Published on 18/03/2015 by admin
Filed under Dermatology
Last modified 22/04/2025
This article have been viewed 2454 times
Sandra A. Kopp and Justin J. Green
Evidence Levels: A Double-blind study B Clinical trial ≥ 20 subjects C Clinical trial < 20 subjects D Series ≥ 5 subjects E Anecdotal case reports
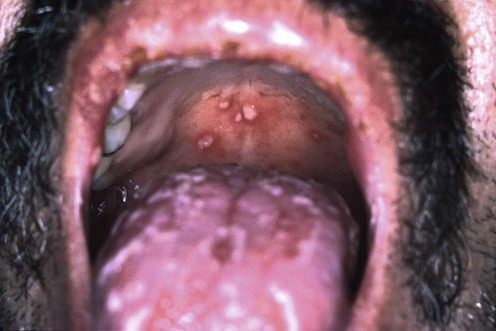
Behçet disease is a chronic inflammatory disorder leading to systemic vasculitis characterized by oral aphthae (at least three episodes in a 12-month period), plus two of the following: genital aphthae, cutaneous lesions (erythema nodosum, pustulosis, acneiform lesions, and pseudofolliculitis), uveitis or retinal vasculitis, or positive pathergy test. Arthritis, gastrointestinal, cardiac and neurologic manifestations may also occur. Many regimens effective for recurrent aphthous stomatitis are used to treat the aphthae of Behçet disease (see chapter on aphthous stomatitis).
In the absence of the multisystem disease, the severity and extent of the mucocutaneous manifestations direct the treatment strategy. First-line therapy for oral and genital aphthae is a high-potency topical corticosteroid in a gel or ointment formulation. Alternatively, intralesional corticosteroids (triamcinolone 5 mg/mL) can be used for major aphthae and severe minor aphthae. Other topical therapies accelerate healing or diminish the pain associated with oral aphthae. These include viscous lidocaine 2–5% applied directly to lesions, amlexanox 5% oral paste, sucralfate, tetracycline suspension and topical tacrolimus 0.1% ointment.
Colchicine 0.6 mg three times daily combined with topical corticosteroid therapy is efficacious in mucocutaneous disease. Dapsone 100–200 mg daily is also effective, but requires more frequent laboratory follow-up.
Those patients who fail the more conservative approaches or have severe mucocutaneous disease may require aggressive therapy. Thalidomide 100–300 mg daily (pediatric dose varies from 1 mg/kg/week to 1 mg/kg daily) is more effective than low-dose methotrexate 7.5–20 mg/week for severe disease. Prednisone taper begun at 1 mg/kg daily can be used for severe mucocutaneous flares, but rebound is a possible complication.
Systemic interferon (IFN)-α2a and anti-tumor necrosis factor (TNF)-α therapies may be best suited for those with severe mucocutaneous lesions and non-ocular systemic manifestations. Etanercept 50 mg weekly by subcutaneous injection, or infliximab 5 mg/kg in single or multiple intravenous infusions, have been shown to be effective in patients with recalcitrant disease and may be used as monotherapy or as an adjunct to conventional immunosuppressive therapy. Notably, infliximab has demonstrated rapid therapeutic effect, which may be useful in patients with vision-threatening posterior uveitis. These patients should be screened for latent tuberculosis infection prior to initiating anti-TNF therapies. Patients with certain extracutaneous signs (e.g., uveitis, aneurysms) may warrant combination therapy with prednisone and an immunosuppressive agent such as cyclosporine, azathioprine, chlorambucil or cyclophosphamide. Mucocutaneous disease alone rarely warrants this therapy; however, these agents have a beneficial effect on skin and mucous membrane lesions.
Pathergy test optional
Culture/polymerase chain reaction of aphthae to exclude herpes simplex virus
Vitamins B1, B2, B6, and B12, folate, zinc, and iron levels
Urinalysis
HLA-B27
Exclude inflammatory bowel disease
Jorizzo JL, Soloman AR, Cavallo T. Arch Pathol Lab Med 1985; 109: 747–51.
