Invariant residues are enclosed in boxes.
FIGURE 6-2 Schematic structure of human preproinsulin. Rapid removal of the first 24 amino acids (signal peptide) gives rise to proinsulin, which then folds to form the native insulin domain stabilized by three disulfide bonds (black bars). The connecting segment is not structurally organized. Cleavage after B30 (T) and before A1 (G) then gives rise to insulin and the free C peptide. (See text for details.)
Morphologic Organization of the Insulin Biosynthetic Machinery
The β cells of the islets of Langerhans share many features with other endocrine and neurosecretory cells. Preproinsulin is the initial translational product of insulin mRNA, as discussed earlier, and is rapidly cleaved to proinsulin in the RER. After folding and disulfide oxidation, proinsulin is transported from the ER to the Golgi apparatus in small coated vesicles20 (Fig. 6-3) in an energy-dependent process that requires about 20 minutes.22,24,25 The biochemical basis of the energy requirement for the intracellular transport of secretory proteins is associated with the budding and/or fusion of small vesicles that transfer secretory products from the ER to and through the cis to trans Golgi cisternae.26,27
FIGURE 6-3 Schematic model of the subcellular transport of preproinsulin after its synthesis on membrane-bound ribosomes of the rough endoplasmic reticulum (RER) and rapid cleavage to proinsulin (within 1 to 2 minutes). Proinsulin is then released into the intracisternal spaces of the RER, where it folds and forms the native disulfide bonds of insulin. It is then transported to the Golgi apparatus by an energy-dependent process. The clathrin-coated early granules budding from the trans-Golgi cisternae are rich in proinsulin and contain the converting proteases, PC2 and PC1/3. Processing occurs mainly, if not exclusively, in the early secretory granules24,30 giving rise to the more condensed mature granules. Fractionation studies have confirmed that the mature granule-dense cores consist almost entirely of insulin, often in crystalline arrays (see Fig. 6-9), whereas the granule-soluble phase that surrounds the inclusion consists mainly of C peptide and small amounts of proinsulin.84 The release of newly synthesized proinsulin and insulin begins only about 1 hour after synthesis in the RER; hence, granules must undergo a maturation process that renders them competent for secretion. There is no evidence for significant nongranular routes of secretion of either proinsulin or insulin in normal islets. Exocytosis of granules is regulated by glucose and many other factors (see section on insulin secretion), and in humans and dogs, it results in the release of insulin and C peptide in approximately equimolar proportions under both basal and stimulated conditions.95 The mechanism of recycling of the granule membrane and its components is not well understood.
The role of the Golgi apparatus in forming insulin-containing secretory vesicles, or β-granules, was first recognized in the 1940s and subsequently confirmed and extended by Munger,28 who used electron microscopy to identify pale, homogeneous-appearing, immature “progranules” near the Golgi body. The structural organization and various functions of this complex organelle continue to engage the attention of cell biologists and biochemists.29 Orci and coworkers demonstrated that these newly formed clathrin-clad vesicles are derived from the trans-Golgi network (TGN) cisternae and contain a high proportion of proinsulin, definitively confirming their proposed role as the major sites of proinsulin processing to insulin.30 Conversion of proinsulin to insulin in vivo behaves kinetically as a pseudo–first-order reaction having a half-time ranging from about 20 minutes to 1 hour.22,31,32 Peak labeling of proteins in the Golgi apparatus occurs 30 to 40 minutes after pulse labeling islets with 3H-amino acids, and relatively little radioactivity remains in this region after 1 hour.33 A similar pattern for proinsulin is observed by means of electron microscopic immunocytochemistry. Thus, although small amounts of proinsulin conversion may be initiated in the TGN, most of it occurs in newly formed secretory vesicles, or progranules, which mature biochemically in the cytosol30 (see Fig. 6-3). Numerous subsequent studies have confirmed that newly synthesized neuroendocrine precursor peptides pass via the Golgi apparatus into secretory granules of the regulated secretory pathway, where they are cleaved and processed to their mature products and then stored until secretion in response to signals such as glucose.10
Structure and Properties of Proinsulin