This method of pathergy testing may be more sensitive than standard techniques devoid of histologic study.
Ghate JV, Jorizzo JL. J Am Acad Dermatol 1999; 40: 1–18.
A review of investigations that should be carried out in a patient with complex aphthosis is presented.
Herpes simplex virus, nutritional deficiencies, neutropenia, lymphopenia, reactive arthritis, and inflammatory bowel disease may simulate the oral aphthae of Behçet disease.
Alexoudi I, Kapsimali V, Vaiopoulos A, Kanakis M, Vaiopoulos G. Clin Rheumatol 2011; 30: 157–63.
An excellent review and guide to treatments of all aspects of Behçet disease.
Hatemi G, Silman A, Bang D, Bodaghi B, Chamberlian AM, Gul A, et al. Ann Rheum Dis 2009; 68: 1528–34.
In this systematic literature review from 1966 to 2006, systemic medications for Behçet disease were analyzed for treating different aspects of the disease.
Yurdakul S, Mat C, Tuzun Y, Ozyazgan Y, Hamuryudan V, Uysal O, et al. Arthritis Rheum 2001; 44: 2686–92.
A prospective, double-blind, controlled trial of 116 patients treated with colchicine vs placebo. Therapy with colchicine 1–2 mg daily was effective for arthritis and erythema nodosum. Orogenital aphthae were more responsive to treatment in females.
Fontes V, Machet L, Huttenberger B, Lorette G, Vaillant L. Ann Dermatol Venereol 2002; 129: 1365–9.
Fifty-four patients were treated with 1–1.5 mg of colchicine daily for at least 3 months. Twelve patients no longer had aphthae and were in complete remission and 22 patients were significantly improved, as the frequency and duration of the lesions had decreased by at least 50%. Treatment failed or tolerance was poor in 20 patients.
Sharquie KE, Najim RA, Al-Dori WS, Al-Hayani RK. J Dermatol 2006; 33: 541–6.
In a prospective, randomized, double-blind, controlled trial, 27 patients with mucocutaneous and/or joint disease and 30 healthy subjects matched for age and gender were treated with zinc sulfate vs placebo. Zinc sulfate 100 mg three times daily was found to be effective in reducing disease severity, and no adverse effects were reported.
Patients with ocular, neurologic, cardiac, or other systemic manifestations were excluded.
Chams-Davatchi C, Barikbin B, Shahram F, Nadji A, Moghaddassi M, Yousefi M, et al. Int J Rheum Dis 2010; 13: 253–8.
This was a randomized, double-blind control trial of pimecrolimus versus placebo in genital aphthous ulcers in patients with Behçet disease. There was a significant decrease in healing time with pimecrolimus and slight improvement of pain.
Sharquie KE, Najim RA, Abu-Raghif AR. J Dermatol 2002; 29: 267–79.
Randomized, double-blind, placebo-controlled, crossover trial of 20 patients treated with either dapsone 100 mg daily or placebo. There was a significant reduction of orogenital ulcers and other cutaneous manifestations in the dapsone group.
Vitamin E 800 IU daily may reduce the hemolysis induced by dapsone. Other studies have confirmed the utility of dapsone for mucocutaneous Behçet disease.
Hamuryudan V, Mat C, Saip S, Ozyazgan Y, Siva A, Yurdakul S, et al. Ann Intern Med 1998; 128: 443–50.
A randomized, double-blind, controlled trial of 96 males evaluated thalidomide 100 mg daily vs 300 mg daily vs placebo for 24 weeks. Both thalidomide dosages led to a significant suppression of oral ulcers at 4 weeks, and genital ulcers and follicular lesions at 8 weeks. Complete responses were observed in 11% of patients treated with thalidomide.
Jorizzo JL, White WL, Wise CM, Zanolli MD, Sherertz EF. J Am Acad Dermatol 1991; 24: 973–8.