Mammalian proinsulins range in size from 81 (cow) to 86 (human, horse, rat) amino acid residues, owing to variations in the length of the C peptide. Various C-peptide sequences are compared in Fig. 6-4. Proinsulin is closely similar to insulin in many properties, including solubility, isoelectric point,22 self-associative properties,34 and reactivity with insulin antisera.11,35 Indeed, the conformation of the insulin moiety in proinsulin is essentially identical to that of insulin itself.36 It is noteworthy that the highly flexible C peptide is much longer than necessary simply to bridge the short 8-Å gap between the ends of the B and A chains as they exist in the folded insulin molecule (see Fig. 6-2). The C-peptide segment overlays a portion of the surface of the insulin monomer, reducing its potency to about 3% to 5% of that of insulin in vitro,22,37 and it undergoes little if any proteolysis or activation in the circulation or tissues.38 The presence of the C peptide in proinsulin also does not prevent the dimerization or hexamerization of its insulin moiety39 (Fig. 6-5). It lies outside the zinc-stabilized globular insulin hexamer in a disordered loop, as assessed by nuclear magnetic resonance (NMR) spectroscopy. Since low levels of proinsulin can form mixed hexamers with insulin, it can be efficiently incorporated into insulin crystals.40 This property accounts for the presence of 1% to 2% of proinsulin in most crystalline preparations of insulin prepared from animal pancreata.41 However, x-ray analysis of crystals of proinsulin have failed to reveal any details regarding the structure of the C-peptide segment.42,43
FIGURE 6-4 Compilation of amino acid sequences of proinsulin C peptides in vertebrates, amphioxus, and in a mollusk, Lymnae stagnalis (for sources see Ref. 96).
FIGURE 6-5 Hypothetical 2-Zn proinsulin hexamer as viewed along the threefold axis. The connecting peptide is shown in light gray and white around the periphery of the darker outline of an insulin hexamer arranged according to the structural data of Blundell et al.88 The central density represents two zinc atoms in coordination linkage to the six (three above and three below hexamer plane) histidine side chains at position 10 in the B chain.
The major intermediate cleavage products of proinsulin consist mainly of forms cleaved at only one site and lacking the exposed basic residues (i.e., des-31,32 or des-64,65 intermediates).12,22,41,44,45 Their relative ratios in the circulation differ among species, depending on differences in the amino acid sequences surrounding the cleavage sites.46 Proinsulin-like proteins having comparably sized C peptides have been found throughout the vertebrates and also in a variety of invertebrates. Although it seems likely that the single-chain insulin-like growth factors, IGF-1 and IGF-2, arose from a single proinsulin-like gene in protochordates,47 some invertebrates, such as Caenorhabditis elegans,48 have numerous insulin-like peptides that lack defined C peptides and typical paired basic residue sites for cleavage by the evolutionarily well-conserved prohormone convertases.
Enzymatic Basis of the Conversion of Proinsulin to Insulin
The major proteolytic cleavages required for converting proinsulin to insulin are summarized in Fig. 6-6. In early studies, it was shown that the joint action of pancreatic trypsin and carboxypeptidase B gives rise to the naturally occurring conversion products—C peptide and native insulin in high yields—quantitatively converting proinsulin to insulin in vitro.49 This model also explains the major intermediate forms found in pancreatic extracts,44,50 and it led to a search for a cellular trypsin-like convertase, assumed to be a serine protease related to trypsin,22 and for carboxypeptidases related to carboxypeptidase B, an exocrine pancreatic exopeptidase with specificity for C-terminal basic residues.49 Early studies with isolated islet secretory granules revealed that these were major sites of proinsulin conversion, and that conversion resulted in the release of insulin along with free arginine, rather than basic dipeptides,51 confirming the likely participation of both trypsin-like and carboxypeptidase B–like proteases in maturing secretory granules.52 Carboxypeptidase E (or H), a homolog of carboxypeptidase B but with a more acidic pH optimum, was first identified in islets52 and subsequently by Fricker in brain.53 Molecular cloning confirmed its structural and evolutionary relationship to the pancreatic carboxypeptidases. Additional processing carboxypeptidases have been uncovered in brain and other tissues.54
FIGURE 6-6 The cleavage of proinsulin to insulin by the combined action of the subtilisin-like prohormone convertases, PC2 and PC1/PC3, and carboxypeptidase E. (See text for details.)