Two female patients with oral and genital aphthae, pyoderma gangrenosum-like lesions, and cutaneous pustular vasculitic lesions cleared with methotrexate 15–20 mg/week.
Yazici H, Barnes CG, Drugs 1991; 42: 796–804.
Intravenous pulse methylprednisolone 1 g on 3 alternate days, or 1 mg/kg daily of prednisone for several weeks with subsequent taper, are recommended for severe or life-threatening major ulcerations.
Green J, Upjohn E, McMormack C, Zeldis J, Prince HM. Br J Dermatol 2008; 158: 197–8.
A case report of improvement of oral ulceration and lethargy in Behçet disease with lenalidomide (10 mg/day).
Of note the patient tolerated the medication well, however did develop a deep vein thrombosis, which can be independently associated with lenalidomide or Behçet disease.
Kötter I, Vonthein R, Zierhut M, Eckstein AK, Ness T, Günaydin I, et al. Semin Arthritis Rheum 2004; 33: 311–19.
An open, uncontrolled study of 50 patients treated with subcutaneous 6 × 106 U rhIFN-α2a daily resulted in a 92% response rate of ocular manifestations, remission of genital ulcerations and skin lesions, and 36% response of oral aphthae.
There are additional reports substantiating the benefits of IFN-α2a in mucocutaneous Behçet disease.
Alpsoy E, Durusoy C, Yilmaz E, Ozgurel Y, Ermis O, Yazar S, et al. Arch Dermatol 2002; 138: 467–71.
A randomized, double-blinded, placebo-controlled trial of 23 patients receiving interferon-α2a three times a week. After 3 months, 15 of 23 patients demonstrated improvement in oral, genital, and papulopustular lesions.
O’Duffy JD, Calamia K, Cohen S, Goronzy JJ, Herman D, Jorizzo J, et al. J Rheumatol 1998; 25: 1938–44.
Seven patients underwent daily subcutaneous injections of 3 million units of interferon-α2a. This open trial reported a substantial reduction in mucocutaneous lesions and joint disease. Flu-like symptoms, leukopenia, psoriasis, seizure, hyperthyroidism, and psychosis were side effects reported.
Akyol M, Dogan S, Kaptanoglu E, Ozcelik S. Clin Exp Rheumatol 2002; 20: S55.
A patient with oral ulcers, acne lesions, and arthritis was treated with 6 months of isotretinoin (total dosage 120 mg/kg). He experienced complete resolution of his acne and arthritis, and reduced severity of oral ulcers, controlled with only topical treatment.
Masuda K, Nakajima A, Urayama A, Nakae K, Kogure M, Inaba G. Lancet 1989; 1: 1093–6.
Cyclosporine (10 mg/kg) was more effective than colchicine (1 mg/kg) in reducing the number and frequency of oral aphthae and cutaneous lesions (erythema nodosum-like, subcutaneous thrombophlebitis and folliculitis-like lesions).
Several case reports and open studies corroborate these findings.
Pacor ML, Biasi D, Lunardi C, Cortina P, Caramaschi P, Girelli D, et al. Clin Rheumatol 1994; 13: 224–7.
Sixteen subjects with Behçet disease received 5 mg/kg of cyclosporine daily for 24 months. A marked improvement of the symptoms was observed after 3 months of therapy, and 14 out of 16 patients obtained a complete clinical remission. Two patients dropped out of the study because of anemia and renal dysfunction, which returned to normal when cyclosporine was withdrawn.
Avci O, Gurler N, Gunes AT. J Am Acad Dermatol 1997; 36: 796–7.
Mucocutaneous lesions were markedly suppressed in an open trial involving 24 patients treated for more than 6 months with cyclosporine 5 mg/kg daily. Most responsive to therapy were genital ulcerations, thrombophlebitis, erythema nodosum-like lesions, and acneiform lesions.
Yazici H, Pazarli H, Barnes CG, Tüzün Y, Ozyazgan Y, Silman A, et al. N Engl J Med 1990; 322: 281–5.
A randomized, controlled, double-blind trial of azathioprine 2.5 mg/kg daily vs placebo in patients with Behçet disease resulted in prevention and decreased frequency of ocular disease. The prevalence of oral ulcers and the incidence of genital ulcers were diminished in the azathioprine group.
Henderson CF, Brodsky RA, Jones RJ, Levine SM. Semin Arthritis Rheum 2011; 41: 301–4.
In this case report, two patients with refractory Behçet disease were successfully treated with cyclophosphamide 200 mg/kg intravenously divided over 4 days with disease-free remission lasting 18–24 months. Both patients had a transient pancytopenia, and one suffered episodes of pneumonia and Clostridium difficile colitis.
Abdalla MI, Bahgat NE. Br J Ophthalmol 1973; 57: 706–11.
Remission of aphthae was achieved in all seven patients treated with oral chlorambucil and corticosteroids, but in a minority of patients treated with corticosteroids alone.
Kaneko F, Oyama N, Nishibu A, Yonsei Med J 1997; 38: 444–54.
In 11 patients treated with minocycline 100 mg daily the frequency of cutaneous symptoms was reduced by 10% for oral aphthae and by 100% for perifolliculitis.
Takeuchi A, Hashimoto T. Int J Clin Pharm Res 1987; 7: 283–9.
In five patients, leg ulcers began to regranulate within 2 weeks of using oral prostaglandin E1 15–30 µg daily.
Matsuda T, Ohno S, Hirohata S, Miyanaga Y, Ujihara H, Inaba G, et al. Drugs R&D 2003; 4: 19–28.
A multicenter, randomized, double-blind, placebo-controlled prospective study of rebamipide 300 mg/kg plus usual therapy (n = 19) vs placebo plus usual therapy (n = 16) showed moderate to marked improvement of oral aphthae in 65% of the treatment group vs 36% in the placebo group.
Melikoglu M, Fresko I, Mat C, Ozyazgan Y, Gogus F, Yurdakul S, et al. J Rheumatol 2005; 32: 98–105.
A randomized, double-blind, controlled trial of 40 Behçet disease patients who received either etanercept 25 mg or placebo subcutaneously twice a week for 4 weeks. Etanercept was found to be effective in suppressing oral ulcers, nodular lesions, and papulopustular lesions.
Almoznino G, Ben-Chetrit E. Clin Exp Rheumatol 2007; 25: S99–S102.
A patient with severe oral lesions refractory to topical and systemic immunosuppressive treatments demonstrated complete recovery and remission after a single administration of infliximab (5 mg/kg). Five more cases of recalcitrant orogenital ulceration in Behçet disease responding to infliximab were also summarized.
Accorinti M, Pirraglia MP, Paroli MP, Priori R, Conti F, Pivetti-Pezzi P. Jpn J Ophthalmol 2007; 51: 191–6.
Twelve patients with Behçet disease and uveitis refractory to conventional immunosuppressive therapy were given infliximab (5 mg/kg at 0, 2 weeks, then monthly for 4 to 6 months and quarterly 1–2 months thereafter). After a median 15-month follow-up, 11 of the 12 showed a significant reduction in the number of ocular relapses, as well as fewer extraocular manifestations, resulting in less systemic corticosteroid use. One patient stopped treatment after 2 months because of the development of pulmonary tuberculosis.
Arida A, Fragiadaki K, Giavri E, Sfikakis P. Semin Arthritis Rheum 2011; 41: 61–70.
In this evidence-based review of the literature regarding anti-TNF agents in Behçet disease, the authors concluded that there is enough evidence to suggest that TNF blockade should be considered in patients with severe or resistant disease.
Lockwood CM, Hale G, Waldman H, Jayne DR. Rheumatology (Oxford) 2003; 42: 1539–44.