The first authentic processing endoprotease, kexin, was identified through studies on the processing of the yeast alpha mating–factor precursor and proved to be a calcium-dependent serine protease related to bacterial subtilisin and the subtilases rather than to trypsin.55–57 This information enabled the discovery of mammalian homologs, which form a family of seven closely related endoproteases with differing points of action within the secretory pathway.10,58,59 These have been designated as subtilisin-like proprotein convertases (SPCs), or more simply as PCs. Two members of this family, PC1/3 and PC2, have proven to be the principal effectors of neuroendocrine peptide precursor processing and are largely confined in their expression to brain and endocrine tissues in most animal species.10 For further information on the extended SPC/PC family and its multiple functions, please see the reviews in Refs. 60 to 62.
Role of PC2 and PC1/3 in Proinsulin Processing
Both PC1/3 and PC2 are required for proinsulin processing and have been shown to be identical with the calcium-dependent type I and type II insulinoma granule-processing activities, respectively, originally described by Davidson et al.63 The importance of calcium for the transport and proteolytic maturation of proinsulin has been well documented.64 The role and order of action of PC2 and PC1/3 in proinsulin conversion has also been carefully studied.65 Rhodes et al.66 demonstrated that the type II convertase (PC2) prefers the proinsulin intermediate that has already been processed at the B chain–C peptide junction (des-31,32 intermediate) as a substrate. This observation has led to the scheme for conversion outlined in (Fig. 6-7), in which PC1/3 acts first to generate the des-31,32 intermediate, which is then cleaved at the C peptide–A chain junction by PC2. This order of cleavage is consistent with observations that PC1/3 achieves an enzymatically active form more rapidly than PC2 and has a somewhat higher pH optimum. Thus, PC1/3 may begin cleaving proinsulin as it is concentrated into immature secretory vesicles in the TGN, whereas PC2 acts only in the maturing granules as the pH decreases to around 5.5. According to this scheme, PC1/3 plays a more important role in proinsulin processing, as born out by observations on islets from mice lacking PC2 or PC1/3.46,67–69
FIGURE 6-7 Routes of processing of proinsulin in the pancreatic β cell. The pathway on the right is probably more dominant under normal conditions because des-31,32 proinsulin is a preferred substrate for PC2,66 and the more acidic pH optimum and slower maturation of this enzyme may delay its action during the initial phases of secretory granule maturation. The C-terminal basic residues are removed by CPE (CPH) after endoproteolytic cleavage by the PCs. (Modified from Rouillé Y, Duguay SJ, Lund K et al: Proteolytic processing mechanisms in the biosynthesis of neuroendocrine peptides: the subtilisin-like proprotein convertases, Front Neuroendocrinol 16:322–361, 1995.)
PC2-null mice are not diabetic, but they exhibit significant hyperproinsulinemia, with plasma proinsulin levels in the range of 60%.67 Pancreatic extracts also show increased proinsulin levels, but only in the homozygous nulls, as indicated in Table 6-1. Pulse-chase studies of insulin biosynthesis in isolated islets, comparing PC2(−/−) mice with wild-type (WT) controls, also confirm significantly slower processing of proinsulin to insulin, with accumulation of significant amounts of des-31,32 intermediate proinsulin.46,67 Approximately a third of the labeled proinsulin remains after a 3- or 4-hour chase, consistent with the levels found by radioimmunoassay in pancreatic extracts. Thus in normal mice, PC2 converts at most about a third of the available proinsulin, while PC1/3 is responsible for processing the remaining two-thirds.69
Table 6-1
Percent Proinsulin-Like Immunoreactivity in Pancreatic Extracts of Wild-Type, PC2-Null, and PC1/3-Null Mice
A human subject with inactivating mutations in both copies of the PC1/3 gene has been reported.70 This 43-year-old woman is obese and had gestational diabetes. Examination of the subject’s serum revealed no detectable circulating insulin, in association with greatly elevated intact proinsulin and significant amounts of des-64,65 intermediate proinsulin but little or no des-31,32 intermediate proinsulin. Similar results with respect to proinsulin processing have been reported for the PC1/3-null mouse, which has very high levels of proinsulin and elevated des-64,65 intermediate proinsulin68,69 (see Table 6-1). This picture is consistent with the conversion scheme outlined in Fig. 6-7 and indicates that the pathway shown on the right side of the diagram is the predominant one. It also confirms the likelihood that PC1/3 is the more important convertase in the processing of proinsulin. The PC1/3-null mice, however, are not obese like the human subject; instead, they exhibit a severe growth defect, most likely due to lack of normally processed growth hormone–releasing hormone.69 These mice are also unable to produce GLP-1 but did not have altered intraperitoneal glucose tolerance. Altered proopiomelanocortin processing and other endocrine abnormalities, as well as intestinal malfunction, also are present in these mice.68
Significance of Circulating Proinsulin and Des-31,32 Intermediate in Man
An important clinical issue concerns observations that in man, des-31,32 intermediate proinsulin is a major intermediate, making up a very significant proportion of the circulating proinsulin-like material.71 It has been suggested that the elevated accumulation of proinsulin and des-31,32 proinsulin in diabetics might be due to defective action of PC2.72 Isolated normal human islets of Langerhans also have been reported to convert proinsulin to insulin with significant accumulation of des-31,32 intermediate, despite the presence of normal levels of PC2.73 However, des-31,32 intermediate only reaches levels of 15% to 20% of total immunoreactive insulin-like material during biosynthetic pulse-chase studies in the islets of PC2-null mice.46 Thus, even the complete absence of PC2 should not in itself give rise to such high levels of this intermediate as are seen in human serum samples. It has recently been clarified that this phenomenon arises due to the faster rate of cleavage by the PCs of substrates having a basic residue in the fourth position upstream (P4 position) of the usual dibasic cleavage sites. Such an upstream basic residue is present at the B chain–C peptide junction in human proinsulin (B29 is Lys) but is lacking at the A chain–C peptide junction (C62 of proinsulin is Leu) (see Fig. 6-6). This causes an imbalance in the relative susceptibility of these two sites to either of the two convertases and tends to favor the accumulation of the des-31,32 intermediate.46 On the other hand, in the dual proinsulins of rats and mice,22 other P4 sequence changes in these cleavage sites produce different ratios of intermediates than those seen in man.46
Studies on the regulation of the biosynthesis of PC2 and PC1/3 in β cells suggest that the rates of translation of both of these enzymes are up-regulated by glucose, similar to that of proinsulin on glucose stimulation.74 Also both PC2 and PC1/3 mRNA levels are equally elevated along with insulin mRNA during more prolonged stimulation of islets with glucose.75 However, under conditions of chronic stimulation, a relative deficiency of PC2 could develop that could exaggerate the abnormalities in circulating proinsulin intermediates seen in diabetics. Accordingly, the normal accumulation of des-31,32 intermediate proinsulin in human subjects merely reflects sequence differences in the two cleavage sites of human proinsulin, whereas increased levels of both proinsulin and des-31,32 proinsulin in prediabetics and diabetics may result from a deficiency of convertase action when islets are stressed by hyperglycemia. Genetic studies have not yet indicated a major role for mutations in the PC1/3,76 PC2,77 or CPE78 genes in susceptibility to any form of diabetes, although common amino acid polymorphisms in PC1/3 may increase risk of obesity.79 However, loss-of-function mutations in the transcription factor PAX6 have recently been described which result in reduced expression of PC1/3 in the islets and are associated with hyperproinsulinemia and mild age-related diabetes.80
Secretory Granule Formation and Maturation