An open prospective study of 18 patients (skin involvement in 67%) treated with 134 mg intravenously of a lymphocyte-depleting anti-CD52 antibody (Alemtuzumab). Three months after treatment, eight patients were in complete remission, seven in partial remission, and two had worsened. Of 13 patients in remission, seven relapsed after an average of 25 months.
Hypothyroidism and prolonged lymphopenia complicated treatment. There was routine prophylaxis against HSV and fungi.
Kanekura T, Gushi A, Fukumaru S, Fukumaru S, Sakamoto R, Kawahara K, et al. J Am Acad Dermatol 2004; 51: S83–7.
A report of successful treatment of orogenital ulceration in two patients who underwent five and eight treatments each.
Mumcu G, Ergun T, Elbir Y, Eksioglu-Demiralp E, Yavuz S, Atalay T, et al. J Oral Pathol Med 2005; 34: 13–16.
In eight patients treated with azithromycin 500 mg three times per week for 4 weeks, faster healing times of pre-existing oral ulcers, as well as complete resolution of folliculitic lesions, were observed.
Calguneri M, Ertenli I, Kiraz S, Erman M, Celik I. Dermatology 1996; 192: 125–8.
A prospective, randomized trial with 60 patients revealed statistically significant decrements in frequency and duration of oral aphthae and erythema nodosum-like lesions and the frequency of genital aphthae with the addition of benzathine penicillin 1.2 million units every 3 weeks to colchicine vs colchicine monotherapy.
This study was apparently non-blinded and not placebo controlled.
Oh SH, Kwon JY, Lee JH, Hand EC, Bang D. Clin Exp Dermatol 2009; 34: e88–90.
In this case report, a patient with Behçet disease had clearing of erythema nodosum and oral and genital ulcers with the administration of oral contraceptives. Upon discontinuation of the oral contraceptive the symptoms returned; however, with re-administration the symptoms resolved.
Ciancio G, Colina M, La Corte R, Lo Monaco A, De Leonardis F, Trotta F, et al. Rheumatology 2010; 49: 501–4.
This case report demonstrated improvement of mucocutaneous lesions with nicotine replacement in ex-smokers with Behçet disease.
This cannot be extrapolated to non-smokers, however represents an interesting therapeutic consideration in those patients who are trying to quit smoking.
Köse O, Simşek I, Pay S. Int J Dermatol 2011; 50: 895–6.
In this open, prospective trial mycophenolate sodium was effective in treating 10 patients with refractory mucocutaneous Behçet disease without major complications.
Treatment of Skin Disease Comprehensive Therapeutic Strategies 4e
WhatsApp us






 Topical/intralesional corticosteroid
Topical/intralesional corticosteroid Topical tacrolimus
Topical tacrolimus Pimecrolimus
Pimecrolimus Amlexanox 5% paste
Amlexanox 5% paste Sucralfate
Sucralfate Tetracycline suspension
Tetracycline suspension Chlorhexidine gluconate
Chlorhexidine gluconate Colchicine
Colchicine Zinc sulfate
Zinc sulfate Dapsone
Dapsone Thalidomide
Thalidomide Methotrexate
Methotrexate Systemic corticosteroid
Systemic corticosteroid IFN-α2a
IFN-α2a Isotretinoin
Isotretinoin Cyclosporine
Cyclosporine Azathioprine
Azathioprine Cyclophosphamide
Cyclophosphamide Chlorambucil
Chlorambucil Minocycline
Minocycline Penicillin
Penicillin Prostaglandin E1
Prostaglandin E1 Rebamipide
Rebamipide Etanercept
Etanercept Infliximab
Infliximab Adsorption apheresis
Adsorption apheresis Azithromycin
Azithromycin Oral contraceptives
Oral contraceptives Nicotine patch
Nicotine patch Mycophenolate sodium
Mycophenolate sodium Lenalidomide
Lenalidomide Alemtuzumab
Alemtuzumab