One of the unsolved puzzles of neuroendocrine and other secretory cells is the nature of the mechanism underlying the efficient sorting of proteins destined for regulated secretion into immature secretory vesicles in the TGN. In the β cell, this process is remarkably efficient, resulting in very low levels of unregulated or “constitutive” release of proinsulin (<1% to 2%). However, the newly formed secretory granules have a clathrin coat (Fig. 6-8) which appears to be involved in some reorganization of the granule contents after and/or during their formation in the TGN.81 This passive sorting presumably occurs via small clathrin-clad vesicles that transport some proteins that are excluded from the condensing granule cores into endosomal pathways that either recycle to the TGN or to the cell surface. As a consequence of this “constitutive-like” pathway, proteins such as furin, procathepsin B, and possibly others briefly pass through the immature granule compartment, where they may play an active albeit transient role (e.g., furin may participate in processing of some prohormones).81,82 Also, small amounts of abundant soluble granule components such as proinsulin and/or C peptide may exit the granules within these vesicles.83 Passive sorting may also play a role in maintaining synchrony between granule membrane area and granule volume as maturation proceeds.81
FIGURE 6-8 A, Clathrin immunolabeling; B, 3-(2,4-dinitroanilino)-3′-amino-N-methyldipropylamine (DAMP) immunolabeling; and C, proinsulin immunolabeling of Golgi areas (G) of B cells (protein A–gold techniques). These electron micrographs show that the population of secretory granules with tightly fitting cores (black arrowheads) is clathrin coated (arrows in A), DAMP poor (B), and proinsulin rich (C). These granules correspond to the maturing coated secretory granules freshly released from the Golgi complex. Conversely, secretory granules with wide, clear halos (white arrowheads) are deprived of clathrin (not shown here), are DAMP rich (B). and are proinsulin poor (C). These correspond to the noncoated mature (storage) insulin-containing secretory granules. Because DAMP immunoreactivity is assumed to represent an indirect measure of intraorganelle acidity, this may indicate a decreasing pH gradient between proinsulin-rich and insulin-rich granules (i.e., between the converting and the storage compartments) (A, ×28,000; B and C, ×27,000). (From Orci L: The insulin cell: its cellular environment and how it processes proinsulin, Diabetes Metab Rev 2:71, 1986.)
Newly formed secretory vesicles in neuroendocrine cells undergo biochemical and morphologic maturation to typical dense-core granules. In β cells, the progranules are larger, less dense, and more uniform in appearance.29,30 The most important biochemical change taking place as these progranules mature is the proteolytic conversion of proinsulin to insulin, accompanied by reorganization of the products.30 Electron microscopic studies indicate that they progressively acquire a crystalline dense central core (Fig. 6-9). High magnification reveals repeat-unit spacings in the cores that are closely similar to those observed in ordinary zinc insulin crystals.84 Thus, as insulin is liberated from proinsulin, it tends to crystallize with zinc that is concentrated by the β cells. Recent studies have demonstrated that zinc is taken up into maturing granules via the ZnT8 zinc transporter, and they do not assume the normal dense-core appearance in its absence, even though proinsulin conversion is not impaired.85 Biochemical fractionation of mature islet secretory granules confirms that the cores contain only insulin, whereas the C peptide liberated in the conversion process remains in solution in the clear fluid space surrounding the dense crystalline core.84 There is no evidence for co-crystallization of the C peptide with insulin under these conditions or in vitro.
FIGURE 6-9 A, Photomicrograph of normal rat β cells (×28,000) showing morphology of mature granules (bar = 1 µm). B, Isolated rat β-granule cores (×17,000)236 (bar = 1 µm). C, High-magnification view (×250,000) showing repeat unit structure of a crystalline core (bar = 0.1 µm). The cores are made up of both rat insulin I and II in approximately equal proportions (Michael J and Steiner DF, unpublished data). Samples were fixed with Karnovsky’s solution and stained with osmium tetroxide. (Electron micrographs courtesy Hewson H. Swift.)
Most of the zinc in islets is present in the β granules and is liberated proportionately to insulin during secretion, in keeping with its role in crystallization of the hormone.86 The insulins of some species, including the guinea pig, coypu, and other hystricomorph rodents42 and the primitive hagfish,87 lack the histidine residue at position 10 of the B chain required for zinc binding during the association of insulin dimers into hexamers.88 A human subject heterozygous for a mutation at B10 (His B10 Asp) had elevated levels of this proinsulin in the blood because it tended to be released constitutively at much higher levels than normal proinsulin.89 However, in a transgenic mouse model of this mutation, there was no defect in its conversion to insulin in the granules, despite its altered self-association and sorting properties.90
The pH of the interior of the mature secretory granule appears to be between pH 5.0 and 6.0,51,91 an optimal pH range for insulin crystallization in vitro. The neutral or slightly alkaline pH in the cisternal spaces of the RER favors proinsulin folding and sulfhydryl oxidation. The pH remains near neutral throughout the Golgi apparatus but becomes more acidic (pH 6.1) in the TGN as the secretory products are sorted into granules and proteolytic processing begins. Vesicular proton pumps may begin to increase the uptake of protons, which then displace the cationic arginine and lysine residues liberated during conversion. As these move out of the granules and are replaced by hydrogen ions, a downward shift in intragranular pH may occur. Thus the initially mildly acidic progranules91 undergo gradual acidification as they mature in the cytosol (see Fig. 6-8), creating appropriate conditions for the conversion and crystallization of the newly formed insulin. One recently discovered role of the constitutive pathway convertase, furin, in the β cell is to activate the proton pump, which acidifies the secretory granules. As a result, proinsulin processing is impaired in β cells lacking furin.92
The cellular processes related to the biosynthesis of insulin via preproinsulin and proinsulin and their intracellular transport, sorting, proteolysis, and ultimate storage in secretory granules are remarkably well integrated, both topologically and biochemically. This delicately poised integration of processes leading to the formation and storage of insulin is disturbed in islet cell tumors, which often show unregulated release of insulin together with large amounts of proinsulin; measurements of the latter can provide a useful diagnostic indicator93 (see Chapter 21).
Biosynthetic Role and Biological Actions of the C Peptide
Because of its cosecretion with insulin in essentially equimolar amounts,22,94,95 the C peptide has been of great value as a marker of insulin secretion in humans under a variety of conditions. Representative vertebrate C-peptide amino acid sequences are compared in Fig. 6-4.96 These peptides exhibit a 15-fold higher rate of mutation acceptance than do the corresponding insulins, a finding that has often been interpreted as indicating that this region in the proinsulin molecule is unlikely to have any specific hormonal function. Nonetheless, several acidic residues are consistently present at certain positions in mammalian C peptides. These offset the added cationic charges due to the pairs of basic residues at the proinsulin cleavage sites, such that the isoelectric pH of proinsulin is nearly the same as that of insulin (i.e., in the range of pH 5.1 to 5.5).22
A large body of evidence supports a biosynthetic role for the C-peptide segment in proinsulin. Primarily, it converts the insulin A and B chain interaction from an inefficient bimolecular reaction to a highly efficient and concentration-independent unimolecular reaction.97 Certain regions of the connecting peptide may also facilitate the folding of the proinsulin polypeptide chain and the formation of the correct disulfide bonds, or guide the enzymatic cleavage of proinsulin to insulin by helping to orient the basic residue pairs for efficient binding and cleavage by the convertases.98,99 Recent molecular modeling studies indicate that the C peptide may assist in achieving important conformational changes in the N-terminal helical region of the A-chain segment, aiding its interaction with the convertases during processing.100
The conservation of a length of 30 to 35 amino acids in almost all proinsulin C peptides, including those of insects, mollusks, and nematodes, despite the short 8- to 10-angstrom separation between A1 and B30 in the native insulin molecule88 (see Fig. 6-2), supports the existence of an important length-related function. Since much shorter connecting segments do not impair sulfhydryl oxidation and formation of the native structure,99,101 it is clear that this is not a critical constraint. A highly plausible reason for the evolutionary retention of a relatively long C peptide in proinsulin may be to facilitate the translocation of proinsulin across the RER membrane during its synthesis. The length of polypeptide chain required to span the large ribosomal subunit and the RER membrane has been estimated to be about 65 residues in extended configuration.102 Moreover, the arrest of translation by the signal recognition particle (SRP) occurs only after synthesis of a nascent chain of about this length and thus may play a role in translational control of insulin biosynthesis.102 Efficient intracellular transport and correct targeting to secretory granules may impose additional demands on the primary (and tertiary) structure of proinsulin and other precursor proteins. However, miniproinsulins with deleted or greatly truncated C peptides do not fold efficiently but are correctly targeted to the regulated secretory pathway.98,101,103
Another very important function of the C-peptide is to facilitate its removal from proinsulin after it has served its biosynthetic functions, as mentioned earlier. The kinetics of insulin metabolism in vivo, as well as its biological activity, are both dependent upon its high affinity for the insulin receptor. Proinsulin’s much lower receptor-binding affinity leads to a much slower rate of clearance, which explains its relatively higher apparent biological activity in vivo. The rapid clearance of insulin is adaptively as important a characteristic as its bioactivity. To assure its interaction with the convertases, proinsulin must present more than just a pair of basic residues to the convertases. Their active sites require an extended β strand that is at least eight residues in length for recognition at each cleavage site. Molecular modeling studies have suggested how the full length of the connecting segment contributes to achieving the necessary conformations for effective proinsulin binding and cleavage by the convertases.100
Since the early 1990s, a number of reports have appeared describing a variety of biological actions of the C peptide and/or smaller peptides derived from it.104 These putative effects include enhancement of glucose transport and utilization; improvements in microcirculation in muscle, skin, retina, and nerve in diabetics; and stimulation of renal tubular Na+,K+-adenosine triphosphatase (ATPase) activity and other parameters of renal function. It has been suggested that tissue receptors for C peptide might exist, and that the circulating C peptide may contribute to improved glycemic control and help to slow the development of the vascular and neural complications of diabetes.105 Further study is needed in this area in view of reports that some C-peptide effects do not follow the usual rules of ligand-receptor chemistry (i.e., chirality does not matter). Thus, a C peptide synthesized entirely of d amino acids is equally as active as a peptide made with l amino acids in reversed order,106 whereas a random sequence of the same amino acids leads to loss of activity. These tantalizing findings suggest the need for a controlled clinical trial of combined insulin and C-peptide therapy in diabetics to fully evaluate its therapeutic potential.
The availability of biosynthetic human proinsulin and insulin, as well as of human C peptide, has opened many new possibilities for studies of the role, metabolism, and antigenicity of these peptides.107–109 In recent years, active insulin analogs with shortened linking peptides have also been developed. Such miniproinsulins and/or “single-chain insulins” appear to readily oxidize to form correct disulfide bridges and either can be used intact or after cleavage to insulin.98,99,101 Other efforts have been directed towards synthesis of modified insulins with improved absorption, aggregation, and/or pharmacokinetic properties for therapeutic use.
Regulation of Insulin Production
Under normal circumstances the pancreatic β cell retains a remarkable state where insulin secreted in response to a stimulus is rapidly replenished at the biosynthetic level in parallel, thus keeping intracellular insulin stores optimal. The vast majority (98% to 99%) of insulin is secreted via the “regulated” secretory pathway110 and only occurs in response to increases in specific nutrients, certain hormones, neuropeptides, and some pharmacologic reagents.111,112 In general, most nutrients that regulate insulin secretion also control proinsulin biosynthesis,111 and the most physiologically relevant of these is glucose.112 Glucose metabolism is required for both glucose-induced insulin secretion and production,111 but the secondary stimulus-coupling mechanisms are quite distinct. For example, stimulated insulin secretion is Ca2+ dependent, but glucose-induced proinsulin biosynthesis is Ca2+ independent.112,113 There are effective inhibitors of glucose-induced insulin secretion, such as diazoxide, epinephrine, and somatostatin, but these have no effect on glucose-induced proinsulin biosynthesis.111 In contrast, effective stimulators of insulin secretion, such as sulfonylureas and fatty acids, have no effect on proinsulin biosynthesis.111,114 This differential regulation of insulin secretion and proinsulin biosynthesis indicates that glucose-induced proinsulin biosynthesis is not regulated by a positive feedback of locally secreted insulin.113 For stimulated insulin secretion, the intracellular secondary signaling mechanisms are relatively well defined,115 but those for glucose-induced proinsulin biosynthesis, other than a need for mitochondrial metabolism and subsequent succinate production,116 are not currently well understood. However, it is known that both glucose-induced insulin secretion and production can be potentiated by an elevation in [cAMP]i secondary to the action of incretins such as GLP-1.117 The glucose-dependent effect of GLP-1 to increase proinsulin biosynthesis is predominately at the translational level, not at the transcriptional level.118
Glucose regulation of proinsulin biosynthesis is relatively specific and occurs independently of the control of general protein synthesis in the β cell.119 There is a minor subset of β-cell proteins (~50) that are also specifically regulated by glucose at the biosynthetic level in parallel to proinsulin, with these mostly being insulin secretory-granule proteins, including the proinsulin-processing endopeptidases, PC1/3 and PC2.74,75,120,121 This provides a mechanism whereby the enzymes that process proinsulin and the intracellular storage compartment in which insulin is stored are coordinately increased in response to the up-regulation of proinsulin biosynthesis.
The mechanism for specific regulation of glucose-induced proinsulin biosynthesis is complex and can occur at multiple levels depending on the temporal exposure to elevated glucose levels. Under normal fluctuations in glucose concentrations that occur over periods of ≤3 hours, glucose-induced proinsulin biosynthesis is entirely mediated at the translational level.111,113,122,123 For periods of ≥12 hours, this translational regulation is supplemented by glucose promoting preproinsulin mRNA stability124 to provide more preproinsulin mRNA template for translational regulation.113 Longer-term increased proinsulin biosynthesis can also be contributed to by increased insulin gene transcription.125,126
However, even under these longer-term circumstances, the primary control of glucose-induced proinsulin biosynthesis is mediated at the translational level. Yet despite glucose-induced translational regulation of proinsulin biosynthesis being known for nearly 40 years,22,122,127 the molecular mechanism remains unresolved. There are adaptive responses of the general translational machinery to up-regulate preproinsulin mRNA translation that have been shown to occur at the initiation and elongation phases of translation,22,123,127 as well as reduction in the duration of SRP signal peptide–mediated arrest of nascent preproinsulin-chain elongation prior to ribosome docking in the early phases of RER translocation.123 However, the specific translational regulation of proinsulin biosynthesis lies in the preproinsulin mRNA itself, particularly in the untranslated regions (UTRs).128 The 3′ UTR of preproinsulin mRNA contains elements such as a polypyrimidine-rich tract (PRT)129 and a highly conserved UUGAA element111,128 that likely control its stability. The control of preproinsulin mRNA stability via the 3′ UTR PRT also involves a glucose-responsive association of the polypyrimidine tract binding protein (PTB).128 The 5′ UTR of preproinsulin mRNA contains a conserved translational control cis element, ppIGE (for preproinsulin glucose element), that is required for glucose-induced translational control of proinsulin biosynthesis.128,130 The ppIGE has a highly conserved ppIGE “mirrored” core of GUCxnCUG or GUUxnUUG (where n ≤ 4 bases). There is a protein trans-acting factor (ppIE-BP) found in islet cytosolic extracts that binds to this translational control ppIGE cis element of preproinsulin mRNA in a glucose-dependent manner that parallels the characteristics of glucose-regulated proinsulin biosynthesis.130 The identity of the ppIGE-BP is currently unknown. Nonetheless, it is intriguing that the ppIGE is also conserved in the 5′ UTR of the mRNA templates of other secretory granule proteins whose biosynthesis is specifically controlled at the translational level in β cells, including PC1/3 and PC2.98,99 As such, the glucose-induced specific translational control of proinsulin biosynthesis and that for other insulin secretory-granule protein is likely via the same mechanism. For the longer-term transcriptional regulation of insulin gene expression by glucose, this is known to be a Ca2+-dependent mechanism131 involving the putative phosphorylation of key β-cell transcription factors, Pdx-1 and MafA.125 Prolonged glucose stimulation can also lead to increased β-cell hyperplasia and hypertrophy,132 which might also be additionally influenced by a wide variety of growth factors and other nutrients.133 Increased numbers of β cells will also increase insulin production, as occurs in nondiabetic obesity and pregnancy.133
Insulin secretory-granule turnover should also be considered in the long-term regulation of insulin production and availability. An insulin secretory granule has an estimated half-life of 3 to 5 days,94 and if it does not undergo exocytosis, it is degraded intracellularly. Degradation of intracellular organelles like insulin secretory granules is mediated via autophagic mechanisms.134,135 Under normal circumstances, autophagy plays a janitorial role, removing aged secretory granules.136
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
